
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMelodinines Y1–Y4, four monoterpene indole alkaloids from Melodinus henryi†
Fa-Lei Zhang,
Juan He,
Tao Feng * and
Ji-Kai Liu*
* and
Ji-Kai Liu*
School of Pharmaceutical Sciences, South-Central University for Nationalities, Wuhan 430074, People's Republic of China. E-mail: tfeng@mail.scuec.edu.cn; jkliu@mail.kib.ac.cn
First published on 22nd December 2020
Abstract
Melodinines Y1–Y4 (1–4), four undescribed alkaloids were isolated from Melodinus henryi. Their structures were elucidated by extensive NMR, mass spectroscopic analyses, theoretical NMR and electronic circular dichroism (ECD) calculations. Compounds 1 and 2 are the first examples of bisindole alkaloids possessing an eburnamine–leuconoxine combination. Compound 3 is a rare 2,3-seco pleiocarpamine type monoterpene indole alkaloid. Compound 1 showed cytotoxic activities against six human cancer cell lines with IC50 values of 0.5–15.2 μM.
Introduction
Monoterpene indole alkaloids (MIAs) are undoubtedly an important class of natural products that have attracted much attention due to their structural diversity and remarkable biological activity.1–4 The genus Melodinus (Apocynaceae) contains 53 species all over the world, 11 of which are mainly distributed in southern China and some of them have been used to treat conditions such as bellyache, dyspepsia, orchitis, and rheumatic heart disease and as a Chinese folk medicine.5 It has been demonstrated that plants of the genus Melodinus are rich in MIAs.6 So far, more than 280 MIAs have been obtained from these plants.6–9 Of them, over 40 bisindole alkaloids, comprised by two MIA units, have been identified, and most of them have been reported to demonstrate cytotoxicity against several human cancer cell lines in vitro.6–9M. henryi is a cane distributed in China, Thailand, and Burma. The fruits of M. henryi is used for treating meningitis and fracture in China.10 Previous chemical investigations on this plant reported a series of MIAs with new carbon skeletons and significant bioactivities.9–17 For instance, melohenine A was isolated as a novel C24 MIA with a rigid eight-ring system,17 melohenine B was isolated as a ketolactam derivative, regarded as a key intermediate from indole to quinolone,17 while melodinine V was a vincanol-eburenine bisindole alkaloids showing selective cytotoxic activities against human HT-29 cells.9 It induced cell cycle arrest at G1 phase and cellular apoptosis by increasing histone-associated DNA fragmentation in the treated HT-29 cells.9 Inspired by the novel structure and remarkable biological activity, we have been engaged in research on the chemical composition of the plant for a long time.11,13,16,17 In the current study, four new MIAs, melodinines Y1–Y4 (1–4), have been isolated from the bark of M. henryi (Fig. 1). Their structures with absolute configurations were elucidated by means of spectroscopic and computational methods. Melodinines Y1 and Y2 (1 and 2) are two novel bisindole alkaloids representing the first example of an eburnamine–leuconoxine combination. Melodinine Y3 (3) is a rare 2,3-seco pleiocarpamine type MIA. All compounds were evaluated for their cytotoxicity against six human cancer cell lines. Herein, the isolation, structural elucidation, and biological activities of these compounds are reported.
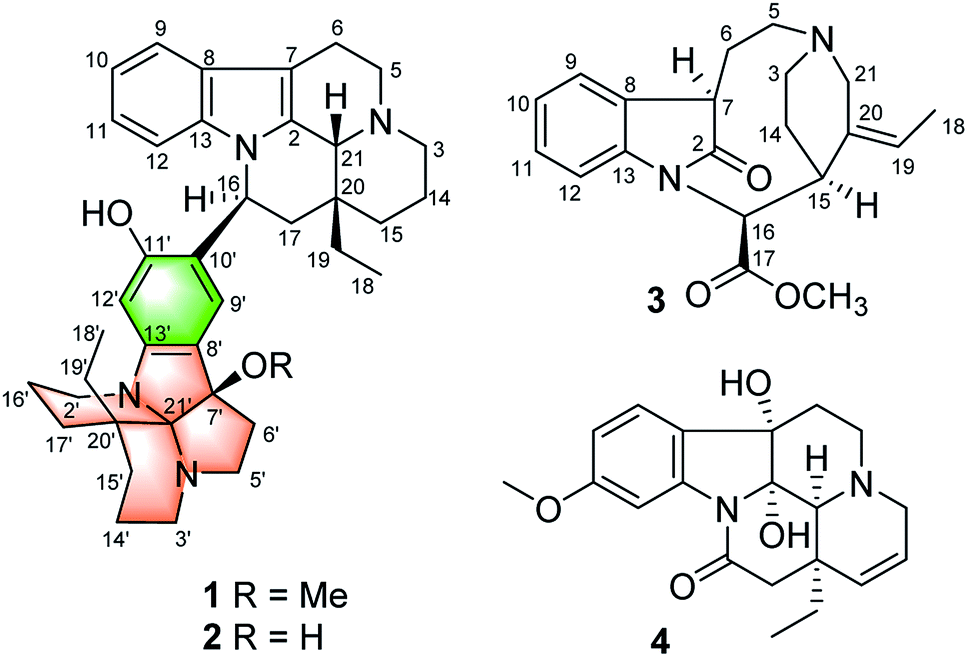 | ||
| Fig. 1 Compounds 1–4 isolated from M. henryi. | ||
Results and discussion
The molecular formula of compound 1 was established as C39H50N4O2 by the positive high resolution (HR) ESI mass spectrum, indicating 17 degrees of unsaturation. It's UV absorption bands at 293 and 207 nm suggesting an indole chromophore,7 and the IR absorption bands at 3418 cm−1 indicated NH and/or OH functionalities. The 1D NMR data revealed that 1 possessed three methyl, 16 methylene, 8 methine, and 12 quaternary carbons (Table 1). Compound 1 was identified preferentially as a bisindole alkaloid with a combination of an eburnamine and a leuconoxine unit, similar to isoeburnamine10 (unit A) and leuconoxine18 (unit B), after analyzed all of the NMR data.| Unit A | Unit B | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | 1 δH | 1 δC | 2 δH | 2 δC | No. | 1 δH | 1 δC | 2 δH | 2 δC |
| 2 | 131.9 s | 132.3 s | 2′ | 2.97 m; 3.26 m | 58.2 t | 3.24 m; 3.63 m | 57.7 t | ||
| 3 | 2.58 t (10.0); 2.80 m | 45.5 t | 2.57 m; 2.77 m | 45.4 t | 3′ | 2.75 m; 3.45 m | 63.6 t | 3.27 m | 61.7 t |
| 5 | 3.46 m | 52.0 t | 3.42 m | 51.9 t | 5′ | 2.91 m; 3.33 m | 64.5 t | 3.02 m; 3.49 m | 63.5 t |
| 6 | 2.82 m; 3.03 m | 17.6 t | 2.81 m; 3.04 m | 17.7 t | 6′ | 2.39 m; 2.48 m | 34.0 t | 2.30 m; 2.60 m | 39.2 t |
| 7 | 105.2 s | 105.1 s | 7′ | 94.3 s | 90.4 s | ||||
| 8 | 129.3 s | 129.2 s | 8′ | 117.4 s | 118.9 s | ||||
| 9 | 7.41 d (7.8) | 119.2 d | 7.42 d (8.0) | 118.9 d | 9′ | 6.78 s | 124.9 d | 6.85 s | 123.1 d |
| 10 | 6.94 t (7.8) | 120.7 d | 6.97 t (8.0) | 120.5 d | 10′ | 122.7 s | 124.3 s | ||
| 11 | 6.81 t (7.8) | 121.9 d | 6.90 t (8.0) | 122.0 d | 11′ | 158.5 s | 157.4 s | ||
| 12 | 6.63 d (7.8) | 113.1 d | 6.79 d (8.0) | 112.7 d | 12′ | 6.48 s | 98.3 d | 6.40 s | 98.3 d |
| 13 | 137.7 s | 137.9 s | 13′ | 150.8 s | 148.1 s | ||||
| 14 | 1.46 m; 1.82 m | 20.6 t | 1.47 m; 1.86 m | 20.7 t | 14′ | 1.70 m | 26.3 t | 1.68 m | 31.4 t |
| 15 | 1.15 m; 1.53 m | 24.4 t | 1.12 m; 1.53 m | 24.5 t | 15′ | 1.91 m; 2.41 m | 26.9 t | 2.14 m; 2.40 m | 29.6 t |
| 16 | 5.55 dd (11.0, 4.5) | 49.9 d | 5.57 dd (10.8, 4.6) | 48.5 d | 16′ | 1.82 m; 2.30 m | 20.0 t | 1.84 m; 2.41 m | 20.7 t |
| 17 | 1.67 m; 2.37 m | 42.7 t | 1.73 m; 2.31 m | 43.5 t | 17′ | 1.40 m; 1.61 m | 33.2 t | 1.61 m; 1.84 m | 31.9 t |
| 18 | 0.93 t (7.2) | 7.7 q | 0.89 t (7.4) | 7.7 q | 18′ | 0.89 t (7.4) | 7.1 q | 0.93 t (7.5) | 7.1 q |
| 19 | 1.54 m; 2.09 m | 29.3 t | 1.54 m; 2.14 m | 29.4 t | 19′ | 1.34 q (7.4) | 34.5 t | 1.30 m | 35.3 t |
| 20 | 36.4 s | 36.5 s | 20′ | 32.8 s | 33.0 s | ||||
| 21 | 4.38 s | 61.2 d | 4.33 s | 61.1 d | 21′ | 103.7 s | 102.1 s | ||
| OMe | 2.68 s | 53.0 q | |||||||
The 1H–1H COSY spectrum revealed nine partial structures a–i as shown in Fig. 2. In the 1H NMR spectrum, four aromatic protons at δH 7.41 (1H, d, J = 7.8 Hz), 6.94 (1H, t, J = 7.8 Hz), 6.81 (1H, t, J = 7.8 Hz) and 6.63 (1H, d, J = 7.8 Hz) were assigned to be the unsubstituted indole moiety in unit A, which were further supported by 1H–1H COSY and the key HMBC correlations of δH 7.41 (1H, d, J = 7.8 Hz, H-9) with δC 137.7 (s, C-13) and δH 6.63 (1H, d, J = 7.8 Hz, H-12) with δC 129.3 (s, C-8). And the key HMBC correlations of δH 3.03 (1H, m, H-6) and 2.82 (1H, m, H-6) with δC 105.2 (s, C-7), δH 4.38 (1H, s, H-21) with δC 131.9 (s, C-2), 45.5 (t, C-3), and 36.4 (s, C-20) suggested the connections of partial structures b and c through N-4 atom, and C-21 with N-4 and the connections of C-21 with C-2 and C-21 with C-20. In addition, the HMBC correlations also revealed the connections of structures c–e and C-21 with C-20. The above data suggested that unit A possessed an eburnan-type skeleton, similar to isoeburnamine.
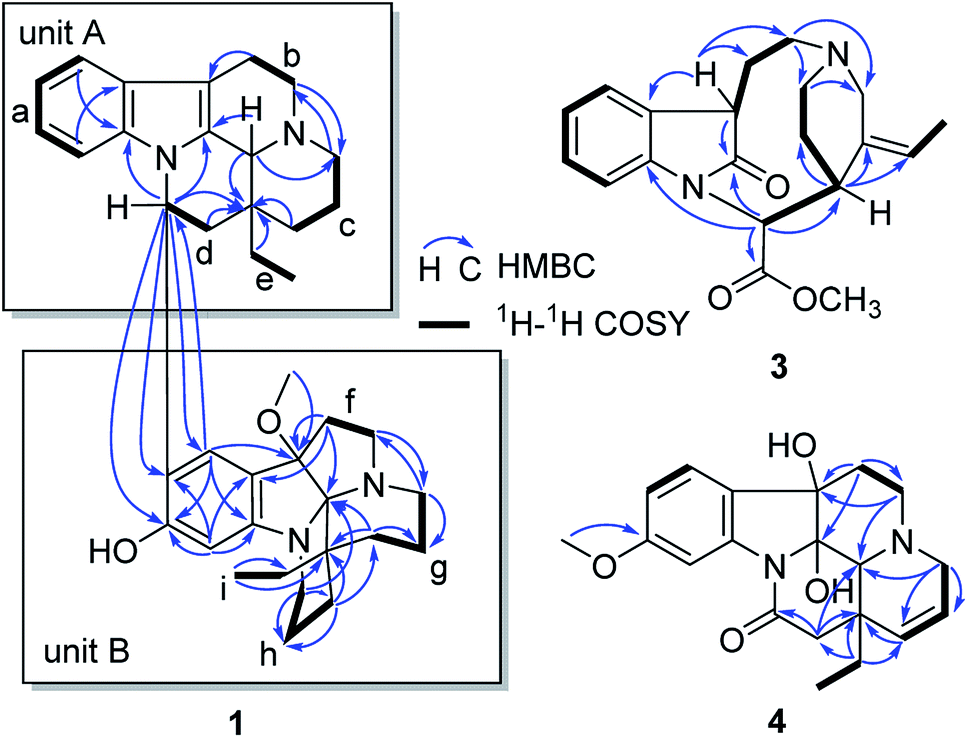 | ||
| Fig. 2 Key HMBC and 1H–1H COSY correlations of 1, 3, and 4. | ||
In the 1H NMR spectrum, two aromatic protons at δH 6.78 (1H, s) and 6.48 (1H, s) were assigned to a disubstituted benzene group in unit B (Fig. 2). In the 13C NMR spectrum, eight signals at δC 158.5 (s), 150.8 (s), 124.9 (d), 122.7 (s), 117.4 (s), 98.3 (d), 103.7 (s), and 94.3 (s) showed characteristics for an indolenine moiety. A methoxy placed at C-7′ was established by the HMBC correlation from δH 2.68 (3H, H3–OMe) to δC 94.3 (s, C-7′). One sp3 quaternary carbon at δC 103.7 (s, C-21′) suggested that unit B might have a structure similar to leuconoxine. The HMBC correlations, associated with the 1H–1H COSY data, established unit B as given in Fig. 2. The linkage of between units A and B by the bond C-16/C-10′ was established by HMBC correlations of δH 5.55 (1H, dd, J = 11.0, 4.5 Hz, H-16) with δC 122.7 (s, C-10′), 158.5 (s, C-11′), and 124.9 (d, C-9′), and of δH 6.78 (1H, s, H-9′) with 49.9 (d, C-16). This data also determined the placement for the OH at C-11′ unambiguously.
The relative configuration of 1 was elucidated by the ROESY spectrum and the 1H–1H coupling constants. The coupling constant of H-16 (dd, J = 11.0, 4.5 Hz) suggested H-16 to be α-oriented by comparison with that of isoeburnamine (dd, J = 11.5, 4.8 Hz).10 The ROESY correlations of H-16/H-15a and H-21/H-19 suggested that H-21 and the ethyl were on the opposite side (Fig. 3).7 The ROESY correlation of H3–OMe/H-15a′ suggested that the methoxy and the ethyl were on the same side (β-orientation) in unit B (Fig. 3), the same as that of leuconoxine.
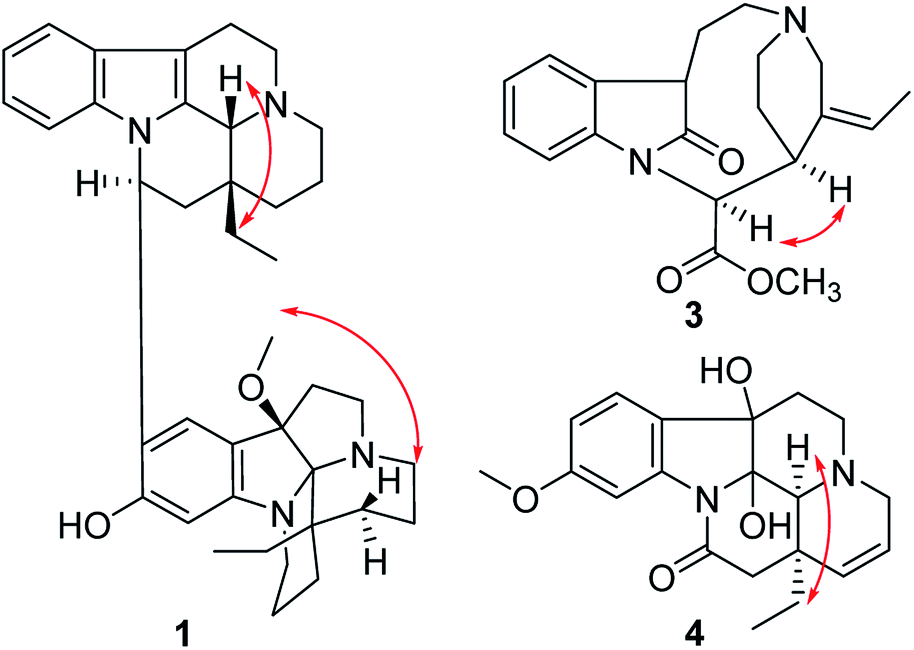 | ||
| Fig. 3 Key ROESY correlations of 1, 3, and 4. | ||
To determine the linkage mode of units A and B, the theoretical NMR calculations and DP4+ probability analyses were employed (see ESI†). The 13C NMR chemical shifts of 1A and 1B were calculated at the B3LYP/6-311+G(d,p) level with the PCM model in methanol. The calculated results for 1A (R2 = 0.9840) were a better match with the experimental data than those of 1B (R2 = 0.9649) (Fig. S3†). Moreover, according to the DP4+ probability analyses, 1A was assigned with a 100% probability (Table S5†). The messages suggested the 1A was the correct relative structure for 1.
The absolute configuration of 1 was determined by ECD calculations. As shown in Fig. 4, the calculated ECD spectrum of (16S,20R,21R,7′R,20′R)-1A matched well with the experimental one, suggesting the absolute configuration of 1 to be 16S,20R,21R,7′R,20′R (the ECD curve was simulated in SpecDis V1.71 using a Gaussian function with sigma/gamma value 0.15 eV and a UV shift of −31 nm). Therefore, the structure of compound 1 was identified and named as melodinine Y1.
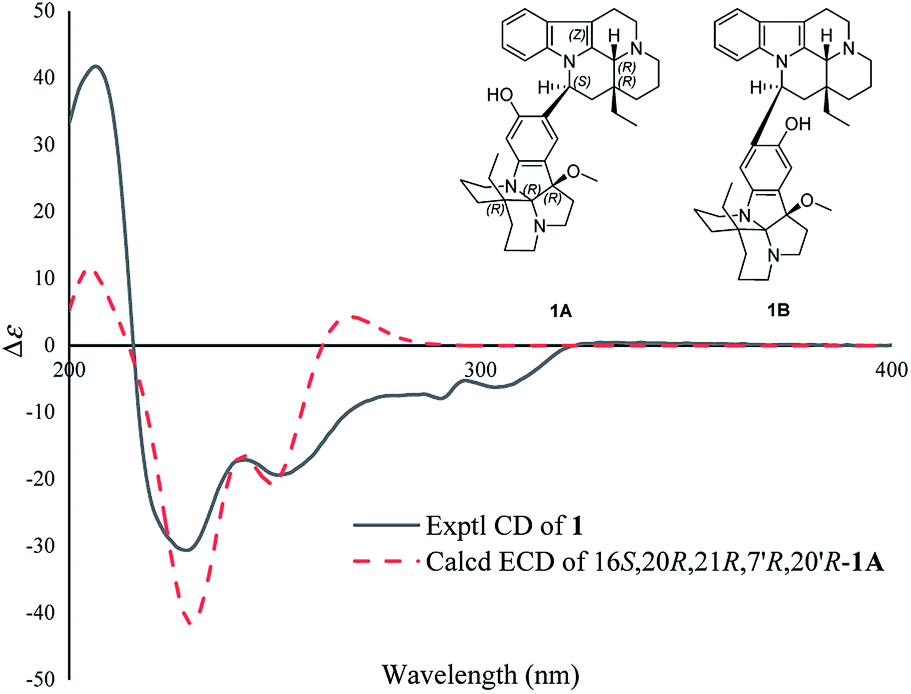 | ||
| Fig. 4 Comparison of the calculated ECD spectra with the PCM model for 1A with the experimental spectrum of 1 in methanol. | ||
Compound 2 was isolated as a white powder, and its molecular formula was established as C38H48N4O2 by HRESIMS data, indicating 17 degrees of unsaturation. With the aid of DEPT and HSQC spectra, the 38 carbon resonances as displayed by the 13C NMR spectrum could be ascribed to two CH3, sixteen CH2, eight CH, and twelve non-protonated carbons (Table 1). Preliminary analyses of these spectroscopic features implied that 2 had the same carbon skeleton as that of 1, except that one hydroxy group at C-7′ in 2 replaced the methoxy group in 1, which was supported by the HRESIMS data. Detailed analysis of 2D NMR data suggested that the other parts of 2 were the same to those of 1. The absolute configuration of 2 was suggested to be 16S,20R,21R,7′R,20′R by comparison with CD curve to that of 1 (see Fig. S32 in the ESI†). Therefore, compound 2 was identified as melodinine Y2.
Compound 3 was isolated as a white powder, and the molecular formula of 3 was established as C20H24N2O3 by HRESIMS data. The 13C NMR and DEPT spectra of 3 displayed 20 carbon resonances ascribed to two CH3 (including a methoxy), five CH2, eight CH, and five C (Table 2). The 1D NMR spectra of 3 were very similar to those of the known compound, secopleiocarpamine A,19 except for the presence of a methylene group rather than a –CN group at C-3 in compound 3. It was confirmed by the key 2D NMR correlations (Fig. 2). The ROESY correlation of H-15/H-16 suggested that H-15 and H-16 were on the same side (Fig. 3). However, the orientation of H-7 could not been deduced from the ROESY correlations. To determine its final structure, the theoretical NMR calculations and DP4+ probability analyses were employed on two possible structures (7R,15S,16S)-3a and (7S,15S,16S)-3b, and the calculations messages suggested that (7R,15S,16S)-3a was the correct relative structure for 3 (Table S11†). As shown in Fig. 5, the absolute configuration of 3 was suggested to be 7R,15S,16S by means of ECD calculation. Therefore, compound 3 was identified as melodinine Y3.
| No. | 3 | 4 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 2 | 184.1 s | 93.1 s | ||
| 3 | 1.96 m; 2.72 m | 46.1 t | 2.88 dt (17.0, 2.0); 3.29 d (4.7) | 55.7 t |
| 5 | 1.94 m; 2.21 m | 51.7 t | 2.59 td (13.0, 3.8); 2.70 dt (11.6, 3.4) | 50.3 t |
| 6 | 2.19 m; 2.55 m | 31.9 t | 1.60 td (13.0, 3.8); 2.01 dt (11.6, 3.4) | 37.5 t |
| 7 | 3.39 d (5.5) | 45.2 d | 79.3 s | |
| 8 | 130.1 s | 130.9 s | ||
| 9 | 7.27 d (7.5) | 125.0 d | 7.26 d (8.5) | 124.0 d |
| 10 | 7.03 t (7.5) | 123.2 d | 6.71 dd (8.5, 2.3) | 111.3 d |
| 11 | 7.05 t (7.5) | 127.3 d | 161.6 s | |
| 12 | 6.68 d (7.5) | 114.3 d | 7.65 d (2.3) | 106.1 d |
| 13 | 145.8 s | 141.2 s | ||
| 14 | 1.67 m; 2.25 m | 31.0 t | 5.77 dd (10.0, 4.7) | 125.2 d |
| 15 | 3.78 d (7.5) | 35.2 d | 5.47 dt (10.0, 2.0) | 135.4 d |
| 16 | 5.43 br s | 63.2 d | 170.2 s | |
| 17 | 172.7 s | 2.35 d (17.3); 2.91 d (17.3) | 45.1 t | |
| 18 | 1.73 dt (7.0, 2.0) | 13.9 q | 0.96 t (7.3) | 9.5 q |
| 19 | 4.86 m | 121.7 d | 1.42 dq (14.5, 7.3); 2.28 dq (14.5, 7.3) | 34.0 t |
| 20 | 136.5 s | 40.4 s | ||
| 21 | 1.60 m; 2.68 m | 59.6 t | 2.79 s | 61.8 d |
| OMe | 3.84 s | 53.0 q | 3.79 s | 56.0 q |
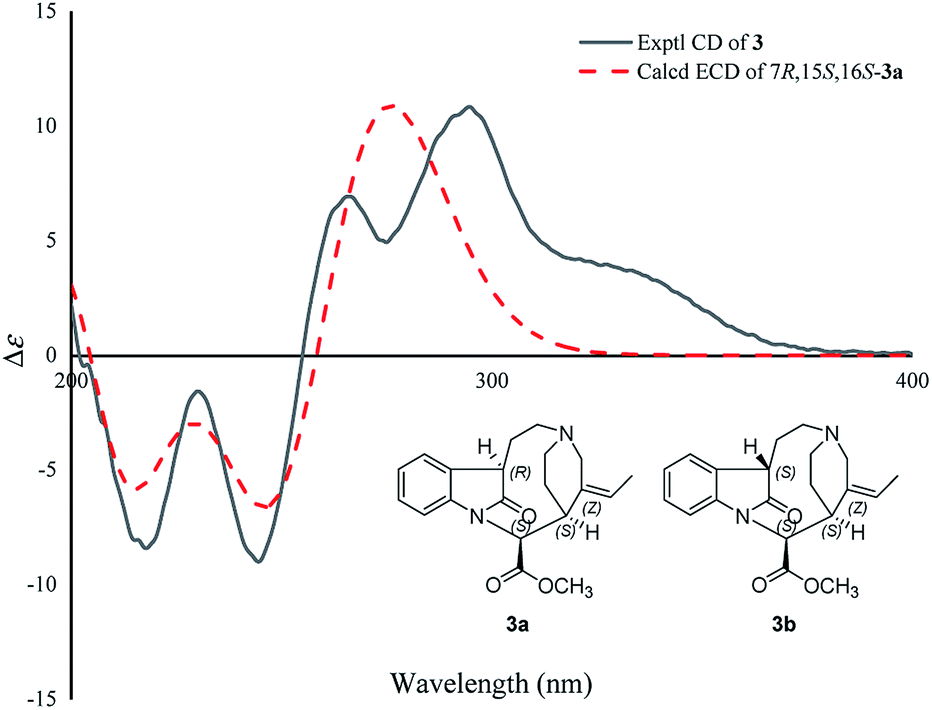 | ||
| Fig. 5 Comparison of the calculated ECD spectra with the PCM model for 3a with the experimental spectrum of 3 in methanol. | ||
The molecular formula of 4, C20H24N2O4, was determined by the positive HR-ESI mass spectrum. According to the comparison of the 13C NMR spectra with that of Δ14-vincinone,20 the difference between them was two hydroxyl groups replaced the double bonds between C-2 and C-7 in 4, which was supported by the HRESIMS data. The relative configuration of 4 was determined by the ROESY correlation of H-19/H-21 (Fig. 3), which allowed the configurations of H-21 and ethyl to be assigned as α. However, the relative configuration of the two hydroxyl groups could not be defined by the ROESY data alone. The theoretical NMR calculations and DP4+ probability were employed to determine its final structure. As shown in Fig. S13,† the 13C NMR chemical shifts of (2R,7R,20R,21S)-4a, (2S,7S,20R,21S)-4b, (2R,7S,20R,21S)-4c and (2S,7R,20R,21S)-4d were calculated at the B3LYP/6-311+G(d,p) level with the PCM model in methanol. The calculated results for 4b (R2 = 0.9980) were a better match with the experimental data than others (Fig. S13†). Moreover, according to the DP4+ probability analyses, 4b was assigned with a 100% probability (Table S20†). The absolute configuration of 4 was suggested to be 2S,7S,20R,21S by means of ECD calculation at the B3LYP/6-31+G(d,p)//B3LYP/6-31G(d) level was compared with the experimental ECD curve (Fig. 6). Therefore, compound 4 was identified as melodinine Y4.
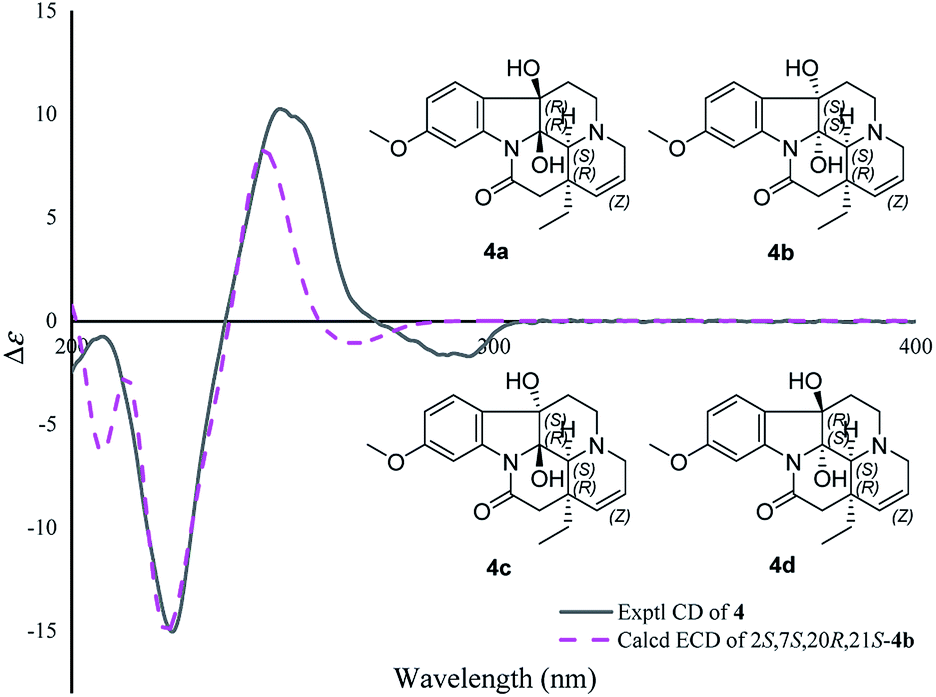 | ||
| Fig. 6 Comparison of the calculated ECD spectra with the PCM model for 4b with the experimental spectrum of 4 in methanol. | ||
All compounds were investigated for their cytotoxicity against SK-BR-3, SMMC7721, HL-60, PANC-1, A549, and MCF-7 in vitro. As a result, compound 1 showed cytotoxicity toward six cancer cell lines, especially strongly inhibited HL-60 cell with an IC50 value of 0.5 μM (Table 3). The other compounds were inactive at the concentration of 40 μM. From the results of compounds 1 and 2, it can be inferred that the methoxy group at position 7′ is the necessary for cytotoxicity.
| No. | SK-BR-3 | SMMC7721 | HL-60 | PANC-1 | A549 | MCF-7 |
|---|---|---|---|---|---|---|
| 1 | 7.8 | 7.4 | 0.5 | 14.5 | 14.7 | 15.2 |
| Cisplatin | 22.5 | 14.1 | 1.8 | 19.5 | 26.5 | 13.8 |
Conclusions
In summary, four new MIAs including two bisindole alkaloids with an unusual eburnamine–leuconoxine combination, and a rare 2,3-seco pleiocarpamine type alkaloid were isolated from the bark of Melodinus henryi. Their structures with absolute configurations were established. Compound 1 showed cytotoxicity toward six cancer cell lines. The new modification of the isolated compounds expand the chemical diversity of the MIAs family. Furthermore, cytotoxicity assays have demonstrated that compound 1 significantly inhibited HL-60 cancer cell line, presenting us with a great opportunity to discover promising natural agents for new antitumor leadings.Experimental section
General experimental procedures
Optical rotations were measured with an Autopol IV polarimeter (Rudolph, Hackettstown, USA). UV spectra were obtained using a Double Beam Spectrophotometer UH5300 (Hitachi High-Technologies, Tokyo, Japan). IR spectra were obtained by a Shimadzu IRTracer-100 spectrometer using KBr pellets. CD spectra were recorded with an Applied Photophysics Chirascan-Plus CD Spectrometer (Chirascan, New Haven, USA). 1D and 2D NMR spectra were run on a Bruker Avance III 600 MHz spectrometer or on a Bruker DRX-500 spectrometer with TMS as an internal standard. Chemical shifts (δ) were expressed in ppm with reference to solvent signals. High resolution electrospray ionization mass spectra (HRESIMS) were recorded on an Agilent 6200 Q-TOF MS system or a LC-MS system consisting of a Q Exactive™ Orbitrap mass spectrometer with a HESI ion source (ThermoFisher Scientific, Bremen, Germany). HREIMS was recorded on a Waters Auto Premier P776 spectrometer or on an API QSTAR Pulsar I spectrometer. Column chromatography (CC) was performed on silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., Qingdao, People's Republic of China), RP-18 gel (20–45 μm, Fuji Silysia Chemical Ltd., Japan), and Sephadex LH-20 (Pharmacia Fine Chemical Co., Ltd., Sweden). Medium Pressure Liquid Chromatography (MPLC) was performed on a Büchi Sepacore System equipping with pump manager C-615, pump modules C-605 and fraction collector C-660 (Büchi Labortechnik AG, Flawil, Switzerland). Preparative High Performance Liquid Chromatography (prep-HPLC) was performed on an Agilent 1260 liquid chromatography system equipped with Zorbax SB-C18 columns (5 μm, 9.4 mm × 150 mm or 21.2 mm × 150 mm) and a DAD detector. Fractions were monitored by TLC (GF 254, Qingdao Haiyang Chemical Co., Ltd. Qingdao), and spots were visualized by Dragendorff's reagent.Plant material
The bark of M. henryi was collected from Mengla County, Yunnan province, P. R. China on August 12th, 2017. The plant was identified by Mr Yu Chen of Kunming Institute of Botany, Chinese Academy of Sciences. A voucher specimen (No. YChen20170812.MHb) has been deposited at School of Pharmaceutical Sciences, Chinese Academy of Sciences.Extraction and isolation
The powdered bark of M. henryi (10.0 kg) were extracted three times with 90% EtOH. The combined extracts were concentrated under reduced pressure, and adjusted to pH = 2–3 with 5‰ HCl. The acidic mixture was defatted with ethyl acetate (EtOAc) and then basified to pH = 9–10 with 10% ammonia solution. The aqueous phase was subsequently extracted with EtOAc to give an alkaloidal extract (201.3 g). The crude alkaloids were then subjected to a silicagel column (200–300 mesh) using CHCl3–MeOH gradient (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0–0:1) to obtain five fractions (A–E). Fraction C (5.1 g) was separated by silica gel CC (petroleum ether–Me2CO, 20:1–0:1) to afford subfractions C-1–C-3. Subfraction C-2 was purified on a preparative C18 HPLC column with a gradient of MeCN–H2O (60:40) to yield 1 (3.0 mg, retention time (tR) = 16.5 min) and 2 (1.5 mg, tR = 22.1 min). Subfraction C-3 was purified on a preparative C18 HPLC column with a gradient of MeCN–H2O (50:50) to yield 3 (2.0 mg, tR = 14.5 min) and 4 (2.2 mg, tR = 26.3 min).
ε) 293 (3.60), 207 (4.49) nm; IR (KBr) vmax 3418, 2926, 1624, 1456, 1384, 1319, 1180 cm−1; 1H NMR (500 MHz) and 13C NMR (100 MHz) data (CD3OD), see Table 1; positive ion HRESIMS m/z 607.4013 [M + H]+ (calcd for C39H51N4O2, 607.4012).ε) 374 (2.69), 294 (3.68), 207 (4.49) nm; IR (KBr) vmax 3422, 3246, 2937, 1626, 1457, 1308, 1136, 745 cm−1; 1H NMR (500 MHz) and 13C NMR (100 MHz) data (CD3OD), see Table 1; positive ion HRESIMS m/z 593.3879 [M + H]+ (calcd for C38H49N4O2, 593.3855).ε) 255 (2.99), 210 (3.49) nm; IR (KBr) vmax 3460, 2090, 1637, 1016, 615 cm−1; 1H NMR (600 MHz) and 13C NMR (150 MHz) data (CD3OD), see Table 2; positive ion HRESIMS m/z 341.18587 [M + H]+ (calcd for C20H25N2O3+, 341.18597).ε) 290 (3.55), 245 (3.79), 220 (4.01), 210 (3.93) nm; IR (KBr) vmax 3350, 2945, 2833, 1666, 1454, 1417, 1114, 1031, 667 cm−1; 1H NMR (600 MHz) and 13C NMR (150 MHz) data (CD3OD), see Table 2; positive ion HRESIMS m/z 357.18048 [M + H]+ (calcd for C20H25N2O4+, 357.18088).
:0–0:1) to obtain five fractions (A–E). Fraction C (5.1 g) was separated by silica gel CC (petroleum ether–Me2CO, 20:1–0:1) to afford subfractions C-1–C-3. Subfraction C-2 was purified on a preparative C18 HPLC column with a gradient of MeCN–H2O (60:40) to yield 1 (3.0 mg, retention time (tR) = 16.5 min) and 2 (1.5 mg, tR = 22.1 min). Subfraction C-3 was purified on a preparative C18 HPLC column with a gradient of MeCN–H2O (50:50) to yield 3 (2.0 mg, tR = 14.5 min) and 4 (2.2 mg, tR = 26.3 min).
ε) 293 (3.60), 207 (4.49) nm; IR (KBr) vmax 3418, 2926, 1624, 1456, 1384, 1319, 1180 cm−1; 1H NMR (500 MHz) and 13C NMR (100 MHz) data (CD3OD), see Table 1; positive ion HRESIMS m/z 607.4013 [M + H]+ (calcd for C39H51N4O2, 607.4012).ε) 374 (2.69), 294 (3.68), 207 (4.49) nm; IR (KBr) vmax 3422, 3246, 2937, 1626, 1457, 1308, 1136, 745 cm−1; 1H NMR (500 MHz) and 13C NMR (100 MHz) data (CD3OD), see Table 1; positive ion HRESIMS m/z 593.3879 [M + H]+ (calcd for C38H49N4O2, 593.3855).ε) 255 (2.99), 210 (3.49) nm; IR (KBr) vmax 3460, 2090, 1637, 1016, 615 cm−1; 1H NMR (600 MHz) and 13C NMR (150 MHz) data (CD3OD), see Table 2; positive ion HRESIMS m/z 341.18587 [M + H]+ (calcd for C20H25N2O3+, 341.18597).ε) 290 (3.55), 245 (3.79), 220 (4.01), 210 (3.93) nm; IR (KBr) vmax 3350, 2945, 2833, 1666, 1454, 1417, 1114, 1031, 667 cm−1; 1H NMR (600 MHz) and 13C NMR (150 MHz) data (CD3OD), see Table 2; positive ion HRESIMS m/z 357.18048 [M + H]+ (calcd for C20H25N2O4+, 357.18088).Cytotoxicity assay
Six human cancer cell lines, human myeloid leukemia HL-60, hepatocellular carcinoma SMMC-7721, lung cancer A-549, breast cancer SK-BR-3, breast cancer MCF-7, and pancreatic cancer PANC-1 cells, were used in the cytotoxic assay. All the cells were cultured in RPMI-1640 or DMEM medium (Hyclone, USA), supplemented with 10% fetal bovine serum (Hyclone, USA) in 5% CO2 at 37 °C. The assays were performed according to the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) method in 96-well microplates.21 Briefly, 100 μL of adherent cells was seeded into each well of 96-well cell culture plates and allowed to adhere for 12 h before drug addition, while suspended cells were seeded just before drug addition with an initial density of 1 × 105 cells per mL. Each tumor cell line was exposed to the test compound at concentrations of 0.064, 0.32, 1.6, 8, and 40 μM in triplicates for 48 h, with cisplatin (Sigma, USA) as a positive control. After each compound treatment, cell viability was detected and a cell growth curve was graphed. IC50 values were calculated by Reed and Muench's method.22Computation methods
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2017YFC1704007) and the National Natural Science Foundation of China (21961142008, 81872762). The authors thank the Analytical & Measuring Center of School of Pharmaceutical Sciences, South-Central University for Nationalities.Notes and references
- J. E. Saxton, Nat. Prod. Rep., 1995, 12, 385–411 RSC.
- T. Beckers and S. Mahboobi, Drugs Future, 2003, 28, 767 CrossRef CAS.
- N. I. Mohd and N. I. Magdy, Mini-Rev. Med. Chem., 2004, 4, 1077–1104 CrossRef.
- S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC.
- Y. Tsiang, in Flora of China, ed P. T. Li, Science Press, Beijing, 1977, vol. 63, pp. 17–30 Search PubMed.
- J. H. Jiang, W. D. Zhang and Y. G. Chen, Trop. J. Pharm. Res., 2015, 14, 2325–2344 CrossRef CAS.
- T. Feng, Y. Li, Y. Y. Wang, X. H. Cai, Y. P. Liu and X. D. Luo, J. Nat. Prod., 2010, 73, 1075–1079 CrossRef CAS.
- Y. P. Liu, Y. L. Zhao, T. Feng, G. G. Cheng, B. H. Zhang, Y. Li, X. H. Cai and X. D. Luo, J. Nat. Prod., 2013, 76, 2322–2329 CrossRef CAS.
- Y. P. Liu, G. G. Yue, J. K. Lee, T. Feng, Y. L. Zhao, Y. Li, C. B. Lau and X. D. Luo, Bioorg. Med. Chem. Lett., 2016, 26, 4895–4898 CrossRef CAS.
- Y. W. Zhang, R. Yang, Q. Cheng and K. Ofuji, Helv. Chim. Acta, 2003, 86, 415–419 CrossRef CAS.
- Q. Shao, R. Ma, X. Wu, F. L. Zhang, Z. H. Li, T. Feng, J. He and J. K. Liu, Phytochem. Lett., 2020, 35, 53–57 CrossRef CAS.
- J. Q. Yu, X. W. Sun, Z. W. Wang, L. Fang and X. Wang, J. Asian Nat. Prod. Res., 2019, 21, 820–825 CrossRef CAS.
- J. He, F. L. Zhang, Z. H. Li, H. X. Yang, Q. Shao, T. Feng and J. K. Liu, Fitoterapia, 2019, 138, 104354 CrossRef CAS.
- L. L. Guo, Y. X. Yuan, H. P. He, S. L. Li, Y. Zhang and X. J. Hao, Phytochem. Lett., 2017, 21, 179–182 CrossRef CAS.
- H. Zhou, H. P. He, Y. H. Wang and X. J. Hao, Helv. Chim. Acta, 2010, 93, 2030–2032 CrossRef CAS.
- T. Feng, X. H. Cai, Y. P. Liu, Y. Li, Y. Y. Wang and X. D. Luo, J. Nat. Prod., 2010, 73, 22–26 CrossRef CAS.
- T. Feng, X. H. Cai, Y. Li, Y. Y. Wang, Y. P. Liu, M. J. Xie and X. D. Luo, Org. Lett., 2009, 11, 4834–4837 CrossRef CAS.
- F. Abe and T. Yamauchi, Phytochemistry, 1993, 35, 169–171 CrossRef.
- A. Ahmed, W. Li, F. F. Chen, J. S. Zhang, Y. Q. Tang, L. Chen, G. H. Tang and S. Yin, Fitoterapia, 2018, 128, 1–6 CrossRef CAS.
- M. Zeches, J. Lounkokobi, B. Richard, M. Plat, L. Le Men-Olivier, T. Sevenet and J. Pusset, Phytochemistry, 1984, 23, 171–174 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- L. J. Reed and H. Muench, Am. J. Hyg., 1938, 27, 493–497 Search PubMed.
- W. J. Hehre, A guide to molecular mechanics and quantum chemical calculations, Wavefunction, Inc., Irvine, CA, 2003, pp. 1–812 Search PubMed.
- Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr, R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Yeh Lin, T. Van Voorhis, S. Hung Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. Min Rhee, J. Ritchie, E. Rosta, C. David Sherrill, A. C. Simmonett, J. E. Subotnik, H. Lee Woodcock Iii, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer Iii, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 RSC.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc.Wallingford CT, 2016 Search PubMed.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 12526–12534 CrossRef CAS.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09819a |
| This journal is © The Royal Society of Chemistry 2021 |