
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromatic nature of neutral and dianionic 1,4-diaza-2,3,5,6-tetraborinine derivatives†
Kei Ota and
Rei Kinjo*
and
Rei Kinjo*
Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371, Singapore. E-mail: rkinjo@ntu.edu.sg
First published on 4th January 2021
Abstract
The aromatically relevant parameters of boron-rich inorganic benzenes—neutral and dianionic 1,4-diaza-2,3,5,6-tetraborinine derivatives (B4N2R6)—have been computationally estimated and evaluated from geometric, electronic, magnetic, and energetic points of view. The majority of the criteria (ASE, NICSzz, ELF, and PDI) indicate that the aromaticity of the neutral B4N2 benzene analogue stabilized by Lewis bases lies in between those of benzene and borazine. On the other hand, the aromaticity of the dianionic B4N2 benzene analogue 4′ is controversial. The pronounced aromatic nature of 4′ is supported by ELFπ, PDI, and NICSπzz, but ASE, the FiPC-NICS plot, and ACID oppose this. These data confirm that even with the same B4N2-skeletal framework of a 6π-system, the aromatic feature varies depending on the overall charge of the B4N2 systems.
Introduction
Since the discovery of the cyclic feature of benzene (CH)6 1 in 1865, “aromaticity” has represented one of the most significant fundamental concepts in modern chemistry.1–3 Nowadays, its implication is not limited to the molecules obeying the [4n + 2] Hückel rule, but extended to those satisfying various criteria—structural, electronic, magnetic and energetic indices can be employed to assess the aromatic nature of a molecule comprehensively. It is well known that benzene 1, an archetypal aromatic molecule, displays a planar geometry, cyclic delocalization of 6π electrons, peculiar magnetic properties and extra thermodynamic stability.4,5Synthetically, incorporation of isolobal heteroatom units into the benzene scaffold is one of the useful strategies to modify the intrinsic electronic property of aromaticity without significant change of the geometric feature. Indeed, a variety of inorganic benzenes have been developed thus far,6 most of which are mainly based on the (XY)3 system, such as borazine (HBNH)3 2, boroxines (RBO)3 and phosphazenes (R2PN)3.7–10 The six-membered ring aromatics based on heavier elements have little been described.11–15
Theoretically, the aromaticity of inorganic benzenes has been evaluated using computational methods such as nucleus-independent chemical shift (NICS), aromatic stabilization energies (ASE) and electron localization function (ELF) analyses.16–26 In 1991, Fink and Richards reported the calculated ASE values of the (XY)3 ring systems (X = BR, AlR; Y = NR, PR),16 and revealed that the ASE values increase in order AlN < BN < BP < CC. In 1997, the group of Schleyer estimated the NICS values of the (XY)3 rings (X = BH, AlH, CH; Y = NH, PH, O, S), as well as, the E6 rings (E = SiH, GeH, N, P).17 The NICS values of the (XY)3 systems are found to be less negative than that of benzene whereas the E6 systems are comparable to benzene. In 1998, Jemmis et al. systematically evaluated the aromaticity of the (XY)3 molecules (X = BH, AlH, GaH, PH2; Y = NH, PH, AsH, N, O, S, Se) using ASE, magnetic susceptibility exaltation (MSE), and NICS analyses. As a result, they concluded that judicious use of all criteria is needed in gauging aromaticity of those systems.18 In 2005, Jenneskens and coworkers reported the electronic structure of the (XY)3 (X = BH, AlH; Y = NH, PH, O, S) and E6 (E = SiH, GeH, N, P) systems by using valence bond (VB) theory and ring-current maps. The authors unveiled that in general, the heteronuclear systems (XY)3 show localization of the lone pair of electrons on the electronegative atoms, and Kekulé-like structures do not contribute.19 In 2010, Phukan and Silvi et al. calculated the NICS, ASE, and ELF values for the substituted (XY)3 systems (X = BR, AlR; Y = NH, PH, AsH, O, S, Se). The results clarified the substituent effect—the bulky electronegative substituents (R) on B or Al atoms dramatically increases the stability and aromaticity.20 Recently, Alvarez-Thon and Tiznado examined the (XYH)3 systems (X = C–Pb, Y = N–Bi) by magnetically induced current density (MICD) and the zz component of NICS, and revealed that most of them are aromatic except for (XNH)3 (X = Si–Pb).21
The extant theoretical study on the aromaticity of inorganic benzenes mainly focuses on the above-mentioned (XY)3 and E6 systems, because those types of analogues have been isolated to date. Recently, we have reported the synthesis of a neutral B4N2-based benzene analogue, namely 1,4-diaza-2,3,5,6-tetraborinine derivative 3,27 being the first examples of the Lewis base-stabilized X4Y2 system.6 Preliminary theoretical investigations including NICS, natural bond orbital (NBO), and anisotropy of the current-induced density (ACID) analysis suggested the pronounced aromatic nature of 3. By contrast, the dianionic B4N2 ring system 4: [(Ph-N)2(Me2N-B)4]2– is found to furnish a non-aromatic twisted structure despite its 6π-electron configuration isoelectronic with 3.28 With the isolable neutral and dianionic B4N2 derivatives in hand, we wondered (i) how the aromatic nature of the B4N2 ring systems differs from the reported (XY)3 systems, and (ii) whether the charge of molecules affects the aromaticity or not. Here, we have investigated the aromaticity of 1,4-diaza-2,3,5,6-tetraborinine derivatives, B4N2 analogues of benzene, by using modern computational aromaticity descriptors involving structure, molecular orbital, electron density, magnetic shielding, and energetics.29 For purpose of comparison, reference molecules—benzene (CH)6 1, and borazine (HBNH)3 2 have also been examined (Scheme 1). The substituent effect on the aromaticity in benzene as well as in borazine has been reported.30–36 According to those prior studies, while the aromaticity of benzene resists substituent influences, that of borazine is significantly affected by the nature of substituents. In borazine, generally the electron-donating group (EDG) on B decreases the aromaticity, although electron-withdrawing group (EWG) on B and both EDG and EWG on N lead to the pronounced aromaticity. Therefore, we employed model compound 3′, and a dianionic B4N2H6 derivative 4′ (Scheme 1).
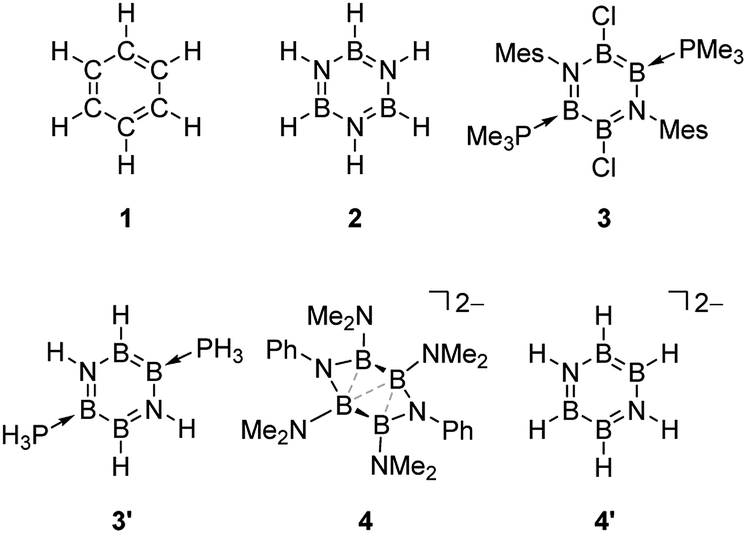 | ||
| Scheme 1 The list of the molecules discussed in this article (Mes = 2,4,6-Me3C6H2). | ||
To evaluate the aromatic nature, five criteria including the harmonic oscillator model of aromaticity (HOMA), ELF, para-delocalization index (PDI), NICS, and ASE have been assessed.16–20 Moreover, we have carried out ACID analysis to visualize the ring current.
Computational methods
All density functional theory calculations have been performed using the Gaussian 16 program package at the B3LYP/6-311+G(d,p) level,37 which is widely employed for the estimation of aromaticity of monocyclic heterobenzenes and inorganic benzenes.17,38–40 Note that when the basis sets as 6-311+G(2df,2pd) and 6-311+G(3df,3pd) in the optimization and NICS calculations for dianionic 4′ were employed, almost no significant improvement was observed (Table S2†). We used the NBO method in the current NBO 7.0 version of the general NBO analysis program.41 PDI, ELF, and atoms in molecules (AIM) studies were generated with the Multiwfn 3.6 program.42Results and discussion
Fig. 1 displays the optimized structures and selected bond lengths of benzene (1), borazine (2), and three B4N2 systems (3, 3′, and 4′). All molecular geometries were fully optimized as closed-shell species and characterized as energy minima by the absence of imaginary vibrational frequencies. The central six-membered ring in compounds 1, 2, 3 and 3′ exhibits a planar geometry. Notably, dianionic 4′ also bears a planar B4N2 ring, which is in stark contrast to the twisted ring framework experimentally observed for 4, implying the impact of substituents at the B and N atoms on the skeletal geometry.30,32–36 The selected π-type molecular orbitals (π-MOs) of 3′ and 4′ are summarized in Fig. 2. Three explicit π-MOs are seen in 3′ and 4′, which are similar to those of benzene 1 and borazine 2. While 1 and 2 have two degenerated highest occupied molecular orbitals (HOMOs), the B4N2 systems (3, 3′, 4′) possess non-degenerate π-MOs.27 In the π-MOs of 3, some contributions from 3p-orbitals of the substituents (Cl and PMe3) are found. To assess the essential nature of the six-membered skeleton without consideration of the significant electronic perturbation from the substituent, 3 is omitted from NICSπ and ELFπ calculation (vide infra).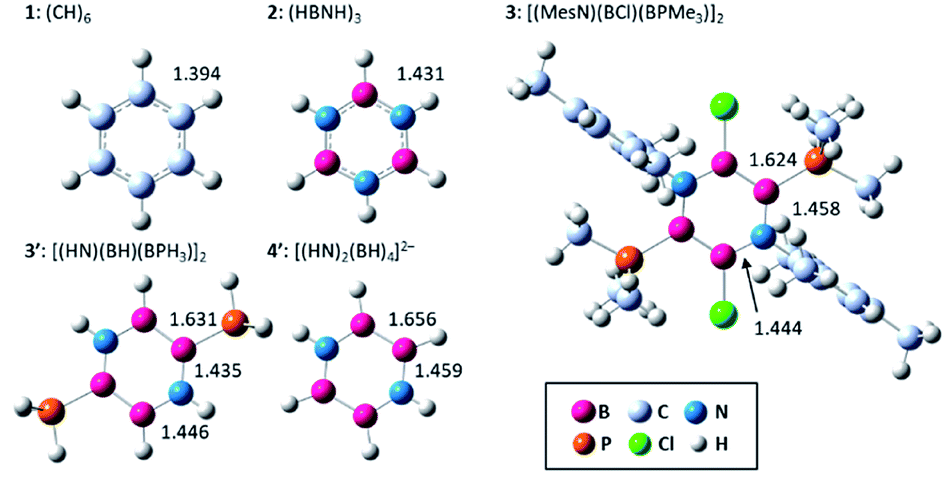 | ||
| Fig. 1 Optimized structures and selected bond lengths (in Å) of 1, 2, 3, 3′ and 4′. | ||
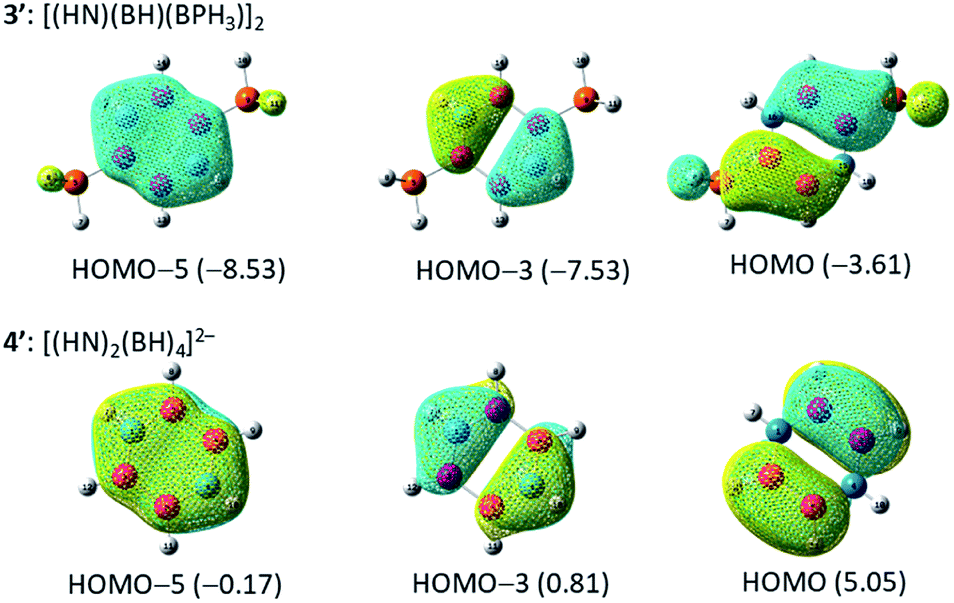 | ||
| Fig. 2 Plots of the π molecular orbitals of 3′ and 4′ (in eV). | ||
Firstly, a structural criterion is evaluated using the HOMA index, which is based on the equalization of the bond lengths and symmetry.43–45 The calculated HOMA values, resonance energy (EN), and bond length alternation (GEO) are summarized in Table 1 (for the details of bond number, see Table S2†).46,47 It should be noted that the HOMA index reflects only the geometrical features and does not consider the polarization effect on electronic distribution. Besides, HOMA values may depend on the choice of reference bond lengths and normalization constant. The reported constants of B–B and B–N bond are calculated in different way.48,49
| HOMA | EN | GEO | ELFave | ELFπ | ELFσ | PDI | |
|---|---|---|---|---|---|---|---|
| 1: (CH)6 | 0.990 | 0.010 | 0.000 | 0.81 | 0.91 | 0.70 | 0.105 |
| 2: (HBNH)3 | 1.000 | 0.000 | 0.000 | 0.56 | 0.72 | 0.40 | 0.041 |
| 3: [(MesN)(BCl)(BPMe3)]2 | 0.698 | 0.189 | 0.113 | — | — | — | 0.068 |
| 3′: [(HN)(BH)(BPH3)]2 | 0.654 | 0.163 | 0.183 | 0.64 | 0.81 | 0.47 | 0.078 |
| 4′: [(HN)2(BH)4]2– | 0.288 | 0.428 | 0.284 | 0.66 | 0.81 | 0.50 | 0.118 |
The HOMA values increase in the order 4′ < 3′ < 3 < 1 ≈ 2, from 0.288 for 4′ up to 1.000 for 2. There is little bond alternation in 1 and 2, in line with their essential D6h and D3h symmetries. For the B4N2 systems (3, 3′, 4′), both EN and GEO values are larger than those for benzene 1 and borazine 2. Particular diagnostic is that the HOMA value of 4′ is mainly dominated by EN term (0.428) rather than GEO term (0.284), indicating elongation of the skeletal bonds. The bond numbers of 3 [1.143 (B–B), 1.444–1.513 (B–N)], and 3′ [1.101 (B–B), 1.504–1.561 (B–N)] indicate partial bond alternation in 3 and 3′, which results in the large GEO values. The average bond numbers of 3 [1.367] and 3′ [1.388] are smaller than that of benzene 1 [1.533] and borazine 2 [1.582], which leads to the increased EN values of 3 and 3′. The bond numbers of 4′ show B–B single bond [0.950] and B–N multiple bond [1.437] characters, suggesting that the π electrons are relatively localized on each B–N–B unit. The average of the bond number of 4′ [1.275] is smaller than other systems, in agreement with the relatively large EN value.
Secondly, as an electronic criterion, the ELF and PDI values have been computed.50–55 The calculated average ELF values together with its σ- and π-contributions and PDI values are listed in Table 1,56–58 and the ELF isosurfaces at the bifurcation points of the σ and π systems are depicted in Fig. 3.59 The ELFave values increase in the order 2 < 3′ < 4′ < 1, from 0.56 for 2 to 0.81 for 1, and the ELFπ values are found to be larger than the ELFσ values for all molecules. The ELFπ values greater than 0.70 indicate that all molecules may have some aromatic character.60 Benzene 1 shows the high ELFπ value of 0.91, in line with the pronounced aromatic nature. Borazine 2 presents an ELFπ value of 0.72 lower than that of benzene 1, implying the less aromatic character. In contrast to highly symmetric benzene 1 (D6h) and borazine 2 (D3h), the B4N2 systems with the lower symmetry exhibit several distinguishable ELFπ values. For the neutral B4N2 system 3′, the separations at the (H3P)B–N bonds, HB–N bonds, and B–B bonds occur at the ELFπ values of 0.68, 0.82, and 0.94, respectively. The dianionic B4N2 system 4′ shows the first separation at the B–N bonds with a relatively high ELFπvalue of 0.75 and the B–B bonds are split at a significantly higher ELFπ value of 0.92. The average ELFπ values of 0.81 for 3′ and 0.81 for 4′ are between those of benzene 1 (0.91) and borazine 2 (0.72), indicating moderate delocalization of π-electrons in those B4N2 systems.
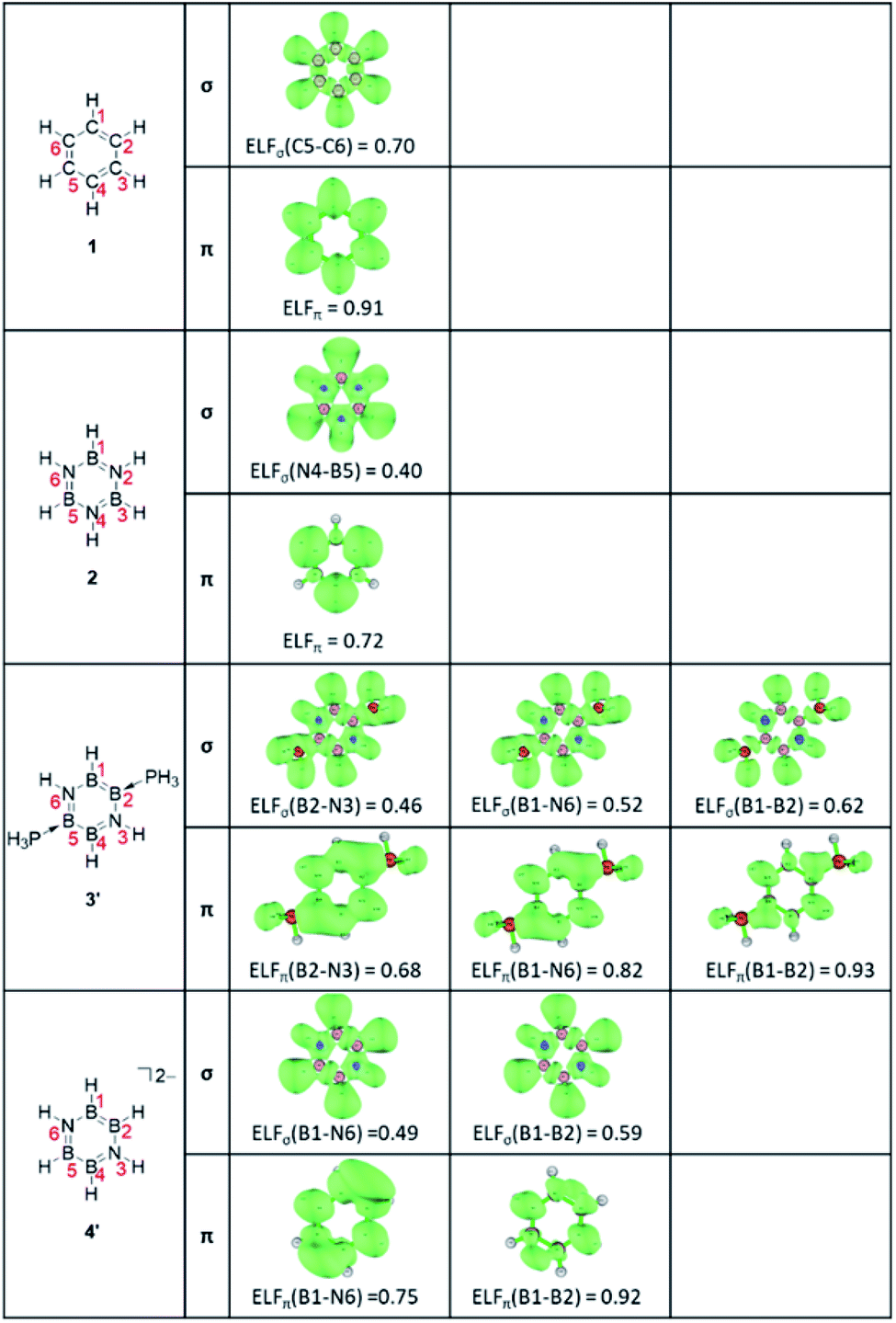 | ||
| Fig. 3 The ELF isosurfaces at the bifurcation points of the σ and π systems of 1, 2, 3′, and 4′. | ||
The ELFσ values of inorganic benzenes [2 (0.40), 3′ (0.47), 4′ (0.50)] are much smaller than that of benzene 1 (0.70), reflecting the difference of electronegativity between B and N atoms. The PDI values increase in the order 2 (0.041) < 3 (0.068) < 3′ (0.078) < 1 (0.105) < 4 (0.118), suggesting the moderate aromaticity of 3 and 3′, which is in agreement with the ELFπ values. However, the PDI value of 4 greater than that of benzene 1, as inconsistent with those ELFπ values, points out that the PDI measures might not be suitable for the evaluation of the relative aromaticity among the differently charged molecules, as well as, compounds consisting of different skeletal atoms.61,62 The calculated ring critical point properties of compounds 1, 2, 3, 3′, 4′ are summarized in Table S4.†
Thirdly, as a magnetic criterion, a series of NICS values have been estimated (Table 2).63–68 Isotropic NICS can be represented by the anisotropic components: NICS = (NICSxx + NICSyy + NICSzz)/3 = NICSin-plane + NICSout-of-plane, in which (NICSxx + NICSyy)/3 = (NICSin-plane) and (NICSzz)/3 = NICSout-of-plane. Free of in-plane component NICS (FiPC-NICS) indicates the NICS value at a point above the molecular ring with zero of NICSin-plane. Fig. 4(a) presents the NICS-profiles of the full-NICSzz and σ- and π-electron contribution to the NICSzz for 3′ and 4′, from the center of the six-membered ring and above up to 5.00 Å, and Fig. 4(b) shows FiPC-NICS plot, respectively.
| NICSzz | NICSπzz | FiPC | |||
|---|---|---|---|---|---|
| Distance (Å) | 0 | 1 | 0 | 1 | — |
| 1: (CH)6 | −14.48 | −29.25 | −35.78 | −29.07 | −9.07 |
| 2: (HBNH)3 | 11.98 | −5.17 | −7.89 | −5.88 | −2.34 |
| 3: [(MesN)(BCl)(BPMe3)]2 | −3.36 | −12.32 | — | — | −4.03 |
| 3′: [(HN)(BH)(BPH3)]2 | −3.75 | −15.18 | −19.73 | −16.54 | −3.85 |
| 4′: [(HN)2(BH)4]2– | −5.49 | −15.42 | −20.73 | −17.02 | — |
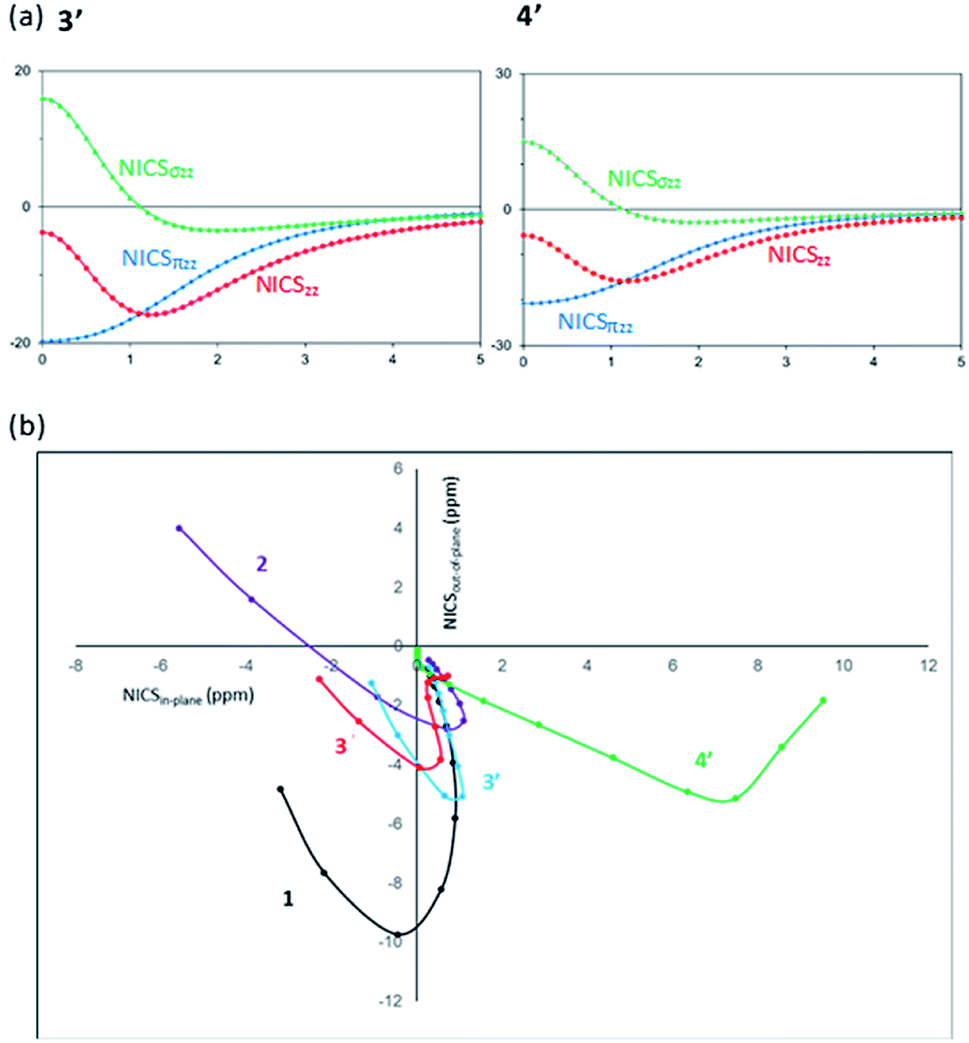 | ||
| Fig. 4 (a) Dissected NICSzz (ppm) vs. distance (Å) of 3′ and 4′. (b) Plots of the NICSin-plane vs. NICSout-of-plane of 1, 2, 3, 3′ and 4′, to identify the FiPC-NICS. | ||
While the NICSzz(0) values of 1 (−14.48), 3 (−3.36), 3′ (−3.75), and 4′ (−5.49) are negative, borazine 2 shows the highly positive NICSzz(0) values of 11.98, which is due to the significantly positive NICSσzz(0) contribution.66 It has been demonstrated that NICSπzz in addition to NICSzz(1) could be the most reliable aromaticity indexes, with statistically good correlation with aromatic stabilization energies.64,69 The NICSzz values of 3 [NICSzz(1) = −12.32], 3′ [NICSzz(1) = −15.18, NICSπzz(0) = −19.73, NICSπzz(1) = −16.54] and 4′ [NICSzz(1) = −15.42, NICSπzz(0) = −20.73, NICSπzz(1) = −17.02] lie in between those of benzene 1 [NICSzz(1) = −29.25, NICSπzz(0) = −35.78, NICSπzz(1) = −29.07] and borazine 2 [NICSzz(1) = −5.17, NICSπzz(0) = −7.89, NICSπzz(1) = −5.88].
The NICSzz profiles of 3′ and 4′ (Fig. 4(a)) confirm that the NICSσzz values are significantly positive near the ring center [NICSσzz(0) = 15.98 (3′), 15.25 (4′)], and they decrease as being apart from the ring and become negative around at 1 Å. On the other hand, the NICSπzz values are significantly negative at the molecular center, and gradually become less negative as being away from the ring. The greatly negative NICSπzz contribution over the NICSσzz component leads to the overall negative NICSzz values. A similar trend of the NICSzz profile is found for benzene.66 The FiPC-NICS value corresponds to the interception on the vertical axis (NICSout-of-plane) in Fig. 4(b). The FiPC-NICS values of −4.03 for 3 and −3.85 for 3′, appear to be between those of benzene 1 (−9.07) and borazine 2 (−2.34), supporting the moderate aromatic character of 3 and 3′. The shape of FiPC-NICS plots of 3 and 3′ is similar to that of 1 but different from that of 2. The positive NICSout-of-plane values are observed near the ring center of 2, which is due to the pronounced σ-contribution but marginal π-contribution in the region.66 The significant deviation of the FiPC-NICS plot of 4′ from others can be rationalized by the largely positive NICSxx values, and therefore, the FiPC-NICS value of 4′ could not be gained.
ACID plot of 4′ is shown in Fig. 5 (For the plots of 1, 2, 3′, see Fig. S1†). While the clear clockwise current–density vectors are confirmed on the ACID isosurface of benzene 1, borazine 2 shows the imperceptible ring current consisting of the three localized circulations on the nitrogen atoms, which is consistent with previous studies.24 The clockwise current–density vectors on the ACID isosurface also observed in 3.27 Despite somehow weak vectors, an explicit diatropic ring current is seen over the B4N2 ring of 4′ with the pronounced stopover at the B–H moieties.
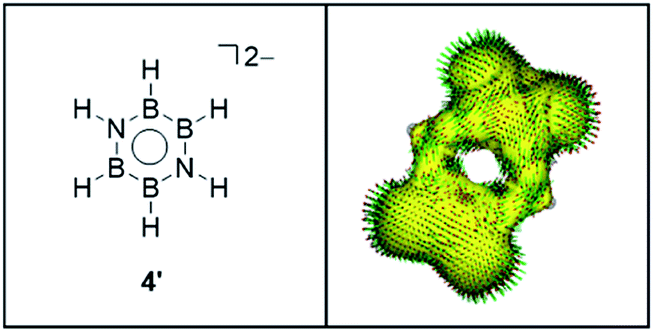 | ||
| Fig. 5 ACID plot of the selected π orbitals of 4′, at an isosurface value of 0.03. | ||
Finally, an energetic criterion is evaluated using the ASE method (Table 3). The ASE values computed by the reactions (a)–(d) (Scheme 2, Table S5–S8†)65,70 decrease in the order 1 (33.4 kcal mol−1) > 3′ (23.8 kcal mol−1) > 4′ (11.0 kcal mol−1) > 2 (10.1 kcal mol−1). Based on the ASE values, it can be estimated that inorganic benzene analogues 2, 3′, 4′ present 30%, 71% and 33% of aromaticity in comparison to benzene, respectively. Note that the significantly smaller ASE value of 4′ compared with that of 3′ is not in line with those NICSπzz(0) values [3′ = −19.73; 4′ = −20.73], indicating that magnetic and energetic criteria are not in good correlation with each other when comparing the differently charged molecules.64
| ASE | |
|---|---|
| 1: (CH)6 | 33.4 (100) |
| 2: (HBNH)3 | 10.1 (30) |
| 3: [(MesN)(BCl)(BPMe3)]2 | — |
| 3′: [(HN)(BH)(BPH3)]2 | 23.8 (71) |
| 4′: [(HN)2(BH)4]2– | 11.0 (33) |
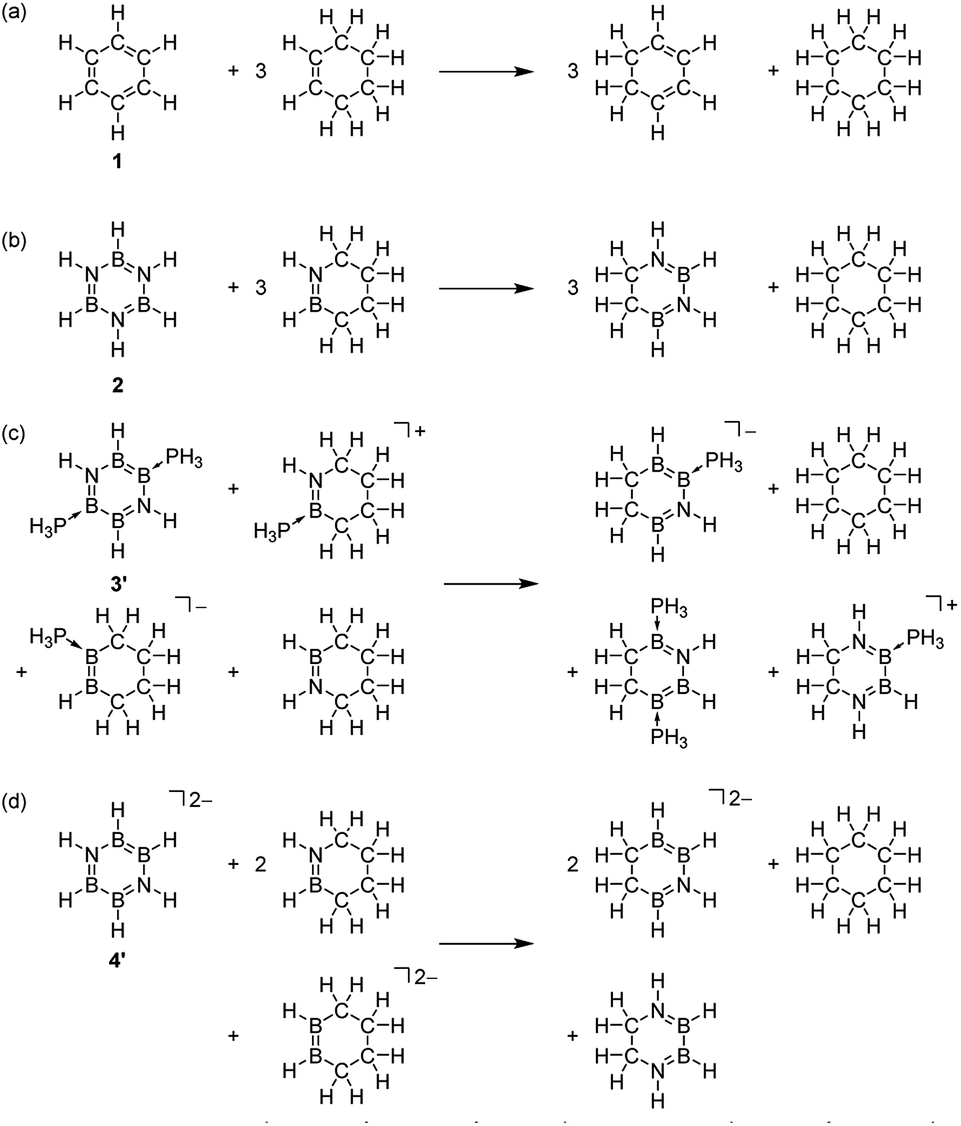 | ||
| Scheme 2 Homodesmotic reaction schemes used to estimate the aromatic stabilization energy (ASE). | ||
Conclusions
In conclusion, we have assessed the aromatics-relevant indices of B4N2-based inorganic benzene analogues together with benzene and borazine, with the aid of HOMA, ELF, PDI, NICS, ACID and ASE methods. It has been demonstrated that the aromaticity of the neutral B4N2 systems 3 and 3′ clearly differs from that of borazine 2 of the (XY)3 system. The major parameters (ELF, PDI, NICSzz, and ASE) other than HOMA suggest that the neutral B4N2 systems 3 and 3′ are less aromatic than benzene and more aromatic than borazine 2. While ELFπ, PDI and NICSπzz values propose the pronounced aromatic nature of the dianionic B4N2 system 4′, the ASE result estimates the aromaticity of 4′ to be smaller than that of 3′ and similar to borazine 2. Moreover, the FiPC-NICS plot of 4′ is different from those observed for the aromatic molecules.65 Besides, ACID plot of 4′ shows that in addition to the ring current over the six-membered ring, localized currents in the RB-BH fragment significantly contributes to magnetic anisotropy. These data indicate that even with the same B4N2-skeletal framework of 6π-system, the index value of each criterion varies depending on the overall charge of the B4N2 systems,32,33,36 giving rise to the different aromatic nature.71,72 This study paves a way for the further design and development of aromatic inorganic benzenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Nanyang Technological University (NTU), Singapore, and the Singapore Ministry of Education (MOE2018-T2-2-048(S)) for R. K. We thank Mr S. H. J. Melvin (NTU) for assistance in the calculation.Notes and references
- P. R. v. Schleyer and J. Haijun, Pure Appl. Chem., 1996, 68, 209–218 CAS.
- P. v. R. Schleyer, Chem. Rev., 2001, 101, 1115–1118 CrossRef CAS.
- A. T. Balaban, P. v. R. Schleyer and H. S. Rzepa, Chem. Rev., 2005, 105, 3436–3447 CrossRef CAS.
- M. Faraday, Phil. Trans. R. Soc. Lond., 1825, 115, 440–466 CrossRef.
- A. Kekulé, Bull. Soc. Chim. Fr., 1865, 3, 98–110 Search PubMed.
- K. Ota and R. Kinjo, Chem.–Asian J., 2020, 15, 2558–2574 CrossRef CAS.
- A. Stock and E. Pohland, Ber. Dtsch. Chem. Ges., 1926, 59, 2215–2223 CrossRef.
- H. R. Allcock, Chem. Rev., 1972, 72, 315–356 CrossRef CAS.
- A. L. Korich and P. M. Iovine, Dalton Trans., 2010, 39, 1423–1431 RSC.
- Y. Tokunaga, Heterocycles, 2013, 87, 991–1021 CrossRef CAS.
- E. W. David, US, US3035095A, 1962 Search PubMed.
- H. V. R. Dias and P. P. Power, Angew. Chem., Int. Ed. Engl., 1987, 26, 1270–1271 CrossRef.
- H. V. R. Dias and P. P. Power, J. Am. Chem. Soc., 1989, 111, 144–148 CrossRef CAS.
- P. P. Power, J. Organomet. Chem., 1990, 400, 49–69 CrossRef CAS.
- A. E. Seitz, M. Eckhardt, A. Erlebach, E. V. Peresypkina, M. Sierka and M. Scheer, J. Am. Chem. Soc., 2016, 138, 10433–10436 CrossRef CAS.
- W. H. Fink and J. C. Richards, J. Am. Chem. Soc., 1991, 113, 3393–3398 CrossRef CAS.
- P. v. R. Schleyer, H. Jiao, N. J. R. v. E. Hommes, V. G. Malkin and O. L. Malkina, J. Am. Chem. Soc., 1997, 119, 12669–12670 CrossRef CAS.
- E. D. Jemmis and B. Kiran, Inorg. Chem., 1998, 37, 2110–2116 CrossRef CAS.
- J. J. Engelberts, R. W. A. Havenith, J. H. van Lenthe, L. W. Jenneskens and P. W. Fowler, Inorg. Chem., 2005, 44, 5266–5272 CrossRef CAS.
- A. K. Phukan, A. K. Guha and B. Silvi, Dalton Trans., 2010, 39, 4126–4137 RSC.
- R. Pino-Rios, A. Vásquez-Espinal, L. Alvarez-Thon and W. Tiznado, Phys. Chem. Chem. Phys., 2020, 22, 22973–22978 RSC.
- P. W. Fowler and E. Steiner, J. Phys. Chem. A, 1997, 101, 1409–1413 CrossRef CAS.
- A. Y. Timoshkin and G. Frenking, Inorg. Chem., 2003, 42, 60–69 CrossRef CAS.
- R. Islas, E. Chamorro, J. Robles, T. Heine, J. C. Santos and G. Merino, Struct. Chem., 2007, 18, 833–839 CrossRef CAS.
- R. Ghiasi, J. Mol. Struct., 2008, 853, 77–81 CrossRef CAS.
- S. Pelloni, G. Monaco, P. Lazzeretti and R. Zanasi, Phys. Chem. Chem. Phys., 2011, 13, 20666–20672 RSC.
- K. Ota and R. Kinjo, Angew. Chem., Int. Ed., 2020, 59, 6572–6575 CrossRef CAS.
- B. Su, K. Ota, K. Xu, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 11921–11925 CrossRef CAS.
- M. Solà, Front. Chem., 2017, 5, 22 Search PubMed.
- J. T. Nelson and W. J. Pietro, Inorg. Chem., 1989, 28, 544–548 CrossRef CAS.
- T. M. Krygowski, K. Ejsmont, B. T. Stepień, M. K. Cyrański, J. Poater and M. Solà, J. Org. Chem., 2004, 69, 6634–6640 CrossRef CAS.
- R. Miao, G. Yang, C. Zhao, J. Hong and L. Zhu, J. Mol. Struct., 2005, 728, 197–202 CrossRef CAS.
- M. Baranac-Stojanović and M. Stojanović, RSC Adv., 2013, 3, 24108–24117 RSC.
- A. K. Srivastava, S. K. Pandey and N. Misra, Theor. Chem. Acc., 2016, 135, 158 Search PubMed.
- A. K. Srivastava, S. K. Pandey and N. Misra, Mol. Phys., 2016, 114, 1763–1770 CrossRef CAS.
- W. A. Rabanal-León, W. Tiznado and L. Alvarez-Thon, Int. J. Quantum Chem., 2019, 119, e25859 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- M. Baranac-Stojanović, Chem.–Eur. J., 2014, 20, 16558–16565 CrossRef.
- M. Stojanović and M. Baranac-Stojanović, J. Org. Chem., 2016, 81, 197–205 CrossRef.
- R. A. Iwaki and T. Udagawa, Chem. Phys. Lett., 2020, 745, 137271–137275 CrossRef.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0., Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- A. Julg and P. François, Theor. Chim. Acta, 1967, 8, 249–259 CrossRef CAS.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- T. M. Krygowski, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS.
- T. M. Krygowski and M. Cyrański, Tetrahedron, 1996, 52, 1713–1722 CrossRef CAS.
- T. M. Krygowski and M. Cyrański, Tetrahedron, 1996, 52, 10255–10264 CrossRef CAS.
- I. D. Madura, T. M. Krygowski and M. K. Cyrański, Tetrahedron, 1998, 54, 14913–14918 CrossRef CAS.
- K. K. Zborowski, I. Alkorta, J. Elguero and L. M. Proniewicz, Struct. Chem., 2013, 24, 543–548 CrossRef CAS.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- A. Savin, A. D. Becke, J. Flad, R. Nesper, H. Preuss and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1991, 30, 409–412 CrossRef.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS.
- J. Poater, X. Fradera, M. Duran and M. Solà, Chem. Eur. J., 2003, 9, 400–406 CrossRef CAS.
- F. Feixas, E. Matito, J. Poater and M. Solà, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- J. C. Santos, W. Tiznado, R. Contreras and P. Fuentealba, J. Chem. Phys., 2004, 120, 1670–1673 CrossRef CAS.
- J. C. Santos, J. Andres, A. Aizman and P. Fuentealba, J. Chem. Theory Comput., 2005, 1, 83–86 CrossRef.
- J. Pilmé, J. Comput. Chem., 2017, 38, 204–210 CrossRef.
- The calculated PDI value (0.041) is larger than that of reported values. We thought the calculated values are depends on the calculation level and software. ref: J. E. Del Bene, M. Yáñez, I. Alkorta and J. Elguero, J. Chem. Theory Comput., 2009, 5, 2239–2247 CrossRef CAS; M. Baranac-Stojanović and M. Stojanović, RSC Adv., 2013, 3, 24108–24117 RSC.
- M. Solà, Aromaticity, John Wiley & Sons, Ltd, 2017 Search PubMed.
- P. Bultinck, Faraday Discuss., 2007, 135, 347–365 RSC.
- F. Feixas, E. Matito, J. Poater and M. Solà, J. Comput. Chem., 2008, 29, 1543–1554 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS.
- J. J. Torres-Vega, A. Vásquez-Espinal, J. Caballero, M. L. Valenzuela, L. Alvarez-Thon, E. Osorio and W. Tiznado, Inorg. Chem., 2014, 53, 3579–3585 CrossRef CAS.
- R. Báez-Grez, L. Ruiz, R. Pino-Rios and W. Tiznado, RSC Adv., 2018, 8, 13446–13453 RSC.
- A. Stanger, Eur. J. Org. Chem., 2020, 2020, 3120–3127 CrossRef CAS.
- R. Gershoni-Poranne and A. Stanger, Chem. Soc. Rev., 2015, 44, 6597–6615 RSC.
- R. Báez-Grez, W. A. Rabanal-León, L. Alvarez-Thon, L. Ruiz, W. Tiznado and R. Pino-Rios, J. Phys. Org. Chem., 2019, 32, e3823 CrossRef.
- E. Osorio, J. K. Olson, W. Tiznado and A. I. Boldyrev, Chem. Eur. J., 2012, 18, 9677–9681 CrossRef CAS.
- P. C. Parambil and R. Hoffmann, J. Am. Chem. Soc., 2018, 140, 12844–12852 CrossRef CAS.
- P. C. Parambil and S. S. R. R. Perumal, Organometallics, 2020, 39, 2951–2955 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09040a |
| This journal is © The Royal Society of Chemistry 2021 |