DOI:
10.1039/D0RA09209F
(Paper)
RSC Adv., 2021,
11, 15-22
Self-reversible mechanofluorochromism of AIE-active C6-unsubstituted tetrahydropyrimidine derivatives†
Received
29th October 2020
, Accepted 4th December 2020
First published on 21st December 2020
Abstract
The mechanofluorochromic properties of three C6-unsubstituted tetrahydropyrimidines (THPs), namely, diethyl 1,2,3-triphenyl-1,2,3,6-tetrahydropyrimidine-4,5-dicarboxylate (1), dimethyl 1,2,3-tri(4-trifluoromethylphenyl)-1,2,3,6-tetrahydropyrimidine-4,5-dicarboxylate (2), and dimethyl 1,2,3-tri(3-trifluoromethylphenyl)-1,2,3,6-tetrahydropyrimidine-4,5-dicarboxylate (3), with aggregation-induced emission (AIE) characteristics were investigated. The blue-green/cyan emissions of the three THPs can be switched reversibly by a grinding–fuming/heating process, with the change in maximum emission wavelength (λem) up to 57 nm and the decrease of fluorescence quantum yields (ΦF). Interestingly, the green or cyan fluorescence of the ground powder (λem is located at 481, 470 and 477 nm for 1b, 2 and 3, respectively) can spontaneously recover to the original blue (λem is located at 434, 442 and 436 nm for 1b, 2 and 3, respectively) in 1–2 d at room temperature without any external stimulation. X-ray single-crystal diffraction, powder X-ray diffraction (XRD) and differential scanning calorimetry (DSC) studies demonstrate that the conversion between the molecular packing modes is the main reason for the mechanofluorochromism and the spontaneously recoverable mechanofluorochromism relates to intermolecular hydrogen bonds. The sensitively and/or spontaneously recoverable mechanofluorochromism of these THPs is expected to have great potential in sensing, optical recording and self-healing fluorescent materials.
1. Introduction
Organic mechanofluorochromic materials are a class of smart materials that exhibit color change upon external physical stimulus (such as pressure, vapor or heat) and have attracted intense attention because of their fundamental importance and potential applications in optical recording, optoelectronic devices, security papers, and sensors.1–7 However, the mechanofluorochromic behavior of organic emissive materials is often difficult to observe because of the notorious aggregation-caused quenching (ACQ) effects in the condensed state, which would also limit the practical application of these materials.8,9 In 2001, Tang et al. discovered a new phenomenon of aggregation-induced emission (AIE),10 and proposed that the restriction of intramolecular motion is the main reason for the AIE effect,11–13 and since then much effort has been devoted to designing and synthesizing propeller-shaped molecules to create solid state luminophores.1,11–15 Interestingly, many AIE luminogens were also found to exhibit mechanofluorochromism.5,16–19 In recent years, a number of mechanofluorochromic and AIE-active organic molecules, including tetraphenylethene-,16,20–23 triphenylethene-,24–26 difluoroboron complex-,18,27–29 anthracene-,30–32 cyanostilbene-19,33,34 and triphenylamine-based35–37 derivatives, have been investigated.1,2 Changes in molecular conformation and packing modes have proven to be the main factors that influencing the switching and alternation of the emissions of the fluorophores.20,25,32,38–42 However, the design and synthesis of new economic mechanofluorochromic organic compounds, and the in-depth understanding of the origin of mechanofluorochromism, still remains to be a challenge.9,25,38,43 Comparing with the external-stimuli-controlled fluorescence switching, the self-recovery of the fluorescence of organic molecules has been seldom reported.26,36,44,45 It will be interesting to have a research about this spontaneous recovery, which may have potential in self-healing materials.
In 2011, we have developed an economic and efficient five-component reaction (5CR) for the synthesis of a novel series of racemic AIE-active C6-unsubstituted tetrahydropyrimidine derivatives.46 These THPs are non-luminescent in dilute solutions, but become highly emissive in the aggregate states. Different from other conventional AIE-active molecules, the solid-state luminescence of the THPs arises from the high conjugation formed by the through-space and through-bond electron coupling and the efficient enantiomeric packing modes in aggregates.47,48 Our research has showed that the THPs could be used as sensitive thermometers in different temperature ranges,49 fluorescent bioprobes for cell imaging,50 and unique fluorescence-turn-on probes for the determination of the critical micelle concentration of ion surfactants.51,52 Excitingly, we recently found that one of the THPs, dimethyl 1,3-bis(4-bromophenyl)-2-phenyl-1,2,3,6-tetrahydropyrimidine-4,5-dicarboxylate (Me-THP-2Br), exhibited highly sensitive and external-stimuli-controlled reversible fluorescence switching between purple and green upon grinding and fuming or heating.38 The research about the three polymorphic crystals and amorphous state with distinctly different fluorescence of Me-THP-2Br indicated that the mechanofluorochromism correlates with conversion between the paired and unpaired packing modes of the R- and S-enantiomers (RS-paired and RR/SS-overlapped packing modes).38 Considering the easy preparation and excellent AIE characteristics of the THPs, we further investigate the mechanofluorochromic behaviors of other compounds, and find that the fluorescence of almost all investigated THPs in ground powder can revert to their original fluorescence in crystals upon fuming or heating. And even more amazing, several THPs (1–3 in Fig. 1) in ground powder can self-recover to their original fluorescence in crystals. In this paper, we investigated the self-reversible mechanofluorochromism in detail to further elucidate the mechanism of the self-reversible mechanofluorochromism of organic AIE compounds.
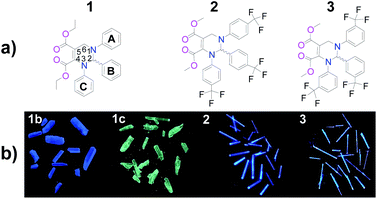 |
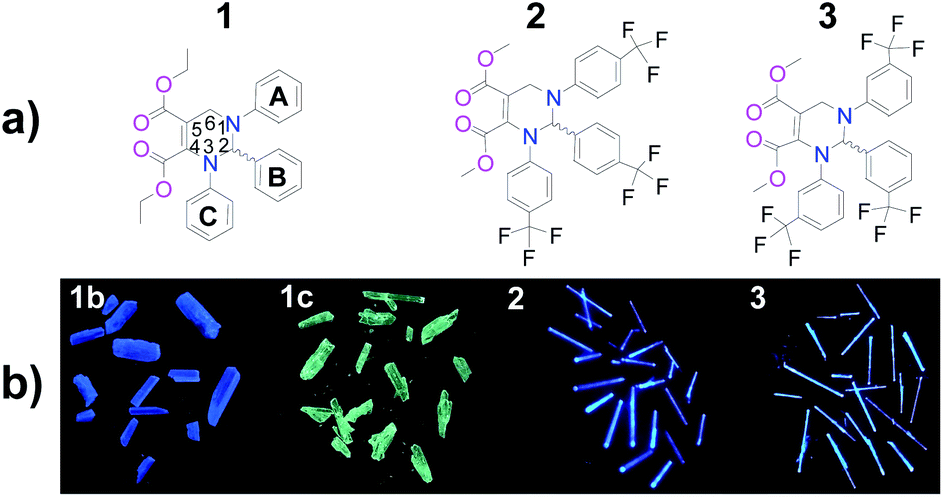
| | Fig. 1 (a) Chemical structures of THPs 1–3 and (b) photos of their as-prepared crystals (under 365 nm UV light). 1b: blue crystals of 1; 1c: cyan crystals of 1. | |
2. Experimental
2.1. Materials and measurements
Photos were taken by MEIZU MX4 under UV 365 nm. IR spectra were obtained as potassium bromide pellets with Nicolet iS50 spectrometer (Thermo Fisher). 1H (400 MHz) and 13C NMR (100.6 MHz) spectra were recorded on Bruker AVANCE III 400 spectrometer with CDCl3 as the solvent. 1H shifts were referenced to CDCl3 at 7.26 ppm. 13C shifts were referenced to CDCl3 at 77 ppm. TLC was performed using commercially available 100–400 mesh silica gel plates (GF254). UV-Vis spectra were measured on UV-5500PC (in solution) and U-3010 (in solid) spectrophotometer respectively. Fluorescence spectra were recorded by Shimadzu RF-5301PC spectrometer. The absolute fluorescence quantum yields and fluorescence lifetimes were measured on Edinburgh FLS 980 fluorescence spectrophotometer. XRD patterns were recorded at room temperature with an X'Pert Pro diffractometer operated at 40 kV voltage and 40 mA current with Cu Kα irradiation at λ = 1.5418 Å. DSC measurements were recorded on a NETZSCH thermal analyzer (NETZSCH 214 or DSC 200 F3) at heating and cooling rates of 10 K min−1 under an N2 atmosphere. The X-ray single-crystal diffraction data were measured by Bruker D8 Venture. HOMO and LUMO were calculated by Gaussian 09 program. The ground powders were prepared by grinding the as-prepared crystals with a pestle in the mortar. The fumed samples were obtained by fuming the ground powder with dichloromethane (DCM) vapor.
Unless otherwise stated, all reagents and solvents were purchased from commercial suppliers and used without further purification.
2.2. Preparation of THPs 1–3
THPs 1–3 were synthesized by adopting the 5CR that we developed.46 The characterization data of 1 and 3 are the same as those reported in our previous work,46,53 and those for 2 see the ESI.† The polymorphic crystals of 1 and the crystals of 2 and 3 were prepared by recrystallization from DCM/n-hexane solution.
3. Results and discussion
3.1. Optical properties of THPs 1–3 in solutions and solid-states
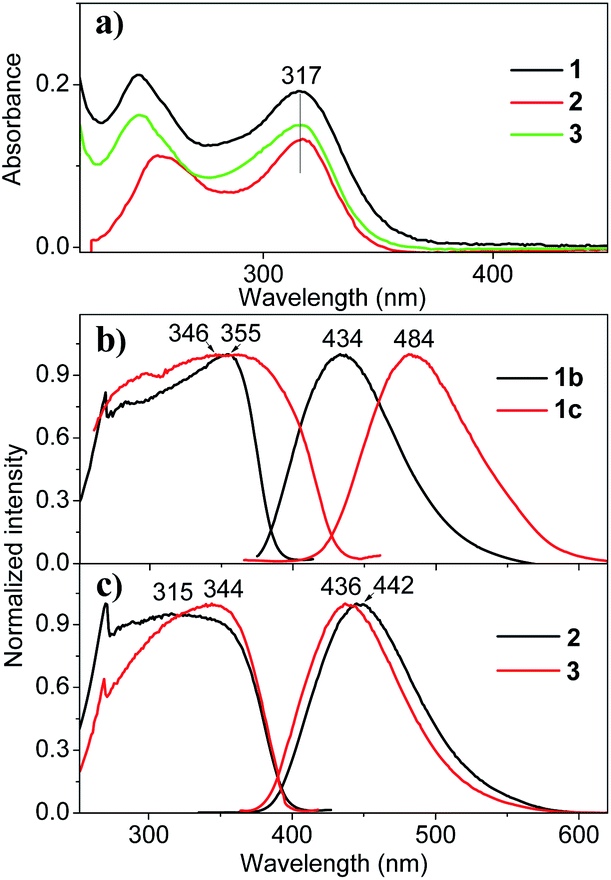
The chemical structures and photos taken under 365 nm UV light of the as-prepared crystals of THPs 1–3 are shown in Fig. 1. The absorption spectra of THPs 1–3 in cyclohexane in Fig. 2a show a peak at 317 nm, corresponding to an n–π* transition. The three THPs are non-emissive in solutions, however, they emit brightly in solid states, with ΦF (excited at 350 nm) equals to 72.0, 93.0, 96.0 and 97.0% for 1b, 1c, 2 and 3, respectively, which means that they are highly AIE-active. The excitation and emission spectra of THPs 1, 2 and 3 are displayed in Fig. 2b and c respectively.
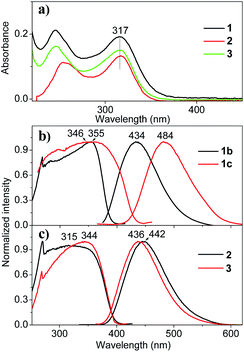 |
| | Fig. 2 Optical properties of THPs 1–3. (a) Absorption spectra of THPs 1–3 in cyclohexane solutions (1.0 × 10−5 M). (b) and (c) Excitation (left) and emission (right) (excited at emission peak marked in the left excitation spectra) spectra of 1b, 1c, 2 and 3, respectively. | |
3.2. Mechanofluorochromic characteristics of THPs 1–3
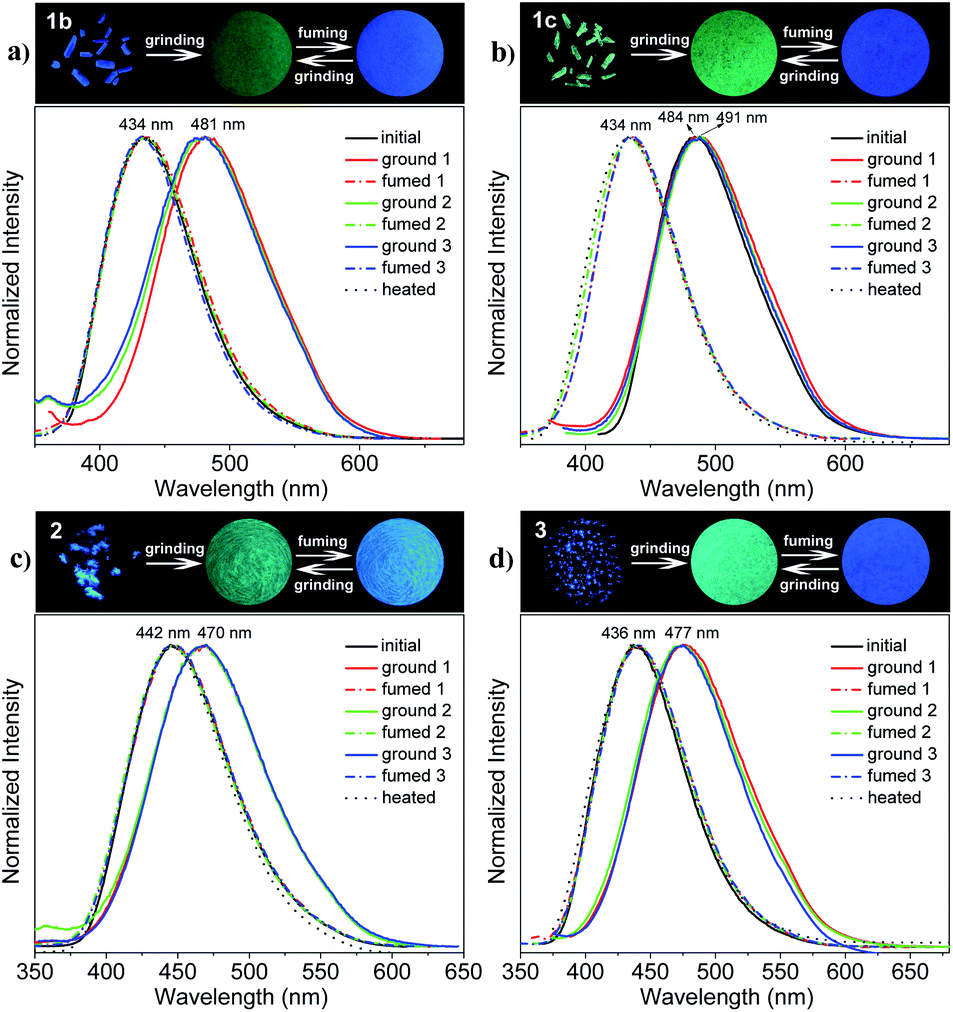
The mechanofluorochromic properties of the polymorphic crystals of THP 1 were investigated through the grinding–fuming/heating process firstly. As shown in Fig. 3a and b (upper), the as-synthesized 1b and 1c crystals emit intense blue and cyan light under UV irradiation, with λem equals to 434 and 484 nm respectively. They change into green-emitting powders with λem locates at 481 and 491 nm respectively, after grinding, showing an obvious mechanofluorochromic effect. Furthermore, the ground powder of 1b could revert back quickly to blue-emitting solid similar to the original 1b crystal, with λem = 434 nm, after fuming with DCM vapor for 2 min or heated under 110 °C for 20 min. But it took more than 1 h of fuming or heating for the ground powder of 1c to recover back to blue-emitting crystal with λem = 434 nm. The blue-green emission could be switched repeatedly by grinding and fuming or heating for many times. The emission spectra of 1b and 1c in the as-synthesized crystals (black solid line), ground powders (solid color lines) and fumed or heated states (dotted color or black lines) in three grinding–fuming cycles are shown in Fig. 3a and b (lower).
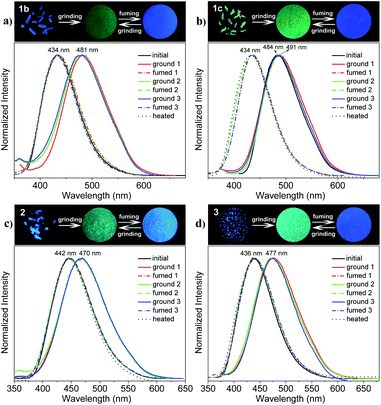 |
| | Fig. 3 Reversible grinding–fuming-controlled fluorescence-color switching of THPs 1–3 respectively. Photos of the initial crystals (left), ground powders (middle) and fumed samples (right) taken under 365 nm light (upper) and their emission spectra (lower) in three grinding–fuming cycles of 1b (a), 1c (b), 2 (c) and 3 (d), respectively. | |
Then, the mechanofluorochromic behaviors of THPs 2 and 3 were investigated. As shown in Fig. 3c and d (upper), the as-synthesized 2 and 3 solids emit blue light with λem equal to 442 and 436 nm under UV irradiation, and their emitting colors turn into cyan with λem locates at 470 and 477 nm, respectively, after grinding. In addition, the cyan-emitting ground powders of 2 and 3 could be transferred into blue-emitting solids similar to the as-synthesized crystals after fuming with DCM for 2 min or heated under 110 °C for 20 min. This blue-cyan emission could also be converted back and forth by grinding and fuming or heating for many times. Fig. 3c and d (lower) show the emission spectra of 2 and 3 in the as-synthesized crystals (black solid line), ground powders (solid color lines) and fumed or heated states (dotted color or black lines) in three grinding–fuming cycles. Furthermore, the ΦF (excited at 380 nm) of the ground powders of 1b, 1c, 2 and 3 are lower (16.0, 55.0, 51.1 and 61.4%, respectively) than those of their original crystals (72.0, 93.0, 96.0 and 97.0%, respectively). It means that grinding can not only switch the emission color but also change the luminescence efficiency of the THPs. The fluorescent lifetimes (τ, excited at 360 nm) of 1b, 1c, 2 and 3 are 7.1, 14.4, 10.1 and 8.3 ns, respectively, and those of the corresponding ground powders of 1b, 1c, 2 and 3 are 5.7, 9.9, 6.3 and 7.1 ns, respectively. The radiative rate constant (kr) and non-radiative rate constant (knr) of THPs 1–3 in crystals and ground powders were calculated (Table 1) according to the ΦF and τ values, that is, kr = ΦF/τ and knr = (1 − ΦF)/τ. It can be seen that the kr values of the crystals are in the range of 0.65–1.17 × 108 s−1, and those of the ground powders are in the range of 0.28–0.86 × 108 s−1. The knr values of the crystals are in the range of 0.04–0.39 × 108 s−1, while those of the ground powders are in the range of 0.45–1.47 × 108 s−1. After grinding, the kr values change slightly except 1b, which means that the changes in molecular conformation and/or packing mode are slightly except 1b. Whereas the knr values of ground powders are much smaller than those of crystals because of the restriction of intramolecular motion (RIM), thus leading to the decrease of the fluorescence. These phenomena indicate that the racemic AIE-active tetrahydropyrimidine derivatives possess sensitive and reversible mechanofluorochromism with the change of λem up to 57 nm. The optical properties of the initial crystals, ground powders, fumed and heated states of THPs 1–3 are summarized in Table 1. The photos of the as-synthesized 1b, 1c, 2 and 3 and their corresponding ground powder under natural light are shown in Fig. S1,† and the absorption spectra of the different solid states of 1b, 1c, 2 and 3 are shown in Fig. S2.† The fluorescence decay profiles are displayed in Fig. S3.† The fluorescent lifetimes of 1b and 1c were reported previously,47 and those of the others were measured in this work.
Table 1 Optical properties of the initial crystals, ground powders, fumed and heated states of THPs 1–3
| THPs |
λem/nm |
Δλem/nm |
ΦF (%) |
τ/ns |
kr/s−1 × 10−8 |
knr/s−1 × 10−8 |
| Initial |
Ground |
Fumed |
Heated |
Initial |
Ground |
Initial |
Ground |
Initial |
Ground |
Initial |
Ground |
| 1b |
434 |
481 |
434 |
434 |
47 |
72.0 |
16.0 |
7.1 |
5.7 |
1.01 |
0.28 |
0.39 |
1.47 |
| 1c |
484 |
491 |
434 |
434 |
57 |
93.0 |
55.0 |
14.4 |
9.9 |
0.65 |
0.56 |
0.05 |
0.45 |
| 2 |
442 |
470 |
442 |
442 |
28 |
96.0 |
51.1 |
10.1 |
6.3 |
0.95 |
0.81 |
0.04 |
0.78 |
| 3 |
436 |
477 |
436 |
436 |
31 |
97.0 |
61.4 |
8.3 |
7.1 |
1.17 |
0.86 |
0.04 |
0.54 |
3.3. Self-reversible mechanofluorochromic characteristics of THPs 1–3
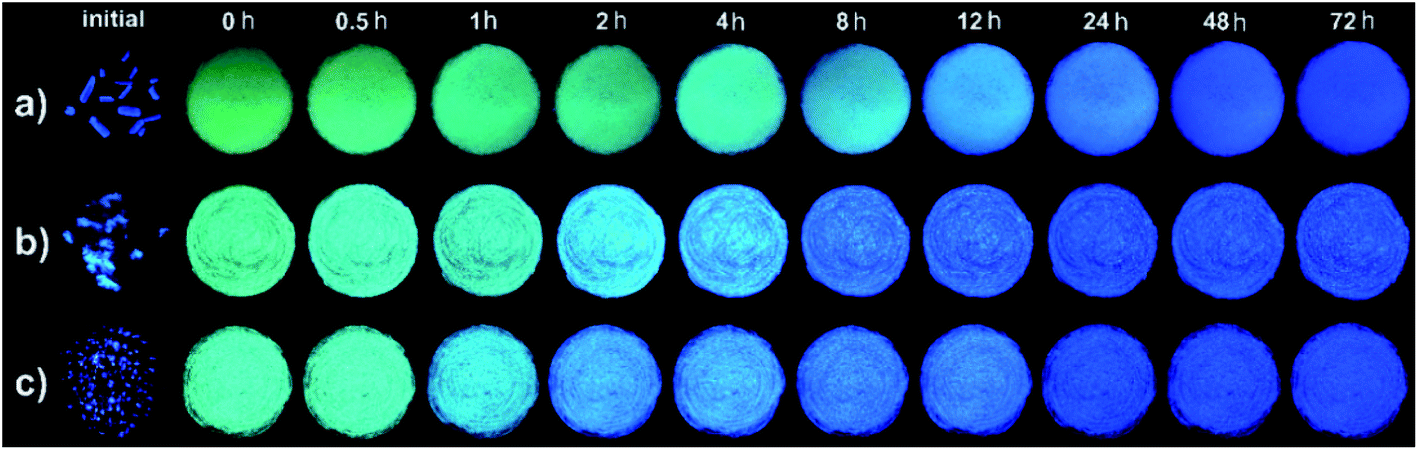
During the above process of mechanofluorochromism investigation, we found that the emitting colors of THPs 1b, 2 and 3 in green ground power could spontaneously recover to blue under room temperature without the exertion of any external stimulation. So we carefully observed and systematically studied the self-reversible characteristics of THPs 1–3. As shown in Fig. 4, the green or cyan-emitting ground powders of 1b, 2 and 3 can all self-recover to blue-emitting solids in 1–2 d under 30 °C, exhibiting wonderful spontaneous fluorescence recovery. But for 1c, the ground powder didn't show fluorescence change when stood still.
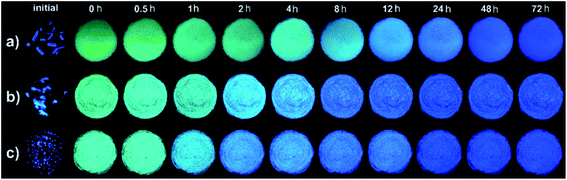 |
| | Fig. 4 Photos of the initial crystals and ground samples kept for different time of 1b (a), 2 (b) and 3 (c) taken under 365 nm light respectively. | |
3.4. Mechanism of the reversible mechanofluorochromism of THPs 1–3
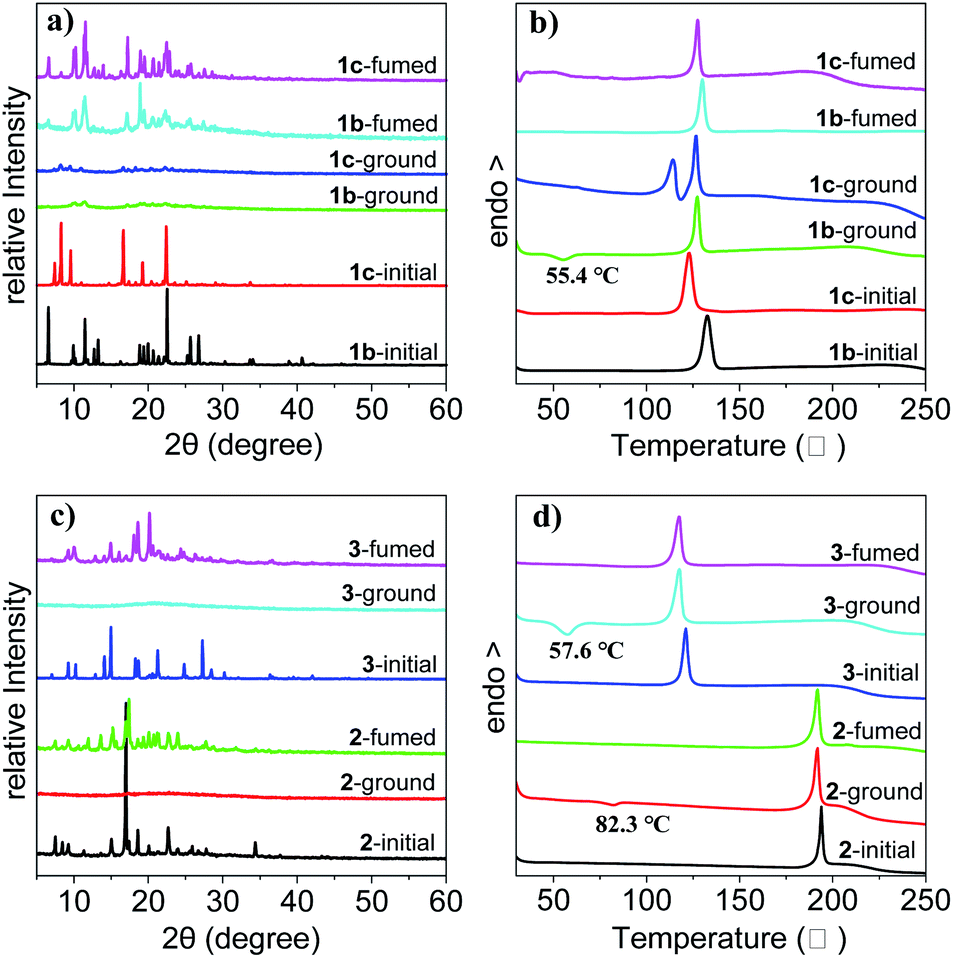
To get insight into the mechanism of the reversible mechanofluorochromic characteristics, the XRD analysis was conducted on the three THPs. As demonstrated in Fig. 5a and c, the as-synthesized crystals of THPs 1–3 (1–3-initial) exhibit sharp and strong reflection peaks, suggesting highly ordered and different molecular arrangements of the crystals. There are only very weak or no reflection peaks for the corresponding ground powders of THPs 1–3, indicating amorphous states with short-range ordered molecular arrangements after grinding. The diffraction patterns of the fumed states of the ground powder of 1b (1b-fumed) and 1c (1c-fumed) samples are almost identical to the as-synthesized 1b, and those of the 2-fumed and 3-fumed samples are almost identical to the as-synthesized 2 and 3 solids respectively. These results show that the ground powders of the THPs can all revert back to their original blue-emitting crystals by fuming, which demonstrates that the as-synthesized crystals are more stable solid states.
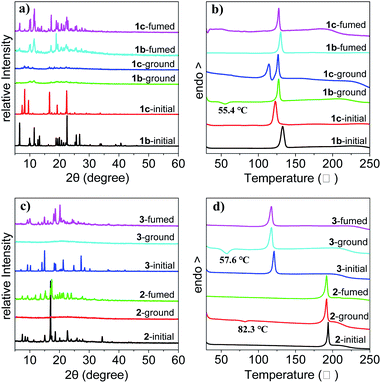 |
| | Fig. 5 XRD patterns of 1b and 1c (a), 2 and 3 (c) and DSC curves of 1b and 1c (b), 2 and 3 (d) in initial, ground and fumed states, respectively. | |
DSC measurements were also conducted on the samples to investigate the phase transition during the mechanofluorochromic process. As shown in Fig. 5b and d, there are no transition peaks before melting points in the heating curves of the as-synthesized 1b, 1c, 2 and 3 crystals. However, it is found that the ground powders of 1b, 2 and 3 exhibit clear exothermic peaks at around 55.4, 82.3 and 57.6 °C in the DSC curves, respectively, corresponding to the cold-crystallization transitions. These results mean that the ground powders of these samples are in metastable phases, and they transform into more stable crystalline phases via an exothermal recrystallization process. But for the ground powder of 1c, there is no clear cold-crystallization peak is observed, instead, there appears another endothermic peak at approximately 114.2 °C before its melting point (126.8 °C) in the heating curve, which is different from the ground powders of other crystals. During the mechanofluorochromic process, we found that 1c was much less responsive to external stimuli than others, and it took much more time for it to transform into green-emitting powder by grinding or change into blue-emitting crystals by fuming. As we have reported in reference,46 the 1c crystals are the kinetically favored form of THP 1 which can be obtained from solutions at lower temperature or higher concentration, while the 1b crystals are the thermodynamically favored form which can be obtained from solutions at higher temperature or lower concentration. During the heating process, the 1c-ground powders first recrystallized partly into kinetically favored 1c crystals at relatively lower temperature, and they might melt with the increasing of temperature and transform into thermodynamically favored 1b crystals, thus leading to the appearance of another endothermic melting peak at 114.2 °C in the DSC curve of the 1c-ground powders. Based on these XRD and DSC results, we can consider that the transformation between the amorphous and crystalline states is responsible for the reversible mechanofluorochromic behaviors of these THPs.
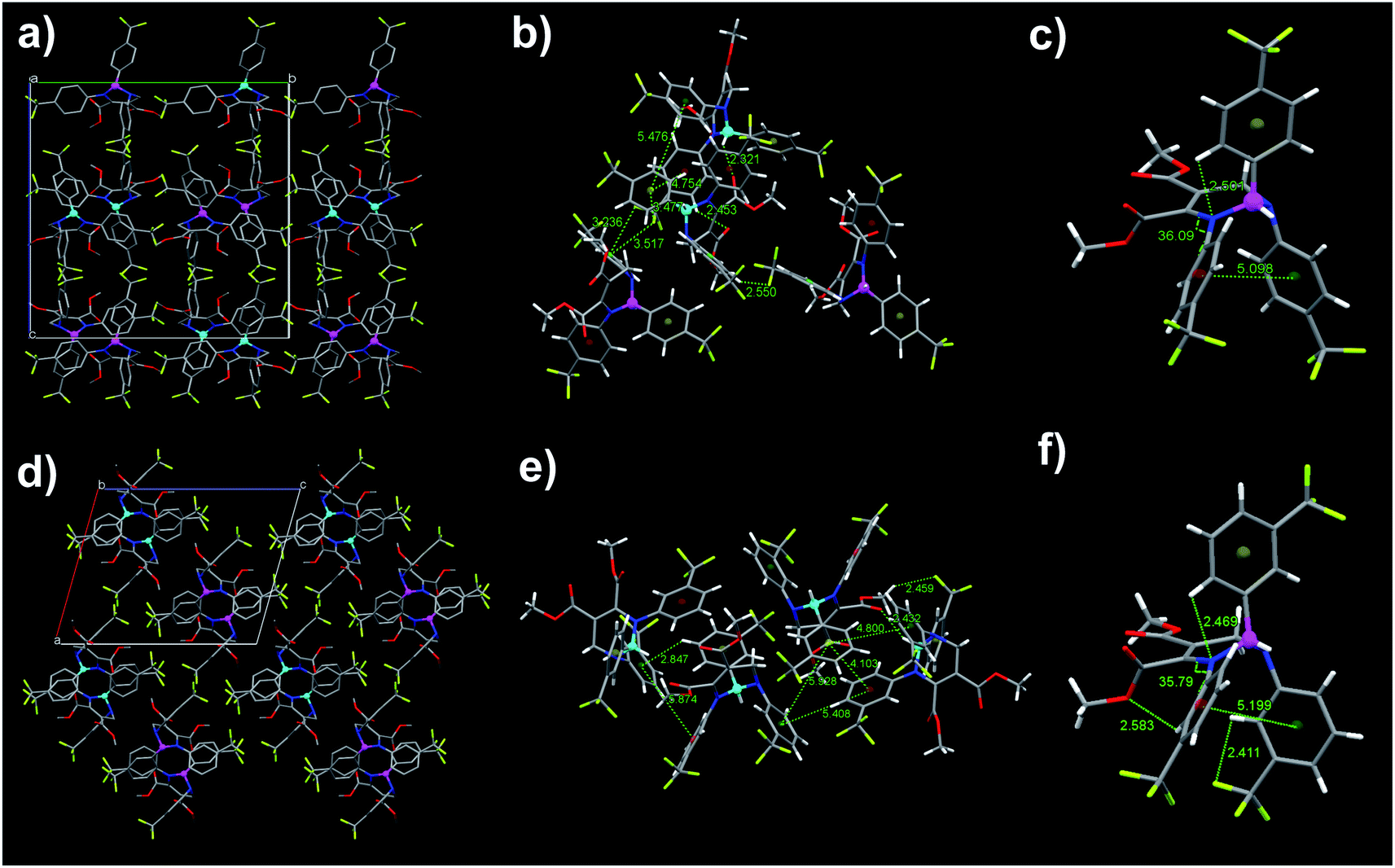
To better understand the reversible mechanofluorochromic behaviors of these THPs, it is necessary to get single crystals. Fortunately, the quality single crystals of THPs 2 and 3 with emission wavelengths 442 and 436 nm, respectively, were obtained in this work, and the polymorphic single crystals 1b and 1c of THP 1 were obtained as we reported previously.46 The crystallographic data of THPs 2 and 3 are listed in Table S1,† it shows that the space groups of 2 and 3 are Pbca and P121/n1, respectively. To better understand their electronic structures, the HOMOs and LUMOs of 2 and 3 were obtained by DFT/B3LYP/6-31G Gaussian calculations based on the molecular conformations in single crystal. The HOMO and LUMO plots are given in Fig. 6. Their HOMOs, LUMOs and through-space conjugations (the area marked in color circles) are similar as those reported in our previous work.47,53 Molecular simulation of THPs 1–3 using DFT/B3LYP/6-31G method was also done and the optimized geometric structures and electron density plots of HOMOs and LUMOs are given in Fig. S4,† and the comparison of the HOMO, LUMO, energy gap and Gibbs free energy of THPs 1–3 in single crystal and optimized geometry by simulation is shown in Table 2. The Gibbs free energies of the conformations in crystals are about 200–300 kcal mol−1 higher than that in their optimized geometries, which suggests that the molecules of THPs 1–3 in crystals exist in the form of non-optimized geometry due to the molecular interactions. 1b is formed under thermodynamically condition, while 1c is formed under kinetically condition. This means the stability of 1b is better than that of 1c crystal, that is, the energy of the former is lower than the latter. However, the energy of the molecular conformation in 1b is a little higher than that in 1c crystal, showing that the decrease of the energy of 1b comes from molecular interactions. These also demonstrate that the self-recover energy mainly originates from molecular interactions. Fig. 7 presents the molecular packing modes, intermolecular interactions and conformations of 2 and 3, respectively, and the intra- and intermolecular short-ring interactions and hydrogen bonds in THPs 1–3 are summarized in Table S2.†
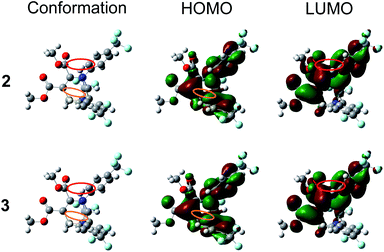 |
| | Fig. 6 Molecular conformations, HOMOs and LUMOs of 2 and 3 in single crystals. | |
Table 2 Comparison of the HOMO, LUMO, energy gap and Gibbs free energy of THPs 1–3 in single crystal and optimized geometry by simulation
| THPs |
Conformation |
HOMO |
LUMO |
ΔEa/eV |
ΔGb/kcal mol−1 |
| Band gap between the HOMO and LUMO. Gibbs free energy. Optimized geometry by simulation. Single crystal. |
| 1 |
1b |
−5.31 |
−0.94 |
4.47 |
−937031 |
| 1c |
−5.31 |
−1.16 |
4.15 |
−937037 |
| 1optc |
−5.31 |
−0.79 |
4.52 |
−937375 |
| 2 |
2cryd |
−6.33 |
−2.02 |
4.31 |
−1522350 |
| 2opt |
−5.94 |
−1.70 |
4.24 |
−1522549 |
| 3 |
3cry |
−6.22 |
−1.89 |
4.33 |
−1522342 |
| 3opt |
−5.84 |
−1.53 |
4.31 |
−1522548 |
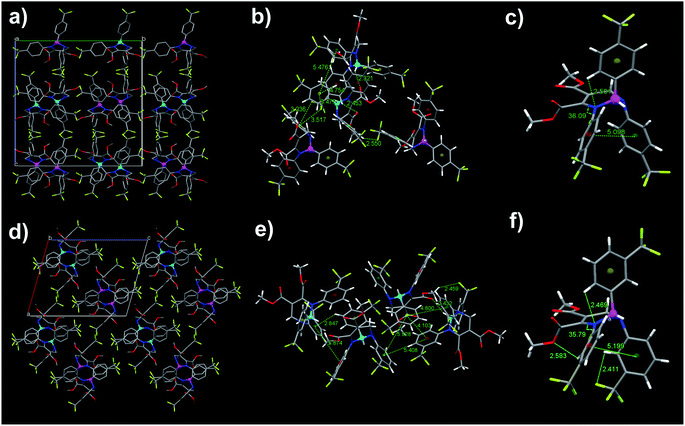 |
| | Fig. 7 The molecular packing modes, intermolecular interactions, and conformations (from left to right) of 2 (a–c) and 3 (d–f) in single crystals with emission wavelengths 442 (2) and 436 nm (3). Cyan and purples balls, respectively, represent the R- and S-configurations of chiral carbon. CCDC 2020055 and 2020056 for 2 and 3, respectively. | |
Fig. 7 shows that both blue-emitting THPs 2 and 3 crystals adopt the unpaired R- and R-enantiomer or S- and S-enantiomer packing mode (RR/SS-overlapped). Our previous investigation47,53 has demonstrated that the blue-emitting 1b polymorph adopted the paired R- and S-enantiomer packing mode (RS-paired) with short-range ring interactions only existing between paired R- and S-enantiomers, while the cyan-emitting 1c polymorph adopted the RR/SS-overlapped packing mode with short-range ring interactions existing among all zigzag-aligned R- or S-enantiomers. The RR/SS-packed and R/S-packed crystals of THPs 1 are formed in kinetically and thermodynamically conditions, respectively.46,47,53 According to these results, together with our previous study, it can be concluded that the reversible mechanofluorochromic behaviors of THPs 1 originate from the conversion of metastable RR/SS-overlapped and stable RS-paired packing modes, similar as the reversible mechanofluorochromic behaviors of Me-THP-2Br that we reported previously.38 We have prepared a series of polymorphs of THPs with different R1–R4 in single crystals, and studied the influence of molecular packing modes on their fluorescence properties.47,53 The research results (see Table S3†) show that the polymorphs with shorter or shortest wavelength emission adopt RS-packing, while polymorphs with longer wavelength emission adopt RR/SS-packing modes. For example, the λem value of 1b with RS-packing mode is shorter than that of its polymorph 1c with RR/SS-packing mode, and the λem value of 5p with RS-packing mode is the shortest among three polymorphs (other two polymorphs are 5b and 5c with RR/SS-packing mode (see Table S3†)). From these, we can deduce that the ground 2 and 3 with longer λem values (470 and 477 nm) than those of their crystals (442 and 436 nm) with RR/SS-packing mode should possess RR/SS-packing mode. This means that the reversible mechanofluorochromism of THPs 2 and 3 originates from the conversion between two kinds of RR/SS-packing modes. The as-prepared crystals packed in RS-paired and RR/SS-overlapped packing modes are stable until they are melted because the tightly packed molecules can't move freely and undergo packing rearrangement. However, the ordered RS- or RR/SS-arrangements in the as-prepared solid states of THPs 1–3 are mostly destroyed upon grinding and hence the kinetically favored RR/SS-packing alignments with more planarized molecular conformations are formed, accompanied by a red shift in emission wavelengths, which can convert into thermodynamically stable RS-paired (THP 1) or RR/SS-overlapped (THPs 2 and 3) packing mode with short emission wavelength upon fuming or heating (for the schematical mechanism of the mechanofluorochromism of THPs, see our previous work38).
Zhang et al. had studied the mechanofluorochromic behaviors of three phenothiazine modified triphenylacrylonitrile derivatives, and proposed that the more twisted conformation is the reason for the fluorescence self-recovery of compound P10TPAN.26 However, our experimental results do not support this conclusion. This is because that the purple-emitting polymorph of Me-THP-2Br (PC) with more twisted conformation (the dihedral angle value is 53.04°) than that of 1b (the dihedral angle value is 49.24°) doesn't have self-reversible mechanofluorochromic characteristics. After carefully analyzing and comparing the molecular interactions in single crystals of 1b, 2 and 3 with Me-THP-2Br in PC, we deduce that the self-reversible mechanofluorochromic characteristics of 1b, 2 and 3 relates to intermolecular hydrogen bonds. There exist eight intermolecular hydrogen bonds in 1b (6 × C–H⋯π, 2.930 Å; 2.837 Å; 2.971 Å; 2 × C–H⋯O, 2.487 Å), six intermolecular hydrogen bonds in 2 (2 × C–H⋯O, 2.453 Å; 2.321 Å; C–H⋯F, 2.550 Å; 3 × C–F⋯π, 3.236 Å; 3.517 Å; 3.477 Å), and three intermolecular hydrogen bonds in 3 (C–H⋯π, 2.847 Å; C–H⋯O, 2.432 Å; C–H⋯F, 2.459 Å), respectively (see Table S2†), while none exist in PC,47 which means that the intermolecular hydrogen bond interaction in 1b, 2 and 3 is much stronger than that in PC. During the grinding process, the stronger intermolecular interaction makes the molecular packing in 1b, 2 and 3 more difficult to be destroyed, and the molecular packing can be easily restored along with the release of grinding. The DSC curve (Fig. 5b) shows that the cold-crystallization temperatures of 1b, 2 and 3 are 55.4, 82.3 and 57.6 °C, respectively, which proves that the ground powder can recrystallize easily, meaning that the ground powders of THP 1b, 2 and 3 packed in metastable RR/SS-mode can spontaneously change into stable blue-emitting RS- or RR/SS-packing mode owing to the relatively stronger hydrogen bond interactions in blue-emitting crystals. Since there is no such relatively stronger hydrogen bond in RS-paired mode, the ground powder of Me-THP-2Br packed in RR/SS-overlapped mode can't spontaneously change into stable RS-paired mode. The self-reversible mechanofluorochromic behavior of these THPs may afford a new kind of smart self-healing fluorescence materials and have great potential in practical applications.
4. Conclusions
In summary, we have studied the reversible mechanofluorochromism of three racemic C6-unsubstituted tetrahydropyrimidine derivatives (THPs 1–3) with AIE characteristics. The three THPs exhibit sensitive and reversible mechanofluorochromic behaviors, the blue-green/cyan emissions can be switched reversibly by grinding and fuming or heating. Furthermore, the ground powders of 1b, 2 and 3 display fluorescence self-recovery without exertion of any external stimuli under room temperature. On the basis of photophysical, XRD, DSC, X-ray single-crystal diffraction results, we suggest that the reversible mechanofluorochromism of THPs originate from the conversion between the metastable RR/SS-packing and stable RS- or RR/SS-packing modes of enantiomers. Furthermore, we propose that the intermolecular hydrogen bond interaction may account for the fluorescence self-recovery of 1b, 2 and 3 at room temperature. After grinding, the ordered RS- or RR/SS-packing can be converted into short-range ordered RR/SS-packing. Upon fuming or heating, the metastable amorphous state transforms into thermodynamically stable packing modes, or self-recovers to its stable initial crystalline state without external stimuli at room temperature. The self-reversible mechanofluorochromic behavior of these THPs may afford a new kind of smart self-healing fluorescence materials and have great potential in practical applications.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported by the Special Funds for the Cultivation of Guangdong College Students' Scientific and Technological Innovation (“Climbing Program” Special Funds) (pdjh2016a0092), and Guangdong Natural Science Foundation (2018A030313976).
Notes and references
- J. Zhao, Z. Chi, Y. Zhang, Z. Mao, Z. Yang, E. Ubba and Z. Chi, J. Mater. Chem. C, 2018, 6, 6327–6353 RSC.
- X. Huang, L. Qian, Y. Zhou, M. Liu, Y. Cheng and H. Wu, J. Mater. Chem. C, 2018, 6, 5075–5096 RSC.
- P. Xue, J. Ding, P. Wang and R. Lu, J. Mater. Chem. C, 2016, 4, 6688–6706 RSC.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS.
- T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS.
- R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
- W. Yuan, Y. Tan, Y. Gong, P. Lu, J. Lam, X. Shen, C. Feng, H. Sung, Y. Lu, I. D. Williams, J. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- G. Feng, R. T. K. Kwok, B. Z. Tang and B. Liu, Appl. Phys. Rev., 2017, 4, 021307 Search PubMed.
- H. Wang, E. G. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365–377 CrossRef CAS.
- X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, W. Zhou, S. Liu, Y. Zhang and J. Xu, Chem.–Asian J., 2011, 6, 808–811 CrossRef CAS.
- H. Li, X. Zhang, Z. Chi, B. Xu, W. Zhou, S. Liu, Y. Zhang and J. Xu, Org. Lett., 2011, 13, 556–559 CrossRef CAS.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS.
- S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS.
- J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS.
- H. Gao, D. Xu, X. Liu, A. Han, L. Zhou, C. Zhang, Z. Li and J. Dang, Dyes Pigm., 2017, 139, 157–165 CrossRef CAS.
- M. Chen, L. Li, H. Nie, J. Tong, L. Yan, B. Xu, J. Z. Sun, W. Tian, Z. Zhao, A. Qin and B. Z. Tang, Chem. Sci., 2015, 6, 1932–1937 RSC.
- J. Tong, Y. Wang, J. Mei, J. Wang, A. Qin, J. Z. Sun and B. Z. Tang, Chem.–Eur. J., 2014, 20, 4661–4670 CrossRef CAS.
- Z. Yang, Z. Chi, T. Yu, X. Zhang, M. Chen, B. Xu, S. Liu, Y. Zhang and J. Xu, J. Mater. Chem. C, 2009, 19, 5541–5546 RSC.
- Y. Zhang, Q. Song, K. Wang, W. Mao, F. Cao, J. Sun, L. Zhan, Y. Lv, Y. Ma and B. Zou, J. Mater. Chem. C, 2015, 3, 3049–3054 RSC.
- G. Zhang, J. Sun, P. Xue, Z. Zhang, P. Gong, J. Peng and R. Lu, J. Mater. Chem. C, 2015, 3, 2925–2932 RSC.
- P. Galer, R. C. Korosec, M. Vidmar and B. Sket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS.
- Z. Zhang, Z. Wu, J. Sun, B. Yao and R. Lu, J. Mater. Chem. C, 2016, 4, 2854–2861 RSC.
- M. Liu, L. Zhai, J. Sun, P. Xue, P. Gong, Z. Zhang, J. Sun and R. Lu, Dyes Pigm., 2016, 128, 271–278 CrossRef CAS.
- X. Zhang, Z. Chi, X. Zhou, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. C, 2012, 116, 23629–23638 CrossRef CAS.
- X. Zhang, Z. Ma, Y. Yang, X. Zhang, Z. Chi, S. Liu, J. Xu, X. Jia and Y. Wei, Tetrahedron, 2014, 70, 924–929 CrossRef CAS.
- Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li and L. Ye, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS.
- H. Zhao, Y. Wang, S. Harrington, L. Ma, S. Hu, X. Wu, H. Tang, M. Xue and Y. Wang, RSC Adv., 2016, 6, 66477–66483 RSC.
- S. J. Yoon and S. Park, J. Mater. Chem., 2011, 21, 8338–8346 RSC.
- M. Chen, H. Nie, B. Song, L. Li, J. Sun, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 2901–2908 RSC.
- P. Hariharan, N. Venkataramanan, D. Moon and S. Anthony, J. Phys. Chem. C, 2015, 119, 9460–9469 CrossRef CAS.
- P. Xue, P. Chen, J. Jia, Q. Xu, J. Sun, B. Yao, Z. Zhang and R. Lu, Chem. Commun., 2014, 50, 2569–2571 RSC.
- Y. Liu, Z. Ye, M. Zhao, Q. Chen, Y. Wang and Q. Zhu, Dyes Pigm., 2017, 145, 391–398 CrossRef CAS.
- Y. Ji, Z. Peng, B. Tong, J. Shi, J. Zhi and Y. Dong, Dyes Pigm., 2017, 139, 664–671 CrossRef CAS.
- Z. He, L. Zhang, J. Mei, T. Zhang, J. W. Y. Lam, Z. Shuai, Y. Q. Dong and B. Z. Tang, Chem. Mater., 2015, 27, 6601–6607 CrossRef CAS.
- M. Yuan, D. Wang, P. Xue, W. Wang, J. Wang, Q. Tu, Z. Liu, Y. Liu, Y. Zhang and J. Wang, Chem. Mater., 2014, 26, 2467–2477 CrossRef CAS.
- R. Li, S. Xiao, Y. Li, Q. Lin, R. Zhang, J. Zhao, C. Yang, K. Zou, D. Li and T. Yi, Chem. Sci., 2014, 5, 3922–3928 RSC.
- Y. Lai, J. Huang, S. Wu, Q. Zhu and Y. Liu, Dyes Pigm., 2019, 166, 8–14 CrossRef CAS.
- X. Luo, W. Zhao, J. Shi, C. Li, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, J. Phys. Chem. C, 2012, 116, 21967–21972 CrossRef CAS.
- X. Luo, J. Li, C. Li, L. Heng, Y. Q. Dong, Z. Liu, Z. Bo and B. Z. Tang, Adv. Mater., 2011, 23, 3261–3265 CrossRef CAS.
- Q. Zhu, L. Huang, Z. Chen, S. Zheng, L. Lv, Z. Zhu, D. Cao, H. Jiang and S. Liu, Chem.–Eur. J., 2013, 19, 1268–1280 CrossRef CAS.
- Q. Zhu, Y. Zhang, H. Nie, Z. Zhao, S. Liu, K. S. Wong and B. Z. Tang, Chem. Sci., 2015, 6, 4690–4697 RSC.
- Q. Zhu, C. Dai, C. Huang, S. Zheng and Y. Tian, J. Phys. Chem. C, 2017, 121, 25503–25508 CrossRef CAS.
- Q. Zhu, W. Yang, S. Zheng, H. H. Y. Sung, I. D. Williams, S. Liu and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 7383–7386 RSC.
- S. Zheng, C. Huang, X. Zhao, Y. Zhang, S. Liu and Q. Zhu, Spectrochim. Acta, Part A, 2018, 189, 231–238 CrossRef CAS.
- X. Cai, W. Yang, L. Huang, Q. Zhu and S. Liu, Sens. Actuators, B, 2015, 219, 251–260 CrossRef CAS.
- Q. Zhu, L. Huang, J. Su and S. Liu, Chem. Commun., 2014, 50, 1107–1109 RSC.
- Q. Zhu, S. Wu, S. Zheng, Z. Ye, C. Huang and Y. Liu, Dyes Pigm., 2019, 162, 543–551 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data, 1H NMR and 13C NMR spectra, fluorescence decay profiles and other data of THPs 1–3. CCDC 2020055 and 2020056. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra09209f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yunhui Liao,
Ziwei Ye,
Lina Chen,
Yun He,
Yifan Huang,
Yingyu Lai,
Junguo Chen and
Qiuhua Zhu
*,
Yunhui Liao,
Ziwei Ye,
Lina Chen,
Yun He,
Yifan Huang,
Yingyu Lai,
Junguo Chen and
Qiuhua Zhu