
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe spatial organization of trace silver atoms on a DNA template
Jinliang Maab,
Huawei Niua and
Shaobin Gu *a
*a
aCollege of Food and Bioengineering, Henan University of Science and Technology, Luoyang, Henan 471023, China. E-mail: shaobingu@haust.edu.cn
bInstitute of Molecular Medicine, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China
First published on 4th January 2021
Abstract
DNA with programmable information can be used to encode the spatial organization of silver atoms. Based on the primary structures of a DNA template containing a controllable base arrangement and number, the surrounding environment and cluster together can induce the folding of the DNA template into an appropriate secondary structure for forming AgNCs with different fluorescence emissions, such as i-motif, G-quadruplex, dimeric template, triplex, monomeric or dimeric C-loop, emitter pair, and G-enhancer/template conjugate. Stimuli can induce the dynamic structural transformation of the DNA template with a recognition site for favourably or unfavourably forming AgNCs, along with varied fluorescence intensities and colours. The array of several or more of the same and different clusters can be performed on simple and complex nanostructures, while maintaining their original properties. By sorting out this review, we systematically conclude the link between the performance of AgNCs and the secondary structure of the DNA template, and summarize the precise arrangement of nanoclusters on DNA nanotechnology. This clear review on the origin and controllability of AgNCs based on the secondary structure of the DNA template is beneficial for exploring the new probe and optical devices.
The high-order molecule provides an exquisite structural domain for the accurate and efficient operation of the living organism. The functionality of the molecule depends on the precise arrangement of atoms on the presupposed structure. Similar to the reaction in nature, in metal materials science, metal atoms aggregate into multiple nanoparticles with a defined composition, size, and shape to implement their functions for different applications, including catalysis,1,2 bioscience,3 photochemistry,4 optoelectronics,5 energy conversion6 and medicine.7 However, a key barrier in the field is the capability to precisely implement the absolute control over the placement and arrangement of each atom in the material framework. Certain biopolymer ligands, as scaffolds, encode a spatial map of atom binding sites and provide confined spaces for the growth of nanoparticles, allowing the tailored formation of metal nanoparticles. The number and arrangement of metal atoms on the coordinating ligand dictate the properties of synthesized nanoparticles. However, the surface chemistry and length of the conventional ligand are difficult to customize for meeting the material diversity requirements in the fields of photochemistry, medicine, and other areas.
Due to their clear thermodynamics and low synthesis cost, the facile DNA structure with controllable base arrangement can be used as a metal nanocluster synthesis scaffold to regulate the spatial position of metal atoms. The predefined DNA scaffold is capable of encoding variously fluorescent ligated silver nanoclusters (AgNCs),8–12 gold nanoclusters,13–15 and copper nanoclusters,16,17 conductive silver18 and palladium19–21 nanowires, and morphologically distinct gold22–24 or silver25 nanoparticles. In particular, the fluorescent atomically DNA scaffolded AgNCs (DNA/AgNCs) are endowed with a high quantum yield. Unlike nanoparticles, the small-size structure endows them with molecule-like non-plasmonic properties. Their tunable emissive colours, ranging from blue to the near-infrared (NIR) region, permit them to become robust candidates for probe design,26,27 biological labelling,28,29 logic devices,30,31 and drug delivery systems.32 The efficient expression and editing of AgNCs depend on not only the programmable primary structure, but also the flexible secondary structure of the DNA template to wield control over the silver atom number and arrangement, achieving tunable fluorescence performance. The previous relevant review articles focused mainly on the analytical applications of DNA/AgNCs from biosensor design to cell imaging,26,28,33–38 such as the detection of nucleic acids, proteins, small biomolecules, and inorganic ions. Meanwhile, they also simply summarized their synthesis process and optical properties. In 2016, the Su group summarized the correlation between the DNA sequence and emission behaviours,39 yet the important role of the secondary structure in regulating the fluorescence performance of AgNCs was not fully embodied and interpreted. In recent years, some approaches revealed that the formation of AgNCs depend largely on the specific secondary structure by interaction of the protonation, base-pairing, and cluster–DNA, further highlighting the role of the secondary structure in regulating the properties of AgNCs.40–44 Consequently, it is necessary to integrate recent progresses with the acknowledged mechanism and summarize again the direct link between the silver atom organization on the DNA secondary structure and the performance of AgNCs, and the stimuli-responsive dynamic performance of AgNCs based on the change of the DNA secondary structure, providing an all-round support for understanding the origin of DNA/AgNCs.
In this review, we sort out the findings from the viewpoints of the silver atom organization on DNA scaffolds with various spatial types based on the base arrangement, including the ordinary and more complex spatial architectures (Fig. 1). To begin with, we discuss how DNA with an alternative base order and number in the primary structure directly regulates the distribution of silver atoms in the core/shell mode to transcribe their sequences into AgNCs. Next, we review the results and show how the secondary structure regulates the twisting state of the AgNC-forming domain to alter the atom position for editing the fluorescence properties of AgNCs, including the i-motif, G-quadruplex, cluster-induced dimer, triplex, base-pairing-induced emitter dimer with two same or different Ag nucleation domains, hairpin-based monomeric or dimeric C-loop, and conjugate with G-rich sequence opposite AgNC-forming dark domain. With the introduction of the stimuli-responsive recognition sites to the DNA template, such as the gene/RNA complementary sequence, ion-binding base, and aptamer, the addition of stimuli induces the dynamic transformation of the DNA template into a reconfigurable structure that is favourable or unfavourable for forming AgNCs, along with the available fluorescence change as a report signal. Finally, we review the two- and three-dimensional arrays of silver atoms on the DNA template contained in duplex DNA, Y/X shaped structures, nanowires, hydrogel networks, tile-based nanotubes and DNA origami. We hope this comprehensive review can help researchers customize precisely personalized AgNCs to meet the requirement for developing intelligent biosensors.
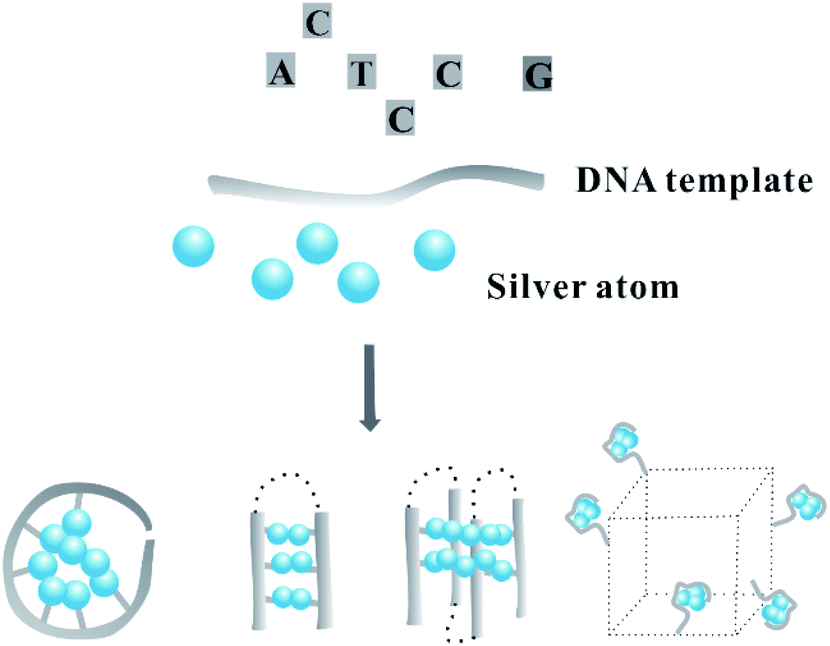 | ||
| Fig. 1 The spatial arrangement of silver atoms on a DNA template with different dimensional secondary structures. | ||
1. The DNA primary structure as a cornerstone for the spatial distribution of silver atoms
Due to the constraints of DNA, the photophysics of the encapsulated AgNCs is extremely different from that of the naked, spherical clusters without a ligand. The formation of AgNCs can be completed by reducing the mixture of Ag+–DNA with freshly prepared borohydride (Fig. 2A). With the redox of the silver cation binding to the N3 position of cytosine on DNA, the final atomic structure of AgNCs has an Ag0-rich core and base-bonded Ag+ shells to conduct their luminescent behaviours45–48 (Fig. 2B). The effective distribution of Ag0 and Ag+ enable the formation of two kinds of species, including the oxidized (Ox) and reduced (Re) structures, and their respective formed species of AgNCs belong to the blue/green emitter and red emitter49 (no. 1 in Table 1). The nucleobases in the DNA template possess different affinities to the silver cation by binding to the endocyclic amines (N7 in purines and N3 in pyrimidines), rather than the phosphate or sugar groups (Fig. 3), and their order is as follows: cytosine > guanine > adenine > thymine.50 The strong interaction between Ag+ and N3 of C depends on their lowest binding energy based on density functional theory (DFT) calculations.51 A DNA template composed of different nucleobases determines the spatial arrangement and distribution of Ag0 and Ag+, leading to a specific neutral core and valence electronic shell of clusters, and thereby producing different emission colours. The bases in completely complementary double-stranded DNA tend to form hydrogen bonds of Watson–Crick base-pairing, leading to the difficulty of binding silver cations for forming AgNCs.52 Due to the necessity of combining Ag and bases, single-stranded DNA is favourable for acting as a template for the formation of AgNCs. The different base stacking interactions in different DNA templates provide different local environment for forming AgNCs, producing the different fluorescence spectra.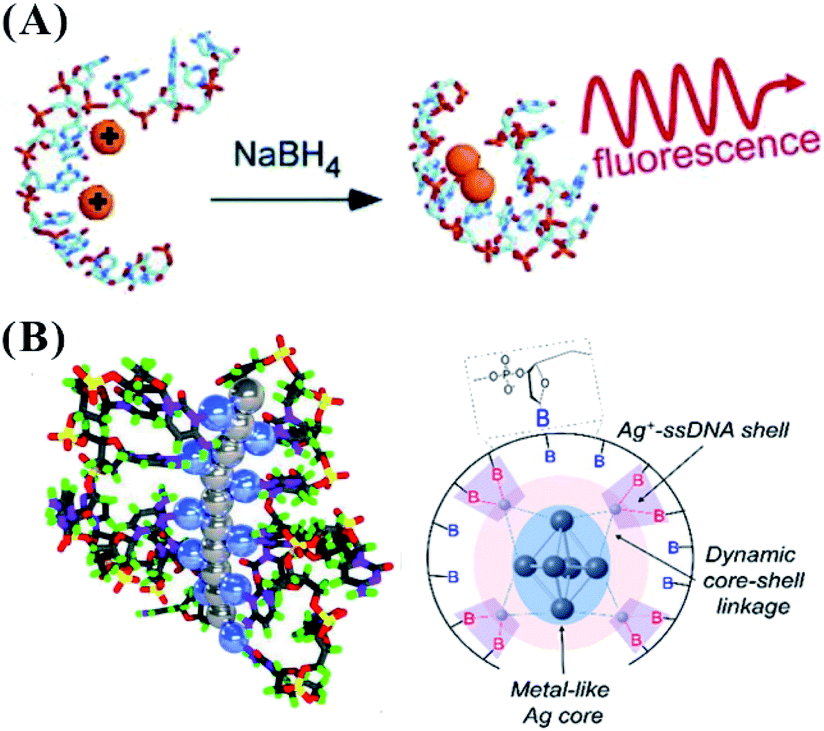 | ||
| Fig. 2 (A) The synthesis of AgNCs.11 (B) The distribution mode of silver atoms and cations on the DNA template. In the left figure, the gray and blue balls represent Ag0 and Ag+, respectively.46,48 | ||
| No. | Type | Sequence | λex/λem (nm) | Reference |
|---|---|---|---|---|
| 1 | Oxidized species | C12 | Ox: 340/485; 440/525 | 49 |
| Reduced species | Re: 280/665; 580/665 | |||
| 2 | The dominant species | Random 10-base DNA with at least three C plus G | 280/540, 280/630 | 53 |
| 3 | Spacer effect | C4–X4–C4, X = A or T | 570–590/615–630 | 55 |
| 4 | Length effect | TGC2T4G4ACG2ATA (intra) | 270/562 | 47 |
| T2C4AC2AC3AG2C4GT2 (intra) | 270/632 | |||
| T2CGC6GC4AG2CGT2 (inter) | 270/644 | |||
| CGC6T2G2CGT (inter) | 270/557 | |||
| C3AC3AC3TC3A (inter) | 270/777 | |||
| 5 | The species with same Ag number | C3AC3AC3TC3A | 750/810 | 58 and 59 |
| C3AC3AC3GC3A | 720/770 | |||
| C3AC3AC3AC3G | 840/870 |
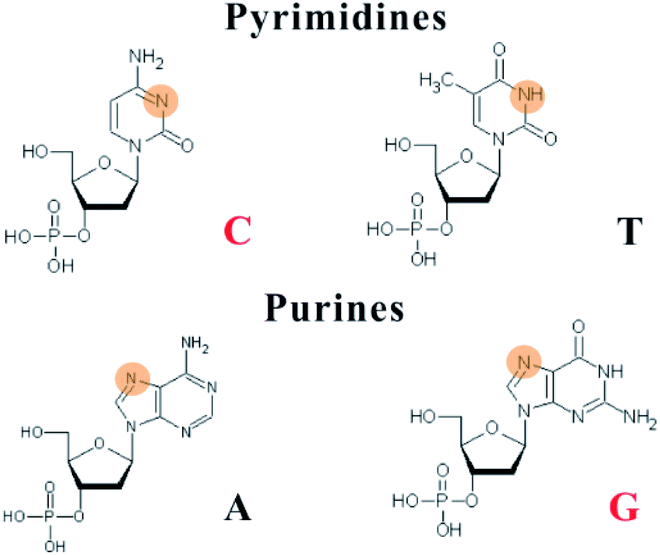 | ||
| Fig. 3 Silver cations interact with the N3 of pyrimidines and the N7 of purines. The binding sites are highlighted in orange red. | ||
The primary structure of the DNA template mainly involves the base composition and template length. In the neural condition, cytosine and guanine with their strong affinity to silver cations are two key bases to synthesize fluorescent AgNCs. In the synthesis of AgNCs, the DNA template needs to have more than two motifs containing more than three cytosine and guanine bases10 (Fig. 4A). In most of the species stabilized by different templates, there are two types of dominant colours and Ag atom numbers, including near 540 nm with four atoms and 630 nm with six atoms53 (no. 2 in Table 1). Because of their low affinities to Ag+, adenine and thymine can only form a fluorescent AgNC species under specific conditions. It is difficult for thymine to form detectable fluorescent AgNCs, except for under highly alkaline conditions.54 The incorporation of A and T hinder the formation of larger size species, possessing a molecule-like structure with smaller size. Using C4–X4–C4 (X containing A and T), the base alignment direction of A and T in the X domain largely affects the homogeneity, quantum yields and brightness of AgNCs, although it has only a limited effect on their excitation and emission55 (no. 3 in Table 1). The thymine in the DNA template enables the required folding of the DNA strand around the silver atoms, promoting the formation of a cluster morphology.41 The cluster–DNA interaction controls the emission wavelengths and intensities of AgNCs. The larger polycytosine length can synthesize AgNCs with high synthesis yield and tolerance to salt conditions.56 The amount of cytosine-based units can control the number of Ag atoms, leading to the altered fluorescence quantum yields and wavelengths of AgNCs57 (the top figure in Fig. 4B). The colours of AgNCs may redden with increased silver numbers.16 Also, the length of the DNA template can control the intra- and inter-strand secondary structure for forming AgNCs (the bottom figure in Fig. 4B, sequence information in no. 4 in Table 1). In the synthesis of AgNCs, some templates tend to form green emitters, while other templates tend to form red emitters. Different near-IR emissive species were directed by DNA templates with similar base numbers and compositions, but they had a similar number of Ag atoms (9–10 atoms)58,59 (no. 5 in Table 1). Therefore, the emission colours of the AgNCs are not directly correlated to their size, but cooperatively interact with the Ag–DNA ligation and Ag–Ag bond as well.60
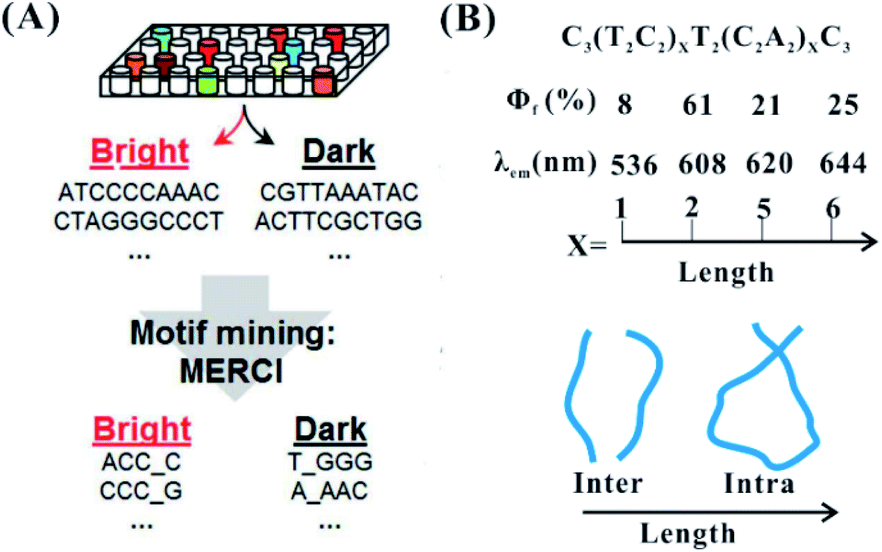 | ||
| Fig. 4 The effect of the base arrangement10 (A) and template length (B) on the performance of AgNCs.47,57 Φf and λem are the fluorescence quantum yield and emission wavelength, respectively. | ||
2. The spatial distribution of silver atoms controlled by the secondary structure of the DNA template
2.1 Secondary-structure-dependent formation of AgNCs
Owing to the cluster–DNA and Ag–cation–DNA interactions, the nucleobase rearrangement triggers the DNA into a specific secondary structure for forming AgNCs with the defined fluorescence colours and intensities.61 The proton-mediated i-motif, as a familiar class of DNA secondary structure, arises from the cytosine-rich sequences under acidic to neutral conditions.62 Consistently, the cytosine-rich domain is the main component in the AgNC template, and the silver cations can fold the sequence (TA2C3)4 into an i-motif structure even at physiological pH63 (Fig. 5A). Using the oligonucleotides, (TA2C4)4 and (C4A2)3C4, with a common C4 i-motif core under a slightly acidic condition, red emitters appear favourably with a proton-dominated i-motif structure. Conversely, in slightly basic conditions, green emitters appear favourably with a cluster–DNA conjugate resembling a compact i-motif structure64 (no. 1 in Table 2). Guanine quadruplexes are formed via stacking interaction of guanine residues in the presence of monovalent cations. The G4T4G4 templates favourably form a G-quadruplex structure to produce AgNCs at 686 nm emission after reduction for 5 h under acidic conditions65 (Fig. 5B) (no. 2 in Table 2). Although the emission wavelength can be tuned from green to NIR, the formation of some secondary structures decreases the affinity of Ag+ and DNA more or less according to the following order: the coiled C-motif > i-motif > duplex strand > G-quadruplex.66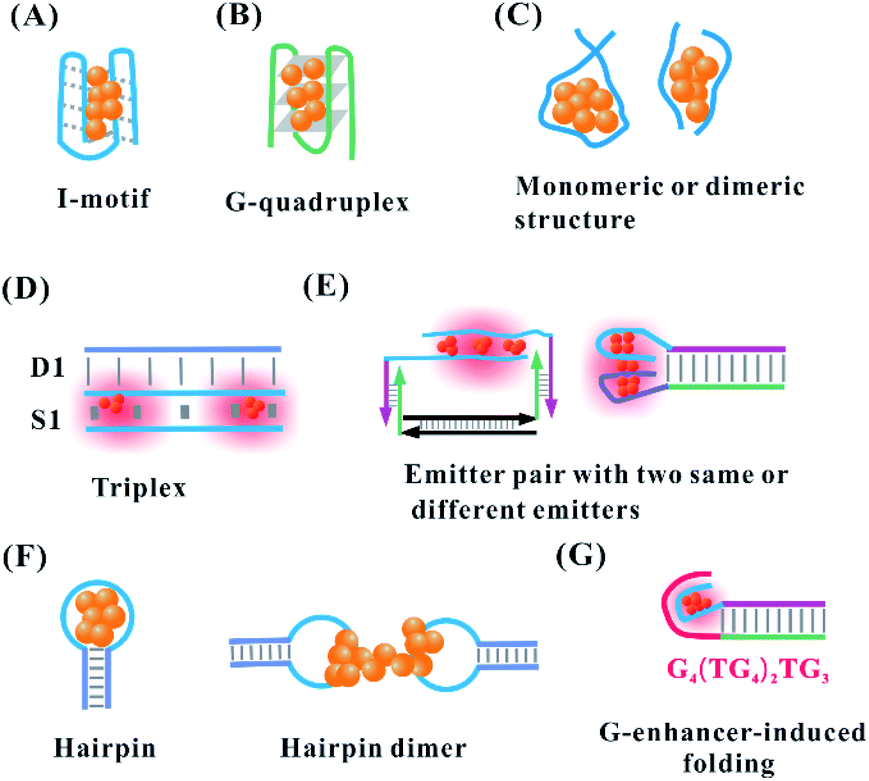 | ||
| Fig. 5 (A) I-Motif,64 (B) G-quadruplex,65 and the (C) length-dependent intra-stand and inter-strand folding of the DNA template in the synthesis of AgNCs. (D) Triplex, and (E) the reorganization of the template structure via the proximity of two identical or different emitters upon hybridization.68,70 (F) Monomeric C-loop52 and dimeric C-loop44 in the hairpin structure. (G) G enhancer-induced folding of the DNA template secondary structure for forming bright AgNCs.74,75 | ||
| No. | Type | Sequence | λex/λem (nm) | Reference |
|---|---|---|---|---|
| a The bold nucleobases are complementary domains. | ||||
| 1 | i-Motif | (TA2C4)4 | 460/560; 560/625 | 64 |
| (C4A2)3C4 | 500/570; 560/625 | |||
| 2 | G-Quadruplex | G4T4G4 | 640/686 | 65 |
| 3 | Inter-strand dimer | A2C4 | 405/540 | 40 |
| CAC2TAGCGA | 525/736 | 41 | ||
| 4 | Parallel-motif triplex | D1: GAGAG2AGAGAGA2GAGG2A2G | 480/534 | 67 |
| 3′-CTCTC2TCTCTCT2CTC2T2C-5′ | ||||
| S1: CTCTC2TCTCTCT2CTC2T2C | ||||
| 5 | Cluster dimer with two identical emitters | C3AC3AC3TC3AT2C3GC2GCTG2A | 730/800 | 68 and 69 |
| TC2AGCG2CG3 | ||||
| 6 | Cluster dimer with two different emitters | C3T2A2TC4T15C3TA2CTC4 | 550/604 | 70–73 |
| 7 | Hairpin dimer | TAGCT2ATCAGACTGATGT2GAC6TCA2CATCAGTCTGATA2GCTA | 480/590 | 43 and 44 |
| 8 | G-Enhancer-induced folding | C3T2A2TC4TATA2TA3T4A3TAT2AT3AT2A2T | 580/636 | 75 |
| AT2A2TA3TA2TAT3A4T3AT2ATAG3TG4TG4TG4 | ||||
In addition to the secondary structure folded by one template, the intermolecular interaction of two identical templates can transform into a specific configuration for the perfect formation of AgNCs via a silver-mediated template dimer (Fig. 5C). The selection of a DNA template in terms of either monomers or dimers to form AgNCs depends on the template length. For example, DNA templates containing more than 19 bases form AgNCs in one strand, while ones with 6, 8, 10, 15, and 16 bases stabilize AgNCs in two identical strands47,53 (no. 4 in Table 1). The atomic structure of AgNCs depends on the interrelationship between the DNA configuration and cluster arrangement. Using A2C4 as a template, AgNCs are ligated on a parallel template dimer, forming a hairpin-like structure containing three cytosine metallo-base pairs in the stem and adenine-rich pocket binding trapezoidal-shaped Ag5 moiety40 (no. 3 in Table 2). Near-infrared emissive Ag16NCs can be formed via silver-atom-mediated interactions in terms of two DNA decamers (CACCTAGCGA)41 (no. 3 in Table 2). The formed horse-shoe-like conformation under the interaction of DNA and AgNCs can be almost fully shielded from the surrounding environment.41 It is difficult to form AgNCs with the full complementary duplex strand due to the lack of binding sites for Ag0 and Ag+. However, when the duplex strand (D1) and single strand (S1) form a stable parallel-motif triplex (D1–S1) containing at least one set of two successive CG·CAg+ via the displacement of the N3 proton of a cytosine with Ag+, the homogenous and bright AgNCs with the Ag2 cluster unit can be formed under acidic to neutral pH conditions67 (Fig. 5D, no. 4 in Table 2).
After adding the complementary sequence for hybridizing with the recognition site, the absorption of the formed AgNCs changes from 400 nm (violet cluster) to 730 nm (near-infrared cluster), which is attributed to the silver-mediated intermolecular antiparallel cluster dimer with twice silver stoichiometry alteration68,69 (no. 5 in Table 2). Also, the same near-infrared cluster arises from the base-pairing-induced intramolecular dimer (the left figure in Fig. 5E). In addition to the cluster dimer with the same nucleation domain, our previous work found that the proximity of the yellow and near-infrared emitters via duplex domain and polyT spacer transforms the original colour into a new orange emitter with excitation and emission at 550 and 604 nm, respectively70–73 (the right figure in Fig. 5E, no. 6 in Table 2).
The hairpin-based cytosine-rich loops enable the formation of AgNCs on the C-loop domain.52 The hairpin-based C-loop forms alternatively red or orange emissive AgNCs by the selection of a monomeric or dimeric structure in the synthesis of AgNCs (Fig. 5F). The lone hairpin C-loop with six cytosines generated red emissive AgNCs, while the speculated dimer of six cytosines via hairpin inter-strand hybridization generated orange emissive AgNCs.42 However, the dimer of six cytosines generated by the two complete complementary strands with 6 cytosines does not form orange-emissive AgNCs, as expected. Benefiting from the important role of the third and fourth cytosines in the C-loop in base flipping, the two six-C-loop structures form head-to-head dimers via the cytosine–Ag–cytosine bridge after flipping base to implement the formation of orange emissive AgNCs43,44 (no. 7 in Table 2). The DNA hairpin containing 9-C loop, as a template, can form two different species, including a green emitter with 11 atoms and red emitter with 13 atoms. Besides the different atoms, they have different DNA conformations. The green emitter adopts a compact structure, while the red emitter has an extended structure similar to the native hairpin conformation.76 In addition to the C-loop-dependent fluorescence,77 the stem in the hairpin can affect the fluorescence of AgNCs. Using a hairpin containing 12-C loop and 10 bp stem with different base composition, the emission of AgNCs can be tuned. This is attributed to the base composition in the stem leading to conformational changes of the DNA-containing hairpin pair.78 The spectrally pure species have two different DNA structures because the stems can be arranged in either a cis or trans orientation.
The proximity of the 12-base cluster-nucleation sequence and guanine-rich enhancer (G4(TG4)2TG3) transforms a dark emitter into a bright emitter with ∼500-fold fluorescence enhancement75 (Fig. 5G, no. 8 in Table 2). A combination of the physical interaction between the N7 of guanine and silver,79 and the electron transfer from guanine to AgNCs, contributes synergistically to the re-organization of AgNCs and results in the strong turn-on fluorescence.75,80 By regulation of the number of cytosines and the base arrangement of the spacer in the cluster-nucleation sequence (Cm–spacer–Cn), AgNCs with a diversity of colours and fluorescence enhancement ratio can be produced.81 The cytosine content can largely affect the emission (140 nm tuning range), while the AT–spacer composition only has a little influence on the emission (40 nm range). The increased cytosines (Cm and Cn) in the DNA template lead to the blue-shift emission wavelength, which is different from the finding that longer polycytosine templates tend to stabilize emitters with longer emission wavelengths using the DNA template without a G-enhancer. In the AT-linker, adding one thymine (T) to the 3′ side of the spacer can red shift the emission peaks of AgNCs by as much as 18 nm. On the contrary, adding one adenine (A) to the 3′ side of the spacer tends to blue shift the emission peaks.74 Using C3–spacer–C4 as a model, the role of the spacer in regulating AgNCs was investigated. This finding showed that the spacer served as a bridge to hold small C3-cluster and C4-cluster with close proximity for the guanine-rich activating strand to contact and fold them upon each other more easily.74 The guanine-rich activating strand then comes into physical contact with the two dark nanoclusters, held together by the spacer, and folds them upon themselves for re-organization into a single ca. six-atom red-emitter.
2.2 Stimuli-induced dynamic structure transformation of DNA template for forming AgNCs
The suitable DNA template provides the binding site for the arrangement of the silver atom and cation, allowing the perfect formation of fluorescent AgNCs. The state of the template directly affects the atomic structure of AgNCs, exhibiting translation of the structural change to fluorescence intensity and colour change. The secondary structure of the DNA template for AgNCs can be regulated dynamically by the addition of stimuli. The binding domain response to stimuli is introduced to the DNA template. The functional domains adjacent to the cluster template involve a DNA/RNA complementary sequence, aptamer, DNAzyme, ion-binding base and so on. The introduction of a stimulus allows the formation and deformation of the secondary structure responsible for the appropriate arrangement of silver atoms on AgNCs, leading to the enhancement or quenching of the fluorescence intensity and/or alteration of the emission wavelength.The attached complementary domain for DNA or miRNA endows the DNA template with a specific secondary structure to favourably or unfavourably form AgNCs. The DNA or miRNA triggers the alteration of the initial structure for the unfavourable82 and favourable83,84 formation of AgNCs. The DNA template containing the miRNA (e.g., miR160) recognition site is hybridized into a mismatch self-dimer to form a red emitter with 620 nm emission.85 The addition of miRNA can disturb the secondary structure of the dimer, leading to decreased emission intensity82,85 (Fig. 6A). The replacement of a recognition site with another sequence endows the probe with the different ability to form a self-dimer, affecting the behaviour of AgNCs and the miRNA response. The extra base is needed to compel the DNA template with the recognition site to form a self-dimer, extending their response to the other stimuli. The different electronic environment occurs before and after adding the stimulus DNA. This leads to a change in the absorption spectra from a 400 nm species containing ∼7 atoms with dark emission to 720 nm species containing ∼11 atoms with bright emission86 (Fig. 6B), where the DNA structure of the former species has a random coil conformation, while the latter species is the stretching state through hybridization with the stimulus. The cluster domain was replaced with the sequence for forming the blue/green cluster. With the addition of the stimulus, the same reaction is produced due to an absorption change from 400 nm to 490 nm and enhanced fluorescence at 550 nm emission.87 The formed species before and after adding the stimulus has the same atom number (11 atoms), which is attributed to the different isomers of the same cluster.87
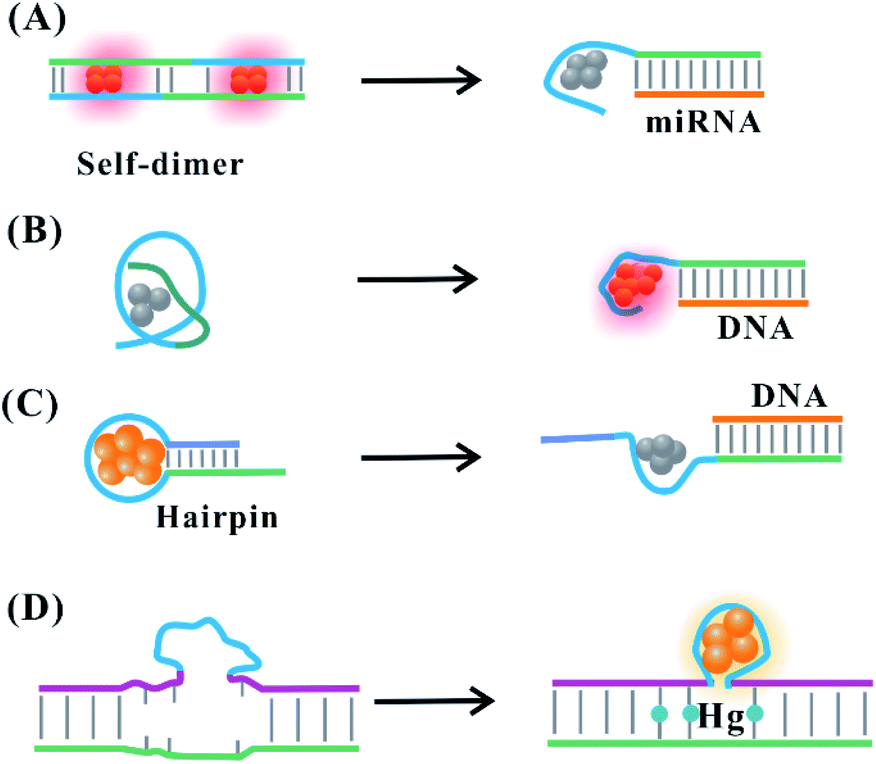 | ||
| Fig. 6 Stimulus-induced dynamic secondary structural transformation. (A) The miRNA-response collapse of the self-dimer.82,85 (B) The DNA-induced transformation of the species resulting from a structural change.86 Stimuli (DNA or Hg)-induced deformation (C)88 and formation (D)89 of the C-loop. | ||
Hairpin-based C-loops vary dynamically with the addition of stimuli, leading to the switch-off and switch-on fluorescence response. The binding of the stimulus (e.g., miRNA) to the stem domain opens the C-loop. Subsequently, the collapse of the C-loop affects the formation of AgNCs, leading to the switch-off fluorescence88 (Fig. 6C). On the contrary, the combination of T–Hg–T promotes the formation of the C-loop for producing more AgNCs, leading to the switch-on fluorescence89 (Fig. 6D). The dark emitter can be transformed into a bright state due to the formation of a G-enhancer/emitter pair with the addition of a stimulus (e.g., DNA)75 (Fig. 7A). The introduction of the stimulus DNA induces the exposure of the domain binding with the G enhancer, and the produced similar proximity of the emitter/enhancer leads to the increased fluorescence of AgNCs90 (Fig. 7B). In addition to AgNCs stabilized by the DNA template with a specific aptamer binding to the targeted nucleus as labels,32,91,92 by combining the aptamer with the cellular receptor and triggering the internalization of the probe, the hybridization of the cellular mRNA and its recognition site enables a structural change of the domain environment surrounding the DNA template, resulting in the fluorescence behavioural transformation of AgNCs from 555/620 nm to 480/595 nm excitation/emission, visualizing the varied cell imaging.93 The specific aptamer binding to the cell,94 protein,95 and ATP96 induces the proximity of the dark emitter and enhancer, leading to the turning on of the fluorescence signal (Fig. 7C). DNAzymes as functional nucleic acids possess protein enzyme-like catalytic activities in the presence of specific metal ions or neutral molecules.97,98 In the presence of Pb2+ or L-histidine, their DNAzyme domain can catalyze the cleavage of the substrate strand combining with the DNA template, favourably binding with the strand containing the G-rich enhancer, leading to a detectable fluorescence enhancement99 (Fig. 7D).
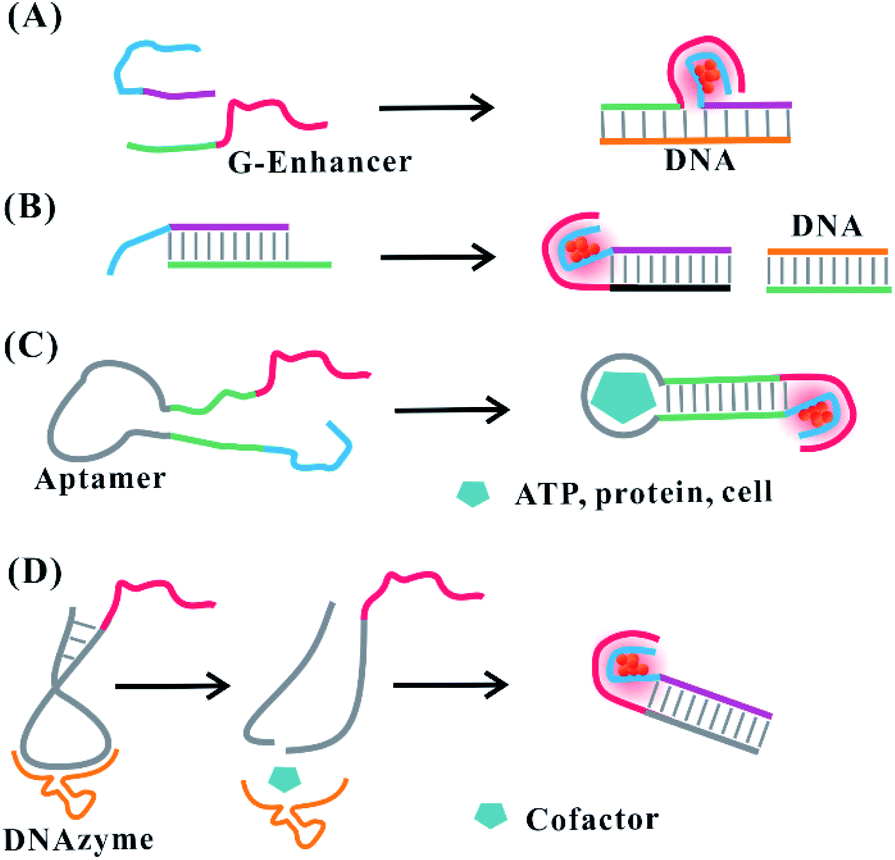 | ||
| Fig. 7 Stimuli-induced formation of the emitter/G-enhancer. (A) DNA (e.g., gene)-induced proximity of the dark emitter and G-enhancer.75 (B) Formation of the emitter/G-enhancer by DNA-induced exposure of the complementary domain binding with the G-enhancer.90. (C) The proximity of the dark emitter and G-enhancer induced by the aptamer structural change in the presence of stimuli (ATP, protein, and cell).94–96 (D) Cofactor-induced cleavage for exposing the complementary domain to form an emitter/G-enhancer pair under the catalysis of DNAzyme.99 | ||
However, some of the stimulus-induced fluorescence changes cannot be explained clearly by a structural change of the DNA template. The combination of the aptamer in the DNA template and targeted thrombin protein induces the turn-off fluorescence of AgNCs, but the fluorescence lifetime, absorbance and morphological characteristic of AgNCs before and after adding protein do not show considerable differences.100 Hg2+ can quench the fluorescence of red emissive AgNCs in the case of no change on the fluorescence lifetime, which is attributed to the Hg2+-induced weakened binding between AgNCs and DNA, rather than the formation of a new species resulting from a structural change.101,102 The stimulus Cu2+ can tune the competitive proportion of two species, promoting the formation of green emissive AgNCs with 480/564 nm excitation/emission at pH 6.0, but precluding the orange or red emissive AgNCs.72,103,104 The mechanism of the Cu2+-induced evolution of AgNCs was not fully verified, which may be due to the tendency of the oxidized species, weakened combination of AgNCs and DNA, and a more rigid structure of the DNA template.
The change of input can alter the secondary structure of the DNA template for switching the performance of AgNCs with the corresponding signal output, which is requisite for the operation of the logic device. Using the 6-C-loop hairpin DNA template containing the G-quadruplex and i-motif domain in the stem domain to build up the molecular logic device, two inputs of K+ and H+ can make the DNA template carry out the structural changes, guiding the fluorescence behaviours of AgNCs.30 In the presence of K+ (1 0), the formation of the G-quadruplex leads to the species with yellow emission, while the formation of the i-motif leads to the species with low orange emission due to the addition of H+ (0 1). The presence of both K+ and H+ (1 1) produces the orange species, while the absence of both (0 0) produces the yellow and red species (Fig. 8A). AgNCs formed in the state of different DNA structures can construct multiple logic operations due to the different fluorescence outputs. At 494 nm excitation, input (1 0) and (0 0) produce yellow emission, leading to the “NOT” gate. At 581 nm excitation, input (0 0) produces red emission, leading to the “NOR” gate. At 537 nm excitation, input (1 1) produces orange emission, leading to the “AND” gate (Fig. 8).
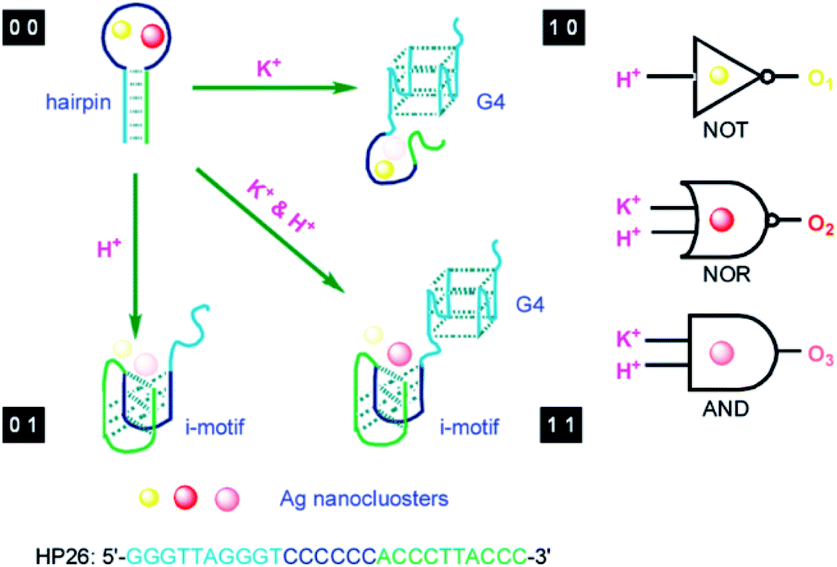 | ||
| Fig. 8 Design of a molecular logic device based on the structural change induced by the stimulus as input. K+ and H+ as two inputs to trigger the transformation of the i-motif and G-quadruplex for regulating the fluorescence output of AgNCs.30 | ||
The opening and closing of the hairpin with DNA hybridization and toehold-mediated strand displacement lead to the change in the respective intensity of AgNCs with dual fluorescence signal (at 565 and 630 nm). The input-induced transformation of two signals are the basis of constructing the concomitant contrary logic gates (CCLGs).31 The fluorescence at 565 nm (F565) and 630 nm (F630) correspond to positive and negative outputs, respectively. The addition of IN1 “1” triggers the F565 increase and F630 decrease, leading to CCLGs with “YES^NOT” and concatenated logic circuits with “YES‖NOT” (Fig. 9A). The high F565 and low F630 were performed in the state of “IN2 IN3” (including “1 0”, “0 1”, and “1 1”), leading to “OR^NOR” and “OR‖NOR” (Fig. 9B). IN4 can disable the opening states of the hairpin due to the formation of duplex structures of IN3/IN4. The high F565 and low F630 appear in the presence of IN3 only, leading to “INHIBIT^IMPLICATION” and “INHIBIT‖IMPLICATION” (Fig. 9C). The presence of X1 or X2 can make the duplex IN2/Xaux release IN2 for opening the hairpin with high F565 and low F630, while the contrary phenomenon occurs in the presence and absence of X1 and X2, leading to “XOR^XNOR” and “XOR‖XNOR” (Fig. 9D). The binding and dissociation of the input strand complementary with the loop induced changes in the dynamic conformation of the hairpin, producing an output with fluorescence intensity alteration at 501/565 and 566/630 nm excitation/emission.
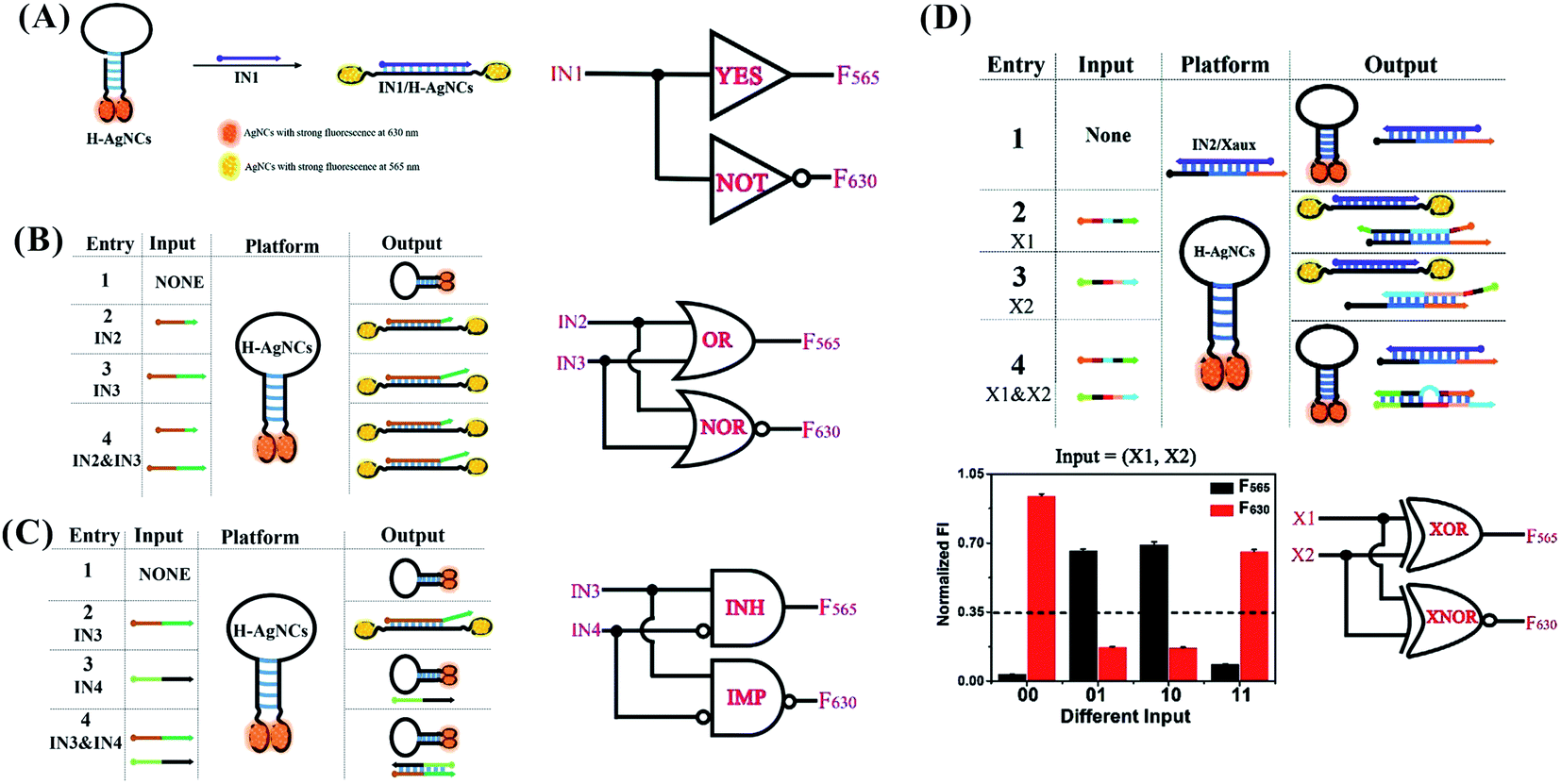 | ||
| Fig. 9 YES^NOT (A), XOR^XNOR (B), INHIBIT^IMPLICATION (C), and XOR^XNOR (D) logic gates, and the concatenated circuits based on the structural-change-induced dual-output by switching the hairpin-AgNCs.31 | ||
3. The spatial distribution of the silver atom on the template anchored to two and three-dimensional architectures
In addition to their efficient distribution on the free single-stranded DNA template, silver atoms can be arranged precisely on the nucleation domain anchored to different DNA structures, such as simple (duplex and Y/X shaped structures) and complex structures (nanowires, DNA hydrogel, and nanotube). To avoid the interaction between each cluster, the distance between the clusters is controlled strictly to maintain their original structures.The proximity of the two clusters tends to drive the structural reorganization to produce a new species with different spectral properties. Due to the favourable intra-strand folding of the DNA template with more than 19 bases to encapsulate AgNCs, the 19-base DNA templates were selected as the nucleation domain of the donors (D) and acceptors (A), respectively. The AT-rich hybridization induced the distance of D and A to approach in the range within 6 nm, producing fluorescence resonance energy transfer (FRET) (Fig. 10A).105 The D and A clusters still maintain their original structure due to no change in the spectra.105 By tuning the spatial structures of the branched DNA scaffold through sticky-end hybridization, the Y-shaped structure by three strands with AgNCs, X-shaped structure by four strands with AgNCs, and (Y–X)-shaped structure by hybridization of Y and X can be formed (Fig. 10B).106 The peak value of the fluorescence spectra of Y and X increased first, and then decreased with increased arm length, regulating the spatial structure for the tunable optical properties of AgNCs.106
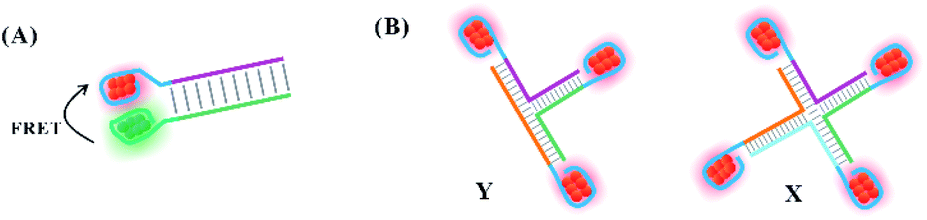 | ||
| Fig. 10 The arrangement of clusters on the simple structures. (A) The arrangement of two different emitters on the duplex DNA.105 (B) The arrangement of emitters on the Y, X and Y–X shaped structures.106 | ||
The hairpin-based C-loop with six or seven cytosines embedded in the nanowires resulting from the hybridization–polymerization of two strands can form yellow or red emissive AgNCs, producing a micrometre-scale luminescent wire107 (Fig. 11A). The C-loop for forming the yellow or red emissive emitters was also embedded in a complex three-dimensional structure. As shown in Fig. 11B, the Y-shaped DNA structures with the C-loop are crosslinked to form a hydrogel network. The yellow or red emissive emitters embedded in the Y-shaped units were formed successfully, producing fluorescent DNA hydrogels.108 The hairpin-based C-loop with nine cytosines outward from the nanotube comprising identical tiles can form AgNCs with nearly identical spectra as a free hairpin containing the same sequence, allowing for the spatial organization of silver atoms and lighting up DNA nanotechnology109 (Fig. 11C). The purified monodisperse AgNCs with a diffusing linker are complementary to the docker extruding spaced by 7.1 nm in the 10-helix tiled DNA nanotubes (NT), being arrayed in a line along specific individual double helices of the NT and realizing the atomically precise nanoscale arrangement of the metal clusters (Fig. 10D).110
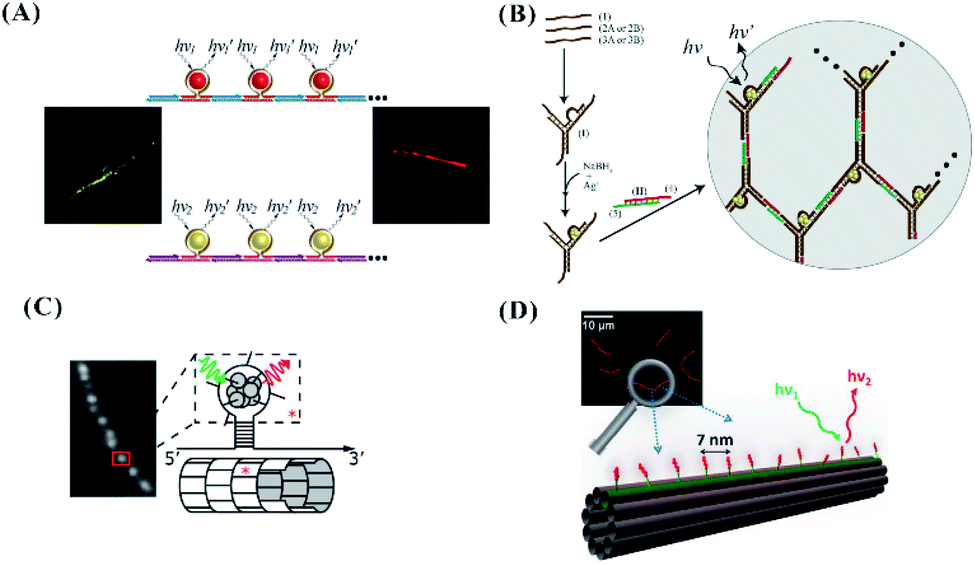 | ||
| Fig. 11 (A) The arrangement of the C-loop/cluster on the nanowire.107 (B) The arrangement of the C-loop/cluster on the hydrogel network.108 The arrangement of the C-loop (C) or ss-DNA (D) template for forming clusters on the tile-based nanotube.109,110 | ||
In addition to the template-guided formation of AgNCs on the origami, DNA modified with sugar moieties111 or thiol groups112 can direct the site-specific arrangement of AgNCs on a DNA origami (Fig. 12). The sugar (galactose)-modified DNA strands on the origami can reduce Ag+ to Ag0 due to the Tollens reaction, forming nucleation sites for AgNCs (Fig. 12A). DNA strands modified by thiol groups with strong affinity for metals can hybridize with extensions on a DNA origami, forming metal and metal oxide nanoclusters (MMONs) (Fig. 12B).
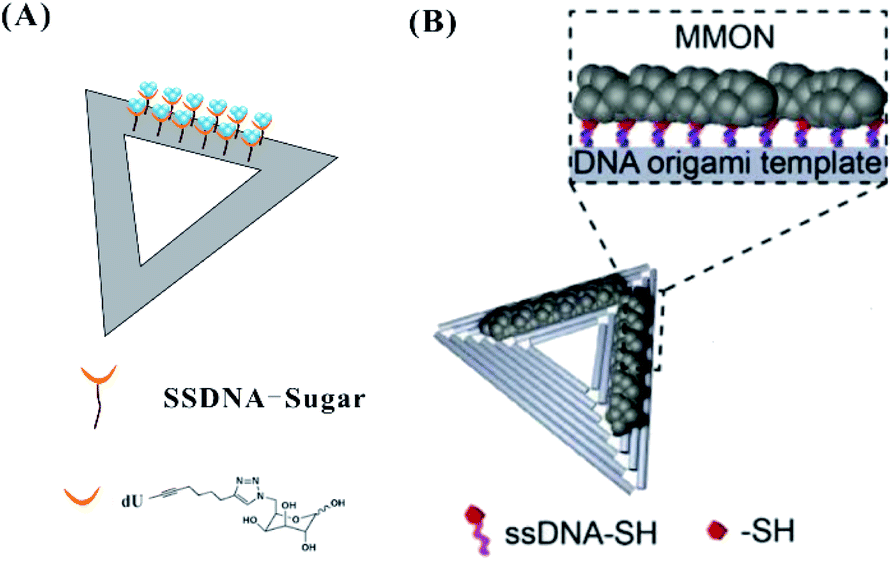 | ||
| Fig. 12 The arrangement of AgNCs on the origami based on DNA with sugar moieties111 (A) and thiol groups112 (B). | ||
4. Conclusions
We have reviewed the relationship between the secondary structure of the DNA template and the fluorescence properties of AgNCs. The base arrangement can control the primary structure to form a specific secondary structure for operating the spatial distribution of Ag0 and Ag+, and the different electronic environment surrounding DNA dictate the fluorescence intensity and colours of AgNCs. Four bases show the differences in the affinity to Ag+. C and G provide excellent binding sites for Ag+ to form a cluster or silver-cation-induced rigid secondary structure, while A and T control the size of the AgNCs and folding of the DNA template. A and T can affect the fluorescence intensity and quantum yield, rather than the wide-range emission colour regulation. Therefore, it is necessary for the DNA template to contain more than two motifs with C and G in the design of AgNCs. The length of the DNA template also controls the number of Ag atom/cations and DNA structure. The longer poly-cytosine can produce AgNCs with higher quantum yield and stability. The strand length can determine the selection of the inter-strand and intra-strand secondary structure for forming AgNCs. Therefore, the base composition and length in the DNA template are cornerstones for forming the DNA secondary structure to produce the controllable performance of AgNCs.The secondary structure of the DNA template provides the spatial position for regulating the fluorescence behaviours of AgNCs. Although some regular DNA secondary structures partially limit the affinities of the Ag cation, the confined spatial binding sites effectively and precisely control the position of Ag on the DNA template. In the specific surrounding condition, the pH-induced i-motif change can form red and green emitters, and the G-quadruplex can form a red emissive species due to the folding of the secondary structure. Different from the conventional speculative schematic mode with one strand surrounding the AgNCs, the short-stranded template can encapsulate AgNCs in terms of the dimer arising from two identical templates. More different from the conventional finding that the duplex strands with a complete complementary domain have difficulty in forming AgNCs due to the base-pairing invading the binding site for Ag+, the triplex can form emissive AgNCs via CG·CAg+. In addition to the cluster-induced DNA template dimer, the complementary hybridization can induce the formation of a DNA template with two identical or different emitters, leading to the transformation of the emitter's brightness from dark to bright, and the colour from original to new. The state of the C-loop can also regulate the properties of AgNCs. The monomeric hairpin-based C-loop can form AgNCs with a red colour, while the C-loop dimer can form orange emissive emitters. In addition to the regulation of the C number in the C-loop to the properties of AgNCs, the stem sequence can control the cluster-induced hairpin structure to affect AgNCs. The G-enhancer can induce the C-rich dark template to fold into the structure for the favourable formation of fluorescent AgNCs.
Due to the significance of the secondary structure to form AgNCs, after designing the recognition site, the addition of the stimuli can induce a structural change along with the fluorescence change of AgNCs, which is the basic scheme for the detection design based on AgNCs. In the absence of stimuli, the DNA template containing the recognition site fold into ordered and un-ordered structures, the stimuli can induce the DNA template from a self-dimer to monomer, from coil to compact, from loop to strand, from strand to loop, from respective to pair emitter/G-enhancer, from close to open, from pair to respective emitter, leading to a fluorescence change. Therefore, by investigating the Ag atom arrangement on the secondary structure of the DNA template, more probes and logic devices can be designed for meeting the requirement of the practical use. By the precise array of the DNA template on the simple and complex structures, the arrangement of the silver atoms can occur in the two- and three-dimensional spaces, which is beneficial for developing the optical device and design.
In the future, using secondary-structure-dependent fluorescence emission/excitation wavelength changes as the signal output, more ratiometric probes and complex logic gates or circuits should be promising areas to be explored, avoiding the disturbance from the practical environment. In addition to the reported site-specific arrangement of AgNCs on the 2-D and 3-D DNA nanotechnology, versatile nano-robots should be promising to be explored using the structure-controlled properties of AgNCs. The comprehensive review can provide some rules for precisely personalized AgNCs, meeting the requirement for developing an intelligent biosensor and molecular machine.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Ding Ding, Institute of Molecular Medicine, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, for providing important advice and proofreading for the Review. This work was financially supported by the National Natural Science Foundation of China (NSFC Grants 21804089).Notes and references
- X. Xia, J. Zhang, N. Lu, M. J. Kim, K. Ghale, Y. Xu, E. Mckenzie, J. Liu and H. Ye, ACS Nano, 2015, 9, 9994–10004 CrossRef CAS.
- C. N. Loynachan, M. R. Thomas, E. R. Gray, D. A. Richards, J. Kim, B. S. Miller, J. C. Brookes, S. Agarwal, V. Chudasama and R. A. Mckendry, ACS Nano, 2017, 12, 279 CrossRef.
- A. Verma, O. Uzun, Y. Hu, Y. Hu, H. Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Nat. Mater., 2008, 7, 588–595 CrossRef CAS.
- R. Jin, Y. C. Cao, E. Hao, G. S. Metraux, G. C. Schatz and C. A. Mirkin, Nature, 2003, 425, 487–490 CrossRef CAS.
- M. A. Noginov, G. Zhu, A. M. Belgrave, R. M. Bakker, V. M. Shalaev, E. E. Narimanov, S. Stout, E. Herz, T. Suteewong and U. Wiesner, Nature, 2009, 460, 1110–1112 CrossRef CAS.
- H. A. Atwater and A. Polman, Nat. Mater., 2010, 9, 205–213 CrossRef CAS.
- R. R. Arvizo, S. Bhattacharyya, R. A. Kudgus, K. Giri, R. Bhattacharya and P. Mukherjee, Chem. Soc. Rev., 2012, 41, 2943–2970 RSC.
- J. T. Petty, J. Zheng, N. V. Hud and R. M. Dickson, J. Am. Chem. Soc., 2004, 126, 5207–5212 CrossRef CAS.
- C. I. Richards, S. Choi, J. C. Hsiang, Y. Antoku, T. Vosch, A. Bongiorno, Y. L. Tzeng and R. M. Dickson, J. Am. Chem. Soc., 2008, 130, 5038–5039 CrossRef CAS.
- S. M. Copp, P. Bogdanov, M. Debord, A. Singh and E. Gwinn, Adv. Mater., 2014, 26, 5839–5845 CrossRef CAS.
- T. Vosch, Y. Antoku, J. C. Hsiang, C. I. Richards, J. I. Gonzalez and R. M. Dickson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12616–12621 CrossRef CAS.
- E. Thyrhaug, S. A. Bogh, M. R. Carro-Temboury, C. S. Madsen, T. Vosch and D. Zigmantas, Nat. Commun., 2017, 8, 15577 CrossRef CAS.
- G. Liu, Y. Shao, K. Ma, Q. Cui, F. Wu and S. Xu, Gold Bull., 2012, 45, 69–74 CrossRef CAS.
- R. Zhou, M. Shi, X. Chen, M. Wang and H. Chen, Chem.–Eur. J., 2009, 15, 4944–4951 CrossRef CAS.
- T. A. C. Kennedy, J. L. Maclean and J. Liu, Chem. Commun., 2012, 48, 6845–6847 RSC.
- A. Rotaru, S. Dutta, E. Jentzsch, K. Gothelf and A. Mokhir, Angew. Chem., Int. Ed., 2010, 49, 5665–5667 CrossRef CAS.
- Z. Qing, X. He, D. He, K. Wang, F. Xu, T. Qing and X. Yang, Angew. Chem., Int. Ed., 2013, 125, 9901–9904 CrossRef.
- E. Braun, Y. Eichen, U. Sivan and G. Ben-Yoseph, Nature, 1998, 391, 775–778 CrossRef CAS.
- J. Richter, R. Seidel, R. Kirsch, M. Mertig, W. Pompe, J. Plaschke and H. K. Schackert, Adv. Mater., 2010, 12, 507–510 CrossRef.
- K. Nguyen, M. Monteverde, A. Filoramo, L. Goux-Capes, S. Lyonnais, P. Jegou, P. Viel, M. Goffman and J. P. Bourgoin, Adv. Mater., 2008, 20, 1099–1104 CrossRef CAS.
- S. Kundu, K. Wang, D. Huitink and H. Liang, Langmuir, 2009, 25, 10146–10152 CrossRef CAS.
- H. T. Li, Y. Yuan, N. S. R. Satyavolu, A. S. Ali, Z. Wang, Y. Wu and Y. Lu, J. Am. Chem. Soc., 2015, 137, 14456–14464 CrossRef.
- X. Ma, J. Huh, W. Park, L. P. Lee, Y. J. Kwon and S. J. Sim, Nat. Commun., 2016, 7, 12873 CrossRef CAS.
- X. Ma, S. Song, S. Kim, M. Kwon, H. Lee, W. Park and S. J. Sim, Nat. Commun., 2019, 10, 836 CrossRef.
- J. Wu, L. H. Tan, K. Hwang, H. Xing, P. Wu, W. Li and Y. Lu, J. Am. Chem. Soc., 2014, 136, 15195–15202 CrossRef CAS.
- J. M. Obliosca, C. Liu and H. C. Yeh, Nanoscale, 2013, 5, 8443–8461 RSC.
- A. Pandya, A. N. Lad, S. P. Singh and R. Shanker, RSC Adv., 2016, 6, 113095–113114 RSC.
- Y. C. Shiang, C. C. Huang, W. Y. Chen, P. C. Chen and H. T. Chang, J. Mater. Chem., 2012, 22, 12972–12982 RSC.
- C. Sungmoon, Y. Junhua, S. A. Patel, T. Yih-Ling and R. M. Dickson, Photochem. Photobiol. Sci., 2011, 10, 109–115 RSC.
- T. Li, L. Zhang, J. Ai, S. Dong and E. Wang, ACS Nano, 2011, 5, 6334–6338 CrossRef CAS.
- M. Lv, W. Zhou, D. Fan, Y. Guo, X. Zhu, J. Ren and E. Wang, Adv. Mater., 2020, 32, 1908480 CrossRef CAS.
- J. Li, W. Wang, D. Sun, J. Chen, P. H. Zhang, J. R. Zhang, Q. Min and J. J. Zhu, Chem. Sci., 2013, 4, 3514–3521 RSC.
- J. Liu, Trends Anal. Chem., 2014, 58, 99–111 CrossRef CAS.
- A. Latorre, R. Lorca and Á. Somoza, J. Chem., 2013, 2013, 1–6 CrossRef.
- Y. Tao, M. Li, J. Ren and X. Qu, Chem. Soc. Rev., 2016, 47, 8636–8663 Search PubMed.
- S. Choi, R. M. Dickson and J. Yu, Chem. Soc. Rev., 2012, 41, 1867–1891 RSC.
- A. Latorre and Á. Somoza, ChemBioChem, 2012, 13, 951–958 CrossRef CAS.
- Z. Yuan, Y. C. Chen, H. W. Li and H. T. Chang, Chem. Commun., 2014, 50, 9800–9815 RSC.
- S. Y. New, S. T. Lee and X. D. Su, Nanoscale, 2016, 8, 17729–17746 RSC.
- D. J. E. Huard, A. Demissie, D. Kim, D. Lewis, R. M. Dickson, J. T. Petty and R. L. Lieberman, J. Am. Chem. Soc., 2019, 141, 11465–11470 CrossRef CAS.
- C. Cerretani, H. Kanazawa, T. Vosch and J. Kondo, Angew. Chem., 2019, 58, 17153–17157 CrossRef CAS.
- P. Shah, S. Choi, R. Nagda, R. Geczy, S. K. Cho, Y. J. Bhang, T. Kim, T. Y. Song, P. H. Lee and J. Kang, Nanoscale, 2018, 10, 20717–20722 RSC.
- R. Geczy, N. J. Christensen, K. K. Rasmussen, I. Kálomista, M. K. Tiwari, P. Shah, S. W. Yang, M. J. Bjerrum and P. W. Thulstrup, Angew. Chem., Int. Ed., 2020, 132, 16225–16231 CrossRef.
- P. Shah, R. Nagda, I. L. Jung, Y. J. Bhang, S. W. Jeon, C. S. Lee, C. Do, K. Nam, Y. M. Kim, S. Park, Y. H. Roh, P. W. Thulstrup, M. J. Bjerrum, T. H. Kim and S. W. Yang, ACS Nano, 2020, 14, 8697–8706 CrossRef CAS.
- E. Gwinn, D. Schultz, S. M. Copp and S. Swasey, Nanomaterials, 2015, 5, 180–207 CrossRef.
- D. Schultz, K. Gardner, S. S. R. Oemrawsingh, N. Markešević, K. Olsson, M. Debord, D. Bouwmeester and E. Gwinn, Adv. Mater., 2013, 25, 2757 CrossRef.
- D. Schultz and E. G. Gwinn, Chem. Commun., 2012, 48, 5748–5750 RSC.
- J. T. Petty, O. O. Sergev, M. Ganguly, I. J. Rankine, D. M. Chevrier and P. Zhang, J. Am. Chem. Soc., 2016, 138, 3469–3477 CrossRef CAS.
- C. M. Ritchie, K. R. Johnsen, J. R. Kiser, Y. Antoku, R. M. Dickson and J. T. Petty, J. Phys. Chem. C, 2007, 111, 175–181 CrossRef CAS.
- S. Shukla and M. Sastry, Nanoscale, 2009, 1, 122–127 RSC.
- J. Wu, Y. Fu, Z. He, Y. Han, L. Zheng, J. Zhang and W. Li, J. Phys. Chem. B, 2012, 116, 1655–1665 CrossRef CAS.
- E. G. Gwinn, P. O'Neill, A. J. Guerrero, D. Bouwmeester and D. K. Fygenson, Adv. Mater., 2008, 20, 279–283 CrossRef CAS.
- S. Copp, D. Schultz, S. Swasey, J. Pavlovich, M. Debord, A. Chiu, K. Olsson and E. Gwinn, J. Phys. Chem. Lett., 2014, 5, 959–963 CrossRef CAS.
- B. Sengupta, C. M. Ritchie, J. G. Buckman, K. R. Johnsen, P. M. Goodwin and J. T. Petty, J. Phys. Chem. C, 2016, 112, 18776–18782 CrossRef.
- T. Li, N. He, J. Wang, S. Li, Y. Deng and Z. Wang, RSC Adv., 2016, 6, 22839–22844 RSC.
- J. Sharma, H. C. Yeh, H. Yoo, J. H. Werner and J. S. Martinez, Chem. Commun., 2010, 46, 3280–3282 RSC.
- G. Y. Lan, W. Y. Chen and H. T. Chang, RSC Adv., 2011, 1, 802–807 RSC.
- J. T. Petty, C. Fan, S. P. Story, B. Sengupta, S. J. I. Ashlee, Z. Prudowsky and R. M. Dickson, J. Phys. Chem. Lett., 2010, 1, 2524–2529 CrossRef CAS.
- J. T. Petty, C. Fan, S. P. Story, B. Sengupta and R. M. Dickson, J. Phys. Chem. B, 2011, 115, 7996–8003 CrossRef CAS.
- M. L. Neidig, J. Sharma, H.-C. Yeh, J. S. Martinez, S. D. Conradson and A. P. Shreve, J. Am. Chem. Soc., 2011, 133, 11837–11839 CrossRef CAS.
- S. M. Swasey, N. Karimova, C. M. Aikens, D. E. Schultz, A. J. Simon and E. G. Gwinn, ACS Nano, 2014, 8, 6883–6892 CrossRef CAS.
- K. Gehring, J. Leroy and M. Gueron, Nature, 1993, 363, 561–565 CrossRef CAS.
- H. A. Day, C. Huguin and Z. A. Waller, Chem. Commun., 2013, 49, 7696–7698 RSC.
- B. Sengupta, K. Springer, J. G. Buckman, S. P. Story, O. H. Abe, Z. W. Hasan, Z. D. Prudowsky, S. E. Rudisill, N. N. Degtyareva and J. T. Petty, J. Phys. Chem. C, 2009, 113, 19518–19524 CrossRef CAS.
- Y. Fu, J. Zhang, X. Chen, T. Huang, X. Duan, W. Li and J. Wang, J. Phys. Chem. C, 2011, 115, 10370–10379 CrossRef CAS.
- W. Li, L. Liu, Y. Fu, Y. Sun, J. Zhang and R. Zhang, Photochem. Photobiol. Sci., 2013, 12, 1864–1872 RSC.
- L. Feng, Z. Huang, J. Ren and X. Qu, Nucleic Acids Res., 2012, 40, e122 CrossRef CAS.
- J. T. Petty, D. A. Nicholson, O. O. Sergev and S. K. Graham, Anal. Chem., 2014, 86, 9220–9228 CrossRef CAS.
- J. T. Petty, B. Giri, I. C. Miller, D. A. Nicholson, O. O. Sergev, T. M. Banks and S. P. Story, Anal. Chem., 2013, 85, 2183–2190 CrossRef CAS.
- J. L. Ma, B. C. Yin and B. C. Ye, RSC Adv., 2015, 5, 98467–98471 RSC.
- J. L. Ma, B. C. Yin, H. N. Le and B. C. Ye, ACS Appl. Mater. Interfaces, 2015, 7, 12856–12863 CrossRef CAS.
- J. L. Ma, B. C. Yin, X. Wu and B. C. Ye, Anal. Chem., 2016, 88, 9219–9225 CrossRef CAS.
- B. C. Yin, J. L. Ma, H. N. Le, S. L. Wang, Z. G. Xu and B. C. Ye, Chem. Commun., 2014, 50, 15991–15994 RSC.
- Y. S. Ang, W. W. E. Woon and L. L. Yung, Nucleic Acids Res., 2018, 46, 6974–6982 CrossRef CAS.
- H. C. Yeh, J. Sharma, J. J. Han, J. S. Martinez and J. H. Werner, Nano Lett., 2010, 10, 3106–3110 CrossRef CAS.
- T. Driehorst, P. R. Oneill, P. M. Goodwin, S. Pennathur and D. K. Fygenson, Langmuir, 2011, 27, 8923–8933 CrossRef CAS.
- P. R. O'Neill, L. R. Velazquez, D. G. Dunn, E. G. Gwinn and D. K. Fygenson, J. Phys. Chem. C, 2009, 113, 4229–4233 CrossRef.
- J. T. D. Bonisodonnell, S. Pennathur and D. K. Fygenson, Langmuir, 2016, 32, 569–576 CrossRef.
- H. Arakawa, J. F. Neault and H. A. Tajmirriahi, Biophys. J., 2001, 81, 1580–1587 CrossRef CAS.
- S. Walczak, K. Morishita, M. Ahmed and J. Liu, Nanotechnology, 2014, 25, 155501 CrossRef.
- J. M. Obliosca, M. C. Babin, C. Liu, Y. L. Liu, Y. A. Chen, R. A. Batson, M. Ganguly, J. T. Petty and H. C. Yeh, ACS Nano, 2014, 8, 10150–10160 CrossRef CAS.
- S. W. Yang and T. Vosch, Anal. Chem., 2011, 83, 6935–6939 CrossRef CAS.
- Y. Zhang, C. Zhu, L. Zhang, C. Tan, J. Yang, B. Chen, L. Wang and H. Zhang, Small, 2015, 11, 1385–1389 CrossRef CAS.
- J. T. Petty, B. Sengupta, S. P. Story and N. N. Degtyareva, Anal. Chem., 2011, 83, 5957–5964 CrossRef CAS.
- P. Shah, A. Rørvig-Lund, S. B. Chaabane, P. W. Thulstrup, H. G. Kjaergaard, E. Fron, J. Hofkens, S. W. Yang and T. Vosch, ACS Nano, 2012, 6, 8803–8814 CrossRef CAS.
- J. T. Petty, S. P. Story, S. Juarez, S. S. Votto, A. G. Herbst, N. N. Degtyareva and B. Sengupta, Anal. Chem., 2011, 84, 356–364 CrossRef.
- J. T. Petty, O. O. Sergev, D. A. Nicholson, P. M. Goodwin, B. Giri and D. R. McMullan, Anal. Chem., 2013, 85, 9868–9876 CrossRef CAS.
- P. Shah, S. W. Choi, H. Kim, S. K. Cho, Y. J. Bhang, M. Y. Ryu, P. W. Thulstrup, M. J. Bjerrum and S. W. Yang, Nucleic Acids Res., 2015, 44, e57 CrossRef.
- L. Deng, Z. Zhou, J. Li, T. Li and S. Dong, Chem. Commun., 2011, 47, 11065–11067 RSC.
- L. Zhang, J. Zhu, Z. Zhou, S. Guo, J. Li, S. Dong and E. Wang, Chem. Sci., 2013, 4, 4004–4010 RSC.
- Z. Sun, Y. Wang, Y. Wei, R. Liu, H. Zhu, Y. Cui, Y. Zhao and X. Gao, Chem. Commun., 2011, 47, 11960–11962 RSC.
- Y. Jinjin, H. Xiaoxiao, W. Kemin, Q. Zhihe, W. Xu, S. Hui and Y. Xiaohai, Nanoscale, 2012, 4, 110–112 RSC.
- J. Li, J. You, Y. Zhuang, C. Han, J. Hu, A. Wang, K. Xu and J. J. Zhu, Chem. Commun., 2014, 50, 7107–7110 RSC.
- J. J. Li, X. Q. Zhong, F. F. Cheng, J. R. Zhang, L. P. Jiang and J. J. Zhu, Anal. Chem., 2012, 84, 4140–4146 CrossRef CAS.
- J. Li, X. Zhong, H. Zhang, X. C. Le and J. J. Zhu, Anal. Chem., 2012, 84, 5170–5174 CrossRef CAS.
- M. Zhang, S. M. Guo, Y. R. Li, P. Zuo and B. C. Ye, Chem. Commun., 2012, 48, 5488–5490 RSC.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948–1998 CrossRef CAS.
- X. Zhang, R. M. Kong and Y. Lu, Rev. Anal. Chem., 2011, 4, 105–128 CrossRef CAS.
- L. Gong, H. Kuai, S. Ren, X. H. Zhao, S. Y. Huan, X. B. Zhang and W. Tan, Chem. Commun., 2015, 51, 12095–12098 RSC.
- J. Sharma, H. C. Yeh, H. Yoo, J. H. Werner and J. S. Martinez, Chem. Commun., 2011, 47, 2294–2296 RSC.
- K. Morishita, J. L. Maclean, B. Liu, H. Jiang and J. Liu, Nanoscale, 2013, 5, 2840–2849 RSC.
- W. Guo, J. Yuan and E. Wang, Chem. Commun., 2009, 11, 3395–3397 RSC.
- G. Y. Lan, C. C. Huang and H. T. Chang, Chem. Commun., 2010, 46, 1257–1259 RSC.
- Y. T. Su, G. Y. Lan, W. Y. Chen and H. T. Chang, Anal. Chem., 2010, 82, 8566–8572 CrossRef CAS.
- D. Schultz, S. M. Copp, N. Markešević, K. Gardner, S. S. Oemrawsingh, D. Bouwmeester and E. Gwinn, ACS Nano, 2013, 7, 9798–9807 CrossRef CAS.
- L. Yang, C. Yao, F. Li, Y. Dong, Z. Zhang and D. Yang, Small, 2018, 14, 1800185 CrossRef.
- R. Orbach, W. Guo, F. Wang, O. Lioubashevski and I. Willner, Langmuir, 2013, 29, 13066–13071 CrossRef CAS.
- W. Guo, R. Orbach, I. Mironi-Harpaz, D. Seliktar and I. Willner, Small, 2013, 9, 3748–3752 CrossRef CAS.
- P. R. O'Neill, K. Young, D. Schiffels and D. K. Fygenson, Nano Lett., 2012, 12, 5464–5469 CrossRef.
- S. M. Copp, D. E. Schultz, S. Swasey and E. G. Gwinn, ACS Nano, 2015, 9, 2303–2310 CrossRef CAS.
- S. Pal, R. Varghese, Z. Deng, Z. Zhao, A. Kumar, H. Yan and Y. Liu, Angew. Chem., 2011, 123, 4262–4265 CrossRef.
- N. Li, Y. Shang, R. Xu, Q. Jiang, J. Liu, L. Wang, Z. Cheng and B. Ding, J. Am. Chem. Soc., 2019, 141, 17968–17972 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |