
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of quinolone derivatives as antimycobacterial agents†
Kun-Lin Liu‡
a,
Fei Teng‡a,
Lu Xionga,
Xiao Lia,
Chao Gao*b and
Luo-Ting Yu *a
*a
aState Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Collaborative Innovation Center, Chengdu, 610041, China. E-mail: yuluot@scu.edu.cn
bLaboratory of Human Diseases and Immunotherapies, West China Hospital, Sichuan University, Chengdu, 610041, China. E-mail: gaochao527@hotmail.com
First published on 8th July 2021
Abstract
Tuberculosis (TB), an infectious disease caused by Mycobacterium tuberculosis (M. tuberculosis), is an important public health issue. Current first-line drugs administered to TB patients have been in use for over 40 years, whereas second-line drugs display strong side effects and poor compliance. Additionally, designing effective regimens to treat patients infected with multi- and extremely-drug-resistant (MDR and XDR) strains of TB is challenging. In this report, we screened our compound library and identified compound 1 with antituberculosis activity and a minimal inhibitory concentration (MIC) against M. tuberculosis of 20 μg mL−1. Structure optimization and the structure–activity relationship of 1 as the lead compound enabled the design and synthesis of a series of quinolone derivatives, 6a1–6a2, 6b1–6b36, 6c1, 6d1–6d14, 7a1–7a2, 7b1–7b2, 7c1, 8a1–8a5, 9a1–9a4 and 10a1–10a6. These compounds were evaluated in vitro for anti-tubercular activity against the M. tuberculosis H37Rv strain. Among them, compounds 6b6, 6b12 and 6b21 exhibited MIC values in the range of 1.2–3 μg mL−1 and showed excellent activity against the tested MDR-TB strain (MIC: 3, 2.9 and 0.9 μg mL−1, respectively). All three compounds were non-toxic toward A549 and Vero cells (>100 and >50 μg mL−1, respectively). In addition, an antibacterial spectrum test carried out using compound 6b21 showed that this compound specifically inhibits M. tuberculosis. These can serve as a new starting point for the development of anti-TB agents with therapeutic potential.
1. Introduction
Tuberculosis (TB) is caused by the pathogen Mycobacterium tuberculosis (M. tuberculosis) and is a leading cause of human fatalities by an infectious microorganism.1 The World Health Organization (WHO) has estimated that approximately one quarter of the total world population is infected with TB. In 2018, the WHO reported that 10 million people were infected with TB and 1.2 million fatalities were caused by this pathogen.2 Moreover, TB control is threatened by the emergence and spread of multi- and extremely-drug-resistant (MDR and XDR) strains of TB.3–8 Approximately half a million people were estimated to be infected by rifampicin-resistant TB (RR-TB) in 2018, among which 78% had MDR-TB.2 The absence of new drugs hampers efforts to improve the treatment outcomes of MDR-TB infection, and the development of novel anti-tuberculosis drugs with new action mechanisms are urgently needed.9–11TB drug discovery follows two main routes: target-based approaches or cell-based approaches. One challenge with target-based approaches is that compounds identified in target-based screens often fail to find useful minimum inhibitory concentrations (MIC) against TB.12–16 Current drugs and drug candidates in clinical trials have been discovered by using whole cell activity assays, thereby highlighting the importance of this approach. From a cell-based screening campaign of commercially available libraries and our laboratory compound collection, the hexahydroisoquinoline compound 1 was identified as a promising anti-tubercular hit with a MIC of 20 μg mL−1 against M. tuberculosis H37Rv.
The structural skeleton of compound 1 was first used in quinolones, which are broad-spectrum antibacterial drugs first discovered in the 19th century. Fluoroquinolones are a class of very potent, broad-spectrum synthetic antimicrobial agents that are in clinical use. Currently, fluoroquinolones such as ciprofloxacin, ofloxacin and levofloxacin are recommended as second-line drugs for the treatment of TB.17 Generally, all fluoroquinolones function by targeting DNA gyrase in Gram-negative bacteria and topoisomerase IV in Gram-positive bacteria.18 In 2014, Panda et al. report a novel pyrazolopyridone class of inhibitors, which were identified from whole cell screening against M. tuberculosis.19 Evaluation of these compounds by an overexpression assay revealed that these compounds are selective against decaprenylphosphoryl-β-D-ribose oxidase (DprE1). Naik et al. reported a novel scaffold of 4-aminoquinolone piperidine amides that were discovered by using a whole cell assay. Approximately 320![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds from the AstraZeneca Corporate collection were screened.20 These compounds were found to bind non-covalently to DprE1 and were reversible DprE1 inhibitors. OPC167832, a dihydroquinolone developed by Otsuka, has entered clinical stage II trials. Thus, the quinolone scaffold shows potential for targeting M. tuberculosis because many antibacterial compounds based on this structure show antibacterial activity. Although the structure of the lead compound 1 we screened is similar to the structure of the compound reported above, the target of compound needs further verification.
000 compounds from the AstraZeneca Corporate collection were screened.20 These compounds were found to bind non-covalently to DprE1 and were reversible DprE1 inhibitors. OPC167832, a dihydroquinolone developed by Otsuka, has entered clinical stage II trials. Thus, the quinolone scaffold shows potential for targeting M. tuberculosis because many antibacterial compounds based on this structure show antibacterial activity. Although the structure of the lead compound 1 we screened is similar to the structure of the compound reported above, the target of compound needs further verification.
In this report, the hit-to-lead campaign for quinolone derivatives was driven by MIC-based structure–activity relationships (SAR). Initially, we explored and simplified the chemical structure of the compound 1 side chain. The SAR between the linker and the parent nucleus was then explored by using the pharmacochemical principles of bioelectronic isosteric, skeletal transition and homologues transformation. Finally, to identify substitutions and establish a link with a target whose inhibition leads to an anti-tubercular effect, a more active lead was required. Herein, we report on the discovery of hexahydroisoquinolines as potential anti-tubercular agents, and synthesize a number of analogues that have improved anti-tubercular potency over the original lead compound 1.
2. Results and discussion
2.1 Chemistry
The key intermediates 3a–3c were prepared as described in Scheme 121 for the synthesis of important intermediates 5a–5d. Compounds 2a–2c were reacted with morpholine in toluene under reflux conditions after adding p-toluenesulfonic acid. Acetylation of the intermediate enamines to the corresponding ketones was then carried out in the presence of triethylamine at −5 °C. | ||
| Scheme 1 General synthesis of acetyl cyclohexanone and its analogues. aReagents and conditions: (i) toluene, morpholine, p-toluenesulfonic, reflux, overnight. (ii) DCM, triethylamine, −5 °C; C2H3ClO, rt, overnight. | ||
The title compounds 6a1–6a2, 6b1–6b36, 6c1 and 6d1–6d13 were synthesized via the pathway shown in Scheme 2. The compounds 4a–4d were synthesized by cyclizing the intermediates 3a–3b using 2-cyanoacetamide and triethylenediamine at 40 °C for 10 h.22 The mixture of 4b and 4d was dissolved in ethanol and stirred and refluxed for more than 1 h. Solids in the reaction mixture were filtered and then washed with hot ethanol while the solution was hot. Pure 4b and 4d were obtain before above solids washed several times. 4a–4d was catalyzed by RANEY®-nickel under 11427 torr H2 to get the intermediates 5a–5d, which reacted with various organic acids in the presence of EDCI and HOBT at room temperature to afford compounds 6a1–6a2, 6b1–6b36, 6c1 and 6d1–6d14. The target compounds 7a1–7a2, 7b1–7b2, 7c1 and 8a1–8a5 were obtained by the route described in Scheme 3.23–25 The butanedione derivatives were cyclized and reduced to amines under H2, which then condensed with carboxylic acid at room temperature to afford target compounds 7a1–7a2, 7b1–7b2, 7c1 and 8a1–8a5.
 | ||
| Scheme 2 | ||
 | ||
| Scheme 3 a,b Reagents and conditions: (i) ethanol, 2-cyanoacetamide, triethylenediamine, 40 °C, 10 hours. (ii) methanol and ammonia (1:1), H2, RANEY®-nickel, 80 °C, 36 h. (iii) DCM, EDCI, HOBT, and 4-methylmorpholine, rt, 10 hours. | ||
The intermediate 9a was synthesized by the reaction of amine transesterification and Knoevenagel between 2-aminoacetophenone and ethyl cyanoacetate.26 Hydrogenation of 9a to its corresponding amine, which condensed with carboxylic acid afforded target compounds 9a1–9a4. Compound 10a1 was obtained by nucleophilic substitution reaction of 5b. Compounds 10a2–10a6 were synthesized as described in Scheme 4.
 | ||
| Scheme 4 a Reagents and conditions: (i) ammonium acetate, 120 °C, 8 hours. (ii) H2, RANEY®-nickel, 80 °C, overnight. (iii) DCM, EDCI, HOBT, and 4-methylmorpholine, rt, 10 hours. (iv) DIEA, 5b, −30 °C, 30 min; 4-(trifluoromethyl)benzyl bromide, −30 °C, over 4 hours. (v) DCM, 0 °C, 4-(trifluoromethyl)benzene-1-sulfonyl chloride slowly drops, triethylamine, 30 min. (vi) Methanol, NaOH, sealed tube, 120 °C, overnight. (vii) DCM, thionyl chloride, N,N-dimethylformamide, 0 °C, 3 hours. (viii) 4-(Trifluoromethyl)benzhydrazide, pyridine 4-dimethylaminopyridine, rt, 6 hours, getting intermediate. (ix) Dried DCM, trifluoromethanesulfonic anhydride, triphenylphosphine oxide, stirred for 5 min, intermediate was added, rt, 7 hours. | ||
2.2 Structure activity relationship (SAR)
The synthesized compounds were tested for antimycobacterial activity against M. tuberculosis H37Rv using the Microplate Alamar Blue Assay and isoniazid was used as the reference drug (Fig. 1). The substituent group was modified at the para- or meta-position of R to explore the SAR (Table 1). The introduction of –F, –OCF3 and N,N-dimethylamino at the para position resulted in a clear loss of activity (compounds 6b1–6b3). However, compound 6b4 had potent activity compared with those of 6b1–6b3. Moreover, 6b4 displayed better potency than 3-methoxy 6b5. All results above indicated that the methoxy group at the para position may be crucial in single substituents. Comparison of 6b4 with 6b7 showed that the addition of one methoxy group at the meta-position of the phenyl ring resulted in lower activity for 6b7. Exploration of dimethoxy substituted groups (6b6–6b9) revealed that compound 6b6 with two methoxy group substitutions at the meta-position yielded greatest activity. 6b8–6b9 gave varying degrees of reduced antimycobacterial activity compared with 6b6. Keeping two methoxy groups like 6b6 and adding a hinge –CH2– or –O– to get 6b10–6b11, which only resulted in a slight reduction in potency compared with that of 6b6, thus revealing the importance of the methoxy groups as mentioned above. Overall, double methoxy substituted compounds possessed better anti-tubercular activity than single substituted (6b5 vs. 6b7 and 6b10), and the activity of the 1,3-dimethoxybenzene derivative 6b6 was 7-fold higher than that of compound 1. Comparison between compounds 6b8 and 6b9 demonstrated that the methoxy group played a key role in improving the anti-tubercular activity. Moreover, compared with 6b6, 6b10 and 6b11 reduced or even abolished the antibacterial activity, indicating 6b6 had a dominant conformation that two methoxy groups may serve as hydrogen bond acceptor interaction with active sites on some key proteins, however, the hinge –O– of 6b11 may act as another hydrogen bond acceptor interaction with inactive sites other than active sites on some key proteins, leading to loss of activity. | ||
| Fig. 1 The design strategy for compound 1. | ||
|
||||
|---|---|---|---|---|
| Compound no. | R | % inhibitiona 25 μg mL−1 | MICb (μg mL−1) | ClogPc |
| a The inhibition rate is the average of the three measurements.b MIC90 is the average value of three measurements.c ClogP is calculated by ChemBioDraw Ultra. | ||||
| 6b1 | 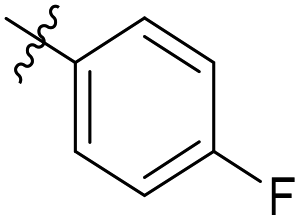 |
NA | NA | 4.26 |
| 6b2 |  |
NA | NA | 4.05 |
| 6b3 |  |
NA | NA | 4.28 |
| 6b4 |  |
98.3% | 25 | 4.05 |
| 6b5 |  |
43.6% | >25 | 4.05 |
| 6b6 | 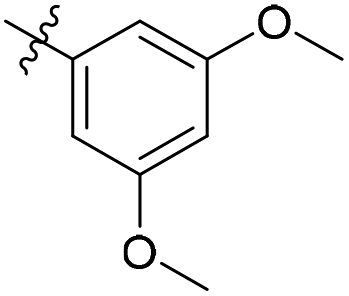 |
99.8% | 3 | 4.11 |
| 6b7 |  |
54.7% | NA | 3.76 |
| 6b8 |  |
36.9% | NA | 5.16 |
| 6b9 |  |
93.7% | 7.2 | 4.55 |
| 6b10 |  |
93.1% | 15 | 4.26 |
| 6b11 |  |
64.3% | NA | 4.29 |
| Isoniazid | — | 98.9% | 0.25 | −0.67 |
We then synthesized compounds 6b12–6b20 to assess the effect of different moieties placed at R on the potency toward M. tuberculosis. Compound 6b12 had the lowest MIC (2.5 μg mL−1) in the single substituted trifluoromethyl compounds. Subsequently, other single substituted compounds 6b15–6b20 were prepared to further explore the SAR of this class of compounds. These compounds were found to lose potency. Only compound 6b12 showed suitable activity based on this SAR investigation, indicating that after simplifying the substituted groups, a single substituted trifluoromethyl group at the para-position of the phenyl was significant for maintaining activity (Table 2).
|
||||
|---|---|---|---|---|
| Compound no. | R | % inhibition 25 μg mL−1 | MIC (μg mL−1) | ClogP |
| 6b12 |  |
99.3% | 2.5 | 3.42 |
| 6b13 | 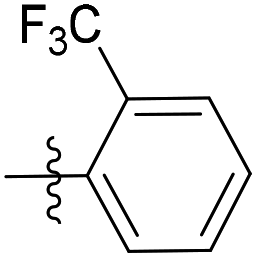 |
33.3% | NA | 2.21 |
| 6b14 |  |
45.1% | NA | 3.42 |
| 6b15 |  |
64.2% | NA | 3.50 |
| 6b16 |  |
13.3% | NA | 2.39 |
| 6b17 |  |
NA | NA | 2.53 |
| 6b18 |  |
26.9% | NA | 2.55 |
| 6b19 | 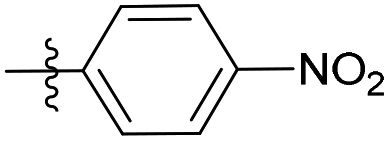 |
12.6% | NA | 2.36 |
| 6b20 | 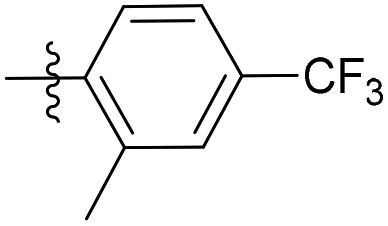 |
34.5% | NA | 3.58 |
| Isoniazid | 98.9% | 0.25 | −0.67 | |

The phenyl ring was replaced with aromatic heterocycles to further explore the anti-tuberculosis activity of different types of aromatic rings in the R group (Table 3). Most of compounds 6b21–6b36 showed better activity than compounds 6b13–6b20. Comparison of compounds 6b21 and 6b22 indicated that the position of the nitrogen atom in the pyridine ring is crucial in defining anti-tuberculosis activity. Based on this limited search, we selected the 2-substituted pyridine as the preferred group. Substitution on the pyridine with an electron-withdrawing or electron donor displayed good potency (6b21, 6b24–6b28). We then introduced a five-membered heterocyclic ring (6b29–6b36) in an effort to identify simpler compounds with relatively good hydrophilic profiles. Compounds 6b29–6b36 generally showed weaker bacteriostatic activity when compared with that of the six-member heterocyclic compounds. Overall, the best substitution group on the pyridine ring and furan ring was trifluoromethyl (6b21, 6b33). The solubility of this series of compounds was greatly improved when compared with compounds with the phenyl group substitution.
|
|||||
|---|---|---|---|---|---|
| Compound no. | R | % inhibition 20 μg mL−1 | MIC (μg mL−1) | Solubility (μg mL−1) | ClogP |
| 6b21 |  |
100% | 1.2 | 132.4 | 2.77 |
| 6b22 |  |
22.3% | NA | 102.2 | 2.42 |
| 6b23 |  |
12.3% | NA | 145.6 | 1.57 |
| 6b24 | 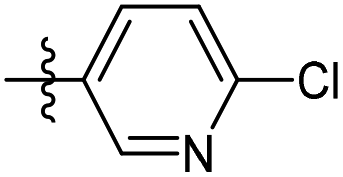 |
98.3% | 4.9 | 213.4 | 2.23 |
| 6b25 |  |
98.9% | 3.2 | 239.0 | 2.37 |
| 6b26 |  |
94.1% | 1.6 | 113.5 | 1.66 |
| 6b27 |  |
98.6% | 1.3 | 184.0 | 1.93 |
| 6b28 |  |
98.9% | 2.3 | 144.2 | 1.32 |
| 6b29 | 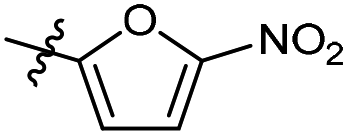 |
100.0% | 2.8 | 203.5 | 1.34 |
| 6b30 |  |
78.2% | NA | 178.3 | 1.54 |
| 6b31 |  |
97.7% | 3.4 | 101.0 | 2.43 |
| 6b32 |  |
96.4% | 5.4 | 129.6 | 2.10 |
| 6b33 |  |
98.3% | 2.0 | 100.2 | 2.60 |
| 6b34 |  |
NA | NA | 97.4 | 1.72 |
| 6b35 |  |
94.6% | 14 | 245.7 | 1.13 |
| 6b36 | 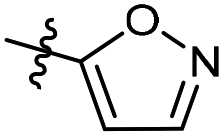 |
25.1% | — | 279.5 | 0.97 |
| Isoniazid | 98.9% | 0.25 | NA | −0.67 | |

In the process of synthesizing 4-(aminomethyl)-1-methyl-5,6,7,8-tetrahydroisoquinolin-3(2H)-one, intermediate 5b, we also obtained the isomer of this compound, i.e., intermediate 5d. Examination of the activity of compounds 6d1–6d4 revealed that the activity of the isomers were mostly lost, indicating that the position of the six-membered ring was important in defining bactericidal activity. Only compounds 6d5–6d7 (Table 4) displayed activity. Alternatives to the piperidine ring were then explored. Replacing the aliphatic ring with an aromatic ring resulted in a clear loss of activity (9a1–9a4). We next explored the effect of replacing the piperidine ring with a five and seven-membered saturated aliphatic ring. These analogues (6a1–6a2, 6c1) all showed reduced or no activity.
|
||||
|---|---|---|---|---|
| Compound no. | R | % inhibition 50 μg mL−1 | MIC (μg mL−1) | ClogP |
| 6d1 |  |
NA | >50 | 2.37 |
| 6d2 |  |
NA | >50 | 1.97 |
| 6d3 |  |
NA | >50 | 1.88 |
| 6d4 |  |
NA | >25 | 4.06 |
| 6d5 |  |
100% | 5.5 | 2.05 |
| 6d6 |  |
100% | 24.1 | 2.37 |
| 6d7 |  |
99% | 26.3 | 2.28 |
| 6d8 |  |
NA | >50 | 3.41 |
| 6d9 |  |
18% | >50 | 3.70 |
| 6d10 |  |
NA | >50 | 3.23 |
| 6d11 | 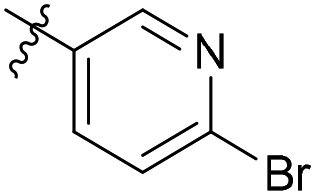 |
NA | >50 | 2.33 |
| 6d12 |  |
NA | >50 | 1.61 |
| 6d13 |  |
NA | >50 | 2.18 |
| 6d14 |  |
NA | >50 | 1.49 |
| 9a1 | 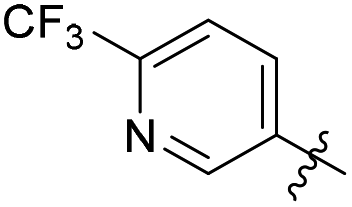 |
2% | >50 | 2.18 |
| 9a2 |  |
15% | >50 | 3.87 |
| 9a3 |  |
13% | >50 | 3.51 |
| 9a4 | 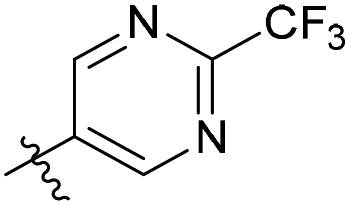 |
3% | >50 | 4.79 |
| 6a1 |  |
53% | >50 | 1.54 |
| 6a2 | 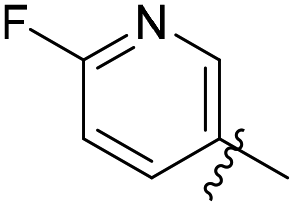 |
14% | >50 | 1.09 |
| 6c1 |  |
33% | >50 | 2.93 |
| Isoniazid | 98.9% | 0.25 | −0.67 | |

We then replaced 4 and 6 positions on the six-membered pyridone ring with alkyl, cyclopropyl or phenyl moiety (Table 5). Substitution of the ternary ring and benzene ring reduced anti-tuberculosis activity (7a1–7a2, 7b1–7b2, 7c1). The reduced activity may be caused by the increasing freedom of rotatable bond making it more difficult for the compounds to enter the cavity of the target. An increase in the activity of compounds (8a3, 8a5 vs. 8a1–8a2) was found to correlate with lipophilicity (ClogP), indicating that substitution with a long alkyl side chain at positions 2 and 4 of the pyridine moiety improves the lipophilicity and activity of the compounds when compared with compounds with short alkyl side chain substitutions. However, compared with alicyclic rings, the activity of compounds after ring opening decreased (8a1–8a2 vs. 6b29; 8a4 vs. 6b12), presumably because of the decrease in lipophilicity of the compounds, making it difficult for the compounds to cross the cell membrane.
|
||||||
|---|---|---|---|---|---|---|
| Compound no. | R | R1 | R2 | % inhibition 20 μg mL−1 | MIC (μg mL−1) | ClogP |
| 7a1 |  |
 |
 |
5% | >20 | 1.74 |
| 7a2 |  |
 |
 |
21% | >20 | 3.43 |
| 7b1 |  |
 |
 |
NA | >12.5 | 2.90 |
| 7b2 |  |
 |
 |
33% | >20 | 4.58 |
| 7c1 |  |
 |
 |
NA | >20 | 2.03 |
| 8a1 |  |
 |
 |
75.3% | NA | 0.41 |
| 8a2 | 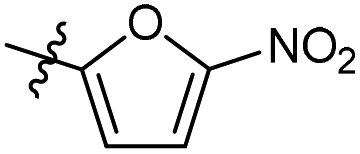 |
 |
 |
98.2% | NA | 0.94 |
| 8a3 |  |
 |
 |
98.1% | 19.8 | 1.47 |
| 8a4 |  |
 |
 |
87.6% | NA | 2.30 |
| 8a5 | 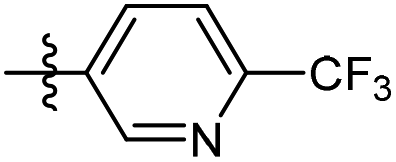 |
 |
 |
94.4% | 6.7 | 2.36 |
| Isoniazid | 99.4% | 0.25 | −0.67 | |||

The influence of the amide bond linker on anti-mycobacterial activity was then investigated. Replacing amide bonds with sulfamide, 1,3,4-oxadiazole and formamyl moieties via an isosteric modification (compounds 10a2, 10a3 and 10a4) resulted in loss of activity to varying degrees. By adding or reducing the carbon chains, a decrease in antibacterial activity was observed, possibly because of the increase in rotatable bonds hampers antibacterial activity. Overall, the SAR for the amide bond revealed that there was limited scope for improving antibacterial activity (Table 6).
|
||||
|---|---|---|---|---|
| Compound no. | % inhibition 20 μg mL−1 | MIC (μg mL−1) | ClogP | |
| 10a1 |  |
65.7% | NA | 2.91 |
| 10a2 | 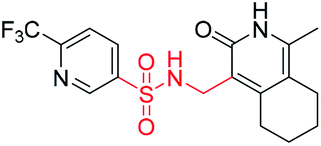 |
92.7% | 3.5 | 2.67 |
| 10a3 |  |
NA | NA | 3.5 |
| 10a4 |  |
36.8% | NA | 3.69 |
| 10a5 |  |
99.3% | 20 | 2.96 |
| 10a6 |  |
NA | >50 | 2.32 |
| Isoniazid | 99.4% | 0.25 | −0.67 | |

Among these compounds, compounds 6b6, 6b12, 6b21 showed potential anti-mycobacterial activity against the tested MDR–MTB strains with MIC ranging 0.9 to 3.0 μg mL−1(Table 7).



2.3 Cytotoxicity assay
The cytotoxicity of compounds with the highest anti-tuberculosis activity was tested to eliminate the possibility that the anti-tuberculosis activity of the synthesized compounds arises from general toxicity. Human lung adenocarcinoma A549 cells and Vero cells from the African green monkey kidney were used to measure cytotoxicity and growth inhibition. Cell activity was measured by the MTT assay. The results showed that these compounds had no in vitro cytotoxicity (Table 8).2.4 Antibacterial in vitro assay
The potential broad-spectrum antibacterial activity of compound 6b21 was tested against Escherichia coli (E. coli) Esbls, Klebsiella pneumoniae Esbls, Pseudomonas aeruginosa, Enterobacter cloacae and E. coli ATCC25922. Levofloxacin was used as the control. The MIC value of compound 6b21 toward other bacteria was >128 μg mL−1, indicating that compound 6b21 is specific toward M. tuberculosis (Table 9).| Bacterial strainsa | MIC (μg mL−1) | Bacteria control+/− | |
|---|---|---|---|
| Compound 6b21 | Levofloxacin tablets | ||
| a Escherichia coli ATCC25922 was the standard strain, and the rest were clinical bacteria. | |||
| Escherichia coli Esbls−16-23 | >128 | 16 | + |
| Escherichia coli Esbls−16-24 | >128 | <0.008 | + |
| Klebsiella pneumoniae Esbls−17-11 | >128 | 0.03 | + |
| Klebsiella pneumoniae Esbls−17-12 | >128 | <0.008 | + |
| Pseudomonas aeruginosa 17-1 | >128 | 0.25 | + |
| Pseudomonas aeruginosa 17-2 | >128 | 16 | + |
| Enterobacter cloacae 17-11 | >128 | <0.008 | + |
| Enterobacter cloacae 17-12 | >128 | 64 | + |
| Escherichia coli ATCC25922 | >128 | <0.008 | + |
3. Conclusion
There have been many articles and patent reports describing the anti-tuberculosis activity of quinolone derivatives. Initially, SAR analysis of compound 1 analogues that had substitutions on the phenyl side chains revealed that the 3,5-dimethoxy substitution (i.e., compound 6b6) gave the lowest MIC of 3 μg mL−1 against M. tuberculosis, which is seven-fold lower than the MIC reported for compound 1. With reference to the active structure of other quinolone compounds, a simple side chain modification to obtain compound 6b21 reduced the MIC to 1.2 μg mL−1, with an MIC of 0.9 μg mL−1 against MDR–MTB. The isomer of the 6b analogue (6d1–6d14) resulted in a decrease in activity. No activity was observed when the cyclohexane group was replaced with a benzene ring (6d10–6d14). Compounds 6a1–6a2 and 6c1 where the cyclohexane group were expanded or reduced did not show an improvement in anti-tuberculosis activity. We also carried out a preliminary SAR of the linker by using the principles of bioisosterism and homologous conversion. Here, activity was highest when the amide bond was used, and any extension or shortening of the junction structure resulted in a significant decrease in anti-tuberculosis activity (10a1–10a6). The activity was not maintained when the doublet ring was replaced or when the ring was opened. Finally, the optimal compound 6b21 was not cytotoxic and displayed specific anti-tuberculosis activity.4. Experimental section
4.1 Experimental
Synthesis of intermediates 4b and 4d. To a solution of cyclohexanone (29.50 g), morpholine (31.45 g), toluene and p-toluenesulfonic acid (0.53 g) were added, and the reaction stirred at 120 °C. The water generated by the reaction and distilled by Dean–Stark apparatus. The reaction was stopped when 6.00 mL of water was distilled. The reaction system was gradually cooled to room temperature, and a dark yellow viscous liquid was obtained by vacuum distillation to remove the solvent. After dissolving the liquid in dichloromethane, 50.00 mL triethylamine was added to the mixture at 0 °C and the mixture stirred at 0 °C for 5 min. Then, 24.00 mL acetyl chloride diluted with 100 mL dichloromethane was dropwise added to the reaction with a large amount of white smoke generated. The reaction was then stirred at 0 °C for 30 min before stirring at room temperature for 10 h. After completion of the enamine reaction, as determined by TLC analysis, 47 mL HCl (concentrated hydrochloric acid/water 1/1) was dropwise added to the reaction at 0 °C with the reaction solution gradually becoming black and cloudy.
Acetyl cyclohexanone (31.23 g) and cyanoacetamide (20.00 g) were dissolved in anhydrous ethanol (50 mL), and triethylenediamine (41.27 g) was added to the mixture. After the addition of triethylenediamine, the reaction was stirred at 40 °C for 10 h with a large amount of white precipitant formed. In TLC analysis, a ferric chloride color indicated that the acetylcyclohexanone reaction was completed. This was immediately followed by filtration and the filter precipitant was washed with anhydrous ethanol (20 mL). After separating the compounds by column chromatography, compounds 4b and 4d were obtained as yellow solids (38.21 g; yield: 75%) and 4d (10.32 g; yield: 20%). Compounds 4a and 4c were prepared by the same procedure described above.
4b. 1H NMR (400 MHz, DMSO-d6) δ 2.73 (d, J = 3.2 Hz, 2H), 2.37 (d, J = 6.0 Hz, 2H), 2.21 (s, 3H), 1.68 (m, J = 3.1 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 160.37, 160.21, 150.39, 116.61, 113.41, 98.34, 29.42, 23.80, 22.18, 21.46, 17.31; ESI-MS m/z 211.1 [M + Na]+.
4d. 1H NMR (400 MHz, DMSO-d6) δ 12.06 (s, 1H), 2.53 (d, J = 5.2 Hz, 2H), 2.40–2.32 (m, 2H), 2.27 (s, 3H), 1.80–1.54 (m, 4H); 13C NMR (101 MHz, DMSO-d6) 160.45, 160.19, 149.07, 116.89, 113.54, 100.71, 27.49, 23.65, 22.11, 21.05, 18.86; ESI-MS m/z 211.1 [M + Na]+.
Synthesis of intermediates 5b and 5d. Compound 4b (38.21 g) or 4d (10.32 g) was mixed with RANEY®-nickel (35.74 g), ethanol and ammonia solution (50.00 mL). The reaction was stirred under 11
427 torr H2 at 40 °C for 10 h. Completion of the reaction was detected by TLC. The reaction was gradually cooled to room temperature and the RANEY®-nickel material was carefully filtered. The pressure was reduced to remove solvent, yielding a dark green oily substance. Compound 5b or 5d (30.46 mg) was obtained by column chromatography as a white solid. ESI-MS m/z 215.1 [M + Na]+.
Synthesis of intermediate 9a. 2-Aminoacetophenone (5 g, 36.99 mmol), ethyl cyanoacetate (4.18 g, 36.96 mmol) and ammonium acetate (2.85, 36.99 mmol) were added into a round-bottom flask. The mixture was stirred at 90 °C for 2 h. The solvent was removed by rotary evaporation and the solid material dissolved in CH2Cl2 and MeOH. The residue was purified by silica gel column chromatography to yield compound 9a. ESI-MS m/z 207.1 [M + Na]+.
Compound 9a was added to a solution of ammonium hydroxide and RANEY®-nickel in MeOH. The mixture was stirred under 11427 torr H2 at 80 °C for 36 h. After cooling, the reaction was filtered to remove white insoluble material. The filtrate was concentrated by rotary evaporation and the reduced product (9a) was obtained by silica gel column chromatography. ESI-MS m/z 211.1 [M + Na]+.
Synthesis of intermediates 7a, 7b and 7c. One equivalent of the corresponding ketone (1-cyclopropyl-1,3-butanedione, 1-phenyl-1,3-butanedione, 1,1,1-trifluoro-2,4-pentanedione) and 1 eq. cyanoacetamide were added to a 25 mL solution of aqueous ethanol (ethanol
:H2O = 4:1) with stirring. One equivalent of triethylenediamine was then added and the reaction was gradually heated to 40 °C for 10 h. A significant amount of white solid was formed, which was filtered and washed with small portions of cold ethanol to remove any remaining residues.
7a yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 12.40 (s, 1H), 5.94 (s, 1H), 2.27 (s, 3H), 1.87 (tt, J = 8.4, 5.0 Hz, 1H), 1.13–1.08 (m, 2H), 0.96–0.91 (m, 2H).
7b yield: 62%. 1H NMR (400 MHz, DMSO-d6) δ 12.56 (s, 1H), 7.84–7.74 (m, 2H), 7.58–7.50 (m, 3H), 6.75 (s, 1H), 2.42 (s, 3H).
7c yield: 82%. 1H NMR (400 MHz, DMSO-d6) δ 13.34 (s, 1H), 6.65 (s, 1H), 2.38 (s, 3H).
Synthesis of intermediate 8a. The corresponding ketone (0.1 mol of either acetylacetone or 3,5-heptanedione) and cyanoacetamide (0.12 mol) were dissolved in 50 mL ethanol/H2O (at a ratio of 4
:1). Potassium carbonate (0.1 mol) was added. The system was refluxed for 3 h and a significant amount of white solid was formed during the reflux. The solid was filtered and washed with petroleum ether. A white solid was produced after drying. The remaining intermediate 3-cyano-4,6-dimethyl-2-hydroxypyridine were acquired from the Bidepharm.8a, n = 0, n = 1 ESI-MS m/z 163.1 [M + H]+.
8a, n = 1, n = 0 ESI-MS m/z 163.1 [M + H]+.
8a, n = 1, n = 1 ESI-MS m/z 177.1 [M + H]+.
Hydrolysis of intermediate 4b. Compound 4b was added to a 25 mL aqueous ethanol (ethanol
:H2O = 5:1) solution saturated with potassium hydroxide and placed in a sealed tube. Under nitrogen protection, the system was sealed and stirred at 120 °C for 24 h. The reaction mixture was cooled to ambient temperature. The reaction solution was poured into a beaker and 5 mL H2O was added. Concentrated HCl was slowly added under agitation and the pH was monitored. A large amount of white precipitant appeared when the pH reached 8–9. The reaction was terminated and the precipitant discarded. Dilute HCl (4 M) was then added dropwise to the filtrate until the pH reached 3. A large amount of precipitant formed, which was washed with excess (three times) H2O to afford a grey solid. Yield: 82%. 1H NMR (400 MHz, DMSO-d6) δ 16.06 (s, 1H), 13.09 (s, 1H), 3.20 (t, J = 5.9 Hz, 2H), 2.46 (d, J = 6.2 Hz, 2H), 2.30 (s, 3H), 1.74–1.55 (m, 4H).
General procedures for the syntheses of compounds 6b1–6b36, 6d1–6d14, 9a1–9a4, 6a1–6a2, 6c1, 7a1–7a2, 7b1–7b2, 7c1 and 8a1–8a5. The intermediates, carboxylic acid analogues, or benzoic acid derivative (190.12 mg), HOBT (202.68 mg) and EDCI (287.55 mg) were weighed and dissolved in 20 mL dichloromethane or DMF. One milliliter N-methyl morpholine was added to the mixture and the reaction system changed to a pale yellow clarified solution. After stirring at room temperature for 30 min, the intermediate 5b or amino analogue (211.34 mg) was dissolved in 10 mL dichloromethane and dropwise added to the reaction. The reaction turned yellow and cloudy, with stirring continued at room temperature for 7 h. The reaction was monitored by TLC. The completed reaction was filtered and the filtrate washed with H2O (20 mL × 1). The aqueous phase was extracted with dichloromethane (10 mL × 2), combined with the organic phase, washed with dilute HCl (1 M; 15 mL × 1), washed with saturated NaCl (15 mL × 1) and dried overnight with anhydrous sodium sulfate. After filtration, the pressure was reduced to remove the solvent, and column chromatography (Vdichloromethane/Vmethanol = 50/1) was used to separate and obtain the target product as a solid powder (6b1, 326.73 mg) with a yield of 80–90%.
4′-Fluoro-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (6b1). Yield 81% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.48 (s, 1H), 8.35 (t, J = 4.8 Hz, 1H), 7.90 (d, J = 8.3 Hz, 2H), 7.69 (d, J = 8.1 Hz, 2H),7.73 (dd, J = 7.6, 1.2 Hz, 2H), 7.70–7.64 (m, 2H), 4.30 (d, J = 4.9 Hz, 2H), 2.71 (s, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.64 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 413.2, found 413.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4'-(trifluoromethoxy)-[1,1′-biphenyl]-4-carboxamide (6b2). Yield 79% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.49 (t, J = 4.7 Hz, 1H), 8.11 (d, J = 1.8 Hz, 2H), 7.91–7.75 (m, 4H), 7.47 (d, J = 8.3 Hz, 2H), 4.36 (d, J = 4.6 Hz, 2H), 2.70 (s, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.63 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 479.2, found 479.1.
4'-(Dimethylamino)-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (6b3). Yield 89% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.37 (t, J = 4.8 Hz, 1H), 7.90 (d, J = 8.3 Hz, 2H), 7.69 (d, J = 8.1 Hz, 2H), 7.27 (t, J = 8.1 Hz, 1H), 7.00–6.91 (m, 2H), 6.76 (dd, J = 8.4, 2.4 Hz, 1H), 4.36 (d, J = 4.7 Hz, 2H), 2.96 (s, 6H), 2.72 (s, 2H), 2.38 (s, 2H), 2.12 (s, 3H), 1.63 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 438.2, found 438.2.
4′-Methoxy-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (6b4). Yield 82% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.46 (s, 1H), 8.77 (d, J = 5.5 Hz, 1H), 7.92 (d, J = 8.5 Hz, 2H), 7.69 (dd, J = 11.2, 8.6 Hz, 4H), 7.04 (d, J = 8.9 Hz, 2H), 4.40 (d, J = 4.8 Hz, 2H), 3.80 (s, 3H), 2.82 (s, 2H), 2.44 (s, 2H), 2.21 (s, 3H), 1.67 (t, J = 3.5 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 425.1, found 425.1.
3′-Methoxy-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (6b5). Yield 80% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 12.43 (s, 1H), 8.44 (d, J = 5.5 Hz, 1H), 7.95 (d, J = 8.5 Hz, 2H), 7.74 (dd, J = 11.2, 8.6 Hz, 2H), 7.44 (d, J = 8.9 Hz, 1H), 7.29 (dd, 1H), 7.13 (m, 2H), 4.40 (d, J = 4.8 Hz, 2H), 3.82 (s, 3H), 2.80 (s, 2H), 2.42 (s, 2H), 2.25 (s, 3H), 1.64 (t, J = 3.5 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 425.2, found 425.1.
3′,5′-Dimethoxy-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (6b6). Yield 84% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.40 (t, J = 4.8 Hz, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H), 6.82 (d, J = 2.2 Hz, 2H), 6.53 (t, J = 2.2 Hz, 1H), 4.35 (d, J = 4.8 Hz, 2H), 3.81 (s, 6H), 2.71 (s, 2H), 2.38 (s, 2H), 2.12 (s, 3H), 1.63 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 455.2, found 455.2; HPLC purity at 254 nm, 97.1%.
3′,4′-Dimethoxy-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (6b7). Yield 82% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.56 (s, 1H), 8.08 (d, J = 4.5 Hz, 1H), 7.87 (m, J = 4.4 Hz, 3H),7.73 (s, J = 4.4 Hz, 1H),7.63 (m, J = 4.4 Hz, 3H), 4.32 (d, J = 4.7 Hz, 2H), 3.62 (s, 3H), 3.54 (s, 3H), 2.67 (s, 2H), 2.37 (d, J = 6.2 Hz, 2H), 2.12 (s, 3H), 1.63 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 455.2, found 455.2.
3′-Methoxy-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5'-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxamide (6b8). Yield 92% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.45 (t, J = 4.8 Hz, 1H), 7.95 (d, J = 8.1 Hz, 2H), 7.83 (d, J = 8.1 Hz, 2H), 7.60–7.52 (m, 2H), 7.27 (s, 1H), 4.36 (d, J = 4.8 Hz, 2H), 3.92 (s, 3H), 2.72 (s, 2H), 2.39 (s, 2H), 2.12 (s, 3H), 1.64 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 493.2, found 493.1.
3′-Methoxy-5′-methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (6b9). Yield 92% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.38 (t, J = 4.8 Hz, 1H), 7.91 (d, J = 8.1 Hz, 2H), 7.71 (d, J = 8.3 Hz, 2H), 7.10 (s, 1H), 7.02 (t, J = 1.9 Hz, 1H), 6.79 (s, 1H), 4.35 (d, J = 4.7 Hz, 2H), 3.31 (s, 1H), 2.71 (s, 2H), 2.37 (d, J = 13.5 Hz, 5H), 2.12 (s, 3H), 1.63 (t, J = 3.5 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 439.2, found 439.2; HPLC purity at 254 nm, 95.7%.
4-(3,5-Dimethoxybenzyl)-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide (6b10). Yield 89% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.40 (t, J = 4.8 Hz, 1H), 7.84 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H), 6.82 (d, J = 2.2 Hz, 2H), 6.53 (t, J = 2.2 Hz, 1H), 4.35 (d, J = 4.8 Hz, 2H), 4.07 (s, 2H), 3.81 (s, 6H), 2.71 (s, 2H), 2.38 (s, 2H), 2.12 (s, 3H), 1.63 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 469.2, found 469.2.
4-(3,5-Dimethoxyphenoxy)-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide (6b11). Yield 75% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.48 (s, 1H), 8.26 (t, J = 4.8 Hz, 1H), 7.91–7.82 (m, 2H), 7.06–6.97 (m, 2H), 6.34 (t, J = 2.2 Hz, 1H), 6.20 (d, J = 2.2 Hz, 2H), 4.32 (d, J = 4.8 Hz, 2H), 3.71 (s, 6H), 2.70 (s, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.63 (t, J = 3.4 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 471.1, found 471.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4-(trifluoromethyl) benzamide (6b12). Yield 90% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H), 8.58 (t, J = 4.8 Hz, 1H), 8.02 (d, J = 8.1 Hz, 2H), 7.80 (d, J = 8.2 Hz, 2H), 4.34 (d, J = 4.7 Hz, 2H), 2.70 (s, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.63 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 387.1, found 387.1; HPLC purity at 254 nm, 100%.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-2-(trifluoromethyl) benzamide (6b13). Yield 84% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 8.07 (dd, J = 5.5, 3.6 Hz, 1H), 7.95–7.89 (m, 1H), 7.81–7.73 (m, 3H), 4.32 (d, J = 5.3 Hz, 2H), 2.61–2.54 (m, 2H), 2.30 (d, J = 6.0 Hz, 2H), 2.05 (s, 3H), 1.63–1.52 (m, 4H). ESI-MS: m/z [M + Na]+ calculated for 387.1, found 387.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-3-(trifluoromethyl) benzamide (6b14). Yield 89% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.66 (s, 1H), 8.20 (s, 1H), 8.15 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 4.35 (d, J = 4.4 Hz, 2H), 2.70 (s, 2H), 2.39 (s, 2H), 2.12 (s, 3H), 1.63 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 387.1, found 387.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4-(trifluoromethoxy) benzamide (6b15). Yield 92% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H), 8.02 (t, J = 4.9 Hz, 1H), 7.64 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 3.51 (s, 2H), 2.54 (t, J = 6.3 Hz, 2H), 2.35 (t, J = 6.3 Hz, 2H), 2.10 (s, 3H), 1.64–1.49 (m, 4H). ESI-MS: m/z [M + Na]+ calculated for 403.1, found 403.1.
4-Methoxy-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide (6b16). Yield 94% (white solid). 1H NMR (400 MHz, chloroform-d) δ 7.16–7.11 (m, 2H), 6.83–6.78 (m, 2H), 4.34 (d, J = 5.9 Hz, 2H), 3.72 (s, 3H), 2.89–2.82 (m, 2H), 2.44 (d, J = 5.9 Hz, 2H), 2.13 (s, 3H), 1.72 (t, J = 3.4 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 349.1, found 349.1.
4-Fluoro-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)benzamide (6b17). Yield 95% (white solid). 1H NMR (400 MHz, chloroform-d) δ 12.10 (s, 1H), 8.29 (s, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.4 Hz, 2H), 4.59 (d, J = 5.6 Hz, 2H), 2.92 (s, 2H), 2.46 (s, 2H), 2.24 (s, 3H), 1.65 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 337.1, found 337.1.
4-(Dimethylamino)-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl) benzamide (6b18). Yield 90% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.48 (s, 1H), 7.94 (t, J = 4.9 Hz, 1H), 7.68 (d, J = 8.6 Hz, 2H), 6.66 (d, J = 8.7 Hz, 2H), 4.29 (d, J = 4.8 Hz, 2H), 2.95 (s, 6H), 2.70 (s, 2H), 2.37 (s, 2H), 2.10 (s, 3H), 1.62 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 362.2, found 362.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4-nitrobenzamide (6b19). Yield 92% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.53 (s, 1H), 8.72 (t, J = 4.7 Hz, 1H), 8.28 (d, J = 2.0 Hz, 2H), 7.81 (d, J = 8.4 Hz, 2H), 4.34 (d, J = 4.7 Hz, 2H), 2.68 (s, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.63 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 364.1, found 364.1.
2-Methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4-(trifluoromethyl)benzamide (6b20). Yield 83% (white solid). 1H NMR (400 MHz, chloroform-d) δ 7.75 (dd, J = 1.5, 0.8 Hz, 1H), 7.50 (d, J = 1.7 Hz, 2H), 4.50 (d, J = 5.7 Hz, 2H), 2.86 (t, J = 6.0 Hz, 2H), 2.41 (s, 3H),2.37 (t, J = 5.9 Hz, 2H), 2.09 (s, 3H), 1.68 (d, J = 4.8 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 401.1, found 401.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-6-(trifluoromethyl) nicotinamide (6b21). Yield 92% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.52 (s, 1H), 8.82–8.76 (m, 1H), 8.52 (t, J = 5.3 Hz, 1H), 8.26 (dd, J = 8.1, 1.4 Hz, 1H), 7.72–7.66 (m, 1H), 4.33 (d, J = 5.3 Hz, 2H), 2.73 (s, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.63 (t, J = 3.5 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 388.1, found 388.1; HPLC purity at 254 nm, 99.4%.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(trifluoromethyl) picolinamide (6b22). Yield 81% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.52 (s, 1H), 8.82–8.76 (m, 1H), 8.52 (t, J = 5.3 Hz, 1H), 8.26 (dd, J = 8.1, 1.4 Hz, 1H), 7.72–7.66 (m, 1H), 4.33 (d, J = 5.3 Hz, 2H), 2.73 (s, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.63 (t, J = 3.5 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 388.1, found 388.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-2-(trifluoromethyl) pyrimidine-5-carboxamide (6b23). Yield 78% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 12.67 (s, 1H), 9.03 (m, 1H), 8.83 (t, J = 4.6 Hz, 1H), 8.68 (dd, J = 6.8, 1.4 Hz, 1H), 4.43 (d, J = 5.4 Hz, 2H), 2.89 (s, 2H), 2.55 (s, 2H), 2.12 (s, 3H), 1.54 (t, J = 3.6 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 389.1, found 389.1.
6-Chloro-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)nicotinamide (6b24). Yield 86% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.42 (s, 1H), 8.67 (s, 1H), 8.58 (s, 1H), 8.38 (s, 1H), 7.24 (d, J = 8.8 Hz, 1H), 4.33 (s, 2H), 2.70 (s, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.63 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.94, 162.03, 150.93, 147.98, 147.82, 141.11, 129.15, 120.81, 112.16, 109.80, 109.43, 35.57, 26.99, 24.60, 22.56, 22.37, 16.43. ESI-MS: m/z [M + Na]+ calculated for 354.1, found 354.1; HPLC purity at 254 nm, 99.6%.
6-Bromo-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)nicotinamide (6b25). Yield 76% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.77 (d, J = 2.5 Hz, 1H), 8.62 (t, J = 4.8 Hz, 1H), 8.11 (dd, J = 8.3, 2.6 Hz, 1H), 7.73 (d, J = 8.3 Hz, 1H), 4.32 (d, J = 4.8 Hz, 2H), 2.68 (d, J = 5.7 Hz, 2H), 2.37 (d, J = 6.1 Hz, 2H), 2.11 (s, 3H), 1.63 (t, J = 3.4 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 164.11, 161.97, 150.91, 149.96, 144.12, 141.13, 138.80, 130.06, 128.15, 120.73, 112.10, 35.61, 27.00, 24.60, 22.57, 22.37, 16.44. ESI-MS: m/z [M + Na]+ calculated for 398.1, found 398.1; HPLC purity at 254 nm, 97.8%.
6-Fluoro-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)nicotinamide (6b26). Yield 75% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H), 8.80 (d, J = 2.4 Hz, 1H), 8.61 (t, J = 4.7 Hz, 1H), 8.23 (dd, J = 8.3, 2.5 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 4.33 (d, J = 4.7 Hz, 2H), 2.69 (s, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.63 (t, J = 3.5 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 164.00, 161.98, 152.73, 150.90, 149.55, 141.12, 139.16, 129.80, 124.37, 120.75, 112.09, 35.61, 27.00, 24.60, 22.57, 22.37, 16.44. ESI-MS: m/z [M + Na]+ calculated for 338.1, found 338.1; HPLC purity at 254 nm, 99.8%.
6-Methyl-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)nicotinamide (6b27). Yield 85% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H), 8.85 (d, J = 2.3 Hz, 1H), 8.45 (t, J = 4.8 Hz, 1H), 8.06 (dd, J = 8.1, 2.4 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 4.33 (d, J = 4.8 Hz, 2H), 2.70 (d, J = 6.0 Hz, 2H), 2.37 (d, J = 6.1 Hz, 2H), 2.11 (s, 3H), 1.63 (t, J = 3.4 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.08, 162.04, 160.93, 150.76, 148.45, 140.99, 135.71, 127.61, 122.96, 120.99, 112.11, 35.50, 27.00, 24.61, 24.44, 22.58, 22.38, 16.43. ESI-MS: m/z [M + Na]+ calculated for 334.1, found 334.1; HPLC purity at 230 nm, 100%.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-6-nitronicotinamide (6b28). Yield 85% (white solid). 1H NMR (400 MHz, chloroform-d) δ 8.78 (d, J = 2.5 Hz, 1H), 8.34 (t, J = 5.6 Hz, 1H), 8.10 (dd, J = 8.3, 2.4 Hz, 1H), 7.36 (d, J = 8.3 Hz, 1H), 4.57 (d, J = 5.4 Hz, 2H), 2.90 (d, J = 8.7 Hz, 2H), 2.44 (d, J = 3.3 Hz, 2H), 2.24 (s, 3H), 1.75 (d, J = 5.8 Hz, 5H). 13C NMR (101 MHz, DMSO-d6) δ 164.00, 161.98, 152.73, 150.90, 149.55, 141.12, 139.16, 129.80, 124.37, 120.75, 112.08, 35.61, 27.00, 24.60, 22.57, 22.37, 16.44. ESI-MS: m/z [M + Na]+ calculated for 365.1, found 365.1; HPLC purity at 230 nm, 99.4%.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-nitrofuran-2-carboxamide (6b29). Yield 75% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.56 (s, 1H), 8.74 (t, J = 5.0 Hz, 1H), 7.71 (d, J = 3.9 Hz, 1H), 7.45 (d, J = 3.9 Hz, 1H), 4.32 (d, J = 5.0 Hz, 2H), 2.68 (d, J = 5.2 Hz, 2H), 2.38 (s, 2H), 2.11 (s, 3H), 1.70–1.58 (m, 4H). ESI-MS: m/z [M + Na]+ calculated for 354.1, found 354.1; HPLC purity at 230 nm, 99.4%.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-4-nitrofuran-2-carboxamide (6b30). Yield 75% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 12.37 (s, 1H), 8.98 (t, J = 5.4 Hz, 1H), 7.83 (d, J = 3.6 Hz, 1H), 7.65 (d, J = 3.6 Hz, 1H), 4.68 (d, J = 5.4 Hz, 2H), 2.76 (d, J = 5.2 Hz, 2H), 2.68 (s, 2H), 2.33 (s, 3H), 1.89–1.75 (m, 4H). ESI-MS: m/z [M + Na]+ calculated for 354.1, found 354.1.
5-Bromo-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)furan-2-carboxamide (6b31). Yield 79% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.55 (s, 1H), 8.23 (t, J = 5.1 Hz, 1H), 7.16 (d, J = 3.5 Hz, 1H), 6.71 (d, J = 3.5 Hz, 1H), 4.27 (d, J = 5.1 Hz, 2H), 2.74–2.62 (m, 2H), 2.36 (d, J = 6.0 Hz, 2H), 2.11 (s, 3H), 1.62 (p, J = 3.2 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.06, 156.86, 150.47, 150.11, 141.07, 124.63, 120.87, 116.09, 114.28, 112.18, 34.83, 26.97, 24.60, 22.55, 22.35, 16.43. ESI-MS: m/z [M + Na]+ calculated for 387.0, found 387.0; HPLC purity at 230 nm, 96.9%.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-nitrothiophene-2-carboxamide (6b32). Yield 81% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.15–8.01 (m, 1H), 7.86 (d, J = 4.5 Hz, 1H), 4.32 (s, 2H), 2.67 (s, 3H), 2.38 (s, 2H), 2.11 (s, 3H), 1.63 (d, J = 3.5 Hz, 5H). ESI-MS: m/z [M + Na]+ calculated for 370.1, found 370.1; HPLC purity at 254 nm, 99.4%.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-5-(trifluoromethyl)furan-2-carboxamide (6b33). Yield 80% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 12.24 (s, 1H), 8.04 (t, J = 4.6 Hz, 1H), 7.32 (d, J = 3.2 Hz, 1H), 6.40 (d, J = 3.2 Hz, 1H), 4.26 (d, J = 5.6 Hz, 2H), 2.74 (m, 2H), 2.34 (d, J = 6.0 Hz, 2H), 2.21 (s, 3H), 1.52 (p, J = 3.4 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 377.1, found 377.1; HPLC purity at 230 nm, 96.1%.
5-(Dimethylamino)-N-((1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)furan-2-carboxamide (6b34). Yield 85% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 7.23 (t, J = 4.5 Hz, 1H), 7.12 (d, J = 4.4 Hz, 1H), 7.04 (d, J = 3.4 Hz, 1H), 3.45 (d, J = 5.8 Hz, 2H), 2.84 (m, 2H), 2.76 (d, 2H), 2.26 (s, 3H), 1.53 (p, J = 3.6 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 352.2, found 352.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-1H-pyrazole-4-carboxamide (6b35). Yield 84% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.13 (s, 1H), 7.81 (s, 1H), 4.25 (s, 2H), 2.64 (s, 2H), 2.37 (s, 2H), 2.10 (s, 3H), 1.61 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 286.1, found 286.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)isoxazole-5-carboxamide (6b36). Yield 85% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.56 (s, 1H), 9.04 (d, J = 1.6 Hz, 1H), 8.52 (s, 1H), 6.89 (d, J = 1.6 Hz, 1H), 4.33 (d, J = 5.2 Hz, 2H), 2.72 (s, 2H), 2.37 (s, 2H), 2.11 (s, 3H), 1.63 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.08, 161.82, 158.44, 158.28, 150.22, 141.19, 120.59, 112.22, 104.57, 35.31, 27.02, 24.61, 22.53, 22.35, 16.44. ESI-MS: m/z [M + Na]+ calculated for 310.1, found 310.1.
N-((4-Methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)-6-(trifluoromethyl) nicotinamide (6d1). Yield 79% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.31 (s, 1H), 9.12 (d, J = 2.1 Hz, 1H), 8.81 (t, J = 4.8 Hz, 1H), 8.45 (dd, J = 8.2, 2.1 Hz, 1H), 7.99 (d, J = 8.2 Hz, 1H), 4.41 (d, J = 4.8 Hz, 2H), 2.46 (s, 2H), 2.35 (s, 2H), 2.11 (d, J = 4.6 Hz, 3H), 1.65 (dd, J = 9.7, 4.0 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.87, 162.13, 150.54, 149.54, 137.92, 133.49, 122.40, 120.91, 112.56, 36.14, 26.99, 24.30, 22.79, 21.55, 15.81. ESI-MS: m/z [M + Na]+ calculated for 388.1243, found 388.1246.
N-((4-Methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)thiophene-2-carboxamide (6d2). Yield 80% (yellow solid). 1H NMR (400 MHz, DMSO-d6) δ 11.27 (s, 1H), 8.34 (t, J = 5.1 Hz, 1H), 7.79 (dd, J = 3.8, 1.2 Hz, 1H), 7.70 (dd, J = 5.0, 1.1 Hz, 1H), 7.09 (dd, J = 5.0, 3.7 Hz, 1H), 4.34 (d, J = 4.9 Hz, 2H), 2.44 (d, J = 5.3 Hz, 2H), 2.37–2.31 (m, 2H), 2.08 (s, 3H), 1.64 (dd, J = 11.4, 4.7 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.23, 161.34, 150.44, 140.98, 140.49, 130.96, 128.50, 128.29, 122.74, 112.74, 35.79, 26.97, 24.29, 22.78, 21.54, 15.77. ESI-MS: m/z [M + Na]+ calculated for 325.0981, found 325.0983.
6-Methyl-N-((4-methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)nicotinamide (6d3). Yield 79% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.31 (s, 1H), 8.86 (d, J = 2.3 Hz, 1H), 8.47 (t, J = 4.8 Hz, 1H), 8.07 (dt, J = 8.1, 2.5 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 4.38 (d, J = 4.8 Hz, 2H), 2.45 (d, J = 4.9 Hz, 2H), 2.34 (d, J = 5.3 Hz, 2H), 2.09 (s, 3H), 1.65 (dq, J = 10.4, 5.0, 4.2 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.04, 162.20, 160.96, 150.34, 148.47, 140.92, 135.70, 127.58, 122.94, 122.74, 112.58, 35.95, 26.98, 24.37 (d, J = 15.5 Hz), 22.80, 21.56, 15.76. ESI-MS: m/z [M + Na]+ calculated for 334.1526, found 334.1541.
3′,5′-Dimethoxy-N-((4-methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (6d4). Yield 71% (orange solid). 1H NMR (400 MHz, DMSO-d6) δ 11.35 (s, 1H), 8.40 (t, J = 4.7 Hz, 1H), 7.92 (dq, J = 8.4, 1.8 Hz, 2H), 7.76–7.69 (m, 2H), 6.83 (d, J = 2.2 Hz, 2H), 6.54 (t, J = 2.2 Hz, 1H), 4.41 (d, J = 4.8 Hz, 2H), 3.82 (s, 6H), 2.46 (d, J = 5.2 Hz, 2H), 2.33 (d, J = 5.8 Hz, 2H), 2.11 (s, 3H), 1.71–1.59 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 166.02, 162.28, 161.37, 150.19, 142.90, 141.87, 140.83, 133.96, 128.29, 127.00, 122.95, 112.64, 105.43, 100.36, 55.79, 36.10, 26.98, 24.31, 22.81, 21.56, 15.75. ESI-MS: m/z [M + Na]+ calculated for 455.1941, found 455.1948.
N-((4-Methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)-5-nitrothiophene-2-carboxamide (6d5). Yield 79% (yellow solid). 1H NMR (400 MHz, DMSO-d6) δ 11.32 (s, 1H), 8.90 (dt, J = 10.8, 4.8 Hz, 1H), 8.08 (d, J = 4.4 Hz, 1H), 7.87 (dd, J = 4.4, 1.8 Hz, 1H), 4.39–4.30 (m, 2H), 2.45 (d, J = 5.2 Hz, 2H), 2.40–2.31 (m, 2H), 2.08 (s, 3H), 1.69–1.60 (m, 4H). ESI-MS: m/z [M + Na]+ calculated for 370.0832, found 370.0835; HPLC purity at 254 nm, 98.3%.
3,5-Dimethoxy-N-((4-methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)benzamide (6d6). Yield 94% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.28 (s, 1H), 8.32 (d, J = 4.6 Hz, 1H), 7.00 (dd, J = 5.2, 2.3 Hz, 2H), 6.60 (q, J = 1.9 Hz, 1H), 4.33 (dd, J = 18.6, 4.7 Hz, 2H), 3.76 (s, 6H), 2.45 (s, 2H), 2.34 (s, 2H), 2.08 (s, 3H), 1.73–1.55 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.90, 160.68, 150.23, 137.00, 105.62, 103.63, 55.87, 36.10, 26.97, 22.81, 21.56, 15.75. ESI-MS: m/z [M + Na]+ calculated for 379.1628, found 379.1628.
2-Chloro-N-((4-methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)-6-(trifluoromethyl)nicotinamide (6d7). Yield 70% (yellow solid). 1H NMR (400 MHz, DMSO-d6) δ 11.29 (s, 1H), 8.72–8.61 (m, 1H), 8.11 (d, J = 7.8 Hz, 1H), 7.97 (d, J = 7.8 Hz, 1H), 4.38 (d, J = 5.0 Hz, 2H), 2.45 (s, 2H), 2.38–2.30 (m, 2H), 2.14 (s, 3H), 1.71–1.62 (m, 4H). ESI-MS: m/z [M + Na]+ calculated for 422.0854, found 422.0853.
N-((4-Methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)-5-phenylfuran-2-carboxamide (6d8). Yield 77% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 8.94–8.83 (m, 2H), 8.50 (tt, J = 7.8, 1.7 Hz, 1H), 8.09 (s, 3H), 8.02–7.94 (m, 2H), 3.85 (d, J = 5.7 Hz, 2H), 2.49 (s, 2H), 2.37 (d, J = 5.4 Hz, 2H), 2.13 (s, 3H), 1.69–1.66 (m, 2H), 1.65–1.61 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 161.84, 151.90, 145.76, 142.72 (d, J = 9.1 Hz), 127.48, 118.74, 117.01, 113.07, 112.80, 35.57, 27.01 (d, J = 11.8 Hz), 24.56, 24.35, 22.66, 22.38, 22.16, 21.38, 16.56, 15.98. ESI-MS: m/z [M + Na]+ calculated for 385.1523, found 385.2598.
N-((4-Methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)-4-(6-(trifluoromethyl) pyridin-3-yl)benzamide (6d9). Yield 70% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.28 (s, 1H), 9.14 (s, 1H), 8.52–8.37 (m, 2H), 8.04–7.86 (m, 5H), 4.41 (s, 2H), 2.35 (s, 4H), 2.11 (s, 3H), 1.66 (s, 4H). 13C NMR (101 MHz, DMSO-d6) δ 165.81, 162.25, 150.30, 148.83, 140.90, 138.34, 136.78, 135.19, 128.68, 127.63, 121.34, 36.14, 26.97, 24.31, 22.80, 21.56, 15.77. ESI-MS: m/z [M + Na]+ calculated for 464.1556, found 464.1562.
4-Methyl-3-(((1-(4-(trifluoromethoxy)benzoyl)piperidin-4-yl)amino)methyl)-5,6,7,8-tetrahydroquinolin-2(1H)-one (6d10). Yield 35% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 7.51 (d, J = 8.2 Hz, 2H), 7.42 (d, J = 8.2 Hz, 2H), 3.60 (s, 2H), 3.04 (s, 2H), 2.68 (d, J = 7.6 Hz, 2H), 2.43 (d, J = 5.4 Hz, 1H), 2.34 (q, J = 7.3, 6.2 Hz, 2H), 2.07 (s, 3H), 1.90 (s, 4H), 1.73–1.55 (m, 4H), 1.25 (d, J = 14.9 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 172.53, 168.05, 162.30, 149.21 (d, J = 13.6 Hz), 140.21, 136.13, 129.36, 125.00, 121.41, 112.55, 53.73, 42.09, 26.93, 24.31, 22.82, 21.66, 21.57, 15.54. ESI-MS: m/z [M + H]+ calculated for 464.2156, found 464.2163.
6-Bromo-N-((4-methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)nicotinamide (6d11). Yield 79% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.27 (s, 1H), 8.77 (d, J = 2.4 Hz, 1H), 8.63 (t, J = 5.0 Hz, 1H), 8.11 (dt, J = 8.3, 2.3 Hz, 1H), 7.73 (d, J = 8.3 Hz, 1H), 4.37 (d, J = 4.8 Hz, 2H), 2.45 (d, J = 5.3 Hz, 2H), 2.33 (d, J = 5.2 Hz, 2H), 2.09 (s, 3H), 1.69–1.61 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 164.06, 162.13, 150.48, 149.95, 144.13, 141.05, 138.80, 130.05, 128.15, 122.49, 112.55, 36.07, 26.98, 24.30, 22.80, 21.55, 15.79. ESI-MS: m/z [M + Na]+ calculated for 398.0475, found 398.0467.
6-Fluoro-N-((4-methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)nicotinamide (6d12). Yield 69% (orange solid). 1H NMR (400 MHz, DMSO-d6) δ 11.28 (s, 1H), 8.67 (d, J = 2.5 Hz, 1H), 8.57 (t, J = 5.0 Hz, 1H), 8.38 (tt, J = 8.0, 2.3 Hz, 1H), 7.24 (dd, J = 8.6, 2.7 Hz, 1H), 4.38 (d, J = 4.8 Hz, 2H), 2.45 (d, J = 5.2 Hz, 2H), 2.39–2.31 (m, 2H), 2.09 (s, 3H), 1.74–1.56 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.86, 150.45, 147.98, 147.82, 141.97, 141.88, 141.02, 122.57, 112.55, 109.80, 109.43, 36.03, 26.99, 24.30, 22.80, 21.56, 15.79. ESI-MS: m/z [M + Na]+ calculated for 338.1275, found 338.1280.
6-Chloro-N-((4-methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)nicotinamide (6d13). Yield 79% (yellow solid). 1H NMR (400 MHz, DMSO-d6) δ 11.30 (s, 1H), 8.81 (d, J = 2.4 Hz, 1H), 8.63 (t, J = 5.1 Hz, 1H), 8.23 (dt, J = 8.3, 2.2 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 4.38 (d, J = 4.8 Hz, 2H), 2.45 (d, J = 5.2 Hz, 2H), 2.35 (q, J = 8.3, 6.6 Hz, 2H), 2.09 (s, 3H), 1.74–1.59 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 163.95, 162.14, 152.75, 150.48, 149.54, 141.05, 139.15, 129.78, 124.37, 122.50, 112.56, 36.07, 26.99, 24.60, 24.30, 22.80, 21.55, 15.79. ESI-MS: m/z [M + Na]+ calculated for 354.0980, found 354.0991.
N-((4-Methyl-2-oxo-1,2,5,6,7,8-hexahydroquinolin-3-yl)methyl)-5-nitrofuran-2-carboxamide (6d14). Yield 79% (yellow solid). 1H NMR (400 MHz, DMSO-d6) δ 11.32 (s, 1H), 8.74 (t, J = 5.1 Hz, 1H), 7.71 (d, J = 3.9 Hz, 1H), 7.45 (d, J = 3.9 Hz, 1H), 4.37 (d, J = 5.1 Hz, 2H), 2.45 (d, J = 4.8 Hz, 2H), 2.35 (d, J = 5.4 Hz, 2H), 2.09 (s, 3H), 1.66 (q, J = 7.5, 6.9 Hz, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.14, 156.28, 150.38, 148.72, 115.82, 113.82, 35.70, 26.99, 24.30, 22.78, 21.53, 15.79. ESI-MS: m/z [M + K]+ calculated for 370.0800, found 370.0802.
N-((4-Methyl-2-oxo-1,2-dihydroquinolin-3-yl)methyl)-6-(trifluoromethyl)nicotinamide (9a1). Yield 49% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.78 (s, 1H), 9.13 (d, J = 2.0 Hz, 1H), 8.92 (t, J = 4.8 Hz, 1H), 8.46 (dd, J = 8.1, 2.1 Hz, 1H), 8.00 (d, J = 8.2 Hz, 1H), 7.81 (dd, J = 8.2, 1.3 Hz, 1H), 7.50 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.32 (dd, J = 8.3, 1.2 Hz, 1H), 7.21 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 4.57 (d, J = 4.8 Hz, 2H), 2.53 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.99, 161.94, 149.55, 146.23, 138.23, 137.94, 133.40, 130.56, 126.88, 125.54, 122.27, 120.94, 120.29, 115.70, 36.30, 15.45. ESI-MS: m/z [M + Na]+ calculated for 384.0930, found 384.0930.
3′,5′-Dimethoxy-N-((4-methyl-2-oxo-1,2-dihydroquinolin-3-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (9a2). Yield 42% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.75 (s, 1H), 8.48 (s, 1H), 7.93 (d, J = 7.7 Hz, 2H), 7.80 (d, J = 8.2 Hz, 1H), 7.73 (d, J = 7.9 Hz, 2H), 7.49 (t, J = 7.7 Hz, 1H), 7.32 (d, J = 8.2 Hz, 1H), 7.21 (t, J = 7.8 Hz, 1H), 6.82 (s, 2H), 6.53 (s, 1H), 4.55 (d, J = 4.8 Hz, 2H), 3.81 (s, 6H), 2.67 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 164.03, 162.07, 152.76, 150.09, 149.53, 139.19, 129.79, 124.39, 36.45, 31.88, 26.99 (d, J = 10.5 Hz), 25.97, 17.00. ESI-MS: m/z [M + Na]+ calculated for 451.1628, found 451.1632; HPLC purity at 254 nm, 96.8%.
N-((4-Methyl-2-oxo-1,2-dihydroquinolin-3-yl)methyl)-4-(6-(trifluoromethyl)pyridin-3-yl)benzamide (9a3). Yield 45% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.75 (s, 1H), 9.14 (s, 1H), 8.57 (s, 1H), 8.42 (d, J = 8.2 Hz, 1H), 8.02 (d, J = 8.1 Hz, 3H), 7.90 (d, J = 8.0 Hz, 2H), 7.81 (d, J = 8.2 Hz, 1H), 7.50 (t, J = 7.8 Hz, 1H), 7.32 (d, J = 8.2 Hz, 1H), 7.21 (t, J = 7.7 Hz, 1H), 4.57 (d, J = 4.8 Hz, 2H), 2.52 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 165.88, 162.04, 148.85, 138.43, 136.79, 130.44, 128.71, 127.66, 125.48, 120.38, 15.42. ESI-MS: m/z [M + Na]+ calculated for 460.1243, found 460.1251; HPLC purity at 254 nm, 98.5%.
N-((4-Methyl-2-oxo-1,2-dihydroquinolin-3-yl)methyl)-2-(trifluoromethyl)pyrimidine-5-carboxamide (9a4). Yield 44% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.80 (s, 1H), 9.37 (s, 2H), 9.07 (t, J = 4.9 Hz, 1H), 7.85–7.80 (m, 1H), 7.50 (ddd, J = 8.3, 7.1, 1.3 Hz, 1H), 7.32 (dd, J = 8.2, 1.2 Hz, 1H), 7.22 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 4.59 (d, J = 4.9 Hz, 2H), 2.54 (s, 3H). ESI-MS: m/z [M + Na]+ calculated for 485.0883, found 485.0883.
N-((1-Methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)-5-nitrothiophene-2-carboxamide (6a1). Yield 44% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.31 (s, 1H), 9.00 (dt, J = 58.9, 5.1 Hz, 1H), 8.09 (t, J = 4.0 Hz, 1H), 7.87 (t, J = 4.2 Hz, 1H), 4.28 (dd, J = 36.4, 5.0 Hz, 2H), 2.77 (dt, J = 38.4, 7.5 Hz, 2H), 2.60 (dt, J = 17.7, 7.3 Hz, 2H), 2.14 (s, 3H), 1.96 (dq, J = 27.8, 7.5 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 163.40, 159.73, 158.72, 153.21, 147.10, 137.56, 130.60, 127.86, 127.81, 119.48, 37.03, 31.65, 31.28, 28.98, 28.34, 25.08, 22.00, 16.87, 16.75. ESI-MS: m/z [M + Na]+ calculated for 356.0657, found 356.0681.
6-Fluoro-N-((1-methyl-3-oxo-3,5,6,7-tetrahydro-2H-cyclopenta[c]pyridin-4-yl)methyl)nicotinamide (6a2). Yield 39% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 7.96 (d, J = 8.5 Hz, 2H), 7.41 (t, J = 6.8 Hz, 2H), 4.33 (d, J = 4.9 Hz, 2H), 2.81 (t, J = 7.5 Hz, 2H), 2.64–2.56 (m, 2H), 2.14 (s, 3H), 1.96 (dt, J = 22.5, 7.4 Hz, 2H). ESI-MS: m/z [M + Na]+ calculated for 324.1119, found 324.1120.
6-Bromo-N-((1-methyl-3-oxo-3,5,6,7,8,9-hexahydro-2H-cyclohepta[c]pyridin-4-yl)methyl)nicotinamide (6c1). Yield 67% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H), 8.78 (d, J = 2.5 Hz, 1H), 8.63 (t, J = 4.8 Hz, 1H), 8.12 (dd, J = 8.3, 2.5 Hz, 1H), 7.73 (d, J = 8.3 Hz, 1H), 4.39 (d, J = 4.7 Hz, 2H), 2.72–2.64 (m, 2H), 2.56–2.52 (m, 2H), 2.15 (s, 3H), 1.75 (p, J = 5.7 Hz, 2H), 1.54 (q, J = 5.4 Hz, 2H), 1.46 (p, J = 5.5 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 164.11, 162.04, 149.99 (d, J = 5.4 Hz), 144.13, 138.83, 130.07, 128.15, 121.65, 36.44, 32.32, 31.90, 27.01 (d, J = 9.9 Hz), 25.98, 17.00. ESI-MS: m/z [M + Na]+ calculated for 412.0631, found 412.0634.
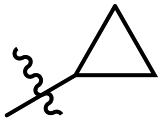
N-((6-Cyclopropyl-4-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-6-(trifluoromethyl) nicotinamide (7a1). Yield 68% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.45 (s, 1H), 9.12 (d, J = 2.1 Hz, 1H), 8.83 (t, J = 5.0 Hz, 1H), 8.45 (dd, J = 8.1, 2.1 Hz, 1H), 7.99 (d, J = 8.1 Hz, 1H), 5.72 (s, 1H), 4.33 (d, J = 5.0 Hz, 2H), 2.18 (d, J = 3.9 Hz, 3H), 1.81 (dtt, J = 13.5, 8.5, 4.2 Hz, 1H), 0.98 (t, J = 7.1 Hz, 2H), 0.79 (td, J = 5.7, 5.1, 2.3 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 163.91, 163.31, 150.57, 149.53, 137.91, 133.51, 120.93, 36.01, 19.53, 12.90, 8.93. ESI-MS: m/z [M + Na]+ calculated for 374.1087, found 374.1092.
N-((6-Cyclopropyl-4-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3′,5′-dimethoxy-[1,1′-biphenyl]-4-carboxamide (7a2). Yield 61% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 8.40 (t, J = 5.0 Hz, 1H), 7.97–7.88 (m, 2H), 7.72 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 2.3 Hz, 2H), 6.53 (d, J = 2.2 Hz, 1H), 5.71 (s, 1H), 4.31 (d, J = 4.8 Hz, 2H), 3.81 (s, 6H), 2.17 (s, 3H), 1.79 (tt, J = 8.4, 5.1 Hz, 1H), 0.99–0.88 (m, 2H), 0.82–0.73 (m, 2H). ESI-MS: m/z [M + Na]+ calculated for 441.1785, found 441.1790.
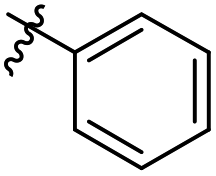
N-((4-Methyl-2-oxo-6-phenyl-1,2-dihydropyridin-3-yl)methyl)-6-(trifluoromethyl) nicotinamide (7b1). Yield 61% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.71 (s, 1H), 9.14 (d, J = 2.1 Hz, 1H), 8.93 (t, J = 4.9 Hz, 1H), 8.47 (dd, J = 8.1, 2.2 Hz, 1H), 8.00 (d, J = 8.2 Hz, 1H), 7.81–7.62 (m, 2H), 7.48 (h, J = 3.4 Hz, 3H), 6.50 (s, 1H), 4.42 (d, J = 4.8 Hz, 2H), 2.32 (s, 3H). ESI-MS: m/z [M + Na]+ calculated for 410.1087, found 410.1094.
3′,5′-Dimethoxy-N-((4-methyl-2-oxo-6-phenyl-1,2-dihydropyridin-3-yl)methyl)-[1,1′-biphenyl]-4-carboxamide (7b2). Yield 65% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.71 (s, 1H), 8.51 (s, 1H), 7.94 (d, J = 8.3 Hz, 2H), 7.80–7.68 (m, 4H), 7.52–7.42 (m, 3H), 6.83 (d, J = 2.2 Hz, 2H), 6.56–6.45 (m, 2H), 4.41 (d, J = 4.9 Hz, 2H), 3.81 (s, 6H), 2.32 (s, 3H). ESI-MS: m/z [M + K]+ calculated for 493.1524, found 493.1533; HPLC purity at 254 nm, 98.3%.
N-((6-Methyl-2-oxo-4-(trifluoromethyl)-1,2-dihydropyridin-3-yl)methyl)-6-(trifluoromethyl)nicotinamide (7c1). Yield 62% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 12.38 (s, 1H), 9.09 (d, J = 2.0 Hz, 1H), 8.79 (t, J = 4.3 Hz, 1H), 8.41 (dd, J = 8.3, 2.1 Hz, 1H), 8.02 (d, J = 8.2 Hz, 1H), 6.31 (s, 1H), 4.44–4.39 (m, 2H), 2.27 (s, 3H). ESI-MS: m/z [M + Na]+ calculated for 402.0648, found 402.0651.
N-((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-nitrofuran-2-carboxamide (8a1). Yield 83% (white solid). 1H NMR (400 MHz, chloroform-d) δ 8.82 (t, J = 5.1 Hz, 1H), 7.71 (d, J = 3.9 Hz, 1H), 7.48 (d, J = 3.9 Hz, 1H), 5.87 (s, 1H), 4.28 (d, J = 5.1 Hz, 2H), 2.16 (s, 3H), 2.12 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.44, 156.32, 151.84, 150.34, 148.72, 143.65, 121.25, 115.90, 113.86, 107.98, 35.48, 19.39, 18.67. ESI-MS: m/z [M + Na]+ calculated for 314.1, found 314.0.
N-((6-Ethyl-4-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-nitrofuran-2-carboxamide (8a2). Yield 81% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.55 (s, 1H), 8.75 (t, J = 5.1 Hz, 1H), 7.71 (d, J = 3.9 Hz, 1H), 7.45 (d, J = 3.9 Hz, 1H), 5.94–5.90 (m, 1H), 4.31 (d, J = 5.2 Hz, 2H), 2.53 (d, J = 7.7 Hz, 2H), 2.14 (d, J = 0.9 Hz, 3H), 1.08 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.75, 156.17, 155.64, 151.86, 148.70, 144.04, 120.53, 115.87, 113.83, 106.19, 35.02, 25.83, 18.79, 14.71. ESI-MS: m/z [M + Na]+ calculated for 328.1, found 328.1.
N-((4,6-Diethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-nitrofuran-2-carboxamide (8a3). Yield 86% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 8.82 (t, J = 5.1 Hz, 1H), 7.73 (d, J = 3.9 Hz, 1H), 7.49 (d, J = 3.9 Hz, 1H), 5.95 (s, 1H), 4.32 (d, J = 5.1 Hz, 2H), 2.55 (d, J = 7.5 Hz, 2H), 2.44 (q, J = 7.5 Hz, 2H), 1.14 (t, J = 7.5 Hz, 3H), 1.09 (t, J = 7.6 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 163.81, 156.19, 155.79, 151.84, 149.53, 148.70, 120.79, 115.91, 113.85, 104.71, 35.05, 25.94, 25.85, 14.72, 13.52. ESI-MS: m/z [M + Na]+ calculated for 342.1, found 342.1.
N-((4,6-Dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-4-(trifluoromethyl)benzamide (8a4). Yield 81% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 8.64 (t, J = 4.9 Hz, 1H), 8.03 (d, J = 8.1 Hz, 2H), 7.80 (d, J = 8.2 Hz, 2H), 4.32 (d, J = 4.9 Hz, 2H), 2.69 (s, 1H), 2.18 (s, 3H), 2.12 (s, 3H). ESI-MS: m/z [M + Na]+ calculated for 347.1, found 347.1.
N-((4,6-Diethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-6-(trifluoromethyl)nicotinamide (8a5). Yield 81% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 1H), 9.12 (d, J = 2.0 Hz, 1H), 8.85 (t, J = 4.9 Hz, 1H), 8.44 (dd, J = 8.0, 2.1 Hz, 1H), 7.99 (dd, J = 8.2, 0.8 Hz, 1H), 5.94 (s, 1H), 4.36 (d, J = 4.9 Hz, 2H), 2.59–2.52 (m, 2H), 2.43 (q, J = 7.5 Hz, 2H), 1.12 (dt, J = 19.6, 7.5 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 163.80, 155.92, 149.52, 149.39, 137.89, 133.51, 122.06, 121.08, 120.90, 104.60, 35.50, 25.96, 25.87, 14.74, 13.50. ESI-MS: m/z [M + Na]+ calculated for 376.1, found 376.1.
Synthesis of compound 10a1. The intermediate 5b was dissolved in 30 mL dichloromethane and stirred and cooled to −30 °C. DIEA (1.8 mL) was added dropwise to the reaction and stirred at −30 °C for 30 min. 4-(Trifluoromethyl)benzyl bromide (2.00 g) was weighed and added to the reaction in three batches at a ratio of 2
:1:1 within 4 h. The reaction was stirred for a further 4 h at −30 °C. TLC was used to monitor the completion of the reaction with 4-trifluoromethyl benzyl bromide. After the reaction was completed, the reaction system was warmed gradually to room temperature. After washing with H2O (15 mL × 1), the aqueous phase was extracted with dichloromethane (10 mL × 2) and combined with the organic phase. The sample was washed with dilute HCl (1 M) (15 mL × 1) and saturated NaCl (15 mL × 1) and dried with anhydrous sodium sulfate overnight. The sample was filtered and the solvent removed by reducing the pressure. This was followed by column chromatography to yield the desired product as a white solid.
1-Methyl-4-(((4-(trifluoromethyl)benzyl)amino)methyl)-5,6,7,8-tetrahydroisoquinolin-3(2H)-one (10a1). Yield 63% (white solid). 1H NMR (400 MHz, chloroform-d) δ 7.55 (d, J = 8.1 Hz, 2H), 7.49 (d, J = 8.1 Hz, 2H), 3.87 (s, 2H), 3.71 (s, 2H), 2.69–2.61 (m, 2H), 2.42 (d, J = 6.0 Hz, 2H), 2.19 (s, 3H), 1.71 (t, J = 3.4 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 373.1, found 373.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-6-(trifluoromethyl)pyridine-3-sulfonamide (10a2). The synthesis method as same as 6b1, yield 50% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.41 (s, 1H), 7.95 (d, J = 8.3 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 7.79 (t, J = 5.5 Hz, 1H), 3.90 (d, J = 5.2 Hz, 2H), 2.51 (s, 2H), 2.34–2.22 (m, 2H), 2.01 (s, 3H), 1.56 (t, J = 3.4 Hz, 4H). ESI-MS: m/z [M + Na]+ calculated for 424.1, found 424.1; HPLC purity at 254 nm, 99.7%.
Synthesis of compound 10a3. The hydrolysate of intermediate 4b was dissolved in 25 mL dichloromethane, cooled to 0 °C, stirred and 3 mL sulfoxide chloride was added dropwise. A large amount of irritating gas was produced. Two drops of N,N-dimethylformamide were added and the reaction was left at room temperature for 3 h. TLC was used to monitor the completion of the reaction. The solvent was removed following completion of the reaction to afford a yellowish oil. Ten milliliters dichloromethane was added and the sample redissolved. 4-Trifluoromethyl benzoyl hydrazine (2.04 g), pyridine (0.5 mL), 4-dimethylaminopyridine (0.5 g) and 20 mL dichloromethane were added dropwise with stirring. The reaction was stirred at room temperature for 6 h with TLC used to monitor completion of the reaction. The sample was washed with dilute HCl (1 M; 15 mL × 1), and the aqueous phase extracted with dichloromethane (5 mL × 2) and combined with the organic phase. The sample was dried with anhydrous sodium sulfate. The solvent was removed by reducing the pressure to afford a pale yellow solid with a yield of 84%.
1-Methyl-3-oxo-N′-(4-(trifluoromethyl)benzoyl)-2,3,5,6,7,8-hexahydroisoquinoline-4-carbohydrazide ESI-MS: m/z 416.1 [M + Na]+. Trifluoromethanesulfonic anhydride (100 μL) was slowly added dropwise to an ultra-dry dichloromethane solution of triphenylphosphine oxide (334.00 mg) at 0 °C with stirring for 5 min. 1-Methyl-3-oxo-N′-(4-(trifluoromethyl)benzoyl)-2,3,5,6,7,8-hexahydroisoquinoline-4 carbohydrazide was then added at room temperature and the reaction stirred for 7 h. After the reaction was completed, the pressure was reduced to remove solvent. Dichloromethane was extracted and the organic phase combined. The desired product as a white solid was obtained by column chromatography.
1-Methyl-4-(5-(4-(trifluoromethyl)phenyl)-1,3,4-oxadiazol-2-yl)-5,6,7,8-tetrahydroisoquinolin-3(2H)-one (10a3). Yield 76% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 12.13 (s, 1H), 8.27–8.21 (m, 2H), 8.00 (d, J = 8.3 Hz, 2H), 2.65 (t, J = 6.3 Hz, 2H), 2.45 (t, J = 6.5 Hz, 2H), 2.24 (s, 3H), 1.70 (t, J = 5.8 Hz, 2H), 1.60 (dd, J = 7.8, 3.7 Hz, 2H). ESI-MS: m/z [M + Na]+ calculated for 398.1, found 398.1.
1-Methyl-3-oxo-N-(4-(trifluoromethyl)phenyl)-2,3,5,6,7,8-hexahydroisoquinoline-4-carboxamide (10a4). The synthesis method as same as 6b1, yield 60% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 12.50 (s, 1H), 11.34 (s, 1H), 8.43 (t, J = 4.8 Hz, 1H), 7.43 (d, J = 8.1 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 2.88 (s, 2H), 2.54 (s, 2H), 2.33 (s, 3H), 1.90 (s, 4H). ESI-MS: m/z [M + Na]+ calculated for 373.1, found 373.1.
N-((1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)methyl)-2-(4-(trifluoromethyl)phenyl)acetamide (10a5). The synthesis method as same as 6b1, yield 51% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 7.98 (t, J = 5.0 Hz, 1H), 7.36 (d, J = 8.7 Hz, 2H), 7.27 (dt, J = 7.8, 1.1 Hz, 2H), 4.09 (d, J = 5.0 Hz, 2H), 3.43 (s, 2H), 2.54 (t, J = 6.2 Hz, 2H), 2.35 (t, J = 6.3 Hz, 2H), 2.10 (s, 3H), 1.64–1.49 (m, 4H). ESI-MS: m/z [M + Na]+ calculated for 401.1, found 401.1.
N-(1-Methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolin-4-yl)-6-(trifluoromethyl)nicotinamide (10a6). Yield 49% (white solid). 1H NMR (400 MHz, DMSO-d6) δ 11.74 (s, 1H), 9.89 (s, 1H), 9.25 (s, 1H), 8.57 (d, J = 8.1 Hz, 1H), 8.08 (d, J = 8.2 Hz, 1H), 2.52 (d, J = 7.3 Hz, 2H), 2.42 (t, J = 6.4 Hz, 2H), 2.15 (s, 3H), 1.71–1.54 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 162.83, 159.32, 149.76, 148.57, 140.67, 138.23, 133.27, 123.22, 121.05, 121.04, 111.86, 55.38, 26.33, 24.36, 22.64, 21.88, 16.35. ESI-MS: m/z [M + Na]+ calculated for 374.1, found 374.1.
Standard sample configuration: 2.00 mg of the compound to be tested was precisely weighed and 1000 μL DMSO was added. The compound to be tested was dissolved completely in DMSO by ultrasonication for 10 min. After centrifugation (6 min, 5 × 104 rpm), 100 μL of the supernatant was taken, and diluted to obtain a 200× working solution, which was filtered through a 0.22 μm filter and added to a U-HPLC sample bottle. Blank samples were configured in parallel.
A Dionex UltiMate 3000 ultra-high performance liquid chromatography system was used (Dionex Corporation, California, USA). The data processing system used was Analyst software (Applied Biosystems, Inc., version 1.5.1). The kinetic solubility of the compound was calculated according to the Beer–Lambert law.
Liquid condition: column: Gmini column, model: 00f-4435-E0, specification: 5 μm C18 110 Å 150 × 4.6 nm.
Mobile phase:
| Analysis time | Mobile phase A (%) | Mobile phase B (%) |
|---|---|---|
| 0.00 | 40 | 60 |
| 10.00 | 40 | 60 |
A: water; B: acetonitrile; column temperature: 40 °C; temperature of automatic sampler: 28 °C; flow rate: 1 mL min−1; injection amount: 20 μL.
Microdilution method to determine the minimal inhibitory concentration of M. tuberculosis H37Rv (ATCC 27294). The test strain (M. tuberculosis H37Rv or the resistant strain) was transferred to liquid medium and, after activation, cultured at 37 °C for 2 weeks. A small amount of the culture was taken and placed in 4 mL liquid medium. Ten to twenty sterile glass beads with a diameter of 2–3 mm were added, oscillated for 20–30 s and allowed to settle for 10–20 min. The supernatant of the bacterial suspension was absorbed and the turbidities were adjusted to 1 McChrystal unit with liquid medium, which was equivalent to 1 × 107 CFU mL−1, for standby use. The drug and the positive control were dissolved in the appropriate amount of DMSO to 1 mg mL−1 and filtered through a 0.22 μm filter. The liquid medium was then diluted to the desired experimental concentration of 0.0000125–64 μg mL−1.
At the time of detection, 100 μL of each of the above drug solutions was added to a 96-well microplate, and then 100 μL of the bacterial solution at a concentration of 104 CFU mL−1 (diluted from 107 CFU mL−1) was added. The sample was cultured at 37 °C. The blank control group was given no drugs, and the same drug dilution series was run in triplicate with parallel controls. The MIC of each drug against M. tuberculosis H37Rv was determined.
One milliliter of the test solution was added to a sterile plate and 14 mL of melted MH(A) medium (∼50 °C) was added. The final concentrations of the drugs in each plate were 128, 64, 32, 16, 8, 4, 2, 1, 0.5, 0.25, 0.125, 0.06, 0.03, 0.015, 0.008 μg mL−1. After cooling, bacteria were inoculated with a multi-point inoculator and the agar plate was covered. The agar plates were incubated at 35–37 °C for 18–24 h. At the end of the incubation period, the minimum concentration of the sample without bacterial growth on the plate was determined to be the minimum bacteriostatic concentration. A control without drugs and a solvent control with DMSO were also prepared and analyzed using the same procedure described above.
Conflicts of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the manuscript submitted.Acknowledgements
This work was supported by the Post-Doctoral Research Project, West China Hospital, Sichuan University (2018HXBH036), the Fundamental Research Funds for the Central Universities (2019SCU12029, the Postdoctoral Foundation of Sichuan University), Natural Science Foundation of China (81703570) and National Mega-Project for Innovative Drugs (2019ZX09721001, 2018ZX09201018).References
- S. T. Cole and G. Riccardi, New tuberculosis drugs on the horizon, Curr. Opin. Microbiol., 2011, 14, 570–576 CrossRef CAS PubMed.
- World Health Organization, World Health Statistics, 2018 Search PubMed.
- C. Dye, Global epidemiology of tuberculosis, Lancet, 2006, 367(9514), 938–940 CrossRef.
- M. C. DeRycker, B. Baragana and S. L. Duce, et al. Challenges and recent progress in drug discovery for tropical diseases, Nature, 2018, 559(7715), 498–506 CrossRef CAS PubMed.
- Beena and D. S. Rawat, Antituberculosis drug research: a critical overview, Med. Res. Rev., 2013, 33(4), 693–764 CrossRef CAS PubMed.
- A. Zumla, P. Nahid and S. T. Cole, Advances in the development of new tuberculosis drugs and treatment regimens, Nat. Rev. Drug Discovery, 2013, 12(5), 388–404 CrossRef CAS PubMed.
- G. B. Migliori, G. DeIaco and G. Besozzi, et al. First tuberculosis cases in Italy resistant to all tested drugs, Eurosurveillance, 2007, 12(20), 5 Search PubMed.
- K. E. Dooley, E. A. Obuku and N. Durakovic, et al. World Health Organization group 5 drugs for the treatment of drug-resistant tuberculosis: unclear efficacy or untapped potential, J. Infect. Dis., 2013, 207(9), 1352–1358 CrossRef CAS PubMed.
- S. T. Cole and G. Riccardi, New tuberculosis drugs on the horizon, Curr. Opin. Microbiol., 2011, 14, 570–576 CrossRef CAS PubMed.
- W. T. Watford and J. R. Wright, et al. Surfactant protein A regulates complement activation, J. Immunol., 2001, 167, 6593–6600 CrossRef CAS PubMed.
- H. Esmail, C. E. Barry and D. B. Young, et al. The ongoing challenge of latent tuberculosis, Philos. Trans. R. Soc., B, 2014, 369, 20130437 CrossRef CAS PubMed.
- A. Zumla, J. Chakaya and R. Centis, et al. Tuberculosis treatment and management-an update on treatment regimens, trials, new drugs, and adjunct therapies, Lancet Respir. Med., 2015, 3, 220–234 CrossRef.
- S. K. Parida, R. Axelsson-Robertson and M. V. Rao, et al. Totally drug-resistant tuberculosis and adjunct therapies, J. Intern. Med., 2015, 277, 388–405 CrossRef CAS PubMed.
- S. T. Cole, R. Brosch and J. Parkhill, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence, Nature, 1998, 393, 537–544 CrossRef CAS PubMed.
- E. O. Johnson, et al. Large-scale chemical-genetics yields new M. tuberculosis inhibitor classes, Nature, 2019, 571, 72–78 CrossRef CAS PubMed.
- S. W. Park, et al. Target-based identification of whole-cell active inhibitors of biotin biosynthesis in Mycobacterium tuberculosis, Chem. Biol., 2015, 22, 76–86 CrossRef CAS PubMed.
- L. E. Ziganshina, A. F. Titarenko and G. R. Davies, Fluoroquinolones for treating tuberculosis (presumed drug-sensitive), Cochrane Database Syst. Rev., 2013, 6(6), CD004795 Search PubMed.
- A. S. Ginsburg, J. H. Grosset and W. R. Bishai, Fluoroquinolones, tuberculosis, and resistance, Lancet Infect. Dis., 2003, 3(7), 432–442 CrossRef CAS PubMed.
- M. Panda, S. Ramachandran and V. Ramachandran, et al. Discovery of pyrazolopyridones as a novel class of noncovalent DprE1 inhibitor with potent anti-mycobacterial activity, J. Med. Chem., 2014, 57, 4761–4771 CrossRef CAS PubMed.
- M. Naik, V. Humnabadkar and S. J. Tantry, et al. 4-Aminoquinolone piperidine amides: noncovalent inhibitors of DprE1 with long residence time and potent antimyco-bacterial activity, J. Med. Chem., 2014, 57, 5419–5434 CrossRef CAS PubMed.
- J.-J. Chanot, J. Mane and C. Plessis, New Bicyclic Dioxanes, Their Preparation And Their Use As Fragrant Compounds, WO2011013077 (A1), 2011.
- L. Zhang, X. Song and N. Wang, et al. Design, synthesis and biological evaluation of novel 1-methyl-3-oxo-2,3,5,6,7,8-hexahydroisoquinolins as potential EZH2 inhibitors, RSC Adv., 2015, 5(33), 25967–25978 RSC.
- T. Gueden-Silber, K. Klein and M. Seitz, 4,4′-Bis(trifluoromethyl)-2,2′-bipyridine-a multipurpose ligand scaffold for lanthanoid-based luminescence/19F NMR probes, Dalton Trans., 2013, 42(38), 13882–13888 RSC.
- N.-Y. Wang, W.-Q. Zuo and Y. Xu, et al. Discovery and structure-activity relationships study of novel thieno[2,3-b]pyridine analogues as hepatitis C virus inhibitors, Bioorg. Med. Chem. Lett., 2014, 24(6), 1581–1588 CrossRef CAS PubMed.
- A. A. Abramov, S. A. Chernenko and A. S. Fisyuk, et al. Synthesis and Photophysical Properties of 3-Amino-4-arylpyridin-2(1 H)-ones, Synthesis, 2020, 52(2), 227–238 CrossRef.
- C.-S. Jia, Ya-W. Dong and S.-J. Tu, et al. Microwave-assisted solvent-free synthesis of substituted 2-quinolones, Tetrahedron, 2007, 63(4), 892–897 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09250a |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |