DOI:
10.1039/D0RA08609F
(Paper)
RSC Adv., 2021,
11, 1164-1171
Design, growth, and characterization of Y2Mo4O15 crystals for Raman laser applications†
Received
9th October 2020
, Accepted 19th November 2020
First published on 4th January 2021
Abstract
A new crystal Y2Mo4O15 with dimensions of 14 × 12 × 5 mm3 was successfully grown via a top-seeded solution growth (TSSG) method. The crystal structure shows that Y2Mo4O15 crystallizes in the monoclinic space group P21/c (No. 14, a = 6.8110(4) Å, b = 9.5833(6) Å, c = 10.5124(7) Å, β = 105.512(7)°, and Z = 2) with Mo4O15 and YO7 polyhedra as basic structural units. Optical transmittance spectra of the Y2Mo4O15 crystal exhibited a broad transmission range from 345 nm to 5575 nm. The group theory calculation and spontaneous Raman spectra show that the Y2Mo4O15 crystal has 63 IR-active modes (32Au + 31Bu) and 60 Raman-active modes (30Ag + 30Bg). The strongest Raman shift is located at 953 cm−1 caused by the asymmetric stretching vibrations of the Mo–O bonds. The spontaneous Raman spectra and the possibility of rare-earth doping to the Y2Mo4O15 crystal indicate the Y2Mo4O15 crystal to be a promising Raman and self-Raman crystal.
Introduction
Stimulated Raman Scattering (SRS) provides an efficient and simple method for broad laser sources. A lot of crystals such as diamond,1 LiIO3,2 Ba(NO3)2,3 and YVO4 (ref. 4) have been studied for Raman laser applications. In order to simplify the laser configuration, the self-Raman laser for rare-earth doped crystals, including Nd:YVO4,5 Nd:GdVO4,6 and Yb:KGd(WO4)2,7 have been applied as laser and SRS crystals simultaneously. Recently, both the molybdate and tungstate, such as CaMoO4,8 SrMoO4,9 BaTeMo2O9,10,11 BaWO4,12 SrWO4,13 and CaWO4,8 have been paid attention owing to their excellent SRS properties. Although the above molybdate and tungstate crystals exhibit excellent Raman property, they are difficult for rare-earth doping due to the mismatch of their cation valence. It is well-known that the Raman spectra are mainly determined by anionic groups. The Raman shifts located at 872–930 cm−1, which are valuable for Raman laser, are due to the Mo–O vibrations. Therefore, molybdate crystals with trivalent cation possess good Raman properties, making them very promising hosts of the active dopants for self-Raman materials.
Y2Mo4O15 belongs to the monoclinic system in the space group P21/c with a = 6.8185(2) Å, b = 9.5913(3) Å, c = 10.5299(3) Å, β = 105.586(2)°, and Z = 2.14 The Y2Mo4O15 and rare-earth doped Y2Mo4O15 powders have been synthesized, and special attention is paid to the investigation of these systems, which are used as luminescent host materials for white LEDs.15–19 It is well known that the Y sites are easily accessible for various rare-earth ion substitutions. Therefore, it allows the production of active solid state self-Raman lasers with effective doping of numerous rare-earth ions.
In this study, for the first time, the Y2Mo4O15 crystal was grown by the top seed solution growth method. The crystal structure, optical properties, and Raman spectra have been studied in detail. The lattice vibrations of the Y2Mo4O15 crystal were analysed by the space group theory. The experimental results and theoretical calculations indicate that the Y2Mo4O15 crystal is a candidate for Raman and self-Raman crystals.
Experimental
Synthesis
Y2Mo4O15 powders were synthesized by a high temperature solid-state reaction method. The stoichiometric amounts of Y2O3 (99.99%, Alfa-Aesar) and MoO3 (99.5%, Alfa-Aesar) were thoroughly mixed in an agate mortar and then pressed into a pellet. The pellet was transferred into a platinum crucible in a programmable muffle furnace. After that, the column was preheated to 650 °C at a rate of 10 °C min−1 and then heated at a rate of 2 °C min−1 from 650 °C to 860 °C gradually with a holding time of 72 h. Several intermittent re-grindings were performed until the pure powders were obtained.
Powder X-ray diffraction (PXRD)
An automated Bruker D8 ADVANCE X-ray diffractometer equipped with a diffracted monochromator set for Cu Kα (λ = 1.5418 Å) radiation was used to measure the XRD patterns. The measurements were performed in the angular range (2θ) from 10° to 90°, with a scanning step width of 0.02° and a scanning speed of 20° min−1.
Thermal stability
A series of thermogravimetric and differential scanning calorimetry (TG-DSC) had been done on ground Y2Mo4O15 polycrystals in a shielded nitrogen environment using a TGA/DSC1/1600HT (Mettler Toledo Instruments).
Single-crystal growth
After several attempts, some Y2Mo4O15 crystal seeds were obtained for the first time by a spontaneous nucleation method using Li2CO3–MoO3 as a flux. In the crystallization region, a reaction mixture of Li2CO3, MoO3, and Y2O3 at the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:2 was adequately ground and then placed in a platinum crucible. The reaction mixture was subsequently heated to 960 °C and maintained at that temperature for 96 h to guarantee an entirely homogeneous solution. A platinum wire was dipped into the surface of the solution at 960 °C, and then at a rate of 5 °C h−1, the temperature was decreased to 776 °C to obtain the millimetre-sized crystals by spontaneous nucleation. A single crystal of Y2Mo4O15 was obtained via the top seeded solution growth (TSSG) technique. In addition, the saturation temperature of the solution was obtained by the testing seed crystal method. A regular and transparent crystal seed was gradually brought into the solution at 2 °C above the saturation temperature, and was maintained for an hour to melt slightly surface impurities. After that, the temperature was rapidly reduced to the saturation temperature, and then a fairly slow cooling program of approximately 0.02 °C d−1 was adopted to carry out the growth procedure. When the sizable Y2Mo4O15 crystal was harvested, the furnace temperature was cooled at a rate ranging from 30 to 50 °C h−1.
:8:2 was adequately ground and then placed in a platinum crucible. The reaction mixture was subsequently heated to 960 °C and maintained at that temperature for 96 h to guarantee an entirely homogeneous solution. A platinum wire was dipped into the surface of the solution at 960 °C, and then at a rate of 5 °C h−1, the temperature was decreased to 776 °C to obtain the millimetre-sized crystals by spontaneous nucleation. A single crystal of Y2Mo4O15 was obtained via the top seeded solution growth (TSSG) technique. In addition, the saturation temperature of the solution was obtained by the testing seed crystal method. A regular and transparent crystal seed was gradually brought into the solution at 2 °C above the saturation temperature, and was maintained for an hour to melt slightly surface impurities. After that, the temperature was rapidly reduced to the saturation temperature, and then a fairly slow cooling program of approximately 0.02 °C d−1 was adopted to carry out the growth procedure. When the sizable Y2Mo4O15 crystal was harvested, the furnace temperature was cooled at a rate ranging from 30 to 50 °C h−1.
Single-crystal X-ray diffraction
Crystallographic data were collected on a Bruker AXS SMART diffractometer with monochromatic Mo Kα radiation (λ = 0.71073 Å) at room temperature.20 The data integration and unit cell refinement were determined by the INTEGRATE program of the standard APEX II software, and absorption corrections were performed with the SCALE program for the area detector.20 The structure was solved directly by Fourier synthesis and then refined according to the full-matrix least square techniques fitting on F02 using the SHELXTL program.21 The details of the crystal structure (CCDC 2027158) can be obtained.
Laue back-reflection measurement
The back-reflection Laue technique is widely used for orientation and crystallinity assessment of a single crystal.22 A crystal facet (100) slice of Y2Mo4O15, mechanically polished on both sides, whose dimensions are 4 × 4 × 1 mm3, was employed for the Laue diffraction measurement to assess the crystalline perfection by a real time back-reflection Laue camera system (Multiwire MWL 120 with Northstar software).
UV-visible diffuse reflectance spectra
UV-vis diffuse reflectance measurements of Y2Mo4O15 were performed on a Shimadzu UV 2550 spectrophotometer equipped with an integrating sphere, and the baseline correction was performed using a calibrated reference sample of powdered barium sulfate (BaSO4) over the wavelength range extending from 200 to 800 nm. The band-gap energy of Y2Mo4O15 was calculated according to the Tauc expression.23 A thin {100} crystal plate (4 × 4 × 1 mm3) was cut from the as-grown Y2Mo4O15 crystal. The optical transmittance spectrum in the visible and near IR range (0.2–2 μm) was collected on a Hitachi U-3500 UV-vis-IR spectrometer, and the spectra in the mid-IR (2–6 μm) region were recorded on a Nicolet NEXUS 670 FTIR spectrometer.
X-ray photoelectron spectra
X-ray photoelectron spectra (XPS) were recorded using a Thermo Fisher ESCALAB 250 X-ray photoelectron spectrometer with monochromatic Al Kα X-ray radiation. The binding energy of the samples was calibrated using the C 1s peak (284.6 eV) as a reference. The spectra were successfully deconvoluted using a Gaussian–Lorentzian curve fitting program after background subtraction.
Spontaneous Raman spectroscopy
The spontaneous Raman spectra of Y2Mo4O15 were recorded at room temperature using 1064 nm radiation as the pump source using a Jobin Yvon T64000 spectrometer with dimensions of 4 mm × 4 mm × 1 mm3 square slice as a sample. Raman spectra with 2 cm−1 spectral resolution in the frequency range of 0 to 1100 cm−1 were collected.
Computational details
Ab initio density functional theory calculations were performed using the Vienna Ab initio simulation package (VASP) code.24 The Perdew–Burke–Ernzerhof (PBE) function25 was employed to calculate the exchange-correlation potential under the generalized gradient approximation (GGA).26 A kinetic energy cut-off of 500 eV was used for the plane wave basis set. The Brillouin zone sampling was carried out with a 7 × 7 × 5 Monkhorst–Pack grid. Convergence criteria employed for both the electronic self-consistent relaxation and ionic relaxation are set to be 10−8 and 10−4 eV Å−1 for energy and force, respectively. Gaussian broadening was implemented with a smearing width of 0.05 eV. The calculations of the phonon spectrum and phonon density of states were performed using a 2 × 2 × 1 supercell in the framework of the density functional perturbation theory (DFPT).27 Force-constants supercell has been obtained using the VASP code. The PHONOPY code28 has been used to calculate the phonon frequencies and phonon density of states.
Results and discussion
Synthesis and characterization of polycrystalline
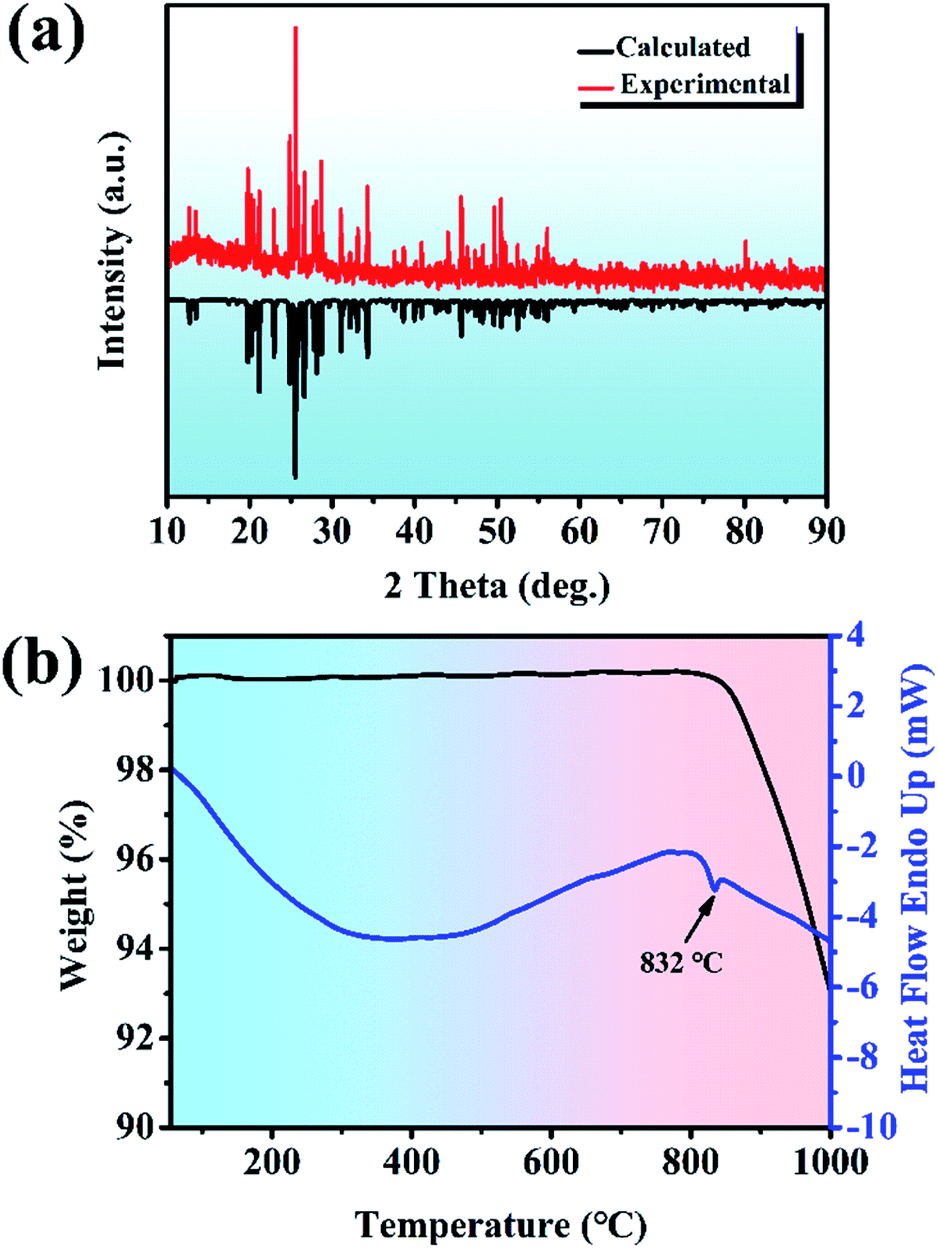
Y2Mo4O15. The Y2Mo4O15 polycrystalline materials were synthesized by the traditional solid-state reaction. XRD patterns of Y2Mo4O15 are depicted in Fig. 1a, together with the reference pattern of Y2Mo4O15 for comparison, which indicates that the as-prepared samples are in the pure phase. DSC and TGA measurements were carried out to measure the thermal stability of the Y2Mo4O15 crystal (shown in Fig. 1b). There is a single evident endothermic peak at 832 °C. It indicates that Y2Mo4O15 potentially decomposes before its melting point. The PXRD detection of the solid residue after heating at 900 °C for an hour revealed that Y2Mo4O15 decomposed to YMoO4. This evidence indicates that Y2Mo4O15 is an incongruent melting compound. Accordingly, large-sized Y2Mo4O15 crystals should be grown via a high-temperature flux method.
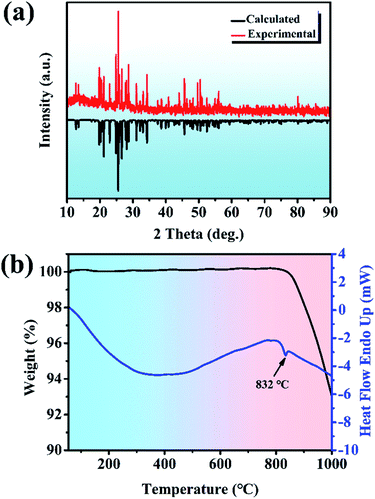 |
| | Fig. 1 (a) Experimental PXRD patterns for polycrystalline Y2Mo4O15 (pure-phase) and calculated PXRD patterns for polycrystalline Y2Mo4O15. (b) DSC and TGA curves for polycrystalline Y2Mo4O15. | |
Single-crystal growth
Flux growth is worthwhile to pursue because it readily allows the crystal growth at a temperature below the melting point. In addition, the crystal grown from flux has a regular morphology and low dislocation density. Therefore, the single crystal Y2Mo4O15 was grown with the TSSG method. The selection of the flux system is extremely crucial to the growth of high-quality crystals. MoO3 with a low melting point (793 °C) and outstanding solubility has been widely used as a self-fluxing agent to grow new oxide crystals.29 Moreover, a special effect of Li+ cations potentially makes the grown crystal surface more visible.30 Using the Li2CO3–MoO3 flux (molar ratio, Li2CO3:MoO3 = 1:8), Y2Mo4O15 was prone to crystallization. Taking advantage of the seed crystals with suitable size and quality obtained by spontaneous crystallization, a colorless and transparent single Y2Mo4O15 crystal with a size of 14 × 12 × 5 mm3 was successfully grown by the TSSG method (see Fig. 2). More importantly, this method is available to achieve the crystal growth at a temperature much lower than its decomposition point.31
 |
| | Fig. 2 Photograph of the as-grown Y2Mo4O15 crystal. | |
Crystal structure of Y2Mo4O15
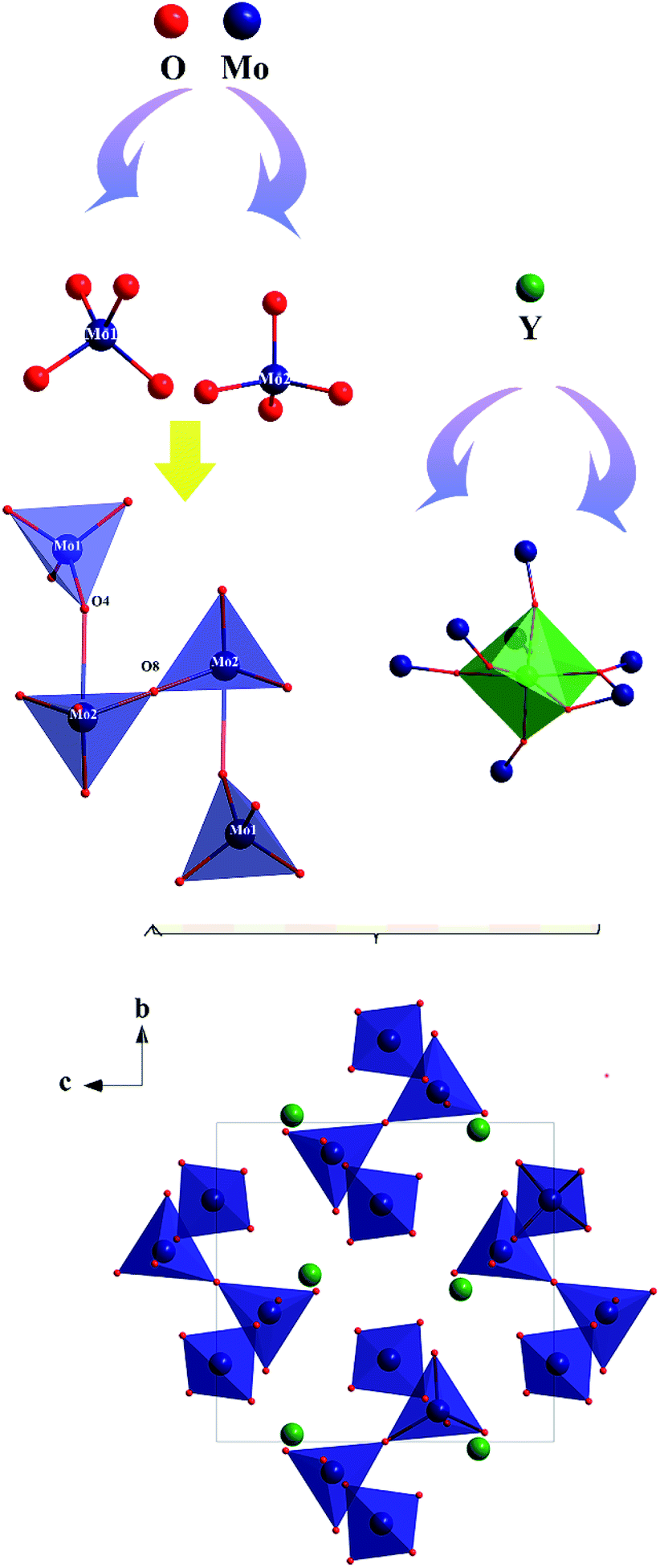
The preliminary crystallographic information on a polycrystalline sample of Y2Mo4O15 is reported. To provide better insights into the crystal structure, single crystal X-ray diffraction data of Y2Mo4O15 were determined at 293 K in detail. It crystallizes in the monoclinic system with the space group of P21/c (No. 14). The refined crystallographic data and details of the experimental conditions of Y2Mo4O15 are given in Tables S1–S3 of the ESI.† According to the structure (see Fig. 3), there are two different molybdate units in the present structure. Mo(1) is surrounded by four oxygen atoms to form a fairly regular Mo(1)O4 tetrahedron. For Mo(2), except for the four nearest coordinated oxygen atoms, two Mo(2)O4 tetrahedra are connected by O(8) to form a Mo(2)2O7 pyromolybdate, in which the bridging O(8) atom between the two vertex-shared MoO4 tetrahedra reside on an inversion center. In addition, Mo(2)2O7 pyromolybdate is loosely connected to two Mo(1)O4 tetrahedra and forms an entire Mo4O15 unit, as shown in Fig. 3. Furthermore, the anionic environment of the crystallographically distinct Y3+ position is coordinated with seven oxygen atoms (CN = 7) to form a singly capped trigonal prism with the Y–O distances ranging from 0.247 nm to 0.355 nm (see Table S2†). The crystal structure is consistent with the result reported by Laufer et al.14 Due to the flexibility of coordination geometry for the Y3+ center,32 the Y2Mo4O15 compounds are isomorphous with the previously reported Ln2Mo4O15 (Ln = Y, Dy, Ho, and Tm),33 which means doping with rare-earth ions is easy for the Y3+ position. In addition, MoO4 tetrahedron and YO7 polyhedron active vibrations laid the theoretical foundation for Raman scattering.
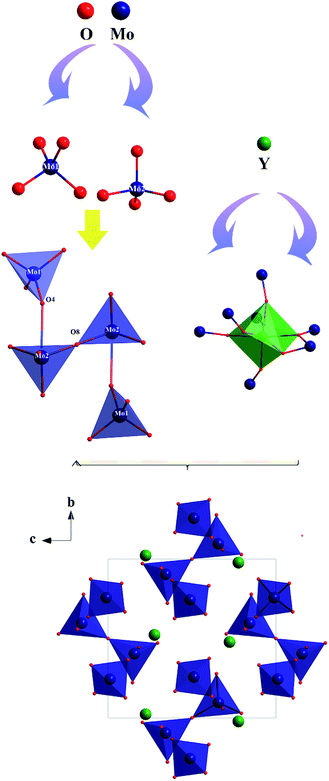 |
| | Fig. 3 Ball-and-stick diagram of the Y2Mo4O15 crystal structure. | |
Laue back-reflection patterns
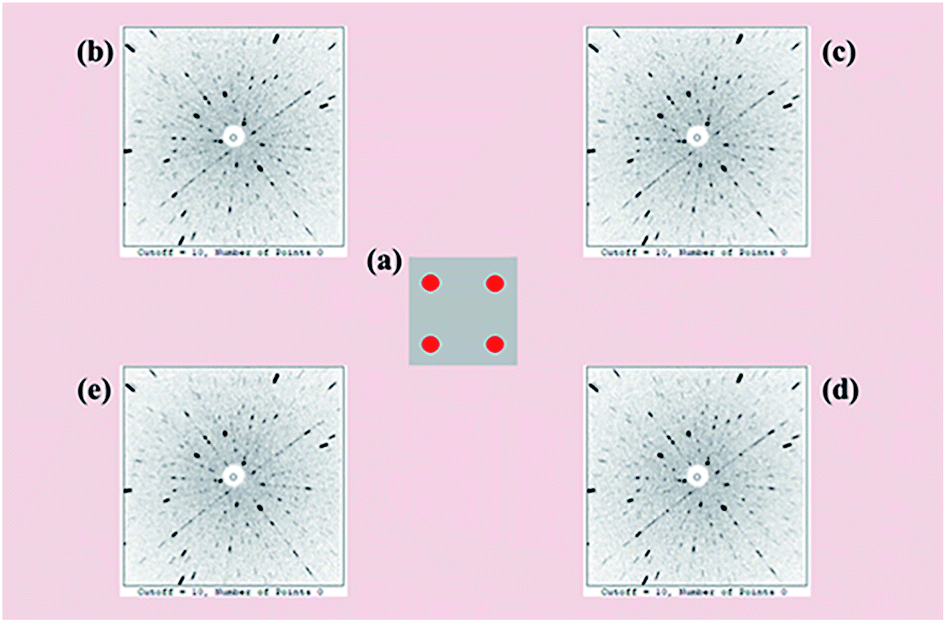
To facilitate a precise investigation of the crystallinity of an as-grown crystal, the Laue back-reflection measurement is broadly exploited.34 The characteristic Laue back-reflection patterns of different positions in the (100) crystal wafer are accordant and distinct, as shown in Fig. 4. It demonstrates that the crystal quality of the Y2Mo4O15 crystal is high enough, which furnishes the basis for performing the measurements and evaluations of intrinsic physical properties.
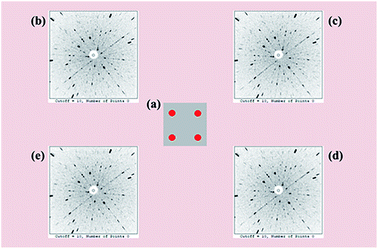 |
| | Fig. 4 (a) Schematic positions (red dots) of the Laue back-reflection measurement on the crystal piece. From clockwise rotation (b–e), characteristic Laue back-reflection patterns at different positions with the X-ray beam hitting the crystal piece. | |
Optical properties
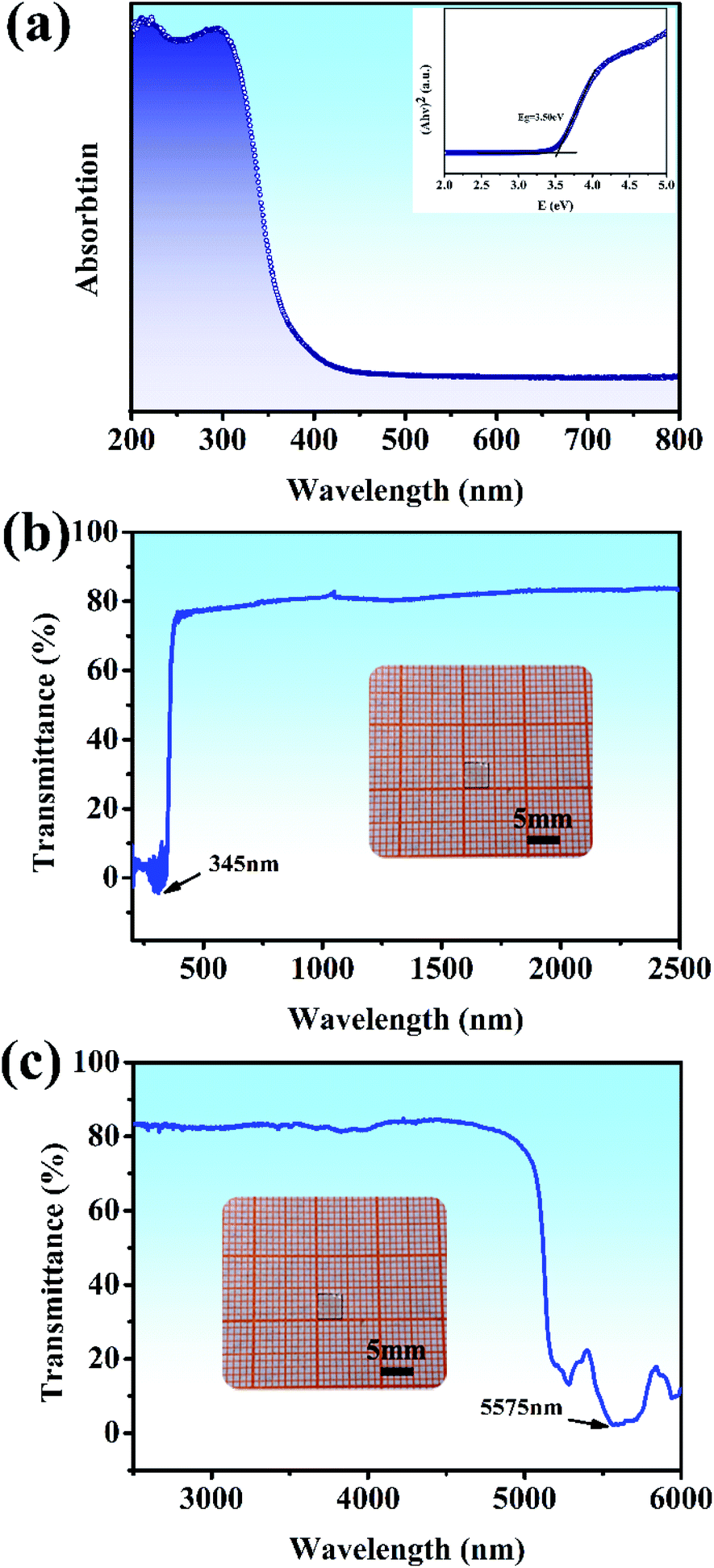
The UV-vis diffuse reflectance spectrum based on the ground powders of the Y2Mo4O15 crystal is illustrated in Fig. 5a. As presented in Fig. 5a (inset), the indirect band gap Eg of Y2Mo4O15 is estimated to be about 3.50 eV by fitting the corresponding Tauc plot of the Kubelka–Munk transformed UV-vis diffuse reflection data,35 which is similar to the previous work.16 The optical transmission spectra (Fig. 5b and c) of the Y2Mo4O15 crystal wafer (Fig. 5b and c, inset) were performed at room temperature. The UV λcut-off of Y2Mo4O15 is located at 345 nm, which is more reliable than that of the powdered Y2Mo4O15 (334 nm).16 In the mid-IR range, the Y2Mo4O15 crystal displays high transparency up to 5.1 μm with an absorption edge of 5.575 μm. The Y2Mo4O15 crystal exhibits a wider transparent window than that of ternary molybdate and tungstate, such as SrMoO4 (0.31–3 μm), BaWO4 (0.26–3.7 μm), and SrWO4 (0.3–2.7 μm). These results indicate that Y2Mo4O15 is a promising alternative for a large variety of Raman applications, which can be extended over a wide spectral range.
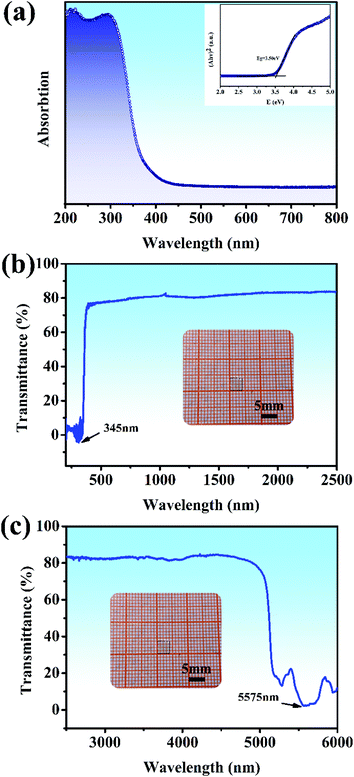 |
| | Fig. 5 (a) UV-visible diffuse reflectance spectra data for ground powders of the Y2Mo4O15 crystal. The inset shows the relationship between K/S and E (eV); (b) and (c) UV-vis and IR transmission spectra of the Y2Mo4O15 single crystal. Inset: Y2Mo4O15 crystal wafer of dimensions 4 × 4 × 1 mm3. | |
Electronic structure
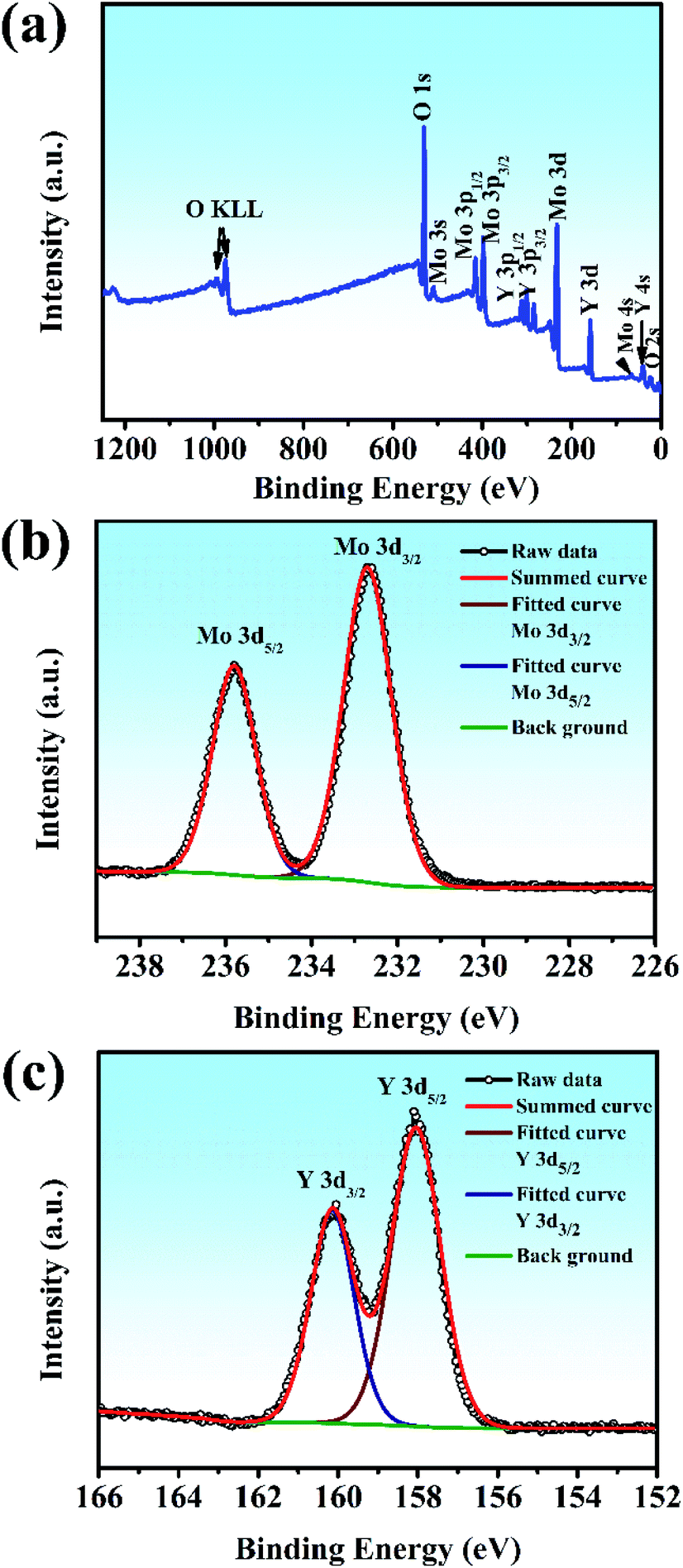
XPS was used to shed light on the surface composition and binding interactions of the Y2Mo4O15 crystal (Fig. 6). All the spectral features from the curves, except for the C 1s core level, can be attributed to the core-levels or Auger lines of the constituent elements of Y2Mo4O15. The high-resolution XPS spectra of Mo 3d and Y 3d core-level spectra correspond to the simple spin–orbit doublets. The binding energy values of the Mo 3d5/2 (235.8 eV), Mo 3d3/2 (232.7 eV) Y 3d3/2 (160.15 eV), and Y 3d5/2 (158.05 eV) core levels in Y2Mo4O15, as obtained by the present XPS measurement, are similar to the previous reports on Y2Mo4O15,36 and correspond to those of Y and Mo in the formal valence states 3+ and 6+. The results show that the Y2Mo4O15 crystal has stable chemical properties.
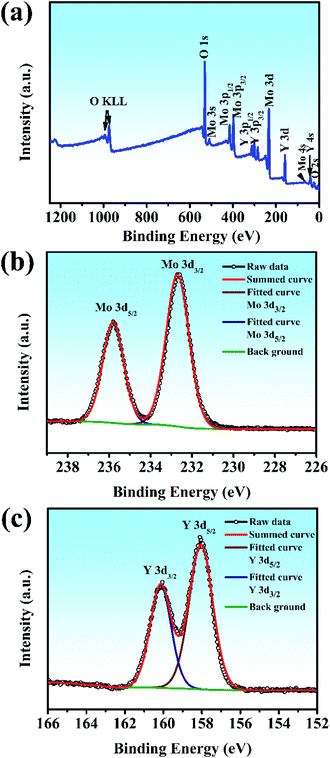 |
| | Fig. 6 (a) Survey X-ray photoelectron spectra of Y2Mo4O15 crystals. The different elements and their associated electronic states are denoted for respective peaks: high-resolution X-ray photoelectron spectra of (b) Mo 3d core level and (c) Y 3d core level. | |
Raman spectra
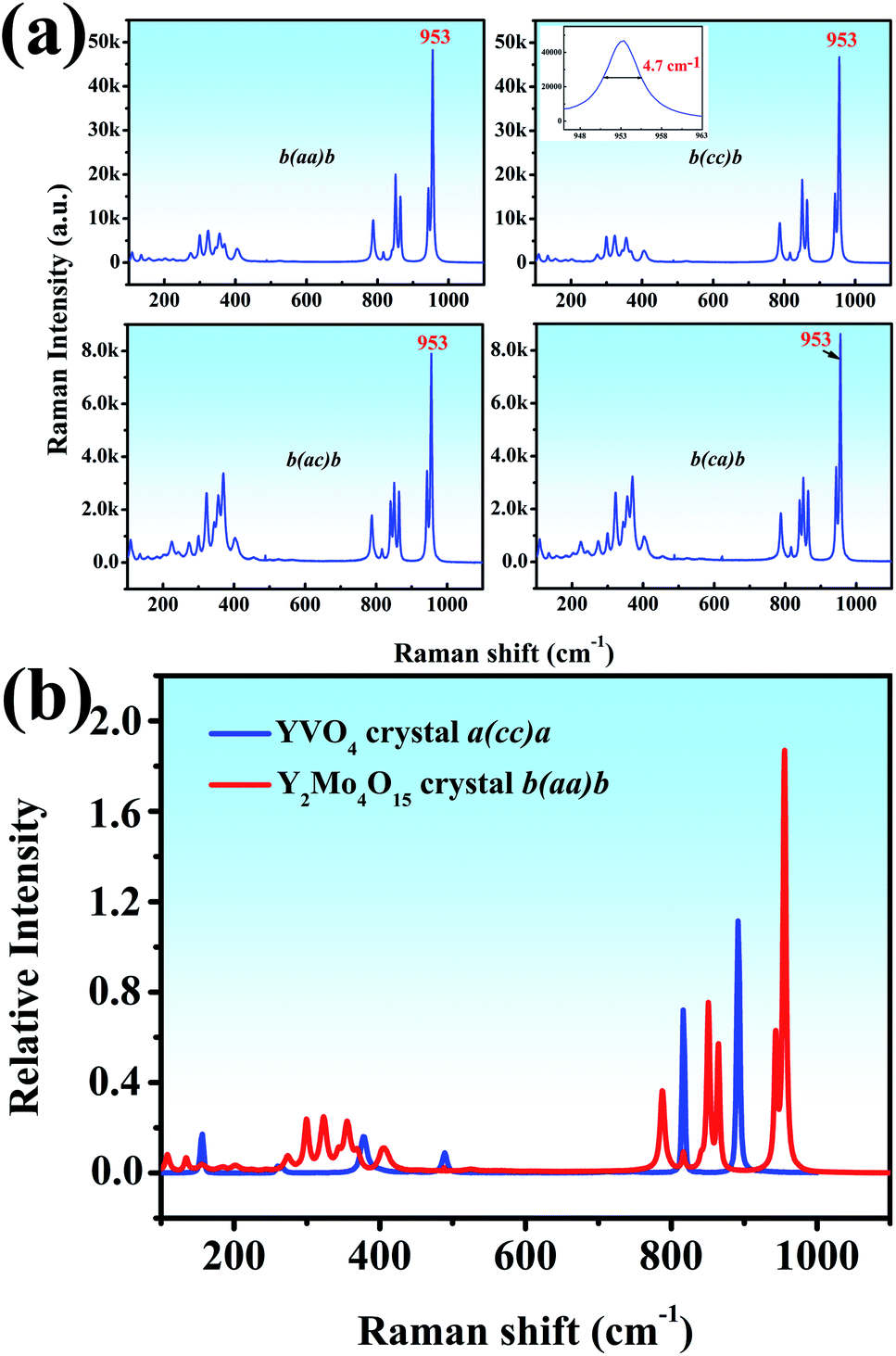
Both the polarized Raman spectrum and spontaneous Raman spectra are beneficial to the analysis of vibrational modes.37 We report the spontaneous Raman spectra of the Y2Mo4O15 single crystal along the b-axis with different Raman configurations, as shown in Fig. 7a. It can be seen that both b(aa)b and b(cc)b Raman configurations exhibit the strongest Raman shifts at 953 cm−1. It is well known that Raman spectra are mainly determined by anionic groups. As is shown in Fig. 7a (inset), the peak linewidth is about 4.7 cm−1, which is shorter than that of CaMoO4 (5.5 cm−1) but wider than that of BaMoO4 (1.85 cm−1).38
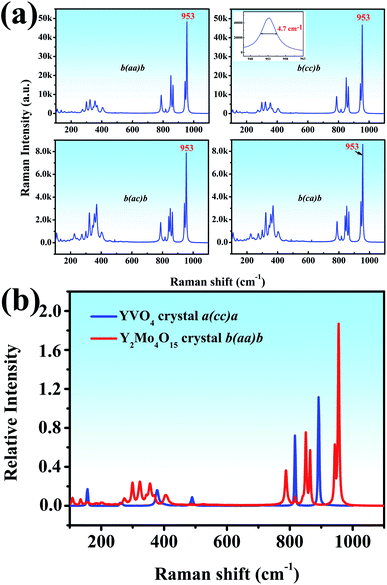 |
| | Fig. 7 (a) The spontaneous Raman spectra of Y2Mo4O15 along the b axis (b) relative spontaneous Raman spectra of YVO4 and Y2Mo4O15. | |
To evaluate the potential Raman property, the Raman shift intensity of Y2Mo4O15 crystal is at 953 cm−1 in the b(aa)b polarization configuration is compared to that of the YVO4 crystal. It is about 1.68 times than that of the Raman shift at 890 cm−1 in YVO4, which indicates excellent Raman scattering properties (Fig. 7b).
Raman spectral characteristics of P21/c symmetry
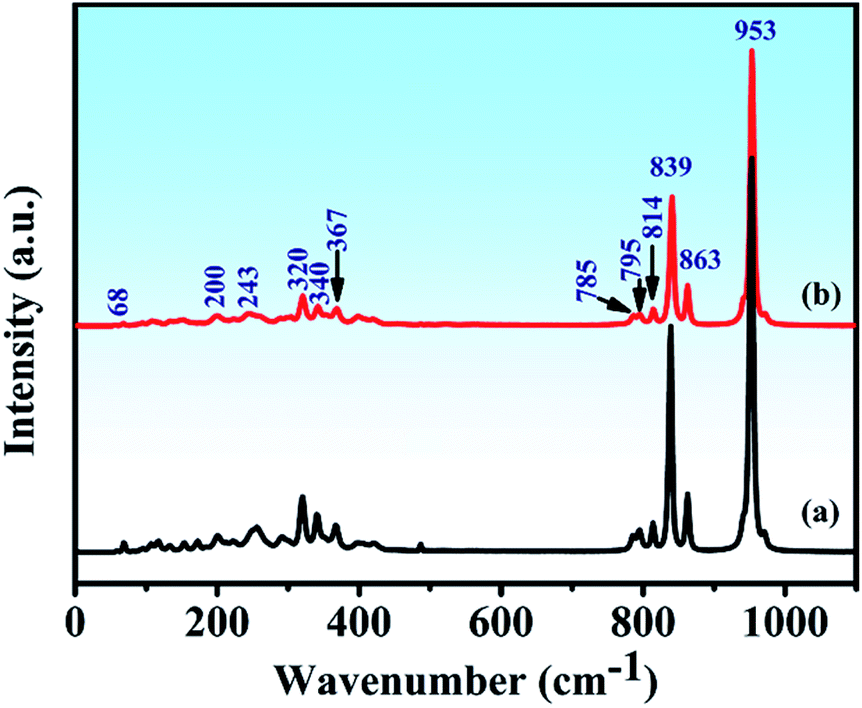
The Raman spectra and assignment of the lattice modes of Y2Mo4O15 have never been reported in previous studies. From space group theoretical considerations, the primitive cell of the P21/c structure of Y2Mo4O15 comprises two formula units and 24 atoms, and it can be described in terms of 72 vibrational degrees of freedom. Therefore, the total Brillouin zone centre vibrational modes can be classified according to the irreducible representation of the C2h point group of this material as Γtotal = Γacoustic + Γoptic = 30Ag + 33Au + 30Bg + 33Bu. The Ag and Bg modes are Raman active, and Au and Bu modes are IR active. It should be remembered, however, that three of these translational motions, Au + 2Bu, correspond to acoustic modes. These modes involve stretching and bending vibrations of the Mo–O bonds as well as librations of the Y–O polyhedral. The spontaneous Raman spectra of Y2Mo4O15 is shown in Fig. 8 (line b). As observed, twelve key Raman shifts are seen, which are 953 cm−1, 863 cm−1, 839 cm−1, 814 cm−1, 795 cm−1, 785 cm−1, 367 cm−1, 340 cm−1, 320 cm−1, 243 cm−1, 200 cm−1, and 68 cm−1. Most of the calculated frequencies in the calculated Raman spectrum (Fig. 8 (line a)) coincide well with the experimental spectra except for some weak peaks, such as 154 cm−1, 172 cm−1, 256 cm−1, and 292 cm−1 peaks. Particularly, the strongest Raman frequency shift is found to be at 953 cm−1, which is assigned to the Bg mode of asymmetric stretching vibration of the Mo–O bonds by the V_Sim software.
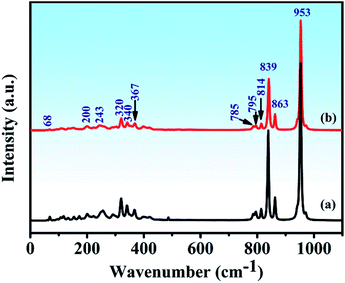 |
| | Fig. 8 (a) Calculated and (b) Experimental Raman spectra of Y2Mo4O15 crystal. | |
Phonon dispersion relations
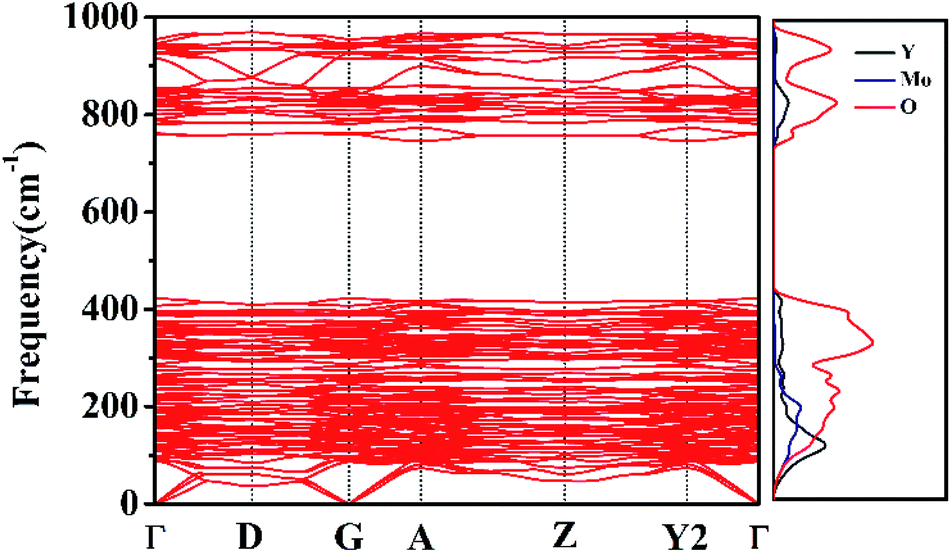
Phonon dispersion calculations were performed by sampling other k-points in the first Brillouin Zone other than Γ (k = 0) using a direct-space approach.39,40 Phonon dispersion curves of the monoclinic system in the path Γ→D→G→A→Z→Y2→Γ are reported in Fig. 9 for the 0–1000 cm−1 range, alongside the calculated total and atom-projected phonon density of states. From Fig. 9 (right), we can see the asymmetric (953 cm−1) and symmetric (863–785 cm−1) stretching regions. The total phonon DOS originates mainly from the Mo–O vibrations in accordance with the assignments for Y2Mo4O15.41 The spectral region between 367 cm−1 and 256 cm−1 corresponds to the bending motions. Y, Mo, and O atoms all take part in the vibrations in spite of the motions of Y–O being relatively small. The low frequency region (ν < 200 cm−1) is characterized by rotational motions. YO7 polyhedron and MO4 tetrahedron all contribute considerably to the total phonon DOS. They can together affect the Raman behaviour.
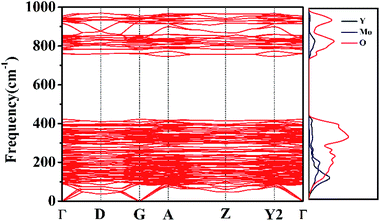 |
| | Fig. 9 Phonon dispersion curves and phonon density of states of Y2Mo4O15. | |
Conclusions
In summary, a new Raman crystal Y2Mo4O15 was grown using the TSSG method. The Y2Mo4O15 crystallizes in the monoclinic space group P21/c (No. 14) with Mo4O15 and YO7 polyhedra as the basic structural units. The experiments and theoretical calculations indicate that the Y2Mo4O15 crystal has the strongest Raman shift at 953 cm−1 caused by the asymmetric stretching vibration of the Mo–O bonds. Moreover, the crystal shows a wide transmission range from 345 nm to 5575 nm. Both the spontaneous Raman spectra and the possibility of rare-earth doping indicate that the Y2Mo4O15 crystal will be a promising Raman and self-Raman crystal.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2016YFB1102201), the 111 Project 2.0 (Grant No: BP2018013), the National Natural Science Foundation of China (Grant No. 51772170, 51572155 and 11504389), the Shandong Provincial Key R&D Program (2018CXGC0411), the Young Scholars Program (2018WLJH67), and the Fundamental Research Funds of Shandong University (2017JC044). We acknowledge Prof. Di Sun for assistance with collecting the single crystal X-ray diffraction data and crystal structure refinement.
Notes and references
- G. Eckhardt, D. P. Bortfeld and M. Geller, Appl. Phys. Lett., 1963, 3(8), 137–138 CrossRef CAS
 .
. - E. O. Ammann and C. D. Decker, J. Appl. Phys., 1977, 48(5), 1973–1975 CrossRef CAS .
- A. S. Eremenko, S. N. Karpukhin and A. Stepanov, Soviet Journal of Quantum Electronics, 1980, 10(1), 113–114 CrossRef .
- A. A. Kaminskii, K. Ueda, H. J. Eichler, Y. Kuwano and J. Lu, Laser Phys., 2001, 11(10), 1124–1133 CAS .
- Y. F. Chen, Appl. Phys. B: Lasers Opt., 2004, 78(6), 685–687 CrossRef CAS .
- P. Dekker, H. M. Pask, D. J. Spence and J. A. Piper, Opt. Express, 2007, 15(11), 7038–7046 CrossRef CAS .
- A. A. Lagatsky, A. Abdolv and N. V. Kuleshov, Opt. Lett., 2000, 25(9), 616–618 CrossRef CAS .
- S. P. S. PortoandJ and F. Scott, Phys. Rev., 1967, 157(3), 716–719 CrossRef .
- H. Yu, Z. Li, A. J. Lee, J. Li, H. Zhang, J. Wang, H. M. Pask, J. A. Piper and M. Jiang, Opt. Lett., 2011, 36(4), 579 CrossRef CAS .
- Z. L. Gao, S. D. Liu, J. J. Zhang, S. J. Zhang, W. G. Zhang, J. L. He and X. T. Tao, Opt. Express, 2013, 21(6), 7821 CrossRef CAS .
- Z. Gao, S. Liu, S. Zhang, W. Zhang, J. He and X. Tao, Appl. Phys. Lett., 2012, 100(26), 501 CrossRef .
- L. Fan, Y. Fan, Y. Li, H. Zhang, Q. Wang, J. Wang and H. Wang, Opt. Lett., 2009, 34(11), 1687–1689 CrossRef CAS .
- H. Yu, D. Hu, H. Zhang, Z. Wang, W. Ge, X. Xu, J. Wang, Z. Shao and M. Jiang, Opt. Laser Technol., 2007, 39(6), 1239–1242 CrossRef CAS .
- S. Laufer, S. Strobel, T. Schleid, J. Cybinska, A. V. Mudringand and I. Hartenbach, New J. Chem., 2013, 37(7), 1919–1926 RSC .
- Y. Wang, X. Liu, L. Jing and P. Niu, Ceram. Int., 2016, 42(11), 13004–13010 CrossRef CAS .
- I. Mackeviciute, A. Linkeviciute and A. Katelnikovas, J. Lumin., 2017, 190, 525–530 CrossRef CAS .
- W. Peng and T. Pang, Phys. B, 2018, 550, 145–153 CrossRef CAS .
- Z. Yu, D. Chen, H. Wang, J. Peng, Y. Cheng, M. Wu and Z. Hu, Rare Met., 2007, 26(5), 426–434 CrossRef CAS .
- H. Deng, N. Xue, Z. Hei, M. He, T. Wang, N. Xie and R. Yu, Opt. Mater. Express, 2015, 5(3), 490–496 CrossRef .
- APEX2. Bruker Analytical X-ray Instruments, Inc., Madison, WI, 2005 Search PubMed .
- G. M. Sheldrick, SHELXTL, version 6.12, Bruker Analytical X-ray Instruments, Inc., Madison, WI, 2001 Search PubMed .
- C. Marín, A. Cintas and E. Diéguez, J. Appl. Crystallogr., 1994, 27(5), 846–852 CrossRef .
- M. Khan, J. Xu, N. Chen and W. Cao, J. Alloys Compd., 2012, 513(1–4), 539–545 CrossRef CAS .
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59(3), 1758–1775 CrossRef CAS .
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS .
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100(13), 246–249 CrossRef .
- S. Baroni, S. De Gironcoli, A. D. Corso and P. Giannozzi, Physics, 2000, 73(2), 515–562 Search PubMed .
- A. Togo, F. Obaand and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78(13), 134106 CrossRef .
- P. Zhao, Q. Wu, Z. Gao, X. Tian, C. Li, Y. Sun, C. Zhang, S. Xia and X. Tao, RSC Adv., 2017, 7(8), 4278–4284 RSC .
- J. Wang, H. Zhang, M. Jiang, M. Nikitina and N. Leonyuk, Cryst. Growth Des., 2009, 9(2), 1190–1193 CrossRef CAS .
- D. E. Bugaris and H. C. Zur Loye, Angew. Chem., Int. Ed., 2012, 51(16), 3780–3811 CrossRef CAS .
- H. Naruke and T. Yamase, J. Solid State Chem., 2003, 173(2), 407–417 CrossRef CAS .
- L. Sebastian, S. Sumithra, J. Manjanna, A. M. Umarji and J. Gopalakrishnan, J. Mater. Sci. Eng. B, 2003, 103(3), 289–296 CrossRef .
- W. Bogdanowicz, J. Krawczyk and R. Paszkowski, et al., Materials, 2019, 12(24), 4126 CrossRef CAS .
- P. Kubelka and F. Munk, Ztschrift Fur Technische Physik, 1931, 12, 593–601 Search PubMed .
- J. You, R. Wang, C. Liu, X. Shi, F. Han, R. Guo and X. Liu, J. Rare Earths, 2018, 36(8), 844–850 CrossRef CAS .
- C. G. Li, Z. L. Gao, X. X. Tian, J. J. Zhang, D. X. Ju and X. T. Tao, CrystEngComm, 2019, 21, 2508–2516 RSC .
- T. T. Basiev, A. A. Sobol, Yu. K. Voronko and P. G. Zverev, Opt. Mater., 2000, 15(3), 205–216 CrossRef CAS .
- C. D. Wallace, Am. J. Phys., 1972, 40(11), 1718–1719 CrossRef .
- K. Parlinski, Z. Q. Li and Y. Kawazoe, Phys. Rev. Lett., 1997, 78(21), 4063–4066 CrossRef CAS .
- L. Wang, F. Wang, P. F. Yuan, Q. Sun, E.-J. Liang, Y. Jia and Z. X. Guo, Mater. Res. Bull., 2013, 48(7), 2724–2729 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures and tables (PDF); X-ray crystallographic data (CIF) available. CCDC 2027158. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra08609f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b and
Xutang Tao
b and
Xutang Tao