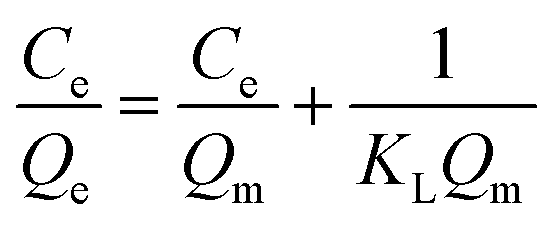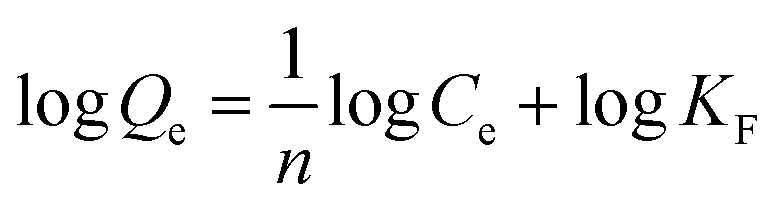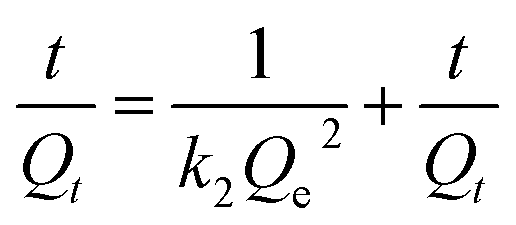DOI:
10.1039/D0RA09190A
(Paper)
RSC Adv., 2021,
11, 4713-4722
Self-assembly preparation of lignin–graphene oxide composite nanospheres for highly efficient Cr(VI) removal†
Received
28th October 2020
, Accepted 19th January 2021
First published on 25th January 2021
Abstract
Recently, research interest in the application of lignin is growing, especially as adsorbent material. However, single lignin shows unsatisfactory adsorption performance, and thus, construction of lignin-based nanocomposites is worth considering. Herein, we introduced graphene oxide (GO) into lignin to form lignin/GO (LGNs) composite nanospheres by a self-assembly method. FTIR and 1H NMR spectroscopy illustrated that lignin and GO are tightly connected by hydrogen bonds. The LGNs as an environmental friendly material, also exhibit excellent performance for Cr(VI) removal. The maximum sorption capacity of LGNs is 368.78 mg g−1, and the sorption efficiency is 1.5 times than that of lignin nanospheres (LNs). The removal process of Cr(VI) via LGNs mainly relies on electrostatic interaction, and it also involves the reduction of Cr(VI) to Cr(III). Moreover, LGNs still have high adsorption performance after repeating five times with the sorption capacity of 150.4 mg g−1 in 200 mg g−1 Cr(VI) solution. Therefore, the prepared lignin–GO composite nanospheres have enormous potential as a low-cost, high-absorbent and recyclable adsorbent, and can be used in wastewater treatment.
1. Introduction
In environmental and ecological pollution, heavy metal pollutants in water have become a crucial issue worldwide due to their strong diffusibility, huge virulence, and non-biodegradability.1–7 Hexavalent chromium (Cr(VI)) is assessed as one of the eight most adverse chemicals to humans. Long-term exposure to Cr(VI) can cause serious diseases such as allergic dermatitis, skin ulcers and bronchial cancer.8 The US Environmental Protection Agency and the Ministry of Ecology and Environment the People's Republic of China also define Cr(VI) as the highest priority of key pollutants.9–11 Numerous remediation techniques are used to remove Cr(VI) including chemical precipitation, biological repair, electrochemical methods, ion exchange, photocatalytic reduction and adsorption.12–20 Among them, adsorption has unique advantages of low cost and ease of handling because no additional equipment is required.21,22 Consequently, it is of importance to design and create an adsorbent with an economic and environmental friendly nature, and excellent adsorption effect.
Lignin is one of the richest green resources that can be used by humans in the 21st century.23,24 However, the utilization of lignin is greatly restricted due to its complex structure. Lignin, a low-cost and environmentally friendly material, has shown good results in the removal of heavy metal pollutions. Fu and co-workers used lignin which grafted by N,N′-methylene-bisacrylamide prepared lignin-based acrylic nanocomposites with acrylic acid. The composite material has good adsorption capacity for Pb2+.25 Wang and his colleagues used lignin to make lignin-based phenolic resin which can reduce the concentration of chromium ions.26 However, the lignin adsorbents invented by these studies have poor mechanical property, stability, and adsorption efficiency. Therefore, further research is urgently needed on reusable lignin-based adsorbents with higher adsorption capacity. In recent years, graphene oxide (GO) materials prepared by self-assembly methods such as graphene hydrogels and aerogels have received comprehensive attention in the field of adsorption.27–29 Suhr and co-workers synthesized GO-based sponges with hierarchical porous structures, which could effectively adsorb dye molecules.30 We can make a prediction that the introduction of GO into lignin will greatly improve the adsorption performance due to the hierarchical porous structures and abundant functional groups on GO.
Generally, adsorbents with a spherical structure are promising materials in wastewater treatment due to their appropriate size, transport capacity, and large surface area. Herein, we combined lignin and GO to prepare lignin/GO composite nanospheres (LGNs) by a facile self-assembly method. The LGNs show high removal effect for Cr(VI). The structure of LGNs was tested by Fourier transform infrared spectroscopy (FTIR), dynamic light scattering (DLS) and 1H nuclear magnetic resonance (1H NMR). Then, the influences of temperature, pH, and other ion concentrations on Cr(VI) adsorption efficiency were also studied. Finally, the isotherm/kinetic analysis was performed on the adsorption process to clarify the possible Cr(VI) repair mechanism. And the recycling capacity of LGNs was determined through adsorption and desorption.
2. Experiments
2.1 Materials
Kraft lignin (KL, the extraction method is shown in ESI†). Tetrahydrofuran (THF, chromatographic grade) was got from Sinopharm Chemical Reagent Co. Ltd. Graphene oxide (Advanced Materials Supplier, Nanjing, China). 2.8286 g potassium dichromate (K2Cr2O7, Superior Grade Pure, Aladdin Reagent Company) was dried at 105 °C for 2 h and cooled to 25 °C in a desiccator. After dissolving K2Cr2O7 in ultrapure water, we transfer it to a 1000 ml volumetric flask, dilute it with water to the mark, and shake it up to acquire a standard solution with a concentration of 1000 mg l−1. Other concentrations of Cr(VI) solutions were acquired by diminishing the above 1000 mg l−1 standard solution to a certain multiple.
2.2 Preparation of lignin nanospheres
Firstly, add 20 mg KL to 20 ml THF and ultrasound for 5 minutes to promote the dissolution of lignin. Then, the above solution was magnetically churned at about 300 rpm at 25 °C. At the same time, the ultrapure water was equably added to the KL/THF solution by a peristaltic pump; it occupied 70% of the total system, and the addition rate was 2 ml min−1. Then, place the nanoparticle suspension in a dialysis bag and immerse it in excess ultrapure water to remove THF. Finally, the prepared lignin nanospheres (LNs) were freeze-dried and collected.
2.3 Preparation of lignin–GO nanospheres
Firstly, add 20 mg KL to 20 ml THF and ultrasound for 5 minutes to promote the dissolution of lignin. Then, add 20 mg GO to 47 ml ultrapure water and ultrasound for 10 minutes to promote the dissolution of GO. Next, the KL/THF solution was magnetically churned at about 300 rpm and 25 °C. At the same time, the solution of GO was evenly added to the KL/THF solution by peristaltic pump to prepare LGNs; it occupied 70% of the total system, and the addition rate was 2 ml min−1. Finally, the obtained samples were freeze-dried and collected until use.
2.4 Characterizations
The shape of LNs and LGNs were performed on a Quanta FEG 250 field emission scanning electron microscope (FE-SEM, FEI, USA). Transmission electron microscopy (TEM) images were attained by an FEI Tecnai G2 F20 TEM (FEI, USA). The chemical structure of LGNs was tested by the FT-IR, (Nicolet iS10 Fourier Infrared Spectrometer, Nicolet, USA). Firstly, the sample was mixed with potassium bromide in a mass ratio of 1/50. Then it was finely ground and pressed into a sheet for testing, and the scanning wave number of the instrument is between 400 and 4000 cm−1. 1H nuclear magnetic resonance (1H NMR) spectra was observed with a Bruker AVIII 600 MHz spectrometer (Bruker, Germany). 30 mg of lignin was dissolved in 1 ml deuterated dimethyl sulfoxide (DMSO-d6), and performed nuclear magnetic resonance spectroscopy at 60 °C. The size and zeta potential of samples were determined by Nano ZS ZEN 3600 Zeta (Malvern, UK). X-ray photoelectron spectroscopy (XPS) was measured on Thermo Scientific Escalab 250Xi (XPS, Thermo, USA). Concentrations of Cr(VI) were analyzed with inductively coupled plasma (ICP, Agilent 720ES, USA).
2.5 Adsorption experiments
10 mg of adsorbent (LNs or LGNs) was added to the conical flask, which contains 10 ml of K2Cr2O7 aqueous solution and the concentration of Cr(VI) was 200 mg l−1. The pH of 200 mg l−1 Cr(VI) solution was regulated to 1–14 by HCl and NaOH solution for exploring the influence of pH. Unless otherwise specified, the adsorption experiments were accomplished under the condition of pH = 2. And the used adsorbent was moved to 20 ml of desorbent and stirred at 300 rpm for 2 hours to perform cyclic adsorption. The Cr(VI) removal efficiency (Qe) was counted by formula (1):| | |
Qe = (C0 × V0 − Ce × Ve)/W
| (1) |
where Qe is the equilibrium capacity (mg g−1); C0 and Ce are the Cr(VI) concentration before and after sorption (mg l−1), respectively; V0 and Ve are the volume of solution before and after sorption (l), respectively; W is the mass of sorbents (g).
2.6 Adsorption kinetics and isotherm
10 mg of adsorbent (LNs or LGNs) was added to the conical flask, which contains 10 ml of K2Cr2O7 solution (pH = 2) and the concentration of Cr(VI) was various (100, 200, 300, 400, 500 mg l−1). Each admixture was sealed and saved in an oscillator at 298 K for 24 h so that the adsorption got the equilibrium, and then leached respectively through a 0.22 μm membrane filter. Kinetic adsorption was tested at 298 K, and 10 mg of adsorbent (LNs or LGNs) was added to conical flask containing 10 ml of Cr(VI) solution. Samples were gained at diverse times and the concentration of Cr(VI) was measured by ICP.
3. Results and discussion
3.1 Morphology, zeta potential and size of LGNs
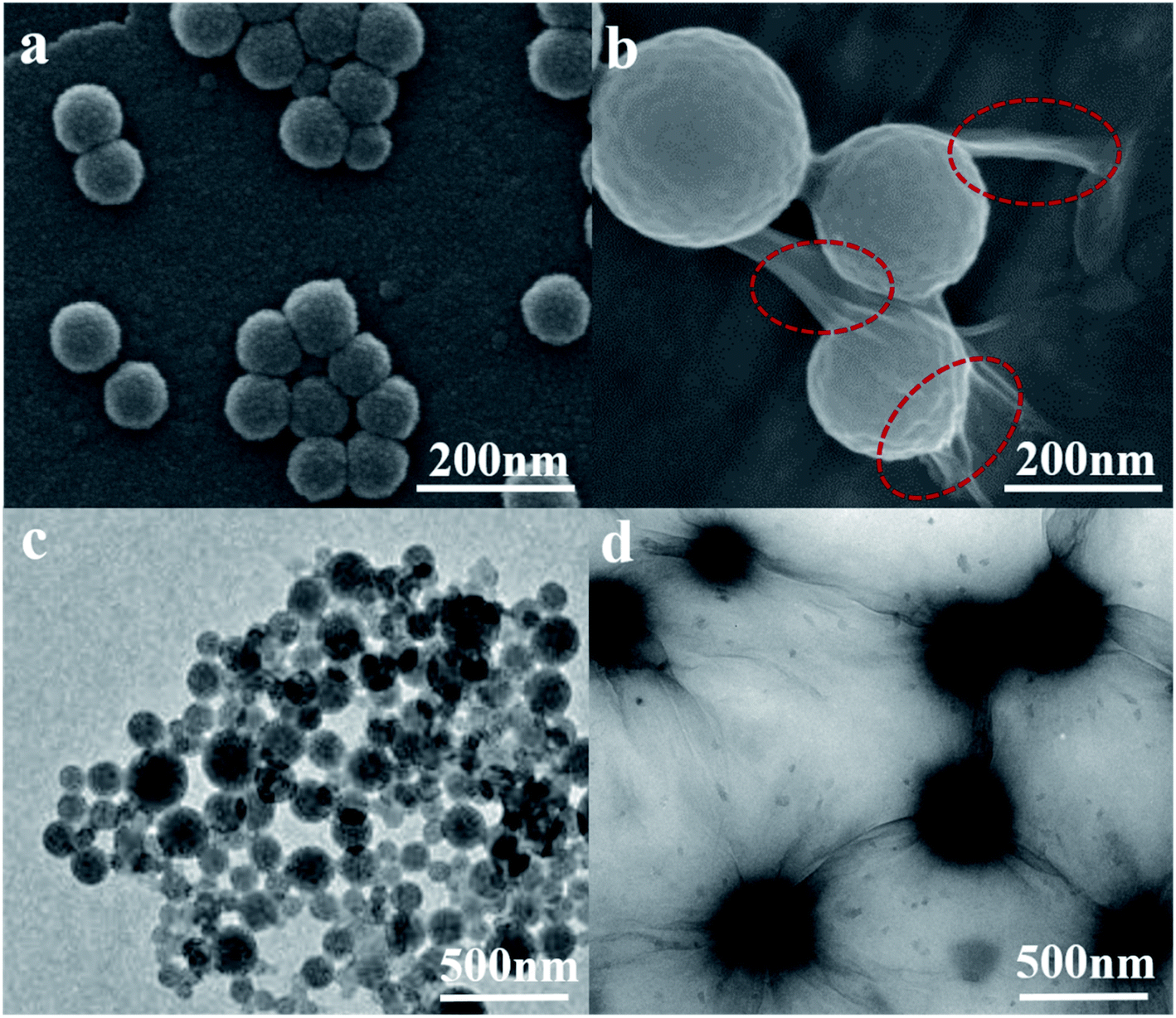
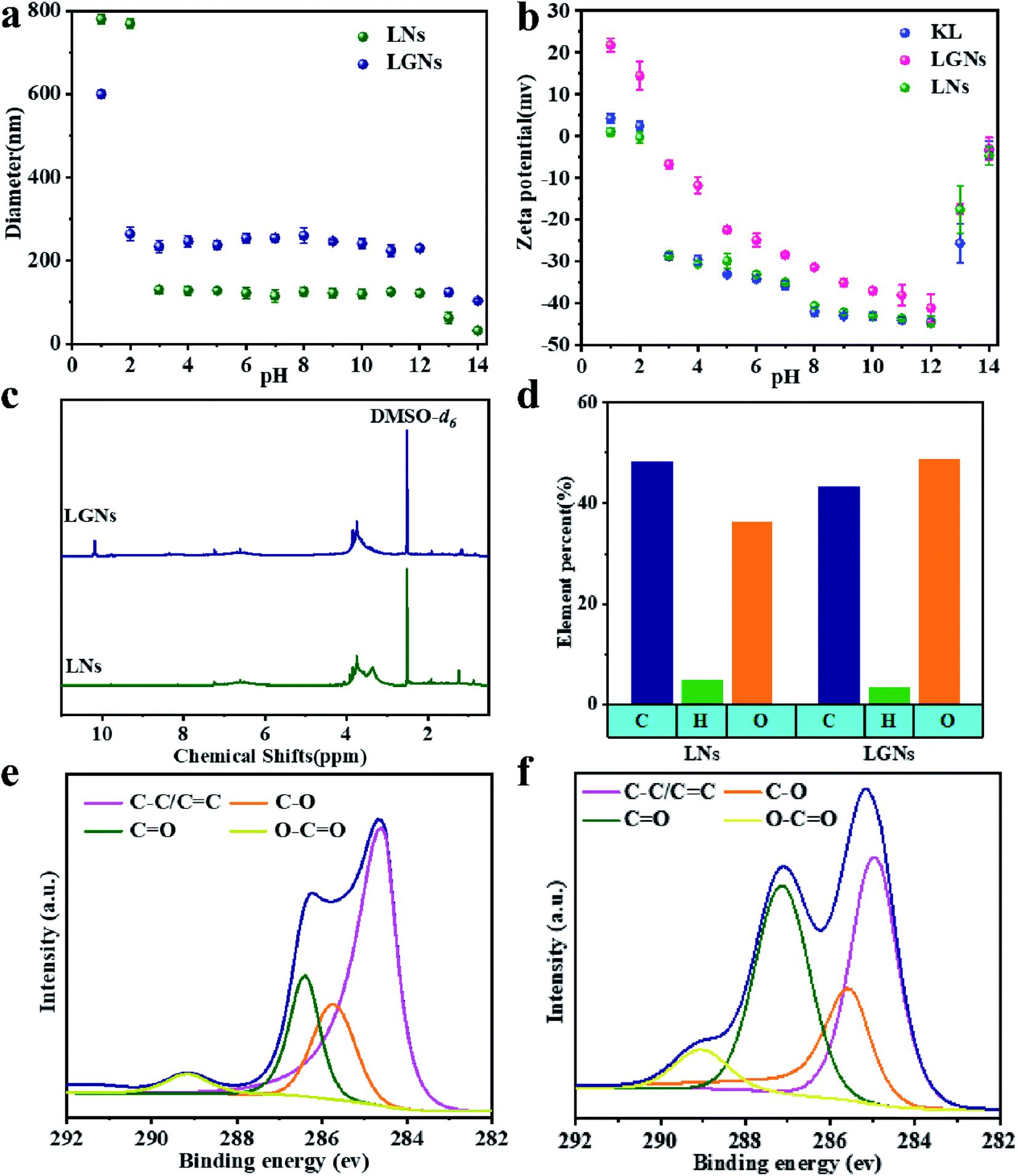
Fig. 1(a–d) are the SEM and TEM figures of LNs and LGNs. These figures show that both LNs and LGNs have good spherical shapes. Moreover, we can find from Fig. 1(b and d) that the surface of LGNs was smoother than that of LNs, and KL can be combined with GO very well. However, the size of LGNs was bigger than LNs. Previous studies have proved that the π–π interaction acted a crucial role in the preparation of lignin nanospheres.31,32 The larger size of LGNs may be due to the formation of hydrogen bonds between lignin and GO, which weaken the π–π interaction between lignin molecules. To evaluate the stability of the adsorbent, we measured the particle size of LNs and LGNs at various pH. In the range of pH 3–12, the size of LNs and LGNs had no apparent changes. And the average size of LNs was 124.1 nm, while the size of LGNs was 242.9 nm. Moreover, the size of LNs increased with the increase of acidity under intensively acidic circumstance (pH 1–2). This is due to the surface charge of LNs was diminished, leading to the conglomeration of LNs.33 The size of particles decreased under strongly alkali conditions (pH 13–14). This may be due to the dissolution of lignin increase in strongly alkali environment. Additionally, the size of LGNs fluctuated less compared with LNs, indicating that the addition of GO can enhance the stability of the adsorbent.
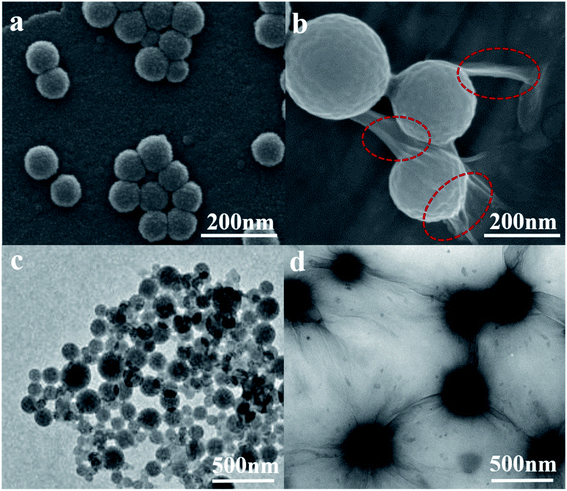 |
| | Fig. 1 SEM images of LNs (a) and LGNs (b). TEM images of LNs (c) and LGNs (d) (the red circle marks the graphene oxide attached to the particle surface). | |
Previous studies have shown that the sorption ability for Cr(VI) is closely correlated with the charge of the adsorbent.34,35 Therefore, the zeta potential at different pH values was analyzed (Fig. 2b). The KL and LNs showed negative charges under all qualifications except strongly acidic environment (pH = 1). This is because some functional groups of KL and LNs can ionize in the solution.34,36 In addition, the negative potential of KL, LNs and LGNs gradually augments with the augment of pH (pH 0–12). It is because the deprotonation of the oxygen-containing functional groups of the materials increases the concentration of negative charges.34,37 However, the surface charges of the LGNs were changed to be more positive through the GO modification process. When the pH of the colloidal fluid increases from 1.0 to 12.0, the zeta potential of the LGNs varies from +23.6 to −41.1 mV. It can be speculated that the GO has interacted with the negatively charged functional groups in lignin during the preparation process, reducing its negative charge.38,39 This result also proved the successful combination of GO and lignin. However, the absolute value of zeta potential decreased under pH 13–14. This is because adding too much alkali will increase the salt ion concentration, thereby compressing the electric double layer.
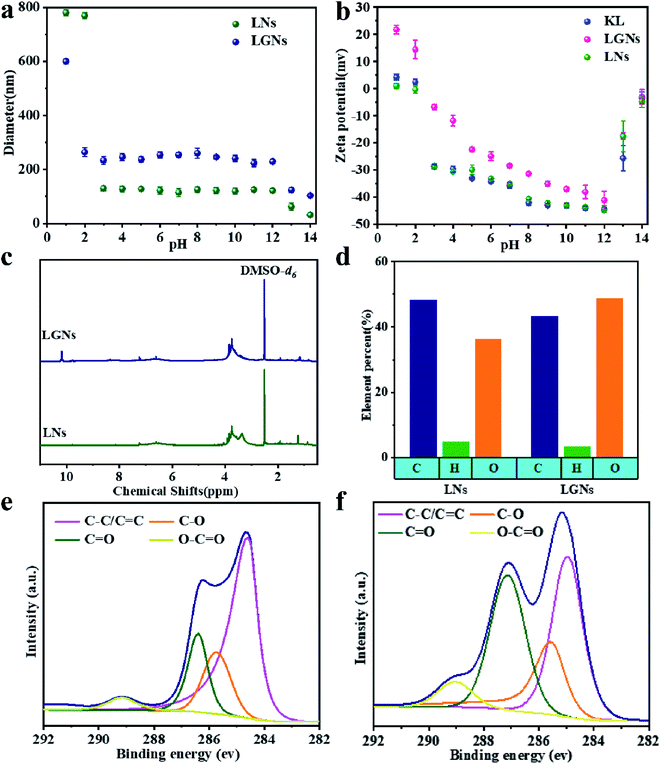 |
| | Fig. 2 (a) Zeta potential of particles and (b) average diameter in aqueous media with different pH values. (c) 1H NMR spectrum of LNs and LGNs. (d) Element contents and (e) C1 XPS peaks of LNs. (f) C1 XPS peaks of LGNs. | |
3.2 Synthesis mechanism of LGNs
In order to explicate the impact of GO modification, the results of FTIR are shown in Fig. S5.† KL showed distinctive peak at 3500 cm−1, and this peak becomes wider and stronger, demonstrating that lignin includes numerous hydroxyl groups, which can form hydrogen bonds. The hydrogen bonds are beneficial for the combination of KL and GO. Nevertheless, the absorption peaks of aromatic rings at 1513.1 cm−1 and 1426.4 cm−1 in the LNs have shifted to low wave number, 1512.2 cm−1 and 1425.4 cm−1 respectively. This is due to the stronger π–π interaction of LNs than KL. Compared with KL and LNs, LGNs have a wider absorption peak at 3600 cm−1, which is caused by the strong hydrogen bonds between KL and GO. It is speculated that KL combined with GO together by hydrogen bonding. The formation of hydrogen bonds also corroborated the trend of zeta potential. The hydrogen from the hydroxyl and the oxygen from the carboxyl could form hydrogen bonds, which promoted the dissociation of hydrogen and led to increase in positive potential.
The results of potentiometric titration show that the phenolic hydroxyl content in LNs is lower and the π–π interaction between LNs molecules are stronger (Fig. S4†). Elemental analysis shows that the O/C ratio of LGNs had a significant increase. It is because of the incorporation of oxygen-containing functional groups from GO. Moreover, nano-fabrication can expose more oxygen-containing functional groups. Fig. 2c shows that both LNs and LGNs contain methoxyl groups (3.48–4 ppm). Furthermore, LGNs have absorption peak at 10.2 ppm, marking the formation of intermolecular hydrogen bonds. Subsequently, the intensity of characteristic peaks such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of LGNs is significantly higher than that of LNs. It is proved that the introduction of GO is beneficial to increasing the effective content of oxygen-containing groups in the adsorbent.
O of LGNs is significantly higher than that of LNs. It is proved that the introduction of GO is beneficial to increasing the effective content of oxygen-containing groups in the adsorbent.
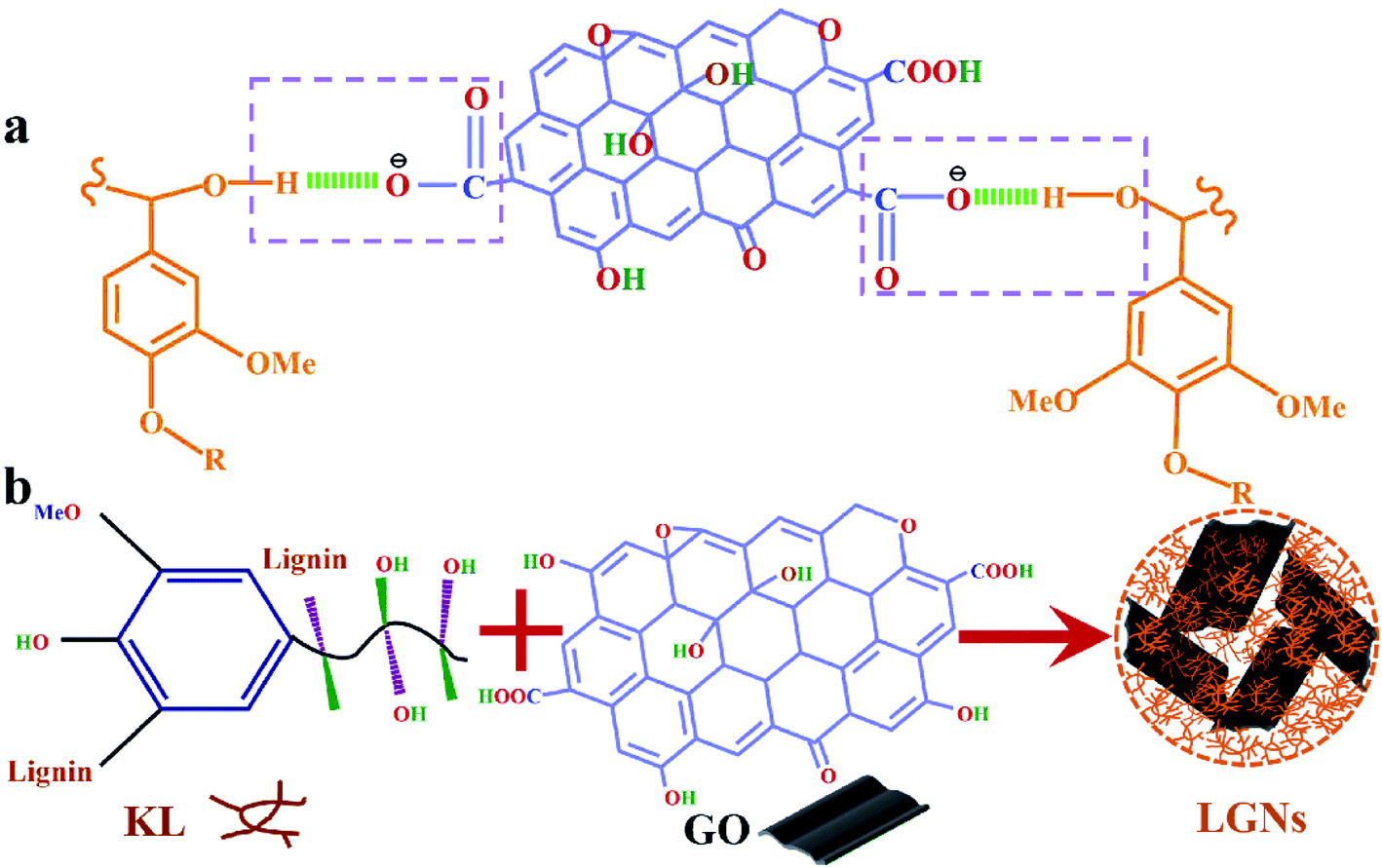
According to the above analysis, the feasible synthesis mechanism of LGNs is described as the following. GO contains a multitude of oxygen-containing functional groups, so that it can be dispersed in water well.40 Additionally, KL can be easily dissolved in THF and formed well-mixed solutions. As the solution of GO was added to KL/THF solution, the active groups (hydroxyl groups) of the lignin molecules were in contact with the active groups (carboxyl groups) of the GO. Then the two materials were combined by hydrogen bonds which formed between the hydroxyl and carboxyl. This is coincident with the experimental data proved by the previous FTIR analysis. Furthermore, as the water content in the mixed solution increased, phase separation occurred between water and mixed solution. Due to the strong hydrophobicity of KL molecules, a film layer was formed at the interface between the mixture solution and water.33,41 With the further increase of water content, GO was also tightly bound to KL through hydrogen bonding and then aggregated on the surface of the membrane. In order to reduce surface energy, the nanoparticles tended to form spherical structures through π–π interaction.32,42 The introduction of oxygen-containing functional groups into LGNs could effectively increase the number of active groups of the adsorbent, conducive to the increase of the adsorption effect (Fig. 3).43
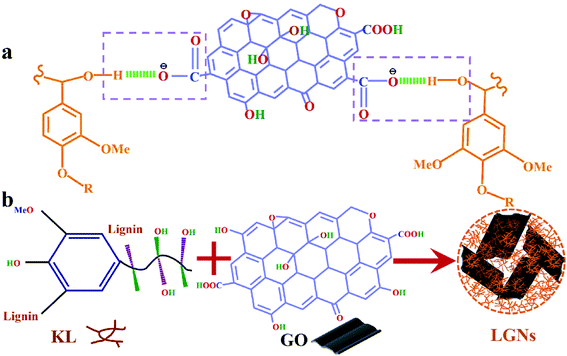 |
| | Fig. 3 (a) KL and GO were combined by hydrogen bonds which formed between the hydroxyl and carboxyl. (b) Synthetic schematic diagram of LGNs. | |
3.3 Cr(VI) adsorption with LGNs
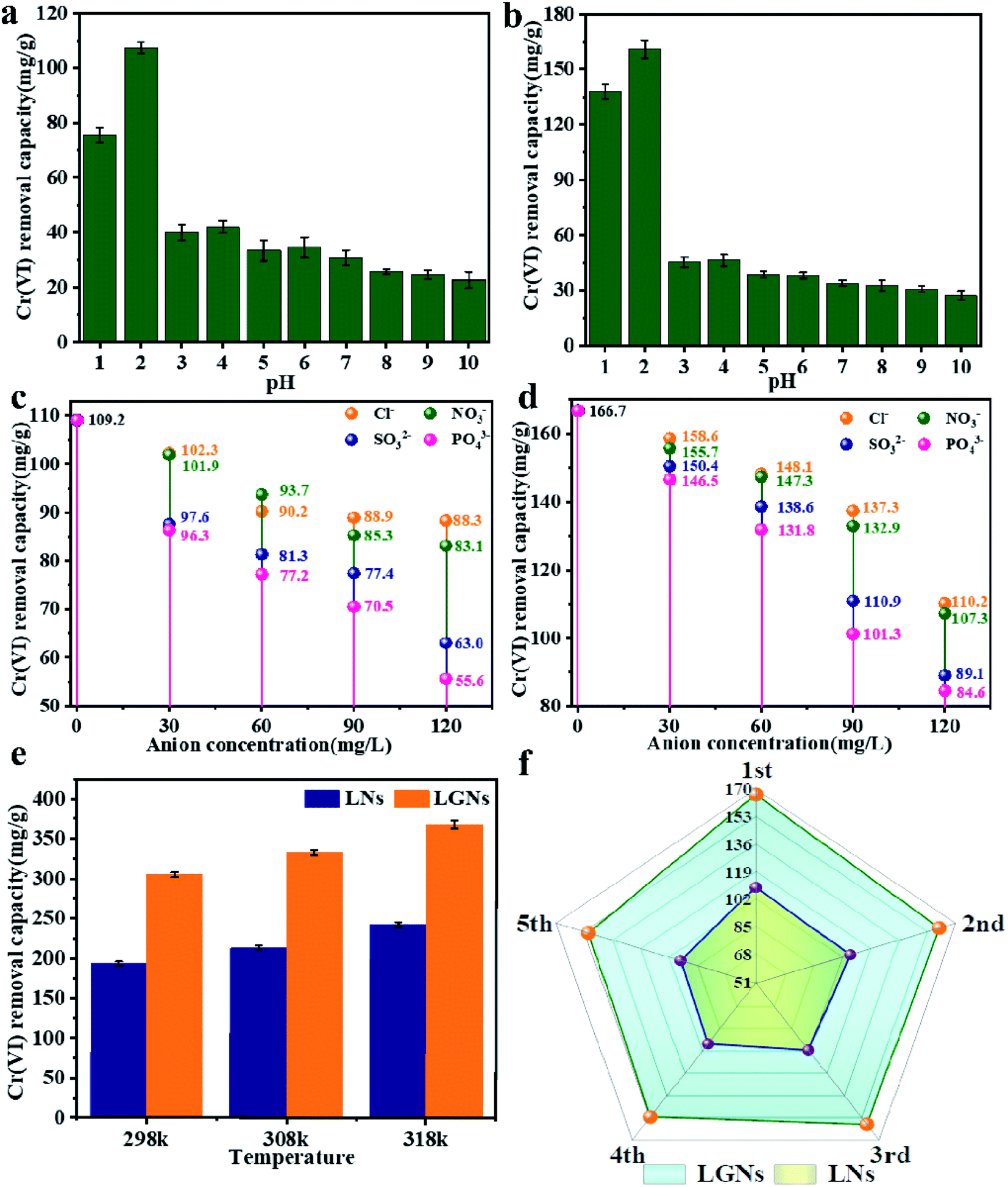
500 mg l−1 Cr(VI) solution was applied to explore the influence of temperature on sorption efficiency (Fig. 4e). The study confirmed that the Cr(VI) removal rate of LGNs increased with temperature. Here, we compared the results of literature whose experimental conditions are close to our work. The maximum adsorption capacity of LGNs is 368.78 mg g−1, higher than many previously reported adsorbents (Table 1). The result proves that lignin and graphene oxide have great value in the sorption of heavy metal ions.
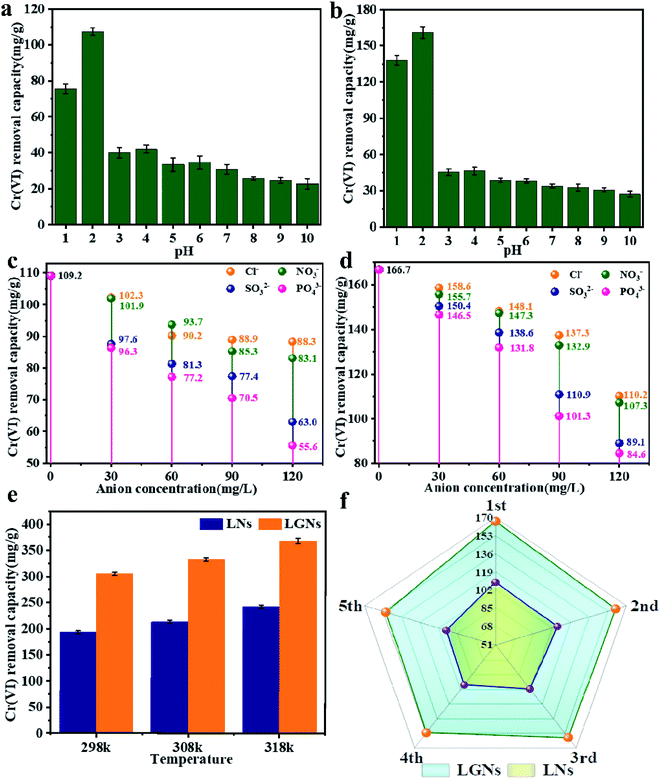 |
| | Fig. 4 Influence of pH on Cr(VI) removal by (a) LNs and (b) LGNs. Effect of other ions on Cr(VI) adsorption capacity by (c) LNs and (d) LGNs. (e) Effect of temperature on Cr(VI) adsorption. (f) Adsorption efficiency after recycling. | |
Table 1 Comparison of efficiency for varied sorbents on Cr(VI) removal
| Sorbent |
Cr(VI) removal efficiency (mg g−1) |
Ref. |
| Lignin |
31.6 |
35 |
| Cellulose–PEI aerogel |
229.1 |
44 |
| PEI–silica nanocomposite |
183.7 |
47 |
| Lignocellulosic substrate |
35 |
48 |
| Coconut coir |
50 |
49 |
| Chitosan-coated sour cherry kernel shell beads |
24.5 |
50 |
| PEI-modified sericin bead |
365.3 |
51 |
| Electrospun chitosan nanofibers |
68.3 |
52 |
| Magnetic nanocomposite |
78.2 |
53 |
| Polymer nanotubes |
147.8 |
54 |
| Lignin–GO nanospheres |
368.8 |
This work |
In fact, Cr(VI) doesn't exist individually in the effluent, but exists with diverse ions (such as Cl−, NO3− and PO43−).44 The coexisting anions may interfere with the sorption of Cr(VI) by competing for adsorption sites. The pH of the sewage can also influence the characters of the materials and the existent species of Cr(VI). Therefore, we evaluated the impact of other anions and different pH on Cr(VI) removal. The results suggest that Cl− and NO3− had no obvious effect on Cr(VI) removal. Nevertheless, the SO32− and PO43− reduced the adsorption rate of Cr(VI) distinctly, and the effect of PO43− was stronger than SO32−. This competitive trend is concordant with other previous studies, suggesting electrostatic interaction plays a crucial role in the sorption process.45,46
The process of Cr(VI) removal is closely correlated to the pH value (Fig. 4a and b), and acidic conditions are profitable to Cr(VI) removal. When pH = 2, the two adsorbents reached the maximum absorption capacity, which was 109.2 and 166.7 mg g−1 respectively. This may be caused by changes in electric potential and the existent species of Cr(VI), with the main species of Cr(VI) being HCrO4− and Cr2O72− at lower pHs.55 Overall, electrostatic attraction occurred between the positively charged adsorbents and the negatively charged anion, and the more positive charge brought the stronger adsorption force.56 Although the zeta potential was the largest at pH = 1, the adsorption effect failed to reach the optimum. It is because the strong acid environment has affected the structure of the adsorbents, and the size changes of LNs and LGNs also confirmed this result.
The adsorption isotherm expounds the equilibrium relationship between adsorbent and adsorbate. Langmuir and Freundlich models (eqn (2) and (3)) were used to fit the adsorption data of LNs and LGNs. Among them, Qe is the adsorption capacity at equilibrium (mg g−1); KL is the Langmuir constant linked with the saturated sorption capacity and energy; KF and 1/n are the Freundlich constants linked with sorption capacity and strength. The fitting curves of isotherm adsorption models and related parameters are revealed in Fig. S8(a and b)† and Table 2. The results of Freundlich model are more similar to the experimental data according to the correlation modulus R2. This Freundlich adsorption behavior often arises in polymers or composite adsorbents because of their uneven surface properties. Similar conclusions were found in previous researches on the removal and treatment of Cr(VI).34,35,57
| |
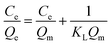 | (2) |
| |
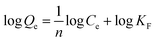 | (3) |
Table 2 Langmuir and Freundlich fittings of Cr(VI) removal by LNs and LGNs
| |
LNs |
LGNs |
| Langmuir model |
Qm (mg g−1) |
263.16 |
333.33 |
| KL (l mg−1) |
0.0076 |
0.0309 |
| R2 |
0.9726 |
0.9782 |
| Freundlich model |
KF (mg g−1) |
9.6783 |
43.2912 |
| 1/n |
0.3687 |
0.5234 |
| R2 |
0.9899 |
0.9938 |
Adsorption kinetics is also a vital factor in the sorption process. The most widely used sorption kinetic models are Lagergren pseudo-first-order kinetic model and pseudo-second-order model, which are described with the following forms respectively (eqn (4) and (5)). k1 and k2 are the rate constants related to the first-order kinetic model and the second-order kinetic model respectively; Qt means the sorption capacity at time t (mg g−1). It could be found that the linear correlation coefficients of the first-order kinetic model and the second-order kinetic model are good by analyzing the data in Fig. S8(c and d)† and Table 3. However, according to the correlation modulus R2, the second-order model is more appropriate for describing the removal process of Cr(VI) on LNs and LGNs than the first-order model. This phenomenon shows that the rate of Cr(VI) sorption on LNs and LGNs are controlled by kinetics.
| |
ln(Qe − Qt) = ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qe − k1t Qe − k1t
| (4) |
| |
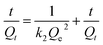 | (5) |
Table 3 Pseudo-first-order and pseudo-second-order kinetics fittings of Cr(VI) removal
| |
LNs |
LGNs |
| Pseudo-first-order |
k1 (min−1) |
0.0045 |
0.0062 |
| Qe (mg g−1) |
176.50 |
275.89 |
| R2 |
0.9891 |
0.9621 |
| Pseudo-second-order |
k2 (g min−1 mg−1) |
1.96 × 10−5 |
1.62 × 10−5 |
| Qe (mg g−1) |
243.90 |
384.62 |
| R2 |
0.9957 |
0.9839 |
The good recycling performance of adsorbent is of immense significance in reducing the cost of sewage treatment and realizing commercial applications. Previous studies have confirmed that the desorption performance under alkaline conditions is better than other conditions.34 Therefore, 0.1 M NaOH was used to achieve the desorption of chromium ions. We can find from the Fig. 4f that as the number of recycling increased, the sorption capacity decreased. When the number of reuse reaches 5 times, LGNs still maintain a high adsorption capacity, which is 150.4 mg g−1 in 200 mg g−1 Cr(VI) solution. Its adsorption efficiency is only reduced by 9.7%. In general, LGNs have great reusability and show excellent potential as effective adsorbents for Cr(VI) removal.
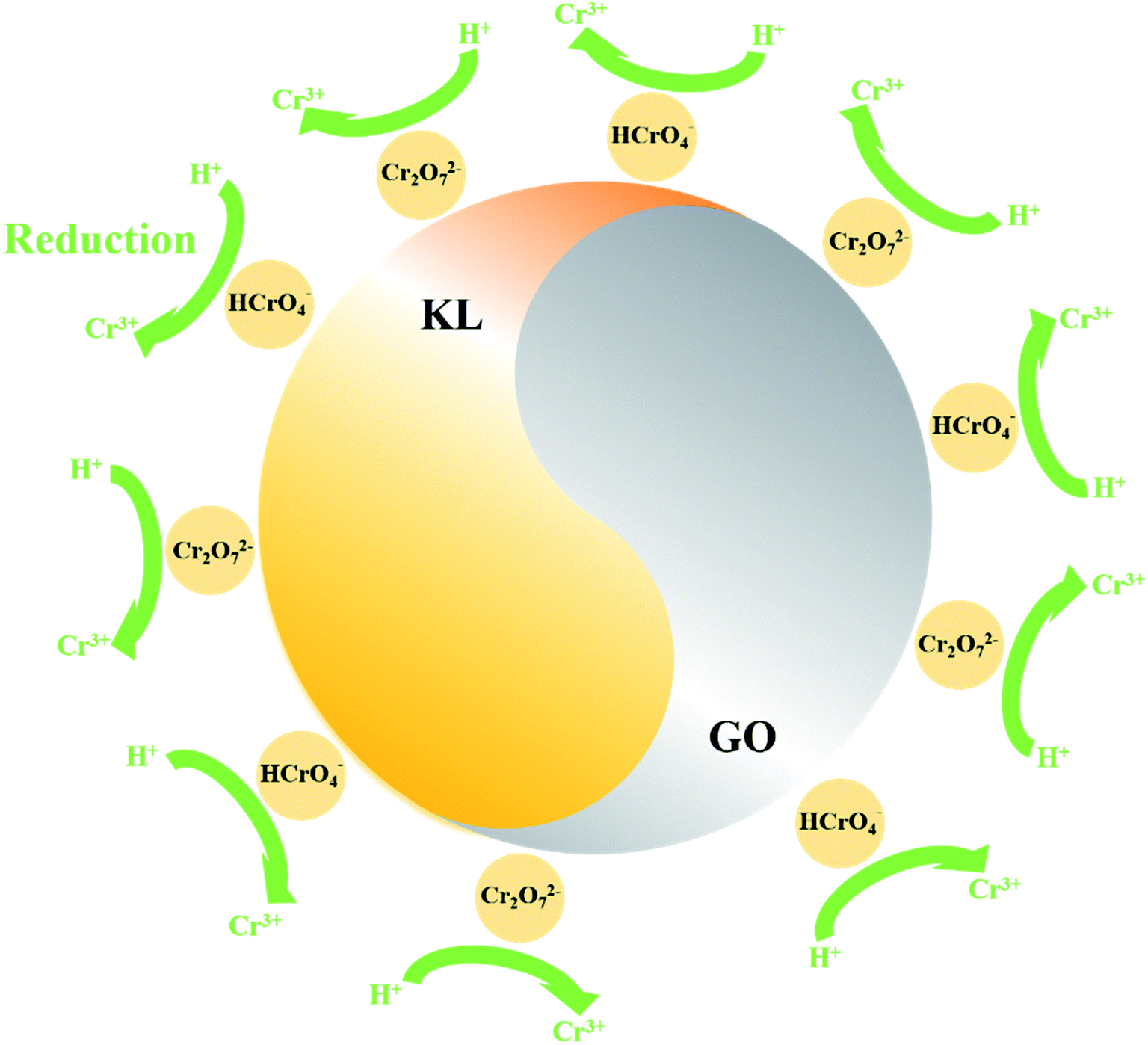
3.4 Mechanism
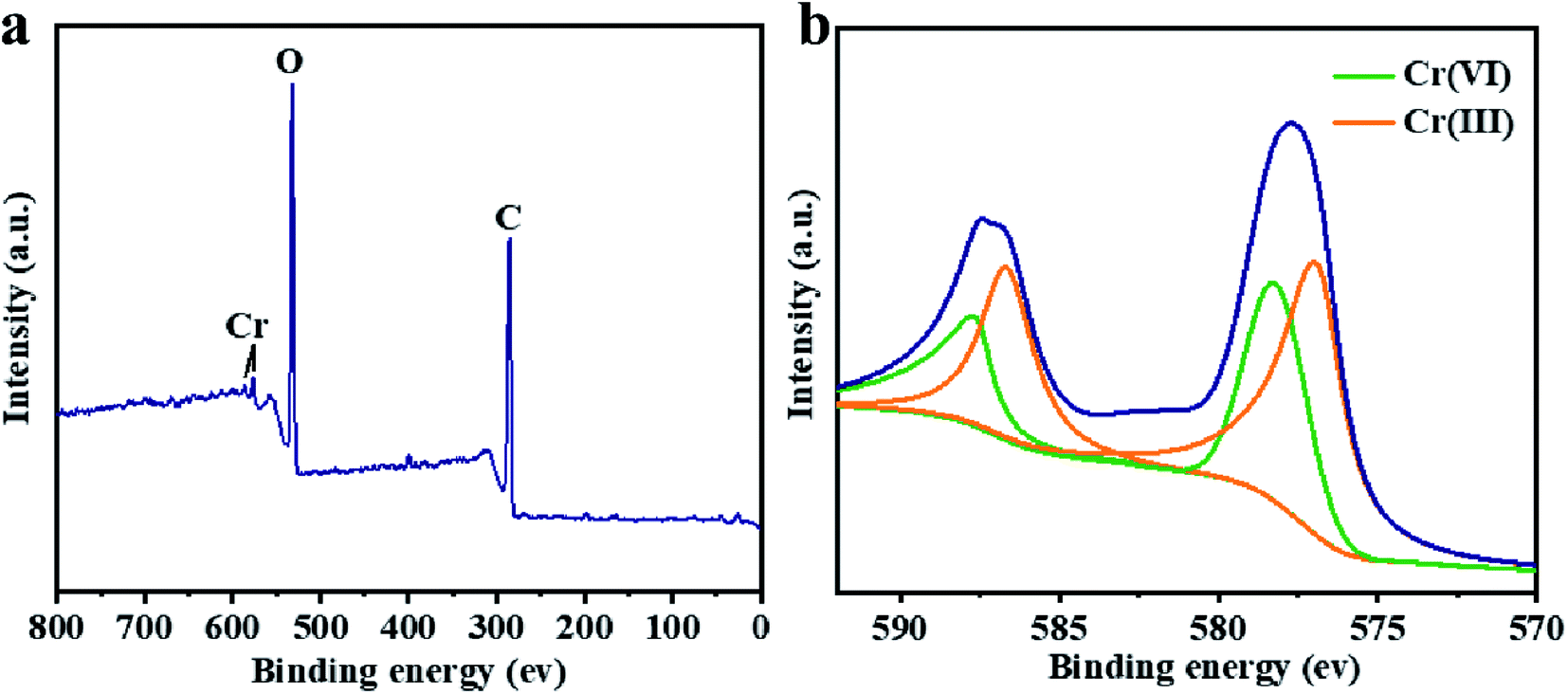
The XPS characterization was used to discover the species of Cr during the Cr(VI) removal process. It can be found from Fig. 5 that peaks appeared at 578 and 587 eV, which correlates to the Cr 2p3/2 and Cr 2p1/2 orbitals respectively. The result indicates that Cr(VI) has been successfully sorbed to the surface of LGNs. According to the Cr 2p spectrum, it can be determined that this region is divided into two characteristic peaks. The peaks at 576.9 eV and 586.5 eV are derived from Cr 2p3/2 and Cr 2p1/2 of Cr(III), and the binding energies of 578.4 and 587.8 eV belong to Cr(VI). This phenomenon indicates that part of Cr(VI) was reduced to Cr(III) during the adsorption process. In the previous content, we have confirmed that electrostatic interaction acts an essential role in Cr(VI) removal. Additionally, XPS results also show that Cr(VI) is reduced to Cr(III) under acidic condition. Therefore, the adsorption of Cr(VI) can be secured through electrostatic attraction between positively charged groups (such as protonated hydroxyl groups, carboxyl groups, etc.) on the surface of adsorbent, or by reducing Cr(VI) to Cr(III) under strongly acidic circumstances. The previous researches have proved Cr(VI) remains in water as oxyanions (HCrO4− and Cr2O72− etc.) and its species is related to the pH.58 Furthermore, that the removal process of Cr(VI) consumes a lot of protons has also been confirmed.48 According to the above mentioned, the feasible Cr(VI) repair mechanism of LGNs may be summarized as the following: firstly, the hydroxyl groups of LGNs could be availably protonated under acidic conditions, and Cr(VI) was availably sorbed onto LGNs through electrostatic attraction.35 Secondly, partial Cr(VI) was reduced to Cr(III) by neighboring electron-donor groups (such as CO and O–CH3).46,59,60 The process of reducing Cr(VI) to Cr(III) is shown as eqn (6) and (7). And then Cr(III) can be sorbed on the surface of LGNs by ion exchange or surface complexation (Fig. 6).61| | |
HCrO4− + 7H+ + 3e− → Cr3+ + 4H2O
| (6) |
| | |
Cr2O72− + 14H+ +6e− → 2Cr3+ + 7H2O
| (7) |
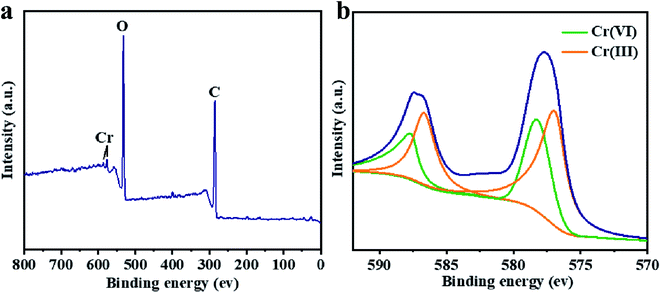 |
| | Fig. 5 XPS spectrum of (a) total spectra and (b) Cr 2p spectra. | |
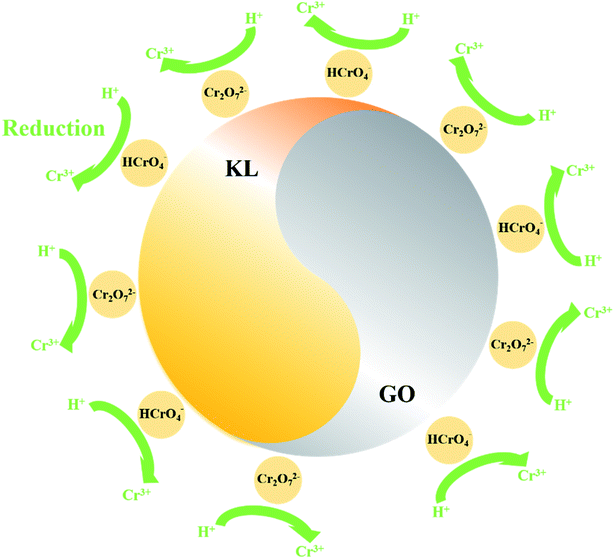 |
| | Fig. 6 Schematic diagram of Cr(VI) repair mechanism. | |
4. Conclusion
Lignin is one of the most abundant biomasses in nature. Herein, we introduced GO into lignin via a simple self-assembly method to prepare LGNs, which can be used as an effective sorbent for Cr(VI). Lignin and GO are tightly fitted together by hydrogen bonds. The addition of GO can significantly improve the stability of nanoparticles. In addition, the number of adsorption sites increased because GO contains numerous oxygen-containing functional groups. As a result, the maximum adsorption capacity of LGNs can reach 368.78 mg g−1, a 50% increase in adsorption efficiency compared with LNs. It is found that the sorption of Cr(VI) by LGNs obeys Freundlich and pseudo-second-order kinetics forms. In the principle analysis, we discovered that the sorption of Cr(VI) by LGNs through electrostatic interaction and Cr(VI) was reduced to Cr(III). And when the number of reuses reaches 5 times, LGNs still maintain a high adsorption capacity. LGNs have the superiority of great adsorption efficiency, convenient formation and outstanding recovery ability, thus being auspicious sorbents for Cr(VI) removal from sewage.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by Fundamental Research Funds of CAF (CAFYBB2018GD002); and Natural Science Foundation of China (No. 31890771), and Foundation of State Key Laboratory of Biobased Material and Green Papermaking (No. KF201904).
References
- S.-H. Ho, S. Zhu and J.-S. Chang, Bioresour. Technol., 2017, 246, 123–134 CrossRef CAS.
- Y. Zhang, M. Xu, H. Li, H. Ge and Z. Bian, Appl. Catal., B, 2018, 226, 213–219 CrossRef CAS.
- G. Liao, L. Zhong, C. S. Cheung, C. Du, J. Wu, W. Du, H. Zheng and H. Gao, Microporous Mesoporous Mater., 2020, 307, 110469 CrossRef CAS.
- G. Liao, W. Zhao, Q. Li, Q. Pang and Z. Xu, Chem. Lett., 2017, 46, 1631–1634 CrossRef CAS.
- T. C. Maponya, K. E. Ramohlola, N. H. Kera, K. D. Modibane, A. Maity, L. M. Katata-Seru and M. J. Hato, Polymers, 2020, 12, 679 CrossRef CAS.
- V. E. Pakade, N. T. Tavengwa and L. M. Madikizela, RSC Adv., 2019, 9, 26142–26164 RSC.
- S. Zhang, H. Zhang, F. Liu, F. Yang, S. Zhou, K. Zheng, C. Chu, L. Liu and M. Ju, RSC Adv., 2019, 9, 31333–31342 RSC.
- D. Pradhan, L. B. Sukla, M. Sawyer and P. K. S. M. Rahman, J. Ind. Eng. Chem., 2017, 55, 1–20 CrossRef CAS.
- B. Nanda, A. C. Pradhan and K. Parida, Chem. Eng. J., 2017, 316, 1122–1135 CrossRef CAS.
- M. Akram, H. N. Bhatti, M. Iqbal, S. Noreen and S. Sadaf, J. Environ. Chem. Eng., 2017, 5, 400–411 CrossRef CAS.
- R. A. Abu-Zurayk, R. Z. Al Bakain, I. Hamadneh and A. H. Al-Dujaili, Int. J. Miner. Process., 2015, 140, 79–87 CrossRef CAS.
- B. Jiang, Y. Gong, J. Gao, T. Sun, Y. Liu, N. Oturan and M. A. Oturan, J. Hazard. Mater., 2019, 365, 205–226 CrossRef CAS.
- Y. Zhang and S.-J. Park, Appl. Catal., B, 2019, 240, 92–101 CrossRef CAS.
- S. Xia, Z. Song, P. Jeyakumar, S. M. Shaheen, J. Rinklebe, Y. S. Ok, N. Bolan and H. Wang, Crit. Rev. Environ. Sci. Technol., 2019, 49, 1027–1078 CrossRef CAS.
- G. Liao, J. Chen, W. Zeng, C. Yu, C. Yi and Z. Xu, J. Phys. Chem. C, 2016, 120, 25935–25944 CrossRef CAS.
- G. Liao, J. Fang, Q. Li, S. Li, Z. Xu and B. Fang, Nanoscale, 2019, 11, 7062–7096 RSC.
- G. Liao, Q. Li, W. Zhao, Q. Pang, H. Gao and Z. Xu, Appl. Catal., A, 2018, 549, 102–111 CrossRef CAS.
- H. Che, C. Liu, G. Che, G. Liao, H. Dong, C. Li, N. Song and C. Li, Nano Energy, 2020, 67, 104273 CrossRef CAS.
- J.-Z. Cheng, L.-L. Liu, G. Liao, Z.-Q. Shen, Z.-R. Tan, Y.-Q. Xing, X.-X. Li, K. Yang, L. Chen and S.-Y. Liu, J. Mater. Chem. A, 2020, 8, 5890–5899 RSC.
- W.-Y. Huang, Z.-Q. Shen, J.-Z. Cheng, L.-L. Liu, K. Yang, X. Chen, H.-R. Wen and S.-Y. Liu, J. Mater. Chem. A, 2019, 7, 24222–24230 RSC.
- N. H. Kera, M. Bhaumik, K. Pillay, S. S. Ray and A. Maity, J. Colloid Interface Sci., 2017, 503, 214–228 CrossRef CAS.
- G. Zhao, X. Huang, Z. Tang, Q. Huang, F. Niu and X. Wang, Polym. Chem., 2018, 9, 3562–3582 RSC.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS.
- Y. Ma, L. Lv, Y. Guo, Y. Fu, Q. Shao, T. Wu, S. Guo, K. Sun, X. Guo and E. K. Wujcik, Polymer, 2017, 128, 12–23 CrossRef CAS.
- S. Chen, G. Wang, W. Sui, A. M. Parvez, L. Dai and C. Si, Ind. Crops Prod., 2020, 145, 112164 CrossRef CAS.
- Z. Wu, W. Huang, X. Shan and Z. Li, Int. J. Biol. Macromol., 2020, 143, 325–333 CrossRef CAS.
- G. Liao, Y. Gong, L. Zhong, J. Fang, L. Zhang, Z. Xu, H. Gao and B. Fang, Nano Res., 2019, 12, 2407–2436 CrossRef CAS.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- Y. Sun, L. Chen, J. Yu, B. Yoon, S. K. Lee, J.-D. Nam, L. Ci and J. Suhr, Carbon, 2020, 160, 54–63 CrossRef CAS.
- Y. Qian, Y. Deng, X. Qiu, H. Li and D. Yang, Green Chem., 2014, 16, 2156–2163 RSC.
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, Ind. Crops Prod., 2017, 100, 146–152 CrossRef CAS.
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, ACS Sustainable Chem. Eng., 2017, 5, 2273–2281 CrossRef CAS.
- H. W. Kwak, H. Lee and K. H. Lee, Chemosphere, 2020, 239, 124733 CrossRef CAS.
- A. B. Albadarin, H. Ala'a, N. A. Al-Laqtah, G. M. Walker, S. J. Allen and M. N. Ahmad, Chem. Eng. J., 2011, 169, 20–30 CrossRef CAS.
- W. Han, L. Luo and S. Zhang, Int. J. Environ. Sci. Technol., 2012, 9, 543–548 CrossRef CAS.
- W. Yang, E. Fortunati, D. Gao, G. M. Balestra, G. Giovanale, X. He, L. Torre, J. M. Kenny and D. Puglia, ACS Sustainable Chem. Eng., 2018, 6, 3502–3514 CrossRef CAS.
- Z. Zhang, R. Yang, W. Gao and K. Chen, Desalin. Water Treat., 2017, 83, 168–175 CrossRef CAS.
- Y. Tang, M. Li, C. Mu, J. Zhou and B. Shi, Chemosphere, 2019, 229, 570–579 CrossRef CAS.
- Z. Zhang and R. Yang, Fibers Polym., 2017, 18, 334–341 CrossRef CAS.
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, ACS Sustainable Chem. Eng., 2018, 6, 9342–9351 CrossRef CAS.
- Z. Yan, G. Liao, X. Zou, M. Zhao, T. Wu, Y. Chen and G. Fang, J. Agric. Food Chem., 2020, 68, 8341–8349 CrossRef CAS.
- S.-T. Yang, Y. Chang, H. Wang, G. Liu, S. Chen, Y. Wang, Y. Liu and A. Cao, J. Colloid Interface Sci., 2010, 351, 122–127 CrossRef CAS.
- D.-M. Guo, Q.-D. An, Z.-Y. Xiao, S.-R. Zhai and Z. Shi, RSC Adv., 2017, 7, 54039–54052 RSC.
- S. Yu, X. Wang, Z. Chen, X. Tan, H. Wang, J. Hu, A. Alsaedi, N. S. Alharbi, W. Guo and X. Wang, Chem. Eng. J., 2016, 302, 77–85 CrossRef CAS.
- R. Zhao, X. Li, B. Sun, Y. Li, Y. Li, R. Yang and C. Wang, J. Mater. Chem. A, 2017, 5, 1133–1144 RSC.
- K. Choi, S. Lee, J. O. Park, J.-A. Park, S.-H. Cho, S. Y. Lee, J. H. Lee and J.-W. Choi, Sci. Rep., 2018, 8, 1438 CrossRef.
- L. Dupont and E. Guillon, Environ. Sci. Technol., 2003, 37, 4235–4241 CrossRef CAS.
- Y.-S. Shen, S.-L. Wang, S.-T. Huang, Y.-M. Tzou and J.-H. Huang, J. Hazard. Mater., 2010, 179, 160–165 CrossRef CAS.
- T. Altun, J. Anal. Sci. Technol., 2019, 10, 14 CrossRef.
- H. W. Kwak, Y. Kim, N. K. Yun and K. H. Lee, Macromol. Res., 2014, 22, 788–795 CrossRef CAS.
- L. Li, Y. Li, L. Cao and C. Yang, Carbohydr. Polym., 2015, 125, 206–213 CrossRef CAS.
- C. Naicker, N. Nombona and W. E. van Zyl, Chem. Pap., 2020, 74, 529–541 CrossRef CAS.
- S. Yu, G. Yuan, H. Gao and Y. Liao, J. Mater. Sci., 2020, 55, 163–176 CrossRef CAS.
- A. Demirbaş, Energy Sources, 2005, 27, 1449–1455 CrossRef.
- N. Tazrouti and M. Amrani, Bioresources, 2009, 4, 740–755 CAS.
- S. S. Baral, S. N. Das and P. Rath, Biochem. Eng. J., 2006, 31, 216–222 CrossRef CAS.
- H. Wang, X. Yuan, Y. Wu, G. Zeng, X. Chen, L. Leng, Z. Wu, L. Jiang and H. Li, J. Hazard. Mater., 2015, 286, 187–194 CrossRef CAS.
- P. Suksabye, P. Thiravetyan, W. Nakbanpote and S. Chayabutra, J. Hazard. Mater., 2007, 141, 637–644 CrossRef CAS.
- Y.-S. Shen, S.-L. Wang, S.-T. Huang, Y.-M. Tzou and J.-H. Huang, J. Hazard. Mater., 2010, 179, 160–165 CrossRef CAS.
- D. Park, Y.-S. Yun, H. W. Lee and J. M. Park, Bioresour. Technol., 2008, 99, 1141–1147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09190a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Miao Rana,
Kuizhong Shena and
Guangfu Liao
*ab,
Miao Rana,
Kuizhong Shena and
Guangfu Liao