
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDoping strategy, properties and application of heteroatom-doped ordered mesoporous carbon
Yuan Gao
 *ab,
Qing Wang
a,
Guozhao Ji
a,
Aimin Li
a and
Jiamin Niu
a
*ab,
Qing Wang
a,
Guozhao Ji
a,
Aimin Li
a and
Jiamin Niu
a
aKey Laboratory of Industrial Ecology and Environmental Engineering (Ministry of Education), School of Environmental Science and Technology, Dalian University of Technology, Linggong Road 2, Dalian 116024, P. R. China. E-mail: gaoyuan1988@dlut.edu.cn
bNational Marine Environmental Monitoring Center, Dalian 116023, P. R. China
First published on 28th January 2021
Abstract
To date, tremendous achievements have been made to produce ordered mesoporous carbon (OMC) with well-designed and controllable porous structure for catalysis, energy storage and conversion. However, OMC as electrode material suffers from poor hydrophilicity and weak electrical conductivity. Numerous attempts and much research interest have been devoted to dope different heteroatoms in OMC as the structure defects to enhance its performance, such as nitrogen, phosphorus, sulphur, boron, and multi heteroatoms. Unfortunately, the “how–why–what” question for the heteroatom-doped OMC has not been summarized in any published reports. Therefore, this review focuses on the functionalization strategies of heteroatoms in OMC and the corresponding process characteristics, including in situ method, post treatment method, and chemical vapor deposition. The fundamentally influencing mechanisms of various heteroatoms in electrochemical property and porous structure are summarized in detail. Furthermore, this review provides an updated summary about the applications of different heteroatom-doped OMC in supercapacitor, electrocatalysis, and ion battery during the last decade. Finally, the future challenges and research strategies for heteroatom-doped OMC are also proposed.
 Yuan Gao | Dr Yuan Gao majored in environmental engineering and received her PhD degree in 2017 at Shandong University, China. She currently works in School of Environmental Science and Technology at Dalian University of Technology in China as an Associate Professor. Her current research interests primary focus on the preparation of activated carbon, ordered mesoporous carbon, and biochar for adsorption and supercapacitor. |
 Qing Wang | Qing Wang received her bachelor degree in 2019 at Nanjing Technology University, China. She currently studies as a master candidate in School of Environmental Science and Technology at Dalian University of Technology under the supervision of Dr Yuan Gao. Her current research interests focus on the preparation of ordered mesoporous carbon and its application in catalysis and supercapacitor. |
 Guozhao Ji | Dr Guozhao Ji majored in chemical engineering and received his PhD degree in 2014 at the University of Queensland, Australia. He currently works in School of Environmental Science and Technology at Dalian University of Technology in China as an Associate Professor. His research interests are computational fluid dynamic application in chemical engineering processes, modelling of gas transport in microporous material, gas separation by inorganic membrane, high temperature CO2 capture, solid waste gasification and kinetic modelling of thermochemical conversions. |
 Aimin Li | Dr Aimin Li majored in engineering thermophysics and received his PhD degree in 1999 at Zhejiang University, China. He currently works in School of Environmental Science and Technology at Dalian University of Technology in China as a Professor. His current research interests are pollution prevention and thermochemical conversion as well as solidification of solid waste. |
 Jiamin Niu | Jiamin Niu is a third-year undergraduate majoring in environmental science at Dalian University of Technology. She currently participates in the innovative practice projects on the reuse of solid waste for carbon-based electrode material under the supervision of Dr Yuan Gao. |
1. Introduction
Nowadays, the global environmental pollution and energy crisis have driven the urgent development of sustainable and renewable energy, such as solar energy, geothermal energy, wind energy, tidal energy, and biological energy.1 To solve their unstable and intermittent problems, it is necessary to develop energy storage and conversion systems with high efficiency and cycling stability.2 Supercapacitors, lithium-ion batteries, and electrocatalysis are three common systems for energy storage and conversion, which can be significantly affected by the electrode materials. The carbon-based materials, including graphene, carbon nanotube, activated carbon, and carbon aerogel, have been paid a great deal of attention due to their cost-effective, chemical stability, and high specific surface area (SSA).3–6 However, most of these carbon materials suffer from disordered and narrow porous structure as well as excess micropores, leading to high mass transfer resistance of electrolyte ions.7 It has been acknowledged that the ordered mesoporous carbon (OMC) with tunable diameters ranging from 2 to 50 nm has been identified as a potential candidate electrode material.8,9 The well-designed mesoporous structure could shorten ion transport pathways and favor the diffusion of ions into the inner pores.10,11 In 1999, Ryoo's group used MCM-48 as template, sucrose as carbon precursor, and sulfuric acid as catalyst to prepare OMC with a SSA of 1380 m2 g−1. Afterwards, the synthesis and application of OMC in contaminant adsorbent, catalyst, lithium battery, supercapacitor, and hydrogen storage have been performed by many outstanding researchers, such as Sheng Dai,12 Nishiyama,13 and Dongyuan Zhao.14Recently, several previous reviews have been conducted on the synthesis and application of carbon-based material. For example, Gang et al. reported the adsorption performance of various modified OMC to inorganic, organic, and emerging water pollutants.15 Kim et al. provided a summary on the layered metal decorated carbon nano-composites for energy storage.16 Liu et al. introduced the recent advances in block copolymer-based porous carbons for supercapacitor.17 However, to the best of our knowledge, a comprehensive summary on unique electrochemical characteristics of doped-OMC is still lacking. Many previous studies indicated that the application of pure OMC as electrode material suffered from some limitations, such as poor hydrophilicity, few activate sites, and weak electrical conductivity. Therefore, more and more attentions have been paid on the investigation of modified OMC via physical or chemical methods. The doping of heteroatoms, nitrogen,18,19 phosphorus,20,21 sulphur,22,23 and boron,24,25 can not only change the surface chemical structure but also affect the electron distribution of the carbon network, thus leading to the improvement of electrochemical properties.26–29
In this review, we systematically summarize the latest progresses in the doping strategies and the involved reaction mechanisms of heteroatom-doped ordered mesoporous carbon. Furthermore, an overview on the effects of different dopants on the electrical properties and porous structures are also been performed. The representative samples about the application of doped-ordered mesoporous carbon in catalysis, energy storage and conversion are also presented. Finally, we highlight the main challenges of future prospects to improve the properties and apply in the industry.
2. Doping strategies
Table 1 summarizes the synthesis parameters and the physicochemical property of the heteroatom-doped OMC. The methods for the heteroatom-doping in OMC mainly include in situ, post treatment, and chemical vapor deposition (CVD). The final objective of each method is to synthesize doped-OMC with ordered crystalline structure, high SSA, homogeneous heteroatom incorporation, and appropriate doping level.| Methods | Precursor | Physical property | Chemical property | Application | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Heteroatom | Carbon | Template | SBET (m2 g−1) | Vtol (cm3 g−1) | Dave (nm) | Heteroatom | Doping content (%) | |||
| In situ | Ammonium peroxydisulfate | Aniline | KIT-6 | 778 | 0.71 | 4.85 | N | 6.90 | SCs | 125 |
| Benzoxazine | Benzoxazine | SBA-15 | 840 | 1.148 | — | N | 3.34 | SCs | 148 | |
| Cyanamide | Phenolic resin | SBA-15 | 1741 | 1.48 | 4.10 | N | 15.1 | SCs | 67 | |
| Ethidene diamine | CCl4 | SBA-15 | 682 | 0.93 | 4.71 | N | 3.90 | ORR | 149 | |
| Gelatin | Gelatin | Silica | 474 | 1.186 | 12.0 | N | 7.40 | SCs | 129 | |
| Honey | Honey | SBA-15 | 1273 | 1.00 | 3.71 | N | 0.93 | ORR | 41 | |
| Melamine | Furfuryl alcohol | KIT-6 | 725 | 0.57 | 3.10 | N | 12.6 | SCs | 124 | |
| Polyvinyl alkanone | Polyvinyl alkanone | KIT-6 | 1403 | 1.29 | — | N | 11.06 | SCs | 150 | |
| Quinoline | Quinoline | SBA-15 | 1420 | 1.21 | — | N | 5.90 | SCs | 52 | |
| Urea | Phenolic resin | F127 | 640 | 0.40 | 2.50 | N | 3.00 | ORR | 151 | |
| Methenamine | M-Aminophenol + methenamine | F127 | 1207 | 0.77 | 11.0 | N | 3.14 | SCs | 152 | |
| Pyrrole + urea | Phenolic resin | F127 | 1386 | 1.63 | 4.73 | N | 5.30 | SCs | 130 | |
| Triphenylphosphine | Phenol | SBA-15 | 1182 | 1.87 | — | P | 1.36 | ORR | 80 | |
| Triphenylphosphine | Furfural | SBA-15 | 1730 | 3.20 | 3.40 | P | 0.89 | ORR | 20 | |
| Phosphoric acid | Phenolic resin | F127 | 438 | 0.42 | 3.80 | P | 2.80 | Catalyst | 21 | |
| Triphenylphosphine | Sucrose | SBA-15 | 1339 | 1.36 | 3.80 | P | 0.64 | Catalyst | 84 | |
| Phosphoric acid | Phenolic resin | Polystyrene | 1137 | 1.11 | — | P | 3.18 | SCs | 131 | |
| Benzyl disulfide | Sucrose | SBA-15 | 1261 | 1.383 | 4.553 | S | 1.505 | ORR | 21 | |
| Toluenesulfonic acid | Acetone toluene sulfonate | SBA-15 | 1030 | — | 5.0 | S | 2.3 | SCs | 153 | |
| Boric acid | Furfural | KIT-6 | 957 | 0.89 | 4.96 | B | 2.9 | SCs | 25 | |
| Boric acid | Phenolic resin | F127 | 690 | 0.49 | 5.00 | B | 1.96 | SCs | 154 | |
| Boric acid | Phenolic resin | F127 | 558 | 0.51 | 3.62 | B | 1.17 | ORR | 47 | |
| Boric acid | Sucrose | SBA-15 | 470 | 0.49 | — | B | 0.60 | SCs | 104 | |
| Boric acid | Phenolic resin | F127 | 462 | 0.36 | 3.7 | B | 1.01 | SCs | 103 | |
| Boric acid | Phenolic resin | F127 | 539 | 0.73 | — | B | 1.78 | SCs | 9 | |
| Hydroxyphenyl boronic acid | Phenol | SBA-15 | 1392 | 1.23 | 3.29 | B | 1.3 | ORR | 102 | |
| Hydroxyphenylboronic acid | Phenolic resin | F127 | 568 | 0.38 | 4.79 | B | 3.96 | SCs | 113 | |
| Hydroxyphenylboronic acid | Sucrose | SBA-15 | 900 | 1.24 | 6.4 | B | 1.3 | ORR | 111 | |
| Urea + phytic acid | Saccharose | KIT-6 | 1968 | 2.47 | 4.1 | N + P | 3.2 + 1.7 | SCs | 7 | |
| Urea + thiourea | Sucrose | SBA-15 | 838 | — | 4.2 | N + P | 20.32 + 0.82 | SIBs | 92 | |
| Ammonium fluoride | Phenolic resin | SBA-15 | 671 | 1.27 | 6.2 | N + F | 2.25 + 0.35 | ORR | 155 | |
| H3BO3 + H3PO4 | Phenolic resin | F127 | 692 | 0.62 | 5.0 | B + P | 2.23 + 0.69 | SCs | 101 | |
| Saccharin | Saccharin | SBA-15 | 743 | — | — | N + S | 3.4 + 5.06 | ORR | 37 | |
| Slphuric acid | Pyrrole | SBA-15 | 1021 | 1.13 | 3.24 | N + S | 7.4 + 0.81 | SCs | 36 | |
| Polypyrrole + polythiophene | CHCl3 | SBA-15 | 602 | — | — | N + S | 5.72 + 5.641 | ORR | 6 | |
| Pyrrole + phosphoric acid | Pyrrole | SBA-15 | 1073 | 1.17 | 11.0 | N + P | 5.18 + 3.42 | SCs | 122 | |
| Glucosamine + p-toluenesulfonic acid | Sucrose | SBA-15 | 1060 | 1.85 | — | N + S | 3.04 + 2.58 | ORR | 116 | |
| Pyrrole + H2SO4 | Pyrrole | KIT-6 | 693 | 0.75 | 3.27 | N + S | 10.08 + 0.94 | SCs | 133 | |
| H3BO3 + H3PO4 | Phenolic resin | F127 | 692 | 0.62 | 5.0 | P + B | 0.69 + 2.23 | SCs | 101 | |
| Post treatment | Melamine | Phenolic resin | F127 | 1166 | 1.37 | 5.5 | N | 2.90 | ORR | 27 |
| Ammonia gas | Phenolic resin | F127 | 1923 | 2.11 | 4.0 | N | 4.0 | ORR | 12 | |
| Sublimed sulfur | Phenolic resin | SiO2 | 450 | 0.7 | — | S | 59.4 | ORR | 140 | |
| Sulfur | Furfuryl alcohol | Silica | 440 | — | — | S | 67 | Li–S | 97 | |
| Sulphur | Sucrose | SBA-15 | 46 | 0.028 | 1.1 | S | 69.3 | Li–S | 142 | |
| Sulphur | Sucrose | SBA-15 | 1013 | 1.26 | — | S | 60.6 | Li–S | 141 | |
| Sulphur | Furfuryl alcohol | SBA-15 | 304 | 0.62 | 3.6 | S | 53 | Li–S | 156 | |
| Sulphur + CS2 | Phenolic resin | SBA-15 + F127 | 119 | 0.50 | — | S | 76.4 | Li–S | 157 | |
| Sulphur + CS2 | Phenolic resin | SiO2 | 94 | 0.40 | — | S | 63.6 | Li–S | 93 | |
| Triphenylphosphine + dicyandiamide | Phenolic resin | SBA-15 | 801 | 0.604 | — | N + P | 3.61 + 0.49 | ORR | 121 | |
| CVD | Pyrrole | Pyrrole | SBA-15 | 661 | 0.75 | 5.1 | N | 5.36 | ORR | 65 |
| Acetonitrile | Acetonitrile | Zeolite | 1322 | 0.84 | — | N | 5.2 | ORR | 34 | |
| Acetonitrile | Acetonitrile | SBA-15 | 885 | 1.70 | 8.47 | N | 11.3 | Catalyst | 63 | |
| Acetonitrile | Acetonitrile | TUD-1 | 533 | 0.4 | — | N | 4.8 | — | 57 | |
| Acetonitrile | Acetonitrile | SBA-15 | 570 | 1.18 | — | N | 8.68 | — | 64 | |
| Pyrimidine + thiophene | Pyrimidine + thiophene | SBA-15 | 680 | 0.70 | 3.2 | N + S | 4.6 + 2.1 | ORR | 62 | |
| In situ + Post treatment | Tri(p-tolyl) phosphine | Phenolic resin | PS-b-PEO | 1090 | — | 43.0 | N + P | 0.69 + 0.24 | ORR | 158 |
| Post treatment + CVD | Dicyandiamide + ammonia gas | Polyfurfuryl alcohol | SBA-15 | 1580 | 2.2 | 2.25 | N | 11.9 | SCs | 127 |
2.1 In situ method
In situ doping method, also called direct synthesis approach, is usually thermal treated under the protection of inert gas, during which the precursor containing natural heteroatoms or the compounds containing heteroatoms are thermally treated with templates to form well-ordered intermediate composite, followed by carbonization at the temperature of 500–900 °C.30 The synthesis procedures are shown in Fig. 1. The intrinsic feature of the in situ method is the synchronous generation of heteroatom doping and graphitization porous structure. Therefore, the heteroatoms prefer to incorporate inside the carbon skeleton in the form of structural type, which are more stable. In addition, the in situ doping method is beneficial for embedding entire carbon matrix with homogeneous foreign atoms.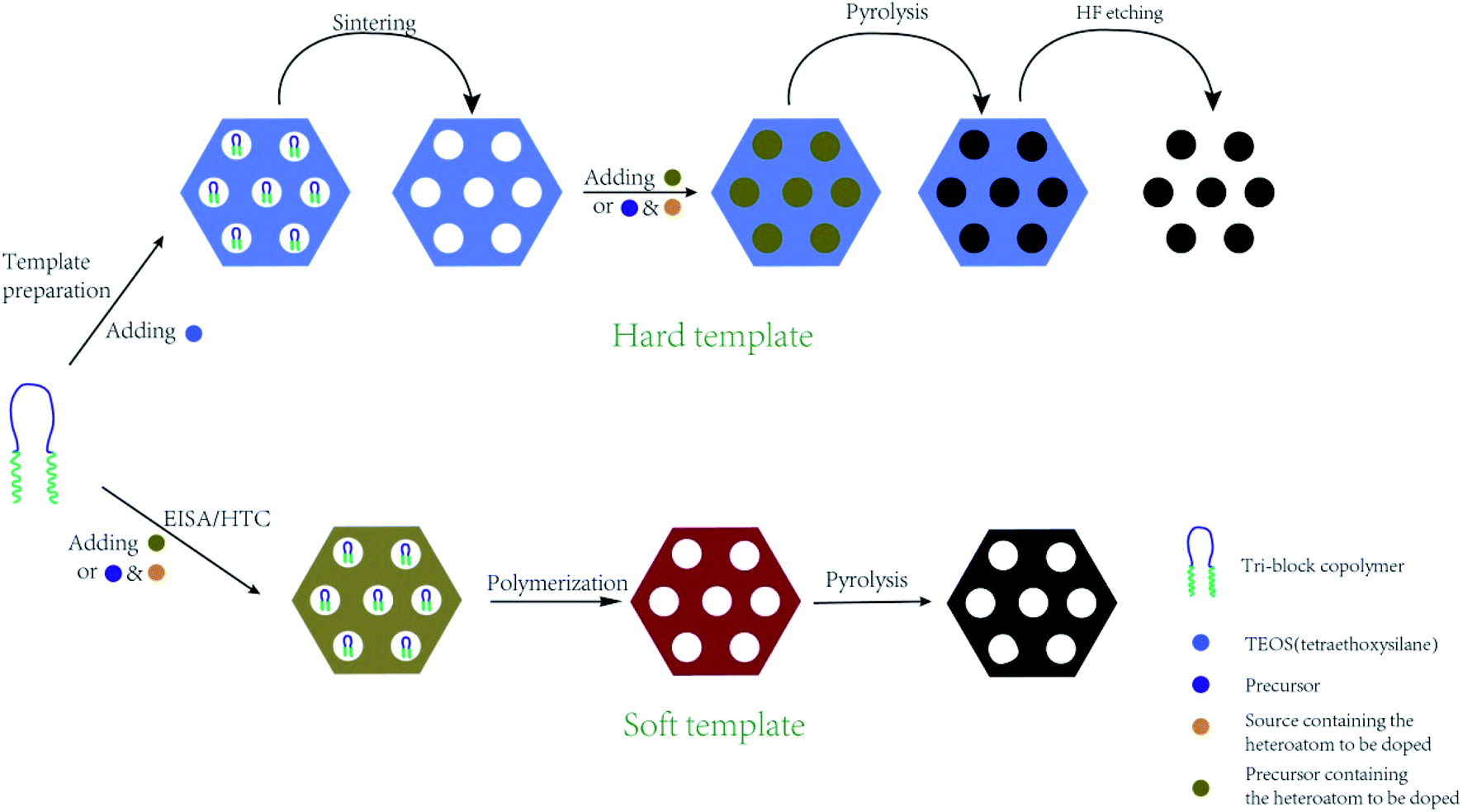 | ||
| Fig. 1 Synthesis process of the in situ method. | ||
Generally, the in situ doping method can be divided into two different synthesis routes, namely hard template and soft template. The hard template route, also named as nanocasting, involves in three steps: (i) first preparing the silica-based, metal oxide-based, or zeolite-based template; (ii) second mixing the template with fluidic carbon precursors; (iii) finally removing the silica-based template through concentrated hydrofluoric acid or hot sodium hydroxide solution.31–34 The characteristic of this method is the perfect replication of hard template accompanied by a controllable and regular ordered structure, but complicated synthesis processes and overlong operation time. For example, Le et al. mixed 3 g of alkali fused kaolin clay, 30 mL of DI water, 30 mL of HCl solution, and 2 g of Pluronic P123 to fabricate hard template SBA-15 through hydrothermal treatment at 100 °C for 24 h. Afterwards, the SBA-15, sucrose, melamine, and sulfuric acid were mixed homogeneously and then heated at 900 °C for 3 h to obtain N-doped OMC.35 Zhang et al. applied the pyrrole as a carbon precursor as well as a nitrogen source and the sulphuric acid as a catalyst as well as a sulfur source to synthesis N, S co-doped OMC. TEM images in Fig. 2 illustrate a stripe-like and hexagonally arranged pore structure with a wall thickness of 6 nm.36 Hua et al. used saccharin as a three-in-one precursor of C, N, and S source, SBA-15 as a hard template, and FeCl3 as a catalyst to prepare N, S co-doped OMC.37 The natural biomasses, such as polydopamine, dopamine, crab shell, and honey, were also applied as low-cost, renewable, and heteroatom-enriched source.38–40 Zhang et al. used honey as the carbon and nitrogen sources to fabricate N-doped OMC via an in situ hard template.40 All the as-prepared N-doped OMC had a SSA over 600 m2 g−1 and a high mesopore to total pore ratio of 93%. The nitrogen content decreased from 4.32 wt% to 1.38 wt% with the increasing carbonization temperature from 700 to 900 °C. Lu et al. also used honey as a precursor and similar synthesis route to produce N-doped OMC.41 On the one hand, the honey was composed of carbohydrates, proteins, enzymes, and aminoacids, which served as in situ carbon and nitrogen source. On the other hand, the honey contained 15–17% of water, which served as a natural solvent for its homogeneous mixing with the template. Furthermore, the honey was hydrophilic and semitransparent, which also contributed to the mixing process. Zhou et al. reported a dual template route to produce OMC nanofiber, during which the crab shell and triblock copolymer F127 were used as the hard and soft template, respectively. Additionally, a mixed solvent of ethanol and water was required to impregnate the templates, followed by a thermal curing at 100 °C for 24 h. The pore size of the as-obtained N-doped OMC by a dual-template (15 nm) was larger than that of traditional EISA method (4.1 nm).42
 | ||
| Fig. 2 SEM and TEM images of N–S co-doped OMC by a hard template method. Reproduced with permission from ref. 35. Copyright 2013, Royal Society of Chemistry. | ||
During the soft template route, the block copolymer (e.g. anionic, cationic, or nonionic-type surfactants) as the template are normally reacted with polymerizable precursors with heteroatoms by using evaporation induced self-assembly (EISA) or hydrothermal carbonization (HTC), during which the ordered intermediate product are formed due to hydrogen bonding. After pyrolysis above 400 °C, the soft template are volatilized and removed from the intermediate product to produce heteroatom-doped OMC.43–45 The template removal process by HF or hot NaOH solution was replaced by the direct volatilization of block copolymer above 400 °C. Therefore, one-step synthesis of soft template route is much simpler than that of hard template route. It should be mentioned that the heating rate should be slow in the range of 1–5 °C min−1 and maintained for 1–5 h in order to avoid the breakdown and shrinkage of the ordered structure. More importantly, the ordered porous structure is highly related to the interaction of the heteroatom precursor, the carbon precursor and the structure directing template during the polymerization process before carbonization.5 For example, Zhang et al. applied phenol as a carbon source, 4-hydroxythiophenol as a sulphur source, triblock copolymer Pluronic F127 as a soft template, and DI water as a solvent to form an order intermediate mixture, which was hydrothermally treated at 130 °C for 24 h and then pyrolyzed at 800 °C for 3 h to produce S-doped OMC nanospheres.3 Wei et al. demonstrated that the hydrogen bonding and electrostatic interaction between resol carbon source, F127 template, and dicyandiamide nitrogen source resulted in the formation of ordered mesostructures with various symmetries and high N content (13.1%).46 Su et al. summarized a three-step synthesis of B-doped OMC by soft template route, including evaporation induced self-assembly, polymerization, and pyrolysis.47 A time-saving sol–gel approach was developed by Sui's group to strengthen the self-assembly between carbon precursor and soft template.10 During the synthesis process, the phenolic resin was used as the carbon precursor, and pyrrole monomer as well as amino trimethylene phosphonic acid served as the N and P sources. A binary template was applied by introducing both F127 and tetraethyl orthosilicate as structural reinforcing agent. Interestingly, the mechanical or grinding mixing of template and precursor without using any solvent has been demonstrated a fast, green and scalable synthesis method for doped-OMC.28,48,49 As shown in Fig. 3, Zhao et al. mechanically ground the mixture of tannin, F127, metal acetates and nitrogen source (e.g. amino acids, urea, melamine, and guanine) in a ball miller for 1 h to generate a metal–organic coordination polymer, followed by vacuum heated at 40 °C for 12 h, and carbonized at 800 °C for 2 h.50 Zhu and co-workers also accomplished the N-doped OMC by using melamine, phenolic resin and F127 as the N source, C source and template via a solvent-free ball-milling method.18
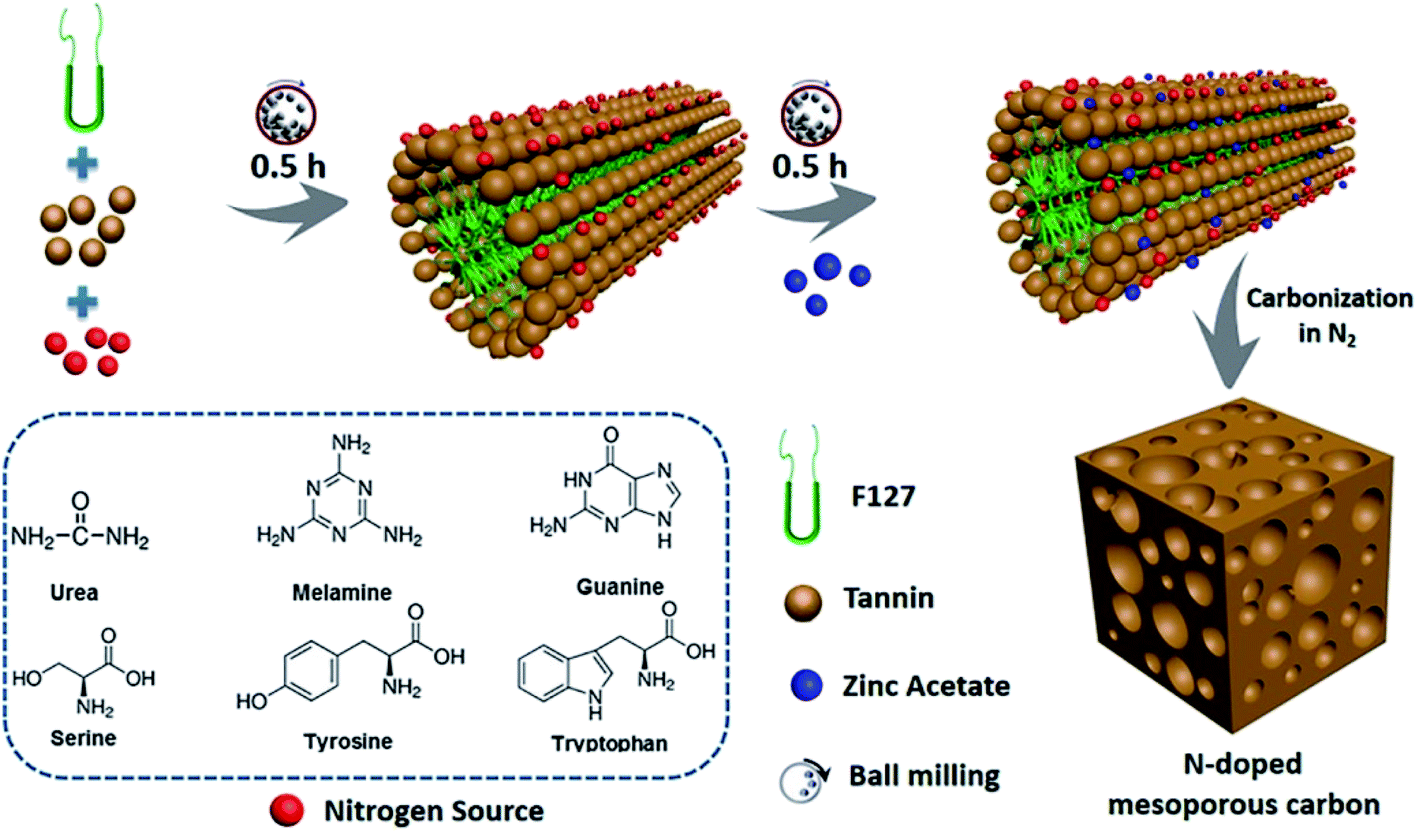 | ||
| Fig. 3 Synthesis process of N-doped OMC via a one-pot mechanochemical assembly. Reproduced with permission from ref. 49. Copyright 2020, Elsevier. | ||
2.2 Post treatment method
During the post treatment process, the heteroatoms are grafted to the original OMC through oxidation reaction, thermal polymerization, and replacement reaction. How to prevent the ordered mesostructural arrangement from destroying due to post treatment has been paid significant attention.51,52 Furthermore, how to embed the heteroatoms throughout the whole bulk rather than the partial surface is also very important. The post treatment method can be classified to dried modification and wet modification (Fig. 4). For the dried modification, OMC is exposed to gas atmosphere containing heteroatoms under high temperature without any solvents. Generally, ammonia gas is used to produce N-doped OMC.53,54 For example, Dai's group synthesized the N-doped OMC via a thermal treatment of 2D hexagonal OMC under a NH3 atmosphere in the temperature range of 950–1050 °C. It was deduced that the substitution of the oxygen species by the nitrogen species and the corrosion of carbon skeleton by the nitrogen-based radicals contributed to the formation of N species.12 For the wet modification, OMC is normally impregnated in the heteroatom-contained chemical solutions for several minutes, followed by drying and heating treatment. For instance, Maluta et al. impregnated the as-prepared OMC nanoparticles with thiophenol at a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.94. After drying at 70 °C, the samples were carbonized at 600 or 900 °C for 7 h under the protection of argon.55 Wu et al. ground the pristine OMC with melamine at a mass ratio of 1:1.5, followed by mixing with 20 mL of methanol solution and evaporating to obtain powder mixture. After pyrolysis at 500–900 °C, the N-doped OMC was obtained.51 Yang et al. used ionic liquid as heteroatom source to produce N–S co-doped OMC. Typically, 70 μl of 1-butyl-1-methyl-pyrrolidinium bisimide was mixed with OMC by grinding for several minutes, followed by carbonizing at 850 °C for 2 h.56
:0.94. After drying at 70 °C, the samples were carbonized at 600 or 900 °C for 7 h under the protection of argon.55 Wu et al. ground the pristine OMC with melamine at a mass ratio of 1:1.5, followed by mixing with 20 mL of methanol solution and evaporating to obtain powder mixture. After pyrolysis at 500–900 °C, the N-doped OMC was obtained.51 Yang et al. used ionic liquid as heteroatom source to produce N–S co-doped OMC. Typically, 70 μl of 1-butyl-1-methyl-pyrrolidinium bisimide was mixed with OMC by grinding for several minutes, followed by carbonizing at 850 °C for 2 h.56
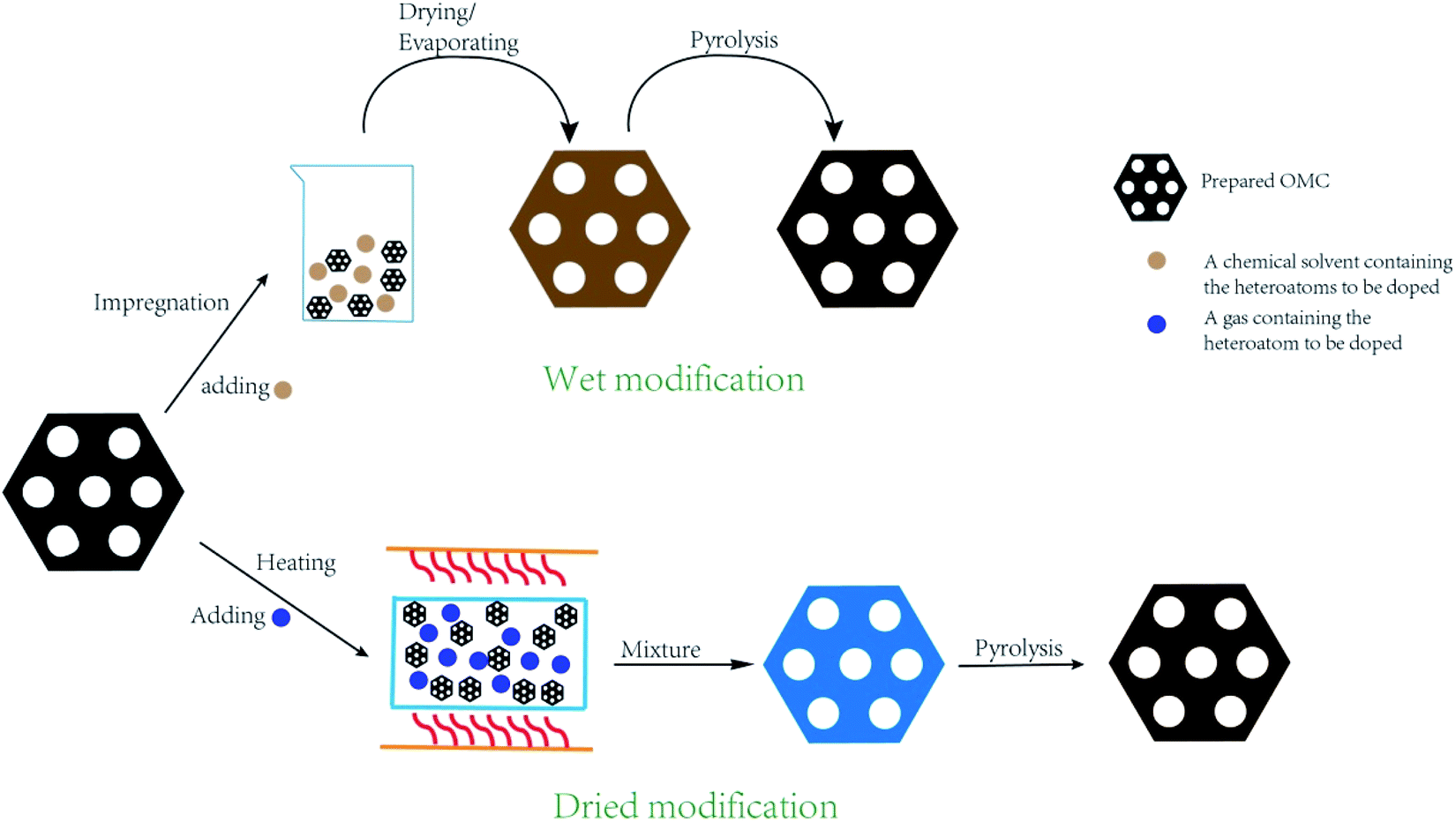 | ||
| Fig. 4 Synthesis process by the post treatment method. | ||
2.3 Chemical vapor deposition
During a typical chemical vapor deposition, the hard template is placed onto a quartz boat in a tube furnace, and heated up to 700–900 °C under an argon/nitrogen gas flow for several minutes. Next, the foreign gases, such as acetonitrile, thiophene, and pyrimidine, are purged into the reactor and replaced the original gas flow in hours (Fig. 5). After cooling down naturally, the template can be filled with carbon and heteroatom elements simultaneously.34,57,58 The doped OMC are produced by the inverse replica of the pore arrangement of hard template. Some previous studies have found that the OMC with high graphitization and heteroatom contents could be prepared by the chemical vapor deposition.59–61 However, the shortcomings are the special requirements for precursors, excessively high reaction temperature, and rigorous operation procedures. Xu et al. used mesoporous silica SBA-15 as template, and steam mixtures of thiophene and pyrimidine as the sulphur and nitrogen source to fabricate 2-D hexagonal P6mm N–S co-doped OMC. The introduction of ferrocene not only improved the graphitization degree, but also facilitated the doping of the foreign atoms into the carbon framework. Besides, the pore blockings were weakened by using low-molecular weight foreign source. The optimal OMC possessed a high SSA of 1100 m2 g−1 with the bimodal peaks at 2.5 and 3.5 nm.62 Ravat et al. selected acetonitrile as the carbon and nitrogen source, and SBA-15 as the template, which were reacted in a quartz reactor at 900 °C for 2 h. The as-made OMC possessed a SSA of 885 m2 g−1 and nitrogen contents of 11.3%.63 Sánchez-Sánchez found that the nitrogen content on the surface and in the bulk of the N-doped OMC by CVD were different, which was 7.26% and 9.42%, respectively. Moreover, the nitrogen and oxygen contents were in an opposite trend.64 Except the aforementioned traditional routes, some modified CVD methods were also developed by the researches.65,66 For example, a novel vaporization–capillary condensation method was developed by Tsiakaras's group.65 In a typical process, the pyrrole/aniline together with SBA-15 was heated at 133/183 °C for 2 h under vacuum to guarantee full evaporation of the liquid monomer around the template. During the cooling down step, the phlegma homogeneously permeated into the mesopores of the template. The SSA of N-doped OMC by this new method was 661 m2 g−1, which was larger than that of traditional wet method (292 m2 g−1). It has also been noticed in this study that the contents and species of heteroatom was negative correlation with the pyrolysis temperature. Li et al. developed a three-step vaporization–condensation–conversion method to incorporate red P into carbon skeleton of OMC.66 The whole synthesis process was carried in a sealed tube under the protection of argon. First, the red P transformed from powder to vapor above the sublimation temperature. Then, the P atoms deposited into the nanopores of OMC in the form of white P. Finally, the amorphous red P was obtained due to the transformation of white P at the temperature of 260 °C. The most outstanding characteristic of this process was that the P element was anchored into the mesopore channels without creating any bonding.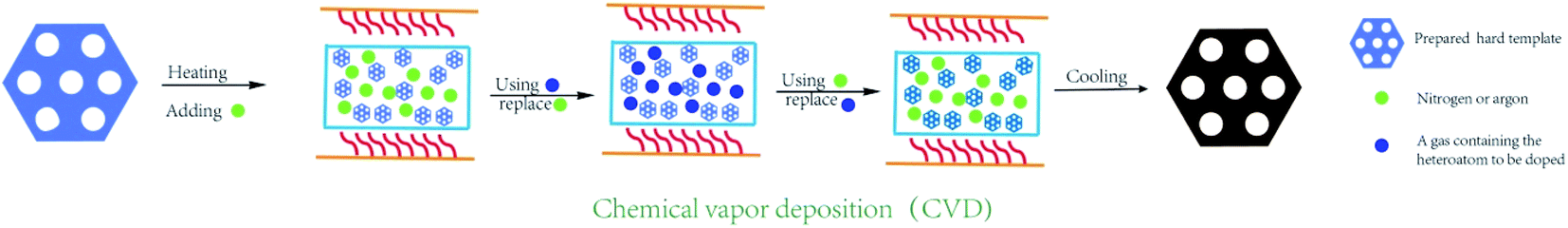 | ||
| Fig. 5 Synthesis process by the chemical vapor deposition. | ||
3. Electrical properties of doped-OMC
The heteroatoms in carbon skeleton, including nitrogen (N), phosphorus (P), sulphur (S), boron (B), and co-doping heteroatoms, could change the electron distribution, electronegativity, and produce structural defects of the OMC.23,40 Fig. 6 summarize the heteroatom species in doped-OMC and their corresponding binding energies in XPS based on literature review.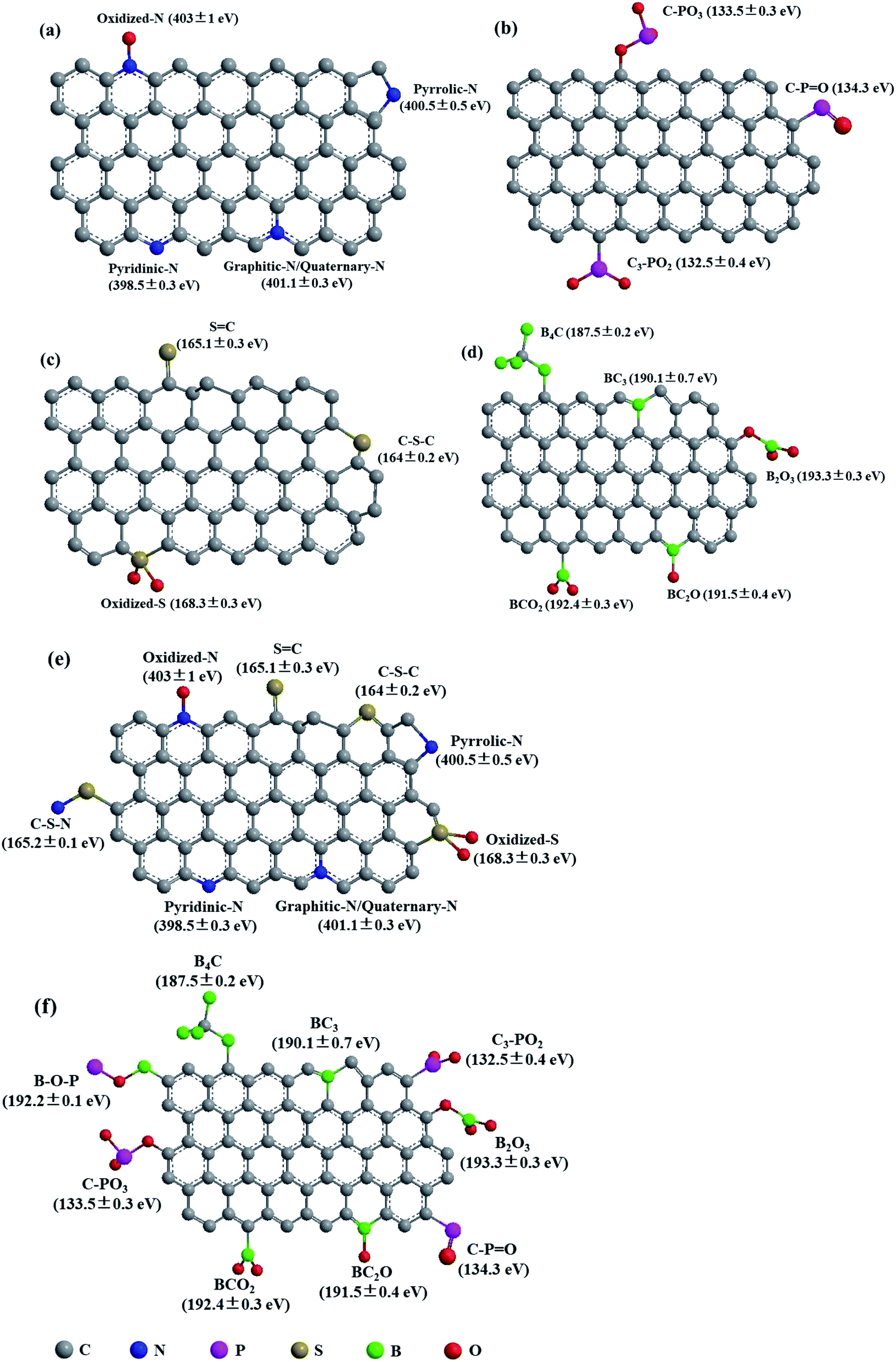 | ||
| Fig. 6 Schematic representation of heteroatom species in (a) N-doped OMC,10,30,35,40,44,52 (b) P-doped OMC,7,21 (c) S-doped OMC,23,90,93 (d) B-doped OMC,25,29,47,111,114,115 (e) N, S co-doped OMC,36,117 and (f) B, P co-doped OMC.101 | ||
3.1 N-Doped OMC
Nitrogen is one of the most popular doped-elements for OMC. Generally, the doped-N exited in four forms, namely pyrrolic N, pyridinic N, graphitic/quaternary N, and oxidized N.29,44 The electrochemical properties of N-doped OMC are highly affected by the types of doped-N species. The pyrrolic N and pyridinic N at the edge site of the carbon framework are negatively charged, which can transfer extra free electrons or delocalized electrons. They can also participate in the pseudo-capacitive reaction, which has a major impact on the improvement of the electrode capacitance.67 On the other hand, the graphitic N and pyridinic N at the bottom of the carbon framework are positively charged, which can activate the electrode surface, and improve the charge transfer rate at high current density.68The reasons why N-doped OMC owns excellent electrochemical performance are summarized as follows. First, the conductivity of the N-doped electrode materials can be enhanced due to the extra lone pair electrons in N atoms, which can provide a negative charge for the sp2 hybridized carbon skeleton of the delocalized π system, and facilitate the transport of the electrons.69 Second, the surface hydrophilicity of the electrode materials is increased owing to the nitrogen doping. This is because the nitrogen functional groups are hydrophilic functional groups, which can facilitate the contact between the electrolyte ions and the electrodes.70,71 Third, the pore structure of OMC after N-doping is increased. The increase of the pore size promotes contact between the electrolyte and the electrode, thereby enhancing electrochemical performance.72 Fourth, the nitrogen atoms have higher electronegativity (3.0) than that of carbon atoms (2.5), leading to positive charge density around the adjacent C atoms.6
The N percentages of four different species and the porous structures are related to the process parameters (e.g. carbonization temperature, carbonization time, heating rate, type of nitrogen source, mass ratio of nitrogen source to carbon precursor).73,74 Wang et al. found that the peaks of pyrrolic N, pyridinic N, graphitic N, and oxidized N located around the binding energy of 398.2, 399.7, 400.8, and 403.2 eV, respectively.10,30,35,40,44,51,52 The total N content of the as-prepared OMC increased from 6.3 to 9.2% with the mass ratio of urea to sucrose ranging from 4 to 16. Interestingly, the dominant N species was graphitic N due to the transformation of pyridinic N and pyrrolic N at high temperatures. However, there was no obvious changing pattern for the SSA and average pore diameter.30
To improve the property of doped-OMC, how to increase the doping dose has gained a lot of attention.75–77 EL-Mahdy et al. found that the direct using of N-containing resol polymer as carbon and inherent nitrogen source was an effective method to yield OMC with high N percentage (up to 13%). It has been also discovered that the nanostructures of the as-obtained N-doped OMC transformed between gyroid and cylinder with the N-containing resol concentration increasing from 50 to 70% due to the hydrogen bonding interaction and induced micro-phase separation.76 Chen et al. produced N-doped OMC with a maximum content of 11.64% by varying the mass ratios of phenolic resin to melamine.77 The weakened hysteresis loop and capillary condensation in the nitrogen adsorption–desorption isotherms of OMC indicated the degradation of ordered mesostructure after N-doping. The N species consisted of 47.1% of pyridinic-N, 33.9% of pyrrolic-N, and 19.0% of quaternary-N. The high percentage of pyridinic-N and pyrrolic-N were attributed to the vulnerable replacement of the carbon atoms around edges or defect sites by the nitrogen atoms.
Another challenge is how to improve the SSA without reducing the nitrogen content and destroying the ordered pore structure.53,67 A one-pot simple method combing carbonization, foreign element doping, and activation was carried out to fabricate N-doped OMC with a desirable SSA of 1415 m2 g−1 and high nitrogen content of 9.3%.53 The synthesis process was performed based on a preferential reaction between the active oxygen sites of the carbon precursor and ammonia gas during the thermal treatment. More importantly, the ordered structure barely changed with the pyrolysis time and temperature, except a slight shrinkage of the unit cell. The nitrogen content increased from 7.1 to 9.3% with the pyrolysis time increasing from 1 to 4 h, and the SSA increased from 643 to 1415 m2 g−1 with the pyrolysis temperature increasing from 700 to 850 °C. The aforementioned results indicated that the nitrogen content was more sensitive to pyrolysis time than temperature. On the contrary, the SSA was more related to the pyrolysis temperature rather than time. Similar results were also found by Ahn et al., in which the acrylonitrile was thermally polymerized to polyacrylonitrile as in situ nitrogen source.78 The SSA increased from 690 to 1092 m2 g−1 along with the decrease of nitrogen content from 22.2 to 5.85% with the pyrolysis temperature increasing from 600 to 900 °C. Shi et al. produced N-doped OMC with a SSA 1741 m2 g−1 and nitrogen content of 15% by the combined modification of ammonia and cyanamide–resol.67 The pyrolysis temperature, cyanamide–resol ratio, and precursor–template ratio all affected the porous structure and nitrogen content as well as species distribution. For example, the SSA and average pore size increased from 1147 to 1741 m2 g−1 and 3.7 to 4.1 nm with the cyanamide-to-resol ratio increasing from 0 to 2, respectively. After that, much higher cyanamide-to-resol ratio led to the collapse of the ordered mesopore structure and the shrinkage of pore volume and average size. The percentage of the pyridine nitrogen species was positively associated with the cyanamide-to-resol ratio. However, the percentage of the quaternary N was negatively connected with the cyanamide-to-resol ratio due to the different anchoring locations of N atoms inside or outside the carbon atoms. A N-doped OMC with a relatively high SSA of 2023 m2 g−1 was prepared with aminophenyl-functionalized polyhedral oligosilsesquioxanes as carbon source by using a soft template route.79 The amino groups involved in the formation of hydrogen bond, which facilitated the following micro-phase separation process. Additionally, the stable Si–C bond from the condensation and the rearrangement of the oligosilsesquioxanes also contributed to the uniform porous structure.
3.2 P-Doped OMC
Phosphorus atoms own same valence electrons with nitrogen atoms, but higher atomic radius and electron-donating nature.7,80 Therefore, the phosphorus doping is another choice to improve the electrochemical properties of OMC, which can be explained in following aspects. First of all, the doped phosphorus atoms can incorporate into the carbon framework in the form of P–C bond. The charge density and spin density of the carbon layer are redistributed owing to the interaction between the phosphorus atoms and surrounding C atoms.81 Besides, the asymmetric spin density in the adjacent carbon atoms will be enhanced due to the effective electron pair in 3p orbitals of the phosphorus atoms. The electron delocalization is increased due to the good electron-donating properties of phosphorus atoms.80 Accordingly, the P-doped OMC produce more active sites for the electrochemical performance. Second, the phosphorus elements can be also doped with carbon layer in the form of P–O bond. The phosphorus elements incorporate around the edge plane sites of the carbon graphitic framework due to its larger atomic size (0.106 nm) than that of carbon elements (0.077 nm), which can increase the electrochemical performance. Third, the changes of the physical structures, including SSA, pore size distribution, and pore volume, also affect the electrochemical performance. It has been proved that the P-doped OMC may possess larger SSA and pore volume than those of N-doped OMC owing to the larger atomic size of phosphorus atoms.7 The interlayer spacing between carbon graphite layers of P-doped OMC are enlarged, which can be attributed to the creation of more vacancies or defects.82 Actually, the electrons transfer much easier and faster in the enlarged carbon interlayer space.83 Last but not least, the induced local defects by P doping in carbon matrix could improve the amount of active sites due to the different bonding length and bonding angle of C and P atoms.84,85The inorganic phosphate acids were commonly used as P source for the modification of OMC. The hydroxyl groups in resol, triblock copolymer, and phosphoric acid favored the homogeneous dispersion of P in the carbon matrix via the polycondensation reaction.21,86 Xue et al. proved that the aromatization and graphitization of the P-doped OMC was promoted due to the cyclization, dehydration and condensation of phosphoric acid.21 The peaks at 1170 and 1430 cm−1 in the FTIR spectra corresponded to the stretching vibration of P–O–C and P–C band, respectively. The previous works also revealed that the doping contents of P element and ordered mesoporous structure were related to the pyrolysis temperature, acid dose, and other additives.21,86,87 Lower pyrolysis temperature generated more P–O–C band through the dehydration of the surface hydroxyl groups, while higher pyrolysis temperature resulted in the formation of more P–C band.86 The small-angle X-ray scattering (SAXS) and transmission electron microscope (TEM) results indicated the overdosed phosphoric acid could destroy the ordered porous structure by breaking the organic–organic self-assembly.21,88
Except the inorganic acid, the organic P-contained chemicals were also used to produce P-doped OMC. Yang et al. produced P-doped OMC with a thin film-type platelet by using triphenylphosphine as P source.20 The P content reduced from 1.41 to 0.23% with the pyrolysis temperature increasing from 700 to 1000 °C. The XPS results displayed that the P element presented in the form of P–C and P–O at 130.6 and 132.3 eV, respectively. Song and Zhou et al. also applied triphenylphosphine as P source to produce P-doped OMC by an in situ method.84,85 They both found that P-doping could improve the SSA of OMC, but lead to a slight deterioration of ordered porous structure. Furthermore, a positive shift of P2p peak about 2.1 ± 0.1 eV occurred, which was ascribed to the interaction of Pt particles, P, O and C atoms. The XPS results in Song's study indicated that the P-doping improved the amounts of oxygen-contained functional groups and zero-valent Pt.
3.3 S-Doped OMC
Sulfur element is also used as a foreign element to improve the electrochemical performance of OMC. The reasons why S-doped OMC has special electrochemical property are concluded in the following aspects. Although the electronegativity value of sulfur (2.58) and carbon element (2.55) is similar, the atomic radius of sulfur element (102 pm) is larger than that of carbon (75 pm) and nitrogen element (77 pm).22 On the one hand, the larger atomic radius of sulfur element and the activation role of sulfur-contained chemical result in a larger interlayer spacing, which could preserve the structural stability during the ions intercalation/extraction process.23 On the other hand, a larger atomic radius leads to a higher SSA and pore volume of the as-obtained OMC, which can improve the contact interface with the electrolyte ions and shorten the electrons/ions diffusion pathway.89 The replacement of carbon atoms by sulfur atoms leads to the increase of more vacancies and defects, which could accommodate more electrolyte ions during the charge–discharge process.90 Second, the sulfur functionalities usually present in the form of C–S–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S, –C–O–S, and SOx bond.90,91 The thiophene-type sulfur element, especially the C–S–C bond can serve as active sites for the oxygen reduction activity, thereby enhancing the electrocatalytic activity and cycle stability.92 Generally, the S 2p3/2 peaks of C–S–C groups correspond to the binding energy of 163.7 ± 0.1 eV. The S 2p1/2 peaks of C–S–C groups are located around the binding energy of 164.7 eV. The SOx peaks is normally around 168.3 ± 0.3 eV.90,93 Third, the larger covalent radius of sulfur atom (0.102 nm) than carbon atom (0.077 nm) contributes to more defects of the S-doped OMC. Furthermore, the lower internal energy of sulfur atom (2.85 eV) than carbon atom (7.37 eV) reduces the ions transfer resistance, but improves the mutual interaction between the electrolyte ions and electrode.94
S, –C–O–S, and SOx bond.90,91 The thiophene-type sulfur element, especially the C–S–C bond can serve as active sites for the oxygen reduction activity, thereby enhancing the electrocatalytic activity and cycle stability.92 Generally, the S 2p3/2 peaks of C–S–C groups correspond to the binding energy of 163.7 ± 0.1 eV. The S 2p1/2 peaks of C–S–C groups are located around the binding energy of 164.7 eV. The SOx peaks is normally around 168.3 ± 0.3 eV.90,93 Third, the larger covalent radius of sulfur atom (0.102 nm) than carbon atom (0.077 nm) contributes to more defects of the S-doped OMC. Furthermore, the lower internal energy of sulfur atom (2.85 eV) than carbon atom (7.37 eV) reduces the ions transfer resistance, but improves the mutual interaction between the electrolyte ions and electrode.94
It should be mentioned that the S-doping may result in the reducing SSA of OMC, especially for the post treatment method.95–97 For example, Wang et al. found that the SSA and pore volume of OMC decreased from 1.21 cm3 g−1 and 1345 m2 g−1 to 0.41 cm3 g−1 and 335 m2 g−1 after 25% sulfur embedding, respectively. In Liu's study, the SSA and pore volume of OMC also reduced from 1.671 cm3 g−1 and 1234 m2 g−1 to 0.97 cm3 g−1 and 867 m2 g−1 after 2.5% sulfur doping, respectively. In Song's work, the SSA and pore volume of S-doped OMC decreased about 14 times and 7 times than those of the original OMC. The changes of SSA before and after doping are highly depended on the precursors and reaction parameters.
Some researchers demonstrated that the doping of sulfur element could enhance the stability of metal nanoparticles through spatial confinement and electron donor–acceptor interaction.98,99 The binding energy of Pt in pure OMC was 71.3 eV, which was positively shifted to 71.6 eV in S-doped OMC.98 Meanwhile, the sulfur peaks exhibited a negative shift of 0.8 eV due to the electron transfer reaction between sulfur element and Pt particles. Furthermore, the particle size of Pt in S-doped OMC was only 1 nm, which was 4 times lower than that in pure OMC.99
3.4 B-Doped OMC
First, the carbon atoms (0.077 nm) and boron atoms (0.082 nm) have similar sizes of atomic radius, leading to a relatively facile replacement of carbon atoms by boron atoms in theory.100 However, the doping content of boron species in OMC is relatively low (<2%) in experiment,101–104 because the boron species volatilize above 500 °C during the pyrolysis and the inevitable competition between boric acid and template toward hydrogen bond of phenolic resin leading to the weakened assemble.105 Another reason is that the boron species prefer to incorporate inside sp2 carbon lattice, which is harder than the bonding with carbon atoms at the defects or edges.106 Second, the electronegativity of carbon atoms is higher than that of boron atoms. Moreover, the boron atoms may play a role as electron acceptor for carbon lattice due to its three valence electrons. Hence, the B atoms in the CB3 structure are positively charged with stronger interaction with electrolyte anions. The redistribution of π-system in the carbon lattice due to the B-doping can improve the van der Waals interactions between electrolyte cations and π-system.107 Third, the graphitization degree and wettability between the electrolyte and the electrode can be improved due to the B-doping, which could enhance the electrical conductivity.108 Fourth, the previous studies indicated that the boron element in OMC usually presents in the form of B–C and B–O, corresponding to the peaks at 188 ± 1 and 191.5 ± 0.1 eV.109,110 Besides, another peak around 192.1 eV is ascribed to the bonding of B–O–C.31,105 The presence of the B–C bond in the form of B4C can reduce the Fermi energy level of graphite carbon at the trigonal sites, thus modifying the electronic structure of OMC.102,110 The B–C bond in the form of B4C or B3C can enhance the charge density and charge carriers of the nearby carbon atoms, producing highly electrical conductivity.47It is worth noting that the binding of boron element can synchronously contribute to the incorporation of more oxygen groups into the carbon skeleton, providing an extra pseudo capacitance.9,25,111,112 This is because the redistribution of π-system in the B-neighbouring carbon lattice boosts the intensity of C–O bond as well as weakens the intensity of C–C bond at the same time.9,104 In addition, the oxygen atoms serve as the bridge between the carbon and boron atoms.113 And the boron atoms nearby the carbon atoms with the lower electronegativity prefer positive charged, promoting the capture of the O2 molecule. The boron oxide could play a role as an oxygen retardant for the pyrolysis process, during which the thermally stable quinones and carbonyl groups are preserved and the thermally active carboxyl groups are burn-off.103 Guo et al. found that the contents of oxygen groups increased from 12.33 to 16.06% with the contents of doped-B increased from 0.64 to 2.11%.111 Wang et al. also discovered that the contents of oxygen groups increased from 8.5 to 10.3% with the mass ratio of boric acid to furfuryl alcohol increasing from 0.02 to 0.08.25
Another characteristic is that the existence of boron atoms could facilitate the uniform dispersion of Pd nanoparticles due to their more defective sites and stronger interaction with Pd nanoparticles.24,33,114 The positive shift of 0.7 eV in the XPS spectra of Pd 3d between the B-doped OMC and the original OMC demonstrated the electron transfer between the Pd nanoparticles and boron atoms.24 The TEM tests by Nsabimana et al. display that the Pd nanoparticles on the original OMC preferred to aggregate into large particle clusters with an average size of 4.36 nm. Nevertheless, the Pd nanoparticles on the B-doped OMC formed more homogeneous and smaller particle with an average size of 2.62 nm.114 Similar results can be also found in Zhou's study.33 The average diameter of Pd on the B-doped OMC was 2.5 nm, which was much lower than that of the original OMC (4.1 nm).
However, some previous reports show that the incorporation of boron atoms into the carbon skeleton may lead to the decline of the SSA and the collapse of ordered mesoporous structure.25,103,114,115 The SSA and pore volume of B-doped OMC decreased from 1258 to 780 m2 g−1 and 1.56 to 0.78 cm3 g−1 with the boron percentage increasing from 0 to 4%, respectively. Meanwhile, the low-angle X-ray diffraction results show that the gradual disappearance of 3D cubic Ia 3d peaks after B-doping. Analogously, the strength weakening of (100) peak in B-doped OMC at the boron content of 3.6% also indicated a deterioration of ordered structure.108 The negative influence of B-doping on the SSA and pore volume were also investigated by Zhang et al., in which the SSA and pore volume decreased about 3-fold and 2.5-fold in comparison with the original OMC as a result of transformation of the exiting micropores.103 Fig. 7 summarized effects of heteroatom doping on the electrical properties of OMC.
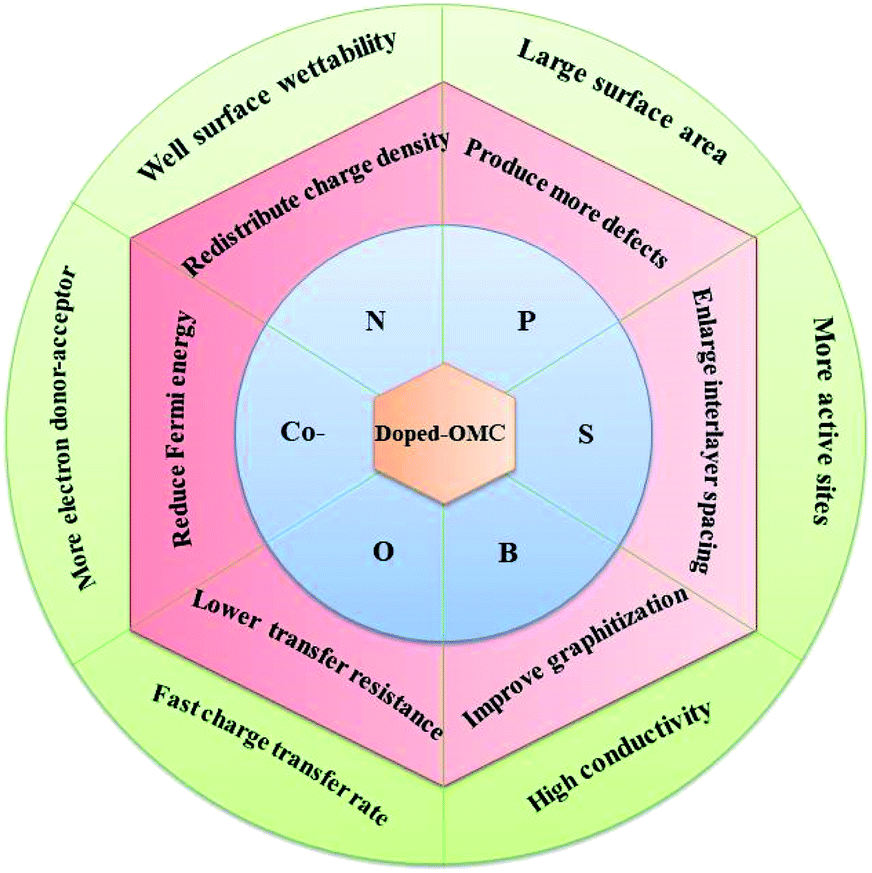 | ||
| Fig. 7 Effects of heteroatoms doping on the electrical properties of OMC. | ||
3.5 Heteroatom co-doped OMC
Many publications have demonstrated that the synergistic effects of co-doping heteroatoms could provide more active sites and additional functional groups as well as promote the surface wettability for OMC compared to the single doping, which create excellent electrochemical behavior.10,11 In most of co-doping cases, the hard template is preferred than the soft template, because the binary or ternary foreign atoms could weaken the driving force and prevent the carbon precursor and soft template from organic–organic self-assembly.6,7,10,29,92,116,117 The remarkable characteristics of co-doping include the increase of structural defects, the change of SSA, and ordered mesoporous structure.The defect degrees due to the doping of various heteroatoms can be revealed by the Raman tests. The Raman spectra of OMC are normally consist of two peaks at approximately 1330 ± 20 and 1600 ± 20 cm−1, corresponding to the D band and G band.10,29,92,118,119 The D band represents the stretching vibration of the lattice distortion, vacancy, topological defects, while the G band stands for the E2g symmetry vibration of interplane sp2-hybridized carbon. Therefore, the defect degree of OMC is commonly evaluated according to the intensity ratio of the D band and G band (ID/IG). In Ye's research, the ID/IG value were 1.08, 1.01, 1.0 for the N–S co-doped OMC, N-doped OMC, and original OMC, respectively.92 In Deng's study, N–P binary-doped OMC (0.95) exhibited higher ID/IG value than that of N-doped OMC (0.92) and P-doped OMC (0.86).119 In Zeng's report, the N–B co-doped OMC possessed the highest ID/IG value (1.31) compared to B-doped OMC (1.25), N-doped OMC (1.20), and original OMC (1.16).29 The above-mentioned data suggested that more defects were generated from the synergistic contribution of various heteroatoms, which may provide more activate sites for electrochemical process.
The co-doping of heteroatoms not only improved the defects in the resultant OMC, but also affected the SSA and average pore size. In some studies, the SSA of OMC increased after doping.7,11,29,119,120 Panja et al. reported the SSA and pore volume of pure OMC and N–P co-doped OMC were 1127, 1968 m2 g−1 and 1.43 and 2.47 cm3 g−1, respectively.7 Almost two-fold increases of the SSA and pore volume were ascribed to the bond cleavage reactions and crosslink condensations between the carbon atoms at the edge-plane sites and phosphate compounds from the pyrolyzation of phytic acid. Additionally, the interlayer spacing of [002] plane peaks expanded from 0.334 to 0.363 nm after N–P doping. However, the SSA of doped-OMC also decreased in some cases.10,56,92,117,121 Zhang et al. demonstrated the corrosion role of sulfuric acid towards carbon channels resulted in a decrease of SSA from 869 to 772 m2 g−1 and an increase of average pore size from 4.1 to 6.4 nm with the increasing amount of sulfuric acid.117 The expansion of the pore size was also shown in Xin's study.10 The unit cell values calculated from small-angle X-ray scattering (SAXS) of OMC and N–P co-doped OMC were 11.16 and 12.07 nm. Ye et al. found that the SSA of N–S co-doped OMC even reduced 2.4-fold at the highest mass ratio of thiourea to OMC by the attack of released pyrolysis gases.92 Feng et al. reported the N-doping with cyanamide as N source lead to a decreasing SSA about 231 m2 g−1. Nevertheless, the P-doping with phosphoric acid as P source resulted in an increasing SSA about 533 m2 g−1. More interestingly, the N–P co-doped OMC had a SSA of 1300 m2 g−1, which was higher than that of the single N-doped OMC, but lower than that of P-doped OMC.11 Deng et al. carried out a study to produce N–P co-doped OMC with ammonia gas and triphenylphosphine as N and P source in a soft-template strategy.119 The as-prepared P-doped OMC (448 m2 g−1) and N-doped OMC (903 m2 g−1) possessed a lower SSA than that of N–P co-doped OMC (1118 m2 g−1). Overall, the most important factor for the improvement or reduction of SSA is heteroatom sources. X-ray diffraction (XRD), small angle X-ray scattering (SAXS), and transmission electron microscopy (TEM) are useful tools to observe the ordered porous structure before and after doping.29,92,117 The broader peaks at 24° and 43° in the XRD spectra, corresponding to (002) and (100) planes of the graphitic carbon, indicated the decrease of ordered degree.29,92 The deteriorated channels in TEM images and weakened peaks in SAXS patterns all reflected the increase of the disordered degree.29,117 Hence, the preservation of ordered mesoporous structure after heteroatoms doping should be paid much attention in the future study.
Except the porous structure, the influences of process parameters, such as reaction temperature, aging time, template amount, acid amount, and pyrolysis temperature, on the doping content and dopant species were observed during the binary or ternary doping process.36,101 Zhao et al. found that the effects of the parameters on the amount of foreign elements were different in solo-case and co-doping case.101 The involved mechanism was deduced according to Fig. 8. The organic–organic self-assembly of carbon precursor, template, and B/P anions were performed based on I+X−S+ coulombic interactions. During this process, the protonation of precursor needed to be motivated under acidic condition. Therefore, the formation of borophosphate anions like [BxPyOz]n− in the co-doping case facilitated the improvement of doping amount in the as-obtained OMC. And the competition of hydrochloric acid and borophosphate anions were responsible for the decrease of doping content under the condition of high hydrochloric acid concentrations. The research confirmed that the co-doping of N and P element not only improved the contents of each foreign element in OMC, but also promoted the development of mesoporous structure. The pyrolysis temperature is one of the most significant parameter for the changing of doping content, which is also changed with the various foreign elements.36,122,123 Zhang et al. found a tremendous reduction of nitrogen content from 10.0 to 4.8% and a slight increase of sulfur content from 0.69 to 0.92% with the pyrolysis temperature increasing from 650 to 950 °C.36 What's more, the relative percentage of quaternary-N enhanced from 75.8 to 95.2%, while the pyridine-N decreased from 24.2 to 4.8%. A study by Zhang et al. shows that the relative percentage of N and P element decreased from 5.18 to 2.06% and 3.42 to 0.88%, respectively. On the contrary, the relative percentage of O element increased from 8.74 to 13.44%.122 In another N–P co-doped case, the increasing of heating temperature from 650 to 950 °C lead to N decrease of 10%, but S increase of 0.63%.117 This is because the thermally unstable C–N groups were easily oxidized to produce small-molecular gases under the simultaneous engagement of high temperature and sulfuric acid. In addition, the sulphonation of sulfuric acid generated sulfur oxides on the carbon surface at high temperature and the capillary effect of pores prolonged the residence time of these oxides. Liu et al. also reported a decrease of C/N ratio from 0.085 to 0.02, but an increase trend of S/C ratio from 0.033 to 0.076 with the pyrolysis temperature increasing from 600 to 1000 °C.123 Zhao et al. found that the doping sequence of N and P source could cause the differences in doping content and dopant species.121 The contents and species percentage of synchronized doping N–P co-doped OMC and first-P-second-N OMC were comparable, which were larger than those of first-N-second-P OMC. For example, the contents of pyridinic-N, pyrrolic-N, graphitic-N, and pyridinic-N in N–P co-doped OMC and first-N-second-P OMC were 38.2, 23.4, 27.6, 10.8%, and 22.1, 11.0, 51.9, 15.0, respectively.
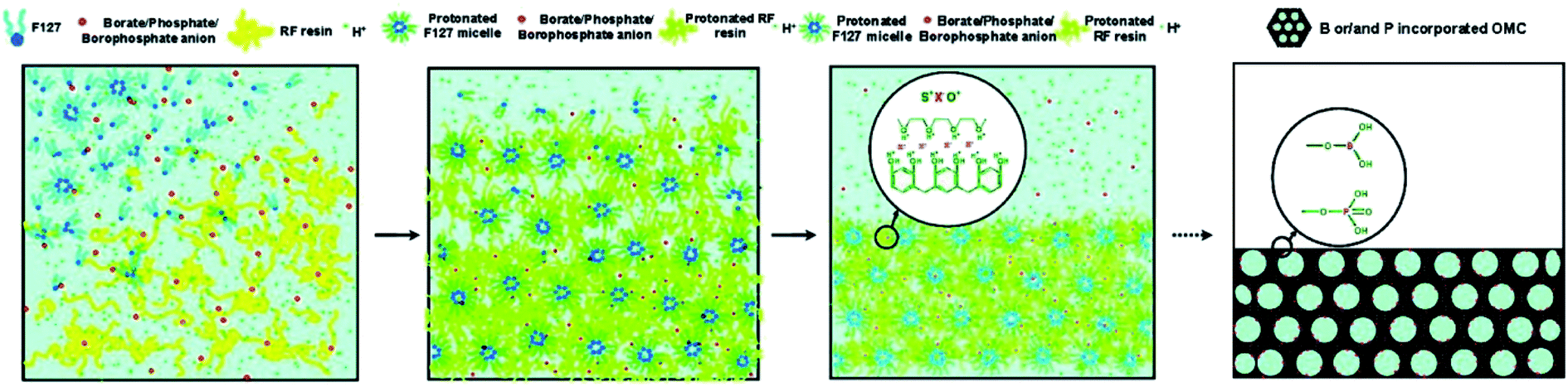 | ||
| Fig. 8 Proposed mechanism of B, P co-doped OMC in a soft template. Reproduced with permission from ref. 100. Copyright 2010, American Chemical Society. | ||
4. Applications as electrodes in supercapacitors
4.1 N-Doped OMC in supercapacitors
The original researches led by Ryoo developed a N-doped OMC for electric double layer capacitor (EDLC) electrodes in 2008.124 In the pioneering studies, the SSA of N-doped OMC and the corresponding specific capacitance were not perfect.124,125 The largest SSA of N-doped OMC by Ryoo and Kim et al. were only 778 and 725 m2 g−1, respectively. The highest capacitance of the electrodes was 182 F g−1 at 10 mV s−1, which was higher than that of pure OMC (147 F g−1). Afterwards, the SSA and pore volume of N-doped OMC were improved to 1420 m2 g−1 and 1.11 cm3 g−1. However, the electrochemical properties were still very poor with a low specific capacitance of 108 F g−1 at 1 mV s−1.126N-Doped OMC with excellent electrochemical properties were gradually synthesized by the scholars in the next decades. Huang et al. applied dicyandiamide and ammonia gas as double N source to prepare N-doped OMC with a large SSA of 1580 m2 g−1 and N content of 11.9% via CVD coupled with dried post treatment method.127 The N-doped electrode exhibited a pronouncedly high specific capacitance of 855 F g−1 at the current density of 1 A g−1, and maintained 615 F g−1 at the high current density of 40 A g−1, which was derived from its high hydrophilicity, low dimensionality, ultrathin carbon layer, and redox reactions of the graphitic nitrogen-associated defects. Zhao et al. produced a N-doped OMC with SSA of 1741 m2 g−1 and N content of 15.1% using cyanamide as N dopant in a simple hard method.67 The resultant electrode displayed a good rate capability with capacitance retention of 77.4% in 6 M KOH electrolyte. They also proved that the N-doped OMC electrode owned comparable capacitance in 1 M H2SO4 electrolyte.46 A hybrid device was assembled with N-doped OMC as negative electrode and MnO2 decorated activated carbon as positive electrode by Qu et al.128 A homogeneous electron–proton hopping due to the additional N-groups and ordered mesoporous structure contributed to a good cycling stability for 150 cycles.
Recently, the synthesis of N-doped OMC with green and environmental friendly sources in a scalable and mass production process is a promising research field. Han et al. prepared gelatin-based N-doped OMC without polymerization and chemical activation procedure. The electrode exhibited a capacitance of 252 F g−1 at the current density of 0.2 A g−1, with a high retention rate of 99.1% after 10000 cycles.129 Sui et al. used pyrrole and carbamide as double N sources to prepare N-doped OMC with a large SSA of 1451 m2 g−1 and high nitrogen content of 5.83%.130 The electrode possessed a capacitance as high as 310 F g−1 at 1 A g−1, and remained 245 F g−1 even at 20 A g−1 in a three-electrode system. The solution resistance and charge-transfer resistance of the N-doped OMC based symmetrical supercapacitor were only 0.55 Ω and 0.72 Ω after 5000 cycles. The synergetic effect of pyrrole and carbamide created more active N species (pyridinic N and pyrrolic N) at the edges of carbon matrix, leading to the enhancement of electrochemical performance.
4.2 P-Doped OMC in supercapacitors
The studies of P-doped OMC are relatively few in comparison with N-doped OMC, especially in the field of supercapacitors. An in situ soft method was used to prepare P-doped OMC with a SSA of 644 m2 g−1 and P content of 2.19% at 800 °C for 2 h. The capacitance of P-doped OMC was 146 F g−1 at 0.2 A g−1 in 6 M KOH electrolyte, which was much higher than that of pure OMC (98 F g−1).101 In another study, an in situ hard method was applied to synthesize P-doped OMC with saccharose as carbon source, KIT-6 as template, and phytic acid as phosphorus source.7 The SSA and capacitance of as-obtained P-doped OMC were 2282 m2 g−1 and 162 F g−1, which were almost 2 times and 1.5 times higher than those of the original OMC. A P-doped three-dimensionally OMC with better electrochemical performance as supercapacitor electrode was produced by using polystyrene sphere as a novel template by an in situ soft method.131 The 3D OMC-based electrode possessed a capacitance of 282 F g−1 at a current density of 0.1 A g−1 with an excellent rate retention capability of 85%.4.3 B-Doped OMC in supercapacitors
As early as in 2008, the B-doped OMC with a doping content of 0.6% and SSA of 470 m2 g−1 was synthesized by co-pyrolysis of sucrose, boric acid, and SBA-15 silica template.131 The capacitance of B-doped OMC electrode was 98.7 and 145.7 F g−1 at 10 mV s−1 in 1 M H2SO4 and 6 M KOH electrolyte, which improved about 1.5 times than those of the pristine OMC. The introduction of B dopants increased the amount of oxygen chemical groups, providing exchangeable proton for redox reaction. Moreover, the B dopants were of great importance for the increase of charge densities and Fermi level density of states. The enhancement of pseudocapacitance and charge transfer by boron doping in OMC was also reported by Enterría.9 In their study, an innovative method combining soft template and hydrothermal carbonization was developed to design the contents and chemical states of boron species by adjusting the mass ratio, hydrothermal temperature, and duration time. The self-generated pressures facilitated the permeation of H3BO3 into the organic phase and transformation of boron oxide to B–C component. More interestingly, the boron doping content was positive correlation with the intensity of redox peaks and the boron species had an effect on the positions of redox peaks. The capacitance of B-doped OMC with a content of 1.78% was 156 F g−1 at 1 A g−1 with a cycling ability of 78% after 10000 cycles. To obtain high-performance electrode materials for supercapacitors, some researchers also investigated the strategy to improve the doping content.25,113 In Zhang's study, the replacement of boric acid by 4-hydroxyphenylboronic acid could prevent the shrinkage of the well-ordered mesoporous structure via strengthening the solubility of the carbon source and hydrogen bond interaction of the reactants.113 The capacitance of as-prepared B-doped OMC electrode with a doping content of 3.96% was 183 F g−1 at 1 A g−1, which was three times more than that of pure OMC. In Gao's research, the B-doped OMC electrode with a doping content of 2.9% delivered a high capacitance 250 F g−1 at 1 A g−1 and a low internal resistance of 1.05 Ω.25
4.4 Heteroatom co-doped OMC in supercapacitors
Many researchers explored the multifunctional effect of dual or ternary dopants on the electrochemical properties. Early in 2010, an in situ EISA approach was used to prepare B or P solo-incorporated and co-incorporated OMC with a SSA ranging from 500 to 700 m2 g−1.101 The capacitance of B–P co-doped OMC was 173 F g−1, which was higher than that of solo B-doped OMC (134 F g−1) and P-doped OMC (146 F g−1). In 2011, a hydrothermal carbonization was used to fabricate B–P co-doped OMC with boric acid and phosphoric acid as heteroatom sources under different synthesis temperatures.132 The boron and phosphorus content of the resultant carbon material increased from 0.77 to 2.15% and 1.20 to 3.30% with the hydrothermal temperature increasing from 50 to 150 °C, respectively. Meanwhile, the capacitance of B–P co-doped OMC electrode for supercapacitor was slightly increased from 158 to 177 F g−1, in spite of the decreasing SSA from 659 to 590 m2 g−1. In the following ten years, the incorporation of foreign atoms in OMC mainly focused on the doping of nitrogen element with other heteroatoms, especially sulphur36,117,133 and phosphorus element.7,10,11,122 Professor Zhang's group synthesized N–S co-doped OMC with a high SSA of 1021 m2 g−1 and capacitance of 180 F g−1 at 1 A g−1 by using pyrrole and sulphuric acid as heteroatom sources.36 Afterwards, they also fabricated a 3D cubic N–S co-doped OMC with much higher capacitance of 320 F g−1 using KIT-6 as hard template.133 After 1000 cycles, the capacitance remained 255 F g−1 at 1 A g−1 with a retention rate of 79.7%. The outstanding performance rooted from synergistic effect of electrical double-layer capacitance and pseudocapacitance of N–S co-doped OMC electrode with super conductivity and hydrophilicity to aqueous electrolyte. This research group also used pyrrole and phosphoric acid as heteroatom sources to obtain N–P co-doped OMC with a highest specific area of 1073 m2 g−1. The maximum capacitance of the electrode reached up to 327 F g−1 at 0.5 A g−1, which was 1.9 times higher than that of the un-doped OMC. The capacitance retention rates of N–P co-doped OMC at pyrolysis temperature of 650, 800, and 950 °C were 93.6, 93.9, and 100% after 1000 cycles. In Xin's study, a N–P dual-doped OMC with much higher capacitance 392 F g−1 at 1 A g−1 and capacitance retention of 95% after 5000 cycles were synthesized using a novel sol–gel strategy.105. Applications as electrocatalyst
The use of metal-free electrocatalyst can avoid the sintering and agglomeration of metal or metal oxide catalysts, reduce production cost, and improve the long term stability.20,80 The well-designed mesoporous structure in OMC can provide short transfer pathway for the electrons or ions, and the defects by heteroatoms incorporation can serve as active sites for the oxidation–reduction process. The onset potential, number of transferred electrons, kinetic current density, and half-wave potential are the important information of the electrocatalyst.5.1 N-Doped OMC as electrocatalyst
It is well known that the N-doped OMC has also been widely applied as promising oxygen reduction catalyst due to its cost-effective and corrosion resistance.134,135 The cyclic voltammetry and linear sweep voltammogram tests in N2 and O2-saturated are usually used to evaluate the ORR performance.26,27,136 The earliest application of N-doped OMC in oxygen reduction reaction (ORR) can be dated from 2009.134 The as-obtained N-doped OMC with a SSA of 1132 m2 g−1 and nitrogen percentage of 6.88% exhibited a 0.88 V onset potential vs. standard hydrogen electrode for oxygen reduction, which was comparable to the commercial Pt/C catalyst (0.996 V). The authors proposed that the pyridinic-N at the edge or vacancy of the carbon framework promoted the electron-donating ability and oxygen chemisorption for ORR. Similar catalyst result was obtained by Dai's group, in which the OMC was post-treated with NH3 at 950–1050 °C.135 More efficient N-doped OMC-based catalyst was successfully synthesized by the later researchers.27,136 In situ doping by melamine associated with post treatment of NH3 at 900 °C was performed to prepare Fe–N co-doped OMC with a SSA of 1643 m2 g−1 and nitrogen percentage of 2.90%. The highest onset potential and diffusion-limited current density were 1.018 V and 5.98 mA cm−2, respectively. Additionally, the attenuation of half-wave potential after 5000 cycles was only 22.3 mV, indicating a favourable durability of the Fe–N co-doped OMC.27 An in situ self-assembly carbonization process was used to prepare Co nanoparticles inserted N-doped OMC with an onset potential located at 0.961 V, a half-wave potential located at 0.815, and a good retention rate of 89.2% after 50000 cycles.136 Han et al. applied KIT-6 as a template and 2-methylimidazole as the C, Fe, and N three-in-one source to fabricate a three-dimensional Fe–N–OMC composite.26 The XPS, XANES, and EXAFS results implied that the Fe atoms coordinated with four adjacent pyridinic-N atoms to form a planar FeN4 structure. The onset and half-wave potential of Fe–N co-doped OMC composite were 0.93 and 1.08 V, which were higher than those of commercial Pt/C. The decrease of onset potential for bare OMC catalyst without FeN4 and the lower Tafel slope of OMC after the removal of template indicated the synergistic effect of FeNx active sites along with 3D ordered mesoporous structure. The theoretical calculation based on density functional theory (DFT) also proved the above-mentioned reaction mechanism (Fig. 9). The free energy diagram in Fig. 9c shows a jump of rate-limiting step along with a decreasing overpotential of 0.11 V after N-doping, demonstrating the intrinsic activity of doping-N towards ORR. Fig. 9f displays an increasing Fermi level of 0.18 eV and the occupation of Fe-dyz and Fe-dzx peaks, implying the accommodation of more charges into electron orbitals after N-doping.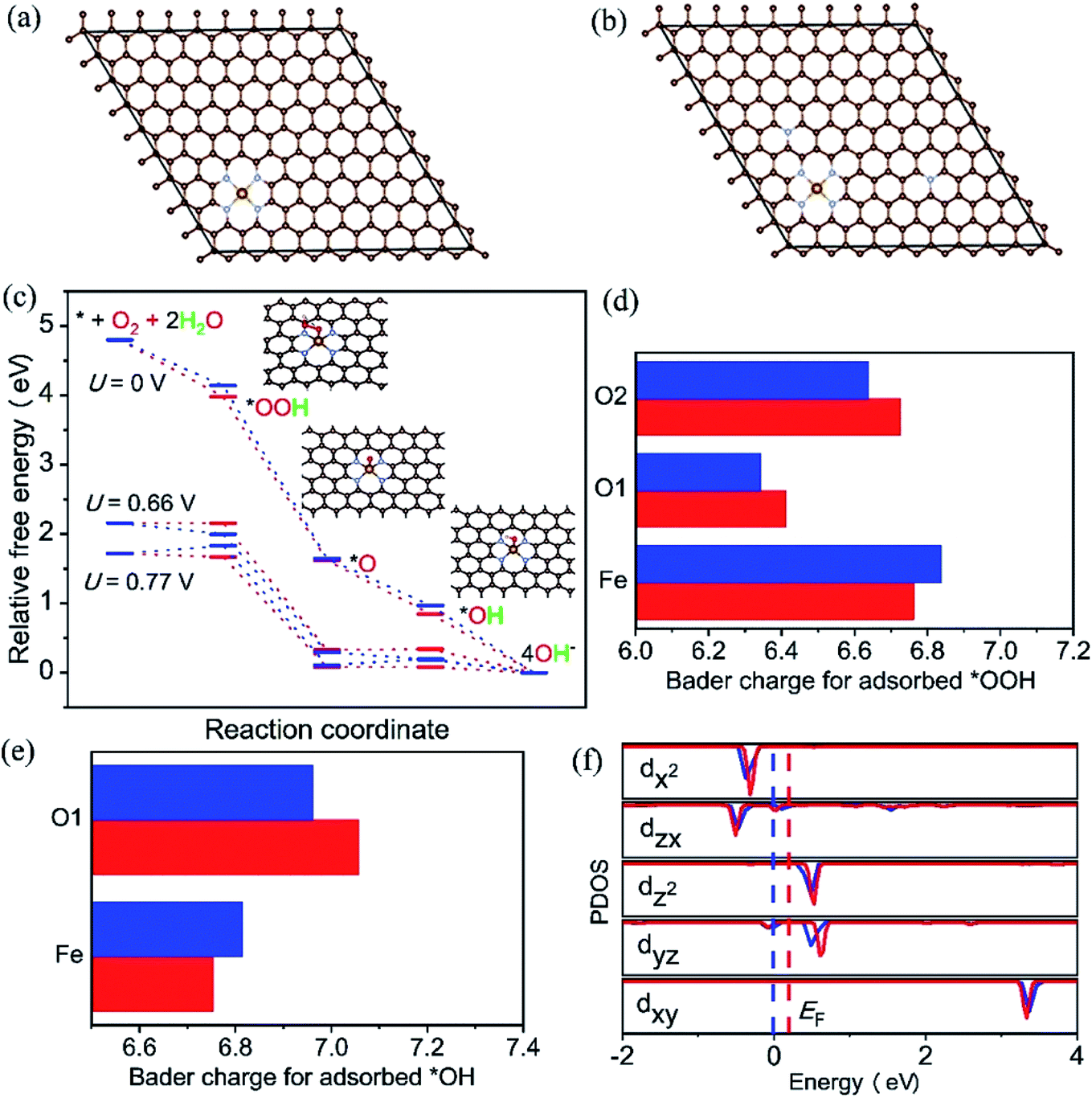 | ||
| Fig. 9 Optimized configuration of (a) Fe–N–C/OMC, (b) Fe–N–C/N–OMC, iron (yellow), nitrogen (blue) and carbon (brown), (c) free energy diagram for the four-electron oxygen reduction pathway on Fe–N–C/OMC (blue) and Fe–N–C/N–OMC (red) in alkaline media, (d) and (e): Bader charge for adsorbed *OOH and *OH on the FeN4 sites of Fe–N–C/OMC (blue) and Fe–N–C/N-OMC (red), (f) density of states and Fermi levels for Fe–N–C/OMC (blue) and Fe–N–C/N-OMC (red). Reproduced with permission from ref. 26. Copyright 2021, Elsevier. | ||
5.2 P-Doped OMC as electrocatalyst
The most important field for the application of P-doped OMC was catalyst, especially for ORR.20,80,84–87 A P-doped OMC was produced by the co-pyrolysis of triphenylphosphine, phenol, and SBA-15 in an in situ hard method.80 It was found that only one-step process with a low onset potential and high current density occurred in the linear sweep voltammetry curves with an O2-saturated 0.1 M KOH solution. Besides, the charge transfer resistance reduced from 29.7 to 10.5 Ω, while the rod length of P-doped OMC shortened from 1.5 to 0.7 μm, resulting in a better ORR performance. Another published literature also displayed the linear relation between rod lengths and the capacitance performance.137 It was proved that the coupling effect of high P content and SSA played a positive role in good activity of P-doped OMC catalyst.20 Specifically, the charge transfer resistance of P-doped OMC at pyrolysis temperature of 800 °C was 2.11 Ω, which was much lower than that of P-doped OMC at pyrolysis temperature of 700 °C with higher P content but lower SSA (9.25 Ω). Other literatures proved that the P-doped OMC could be also used a solid acid catalyst for the esterification of oleic acid and methanol86 and electrooxidation of methanol.845.3 S-Doped OMC as electrocatalyst
The reports on the S-doped OMC as oxygen reduction catalyst were few, and most of the studies were focused on its application in Li–S batteries. Wang et al. used an in situ hard method to prepare S-doped OMC with different doses of sucrose and benzyl disulfide as carbon and sulphur sources.138 The S-doped OMC with a lower SSA of 1099 m2 g−1 showed a higher current of 376.4 μA than that of original OMC with a higher SSA of 1357 m2 g−1 (158.7 μA), indicating the dominating role of sulphur dopants, especially the C-SOx–C species. During the tolerance tests, the relative current of S-doped OMC electrocatalyst maintained 81.56% after 10000 s, which was higher than that of commercial Pt–C electrocatalyst (27.61%). Titirici et al. prepared the S-doped OMC via an all-in-one hydrothermal carbonization of the mixture of glucose, thiophenecarboxy, TEOS and P123 block copolymer.139 The hydrothermal carbonization not only increased the doping content of tenfold by preventing the volatilization of S dopants into gases, but also avoided the traditional pre-step for the formation of silica template. The high electron transfer numbers calculated from Koutecky–Levich equation implied the four-electron reaction mechanism of S-doped OMC during the ORR process.
5.4 B-Doped OMC as electrocatalyst
The incorporation of boron could act as a modifier of the electronic structure and capture assistant for oxygen molecule. Bo et al. produced B-doped OMC with 4-hydroxyphenylboronic acid and sucrose as the boron and carbon source in a hard template method.111 At the mass ratio of 0.15, the obtained B-doped OMC had a SSA of 900 m2 g−1 and boron content of 1.3%. The current decaying of B-doped OMC electrode was only 16% after 10000 s, which was lower than that of commercial Pt/C catalyst (42%). Furthermore, the B-doped OMC electrode also exhibited better methanol tolerance than commercial Pt/C catalyst. Zhou et al. reported that the initial catalytic rates of Pd supported B-doped OMC over 2,4-dichlorophenol enhanced about 3 times compared with that without B-doping.33 Nsabimana et al. reported the onset potentials of H2O2 on OMC and B-doped OMC were 0.45 and 0.38 V.114 Besides, the oxidation peak potentials and current densities towards the electrocatalytic oxidation of methanol by Pd supported OMC and B-doped OMC were 0.71, 0.658 V, and 0.360, 0.511 A mg−1, respectively. These results implied a better electrochemical activity of B-doped OMC electrode due to more active sites and defects after B-doping.
5.5 Heteroatom co-doped OMC as electrocatalyst
Interestingly, the Kelvin probe force microscopy and linear sweep voltammetry (LSV) tests were used to investigate the relationship of heteroatom doping and reaction kinetics for ORR by Cheon.116 Based on LSC curves, the calculated electron number of N–O–S tri-doped OMC was 3.5, which was higher than that of S–O dual-doped OMC (2.7) and O solo-doped OMC (2.5), indicating that the ORR proceeded via quasi-four-electron reduction pathway. Fig. 10a and b clearly show the different values of contact potentials of various OMCs. Fig. 10c and d exhibit the linear relationship of kinetic current densities and electron transfer numbers with the work function, indicating that the lower energy barrier in N–O–S tri-doped OMC promoted the donation of electrons to the adsorbed oxygen. Over the last five years, the density functional theory (DFT) calculations were widely used to discuss the electrocatalysis properties of doped-OMC.106,121 In Daems' research, the N–O co-doped OMC displayed the electron numbers of 6 and higher current density than OMC, indicating a good selectivity to aniline.106 The calculated adsorption energy of graphitic-N and pyridinic-N sites were −0.54 and −0.52 eV, which was higher than that of un-doped OMC (−0.38 eV), suggesting that the graphitic-N and pyridinic-N could act as catalytic sites for the reduction of nitrobenzene. As illustrated in Fig. 11, the lowest energy conjugation occurred under the parallel arrangement between nitrobenzene and carbon plane. In Zhao's study, the DFT results showed that the formation energy of graphitic–N bond nearby P sites was lower than that of un-doped OMC, whether the P embedded inside or outside the carbon framework. And the adsorption energy of N on nearby P sites was 5.68 eV, which was higher than that of un-doped OMC (3.68 eV). The formation of C–N bond was at the cost of C–P bond. The above-mentioned information explained why the former P-doping could affect the later N-doping. Furthermore, the adsorption energy of oxygen on the graphitic-N doped-OMC (0.25 eV) was higher than that of un-doped OMC (0.16 eV), leading to elongation of O–O bond in Yeager mode. Finally, a better activity was performed in a four electron pathway for ORR.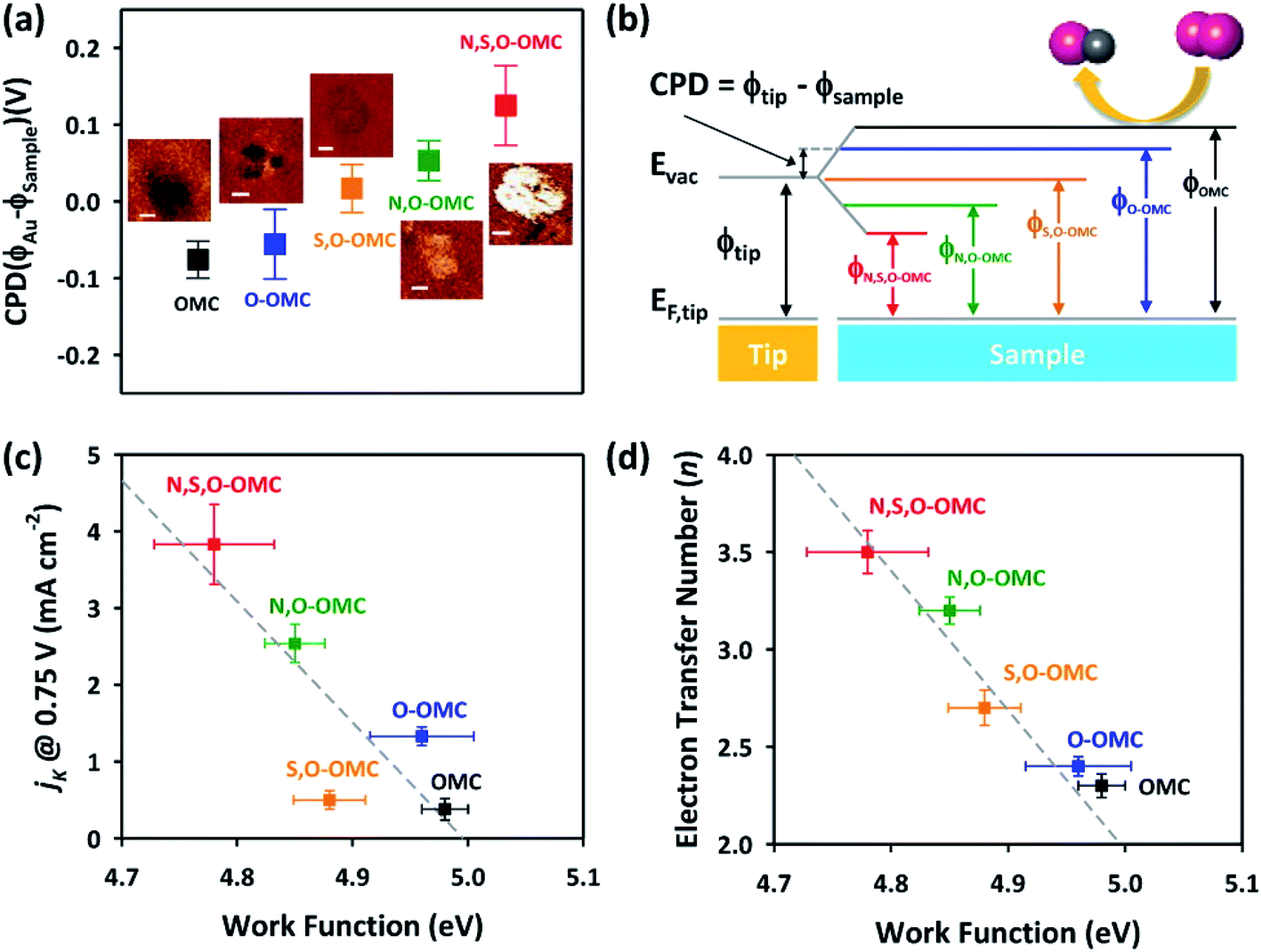 | ||
| Fig. 10 (a) Contact potential difference images, (b) energy diagram, (c) correlation of ORR activity, and (d) four-electron selectivity of the doped-OMC with their work function values. Reproduced with permission from ref. 115. Copyright 2014, American Chemical Society. | ||
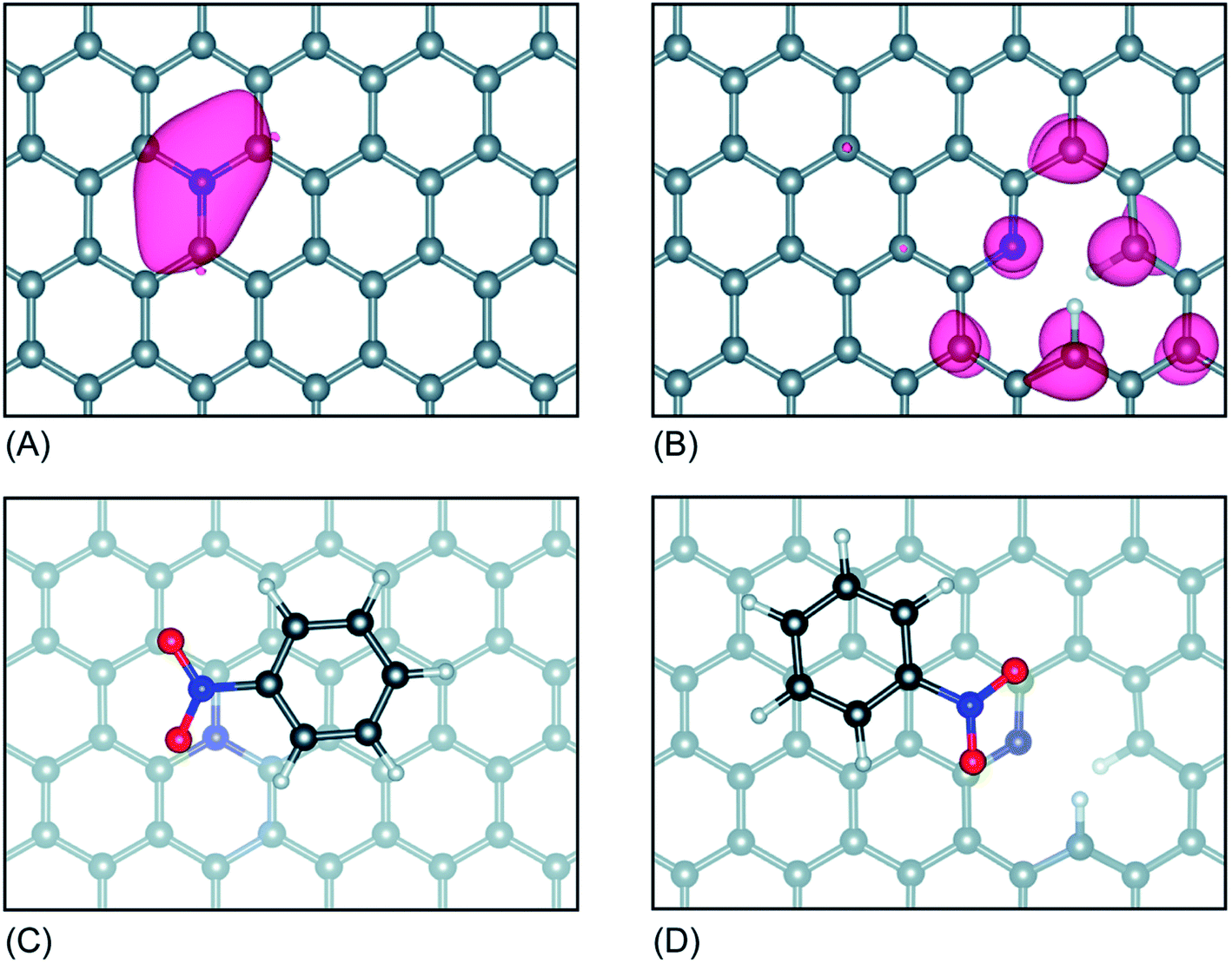 | ||
| Fig. 11 (A) Optimized configuration of graphitic-N OMC, and (B) pyridinic-N OMC, (C) Sable adsorption geometries of nitrobenzene on graphitic-N OMC, and (D) pyridinic-N OMC (grey spheres represent C atoms, red spheres O atoms, blue spheres N atoms and white spheres H atoms). Reproduced with permission from ref. 105. Copyright 2018, Royal Society of Chemistry. | ||
6. Applications in ion batteries
6.1 N-Doped OMC in ion batteries
N-Doped OMC has applied as an attractive alternative for ion batteries, including Li+, Na+, and K+, due to its ordered mesoporous structure, short diffusion pathway, rich edge defects, large interaction interface for the insertion and extraction of the electrolyte ions.18,35,40,71,127 Zhu et al. prepared N-doped OMC with a high N content from 6.3 to 31.7% by adding different doses of melamine.18 Remarkably, the N-doped OMC with the highest N content of 31.7%, but low SSA of 33 m2 g−1 didn't show the best battery performance, suggesting the battery performance was affected by both surface chemistry and porous structure. The weakened solid–electrolyte-interface resistance and charge transfer resistance of N-doped OMC indicated the improvement of interfacial properties after N-doping. The highest reversible capacity was 506 mA h g−1 with a coulombic efficiency of 99.4% after 300 cycles. Qiu et al. produced N-doped OMC by using a post treatment method under 20% of NH3 at 700 °C for 5 h. The reversible charge capacity of the N-doped electrode was higher than that of the initial one after 450 cycles due to the compensation of released K+ ions from the active sites of defects and edges.71 Le et al. produced N-doped OMC with a SSA of 971–1054 m2 g−1 and nitrogen content of 2.84 to 8.57% by using melamine as dopant source.35 The highest discharge capacity of 1887 mA h g−1 at 0.1 A g−1 was rooted from the favourable wettability and conductivity as well as more active sites by N-doping. Furthermore, the associated increasing O element provided more defects and vacancies, leading to the boost of Li insertion and extraction behaviors. Much stronger cathodic peaks appeared at 0.6 V and 1.6 V for N-doped OMC represented the formation of solid electrolyte interphase layer, which could prohibit electrolyte ions from corroding the carbon electrode. It should be noted that the sodium ion batteries (NIBs) have been developed in the battery market due to the widespread and cheap sodium sources.40 The optimal N-OMC displayed a reversible capacity of 1359 mA h g−1 at 0.1 A g−1 and a good cycling stability of 722 mA h g−1 after 200 cycles. Much stronger interaction of Na+ ions and N-doped OMC was observed from XPS analysis as a result of the heterozygosis of lone pair electrons in N atoms and π electrons in C atoms. Additionally, the presence of approximately 2% of hydrogen element played a beneficial role in Li storage through binding the adjacent Li ions. The large interlayer spacing of 0.387–0.395 nm and abundant mesopore pathways also played a positive effect on the electrolyte diffusion.6.2 P-Doped OMC in ion batteries
A red P incorporated OMC was used as an anode electrode for both lithium-ion and sodium-ion batteries.66 Although the as-obtained composite material only had an extraordinarily low specific area of 14 m2 g−1, a high initial reversible capacity of 2250 mA h g−1 for lithium ion batteries (LIBs) and a good reversible capacity of 2591 mA h g−1 for NIBs were obtained, owing to the high electronic conductivity and short diffusion pathway via confining the P nanoparticles in the channels. Three cathodic peaks located at 0.65, 0.33, 0.01 V and 0.52, 0.7, 1.42 V were observed for LIBs and NIBs, respectively, which were due to the insertion of lithium/sodium ions into the mesoporous channels and formation of LixP or NaxP. The rate capacity retentions for both LIBs and NIBs were 40.26% and 27.88% with the current density increased from 0.6 to 9.8 A g−1.6.3 S-Doped OMC in ion batteries
The combining of OMC and sulphur can improve the conductivity of electrons/ions, and mitigate the polysulfide shuttle phenomenon of sulphur cathode through shortening the diffusion pathway of ions and reducing the ion-transport resistance.140,141 It should be noticed the post-thermal treatment was widely applied for the synthesis of S-doped OMC for lithium-ion battery.92,93,96 A melt-diffusion method was used to synthesize S-doped OMC with high sulphur element by heating the pristine OMC and sulphur above the melting point of sulphur. The melt sulphur homogeneously filled inside the pore structures through the capillary forces as well as hydrophobicity of both materials, and then precipitated after cooling down.140–143 In Nazar’ study, the comparison of pore structures before and after sulphur incorporation revealed that about 9% of empty pore volume remained, providing effective space for the accommodation of Li ions.142 Similar phenomenon was also observed by other researchers.140 In additional, the electrical conductivity of S-doped OMC maintained as same as that of the pristine one (0.21 S cm−1), indicating that electron transfer pathways were not blocked by S-doping. The obtained electrode showed a reversible capacity of 1400 mA h g−1 with a low capacity fading of 20%. Another report also used the melt-diffusion strategy to produce S-doped OMC with a high sulfur doping of 60% and an excellent cycling stability of 54.5% after 400 cycles.140 It was established that the formation of bulk crystalline sulfur and the excessive coverage of mesopores above the sulfur doping of 60% could weaken the reaction activity and hinder the transfer of Li ions. Moreover, a novel ordered meso–microporous core–shell carbon assembled with sulfur was developed as a cathode of Li–S battery.141 The mesoporous “cores” served as a container for the loading of high-molecular sulphur (S8), and the microporous “shell” played a role as physical barrier to retard the severe capacity fading over cyclings by anchoring sulphur in the form of small-molecular (S2–4) (Fig. 12). The as-prepared cathode delivered a quite low capacity decay of 19% after 200 cycles. Zheng et al. also investigated the performance of S-doped hierarchical hybrid with ordered mesoporous cores and microporous shells for Li–S batteries.144 It has been confirmed that the space confinement effect of micropores facilitated the accommodation of S2–4 molecular and transformation of short-chain polysulfides, thus prohibiting the migration of long-chain polysulfides from inside pores to external surface during the discharge process. The obtained cathode delivered a reversible capacity as high as 1122 mA h g−1 with a cycling retention of 92% after 300 cycles. Another study was performed to fabricate spherical S-doped OMC with polyaniline as a protective layer, therefore avoiding the dissolution of polysulfides into electrolyte.143 The as-made cathode delivered a capacity of 1338 mA h g−1 with a good coulombic efficiency of 98% after 100 cycles due to the synergistic effect of ordered mesopores and the conductive polyaniline. | ||
| Fig. 12 (a) XPS spectra of S-OMC, (b) XPS spectra of S-MMCS, (c) the proposed charge–discharge process, (d) the discharge profile of a typical electrochemical reaction, (e) TEM images and cycling curves. Reproduced with permission from ref. 140. Copyright 2014, American Chemical Society. | ||
6.4 Heteroatom co-doped OMC in ion batteries
The previous reports confirmed that the better performance of co-doped OMC was derived from the improved active sites and conductivity from higher SSA and more defects.145–147 In Ye's study, the N–S co-doped OMC electrode delivered a reversible capacity of 419 mA h g−1 with a retention rate of 91.8% after 150 cycles.92 Moreover, N–S co-doped OMC also exhibited lower charge transfer resistance and superior rate capability than the solo-doped and un-doped OMC. DFT calculation was used to investigate the charge transfer and adsorption capability. The adsorption energy of N–S co-doped OMC, N-doped OMC, and original OMC was −2.54, −1.90, and −1.08 eV, respectively. Fig. 13d shows the increased valence and conduction bands Fermi level, indicating the improved conductivity after heteroatoms doping. In Li's research, a metal–N (metal = Fe, Co, Ni, and Cu) co-doping graphitic OMC was designed to alleviate the shuttle effect of polysulfide anions.4 The highest adsorption energies of Li2S on Fe–N co-doped OMC (−3.01 eV) based on the DFT calculations explained the reason why the Fe–N co-doped OMC owned the strongest binding ability towards long-chain polysulfides, thus preventing active sulfur decaying. The Fe–N co-doped OMC electrode delivered an initial capacity as high as 1473 mA h g−1 with a slight attenuation of 0.075% after 500 cycles. | ||
| Fig. 13 (a–c) First-principles calculations for Na+ adsorption behavior on surface of OMC and (d) corresponding density of states. Reproduced with permission from ref. 91. Copyright 2019, Elsevier. | ||
7. Summary and prospects
Over the last two decade, numerous investigations have been carried out on the synthesis of different heteroatoms-doped OMC from various dopants, such as nitrogen, sulphur, phosphorus, boron, oxygen, and co-doping of more than one heteroatom. In this review, we have discussed the recent methods for the fabrication of heteroatom-functionalized OMC, including in situ, post treatment, and chemical vapor deposition. Among them, the in situ method with a hard template is the most common due to its contribution to the reservation of well-ordered mesoporous structure. Additionally, we also summarized how the heteroatoms affected the electrochemical properties and the corresponding performances in various applications, such as supercapacitor, electrocatalysis, and ion battery. The previous findings revealed that the dopant contents and species were highly related to the synthesis parameters, such as the types of heteroatom source, the mass ratio of carbon source to foreign source, reaction temperature, pyrolysis temperature, and retention time. More importantly, the specific surface area and doping content as well as the dopant species played synergistic effect on the electrochemical properties. In summary, the doping of heteroatoms into the carbon network could reduce Fermi energy, redistribute charge density, produce more defects, enlarge interlayer spacing, improve graphitization degree, and lower transfer resistance, thereby providing more active sites, faster electron transfer rate, higher conductivity, and better surface wettability during the electrochemical behaviors.Although the doped-OMC has displayed promising potential in many fields, some major questions are still required to be addressed: (i) how to simply the complicated synthesis processes without producing any environmental pollutants. According to this review, it could be easy to realize that the silica-based hard template was needed to fabricate firstly, followed by the nanocasting of carbon and heteroatom source, and finally the removal of silica by highly corrosive HF or hot NaOH in a hard-template process. The escaping gases, low-temperature waste heats, and chemical residues should be recycled and harmless disposal in practice. (ii) How to reduce the production cost. To accomplish the large scale application in industry of doped OMC, a cost-effective and economically feasible strategy should be developed. The replacement of traditional phenolic resin carbon source and heteroatom source by environmentally friendly biomass waste with in situ dopants is a feasible choice. There is no doubt that the optimization of technology is also a good choice. (iii) How to avoid the decrease of SSA as well as improve the doping content without any destruction of the ordered mesoporous structure, especially for boron and phosphorus dopants. (iv) How to accurately control the dopant species at the molecular level during the preparation process, and adjust the distribution of different species according to the specific requirement and target. Limited research work has been performed on the investigation of the interaction mechanism of carbon precursor, heteroatom source, and template, which could be provided by applying the in situ detection instruments.
Authors' contribution
Yuan Gao: Writing the manuscript and drawing the figures. Qing Wang: Preparing the draft and collecting the primary data; Guozhao Ji: Revising the language and grammar. Aimin Li: Assisting in the design of the manuscript. Jiamin Niu: Preparing the related references.Conflicts of interest
The authors declare that there are not any competing financial interests or personal relationships that could have appeared to affect the reported work in this paper.Acknowledgements
This work was financial supported by the National Natural Science Foundation of China (51802035), the China Postdoctoral Science Foundation (2019M661102), and the National Key Research and Development Program of China (2020YFC1807800).References
- S. Yu, V. M. Hong Ng, F. Wang, Z. Xiao, C. Li, L. B. Kong, W. Que and K. Zhou, J. Mater. Chem., 2018, 6, 9332–9367 RSC.
- M. Xu, Q. Yu, Z. Liu, J. Lv, S. Lian, B. Hu, L. Mai and L. Zhou, Nanoscale, 2018, 10, 21604–21616 RSC.
- F. Zhang, X. Liu, M. Yang, X. Cao, X. Huang, Y. Tian, F. Zhang and H. Li, Nano Energy, 2020, 69, 104443 CrossRef CAS.
- H. Li, D. Liu, X. Zhu, D. Qu, Z. Xie, J. Li, H. Tang, D. Zheng and D. Qu, Nano Energy, 2020, 73, 104763 CrossRef CAS.
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680–2721 RSC.
- T. Jiang, Y. Wang, K. Wang, Y. Liang, D. Wu, P. Tsiakaras and S. Song, Appl. Catal., B, 2016, 189, 1–11 CrossRef CAS.
- T. Panja, D. Bhattacharjya and J. Yu, J. Mater. Chem. A, 2015, 3, 18001–18009 RSC.
- W. Libbrecht, A. Verberckmoes, J. W. Thybaut, P. Van Der Voort and J. De Clercq, Carbon, 2017, 116, 528–546 CrossRef CAS.
- M. Enterría, M. F. R. Pereira, J. I. Martins and J. L. Figueiredo, Carbon, 2015, 95, 72–83 CrossRef.
- X. Xin, H. Kang, J. Feng, L. Sui, H. Dong, P. Zhao, B. Pang, Y. Chen, Q. Sun, S. Ma, R. Zhang, L. Dong and L. Yu, Chem. Eng. J., 2020, 393, 124710 CrossRef CAS.
- J. Feng, W. Song, L. Sun and L. Xu, RSC Adv., 2016, 6, 110337–110343 RSC.
- X. Wang, J. S. Lee, Q. Zhu, J. Liu, Y. Wang and S. Dai, Chem. Mater., 2010, 22, 2178–2180 CrossRef CAS.
- F. H. Simanjuntak, J. Jin, N. Nishiyama, Y. Egashira and K. Ueyama, Carbon, 2009, 47, 2531–2533 CrossRef CAS.
- J. Wei, D. Zhou, Z. Sun, Y. Deng, Y. Xia and D. Zhao, Adv. Funct. Mater., 2013, 23, 2322–2328 CrossRef CAS.
- D. Gang, Z. Uddin Ahmad, Q. Lian, L. Yao and M. E. Zappi, Chem. Eng. J., 2021, 403, 126286 CrossRef CAS.
- Y. Kim, T. Park, J. Na, J. W. Yi, J. Kim, M. Kim, Y. Bando, Y. Yamauchi and J. Lin, Nanoscale, 2020, 12, 8608–8625 RSC.
- T. Y. Liu and G. L. Liu, J. Mater. Chem. A, 2019, 7, 23476–23488 RSC.
- J. Zhu, J. Yang, R. Miao, Z. Yao, X. Zhuang and X. Feng, J. Mater. Chem. A, 2016, 4, 2286–2292 RSC.
- Y. Qu, M. Guo, X. Wang and C. Yuan, J. Alloys Compd., 2019, 791, 874–882 CrossRef CAS.
- D. Yang, D. Bhattacharjya, M. Y. Song and J. Yu, Carbon, 2014, 67, 736–743 CrossRef CAS.
- H. Xue, T. Wang, J. Zhao, H. Gong, J. Tang, H. Guo, X. Fan and J. He, Carbon, 2016, 104, 10–19 CrossRef CAS.
- W. Cha, I. Y. Kim, J. M. Lee, S. Kim, K. Ramadass, K. Gopalakrishnan, S. Premkumar, S. Umapathy and A. Vinu, ACS Appl. Mater. Interfaces, 2019, 11, 27192–27199 CrossRef CAS PubMed.
- S. Liu, C. Lai, B. Li, C. Zhang, M. Zhang, D. Huang, L. Qin, H. Yi, X. Liu, F. Huang, X. Zhou and L. Chen, Chem. Eng. J., 2020, 384, 123304 CrossRef CAS.
- R. Li, J. Zhao, D. Han and X. Li, Catal. Commun., 2017, 97, 116–119 CrossRef CAS.
- J. Gao, X. Wang, Y. Zhang, J. Liu, Q. Lu and M. Liu, Electrochim. Acta, 2016, 207, 266–274 CrossRef CAS.
- J. Han, H. Bao, J. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS.
- H. Tan, Y. Li, X. Jiang, J. Tang, Z. Wang, H. Qian, P. Mei, V. Malgras, Y. Bando and Y. Yamauchi, Nano Energy, 2017, 36, 286–294 CrossRef CAS.
- P. Zhang, L. Wang, S. Yang, J. A. Schott, X. Liu, S. M. Mahurin, C. Huang, Y. Zhang, P. F. Fulvio, M. F. Chisholm and S. Dai, Nat. Commun., 2017, 8, 15020 CrossRef PubMed.
- K. Zeng, J. Su, X. Cao, X. Zheng, X. Li, J. Tian, C. Jin and R. Yang, J. Alloys Compd., 2020, 824, 153908 CrossRef CAS.
- Z. Wang, W. Han, H. Tang, Y. Li and H. Liu, Microporous Mesoporous Mater., 2019, 275, 200–206 CrossRef CAS.
- Y. Liu, Y. Xiong, P. Xu, Y. Pang and C. Du, Sci. Total Environ., 2020, 708, 134918 CrossRef CAS PubMed.
- J. R. Maluta, S. A. S. Machado, U. Chaudhary, J. S. Manzano, L. T. Kubota and I. I. Slowing, Sens. Actuators, B, 2018, 257, 347–353 CrossRef CAS.
- J. Zhou, K. Wu, W. Wang, Z. Xu, H. Wan and S. Zheng, Appl. Catal., A, 2014, 470, 336–343 CrossRef CAS.
- H. Zhao, L. Li, Y. Liu, X. Geng, H. Yang, C. Sun and B. An, Appl. Surf. Sci., 2020, 504, 144438 CrossRef CAS.
- H. T. T. Le, T. Dang, N. T. H. Chu and C. Park, Electrochim. Acta, 2020, 332, 135399 CrossRef CAS.
- D. Zhang, Y. Hao, L. Zheng, Y. Ma, H. Feng and H. Luo, J. Mater. Chem. A, 2013, 1, 7584 RSC.
- Y. Hua, T. Jiang, K. Wang, M. Wu, S. Song, Y. Wang and P. Tsiakaras, Appl. Catal., B, 2016, 194, 202–208 CrossRef CAS.
- D. S. Yuan, F. L. Zeng, J. Yan, X. L. Yuan, X. J. Huang and W. J. Zou, RSC Adv., 2013, 3, 5570–5576 RSC.
- D. Zhou, W. Li, X. Dong, Y. Wang, C. Wang and Y. Xia, J. Mater. Chem. A, 2013, 1, 8488 RSC.
- Y. Zhang, L. Chen, Y. Meng, J. Xie, Y. Guo and D. Xiao, J. Power Sources, 2016, 335, 20–30 CrossRef CAS.
- J. Lu, X. Bo, H. Wang and L. Guo, Electrochim. Acta, 2013, 108, 10–16 CrossRef CAS.
- D. Zhou, W. Li, X. Dong, Y. Wang, C. Wang and Y. Xia, J. Mater. Chem. A, 2013, 1, 8488 RSC.
- X. Hu, C. Wang, M. Zhang, F. Zhao and B. Zeng, Talanta, 2020, 217, 121032 CrossRef CAS PubMed.
- J. Wang, H. Liu, H. Sun, W. Hua, H. Wang, X. Liu and B. Wei, Carbon, 2018, 127, 85–92 CrossRef CAS.
- S. Tanaka, H. Fujimoto, J. F. M. Denayer, M. Miyamoto, Y. Oumi and Y. Miyake, Microporous Mesoporous Mater., 2015, 217, 141–149 CrossRef CAS.
- J. Wei, D. Zhou, Z. Sun, Y. Deng, Y. Xia and D. Zhao, Adv. Funct. Mater., 2013, 23, 2322–2328 CrossRef CAS.
- J. Su, X. Cao, J. Wu, C. Jin, J. Tian and R. Yang, RSC Adv., 2016, 6, 24728–24737 RSC.
- S. Liang, J. Mi, F. Liu, Y. Zheng, Y. Xiao, Y. Cao and L. Jiang, Chem. Eng. Sci., 2020, 221, 115714 CrossRef CAS.
- Y. Z. Zhang, Y. Meng, L. Chen, Y. Guo and D. Xiao, J. Mater. Chem. A, 2016, 4, 17491–17502 RSC.
- J. Zhao, W. Shan, P. Zhang and S. Dai, Chem. Eng. J., 2020, 381, 122579 CrossRef CAS.
- Z. Wu, P. A. Webley and D. Zhao, J. Mater. Chem. A, 2012, 22, 11379 RSC.
- W. Kim, M. Y. Kang, J. B. Joo, N. D. Kim, I. K. Song, P. Kim, J. R. Yoon and J. Yi, J. Power Sources, 2010, 195, 2125–2129 CrossRef CAS.
- X. Wang, C. Liu, D. Neff, P. F. Fulvio, R. T. Mayes, A. Zhamu, Q. Fang, G. Chen, H. M. Meyer, B. Z. Jang and S. Dai, J. Mater. Chem. A, 2013, 1, 7920 RSC.
- N. D. Kim, W. Kim, J. B. Joo, S. Oh, P. Kim, Y. Kim and J. Yi, J. Power Sources, 2008, 180, 671–675 CrossRef CAS.
- J. R. Maluta, S. A. S. Machado, U. Chaudhary, J. S. Manzano, L. T. Kubota and I. I. Slowing, Sens. Actuators, B, 2018, 257, 347–353 CrossRef CAS.
- W. Yang, X. Yue, X. Liu, J. Zhai and J. Jia, Nanoscale, 2015, 7, 11956–11961 RSC.
- B. Wang, T. P. Ang and A. Borgna, Microporous Mesoporous Mater., 2012, 158, 99–107 CrossRef CAS.
- Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 1553–1558 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS.
- P. F. Fulvio, M. Jaroniec, C. Liang and S. Dai, J. Phys. Chem. C, 2008, 112, 13126–13133 CrossRef CAS.
- J. Xu, Y. Zhao, C. Shen and L. Guan, ACS Appl. Mater. Interfaces, 2013, 5, 12594–12601 CrossRef CAS PubMed.
- V. Ravat, I. Nongwe and N. J. Coville, Microporous Mesoporous Mater., 2016, 225, 224–231 CrossRef CAS.
- Á. Sánchez-Sánchez, F. Suárez-García, A. Martínez-Alonso and J. M. D. Tascón, J. Colloid Interface Sci., 2015, 450, 91–100 CrossRef PubMed.
- K. Wan, G. Long, M. Liu, L. Du, Z. Liang and P. Tsiakaras, Appl. Catal., B, 2015, 165, 566–571 CrossRef CAS.
- W. Li, Z. Yang, M. Li, Y. Jiang, X. Wei, X. Zhong, L. Gu and Y. Yu, Nano Lett., 2016, 16, 1546–1553 CrossRef CAS PubMed.
- Q. Shi, R. Zhang, Y. Lv, Y. Deng, A. A. Elzatahrya and D. Zhao, Carbon, 2015, 84, 335–346 CrossRef CAS.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS.
- X. Sheng, N. Daems, B. Geboes, M. Kurttepeli, S. Bals, T. Breugelmans, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B, 2015, 176–177, 212–224 CrossRef CAS.
- J. Du, L. Liu, Y. Yu, Y. Zhang, H. Lv and A. Chen, J. Mater. Sci. Technol., 2019, 35, 2178–2186 CrossRef.
- Z. Qiu, K. Zhao, J. Liu and S. Xia, Electrochim. Acta, 2020, 340, 135947 CrossRef CAS.
- P. Matter, L. Zhang and U. Ozkan, J. Catal., 2006, 239, 83–96 CrossRef CAS.
- J. Park, Y. Nabae, T. Hayakawa and M. Kakimoto, ACS Catal., 2014, 4, 3749–3754 CrossRef CAS.
- X. H. Li, K. Wan, Q. B. Liu, J. H. Piao, Y. Y. Zheng and Z. X. Liang, Chin. J. Catal., 2016, 37, 1562–1567 CrossRef CAS.
- J. H. Lee, H. J. Lee, S. Y. Lim, B. G. Kim and J. W. Choi, J. Am. Chem. Soc., 2015, 137, 7210–7216 CrossRef CAS PubMed.
- A. F. M. EL-Mahdy, T. Liu and S. Kuo, J. Hazard. Mater., 2020, 391, 122163 CrossRef CAS PubMed.
- H. Chen, M. Zhou, Z. Wang, S. Zhao and S. Guan, Electrochim. Acta, 2014, 148, 187–194 CrossRef CAS.
- Y. Liu, X. Zhao, G. S. Chauhan and J. Ahn, Appl. Surf. Sci., 2016, 380, 151–158 CrossRef CAS.
- D. Liu, G. Cheng, H. Zhao, C. Zeng, D. Qu, L. Xiao, H. Tang, Z. Deng, Y. Li and B. Su, Nano Energy, 2016, 22, 255–268 CrossRef CAS.
- D. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- F. Wu, R. Dong, Y. Bai, Y. Li, G. Chen, Z. Wang and C. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 21335–21342 CrossRef CAS PubMed.
- J. Wang, Y. Xia, Y. Liu, W. Li and D. Zhao, Energy Storage Mater., 2019, 22, 147–153 CrossRef.
- F. Wu, R. Dong, Y. Bai, Y. Li, G. Chen, Z. Wang and C. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 21335–21342 CrossRef CAS PubMed.
- P. Song, L. Zhu, X. Bo, A. Wang, G. Wang and L. Guo, Electrochim. Acta, 2014, 127, 307–314 CrossRef CAS.
- Y. Zhou, L. Tang, G. Yang, G. Zeng, Y. Deng, B. Huang, Y. Cai, J. Tang, J. Wang and Y. Wu, Catal. Sci. Technol., 2016, 6, 1930–1939 RSC.
- L. Wang, X. Dong, H. Jiang, G. Li and M. Zhang, Catal. Commun., 2014, 56, 164–167 CrossRef CAS.
- P. F. Fulvio, R. T. Mayes, J. C. Bauer, X. Wang, S. M. Mahurin, G. M. Veith and S. Dai, Catal. Today, 2012, 186, 12–19 CrossRef CAS.
- Z. Qiang, Y. Xia, X. Xia and B. D. Vogt, Chem. Mater., 2017, 29, 10178–10186 CrossRef CAS.
- J. R. Maluta, S. A. S. Machado, U. Chaudhary, J. S. Manzano, L. T. Kubota and I. I. Slowing, Sens. Actuators, B, 2018, 257, 347–353 CrossRef CAS.
- W. Li, M. Zhou, H. Li, K. Wang, S. Cheng and K. Jiang, Energy Environ. Sci., 2015, 8, 2916–2921 RSC.
- D. Saha, S. Barakat, S. E. Van Bramer, K. A. Nelson, D. K. Hensley and J. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 34132–34142 CrossRef CAS PubMed.
- J. Ye, H. Zhao, W. Song, N. Wang, M. Kang and Z. Li, J. Power Sources, 2019, 412, 606–614 CrossRef CAS.
- Y. Song, J. Ren, G. Wu, W. Zhang, C. Zhang and F. Yin, J. Mater. Res., 2019, 34, 600–607 CrossRef CAS.
- L. Wang, W. Jia, X. Liu, J. Li and M. M. Titirici, J. Energy Chem., 2016, 25, 566–570 CrossRef.
- S. Liu, C. Lai, B. Li, C. Zhang, M. Zhang, D. Huang, L. Qin, H. Yi, X. Liu, F. Huang, X. Zhou and L. Chen, Chem. Eng. J., 2020, 384, 123304 CrossRef CAS.
- H. Wang, C. Zhang, Z. Chen, H. K. Liu and Z. Guo, Carbon, 2015, 81, 782–787 CrossRef CAS.
- X. Liang, Z. Wen, Y. Liu, H. Zhang, L. Huang and J. Jin, J. Power Sources, 2011, 196, 3655–3658 CrossRef CAS.
- H. I. Lee, S. H. Joo, J. H. Kim, D. J. You, J. M. Kim, J. Park, H. Chang and C. Pak, J. Mater. Chem., 2009, 19, 5934 RSC.
- J. G. Kim, B. Lee, N. N. T. Pham, S. G. Lee and C. Pak, J. Ind. Eng. Chem., 2020, 89, 361–367 CrossRef CAS.
- D. D. Chen, Z. Q. He, M. Wang, D. Wu, X. Y. Chen and Z. J. Zhang, J. Solid State Electrochem., 2020, 24, 641–654 CrossRef CAS.
- X. Zhao, A. Wang, J. Yan, G. Sun, L. Sun and T. Zhang, Chem. Mater., 2010, 22, 5463–5473 CrossRef CAS.
- X. Zhai, Y. Song, J. Liu, P. Li, M. Zhong, C. Ma, H. Wang, Q. Guo and L. Zhi, J. Electrochem. Soc., 2012, 159, 177–182 CrossRef.
- X. Zhao, Q. Zhang, B. Zhang, C. Chen, J. Xu, A. Wang, D. S. Su and T. Zhang, RSC Adv., 2013, 3, 3578 RSC.
- D. Wang, F. Li, Z. Chen, G. Q. Lu and H. Cheng, Chem. Mater., 2008, 20, 7195–7200 CrossRef CAS.
- J. Song, Y. Zhang and Y. Liu, RSC Adv., 2015, 5, 20734–20740 RSC.
- N. Daems, F. Risplendi, K. Baert, A. Hubin, I. F. J. Vankelecom, G. Cicero and P. P. Pescarmona, J. Mater. Chem. A, 2018, 6, 13397–13411 RSC.
- F. Sun, Z. Qu, J. Gao, H. B. Wu, F. Liu, R. Han, L. Wang, T. Pei, G. Zhao and Y. Lu, Adv. Funct. Mater., 2018, 28, 1804190 CrossRef.
- T. Wang, C. Zhang, X. Sun, Y. Guo, H. Guo, J. Tang, H. Xue, M. Liu, X. Zhang, L. Zhu, Q. Xie and J. He, J. Power Sources, 2012, 212, 1–12 CrossRef CAS.
- J. Zhou, K. Wu, W. Wang, Z. Xu, H. Wan and S. Zheng, Appl. Catal., A, 2014, 470, 336–343 CrossRef CAS.
- S. Ding, S. Zheng, M. Xie, L. Peng, X. Guo and W. Ding, Microporous Mesoporous Mater., 2011, 142, 609–613 CrossRef CAS.
- X. Bo and L. Guo, Phys. Chem. Chem. Phys., 2013, 15, 2459–2465 RSC.
- D. Wang, F. Li, Z. Chen, G. Q. Lu and H. Cheng, Chem. Mater., 2008, 20, 7195–7200 CrossRef CAS.
- Y. Zhang, F. Qi and Y. Liu, RSC Adv., 2020, 10, 11210–11218 RSC.
- A. Nsabimana, X. Bo, Y. Zhang, M. Li, C. Han and L. Guo, J. Colloid Interface Sci., 2014, 428, 133–140 CrossRef CAS PubMed.
- N. Wickramaratne and M. Jaroniec, RSC Adv., 2012, 2, 1877 RSC.
- J. Y. Cheon, J. H. Kim, J. H. Kim, K. C. Goddeti, J. Y. Park and S. H. Joo, J. Am. Chem. Soc., 2014, 136, 8875–8878 CrossRef CAS PubMed.
- D. Zhang, L. Lei, Y. Shang, K. Wang and Y. Wang, Appl. Surf. Sci., 2016, 360, 807–815 CrossRef CAS.
- U. Byambasuren, Y. Jeon, D. Altansukh and Y. Shul, J. Solid State Electrochem., 2016, 20, 645–655 CrossRef CAS.
- C. Deng, H. Zhong, X. Li, L. Yao and H. Zhang, Nanoscale, 2016, 8, 1580–1587 RSC.
- Y. Chang, F. Hong, J. Liu, M. Xie, Q. Zhang, C. He, H. Niu and J. Liu, Carbon, 2015, 87, 424–433 CrossRef CAS.
- G. Zhao, L. Shi, J. Xu, X. Yan and T. S. Zhao, Int. J. Hydrogen Energy, 2018, 43, 1470–1478 CrossRef CAS.
- D. Zhang, L. Lei and Y. Shang, J. Mater. Sci.: Mater. Electron., 2016, 27, 3531–3539 CrossRef CAS.
- K. Li, S. Jie, Y. Li, X. Lin and Z. Liu, Catal. Commun., 2018, 112, 39–42 CrossRef CAS.
- J. Kim, M. Choi and R. Ryoo, Bull. Korean Chem. Soc., 2008, 29, 413–416 CrossRef CAS.
- Y. Kim, I. Kim, B. Min and S. Park, Res. Chem. Intermed., 2010, 36, 703–713 CrossRef CAS.
- W. Kim, M. Y. Kang, J. B. Joo, N. D. Kim, I. K. Song, P. Kim, J. R. Yoon and J. Yi, J. Power Sources, 2010, 195, 2125–2129 CrossRef CAS.
- T. Lin, I. W. Chen, F. Liu, C. Yang, H. Bi, F. Xu and F. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS PubMed.
- D. Qu, J. Wen, D. Liu, Z. Xie, X. Zhang, D. Zheng, J. Lei, W. Zhong, H. Tang, L. Xiao and D. Qu, ACS Appl. Mater. Interfaces, 2014, 6, 22687–22694 CrossRef CAS PubMed.
- L. Fan, P. Sun, L. Yang, Z. Xu and J. Han, Korean J. Chem. Eng., 2020, 37, 166–175 CrossRef CAS.
- X. Xin, Z. Wang, R. Jia, C. Gao, L. Sui, H. Dong, J. Feng, S. Ma, B. Pang, Y. Chen, L. Dong and L. Yu, J. Alloys Compd., 2020, 822, 153627 CrossRef CAS.
- Z. Liu, J. Mi, Y. Yang, X. Tan and C. Lv, Electrochim. Acta, 2014, 115, 206–215 CrossRef CAS.
- X. Zhao, Q. Zhang, B. Zhang, C. Chen, A. Wang, T. Zhang and D. S. Su, J. Mater. Chem., 2012, 22, 4963 RSC.
- D. Zhang, L. Zheng, Y. Ma, L. Lei, Q. Li, Y. Li, H. Luo, H. Feng and Y. Hao, ACS Appl. Mater. Interfaces, 2014, 6, 2657–2665 CrossRef CAS PubMed.
- G. Liu, X. Li, P. Ganesan and B. N. Popov, Appl. Catal., B, 2009, 93, 156–165 CrossRef CAS.
- X. Wang, J. S. Lee, Q. Zhu, J. Liu, Y. Wang and S. Dai, Chem. Mater., 2010, 22, 2178–2180 CrossRef CAS.
- H. Wang, W. Wang, M. Asif, Y. Yu, Z. Wang, J. Wang, H. Liu and J. Xiao, Nanoscale, 2017, 9, 15534–15541 RSC.
- N. Liu, H. Song and X. Chen, J. Mater. Chem., 2011, 21, 5345 RSC.
- H. Wang, X. Bo, Y. Zhang and L. Guo, Electrochim. Acta, 2013, 108, 404–411 CrossRef CAS.
- L. Wang, W. Jia, X. Liu, J. Li and M. M. Titirici, J. Energy Chem., 2016, 25, 566–570 CrossRef.
- S. Chen, Y. Zhai, G. Xu, Y. Jiang, D. Zhao, J. Li, L. Huang and S. Sun, Electrochim. Acta, 2011, 56, 9549–9555 CrossRef CAS.
- Z. Li, Y. Jiang, L. Yuan, Z. Yi, C. Wu, Y. Liu, P. Strasser and Y. Huang, ACS Nano, 2014, 8, 9295–9303 CrossRef CAS PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- Z. Ding, D. Zhao, R. Yao, C. Li, X. Cheng and T. Hu, Int. J. Hydrogen Energy, 2018, 43, 10502–10510 CrossRef CAS.
- J. Zheng, G. Guo, H. Li, L. Wang, B. Wang, H. Yu, Y. Yan, D. Yang and A. Dong, ACS Energy Lett., 2017, 2, 1105–1114 CrossRef CAS.
- Y. Qu, Z. Zhang, X. Zhang, G. Ren, Y. Lai, Y. Liu and J. Li, Carbon, 2015, 84, 399–408 CrossRef CAS.
- Y. Fang, R. Liu, L. Zeng, J. Liu, L. Xu, X. He, B. Huang, Q. Chen, M. Wei and Q. Qian, Electrochim. Acta, 2019, 318, 737–745 CrossRef CAS.
- X. Sheng, N. Daems, B. Geboes, M. Kurttepeli, S. Bals, T. Breugelmans, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B, 2015, 176–177, 212–224 CrossRef CAS.
- L. Wang, J. Sun, H. Zhang, L. Xu and G. Liu, J. Electroanal. Chem., 2020, 868, 114196 CrossRef CAS.
- K. K. R. Datta, V. V. Balasubramanian, K. Ariga, T. Mori and A. Vinu, Chem.–Eur. J., 2011, 17, 3390–3397 CrossRef CAS PubMed.
- K. Liang, C. Ma, L. Liu, Y. Yu, H. Lv, Y. Zhang and A. Chen, J. Alloys Compd., 2019, 805, 859–867 CrossRef CAS.
- H. Guo, D. Wen, T. Wang, X. Fan, L. Song, H. Gong, W. Xia, B. Gao, L. Li and J. He, J. Porous Mater., 2019, 26, 371–379 CrossRef CAS.
- S. Liu, P. Zuo, Y. Wang, X. Li, W. Zhang, S. Xu, P. Huo and Z. Li, Microporous Mesoporous Mater., 2018, 259, 54–59 CrossRef CAS.
- K. Kwon, S. Jin, C. Pak, H. Chang, S. H. Joo, H. I. Lee, J. H. Kim and J. M. Kim, Catal. Today, 2011, 164, 186–189 CrossRef CAS.
- Y. Zhang, W. W. Dai, Y. J. Liu and B. G. Ma, RSC Adv., 2017, 7, 8250–8257 RSC.
- H. Wang, J. Ding, J. Zhang, C. Wang, W. Yang, H. Ren and A. Kong, RSC Adv., 2016, 6, 79928–79933 RSC.
- C. Weinberger, S. Ren, M. Hartmann, T. Wagner, D. Ş. Karaman, J. M. Rosenholm and M. Tiemann, ACS Appl. Nano Mater., 2017, 1, 455–462 CrossRef.
- C. Zhang, Z. Zhang, D. Wang, F. Yin and Y. Zhang, J. Alloys Compd., 2017, 714, 126–132 CrossRef CAS.
- Y. E. Song, S. Lee, M. Kim, J. Na, J. Lee, J. Lee and J. R. Kim, J. Power Sources, 2020, 451, 227816 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |