
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1H and 195Pt NMR prediction for inclusion compounds formed by cisplatin and oxidized carbon nanostructures†
Leonardo A. De Souza *a,
Eduardo R. Almeidab,
Joyce H. Cunha e Silvac,
Diego F. S. Paschoalc,
Jadson C. Belchiora,
Hélio F. Dos Santosb and
Wagner B. De Almeidad
*a,
Eduardo R. Almeidab,
Joyce H. Cunha e Silvac,
Diego F. S. Paschoalc,
Jadson C. Belchiora,
Hélio F. Dos Santosb and
Wagner B. De Almeidad
aDepartamento de Química, ICEx, Universidade Federal de Minas Gerais, Campus Universitário, Pampulha, Belo Horizonte, MG 31270-901, Brazil. E-mail: desouza.leonardo.chem@gmail.com
bNúcleo de Estudos em Química Computacional (NEQC), Departamento de Química, ICE, Universidade Federal de Juiz de Fora, Campus Universitário, Martelos, Juiz de Fora, MG 36036-330, Brazil
cNúcleo de Química Teórica e Computacional de Macaé (NQTCM), Polo Ajuda, Universidade Federal do Rio de Janeiro, Campus UFRJ-Macaé, 27.971-525, Macaé, RJ, Brazil
dLaboratório de Química Computacional e Modelagem Molecular (LQC-MM), Departamento de Química Inorgânica, Instituto de Química, Universidade Federal Fluminense, Campus do Valonguinho, Centro, Niterói, RJ 24020-141, Brazil
First published on 24th December 2020
Abstract
Prediction of NMR chemical shifts can assist experimentalists in the characterization of drug delivery systems based on carbon nanocomposites. Chemical shifts are strongly correlated to the nucleus position and its chemical neighborhood. Therefore, to predict structures and NMR properties of complex chemical models, choosing a more consistent theoretical level capable of providing more realistic results and moderate computational demand is a major challenge. In this work, we predicted the NMR spectra of inclusion compounds formed by cisplatin (cDDP) and an oxidized carbon nanotube (CNTox) and nanocone (CNCox) considered by specialists as potential drug delivery systems. The 195Pt NMR chemical shifts calculated at the DFT level with the new relativistic NMR-DKH basis set were −2314 ppm and −2192 ppm for cDDP@CNTox and cDDP@CNCox complexes, respectively, which are both high-field shifted relative to the free cDDP (−2110 ppm). 1H NMR chemical shifts are also sensitive to the inclusion process. The H (NH3) signals are found on average at +4.3 (cDDP), −5.1 (cDDP@CNTox) and +6.6 ppm (cDDP@CNCox). Interestingly, despite the similar inclusion modes in CNTox and CNCox cavities, the 1H NMR shifts were in opposite directions. A possible reason might be the higher stability of cDDP@CNTox (ΔEF = −19.9 kcal mol−1) than that of cDDP@CNCox (ΔEF = −5.7 kcal mol−1), which suggests a short guest–host contact in the former and consequently, a more efficient shielding of hydrogen atoms due to the electron-rich carbon structure. These results may be helpful as comparison data in the NMR spectra assignment in solution and the inclusion compounds' structural elucidation.
Introduction
Antitumor agent cisplatin1 and its derivatives,2,3 when used alone or in combination with other chemotherapeutics4,5 can be effective against the advancement of several clinical cases of cancer. However, there are several known factors such as low selectivity, resistance and consequently, high toxicity that are limiting for their clinical use.6 Thus, alternative formulations of platinum compounds have been studied, in particular, drug delivery systems (DDSs) formed by organic macromolecules,7–9 liposomes,10,11 nanoparticles12,13 and oxidized carbon nanocomposites.14–20 Once in the bloodstream, it is expected that these systems are able to protect the drug against undesirable side reactions as well as provide efficient release in a controlled biological target. Therefore, the understanding of how these systems are provided as inclusion compounds can contribute to design improved complexes with desirable properties.Nuclear magnetic resonance (NMR) spectroscopy is an important tool for investigating the structural and dynamic aspects of many bioactive compounds. Ravera et al.21 showed the synthesis and characterization by 1H, 13C and 195Pt NMR of Pt(IV)–bis(benzoato) complexes with potential anticancer activity. The authors observed that the lipophilicity, cellular accumulation and cytotoxicity on chemoresistant MPM cell lines were considerably increased in relation to their Pt(II) analogs, cisplatin and oxaliplatin. However, it was observed that the structure–activity relationship of Pt(IV) complex derived-oxaloplatin is possibly affected by the interaction of the aromatic ligand with intracellular components. In order to minimize these undesirable reactions, the authors studied the inclusion of the drug in cyclodextrins (CDs) and the inclusion complex formed was characterized by 1H NMR spectra. The results showed that the signals of protons H-3, H-5, and H-6, located inside the β-CD cavity, are shifted, supporting the identification of inclusion compound. Levet et al.22 developed a dry powder type formed by liposomes and/or PEGylated nanoparticles with cisplatin for controlled release of the drug after inhalation during adjuvant chemotherapy in the treatment of lung cancer. The integrity and quantification of cisplatin, reduced to submicron sizes, were assessed by electrothermal atomic absorption spectrometry (ETAAS) and 195Pt NMR spectroscopy. Through the integration of the 195Pt NMR signal, it was observed a slightly larger amount of the drug in the final product compared to the amount found by the ETAAS technique.
Our research group23–27 has reported theoretical calculations of structure, stability and characterization by 1H, 13C and 15N NMR spectroscopy of inclusion nanocomposites. Recently, we used density functional theory (DFT) methods to study inclusion complexes formed between cisplatin and oxidized carbon nanotube and nanocone molecules.26 Our results showed through the comparison between the B3LYP and B97-D density functional that the dispersion correction significantly improves the stabilization energy and thermodynamic parameters of the compounds studied. 1H NMR spectra calculations in aqueous solution showed that experimental detection of the inclusion compounds can be promptly attained through analysis of chemical shifts for cisplatin NH3 protons, with variations around −12 and −5 ppm due to the formation of complexes, compared to the free cisplatin molecule. Hosni et al.20 used the gauge invariant atomic orbitals (GIAO) method to calculate the 195Pt chemical shielding tensors in inclusion complex models formed by various types of carbon nanotube and cisplatin. The authors used the Hartree–Fock (HF) method with aug-cc-pVDZ-PP and STO-3G basis set for the Pt and H, C, N, Cl atoms, respectively, for the geometries optimization and 195Pt NMR chemical shifts calculations in gas phase. Their results showed that the δ195Pt is able to inform about the CNT diameter and also to lead directly to confinement energy of drug. A decreasing variation from 533 to 38 ppm were observed for the δ195Pt of the drug into nanotubes models with approximately 6 to 12 Å in diameter. Even though ECP was used for Pt in ref. 20, it is of great importance for the computational study proposed for this type of system. Moreover, to date, this has been one of the most current studies in attempt to perform theoretical calculations of 195Pt NMR chemical shifts in inclusion compounds formed by carbon nanotubes and cisplatin.
Theoretically, the accurate prediction of NMR parameters involving heavy nuclei must take into account several aspects such as structure, solvent and relativistic effects, basis sets and electronic correlation, as can be seen in the works of Vícha et al.,28,29 Truflandier et al.30 and Pawlak et al.31 However, to study very large systems, such as carbon nanocomposites, the inclusion of relativistic effects can become very expensive and the use of non-relativistic protocols might be viewed as an affordable alternative. In this sense, Paschoal et al.32 performed non-relativistic and relativistic calculations to analyze the 195Pt NMR chemical shifts of 258 Pt(II) complexes, including the anticancer drug cisplatin. The authors proposed a protocol that includes all-electron Gaussian basis sets to describe the δ195Pt and all elements commonly found as Pt-ligands. The new basis sets, named NMR-DKH, were partially contracted as a triple-zeta doubly polarized with all coefficients obtained from a Douglas–Kroll–Hess (DKH) second-order scalar relativistic calculation. Accordingly, the chemical shifts of 195Pt were calculated through empirical models fitted to reproduce experimental NMR data ranging from −1000 to −6000 ppm. Among other theoretical protocols proposed, the best one employed the B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF) level for the geometry optimization and PBEPBE/NMR-DKH/IEF-PCM(UFF) level for δ195Pt calculation. The authors obtained results comparable with relativistic DFT calculation with the mean absolute deviation (MAD) and the mean relative deviation (MRD) around to 168 ppm and 5%, respectively. Very recently, Carvalho et al.33 applied the same protocol to predict the 1J(195Pt–15N) coupling constant in 71 Pt(II) complexes. For a total of 98 coupling constants, the authors found a MAD and MRD of only 36 Hz and 10.4%, respectively, showing the quality of the NMR-DKH basis set and theoretical protocol employed.
In the present work, we used the very same protocol proposed by Paschoal et al.32 for prediction of the 195Pt NMR spectrum for the inclusion complexes formed between cisplatin (cDDP) and oxidized carbon nanotubes (CNTox)/nanocones (CNCox) as potential DDS. Our results provide relevant spectroscopic data of 1H and 195Pt NMR that can assist experimentalists interested in the preparation and characterization of these systems.
Theoretical methodology
The calculations were carried out using the GAUSSIAN 09 package.34 The methodology used for the prediction of the NMR chemical shifts follows the computational protocol proposed by Paschoal et al.32 The geometries of the cDDP@CNTox and cDDP@CNCox inclusion complexes were fully optimized at DFT35 B3LYP36 level using the LANL2DZ37 effective core potential for Pt atom and the def2-SVP38 basis set for the other atoms. The solvent effect was accounted for within the continuum aqueous solution approach using the IEF-PCM39 formalism with dielectric constant adjusted for water solvent (ε = 78.3553) and UFF atomic radii, namely, B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF) level. The complexes formation energies (ΔEF) were calculated as| ΔEF = Ecomplex − (ECNox + EcDDP) | (1) |
The NMR spectra of inclusion complexes were calculated using the GIAO40 method at DFT level with the GGA PBE41,42 functional with the NMR-DKH32 basis set, namely, PBEPBE/NMR-DKH/IEF-PCM(UFF) level, with the fully optimized geometries at B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF) level. For the 1H and 15N NMR chemical shifts, the tetramethylsilane (TMS) and NH4Cl compounds were used as internal reference, respectively. NMR chemical shifts were obtained by eqn (2), where the σref is the calculated shielding constant for a reference compound and the σcalc is the shielding constant calculated for the nuclei of interest.
| δcalc (ppm) = σref(TMS) − σcalc | (2) |
The NMR chemical shift for 195Pt nucleus was calculated using eqn (3), where the σcalc is the 195Pt shielding constant calculated with the NMR-DKH basis set, as proposed by Paschoal et al.32
| δcalc (ppm) = −2065.7558 − 0.9250σcalc | (3) |
The explicit solvent effect on the calculation of NMR chemical shifts was also investigated by analyzing a set of 20 solvated structures generated from molecular dynamics (MD) simulations of the cDDP@CNTox inclusion complex. Regarding the parameterization of the CNTox model, while both intramolecular and Lennard-Jones parameters (LJ 12-6) were selected from the General Amber Force Field (GAFF),43 the atomic charges were computed at HF/6-31G(d,p) level in aqueous solution (PCM) with the ChelpG fitting procedure.44 In relation to the cDDP molecule, the intramolecular parameters were extracted from its optimized geometry in aqueous solution (IEF-PCM) at MP2(FC)/LANL2DZ/6-31G(d,p) level.45–47 In particular, the force constants of both bond stretching (Kb) and bending angle (Ka) were derived by using the Visual Force Field Toolkit (VFFDT).48 With regard to the intermolecular parameters, we used the LJ 12-6 parameters set for the Pt atom reported by our group,49 the LJ 12-6 parameters for the nonmetal atoms (Cl, N, and H) included in the GAFF, and the atomic charges calculated at HF/LANL2DZ/6-31G(d,p) level in aqueous solution (IEF-PCM) with the ChelpG scheme.
When it comes to the computational details of this MD simulation, we firstly placed our the cDDP@CNTox complex at the center of a truncated octahedral box (average length of 71.2 Å) containing water molecules described by the TIP3P model50 and the ff99SB force field.51 By using periodic boundary conditions with the isothermal–isobaric ensemble (NPT), the density was equilibrated aiming to achieve an average value of 0.993 g cm−3 at T = 310.15 K and a pressure of 1.0 bar. The Langevin thermostat52 and the Berendsen barostat53 were applied in order to keep temperature and pressure constant. While the van der Waals interactions were computed within a cutoff of 10 Å, the long-range electrostatic interactions were treated by means of the particle mesh Ewald method.54 The Leap Frog algorithm was used to numerically integrate the Newtonian equations of motion considering a time step of 2.0 fs and the SHAKE algorithm55 to constrain all bonds involving hydrogen atoms.
At last, the simulation protocol firstly involved the energy minimization of the solvent molecules followed by the entire system. While the first 1000 cycles of this stage were performed by using the steepest descent method,56 the last 1000 cycles were computed with the conjugate gradient method.57 Next, the system was heated from 0 to 310 K with a weak restraint (10 kcal mol−1 Å−2) and equilibrated for 540 ps. Finally, the production dynamics was conducted for 100 ns with the NPT ensemble. By selecting a snapshot of the cDDP@CNTox model every 5 ns from this trajectory (100 ns), we organized a set of 20 solvated structures of this inclusion complex that was employed to evaluate the explicit solvent effect on the calculation of NMR chemical shifts. This MD simulation was carried out with the AMBER 12 software58 and the trajectory analysis were carried out with the CPPTRAJ tools.59
Results and discussions
In this work we re-optimize the geometries of the molecular models of cDDP@CNTox or complex I (C168H24O15Cl2N2Pt) and cDDP@CNCox or complex II (C250H38O20Cl2N2Pt) inclusion complexes reported in our previous studies26 at a higher level of theory: B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF). The oxidized nanostructures topologies correspond to the final geometries obtained by De Souza et al.60 in a study of the oxidation mechanism of the surfaces of carbon nanotubes and nanocones. Thus, here we shall describe the changes in the geometry of each inclusion complex and the 195Pt and 1H NMR chemical shifts, not reported in the literature yet.Fig. S1 (ESI)† and 1 show the potential energy curve for the geometry optimization process of inclusion complexes and their fully optimized geometries, respectively. It can be seen that the energy difference between the starting structure and fully optimized geometries is only 2.44 kcal mol−1 (cDDP@CNTox) and about 20 kcal mol−1 (cDDP@CNCox). In complex I the cisplatin molecule is further from the oxidized region of CNTox, and this carbon structure is more rigid structurally than the CNCox model. Thus, in Fig. S1a,† ΔE reflects the slight adjustment of the structural parameters for the complex I. In complex II we observed that the drug molecule is closer to the oxidized region of CNCox and a larger ΔE (Fig. S1b†) is found between the initial and final geometries. CNTox (Fig. 1a) and CNCox (Fig. 1c) structures are about 13.4 Å and 18 Å in length, respectively, and represent the region where the drug is located in the nanostructure cavity. According to the literature, isolated units of carbon nanotubes15 and nanocones16 are obtained in the range of 40–800 nm in length. Free cisplatin geometry and its structure in the inclusion complexes were found to be square planar, as expected, with average Pt–Cl and Pt–N bond lengths of 2.35 Å and 2.07 Å, respectively, which compare well with the X-ray data: Pt–Cl equal to 2.33(9) Å and Pt–N equals to 1.95(4)/2.05(4) Å.61 Some hydrogen-bond interactions can contribute to the inclusion complexes stabilization. Both inclusion complexes have a short intramolecular hydrogen-bond interaction between hydroxyls of carboxyl groups (cDDP@CNTox) and, between a hydroxyl of carboxyl group and vicinal carbonyl (cDDP@CNCox), as shown in Fig. 1b and d. The intramolecular interaction in the complex I (Fig. 1b) is not present in the isolated CNTox (Fig. 1a). Comparing Fig. 1c and d we observe that after the formation of complex II the intramolecular interaction between two neighboring hydroxyl groups of CNCox is broken. The intermolecular hydrogen-bonds in the two inclusion complexes are assigned as medium to long-distance, on average 1.92 Å (see Fig. 1b and d). They are between the NH3 group (cDDP molecule) and the carbonyl group (carboxyl) of oxidized nanostructures.
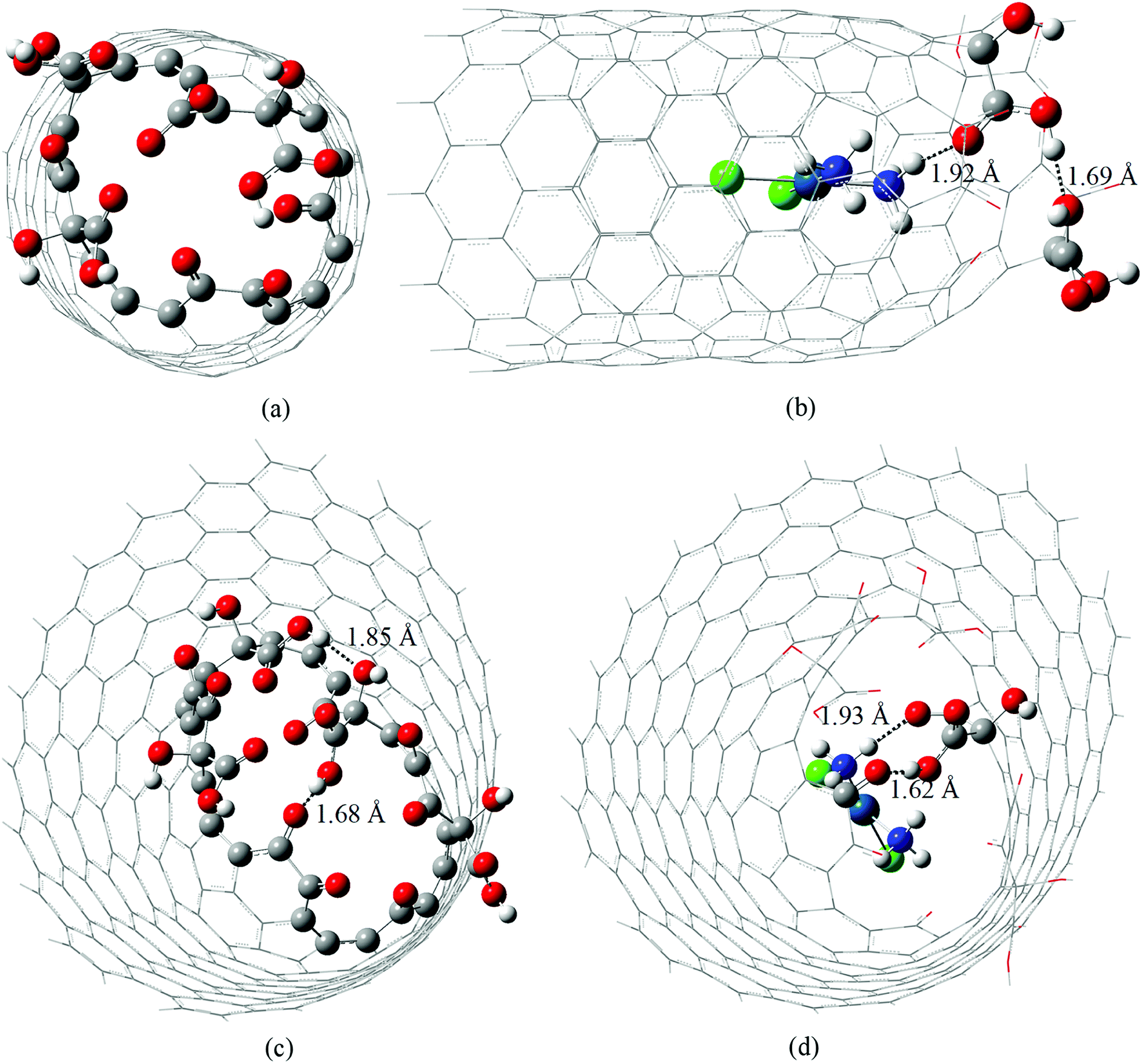 | ||
| Fig. 1 B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF) fully optimized structures of CNTox (a), cDDP@CNTox – complex I (b), CNCox (c) and cDDP@CNCox – complex II (d). Intra and intermolecular hydrogen interactions are highlighted as dotted lines. | ||
Local ChelpG charges44 in the interaction region between cisplatin and carbon atoms of the oxidized nanostructures are highlighted in Fig. 2. The scales represented by the color range show that the cisplatin molecule causes a more pronounced perturbation in the local charge distribution on the CNTox surface than on the CNCox surface when compared to the isolated monomers. The interatomic distances and local ChelpG charge analysis for Pt atom (cisplatin) and the nearest C atoms of the oxidized nanostructures shown in Fig. 2 are given in Table S1 (ESI).† The calculated average distance between the Pt atom and the CNTox surface is slightly smaller than that of the CNCox. The sum of the charge of carbon atoms closer to cisplatin molecule shows a negative value of ∼ −0.2e (complex I) and ∼ −0.03e (complex II) which represents an equivalent local charge accumulation of ∼ −0.14e and ∼ −0.06e in relation to the isolated CNTox and CNCox monomers, respectively. Table S1† also shows that the negative charge concentration on the Pt atom in complex I is increased and in complex II is slightly decreased when compared to the calculated charge for the Pt atom in the isolated cisplatin molecule. These results suggest that the implicit effect of solvent (PCM–water), besides inferring an electrostatic interaction that stabilizes the inclusion complexes, it may also induce an extra interaction represented by stronger host–guest charge transfer from CNTox to metal in complex I than that from CNCox to metal in complex II.
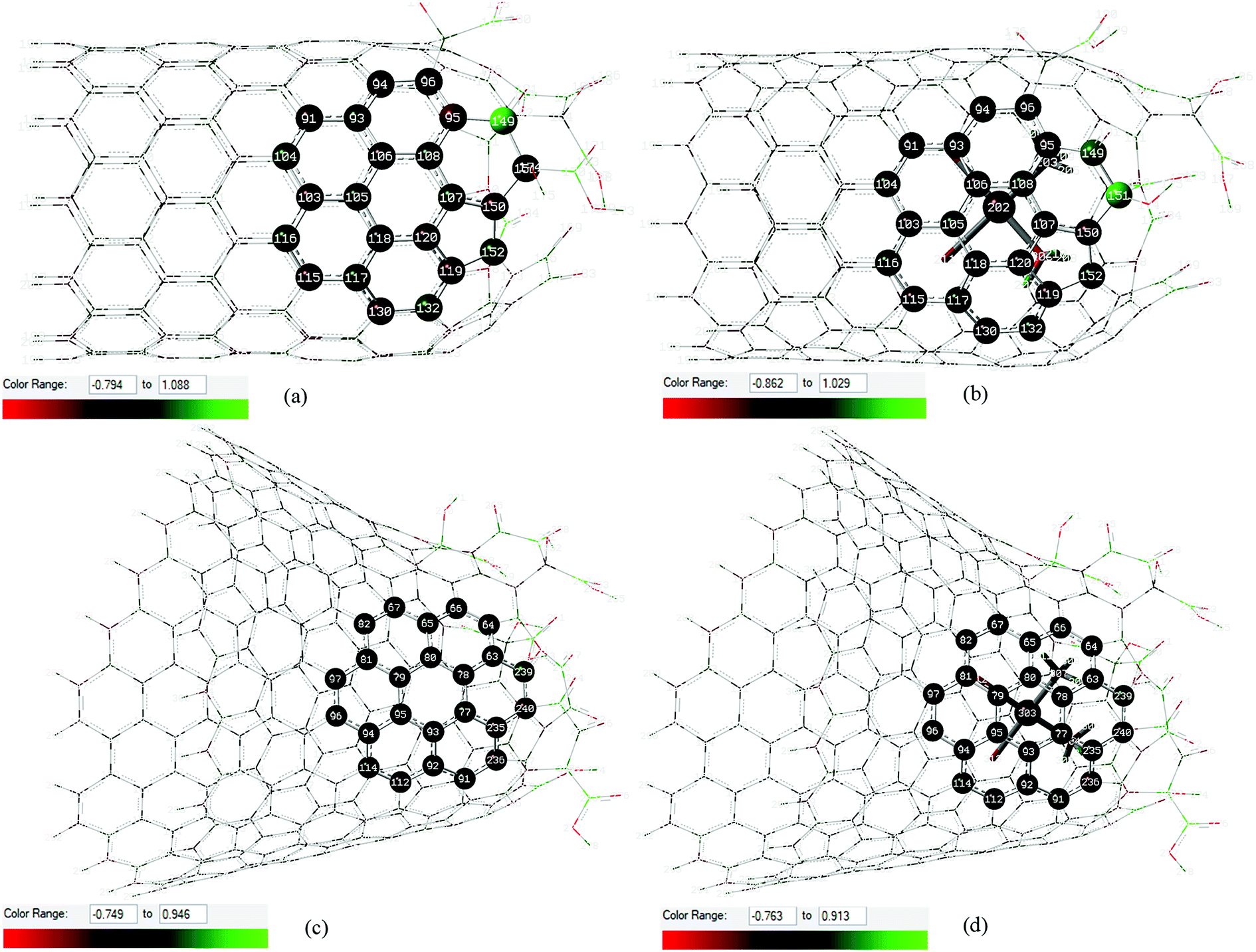 | ||
| Fig. 2 Local ChelpG charges distribution calculated at B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF) level in the interaction region between cisplatin molecule and carbon atoms of the oxidized nanostructures: CNTox (a) and inclusion complex cDDP@CNTox – complex I (b), CNCox (c) and inclusion complex cDDP@CNCox – xomplex II (d). Green and red represent positive and negative atoms according to the scale shown. | ||
Inclusion complexes formation energies (ΔEF) are given in Table 1. We compared the values obtained in this work with those for the same complexes reported in ref. 26. B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF) energies of formation showed that complex I is more strongly bound than complex II. For the cDDP@CNTox complex I, it was observed that the ΔEF calculated in this work (−19.9 kcal mol−1) is similar as that calculated in ref. 26 where the BSSE was considered for the correction of ΔEF with the 6-31G basis set. For the cDDP@CNCox complex II, the difference between ΔEF values is more significant when we consider these two levels of theory (−5.7 kcal mol−1 in the present work – see Table 1). Kruse et al.62 showed examples for geometry optimizations calculations of larger systems with inter- and intramolecular BSSE using DFT calculations and def2-SVP basis sets, where the BSSE is typically below 10% of the interaction energy. Here, we do not perform the BSSE calculation since we believe that it is small for def2-SVP basis sets. We also believe that using a post-Hartree–Fock level of theory, which different from the DFT calculations includes explicitly electron correlation effects, such as MP2 calculation,63 and the use of larger basis sets with highly polarized and diffuse functions are unfeasible from a quantum and computational point of view for large molecular systems with 250–350 atoms, such as those studied in this work. However, we performed the geometry optimization of complex I with dispersion corrected including the D3 Grimme's empirical dispersion64 to GGA PBE functional.36,37 In addition, through single point (SP) calculations, the PBEPBE-D3 and B97D65 functional, it was used to evaluate the effect of the level of theory on the both inclusion complexes formation energies. A comparison of the B3LYP with PBEPBE-D3 and B97D SP results showed that the dispersion correction leads a much larger stabilization of the complexes. Table 1 show that the both PBEPBE-D3 and B97D ΔEF is about 75% (cDDP@CNTox) and 86% (cDDP@CNCox) lower, respectively, than the B3LYP values. ΔEF of the cDDP@CNTox complex optimized at PBEPBE-D3/LANL2DZ/def2-SVP/IEF-PCM(UFF) level is even lower (80%) than the B3LYP level. The small difference between the PBEPBE-D3 values (5%) suggests that the calculation of geometry optimization may not compensate the computational cost obtained. There is a slight change in the geometry of the oxidized region of CNTox when comparing the calculated structures with the B3LYP and PBEPBE-D3 functional. The equilibrium point for the cisplatin molecule in the cDDP@CNTox complex is practically the same at both levels of theory. The dispersion terms that describe better the van der Walls interactions and hydrogen-bonds, which are important for the stabilization of inclusion complexes. In spite of the effect of dispersion corrections, our results suggest that both inclusion complexes are energetically favorable. The higher stability of complex I might be related to the formation of intramolecular hydrogen-bonds in complex I and the disappearance of this type of interaction between neighboring hydroxyl groups in the oxidized region of CNCox in complex II geometry (see Fig. 1c and d). Moreover, the possibility of charge transfer occurring between the CNTox surface and the Pt atom (cDDP) might strength the interaction between monomers in complex I. Harmonic frequency calculations were not attempted due to the size of the molecular system and high level of theory used, since it is a quite demanding computational task. Discussions about the thermodynamic parameters of these inclusion complexes and similar ones can be seen in our previous works.23–27,32
| Theory of level | cDDP@CNTox | cDDP@CNCox |
|---|---|---|
| a Calculated from eqn (1) in this work.b Results from ref. 26.c Calculated using the B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF) fully optimized structures of inclusion complexes. | ||
| B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF)a | −19.9 | −5.7 |
| PBEPBE-D3/LANL2DZ/def2-SVP/IEF-PCM(UFF)a | −99.9 | — |
| B3LYP/LANL2DZ/6-31G/gas-phase – BSSEb | −19.8 | −19.2 |
| PBEPBE-D3/LANL2DZ/def2-SVP/IEF-PCM(UFF) (single point)c | −73.2 | −42.0 |
| B97D/LANL2DZ/def2-SVP/IEFPCM(UFF) (single point)c | −82.0 | −38.0 |
Fig. 3a–c show the PBEPBE/NMR-DKH/IEF-PCM(UFF) 1H NMR spectra for free cDDP, and cDDP@CNTox and cDDP@CNCox inclusions complexes, respectively. The solvent effect (water) was accounted for using single point NMR calculations on the fully optimized geometries following the computational protocol proposed by Paschoal et al.32 Our calculations show that the chemical shifts (δ) for the two distinct hydrogen atoms of cisplatin molecule, opposite (Ha) and near (Hb) the chlorine atoms, are 4.5 and 4.1 ppm (4.3 ppm on average – Table 2), respectively. The experimental value,66 measured in a 5 mmol dm−3 solution containing cisplatin in 95% H2O and 5% D2O, pH 4.72, is 4.06 ppm, which is a fair agreement. The calculated 1H Δδ values may be trusted to ±0.5 ppm, which is adequate for this work in what changes due to complex formation is concerned. Fig. 3b and c show that in the two inclusion complexes formed, the hydrogen atoms of cisplatin (NH3 groups) exhibit a different behavior for the chemical shift variation (Δδ). When the cisplatin spectrum (Fig. 3a) is compared to that of complex I (Fig. 3b), the 1H NMR signals are shifted to high-field region (more shielded), −5.1 ppm on average (see Table 2). The electron-rich carbon nanostructure and the oxygen groups on the CNTox surface, in addition to the close proximity of the drug molecule to the inner wall of the tube, increase the shielding of the hydrogens of cDDP in complex I; therefore, these protons tend to absorb energy on higher intensity fields. These shifts reach up to −6.0 ppm for Ha and −8.2 ppm for Hb nuclei (see definition in Fig. 3a). This result can also be discussed in terms of the ring current effect that affects the chemical NMR shifts of molecules adsorbed on carbon nanostructures. Forse et al.67 carried out a theoretical-experimental study of the nuclei-independent chemical shift (NICS) calculations for molecular species adsorbed on sp2-hybridized carbon systems. The authors observed that the larger graphene-like systems and their derived concave surfaces result in greater chemical shielding of atomic nuclei, even at distances relevant for molecular adsorption. According to Casabianca,68 the largest ring current effect on concave carbon surfaces (such as carbon nanotubes) on NICS, is due to two competing factors: (i) increased curvature breaks ring aromaticity and decreases the magnitude of the NICS, concomitantly; (ii) the proximity of the rings faces increases the shielding overcoming the decrease due to loss of surface aromaticity. Thus, the proximity of the cisplatin molecule to the concave surface of the nanotube in the cDDP@CNTox model, more strongly shields the 1H nuclei shifting them to high-field region in the NMR spectrum (Fig. 3b). On the other hand, we observed the inverse effect for the protons of cDDP in complex II. In complex II, Ha nuclei have +1.5 < Δδ < +4.3 ppm and Hb nuclei have a symmetric shift of only +0.1 ppm (see Table 2), both for low-field region. The average hydrogen chemical shift was +6.6 ppm. This result shows that cisplatin protons are less shielded in complex II. The reason of the distinct trends for complexes I and II might be in the interaction mode o cDDP guest. For the narrower CNTox, the cDDP is closer to the electron-rich carbon surface increasing the shielding of hydrogen atoms (high-field shift), whereas, for CNCox the cDDP is closer to the oxygen groups at the oxidized tip, interacting through hydrogen bond and de-shielding the H nuclei (low-field shift). These results agree with the ChelpG charge analysis and the degree of interaction between the monomers in the two inclusion complexes studied.
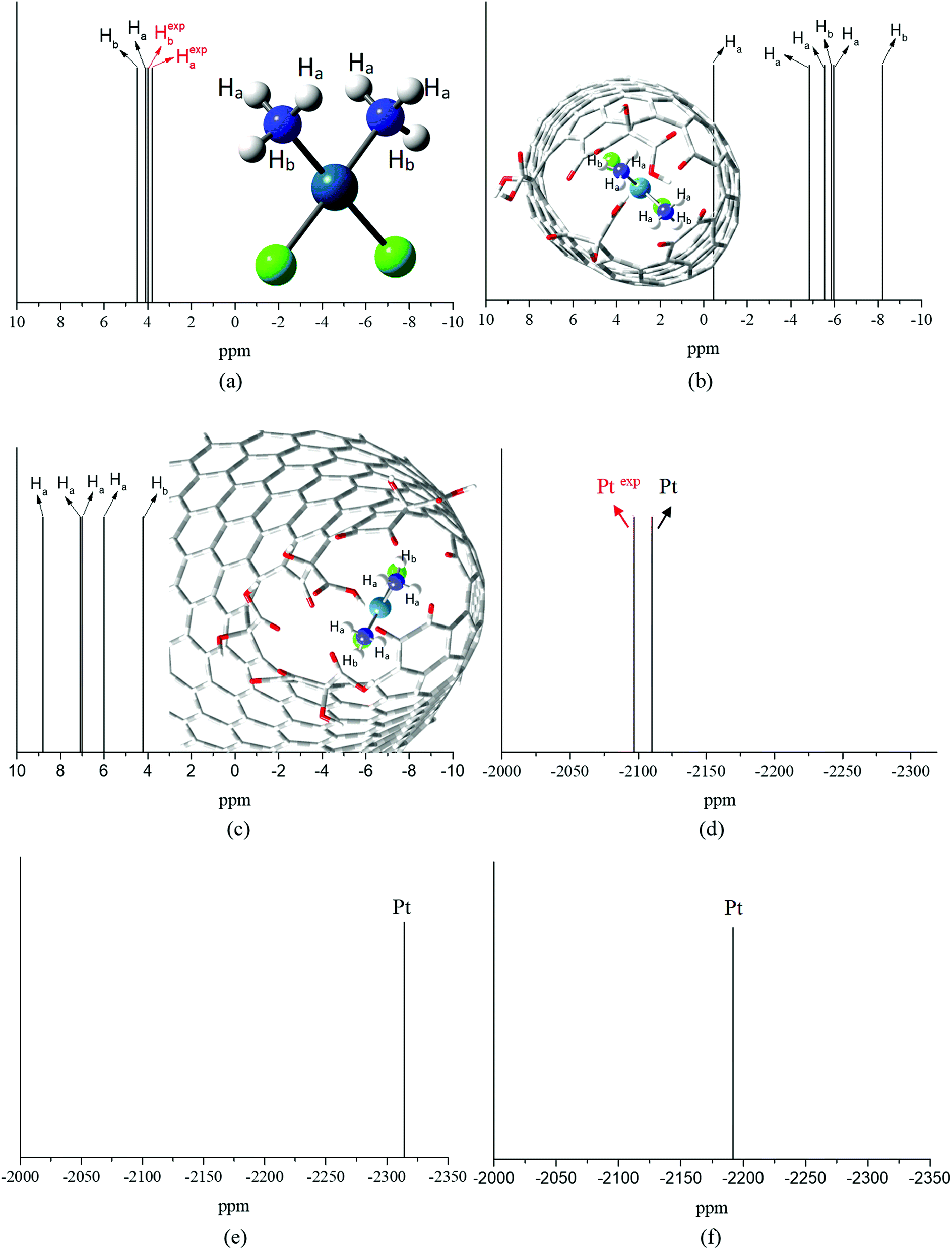 | ||
| Fig. 3 PBEPBE/NMR-DKH/IEF-PCM(UFF) 1H and 195Pt NMR spectra for free cDDP (a and d), cDDP in the cDDP@CNTox – complex I (b and e) and in the cDDP@CNCox – complex II (c and f) inclusion complexes. All values in ppm. | ||
| Structures | Chemical shifts (δ)/ppm | |||||
|---|---|---|---|---|---|---|
| Carboxyls | Hydroxyls | |||||
| Hc | Hd | He | Hf | Hg | Hh | |
| CNTox | 8 | 8.7 | 8.4 | 5.2 | 7.7 | 6.2 |
| cDDP@CNTox | 14.2 | 10.7 | 10.7 | 7.1 | 9.7 | 8.1 |
| Structures | Chemical shifts (δ)/ppm | |||||||
|---|---|---|---|---|---|---|---|---|
| Carboxyls | Hydroxyls | |||||||
| Hi | Hj | HK | HL | Hm | Hn | Ho | Hp | |
| CNCox | 9.8 | 15.2 | 14.7 | 9.4 | 5.7 | 8.2 | 9.2 | 7 |
| cDDP@CNCox | 10 | 16.6 | 13.5 | 9.5 | 5.6 | 8 | 7.9 | 6.5 |
The experimental 15N shifts are referenced to 1.5 mol dm−3 NH4Cl in 1 mol dm−3 HCl and equal to −68.7 ppm.66 In this work, PBEPBE/NMR-DKH/IEF-PCM(UFF) 15N NMR spectra for free cDDP (Table 2) is −63.3 ppm. Our interest is to observe the Δδ15N of cDDP molecule into carbon nanostructures which is much larger than the deviation (∼7%) between theoretical and experimental values for free cDDP. Table 2 shows large variations upon complex I formation, on average −18.6 ppm for high-field region; for complex II, an average shift about 7 ppm is observed for the low-field region. These results show that in the cDDP@CNTox complex, the 15N nuclei (cisplatin NH3 groups) are also more shielded due to the ring current effects.
Fig. 3d–f show PBEPBE/NMR-DKH/IEF-PCM(UFF) 195Pt NMR spectra for free cDDP, and the cDDP@CNTox and cDDP@CNCox inclusions complexes, respectively. Table 2 shows that the use of the relativistic NMR-DKH Gaussian basis sets32 for Pt and other atoms, commonly used as Pt-ligands, leads to δ (calculated by eqn (3)) equals to −2110 ppm for 195Pt nucleus of free cisplatin. This result is in excellent agreement with the experimental69 value for cisplatin in aqueous solution (D2O) that is equals to −2168 ppm, a deviation of only 2.7%. Upon the formation of inclusion complexes, we observed a significant 195Pt signal shift to high-field region for the cDDP@CNTox, found at −2314 ppm. For cDDP@CNCox, the 195Pt chemical shift was at −2192 ppm, also found at high-field region in relation to free cisplatin. These results shown that the cDDP molecule is in closer contact with electron-rich tube surface in complex I than in complex II. Thus, 195Pt nucleus in complex I is more shielded by the ring current effects of the concave surface of the nanotube than in complex II where the internal surface of the nanocone is less concave.
Fig. S2 (ESI)† show the PBEPBE/NMR-DKH/IEF-PCM(UFF) 1H NMR spectra calculated for the organic groups on the oxidized tip of CNTox (Fig. S2a†) and CNCox (Fig. S2b†) and in the inclusion complexes geometries, Fig. S2c and d,† respectively. Analyzing the Fig. S2a and c,† we note that the proton signals of CNTox shift to low-field region (less shielded) when the complex I is formed (Table 2). According to Fig. S2b and d,† due to the proximity of cisplatin to the oxidized surface, most proton signals (Hk, Hm, Hn, Ho and Hp) of CNCox slightly shift to high-field region upon the complex II formation, with average values for both species around 9.8 ppm (Table 2).
The solvent effect on the calculation of NMR chemical shifts is discussed here. As has been pointed out by Benzi et al.70 the continuum solvation models may not be very adequate for the description of solvent effect on DFT calculations of chemical shifts, in what N–H and O–H protons are concerned, due to hydrogen-bond and other solute–solvent interactions. It has been shown recently by Da Silva and De Almeida71 that N–H chemical shifts calculated using the GIAO method and TMS as reference for a series of amine compound (in chloroform) at DFT level using the PCM is systematically underestimated independent of the specific functional employed. The agreement with experimental N–H chemical shifts (in CHCl3) is significantly improved when explicit CHCl3 solvent molecules are included in the NMR calculations, reaching a deviation of −0.1 to −0.2 ppm. The same result was found for cisplatin molecule71 with deviation around 2 ppm being predicted at B3LYP/LANL2DZ/6-31G(d,p)/IEF-PCM(UFF) and PBEPBE/LANL2DZ/6-31G(d,p)/IEF-PCM(UFF) levels of calculation. In the present work using the PBEPBE/NMR-DKH/IEF-PCM(UFF) level the deviation from experiment for N–H cisplatin protons drops to approx. 0.5 ppm, which is a good improvement over the corresponding DFT calculations with the standard 6-31G(d,p) basis set. We expect a similar behavior for the O–H (organic groups on the oxidized surface) chemical shifts, and so O–H Δδ values can be trusted to ±0.5 ppm.
In order to analyze the explicit solvent effect on the calculation of NMR chemical shifts, we performed a MD simulation of the cDDP@CNTox complex in explicit solvent (water molecules). Specifically, the cDDP@CNTox model was selected as a prototype for this analysis which involved the same parameterization procedure and simulation protocol recently employed in our works27,72 and, moreover, in this work it represents the most energetically favorable model.
The simulation protocol proved to be effective in providing the equilibration of the complex I by verifying a convergence in the temporal variation plots of the temperature, kinetic and potential energy, density, volume and pressure through the 100 ns (Fig. S3, ESI†), as also concluded in our recent works with similar systems.27,72 The RMSD value of 0.32 ± 0.05 Å suggests a structural stability of this complex.
Since the NMR chemical shift is sensitive to the chemical environment, we calculated the radial distribution function (RDF) designated as g(r) CM–Ow (see Fig. 4a), which were defined between the center of mass (CM) of the CNTox model and the oxygen atom (Ow) of the water molecules, aiming to define the solvent molecules distribution closest to the encapsulated cDDP drug. In Fig. 4a, the first peak at 2.55 Å indicated the presence of water molecules inside the cavity of nanotube forming an inner solvation shell as previously reported by our group for the carbon nanocones models.27,72 By computing the integral over this first peak, we found a coordination number equals to 2 from the analysis of the red dot line. This means that, on average, there are 2 water molecules distributed around the CM of CNTox. Although this number seems to be small compared to the 13 water molecules found in our previous cDDP@CNHox model,72 it is worth to emphasize that the model studied here (C168H24O15Cl2N2Pt) contains a substantially small nanostructure than that model (C360H38O20Cl2N2Pt) recently studied by our group. When it comes to the solvation structure referring to the oxidized region of the CNTox model, we also calculated the RDF named as g(r) OCNT–OW (see Fig. 4b, which was defined between the oxygen atoms (OCNT) of the functional groups inserted on the CNTox structure and the Ow of the water molecules. By evaluating Fig. 4b, we noticed that while the first solvation shell was centered at 2.85 Å involving, on average 1 water molecule, the third solvation shell was located at 6.45 Å (third peak in Fig. 4) including, on average, 27 solvent molecules.
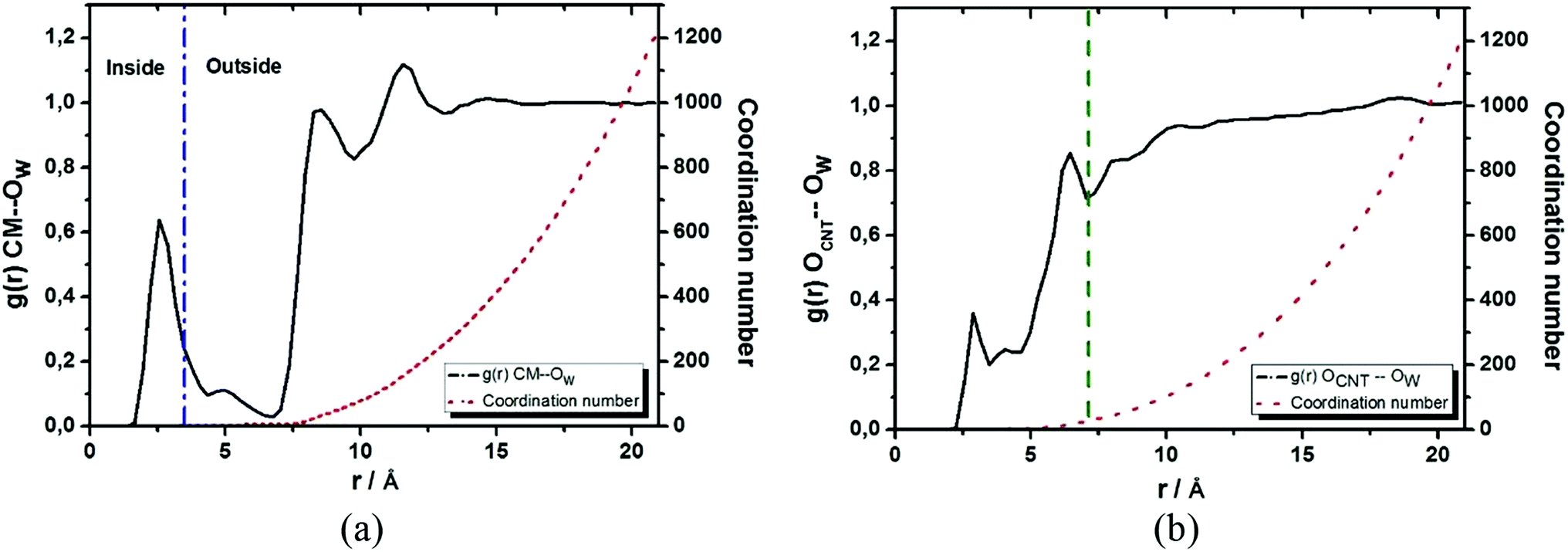 | ||
| Fig. 4 Radial distribution functions: g(r) CM–Ow defined between the center of mass of the CNTox structure and the oxygen atom of the water molecules (a) and g(r) OCNT–OW referring to the oxygen atoms inserted in the CNT structure with respect to the water molecules (b). While the red dot line indicates the coordination number, the blue dash dot line demarcates the water molecules presented in the internal and external region of the CNTox model, and the green dash line indicates the third solvation shell with respect to the oxidized region of the carbon nanostructure. | ||
The temporal variation of the three species (cDDP molecule and two water molecules) encapsulated inside the CNTox cavity (Fig. 4) is presented in Fig. 5a–d, which involves the overlay of 200 frames collected from the 100 ns production run. From Fig. 5a–d, it was possible to conclude that, on average, the plane defined by the square planar geometry of the cDDP molecule remained parallel to the main axis of the carbon nanostructure, as well reported in our recent MD simulations results.27,72 In addition, NH3 groups of cisplatin also remained close to the oxidized region of the CNTox. The same spatial configuration was also observed in our inclusion complexes involving pristine,27 oxidized, and reduced CNH topologies.72 Regarding the inner solvation shell, we observed that the water molecules did not access the space between the cDDP molecule and the oxidized region of the CNTox structure, probably due to steric hindrance of the van der Waals radii. In spite of involving the same overlay of 200 frames selected from the production trajectory (100 ns), Fig. 5e andf focus on the temporal variation of the 10 water molecules closest to the oxidized region of the cDDP@CNTox. These solvent molecules integrated a part of the third solvation shell described by the g(r) OCNT–OW (see Fig. 4b). Fig. 5e and f clearly depicted the almost spherical distribution of the water molecules around the oxidized region of the CNTox model during the 100 ns simulation.
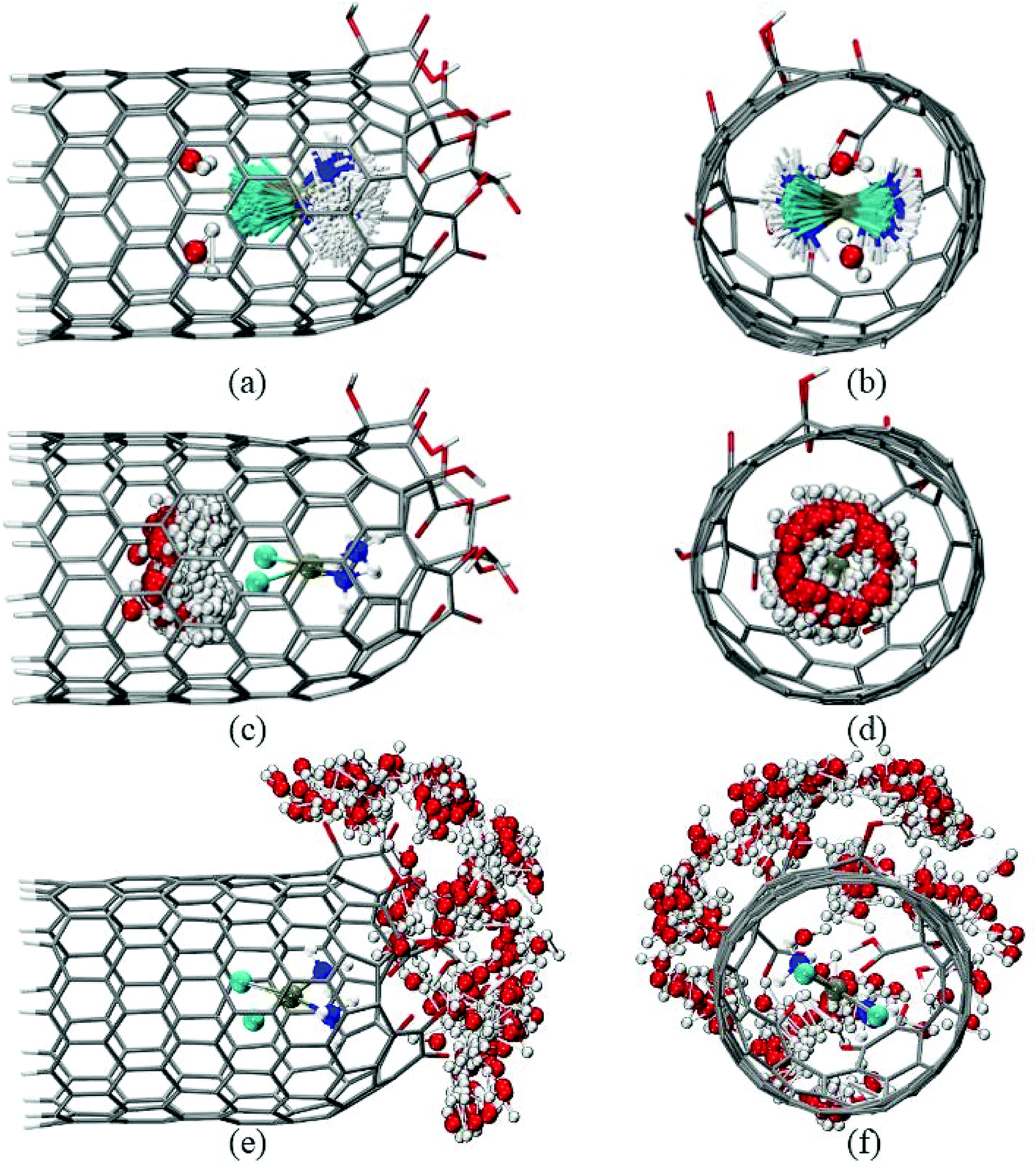 | ||
| Fig. 5 Temporal variation of the encapsulated cDDP molecule, the two encapsulated water molecules, and the ten water molecules around the oxidized region of the CNTox: side view (a, c, and e) and front view (b, d, and f), respectively. | ||
With the inner solvation shell data of the cDDP@CNTox complex, a set of 20 structures (frames) of this model was collected from the 100 ns MD simulation taking into account only the two explicit water molecules. Specifically, these topologies (see the snapshots in Fig. S4–S7, ESI†), named as Structure N (with N in the range of 0 to 19), were selected at 5 ns intervals from the total simulation time. It is worthy to mention that the 20 geometries of the cDDP@CNTox model were not reoptimized at the DFT level of theory before the aforementioned NMR calculations, since the Pt-ligand distances were, on average, close to the experimental results.61
Table S2 (ESI)† shows the PBEPBE/NMR-DKH/IEF-PCM(UFF) 1H, 15N and 195Pt NMR chemical shifts for selected structures (cDDP@CNTox) obtained from the MD simulation. Some dynamic effects can be considered when discussing the chemical shifts, such as: the temporal variation of cisplatin (Fig. 5a and b) and the two water (Fig. 5c and d) molecules inside the nanotube cavity and, the organic groups of the CNTox oxidized region (see Fig. 5e and f). 1H δ values of cDDP (NH3 groups) are more sensitive with respect to temporal variation of the drug itself and organic groups of the CNTox. Among the selected frames, 1H signals can be observed at −188 ppm (Structure 12) and −110.3 ppm (Structure 17), both high-field shifted, respectively. For Structure 3, for example, Ha and Hb signals can be observed at 115 ppm and 96 ppm, both low-field shifted, respectively. From Fig. S8 to S10 (ESI),† the dispersion of the 1H signals of the cisplatin are observed. 1H chemical shifts are influenced by the rotations of the NH3 groups around the Pt–N bond axis and also by the temporal variation of the organic groups of the CNTox model during the MD simulation. 1H nuclei are more strongly shielded (high-field shifted) when the organic groups are closer to them; by contrast, the shielding effect is less (low-field shifted) when such groups are more distant from the cisplatin protons. Thus, considering the dynamic nature of these rotations and the selected frames, our NMR calculations revealed that the protons (NH3 groups) of cisplatin can be identified in the range 115 to −188 ppm. 15N chemical shifts (Table S3†) appear to be more dependent on the temporal variation of the cisplatin molecule that is described by incomplete rotations around the center of mass of the drug in the tube cavity. The 15N δ values are found in the 272–102 ppm range (low-field region). Fig. 5a and b shows that for the selected frames, the translational movement of cDDP molecule inside the CNTox cavity is limited. The intermolecular distances between the two water molecules revealed by the RDF analysis and the drug molecule vary in the 3.86–4.84 Å range (see Table S3, ESI).† Fig. 6 shows the 195Pt NMR spectra for the 20 selected structures obtained from MD trajectory (see also Table S2†). The average chemical shift (−2086 ppm) in the low-field region compared to the experimental value (−2168 ppm, see Table 2) suggests that the two water molecules have no direct influence on the shielding of the 195Pt nucleus. In relation to the cDDP@CNTox model optimized at B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF) level, a difference of approximately +228 ppm for the low-field region can be estimated. Thus, as observed for the hydrogens, the 195Pt NMR chemical shift is also sensitive to the dynamics of the cDDP molecule inside the CNTox cavity. We selected a structure from the MD simulation to investigate the effect of 10 water molecules on the 1H NMR spectrum of the CNTox oxidized region, the 1H δ are shown in the Table S4 (ESI).† For the isolated CNTox in the inclusion complex geometry, all 1H δ values are shifted to the low-field region, on average 8.4 ppm (carboxyls groups) and 9.4 ppm (hydroxyls groups). For the cDDP@CNTox, all 1H δ values of carboxyls groups are shifted to the high-field region while the majority of proton signals of the hydroxyl groups are shifted to the low-field region. These results show that the field effect due to the presence of the cDDP molecule is greater than that caused by the 10 water molecules.
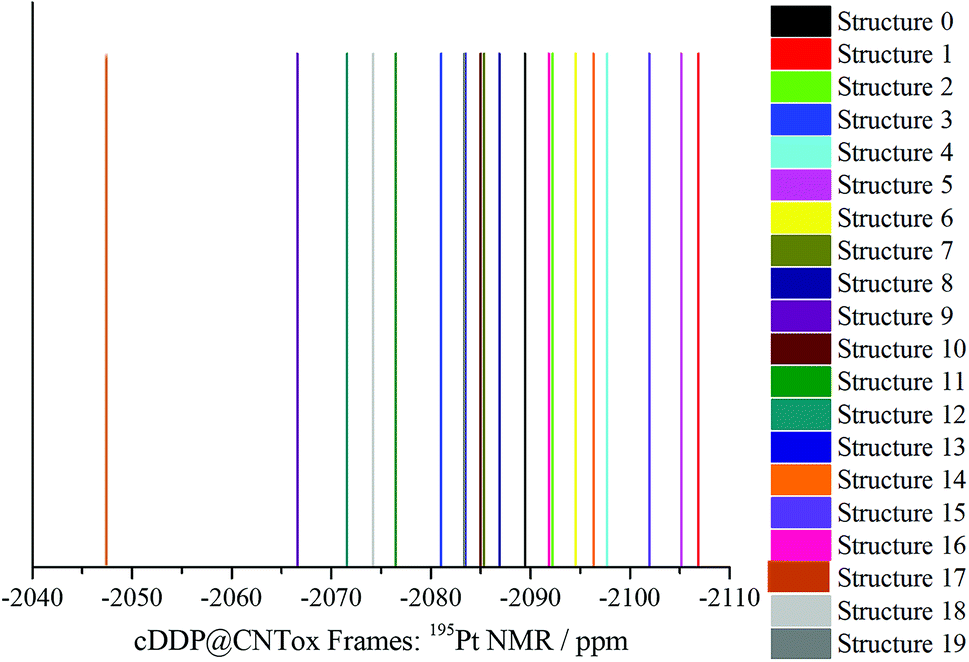 | ||
| Fig. 6 PBEPBE/NMR-DKH/IEFPCM(H2O) 195Pt NMR spectrum of structures 0 to 19 (frames) obtained by MD simulation is shown in Fig. 5a and b. | ||
In light of the reported results, we recommend the new NMR-DKH basis sets and the computational protocol proposed by Paschoal et al.32 as a good alternative to predict the 195Pt and 1H NMR spectra of inclusion complexes formed by cisplatin and carbon nanostructures. Our results combined with experimental NMR investigations can demonstrate an efficient and promising strategy for the application of oxidized carbon nanostructures (CNTox and CNCox) in the area of drug delivery and controlled release systems.
Conclusions
In this work we used the relativistic NMR-DKH basis sets for the calculation of the 1H, 15N and 195Pt NMR spectra of inclusion complexes formed between cisplatin and oxidized carbon nanotube and nanocone molecules. The structural results at B3LYP/LANL2DZ/def2-SVP/IEF-PCM(UFF) level showed that intermolecular hydrogen-bonds contribute to the complexes stabilization. The complex I formed by cDDP and CNTox was around 70% more favorable than that with CNCox. According to the reported results, the experimental detection of the inclusion compound could be attained through the analysis of the 1H and 15N (NH3 groups), and 195Pt NMR spectra for cisplatin guest molecule. 1H NMR results showed a shift in the NMR signals from 4.3 ppm (cDDP) to −5.1 and +6.6 ppm (cDDP@CNTox and CDDP@CNCox complexes, respectively). Our 195Pt NMR also shifted upon inclusion, found at −2314 and −2192 ppm for inclusion complexes with CNTox and CNCox nanostructures, respectively compared to the free CDDP (−2110 ppm). We investigated the explicit solvent effect (water molecules) on the calculation of 1H, 15N and 195Pt NMR chemical shifts by MD simulation of the cDDP@CNTox complex. The results showed that the two water molecules inside the nanotube cavity do not directly influence the 1H, 15N and 195Pt chemical shifts of the cisplatin molecule. The calculated spectra are more sensitive to the dynamics of the drug relative to the inner surface of the tube. Lastly, in addition to the chemical evidences which might guide the experimentalists to characterize the cDDP@CNTox (or CNCox) DDS, the computational protocol used here including the NMR-DKH basis sets might be useful to predict 195Pt NMR chemical shift of big molecules such as the inclusion complexes formed by cisplatin and carbon nanostructures with a reasonable computational cost.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. A. De Souza thanks the CAPES for a Post-Doctoral scholarship (Proc. No. 88882.306139/2018-01) during the period 2017–2018 at Universidade Federal Fluminense and support provided by Professor J. C. Belchior for the continuity of this work at Universidade Federal de Minas Gerais. L. A. De Souza also thanks the Capes for a new Post-Doctoral scholarship (Proc. No. 88887.363111/2019-00) at Universidade Federal de Minas Gerais. E. R. Almeida thanks FAPEMIG for his current PhD scholarship at Universidade Federal de Juiz de Fora. H. F. Dos Santos thanks CNPq and FAPEMIG for providing continuous support to his laboratory. W. B. De Almeida would like to thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for a research fellowship (Proc. No. 310102/2016-2) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) for support (Proc. No. 233888.). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.References
- R. A. Alderden, M. D. Hall and T. W. Hambley, J. Chem. Educ., 2006, 83, 728–734 CrossRef CAS.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS.
- J. Zhang, D. S. Thomas, M. S. Davies, S. J. Berners-Price and N. Farrell, J. Biol. Inorg Chem., 2005, 10, 652–666 CrossRef CAS.
- J. Valle, H. Wasan, D. H. Palmer, D. Cunningham, A. Anthoney, A. Maraveyas, S. Madhusudan, T. Iveson, S. Hughes, S. P. Pereira, M. Roughton and J. Bridgewater, N. Engl. J. Med., 2010, 362, 1273–1281 CrossRef CAS.
- R. Pinto-Leite, R. Arantes-Rodrigues, C. Palmeira, B. Colaço, C. Lopes, A. Colaço, C. Costa, V. M. Da Silva, P. Oliveira and L. Santos, Biomed. Pharmacother., 2013, 67, 116–121 CrossRef CAS.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS.
- S. Duggan, W. Cummins, O. O. '. Donovan, H. Hughes and E. Owens, Eur. J. Pharm. Sci., 2017, 100, 64–78 CrossRef CAS.
- J.-J. Yi, S. Sharma, S. P. Shumyak, Z.-X. Wan, Z.-W. Zhou, Y. Zhang, P. Guo, C.-Z. Li, J. R. Kanwar, T. Yang, S. S. Mohapatra, W. Liu, W. Duan, J.-C. Wan, Q. Li, X. Zhang, J. Tan, L. Jia, J. Liang, M. Q. Wei, X. Li and S.-F. Zhou, PLoS One, 2013, 8, 1–21 Search PubMed.
- A. Dag, M. Callari, H. Lua and M. H. Stenzel, Polym. Chem., 2016, 7, 1031–1036 RSC.
- A. Bunker, A. Magarkar and T. Viitala, Biochim. Biophys. Acta, 2016, 1858, 2334–2352 CrossRef CAS.
- M. Cui, W. Wua, L. Hovgaard, Y. Lu, D. Chen and J. Qi, Inter. J. Pharm., 2015, 489, 277–284 CrossRef CAS.
- C.-Y. Yeh, J.-K. Hsia, Y.-P. Wang, C.-H. Lan and H.-C. Wu, Biomaterials, 2016, 99, 1–15 CrossRef CAS.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS.
- S. K. Vashist, D. Zheng, G. Pastorin, K. Al-Rubeaan, J. H. T. Luong and F.-S. Sheu, Carbon, 2011, 49, 4077–4097 CrossRef CAS.
- A. Kazemi-Beydokhti, S. Z. Heris and M. R. Jaafari, Micro Nano Lett., 2015, 10, 241–247 CrossRef CAS.
- M. Horie, L. K. Komaba, H. Fukui, H. Kato, S. Endoh, A. Nakamura, A. Miyauchi, J. Maru, E. Miyako, K. Fujit, Y. Hagihara, Y. Yoshida and H. Iwahashi, Carbon, 2013, 54, 155–167 CrossRef CAS.
- J. Li, S. Qi Yap, S. L. Yoong, T. R. Nayak, G. W. Chandra, W. H. Ang, T. Panczyk, S. Ramaprabhu, S. K. Vashist, F.-S. Sheu, A. Tan and G. Pastorin, Carbon, 2012, 50, 1625–1634 CrossRef CAS.
- K. Werengowska-Ciecwierz, M. Wisniewski, A. P. Terzyk, N. Gurtowska, J. Olkowska, T. Kloskowski, T. A. Drewa, U. Kiełkowska and S. Druzynski, Carbon, 2014, 70, 46–58 CrossRef CAS.
- R. Bessrour, Y. Belmiloud, Z. Hosni and B. Tangour, AIP Conf. Proc., 2012, 1456, 229–239 CrossRef CAS.
- Z. Hosni, R. Bessrou and B. Tangour, J. Comput. Theor. Nanosci., 2014, 11, 318–323 CrossRef CAS.
- M. Ravera, E. Gabano, S. Bianco, G. Ermondi, G. Caron, M. Vallaro, G. Pelosi, I. Zanellato, I. Bonarrigo, C. Cassino and D. Osella, Inorg. Chim. Acta, 2015, 432, 115–127 CrossRef CAS.
- V. Levet, R. Rosière, R. Merlos, L. Fusaro, G. Berger, K. Amighi and N. Wauthoz, Int. J. Pharm., 2016, 515, 209–220 CrossRef CAS.
- L. A. De Souza, C. A. S. Nogueira, J. F. Lopes, H. F. Dos Santos and W. B. De Almeida, J. Inorg. Biochem., 2013, 129, 71–83 CrossRef CAS.
- L. A. De Souza, C. A. S. Nogueira, J. F. Lopes, H. F. Dos Santos and W. B. De Almeida, J. Phys. Chem. C, 2015, 19, 8394–8401 CrossRef.
- L. A. De Souza, C. A. S. Nogueira, P. F. R. Ortega, J. F. Lopes, H. D. R. Calado, R. L. Lavall, G. G. Silva, H. F. Dos Santos and W. B. De Almeida, Inorg. Chim. Acta, 2016, 447, 38–44 CrossRef CAS.
- L. A. De Souza, H. F. Dos Santos, L. T. Costa and W. B. De Almeida, J. Inorg. Biochem., 2018, 178, 134–143 CrossRef CAS.
- E. R. Almeida, L. A. De Souza, W. B. De Almeida and H. F. Dos Santos, J. Mol. Graphics Modell., 2019, 89, 167–177 CrossRef CAS.
- J. Vícha, M. Patzschkec and R. Marek, Phys. Chem. Chem. Phys., 2013, 15, 7740–7754 RSC.
- J. Vícha, J. Novotný, M. Straka, M. Repisky, K. Ruud, S. Komorovsky and R. Marek, Phys. Chem. Chem. Phys., 2015, 17, 24944–24955 RSC.
- L. A. Truflandier, K. Sutter and J. Autschbach, Inorg. Chem., 2011, 50, 1723–1732 CrossRef CAS.
- T. Pawlak, M. L. Munzarová, L. Pazdersk and R. Marek, J. Chem. Theory Comput., 2011, 7, 3909–3923 CrossRef CAS.
- D. Paschoal, C. F. Guerra, M. A. L. De Oliveira, T. C. Ramalho and H. F. Dos Santos, J. Comput. Chem., 2016, 37, 2360–2373 CrossRef CAS.
- J. Carvalho, D. Paschoal, C. Fonseca Guerra and H. F. Dos Santos, Chem. Phys. Lett., 2020, 745, 137279 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Li, X. Caricato, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Wallingford CT, 2009 Search PubMed.
- R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- K. Wolinski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- W. L. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- S. Zheng, Q. Tang, J. He, S. Du, S. Xu, C. Wang, Y. Xu and F. Lin, J. Chem. Inf. Model., 2016, 56, 811–818 CrossRef CAS.
- J. F. Lopes, W. R. Rocha, H. F. Dos Santos and W. B. De Almeida, J. Chem. Phys., 2008, 128, 165103–165117 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar and J. D. Madura, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins, 2006, 65, 712–725 CrossRef CAS.
- B. P. Uberuaga, M. Anghel and A. F. Voter, J. Chem. Phys., 2004, 120, 6363–6374 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Gunsteren, A. Dinola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- F. Jensen, Introduction to computational chemistry, John Wiley & Sons, Chichester, 2nd edn, 2007 Search PubMed.
- M. R. Hestenes and E. Stiefel, J. Res. Natl. Bur. Stand., 1952, 49, 409–436 CrossRef.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS.
- L. A. De Souza, A. M. Da Silva Jr., H. F. Dos Santos and W. B. De Almeida, RSC Adv., 2017, 7, 13212–13222 RSC.
- G. H. W. Milburn and M. R. Truter, J. Chem. Soc. A, 1966, 1609–1616 RSC.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef.
- A. Szabo and N. S. Ostlund, Modern Quantum Chemistry, Introduction to Advanced Electronic Structure Theory, Dover Publications Inc., New York, 1996 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS.
- S. J. Berners-Price, T. A. Frenkiel, U. Frey, J. D. Ranford and P. J. Sadler, J. Chem. Soc., Chem. Commun., 1992, 789–791 RSC.
- A. C. Forse, J. M. Griffin, V. Presser, Y. Gogotsi and C. P. Grey, J. Phys. Chem. C, 2014, 118, 7508–7514 CrossRef CAS.
- L. B. Casabianca, J. Phys. Chem. A, 2016, 120, 7011–7019 CrossRef CAS.
- F. D. Rochon and A. Morneau, Magn. Reson. Chem., 1991, 29, 120–126 CrossRef CAS.
- C. Benzi, O. Crescenzi, M. Pavone and V. Barone, Magn. Reson. Chem., 2004, 42, 57–67 CrossRef.
- H. C. Da Silva and W. B. De Almeida, Chem. Phys., 2020, 528, 110479 CrossRef CAS.
- E. R. Almeida, L. A. De Souza, W. B. De Almeida and H. F. Dos Santos, J. Chem. Inf. Model., 2020, 60, 500–512 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra06044e |
| This journal is © The Royal Society of Chemistry 2021 |