
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew poly-imidazolium–triazole particles by CuAAC cross-linking of calix[4]arene bis-azide/alkyne amphiphiles – a prospective support for Pd in the Mizoroki–Heck reaction†
Vladimir Burilov *a,
Ramilya Garipovaa,
Diana Mironovaa,
Elza Sultanovaa,
Ilshat Bogdanova,
Evgeny Ocherednyuka,
Vladimir Evtugync,
Yuri Osinc,
Ildar Rizvanovb,
Svetlana Solovievab and
Igor Antipina
*a,
Ramilya Garipovaa,
Diana Mironovaa,
Elza Sultanovaa,
Ilshat Bogdanova,
Evgeny Ocherednyuka,
Vladimir Evtugync,
Yuri Osinc,
Ildar Rizvanovb,
Svetlana Solovievab and
Igor Antipina
aKazan Federal University, 18 Kremlevskaya st, Kazan, 420008, Russian Federation. E-mail: ultrav@bk.ru; Fax: +7-843-238-79-01; Tel: +7-843-2337344
bArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center of RAS, 8 Arbuzov str., Kazan, 420088, Russian Federation
cInterdisciplinary Centre for Analytical Microscopy Kazan Federal University, 18 Kremlevskaya st, Kazan, 420008, Russian Federation
First published on 24th December 2020
Abstract
A new imidazolium amphiphilic calix[4]arene with terminal acetylene fragments in the polar region was synthesized according to a two step scheme including regioselective chloromethylation of distal di-O-butyl calix[4]arene and subsequent interaction with 1-(hex-5-yn-1-yl)-1H-imidazole. The aggregation properties (CAC, the size and zeta potential of aggregates) of alkynyl calix[4]arene as well as of previously synthesized azidopropyl calix[4]arene and their 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture were disclosed. Macrocycles with azide and alkyne fragments in the polar region were covalently cross-linked under CuAAC conditions in water. Successful cross-linking of molecules has been proven by IR spectroscopy and MALDI-TOF spectrometry. The obtained polymeric particles were studied both in solution and the solid state and the presence of submicron (∼200 nm) and micron (∼1–5 μm) particles with the prevalence of the latter was found. The average molecular weight of the polymer according to the static light scattering data was found to be 639 ± 44 kDa. The obtained polymeric imidazolium–triazole particles were tested as a support for Pd(OAc)2 in the Mizoroki–Heck reaction carried out in both organic and water media. In both solvents (especially in water) the addition of imidazolium–triazole particles to Pd(OAc)2 increased the conversion of 4-iodanisole. It was found that the ratio between the products (1,1 and 1,2-substituted ethylenes) changes drastically on going from DMF to water from 1:5 to 1:40 when using supported Pd(OAc)2.
:1 mixture were disclosed. Macrocycles with azide and alkyne fragments in the polar region were covalently cross-linked under CuAAC conditions in water. Successful cross-linking of molecules has been proven by IR spectroscopy and MALDI-TOF spectrometry. The obtained polymeric particles were studied both in solution and the solid state and the presence of submicron (∼200 nm) and micron (∼1–5 μm) particles with the prevalence of the latter was found. The average molecular weight of the polymer according to the static light scattering data was found to be 639 ± 44 kDa. The obtained polymeric imidazolium–triazole particles were tested as a support for Pd(OAc)2 in the Mizoroki–Heck reaction carried out in both organic and water media. In both solvents (especially in water) the addition of imidazolium–triazole particles to Pd(OAc)2 increased the conversion of 4-iodanisole. It was found that the ratio between the products (1,1 and 1,2-substituted ethylenes) changes drastically on going from DMF to water from 1:5 to 1:40 when using supported Pd(OAc)2.
Introduction
In the last thirty years, interest in metal complexes based on N-heterocyclic carbene (NHC) ligands has been explosively growing.1–3 Firstly obtained in 1991 by Arduengo,4 this class of ligands can be easily generated through deprotonation of appropriate precursors (most often imidazolium salts are used) and the steric/electron donor properties of the ligand can be easily regulated by substituents on the heterocycle precursor core. Thus, due to the easy synthesis of the starting imidazolium salts and the stability of the final complexes to moisture and air oxygen, NHC ligands now successfully displace traditional phosphine systems in many reactions including green ones in aqueous media.5–7An important trend in today's catalysis is the creation of heterogeneous catalysts based on well-proven homogeneous ones. It is known that homogeneous catalyst systems suffer from problems with separation and recycling of catalysts. In addition, homogeneous catalysts usually cause contamination of products with residual ligands. Thus, the heterogenization of ligands attracts much attention. Such heterogeneous catalysts have several advantages, the main one is the easy recovery and fast recycling of catalysts by filtration with the same activity as of homogeneous catalytic systems and their high surface area.8–10 For the synthesis of such catalysts, the necessary anchor groups are introduced into the metal complex or NHC-ligand for immobilization on a organic/inorganic support (Scheme 1a).11,12
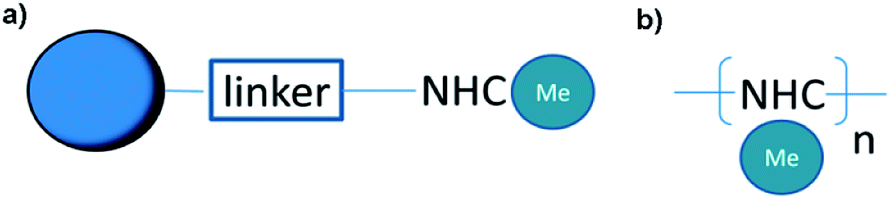 | ||
| Scheme 1 Schematic representation of heterogenic NHC catalysts based on (a) NHC supported on a carrier and (b) self-supported NHC polymers. | ||
An alternative approach is to include NHC fragments into the main polymer chain, which acquires both the function of the carrier and ligand (Scheme 1b). Such approach was successfully realized using cross-linking of imidazole derivatives with di/tri haloalkyl linkage,13,14 Sonogashira coupling of halo-NHC monomer and triacetylene linker15 or metal as linker in metal–organic frameworks (MOFs)16 etc. In this case, the entire volume of the system acts as a catalyst, and the properties (porosity, morphology) can be easily tuned by making appropriate adjustments in the structure of the NHC monomer. In turn, the incorporation of NHC metal complexes into porous structures often affects not only the catalytic activity, but also selectivity due to the specific structure of the reaction “pocket”.17
Thus, organic and coordination chemistry approaches are successfully used for the formation of the self-supported carriers. However, for the best of our knowledge there are no examples of NHC-catalytic systems made by preliminary supramolecular assembly of NHC ligand monomers with their subsequent covalent fixation, which nevertheless has a great advantage in the formation of functional nanomaterials with controlled morphology. NHC-complexes on the calix[4]arene scaffold attracts a great attention18 since it is possible to anchor one or more NHC fragments at one molecular platform, which plays role both as bulky ligand and molecular cavity contribute to catalytic transformations. Recently we demonstrated that amphiphilic Pd(II) bis-NHC complexes on the thiacalix[4]arene backbone19 or in situ made Pd(II) bis-NHC complexes on the calix[4]arene backbone20 can be successfully used in coupling and hydrogenation reactions, performed in organic or water media.
Herein we propose new approach to the formation of NHC carriers, based on sequential self-assembly of amphiphilic calix[4]arenes containing azidoalkyl/alkynyl fragments on the polar region of macrocycles into aggregates in an aqueous solution, followed by foregoing cross-linking of macrocycles using copper-catalyzed azide–alkyne cycloaddition (CuAAC)21 reaction (Scheme 2).
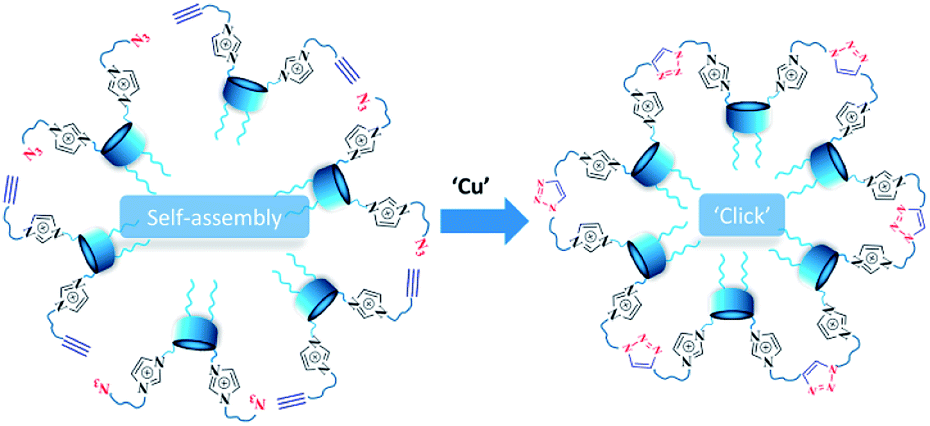 | ||
| Scheme 2 Self-assembly + CuAAC approach to the formation of NHC carriers. | ||
Results and discussion
Synthesis
For the synthesis of target macrocycles containing azidoalkylimidazolium/alkynylimidazolium groups, we used our previously reported approach22 – alkylation of the corresponding N-substituted imidazoles with dichloromethyl calix[4]arene derivative 3 (Scheme 3). Foregoing synthetic strategy allows in just three steps to introduce any alkyl/imidazolium fragments into the lower/upper rims of calix [4]arene macrocyclic platform with high yields. p-H-calix[4]arene 1, pre-synthesized from p-tert-butyl-calix[4]arene using de-tert-butylation with AlCl3 in the presence of phenol,23 was treated by butyl bromide to give bis-O-butyl derivative 2, which was then involved in the Blanc chloromethylation22 to give calix[4]arene 3 with nearly qualitative yield (Scheme 3). The final step was reaction of calix[4]arene 3 with 1-(3-azidopropyl)-1H-imidazole or 1-(hex-5-yn-1-yl)-1H-imidazole. 1-(3-Azidopropyl)-1H-imidazole was synthesized according to literature method.24 Considering the lack of information on the synthesis of 1-(hex-5-yn-1-yl)-1H-imidazole, it was successfully synthesized under reaction of hex-5-yn-1-yl 4-methylbenzenesulfonate with 1H-imidazole in the presence of NaH. The structure of 1-(hex-5-yn-1-yl)-1H-imidazole was proven using NMR 1H and 13C, IR and EI mass-spectrometry. The presence of signals of imidazole protons as two broad singlets at 7.06 and 6.92 ppm as well as singlet of N–CH–N proton at 7.47 and typical set of proton signals of hex-5-yn-1-yl fragment in NMR 1H confirms the structure. Reaction of calix[4]arene 3 with three equivalents of 1-(3-azidopropyl)-1H-imidazole or 1-(hex-5-yn-1-yl)-1H-imidazole in boiling acetonitrile with following precipitation of products from diethyl ether gave alkyne 4 and azide 5. All spectral data for product 5 matched previously published one.22 In the 1H NMR spectra of compound 4, new signals of the protons of the imidazolium fragment appear as two broadened singlets at 7.80 and 9.39 ppm, the protons of hex-5-yn-1-yl fragment are presented as two multiplets at 1.88, 1.42 ppm, –CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) proton signal appears as doublet of triplets with J = 7.2 and 2.4 Hz, whereas terminal CCH proton signal appears as triplet with J = 2.4 Hz. The structure of macrocycle 4 was additionally investigated by 2D 1H–1H NOESY NMR spectroscopy (Fig. 1). The presence of cross peaks between protons of two neighboring benzene rings (δ = 7.31 and 7.04 ppm), cross peaks between protons of benzene rings with methylene protons of the imidazolium fragment, attached to neighboring benzene rings (δ = 7.05 and 5.22 ppm) as well as cross peaks between –OH protons and protons of the O–CH2–CH2- fragments (δ = 8.70 and 3.98/1.96 ppm) clarify that 4 is in “cone” stereoisomeric form. In the MALDI-TOF mass-spectrum of the 4 there is molecular ion peak [M-2HCl + DBH]+ with m/z = 1010 and two peaks correspond to repulsion of one or two fragments of 1-(hex-5-yn-1-yl)-1H-imidazole ([M-2HCl–C9H12N2]+ with m/z = 709 and ([M-2HCl–2C9H12N2]+ with m/z = 561]. Expulsion of substituents in benzyl position of phenol ring was observed earlier in our previous works20,22 and is a result of easy formation of p-quinone methide structures during ionization.25
proton signal appears as doublet of triplets with J = 7.2 and 2.4 Hz, whereas terminal CCH proton signal appears as triplet with J = 2.4 Hz. The structure of macrocycle 4 was additionally investigated by 2D 1H–1H NOESY NMR spectroscopy (Fig. 1). The presence of cross peaks between protons of two neighboring benzene rings (δ = 7.31 and 7.04 ppm), cross peaks between protons of benzene rings with methylene protons of the imidazolium fragment, attached to neighboring benzene rings (δ = 7.05 and 5.22 ppm) as well as cross peaks between –OH protons and protons of the O–CH2–CH2- fragments (δ = 8.70 and 3.98/1.96 ppm) clarify that 4 is in “cone” stereoisomeric form. In the MALDI-TOF mass-spectrum of the 4 there is molecular ion peak [M-2HCl + DBH]+ with m/z = 1010 and two peaks correspond to repulsion of one or two fragments of 1-(hex-5-yn-1-yl)-1H-imidazole ([M-2HCl–C9H12N2]+ with m/z = 709 and ([M-2HCl–2C9H12N2]+ with m/z = 561]. Expulsion of substituents in benzyl position of phenol ring was observed earlier in our previous works20,22 and is a result of easy formation of p-quinone methide structures during ionization.25
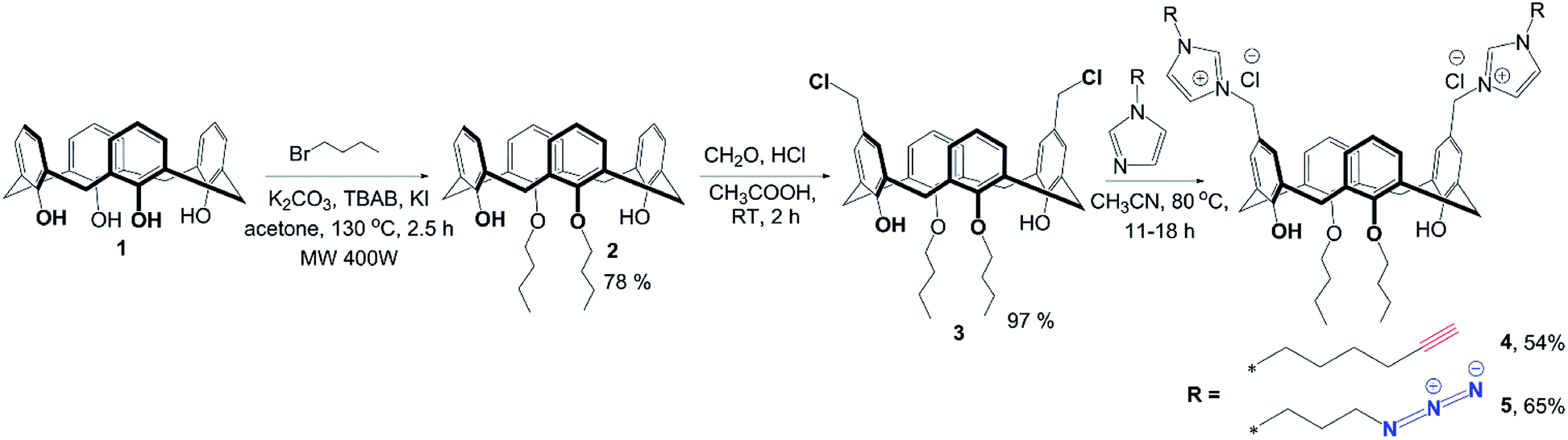 | ||
| Scheme 3 Synthetic pathway for macrocycles 4 and 5 containing azidoalkylimidazolium/alkynylimidazolium groups on the upper rim. | ||
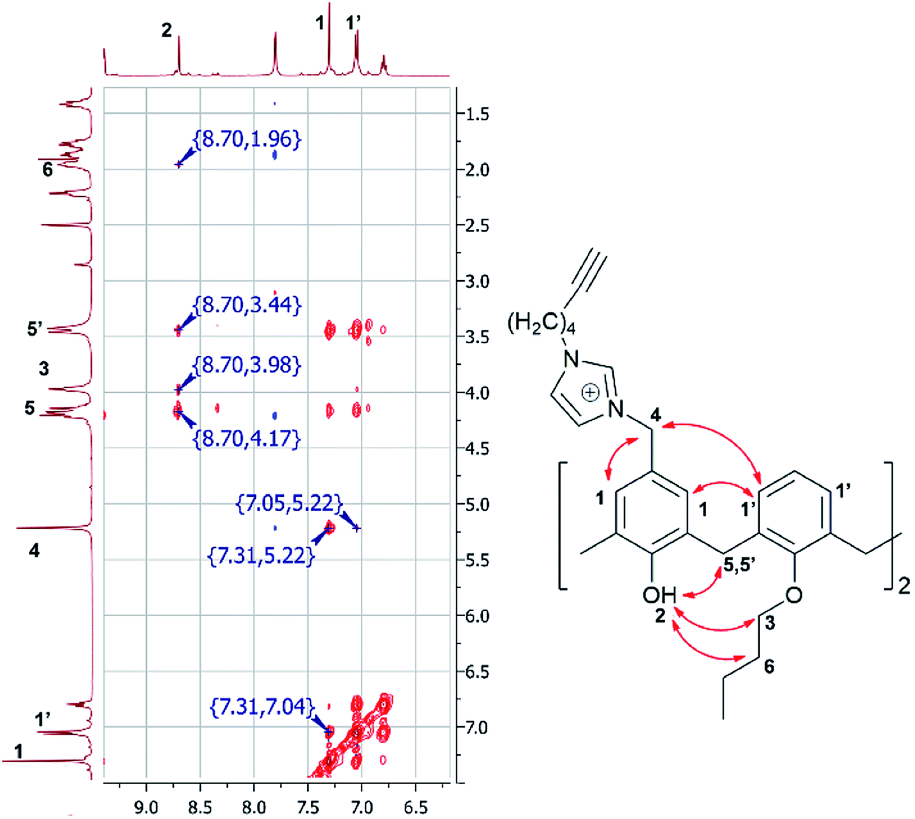 | ||
| Fig. 1 Fragment of the 2D 1H–1H NOESY NMR spectra of 4 (DMSO-d6). | ||
Aggregation behavior of 4, 5 and their mixture in aqueous solutions and their cross-linking using CuAAC
The preliminary task before covalent cross-linking of calixarenes under CuAAC conditions was to find the optimal concentration conditions for the formation of stable colloidal systems in aqueous solutions for each macrocycle undividually as well as for their mixtures in an 1:1 equimolar ratio. Previously we have shown20 that imidazolium macrocycles with similar architecture form submicron vesicle-like particles with high positive electrokinetic potential (around +50 mV) in aqueous solutions. To estimate amphiphilic properties of calixarenes 4, 5 and their mixture, their critical aggregation concentration (CAC) values were measured using fluorescent method with pyrene as a probe. The intensity ratio of the first and third peaks in pyrene fluorescence spectra (polarity index) changes under a decrease in the pyrene polarity environment, caused by its solubilization in the hydrophobic zone of the calixarene aggregates. Corresponding CAC values are given in Table 1. Interestingly, macrocycle 5 with 3-azidopropyl fragments have quite significant CAC value, close to that for N-methylbenzimidazolium derivative (CAC = 120 μM), presented in our recent study.20 This may be a result of increase of the volume of the hydrophilic headgroup of the macrocycle, thanks to the spreading alkylazide groups, oriented to the water phase26 and preventing thus effective aggregation at lower concentrations. In the case of hex-5-yn-1-yl-substituted 4 CAC dramatically decreases by two orders of magnitude. In this case, the more hydrophobic hex-5-yn-1-yl substituents can be oriented towards the alkyl substituents of the calixarene core, increasing thus the whole lipophilicity of the molecule and facilitating its aggregation. Combining two macrocycles together leads to a certain averaging of the CAC, which nevertheless becomes much closer to 4 than to 5. According to dynamic and electrophoretic light scattering (DLS and ELS, Table 1) macrocycles 4, 5 and their mixtures give submicron particles with a hydrodynamic diameter in the range of 200 nm and electrokinetic potential around +50 mV, referring them to the stable colloids. Covalent cross-linking of calixarenes 4 and 5 was performed using 25 mol% of CuCl2 in the presence of 50 mol% of sodium ascorbate in deionized water using 0.2 mM of 4 and 5 during 12 hours at 25 °C under virgious stirring. During the reaction, a significant increase in the turbidity of the solution was observed, indicating thus the formation of large particles (Fig. 2). The resulting particles were purified from copper and sodium ascorbate by dialysis against a solution of Trilon B (0.01 M) and then against deionized water. The resulting particles were then dried and characterized by IR spectroscopy. Intense band at 2096 cm−1, related to the stretching vibrations of the azide group in the spectrum of the initial azide 5 (Fig. S4,† curve b), completely disappeared in the spectra of cross-linked polymer (Fig. S4,† curve c). The same changes were found when comparing to the initial IR spectra of alkyne 4: the shoulder at 3300 cm−1, related to the terminal alkynyl C–H bond stretching vibrations (Fig. S4,† curve a), disappeared in the polymer spectra. According to the transmission electron microscopy (TEM) (Fig. 3a and b), obtained polymeric particles have spherical shape with an average diameter around 50 nm. Polymer nanoparticles also aggregate into larger submicron particles with the size up to 500 nm. The same sample was studied using scanning electron microscopy (SEM) (Fig. 3c). According to SEM data, in addition to small nanoparticles, there are also larger formations with an average diameter closed to 1 μm. Particles were also analyzed using the method of energy-dispersive X-ray spectroscopy (EDX). It is seen (Fig. 4d) that in addition to copper from the copper grid, used in TEM, as well as silicon from the glass chemical vessel, the sample contains signals of carbon, nitrogen and oxygen in a full accordance with the composition of 4 and 5. According to DLS data (Table 1) in aqueous solutions after polymerization of the mixture of 4 and 5 the size of particles dramatically changes: the main particles (60%) presented in solution have the size around 980 nm, but there are also smaller 200 nm particles (20%) and lager micron particles (20%). Thus, the DLS data is in a good accordance with microscopy data. According to ELS data (Table 1), electrokinetic potential of the mixture of 4 and 5 after polymerization undergoes a threefold decrease, which can be attributed to decrease of electrophoretic mobility of large particles27 compared to non-polymeric ones. Additionally polymeric particles were studied using MALDI-TOF mass-spectrometry techniques. The best results were obtained using 2,5-dihydroxybenzoic acid (DHB) matrix. The most intense peak (1177.6 Da) was interpreted as a simplest polymer fragment with two imidazole and triazole moieties on the base of 5 skeleton, containing butylimidazole fragments from two neighboring alkynes 4 (Fig. 4). Also were found peaks corresponding to three calixarene fragments (2258.5 Da), four calixarene fragments and one DHB (3398.0 Da) and a maximum found peak (4540.5 Da) was referred to a fragment of pentamer with two DHB molecules. In all the peaks found, the elimination of hydrogen chloride molecules was observed. Previously,28 similar observations have already been found in the MALDI-TOF spectra of polyimidazole-containing polymers and were explained as a result of conversions of imidazolium ion to neutral carbene by deprotonation of the most acidic N–CH–N proton owing to applied energy of the laser beam. It is important to note that, in contrast to the initial macrocycles, where the MALDI-TOF spectra included only fragmentation of N-alkylimidazolium fragments with the formation of a stable quinone-methide calixarene structure, fragmentation in the polymer includes the cleavage on the “other side” of the imidazolium fragment. As a result, fragment ions with propyl groups are observed. Taking into account that in MALDI-TOF multiply charged molecules usually tend to fragment into a series of singly charged ions and the main possible pathway of fragmentation is the cleavage of a chemical bond to the charged centers,29 this fragmentation pathway is expected. Taking into account the DLS and microscopy data, as well as the MALDI-TOF, it can be assumed that in addition to small structured polymer particles, which are clearly visible in TEM images, linear “open chain” polymers are also formed. The driving force of their formation is the unwrapping of calixarene aggregates during CuAAC cross-linking, which results in uncontrollable increase of the degree of polymerization (Scheme 4). Static light scattering data fully confirm this assumption. According to the results obtained, the cross-linked particles of 4 and 5 have an average molecular weight of 639 ± 44 kDa (ESI, Fig. S3†). Thus, a polymer has at least 600 macrocyclic units, which nevertheless can have completely different packing, including formation of compact particles, which are presented in microscopy photographs. In the future, the problem of formation of open-chain polymers can be solved by increasing the hydrophobicity of both macrocycles and admixing macrocycles containing four azide (or alkyne) fragments on the polar region to ensure crosslinking, that is what we are working on now.
 | ||
| Fig. 2 Photography of 10 mm cuvette containing mixture of 4 and 5 in water before (a) and after (b) CuAAC cross-linkage (C(4) = C(5) = 0.2 mM). | ||
 | ||
| Fig. 3 TEM (a and b), SEM (c) images and EDX spectra (d) of mixture of 4 and 5 after cross-linkage. | ||
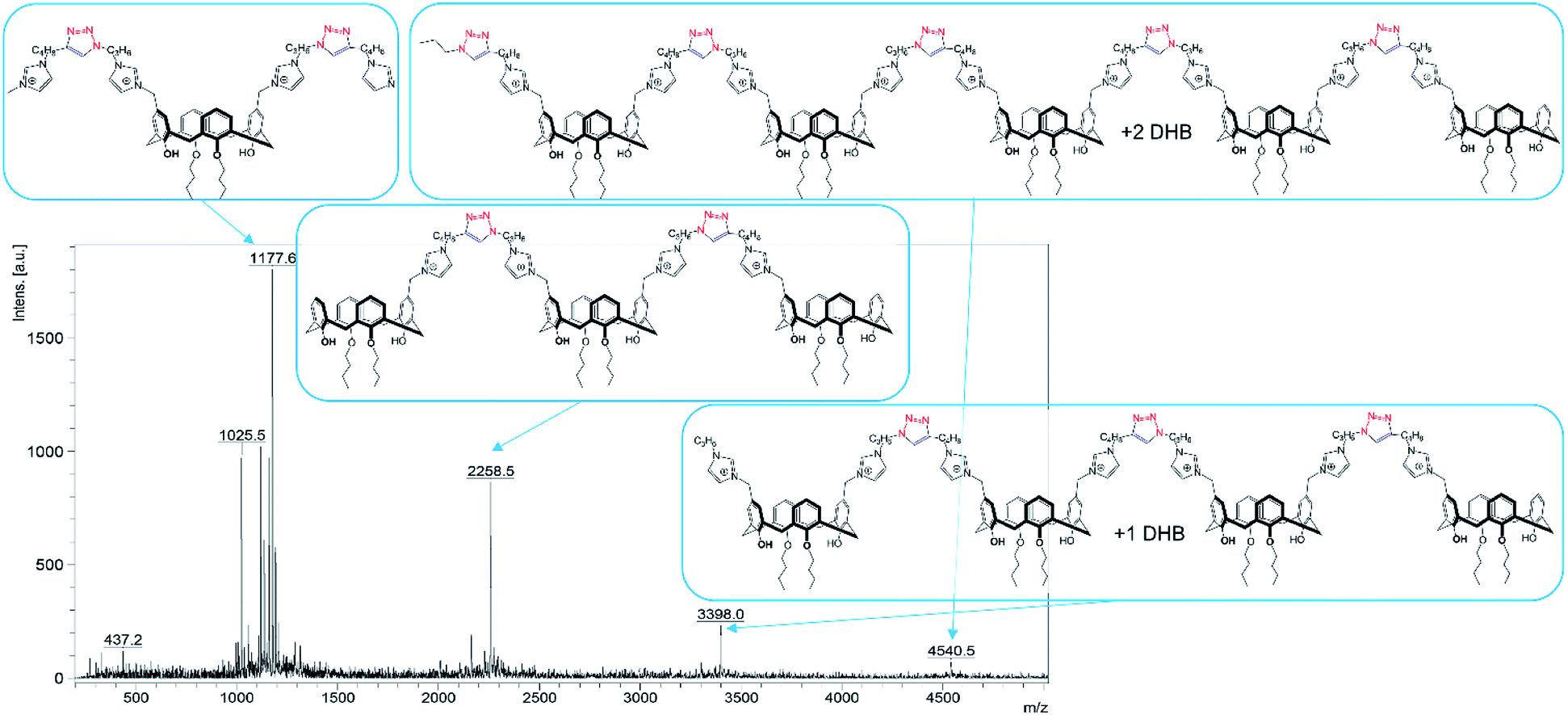 | ||
| Fig. 4 MALDI-TOF spectra of mixture of 4 and 5 after cross-linkage (DHB matrix). | ||
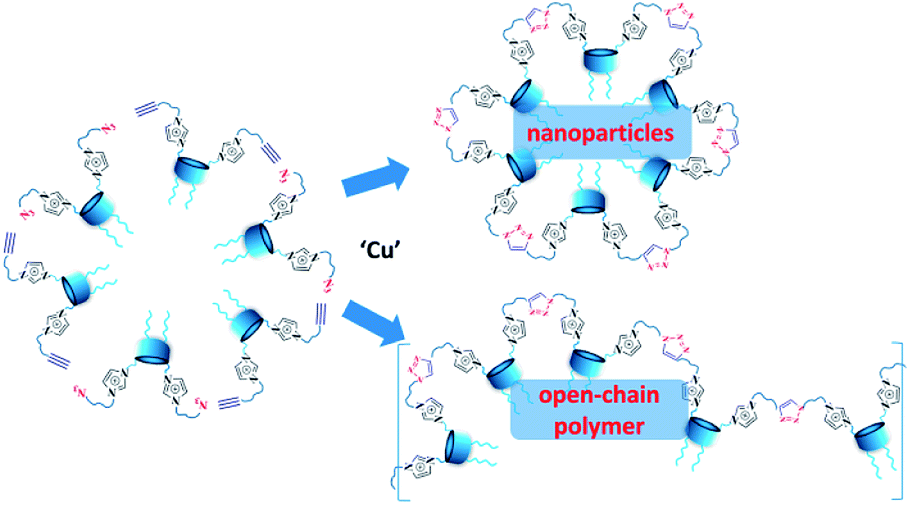 | ||
| Scheme 4 Two competitive paths of CuAAC cross-linking of 4 and 5. | ||
Catalytic activities of cross-linked polymer in Mizoroki–Heck reaction
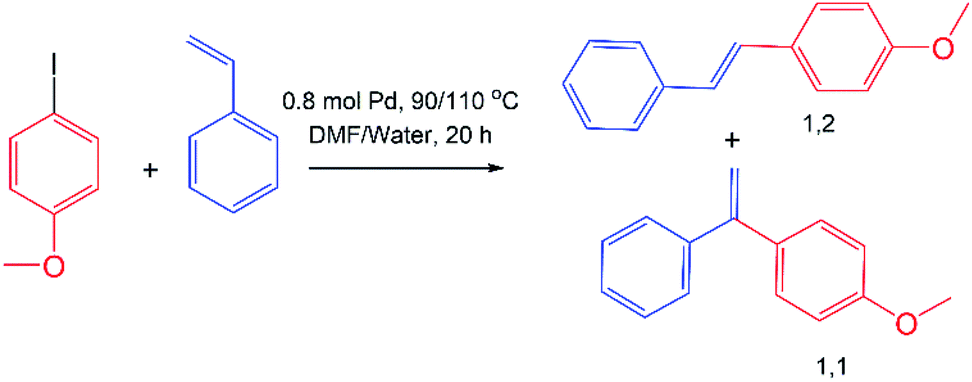
The Mizoroki–Heck reaction is a palladium-catalyzed cross-coupling reaction between aryl (vinyl) halides and alkenes and it occupies a special place in the formation of C–C bonds, and is called as a “sharpening stone of palladium” according to the expression of Irina Beletskaya.30 Traditionally, Mizoroki–Heck reaction was carried out in toxic organic solvents with air-sensitive phosphines as the ligands for Pd. Stability of NHC ligands to moisture and air oxygen allows to make Mizoroki–Heck reaction in water.31 Thus, we carried out a model Mizoroki–Heck reaction between 4-iodanisole and styrene both in traditional organic solvent (DMF) and in water using in situ generated catalyst by mixing cross-linked 4 and 5 with Pd(OAc)2 (Table 2). The conversion and products distribution was estimated using GCMS with internal standard (dodecane). In both solvents addition of polymeric support for Pd(OAc)2 increased the conversion of 4-iodanisole, especially in water. In all cases selectivity of Mizoroki–Heck products formation was closed to 100% (when reaction was performed in DMF, 2% of Ullmann side-product, 4,4′-dimethoxy-1,1′-biphenyl, was found). It is noteworthy that the ratio between the products (1,1 and 1,2-substituted ethylenes) changes drastically on going from DMF to water. So, if in DMF the ratio of products 1,1 to 1,2 is 1:5, which agrees with the literature data,30 upon going to pure water, even when using only Pd(OAc)2, the ratio increases to 1:19, growing to 1:40 when using polymer support. Previously, the same observations with increase of 1,2 product going from organic to water media were observed in intramolecular Mizoroki–Heck reaction.32
|
|||
|---|---|---|---|
| Catalyst | Solvent | Conversion | Selectivity (ratio of 1,1 and 1,2 products) |
| a ν(4-Iodanisole) = 47 μmol, ν(styrene) = 70 μmol, ν(K2CO3) = 100 μmol, ν(polymer) = ν(Pd(OAc)2) = 0.38 μmol, DMF/H2O, 90/110 °C, 20 h. | |||
| Pd(OAc)2 | DMF | 76 | 98 (1:6) |
| Pd(OAc)2 + polymer | 95 | 98 (1:5) |
|
| Pd(OAc)2 | Water | 39 | 100 (1:19) |
| Pd(OAc)2 + polymer | 99 | 100 (1:40) |
|
Methods
Dynamic, static and electrophoretic light scattering (DLS, SLS and ELS)
DLS, SLS and ELS experiments were carried out on Zetasizer Nano ZS instrument (Malvern Instruments, USA) with 4 mW 633 nm He–Ne laser light source and the light scattering angle of 173°. The data were treated with DTS software (Dispersion Technology Software 5.00). The solutions were filtered through 0.8 μM filter before the measurements to remove dust. The experiments were carried out in the disposable plastic cells DTS 0012 (size), standard glass cells (10 mm, molecular weight) or in the disposable folded capillary cells DTS 1070 (zeta potential) (Sigma-Aldrich, USA) at 298 K with at least three experiments for each system. Statistical data treatment was done using t-Student coefficient and the particle size determination error was <2%. The prepared samples were ultra-sonicated within 30 min at 25 °C before measurements.Transition electron microscopy (TEM)
TEM was performed on Hitachi HT7700 ExaLens (Japan) in Interdisciplinary Center for Analytical Microscopy of Kazan Federal University. The images were acquired at an accelerating voltage of 100 kV. Samples were ultrasonicated in water for 10 min, dispersed on 200 mesh copper grids with continuous formvar support films and then dried during 3 hours. Energy dispersive X-ray spectroscopy was performed using an Oxford Instruments X-MaxN 80T detector.Scanning electron microscopy (SEM)
SEM was performed on field-emission high resolution scanning electron microscope, Merlin Carl Zeiss Merlin (Germany). To minimize the impact on the object, the morphology was observed in the secondary electron (SE) mode with an accelerating voltage of 5 kV primary electrons and a probe current of 300 pA.Critical aggregation concentration (CAC) determination
CAC values were measured using pyrene fluorescent probe and calculated from the dependence of the intensity ratio of the first (373 nm) and third (384 nm) bands in the emission spectrum of pyrene vs. calixarene concentration. Fluorescence experiments with pyrene were performed in 10.0 mm quartz cuvettes and recorded on a Fluorolog FL-221 spectrofluorimeter (HORIBA Jobin Yvon) in the range of 350 to 430 nm and excitation wavelength 335 nm with 2.5 nm slit. All studies were conducted in buffered aqueous solution (TRIS buffer, pH 7.4) at 298 K.Gas chromatography mass-spectrometry (GCMS)
GCMS was performed on a GCMS-QP2010 Ultra gas chromatography mass-spectrometer (Shimadzu) equipped with an HP-5MS column (the internal diameter was 0.25 μm and the length was 30 m). The parameters were as follows: helium A was as carrier gas, the temperature of an injector was 250 °C, the flow rate through the column was 2 ml min−1, the thermostat temperature program was a gradient temperature increase from 70 to 250 °C with a step of 10 °C min−1. The range of scanned masses was m/z 35 ÷ 400. The internal standard method using dodecane was used for the quantitative analysis of Mizoroki–Heck products.Refractometry
The dependence of the refractive index of the polymer on the concentration (dn/dc) required for determining the molecular weight by the method of static light scattering was determined on an automatic digital refractometer Atago RX-7000i (Japan) at 25 °C.Mizoroki–Heck model reaction
Reactions were performed in 2 ml Pyrex vials in IKA heating block with vigorous stirring. DMF or aqueous dispersion (0.5 ml) containing 47 μmol of 4-iodanisole, 70 μmol of styrene, 100 μmol of K2CO3, 0.38 μmol of polymer, 0.38 μmol of Pd(OAc)2 and 50 μmol of dodecane, was degassed with N2 during 10 minutes by piercing the septum of the vial with two needles for supplying and discharging gas. The mixture was stirred at 90 °C (DMF) or 100 °C (water). After 20 hours mixture was extracted with chloroform (3 × 0.5 ml) and analyzed by GCMS.Synthesis
TLC was performed on Merck UV 254 plates with Vilber Lourmat VL-6.LC UV lamp (254 nm) control. Elemental analysis of synthesized compounds was done on the PerkinElmer PE 2400 CHNS/O Elemental Analyzer. NMR spectra were recorded on Bruker Avance 400 Nanobay with signals from residual protons of deuterated solvents (CDCl3 or DMSO-d6) as internal standard. MALDI mass-spectra were measured on UltraFlex III TOF/TOF with DHB matrix, laser Nd:YAG, λ = 355 nm. The IR spectra were recorded on a Bruker Vector-22 spectrometer. Powdered samples were mixed with KBr and then pressed in a press form to give KBr tablet. The melting points were measured using the Stuart SMP10.All reagents were purchased from either Acros or Sigma-Aldrich and used without further purification. Solvents were purified according to standard methods.33 hex-5-yn-1-yl 4-methylbenzenesulfonate,34 11,23-bis(chloromethyl)-25,27-dihydroxy-26,28-dibutoxycalix[4]arene 3 (ref. 22) and 11,23-bis[3-(1-(3-azidopropyl))-1H-imidazolium)methyl]-25,27-dihydroxy-26,28-dibutoxycalix[4]arene dichloride 5 (ref. 22) were synthesized by previously reported methods.
1H NMR (400 MHz, CDCl3, 25 °C) δH ppm: 1.47–1.57 m (2H, CH2), 1.87–2.00 m (3H, CH2 + CH), 2.23 brdtd (2H, C–CH2–), 3.97 t (2H, CH2, J = 7.0), 6.92 s (1H, H-Im), 7.06 s (1H, H-Im), 7.47 s (1H, NCHN). 13C{1H} NMR: (100.6 MHz, CDCl3, 25 °C) δc ppm: 18.07, 25.30, 30.08, 46.86, 69.36, 83.42, 118.97, 129.17, 137.08. EI (m/z): 148 [M]+. Elemental analysis calcd for C9H12N2: C, 72.94; H, 8.16; N, 18.90, found: C, 72.9; H, 8.11; N, 18.94.
Melting point: 163 °C (decomp). 1H NMR (400 MHz, DMSO-d6, 25 °C) δH ppm: 1.06 t (6H, CH3, J = 7.2 Hz), 1.35–1.47 m (4H, CH2), 1.71–1.82 m (4H, CH2), 1.91–1.83 m (4H, CH2), 1.92–2.02 m (4H, CH2), 2.22 td (4H, C–CH2–, J = 7.2, 2.4 Hz), 2.86 t (2H, CHC, J = 2.4 Hz), 3.45 d (4H, Ar–CH2–Ar, J = 12.8 Hz), 3.97 t (4H, CH2O, J = 5.2 Hz), 4.11–4.26 m (8H, CH2 + –CH2–) 5.21 s (4H, Ar–CH2-Im), 6.81 t (2H, HAr, J = 7.5), 7.05 d (4H, HAr, J = 7.6 Hz), 7.30 s (4H, HAr), 7.80 brs (4H, HIm), 8.70 s (2H, OH), 9.39 s (2H, NH-Im). 13C{1H} NMR: (100.6 MHz, DMSO-d6, 25 °C) δc ppm: 13.98, 15.16, 17.23, 18.87, 24.64, 28.66, 30.41, 31.83, 48.46, 51.82, 71.80, 76.41, 83.95, 122.52, 122.72, 125.09, 125.51, 128.31, 129.02, 133.49, 135.77, 151.85, 153.24. IR (KBr) νmax cm−1: 1460 (CAr![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CAr), 1485 (CAr–CAr), 2116 (CC), 2933 (–CH2–), 2959 (CH3), 3300 (C–H). MALDI-TOF (m/z): 709 [M-2HCl–C9H12N2]+, 561 [M-2HCl–2C9H12N2]+. Elemental analysis calcd for C56H56Cl2N4O4: C, 72.32; H, 7.15; N, 6.02, found: C, 72.37; H, 7.11; N, 6.11.
CAr), 1485 (CAr–CAr), 2116 (CC), 2933 (–CH2–), 2959 (CH3), 3300 (C–H). MALDI-TOF (m/z): 709 [M-2HCl–C9H12N2]+, 561 [M-2HCl–2C9H12N2]+. Elemental analysis calcd for C56H56Cl2N4O4: C, 72.32; H, 7.15; N, 6.02, found: C, 72.37; H, 7.11; N, 6.11.
Conclusions
New imidazolium amphiphilic calix[4]arene with terminal acetylene fragments in the polar region was synthesized for the first time. Obtained macrocycle was cross-linked with previously synthesized azidopropyl calix[4]arene using CuAAC approach in water using the concentration at which stable compact aggregates are formed. Obtained polymeric particles were studied both in solution and in solid state and the presence of submicron (∼200 nm) and micron (∼1–5 μm) particles with the prevalence of the latter with the average molecular weight of 639 ± 44 kDa was found. Obtained polymeric imidazolium–triazole particles were found effective support for Pd(OAc)2 in Mizoroki–Heck reaction in DMF or water and significantly increased the conversion and selectivity (in water) of 1,2-product formation. Demonstrated approach opens up new possibilities for the target design of structured functional materials using supramolecular templating.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Russian Science Foundation for the financial support of this work (grant No. 19-13-00095).Notes and references
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS.
- I. P. Beletskaya, F. Alonso and V. Tyurin, Coord. Chem. Rev., 2019, 385, 137 CrossRef CAS.
- M. Hopkinson, C. Richter and M. Schedler, Nature, 2014, 510, 485 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- E. Levin, E. Ivry, Ch. E. Diesendruck and N. G. Lemcoff, Chem. Rev., 2015, 115, 4607 CrossRef CAS.
- L. A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2013, 52, 270 CrossRef CAS.
- S. De, A. Udvardy, C. E. Czegeni and F. Joo, Coord. Chem. Rev., 2019, 400, 213038 CrossRef CAS.
- W. J. Sommer and M. Weck, Coord. Chem. Rev., 2007, 251, 860 CrossRef CAS.
- Y. Lei, M. Fan, G. Lan and G. Li, Appl. Organomet. Chem., 2020, 34, e5794 CAS.
- S. Shylesh, V. Schunemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428 CrossRef CAS.
- R. Lambert, A. L. Wirotius, J. Vignolle and D. Taton, Polym. Chem., 2019, 10, 460 RSC.
- N. Kaeffer, H. J. Liu, H. K. Lo, A. Fedorov and C. Copéret, Chem. Sci., 2018, 9, 5366 RSC.
- E. Herzog, H. J. Byrne, A. Casey, M. Davoren, A. G. Lenz, K. L. Maier, A. Duschl and G. J. Oostingh, Toxicol. Appl. Pharmacol., 2009, 234, 378 CrossRef CAS.
- M. Lin, S. Wang, J. Zhang, W. Luo, H. Liu, W. Wang and C.-Y. Su, J. Mol. Catal. A: Chem., 2014, 394, 33 CrossRef CAS.
- W. Wang, A. Zheng, P. Zhao, C. Xia and F. Li, ACS Catal., 2014, 4, 321 CrossRef CAS.
- M. Mondal, J. Joji and J. Choudhury, Chem. Sci., 2018, 130, 83 CrossRef.
- S. Roland, J. M. Suarez and M. Sollogoub, Chem.–Eur. J., 2018, 24, 12464 CrossRef CAS.
- J. Yang, J. Liu, Y. Wang and J. Wang, J. Inclusion Phenom. Macrocyclic Chem., 2018, 90, 15 CrossRef CAS.
- V. A. Burilov, B. Kh. Gafiatullin, D. A. Mironova, E. D. Sultanova, V. G. Evtugyn, Yu. N. Osin, D. R. Islamov, K. S. Usachev, S. E. Solovieva and I. S. Antipin, Eur. J. Org. Chem., 2020, 23, 2180 CrossRef.
- V. Burilov, R. Garipova, E. Sultanova, D. Mironova, I. Grigoryev, S. Solovieva and I. Antipin, Nanomaterials, 2020, 10, 1143 CrossRef CAS.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933 CrossRef CAS.
- V. A. Burilov, R. I. Garipova, S. E. Solovieva and I. S. Antipin, Dokl. Chem., 2020, 490, 1 CrossRef CAS.
- D. C. Gutsche and J. A. Levine, J. Am. Chem. Soc., 1982, 104, 2653 CrossRef.
- M. Wijtmans, Ch. de Graaf, G. de Kloe, E. P. Istyastono, Ju. Smit, H. Lim, R. Boonnak, S. Nijmeijer, R. A. Smits, A. Jongejan, O. Zuiderveld, I. J. P. de Esch and R. Leurs, J. Med. Chem., 2011, 54, 1693 CrossRef CAS.
- J.-B. Regnouf-de-Vains, S. Berthalon and R. Lamartine, J. Mass Spectrom., 1998, 33, 968 CrossRef CAS.
- J. -F. Lutz, S. Pfeifer and Z. Zarafshani, QSAR Comb. Sci., 2007, 26, 1151 CrossRef CAS.
- G. V. Lowry, R. J. Hill, S. Harper, A. F. Rawle, Ch. O. Hendren, F. Klaessig, U. Nobbmann, Ph. Sayreh and J. Rumblei, Environ. Sci.: Nano, 2016, 3, 953 RSC.
- M. Suckow, M. Roy, K. Sahre, L. Häußler, N. K. Singha, B. Voit and F. Böhme, Polymer, 2017, 111, 123 CrossRef CAS.
- X. Lou, B. Li, B. F. M. de Waal, L. Schill, M. B. Baker, R. A. A. Bovee, J. L. J. van Dongen, L. -G. Milroy and E. W. Meijer, J. Mass Spectrom., 2018, 53, 39 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS.
- F. Christoffel and Th. R. Ward, Catal. Lett., 2018, 148, 489 CrossRef CAS.
- S. Lemaire-Audoire, M. Savignac, C. Dupuis and J. P. Genet, Tetrahedron Lett., 1996, 37, 2003 CrossRef CAS.
- W. L. F. Armarego and C. Chai, Purification of Laboratory Chemicals, Elsevier, New York, USA, 2009 Search PubMed.
- J. Deschamps, M. Balog, B. Boury, M. Ben Yahia, J.-S. Filhol, A. van der Lee, A. Al Choueiry, T. Barisien, L. Legrand, M. Schott and S. Dutremez, Chem. Mater., 2010, 22, 3961 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09740c |
| This journal is © The Royal Society of Chemistry 2021 |