
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalcination-free production of calcium hydroxide at sub-boiling temperatures†
Sara Vallejo Castaño ab,
Erika Callagon La Planteacd,
Sho Shimodaa,
Bu Wange,
Narayanan Neithalathf,
Gaurav Sant*acgh and
Laurent Pilon*bc
ab,
Erika Callagon La Planteacd,
Sho Shimodaa,
Bu Wange,
Narayanan Neithalathf,
Gaurav Sant*acgh and
Laurent Pilon*bc
aLaboratory for the Chemistry of Construction Materials (LC2), Department of Civil and Environmental Engineering, University of California, Los Angeles, CA 90095, USA. E-mail: gsant@ucla.edu; Tel: +1 310 206 3084
bDepartment of Mechanical and Aerospace Engineering, University of California, Los Angeles, CA 90095, USA. E-mail: pilon@seas.ucla.edu; Tel: +1 310 206 5598
cInstitute for Carbon Management, University of California, Los Angeles, CA 90095, USA
dDepartment of Materials Science and Engineering, University of Texas, Arlington, TX 76019, USA
eDepartment of Civil and Environmental Engineering, University of Wisconsin, Madison, WI 53706, USA
fSchool of Sustainable Engineering and the Built Environment, Arizona State University, Tempe, AZ 85287, USA
gDepartment of Materials Science and Engineering, University of California, Los Angeles, CA 90095, USA
hCalifornia Nanosystems Institute, University of California, Los Angeles, CA 90095, USA
First published on 6th January 2021
Abstract
Calcium hydroxide (Ca(OH)2), a commodity chemical, finds use in diverse industries ranging from food, to environmental remediation and construction. However, the current thermal process of Ca(OH)2 production via limestone calcination is energy- and CO2-intensive. Herein, we demonstrate a novel aqueous-phase calcination-free process to precipitate Ca(OH)2 from saturated solutions at sub-boiling temperatures in three steps. First, calcium was extracted from an archetypal alkaline industrial waste, a steel slag, to produce an alkaline leachate. Second, the leachate was concentrated using reverse osmosis (RO) processing. This elevated the Ca-abundance in the leachate to a level approaching Ca(OH)2 saturation at ambient temperature. Thereafter, Ca(OH)2 was precipitated from the concentrated leachate by forcing a temperature excursion in excess of 65 °C while exploiting the retrograde solubility of Ca(OH)2. This nature of temperature swing can be forced using low-grade waste heat (≤100 °C) as is often available at power generation, and industrial facilities, or using solar thermal heat. Based on a detailed accounting of the mass and energy balances, this new process offers at least ≈65% lower CO2 emissions than incumbent methods of Ca(OH)2, and potentially, cement production.
Introduction
Calcium hydroxide (Ca(OH)2), also known as portlandite, slaked- or hydrated-lime finds wide use in industries and applications such as: soil stabilization in road and building construction,1,2 bleaching in the Kraft paper process,3 and as a flocculant in water treatment.4,5 According to the U.S. Geological Survey (USGS), global lime (Ca(OH)2 and CaO) production was 420 million metric tons (t) in 2018.6 Moreover, Ca(OH)2 is of interest in the climate change context because it can uptake/sequester CO2 to form calcium carbonate (CaCO3).7,8 For example, 1 t of Ca(OH)2 can uptake 0.59 t of CO2. Thus, any process that emits less than 0.59 t of CO2 over the course of Ca(OH)2 production has the potential to achieve a “carbon-negative” outcome. Carbonation reactions can also result in cementation.9,10 This is significant as the ability to utilize Ca(OH)2 as a cementation agent,11 or indeed as a feedstock (for CaO) could greatly diminish the CO2 emissions associated with traditional cement (“ordinary portland cement”: OPC) production. Today, however, the production of Ca(OH)2 features a CO2 footprint similar to OPC production (∼1 t of CO2 per t of Ca(OH)2 produced). This is because Ca(OH)2 production, similar to OPC production is based around the age-old process of limestone calcination (i.e., thermal decomposition, which results in the desorption of the “mineralized CO2” bound within the limestone and accounts for ≈65% emissions associated with the process; the remainder being from the combustion of fossil fuels to power kiln operations) at ≈900 °C to produce CaO,12,13 the precursor of Ca(OH)2; that is subsequently granulated and hydrated (i.e., contacted with water) to produce Ca(OH)2. In addition, the process requires quarried limestone that not only consumes nonrenewable mineral resources, but also disrupts ecosystems, and causes pollution and biodiversity destruction.14,15Ca-Containing alkaline wastes accrue as a waste-/by-product of numerous large-scale industries. This includes, for example, slags from iron and steel production, and fly ashes from coal combustion. These materials are not only produced in large amounts on an ongoing basis, but there exist tremendous historical reservoirs of these materials in the U.S. and across the world.16–18 For example, even now the U.S. produces greater than 17 million t of slag19,20 and 91 million t of coal combustion residuals. Of the latter, only 58% are beneficially utilized.21 And, global waste reservoirs host well over a billion tons of slag,16 and tens of billions of tons of fly ash,22–24 based on broad estimates. Underutilized quantities of alkaline wastes, i.e., fly ashes and slags, are routinely disposed in landfills, which not only implies a waste disposal cost (“tipping fees”, NB: the average tipping fee in the U.S. is on the order of $50 per t),25 but can also result in environmental damage.26–29 Therefore, with a specific goal of enhancing waste utilization, resource recovery and reutilization, and circular economy, we demonstrate an approach to extract Ca-from alkaline industrial wastes,30,31 and produce Ca(OH)2 via a calcination-free process. While herein we focus on alkaline industrial wastes, of course, the broad contours of this process are also applicable to alkaline rock species, which although harder to solubilize unless externally stimulated,32,33 offer the potential to offer a near-limitless source for alkaline element extraction.
New concept
The conceptual basis of our new process involves the leaching/dissolution of Ca-bearing wastes to mobilize Ca-species in solution (see process flow diagram, Fig. S1†). This results in an aqueous Ca-abundance that is typically substantively lower than the saturation concentration of Ca(OH)2 (∼21.2 mmol L−1 at pH = 12.475 and T = 25 °C (ref. 34)).30,31,35 Therefore, it is necessary to concentrate the leachate by a factor of 2 or more times depending on the initial Ca-content of the leachate. Such concentration is achieved by reverse osmosis (RO) based ion-separations wherein by means of size and/or charge exclusion;36 divalent cations (e.g., Ca2+, Mg2+) can be enriched in the retentate stream. RO is used to achieve large production rates of saturated solution and to improve the energy efficiency of the concentration process. The retentate is then subjected to a temperature swing, e.g., using low-grade waste heat (≤100 °C) as derived from the flue gas of a power plant or another heavy industry operation, wherein the retrograde solubility of Ca(OH)2 (ref. 37) ensures its precipitation to an increasing extent with increasing temperature. The advantage of such a low-temperature approach is straightforward: it obviates the need for limestone's decarbonation and eliminates the associated CO2 emissions. Moreover, the precipitation step can be accomplished at ambient pressure and at sub-boiling temperatures, enabling its deployment using a wide range of potential waste heat streams including coal power plants and combined cycle natural gas power plants.38 Besides, operating at temperatures above 100 °C would be energetically inefficient due to the low solubility of Ca(OH)2 in water39 and the large heat of vaporization of water. These general characteristics facilitate process intensification (PI) by renewable electrification and/or renewable heat (solar thermal) integration. Towards this end, we establish the feasibility of the low-temperature process, assess its limitations, and finally compare its energy and CO2 intensity to incumbent methods.Materials and methods
Slag leaching
Slag leachates were prepared by contacting 1, 3, 5, and 10 g of a technical basic oxygen furnace (BOF) slag (i.e., a crystalline slag‡) with 100 mL DI water to achieve solid to liquid mass fractions (s/l) of 0.01, 0.03, 0.05, and 0.1, respectively. To prepare the slag for leaching, slag fines (<9.5 mm) were dried at 70 °C for 24 h, ground for 1 hour in a planetary ball mill (MSK-SFM-1), and manually sieved to retain particles with a size lower than 106 μm. Batch leaching was conducted in 200 mL plastic bottles under either unstirred or stirred conditions at room temperature. Stirring was controlled using a Corning LSE orbital shaker (6780 NP) at 200 rpm. Solution aliquots of 2 mL were extracted after 0, 5, 10, 30, 60, 180, 360 and 720 minutes for analysis of total dissolved calcium, silicon, sodium, aluminum, and magnesium analysis using a PerkinElmer Avio 200 inductively coupled plasma – optical emission spectrometer (ICP-OES). Prior to elemental analysis, the samples were filtered through 0.2 μm syringe filters and then diluted in 5 vol% HNO3. The ICP-OES was calibrated using standard solutions containing the elements of interest in concentrations of 0, 0.1, 1, 10, 25, 50, and 100 ppm which were prepared using analytical (1000 ppm) standards purchased from Inorganic Ventures.The chemical composition (in mass%) of the simple oxide constituents of the slag was measured using X-ray fluorescence (XRF). The BOF slag was dominantly composed of CaO (38 mass%), followed by Fe2O3 (31%), SiO2 (13%), MgO (6%), and Al2O3 (4%). This information was used to quantify calcium extraction (XCa) according to:  where nCa,sln(t) are the moles of calcium in the solution at time t, and nCa,slag are the moles of calcium in the solid slag as measured by XRF. The calcium leaching rate ṅCa (in mmolCa/ h−1) at time t was defined as:
where nCa,sln(t) are the moles of calcium in the solution at time t, and nCa,slag are the moles of calcium in the solid slag as measured by XRF. The calcium leaching rate ṅCa (in mmolCa/ h−1) at time t was defined as: 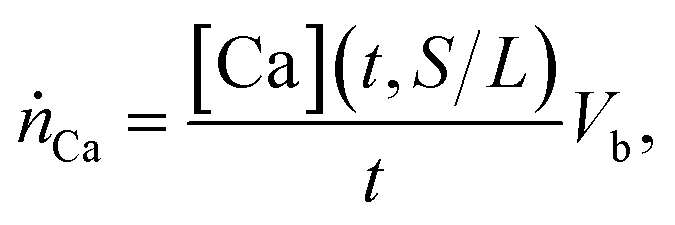 where Vb is the solution volume and t is the leaching time. The leaching rate affects the production rate (“yield”) of Ca(OH)2, and the efficiency of slag valorization because, from stoichiometry, one mole of Ca-leached from the slag should produce one mole of Ca(OH)2 as per the reaction: Ca2+ + 2OH− ↔ Ca(OH)2.
where Vb is the solution volume and t is the leaching time. The leaching rate affects the production rate (“yield”) of Ca(OH)2, and the efficiency of slag valorization because, from stoichiometry, one mole of Ca-leached from the slag should produce one mole of Ca(OH)2 as per the reaction: Ca2+ + 2OH− ↔ Ca(OH)2.
Reverse osmosis (RO) concentration
RO concentration was carried out using two calcium-rich alkaline solutions, namely a reagent grade Ca(OH)2 solution and a slag leachate solution. Reagent grade Ca(OH)2 was added to 1 L deionized (DI) water (resistivity ≥ 18.2 MΩ cm) to produce 5 and 10 mmol L−1 (mM) reagent grade Ca(OH)2 solutions. On the other hand, the slag leachate was prepared by leaching, over time, 150 g of as-received basic oxygen furnace (BOF) slag into 3 L of DI water (s/l = 0.05) at 25 °C. The concentration of total dissolved calcium [Ca] in the solution after leaching was generally in excess of 14 mM, whereas sodium, potassium, silicon, and aluminum impurities were present in concentrations lower than 0.1 mM, as measured by ICP-OES. The concentration of [Ca] in the leachate was adjusted to 5 mM and 10 mM by dilution with DI water to obtain 1 L of solution. Thereafter, the solutions were concentrated using a cross-flow flat-sheet membrane cell (FR42 Sterilities Corp.) fitted with thin film composite (TIC) polyamide RO membranes sourced from Dow Filmtec® (BW30XFR) with an active area of 42 cm2. Prior to each experiment, the membranes were pre-treated by soaking them in DI water for at least 24 hours. A new membrane was used for each experiment to ensure reproducibility. Additionally, a gear pump (GJ Series Micropump®) was used to pressurize the Ca-containing solutions through the membrane. Furthermore, 99% pure N2(g) at atmospheric pressure was continuously bubbled into the feed tank at a flowrate of 0.02 L min−1 to minimize the presence of atmospheric CO2 and to avoid the carbonation of the alkaline Ca-rich solution, and the resulting (undesirable) formation of CaCO3.The feed stream, with an initial Ca-concentration [Ca]i of 5 or 10 mM, was pressurized through the RO membrane at constant flow rate of 120 mL min−1 and gauge pressure of 413 kPa. A by-pass line was used to maintain a constant feed pressure throughout the duration of the experiment. The retentate was recirculated back to the feed tank for further concentration while the solution that permeated through the membrane was collected in the permeate tank. The concentration of calcium as a function of time was measured by taking 5 mL aliquots from both the feed [Ca]f and the permeate [Ca]p tanks every hour. Additionally, the permeate mass flow rate was monitored using an analytical balance (mass resolution: 1 mg) and a timer. A schematic of the RO experimental setup is shown in Fig. S2.† The performance of the RO system was assessed based on three typical metrics: (i) the water recovery Y, (ii) the salt rejection R, and (iii) the concentration factor CF. The water recovery Y (in %), represents the ratio of water volume recovered as permeate Vp (in L), to the initial volume of solution Vi (in L), i.e.,40  . Note that in industrial systems, the water recovery Y ranges typically between 35–85%.40–43 In addition, salt rejection R (in %) represents the fraction of dissolved species that did not pass to the permeate side i.e., the percentage of Ca-rejected by the membrane which is expressed as:44
. Note that in industrial systems, the water recovery Y ranges typically between 35–85%.40–43 In addition, salt rejection R (in %) represents the fraction of dissolved species that did not pass to the permeate side i.e., the percentage of Ca-rejected by the membrane which is expressed as:44  where [Ca]f and [Ca]p are the Ca-concentration in the feed and permeate solutions, respectively. For reference, NaCl rejection ranges from 95–99% for the membrane used herein. Finally, the retentate concentration factor CFr and the permeate concentration factor CFp were respectively defined as the ratio of [Ca]f and [Ca]p to the initial calcium concentration in solution [Ca]i.45
where [Ca]f and [Ca]p are the Ca-concentration in the feed and permeate solutions, respectively. For reference, NaCl rejection ranges from 95–99% for the membrane used herein. Finally, the retentate concentration factor CFr and the permeate concentration factor CFp were respectively defined as the ratio of [Ca]f and [Ca]p to the initial calcium concentration in solution [Ca]i.45
Following concentration, scaling was observed on the RO membranes. To analyze the mineral scale formed, the membranes were dried for 24 h under vacuum conditions. Thereafter, carbon tape was used to remove the scale and to mount it on a metallic support. The morphology and elemental composition of the dried solids were evaluated using a FEI Nova 230 Scanning Electron Microscope fitted with an energy dispersive (SEM-EDS) X-ray analyzer under high vacuum conditions and at an accelerating voltage of 15 kV.
Ca(OH)2 precipitation by temperature swing
Ca(OH)2 precipitation was carried out by adding reagent grade Ca(OH)2 to 100 mL DI water to obtain solutions saturated in Ca(OH)2 at 25 °C (∼21 mM).37 In addition, the actual slag leachate obtained following RO concentration was also utilized in the precipitation experiments. The experiments were carried out by placing the solutions in a stirred jacketed glass reactor, which was connected to a circulating water bath (Polyscience AD07R-20) that was thermally equilibrated at 25 °C. The reactor was sealed with Parafilm™ to minimize evaporation and N2(g) was bubbled through the solution to inhibit CO2 dissolution in water and the subsequent carbonation of Ca(OH)2. To exploit the (inverse) solubility dependence of Ca(OH)2 with temperature and to induce precipitation, the solution temperature was increased by circulating hot water through the jacketed reactor. Reagent grade Ca(OH)2 solutions were heated to 85 °C at (actual) heating rates (dT/dt) of 10 °C h−1, 27 °C h−1, and 54 °C h−1. The solution's electrical conductivity σexp, pH, and temperature T were measured continuously using a ThermoScientific Orion Versa Star meter. These measurements were used to determine the onset of Ca(OH)2 precipitation and the [Ca] concentration in solution according to:,46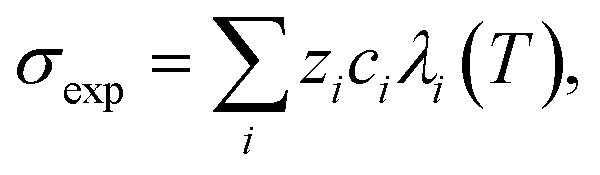 where λi is the equivalent conductivity, zi is the valence, and ci is the concentration of each ionic species (i = Ca2+, OH−). The equivalent conductivity of any ionic species λi was calculated from the Onsager relations expressed as:47 λi(T) = λi(T) − Si(T)Γ1/2, where λi is the ionic conductivity of an ionic species at infinite dilution, S is a parameter dependent on the temperature, viscosity, and dielectric constant of the medium, and Γ is the ional concentration of the solution (see ESI†).47 In the case of precipitation using the concentrated slag leachate, the temperature was increased to 70 °C at a fixed rate of 27 °C h−1.
where λi is the equivalent conductivity, zi is the valence, and ci is the concentration of each ionic species (i = Ca2+, OH−). The equivalent conductivity of any ionic species λi was calculated from the Onsager relations expressed as:47 λi(T) = λi(T) − Si(T)Γ1/2, where λi is the ionic conductivity of an ionic species at infinite dilution, S is a parameter dependent on the temperature, viscosity, and dielectric constant of the medium, and Γ is the ional concentration of the solution (see ESI†).47 In the case of precipitation using the concentrated slag leachate, the temperature was increased to 70 °C at a fixed rate of 27 °C h−1.
To confirm the composition of the precipitates, following precipitation, the solution was vacuum filtered using Whatman filters (3–5 μm pore size) and a heated ceramic funnel to minimize the redissolution of the precipitated Ca(OH)2. The crystals were then dried in an initially N2(g)-purged desiccator for 24 h under vacuum at room temperature to remove water and minimize the carbonation of Ca(OH)2. The dried solids were placed on a carbon tape for morphology and elemental analysis using SEM-EDS, as previously discussed. Further, a small amount (∼3 mg) of dried Ca(OH)2 crystals were characterized using thermogravimetric analysis (TGA) using a PerkinElmer STA 6000. To do so, the sample was initially equilibrated at 35 °C for 5 min to establish a mass baseline and then heated gradually from 35 °C-to-900 °C at a rate of 10 °C min−1 while continuously monitoring the mass. Ultra-high purity N2(g) was circulated through the sample chamber at a flow rate of 20 mL min−1. The mass change versus temperature revealed the presence and quantity of Ca(OH)2 and CaCO3 present on account of their characteristic thermal decomposition at temperatures in excess of ∼400 °C48,49 and ∼600 °C,50 respectively.
Results and discussion
Slag leaching
Fig. 1 shows the calcium concentration [Ca] as a function of time (in log scale) for slag particles smaller than 106 μm leached in DI water under (a) unstirred and (b) stirred conditions. It indicates that [Ca] concentration increased, approximately by a factor of 2, from unstirred to stirred conditions during the first hour of reaction. Stirring enhanced the rate of slag dissolution because calcium transport from the solid/liquid interface to the bulk solution was aided by convective mass transfer, as opposed to unstirred conditions wherein dissolution is transport (diffusion) limited.51 Increasing s/l resulted in larger [Ca] concentrations since adding more slag increased the amount of calcium available for leaching. Fig. 1(b) indicates that the maximum [Ca] concentration obtained was on the order of 17.3 ± 3.1 mM for s/l = 0.1 after 6 h, under stirred conditions, which is around 75% of the saturation concentration of Ca(OH)2 at 20 °C (22 mM).34 The decrease in the [Ca] concentration after achieving its peak value is indicative of secondary precipitation that consumed some of the [Ca] in solution. Such secondary precipitation is consistent with the reduction in the [Si] concentration in the leachate at similar times (see ESI: Fig. S3†), suggesting the precipitation of a calcium silicate hydrate (C–S–H) phase.52 As such, although calcium leaching is enhanced and somewhat accelerated by increasing s/l, the amount of [Ca] that can be sustained in solution is limited by the equilibrium solubility of one or more phases, including Ca(OH)2.30,53,54 In general, Fig. 1(a) and (b) highlight that reducing s/l reduced the amount of Ca-extracted into solution, at shorter and longer reaction times, and independent of the stirring condition. It should be noted, however, that the amount of Ca-leached, depends strongly on the type of slag used (see Fig. S4†).20,54–56 The implication: that certain slags may be more suitable than others for Ca-extraction, and that depending on the slag used, multiple (2 or more) concentration cycles may need to be carried out to sufficiently enrich the Ca-concentration in solution with obvious energy intensity implications. This issue while also applicable to fly ashes, is less relevant, since fly ashes, especially the CaO-rich variants rapidly mobilize their Ca-species in solution.18,57,58 | ||
| Fig. 1 The aqueous [Ca]-concentration as a function of time during batch leaching of a BOF slag for s/l = 0.01, 0.03, 0.05, and 0.1 under (a) unstirred, and (b) stirred conditions. In general, increasing the s/l resulted in larger [Ca] concentrations since the enhanced quantity of slag increased the surface area, and amount of Ca-available for leaching in the solid phase. (c) The calculated calcium conversion XCa, i.e., the ratio (fraction) of the amount of Ca-in solution to the Ca-in the slag solids, and (d) the calcium leaching rate as a function of time for different s/l under stirred conditions. | ||
Fig. 1(c) and (d) shows the calcium extraction XCa and the calcium leaching rate ṅCa as functions of time for different s/l for stirred conditions. Fig. 1(c) shows that XCa decreased with increasing s/l; i.e., suggesting reducing efficiency of Ca-extraction from the slag with increasing s/l;17 albeit offering meaningfully faster Ca-mobilization into solution (see Fig. 1(d)). Taken together, the data in Fig. 1 indicates: (a) larger s/l maximizes the amount of [Ca] leached and a smaller s/l maximizes XCa (b) with short duration leaching (in a single cycle), in the best case (s/l = 0.01, 24 hours leaching) no more than 8 mol% of the Ca-content of slag was extracted.59 Finally and unsurprisingly, with increasing leaching time and a decreasing undersaturation with respect to the dissolving/leaching slag, the Ca-leaching rate ṅCa decreased exponentially with time. Thus, practically, the leaching time should be reduced as much as possible60 (i.e., ≤6 hours) to achieve a critical (minimal) Ca-content in solution as would be appropriate for the follow-on RO concentration step.
RO concentration
RO concentration was carried out using reagent grade Ca(OH)2 solutions and slag leachate solutions. The processing of 1 L of feed solution took between 8 and 10 h for an average feed flow rate around 120 cm3 min−1 and gauge pressure of 413 kPa. The calcium concentration in the feed tank [Ca]f increased throughout the duration of the experiment while the calcium concentration in the permeate tank [Ca]p remained below 1 mM. The final calcium concentration in the feed tank [Ca]f readily approached the saturation level of Ca(OH)2 (∼21 mM at 25 °C)37 in both reagent grade Ca(OH)2 and slag leachate solutions for an initial calcium concentration [Ca]i of 10 mM.Fig. 2 shows the permeate concentration factor CFp and the retentate concentration factor CFr as functions of time for: (a) reagent-grade Ca(OH)2 solutions and (b) slag leachate solutions for initial feed calcium concentrations [Ca]i of 5 mM and 10 mM. It indicates that the retentate concentration factor CFr was always smaller for the solutions with larger initial feed concentration [Ca]i. This difference is on account of the osmotic pressure π, which is larger for solutions with a larger initial feed concentration [Ca]i according to the Van't–Hoff relation:61 π = RUTf([Ca]f + [OH−]f), where RU is the universal gas constant, Tf is the feed temperature and [Ca]f and [OH−]f are the calcium and hydroxide concentrations in the feed solution, respectively. Since larger concentration leads to larger osmotic pressure π, the flow rate of permeate solution ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) p crossing the membrane decreased according to:61 p = AmLp(ΔP − Δπ), where Am is the membrane area, Lp is the membrane permeability, ΔP is the pressure difference and Δπ is the osmotic pressure difference, between the feed and permeate solutions. Since the experiments were performed at a constant feed pressure, ΔP was constant. On the other hand, increasing the initial calcium concentration [Ca]i from 5 to 10 mM increased Δπ, thus reducing the driving force (ΔP − Δπ) and decreasing the permeate flow rate p. Consequently, the retentate concentration factor CFr as a function of time was slightly lower for solutions with [Ca]i of 10 mM compared to 5 mM.
p crossing the membrane decreased according to:61 p = AmLp(ΔP − Δπ), where Am is the membrane area, Lp is the membrane permeability, ΔP is the pressure difference and Δπ is the osmotic pressure difference, between the feed and permeate solutions. Since the experiments were performed at a constant feed pressure, ΔP was constant. On the other hand, increasing the initial calcium concentration [Ca]i from 5 to 10 mM increased Δπ, thus reducing the driving force (ΔP − Δπ) and decreasing the permeate flow rate p. Consequently, the retentate concentration factor CFr as a function of time was slightly lower for solutions with [Ca]i of 10 mM compared to 5 mM.
 | ||
| Fig. 2 The permeate concentration factor CFp (blue) and retentate concentration factor CFr (orange) as a function of time for initial calcium concentration [Ca]i of 5 and 10 mM for: (a) reagent-grade Ca(OH)2 solution, and (b) slag leachate solutions. RO concentration data showing permeate concentration factor CFp (blue) and retentate concentration factor CFr (orange) as a function of water recovery Y for: (c) reagent grade Ca(OH)2 solution, and (d) slag leachate solutions. The dashed line corresponds to maximum theoretical CFr assuming 100% calcium rejection by the RO membrane. | ||
In addition, Fig. 2 also shows the permeate concentration factor CFp and the retentate concentration factor CFr as a function of the water recovery Y for reagent grade Ca(OH)2 solutions and slag leachate solutions. Assuming that all the calcium in the feed solution was rejected by the membrane (R = 100%, CFp = 0), the maximum theoretical value of CFr can be calculated from a mass balance as:62  . Fig. 2(c) and (d) indicates that the calculated retentate concentration factor CFr follows the trend corresponding to 100% rejection for both reagent grade Ca(OH)2 and slag leachate solutions. The slight difference might be due to the uncertainties in calculating the permeate volume, which was not measured directly but estimated from permeate flow rate measurements. Moreover, Fig. 2 reveals that the difference between the retentate concentration factor CFr calculated for the two initial feed concentrations ([Ca]i = 5 mM, 10 mM) was somewhat larger for the slag leachate solutions than reagent grade Ca(OH)2 solutions. This observation could be due to either membrane degradation or membrane scaling. For example, membrane degradation may result in a decrease in the calcium rejection R which would increase the Ca-concentration in the permeate [Ca]p.63 On the other hand, a typical sign of membrane scaling is a decrease in the permeate flow rate p.64–67 We observed that calcium rejection R remained above 90% for both reagent grade Ca(OH)2 and slag leachate solutions. This implies that R was not affected by the presence of impurities in the slag leachate, and that membrane degradation was not significant (N.B.: This was also confirmed using longer duration testing carried out using a pilot-scale production system). On the other hand, significant differences were observed in the permeate flow rate p. In fact, p remained constant at 0.9 ± 0.1 mL min−1 for both 5 and 10 mM reagent grade Ca(OH)2 solutions, whereas it declined when water recovery reached 40% in the case of the 10 mM slag leachate solutions. This decline in p is attributed to scale formation on the membrane surface, e.g., due to the formation of Ca(OH)2, and/or hydrated calcium phases. Based on the retentate composition at 6 hours, Ca(OH)2 and 11 Å Tobermorite have saturation indexes of 0.03 and 2.35, respectively (see Fig. S5†) – suggesting their precipitation.68,69 The results of the RO concentration and the equilibrium calculations also highlight that the solubility of portlandite decreased in comparison with reagent grade Ca(OH)2 solutions, i.e., on account of the common-ion effect, because the presence of other alkaline species (e.g., Na+, K+) in solution – extracted during leaching – alkalinizes the solution shifting the equilibrium towards Ca(OH)2 precipitation.
. Fig. 2(c) and (d) indicates that the calculated retentate concentration factor CFr follows the trend corresponding to 100% rejection for both reagent grade Ca(OH)2 and slag leachate solutions. The slight difference might be due to the uncertainties in calculating the permeate volume, which was not measured directly but estimated from permeate flow rate measurements. Moreover, Fig. 2 reveals that the difference between the retentate concentration factor CFr calculated for the two initial feed concentrations ([Ca]i = 5 mM, 10 mM) was somewhat larger for the slag leachate solutions than reagent grade Ca(OH)2 solutions. This observation could be due to either membrane degradation or membrane scaling. For example, membrane degradation may result in a decrease in the calcium rejection R which would increase the Ca-concentration in the permeate [Ca]p.63 On the other hand, a typical sign of membrane scaling is a decrease in the permeate flow rate p.64–67 We observed that calcium rejection R remained above 90% for both reagent grade Ca(OH)2 and slag leachate solutions. This implies that R was not affected by the presence of impurities in the slag leachate, and that membrane degradation was not significant (N.B.: This was also confirmed using longer duration testing carried out using a pilot-scale production system). On the other hand, significant differences were observed in the permeate flow rate p. In fact, p remained constant at 0.9 ± 0.1 mL min−1 for both 5 and 10 mM reagent grade Ca(OH)2 solutions, whereas it declined when water recovery reached 40% in the case of the 10 mM slag leachate solutions. This decline in p is attributed to scale formation on the membrane surface, e.g., due to the formation of Ca(OH)2, and/or hydrated calcium phases. Based on the retentate composition at 6 hours, Ca(OH)2 and 11 Å Tobermorite have saturation indexes of 0.03 and 2.35, respectively (see Fig. S5†) – suggesting their precipitation.68,69 The results of the RO concentration and the equilibrium calculations also highlight that the solubility of portlandite decreased in comparison with reagent grade Ca(OH)2 solutions, i.e., on account of the common-ion effect, because the presence of other alkaline species (e.g., Na+, K+) in solution – extracted during leaching – alkalinizes the solution shifting the equilibrium towards Ca(OH)2 precipitation.
Fig. 3(c) shows SEM images of the mineral scale formed on the RO membranes after 10 h of concentration of a slag leachate solution with an initial calcium concentration [Ca]i of 10 mM. Here (and in the case of Ca(OH)2 solutions), the crystallite size ranged from 100 μm to 300 μm. However, a higher area density of precipitates was observed on the membrane used for concentrating slag leachate solutions compared to that used with reagent grade Ca(OH)2 solution. This suggests that the presence of impurities in the slag leachate enhanced scale formation; once again, on account of the common-ion effect. This also explains why the permeate flow rate decline was stronger with slag leachate solutions than with reagent grade Ca(OH)2 solutions with the same initial calcium concentration. Nevertheless, scaling could be avoided through precautionary measures. For example, stopping RO concentration before reaching Ca(OH)2 saturation, or reversing the feed flow direction periodically to change the saturation index of precipitate phases within the RO cell.66,70 Table 1 summarizes EDS elemental analysis (in atom%) of crystallites deposited on the membrane surfaces (the sampling points are shown in the inset of Fig. 3(c)). The O–Ca ratio was noted as being 2 ± 0.3 across all solutions considered (see Table 1), whereas carbon, aluminum, and silicon were detected only in small quantities suggesting compositions consisting of mainly Ca(OH)2, and small quantities of CaCO3 and/or low-rank hydrated calcium-silicate compounds.71
 | ||
| Fig. 3 The characterization of precipitated Ca(OH)2 crystals obtained from saturated slag leachate solutions using: (a) thermal analysis showing evidence of Ca(OH)2 and CaCO3, respectively, (b) micrographs confirming the presence of Ca(OH)2 and CaCO3, the former of which are identified by the hexagonal structure, and the latter by their equiaxed form, and (c) micrographs of membrane scale obtained from slag leachate solutions. The yellow squares indicate the location of SEM-EDS sampling points. | ||
| Element (atom%) | Ca(OH)2 solution | Slag leachate solution |
|---|---|---|
| Ca | 32.55 ± 0.94 | 28.80 ± 2.68 |
| O | 60.89 ± 1.99 | 59.48 ± 7.48 |
| C | 6.47 ± 1.18 | 11.32 ± 9.74 |
| Al | 0.03 ± 0.01 | 0.14 ± 0.03 |
| Si | 0.06 ± 0.03 | 0.03 ± 0.02 |
Precipitation of portlandite from RO-concentrated solutions
Following concentration, precipitation experiments were carried out using the reagent grade Ca(OH)2 and slag leachate solutions. Temperature-swing induced precipitation was examined using concentrated slag leachates which featured a [Ca] concentration around ∼19 mM (i.e., wherein the starting Ca-concentration was 10 mM). Herein, at 45 °C a change in the solution's appearance occurred (i.e., from transparent to translucent) corresponding to the formation of colloidal-scale and/or hyper-branched precipitates (e.g., of C–S–H) that induced strong Tyndall scattering effects.72,73 At the end of the temperature ramping process, ∼3 mg of precipitates were collected (via filtration) from the saturated slag leachate solution. Fig. 3 shows the characterization of precipitates obtained from the slag leachate using (a) TGA and (b) SEM. Herein, TGA (Fig. 3(a)) shows that 66 mass% of the analyzed precipitates corresponding to Ca(OH)2 started to decompose at 400 °C.49,74 An additional 15 mass% of the precipitates started to decompose around 600 °C, indicating the presence of impurity CaCO3 that formed due to: (a) the presence of dissolved CO2 in the feed water.50 and/or, (b) brief exposure of the wet Ca(OH)2 precipitates to air during drying wherein they readily carbonate on contact with atmospheric CO2.75 The remaining 19 mass% impurities appear to be composed of slag particulates. As such, although the formation of Ca(OH)2 was confirmed, it is important to control the ambient conditions during precipitation, separation and drying to avoid Ca(OH)2 carbonation. Fig. 3(b) shows representative SEM images of the precipitates whose particle size ranged between 2 and 10 μm. While it is indeed anticipated that particles smaller than 2 μm were present, they were not collected due to the large pores of the filter used for separation (>3 μm). Moreover, the precipitated crystals featured two differentiated morphologies: (i) small hexagonal platelet aggregates corresponding to Ca(OH)2 (ref. 76 and 77) and (ii) individual crystals with rhomb-like structures resembling CaCO3.78,79Finally, Fig. 4 shows (a) the measured ionic conductivity and (b) the corresponding [Ca] concentration [i.e., calculated using eqn (S1)–(S10)]† as functions of temperature for different heating rates ranging from 10 to 50 °C h−1 for near-saturated reagent grade Ca(OH)2 solutions. Also shown is the theoretical solution conductivity σcalc and estimated [Ca] concentration for a solution with an initial Ca-concentration of 21.2 mM: (i) in the absence of precipitation and (ii) during ongoing Ca(OH)2 precipitation which acts to remove calcium and hydroxyl species from solution thereby resulting in a reduction of the solution conductivity. The calculated saturation index (SI) of Ca(OH)2 as a function of temperature for [Ca]initial = 21.2 mM is shown in Fig. 4(c). In qualitative compliance with classical nucleation theory (CNT), Fig. 4 demonstrates that the rate of nucleation is negligible until a critical supersaturation, here 0.25 corresponding to the SI at T = 65 °C, is reached. It is interesting to note that the onset of precipitation occurred at the same critical supersaturation across all heating rates; i.e., as indicated by the reduction of the measured conductivity σexp vis-à-vis the calculated conductivity σcalc and the [Ca] concentration in solution. Nonetheless, the heating rate may affect the precipitation kinetics80,81 and final properties of the precipitates as shown, for example, for the aqueous precipitation of TiO2 from TiOCl4 solutions.82 Overall, across all ramp rates, ∼5 millimoles of Ca(OH)2 per liter of solution were precipitated while heating to 85 °C, consistent with the calculated equilibrium precipitation yield based on the (retrograde) solubility of Ca(OH)2.
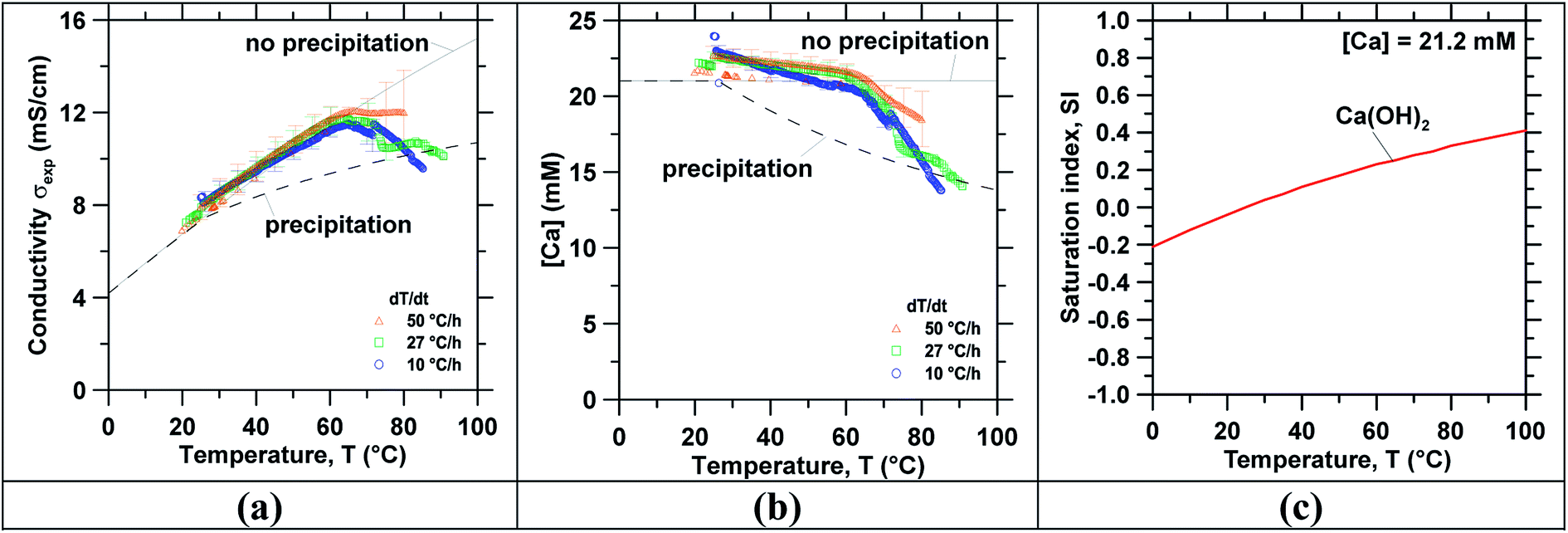 | ||
| Fig. 4 The measured: (a) solution's electrical conductivity as a function of temperature, (b) the corresponding [Ca] concentration (calculated from the measured conductivity), and (c) the saturation index (SI) (calculated using PHREEQC with minteq v4 database) as a function of the temperature for a solution with [Ca] = 21.2 mM, added as Ca(OH)2. This data was obtained during the precipitation of reagent-grade Ca(OH)2 solutions at different temperature ramp rates. | ||
Energy and CO2 balances associated with traditional and novel methods of Ca(OH)2 production
The traditional pathway for Ca(OH)2 production involves the thermal decomposition of limestone (CaCO3) to lime (CaO) at 900 °C; following which the lime is slaked (hydrated) to produce Ca(OH)2.83,84 Here, high-grade heat is sourced from the combustion of fossil fuels. The enthalpic energy consumption QT to produce 1 kg of Ca(OH)2 can be estimated (i.e., without considering thermal losses, or inefficiency) as where MCa(OH)2 = 0.0741 kg mol−1 is the molar mass of Ca(OH)2, ΔHrxn = 179.2 kJ mol−1 CaCO3 is the enthalpy of the calcination reaction CaCO3 → CaO + CO2.49 The thermal efficiency
where MCa(OH)2 = 0.0741 kg mol−1 is the molar mass of Ca(OH)2, ΔHrxn = 179.2 kJ mol−1 CaCO3 is the enthalpy of the calcination reaction CaCO3 → CaO + CO2.49 The thermal efficiency  of a typical vertical shaft kiln – the lime industry standard – is around 60%.84 Thus, the actual energy consumption of the traditional thermal process is on the order of 4 MJ kg−1 Ca(OH)2,85 and can be substantively larger for rotary lime kilns.12,84,86
of a typical vertical shaft kiln – the lime industry standard – is around 60%.84 Thus, the actual energy consumption of the traditional thermal process is on the order of 4 MJ kg−1 Ca(OH)2,85 and can be substantively larger for rotary lime kilns.12,84,86
The energy required to produce 1 kg of Ca(OH)2 via the low-temperature precipitation process can be calculated based on mass and energy conservation principles, and the data from the prior sections (see Fig. S1†). For example, the mechanical work WRO necessary to pressurize the water through the RO system with a high-pressure pump was calculated to be 2.23 MJ kg−1 Ca(OH)2 according to WRO = VΔP, where V = 5.4 m3 is the total volume of solution and ΔP = 414 kPa is the pressure difference between the pump inlet and outlet for the process conditions summarized in Fig. S1.† Similarly, the mechanical work of the low-pressure pumps WP can also be calculated as  , where, ΔP is the pressure drop due to friction losses in pipes,87 D is the pipe diameter (D = 0.009 m), L = 1 m is the length of the pipe, ρ = 1000 kg m−3 is the solution density, and f = 0.041 is Darcy's friction factor corresponding to the solution volumetric flow rate necessary to produce 1 kg of Ca(OH)2 per day ( = 3.1 × 10−5 m3 s−1). Under these assumptions, ΔP is equal to 474 Pa. The mechanical work of the low-pressure pumps WP (is multiplied by 2 to account for the number of pumps) was 5.1 kJ kg−1 of Ca(OH)2. This number is nearly three orders of magnitude lower than the work of the RO pump WRO.
, where, ΔP is the pressure drop due to friction losses in pipes,87 D is the pipe diameter (D = 0.009 m), L = 1 m is the length of the pipe, ρ = 1000 kg m−3 is the solution density, and f = 0.041 is Darcy's friction factor corresponding to the solution volumetric flow rate necessary to produce 1 kg of Ca(OH)2 per day ( = 3.1 × 10−5 m3 s−1). Under these assumptions, ΔP is equal to 474 Pa. The mechanical work of the low-pressure pumps WP (is multiplied by 2 to account for the number of pumps) was 5.1 kJ kg−1 of Ca(OH)2. This number is nearly three orders of magnitude lower than the work of the RO pump WRO.
The sensible heat required to induce the temperature-swing, and in turn, portlandite precipitation, Qs,p was estimated as follows. Previously, it was demonstrated that 5 millimoles of Ca(OH)2 precipitate when heating near-saturated reagent grade Ca(OH)2 solutions from 25 °C to 85 °C (ΔT = 60 °C). As such, following a simple mass balance, the mass of water to produce 1 kg of Ca(OH)2 is m = 2700 kg H2O per kg Ca(OH)2. Assuming >90 mass% recovery of water, this translates into a consumable water demand of less than 270 kg H2O per kg Ca(OH)2. Thus, the sensible heat demand was calculated as Qs,p = mcpΔT = 677.15 MJ kg−1 Ca(OH)2, where cp = 4.18 kJ kg−1 is the specific heat of water.88 This indicates that the thermal energy demand for precipitation is two orders of magnitude larger than the mechanical energy consumption of the RO pump because only 0.37 g of Ca(OH)2 is precipitated per liter of water. However, the thermal energy consumption could be reduced by more than 50%, e.g., by implementing heat recovery loops. Furthermore, since the operating temperature for precipitation is below 90 °C, this energy could be sourced simply in the form of low-grade (e.g., flue gas borne) waste heat from industrial facilities such as thermal plants fired by coal, gas or nuclear power, steel processing facilities, and renewable heat sources like geothermal or concentrated solar power. For example, a mid-sized coal-fired power plants produces up to 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 GJ of low-grade thermal energy per year corresponding to exhaust temperatures up to 128 °C (at the stack).38
000000 GJ of low-grade thermal energy per year corresponding to exhaust temperatures up to 128 °C (at the stack).38
Table 2 compares the energy demand to produce 1 kg of Ca(OH)2 via the traditional and a novel low-temperature precipitation process. It is noted that although the low-temperature process features a substantial energy demand – it is still attractive as it allows valorization of low-grade heat; that would, otherwise, be discarded. As such, if we focus on the “practical demand”, the energy consumption of the novel process is ∼44% lower than that of the traditional process.39,89,90 By contrast, if solution concentration was carried out through thermal evaporation instead of RO – although the electrical energy demand of 2.2 MJ kg−1 of Ca(OH)2 would vanish – 6338 MJ of waste heat would be required to produce 1 kg of Ca(OH)2, reducing by one order of magnitude the maximum Ca(OH)2 production rate for a fixed amount of waste heat (see ESI† for detailed calculations), making this option unpractical. Other alternatives such as evaporating water from solution at ambient conditions (e.g., using solar evaporation) would favor the precipitation of CaCO3 instead of Ca(OH)2 because highly alkaline solutions readily absorb CO2 present in the air.39 Thus, concentration and precipitation of Ca(OH)2 solely by heat (or waste heat) would introduce technical challenges such as the use of an inert atmosphere further complicating the process.
| Energy consumption (kJ kg−1 Ca(OH)2) | Process | ||
|---|---|---|---|
| Traditional | Novel | ||
| Total thermal energy | QT | 4000 | 677150 |
| Total work | WT = WRO + WP | — | 2241 |
| Total energy | ET = QT + WT | 4000 | 679391 |
| Total high-grade energy | ET − Qs,alt | 4000 | 2241 |
The main advantage of the alternative process, however, is that it obviates CO2 emissions associated with limestone's decomposition, while allowing alkaline waste utilization. The alternative process requires 2241 kJ (0.62 kW h) of electrical energy per kg of Ca(OH)2. Hence, assuming that 0.43 kg of CO2 are emitted per kW h of electricity consumed (e.g., corresponding to natural gas combustion, or 0.03 kg of CO2 per kW h for renewable solar power)91–93 and neglecting CO2 emissions associated with low grade waste heat, the CO2 emissions of the alternative process are on the order of ∼0.27 kg CO2 per kg of Ca(OH)2. On the other hand, conservatively, 0.74-to-0.86 kg of CO2 are produced per kg of Ca(OH)2 in the traditional production process depending on the type of fuel utilized.86 This estimate includes CO2 released on account of the thermal decomposition of CaCO3 [0.59 kg CO2 per kg of Ca(OH)2] and the CO2 emitted from natural gas combustion [0.15 to 0.27 kg CO2 per kg of Ca(OH)2],85,86 but these numbers could be larger for less efficient kilns.12 Thus, the CO2 emissions from the alternative process are only a third that of the traditional process. For these reasons, although more work remains, this style of low-temperature precipitation pathway, for portlandite production, is worthy of further work and consideration.
Summary and conclusions
This study has demonstrated a calcination-free pathway to produce Ca(OH)2 using industrial alkaline wastes as a feedstock. The process encompasses discrete unit operations including: leaching, RO concentration, and temperature-swing precipitation. In general, slag leaching in water can produce Ca-concentrations ranging from 2 to 17 mM depending on the slag, s/l, the presence of mixing (or not), and the leaching duration. The Ca-concentration in solution can be systematically enhanced using RO membrane filtration. Finally, the retrograde solubility of portlandite was exploited to precipitate it from near-saturated solutions of itself, by imposing a temperature swing. The quantity of portlandite precipitated in this manner was congruent with estimates from equilibrium thermodynamic calculations. Taken together, this process presents a specific energy demand (kJ per kg of Ca(OH)2) that is nearly 44% lower than traditional thermal processes; and importantly obviates the need to decarbonize limestone. As a result, this low temperature process presents a specific CO2 intensity that is 65% smaller than conventional methods. This is significant as Ca(OH)2 produced in this manner can uptake more CO2 than it associated with its own production forming a basis for a truly CO2-negative material.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support for this research from the Department of Energy via: (a) The Office of Fossil Energy's National Energy Technology Laboratory (NETL:DE-FE0029825), (b) The Advanced Research Projects Agency-Energy (ARPA-E:DE-AR-0001147) and (c) TRANSCEND: a UCLA-NIST Consortium that is supported by its Industry and Agency partners. This research was conducted in the Laboratory for Chemistry of Construction Materials (LC2) and the Molecular Instrumentation Center at UCLA. As such, the authors gratefully acknowledge the support that has made these laboratories and their operations possible. The contents of this paper reflect the views and opinions of the authors, who are responsible for the accuracy of the datasets presented herein, and do not reflect the views and/or polices of the agency, nor do the contents constitute a specification, standard or regulation. The authors would also like to acknowledge: Michael Moore (TMS International) for the supply of the different slags, Prof. Zongsu Wei (Aarhus University) for his assistance with the Reverse Osmosis (RO) processing, and Boral Resources for their assistance with the XRF analysis of the slags.
References
- J. L. Eades and R. E. Grim, A quick test to determine lime requirements for lime stabilization, Highw. Res. Rec., 1966, 62–72 Search PubMed.
- K.-H. Yang, A.-R. Cho, J.-K. Song and S.-H. Nam, Hydration products and strength development of calcium hydroxide-based alkali-activated slag mortars, Constr. Build. Mater., 2012, 29, 410–419 CrossRef.
- A. Strand, E. Korotkova, S. Willför, J. Hakala and E. Lindstedt, The use of calcium hydroxide as alkali source in peroxide bleaching of kraft pulp, Nord. Pulp Pap. Res. J., 2017, 32, 444–451 CAS.
- L. Semerjian and G. M. Ayoub, High-pH–magnesium coagulation–flocculation in wastewater treatment, Adv. Environ. Res, 2003, 7, 389–403 CrossRef CAS.
- J. Leentvaar and M. Rebhun, Effect of magnesium and calcium precipitation on coagulation-flocculation with lime, Water Res., 1982, 16, 655–662 CrossRef CAS.
- U.S. Geological Survey, Lime statistics and information, https://prd-wret.s3-us-west-2.amazonaws.com/assets/palladium/production/atoms/files/mcs-2019-lime.pdf, accessed 26 October 2019.
- S.-J. Han, M. Yoo, D.-W. Kim and J.-H. Wee, Carbon dioxide capture using calcium hydroxide aqueous solution as the absorbent, Energy Fuels, 2011, 25, 3825–3834 CrossRef CAS.
- K. S. Lackner, C. H. Wendt, D. P. Butt, E. L. Joyce and D. H. Sharp, Carbon dioxide disposal in carbonate minerals, Energy, 1995, 20, 1153–1170 CrossRef CAS.
- K. Vance, G. Falzone, I. Pignatelli, M. Bauchy, M. Balonis and G. Sant, Direct carbonation of ca(oh)2 using liquid and supercritical CO2: Implications for Carbon-Neutral Cementation, Ind. Eng. Chem. Res., 2015, 54, 8908–8918 CrossRef CAS.
- Z. Wei, B. Wang, G. Falzone, E. C. La Plante, M. U. Okoronkwo, Z. She, T. Oey, M. Balonis, N. Neithalath, L. Pilon and G. Sant, Clinkering-free cementation by fly ash carbonation, J. CO2 Util., 2018, 23, 117–127 CrossRef CAS.
- I. Mehdipour, G. Falzone, E. C. La Plante, D. Simonetti, N. Neithalath and G. Sant, How microstructure and pore moisture affect strength gain in portlandite-enriched composites that mineralize CO2, ACS Sustainable Chem. Eng., 2019, 7, 13053–13061 CrossRef CAS.
- M. Kenny and T. Oates, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007 Search PubMed.
- A. Dowling, J. O'Dwyer and C. C. Adley, Lime in the limelight, J. Cleaner Prod., 2015, 92, 13–22 CrossRef CAS.
- A. I. Aigbedion and S. E. Iyayi, Environmental effect of mineral exploitation in Nigeria, Int. J. Phys. Sci., 2007, 2, 33–38 Search PubMed.
- J. Vermeulen and T. Whitten, Biodiversity and cultural property in the management of limestone resources - lessons from East Asia, The World Bank, Washington, DC, USA, 1999 Search PubMed.
- D. B. Muller, T. Wang, B. Duval and T. E. Graedel, Exploring the engine of anthropogenic iron cycles, Proc. Natl. Acad. Sci., 2006, 103, 16111–16116 CrossRef.
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, A review of mineral carbonation technologies to sequester CO2, Chem. Soc. Rev., 2014, 43, 8049–8080 RSC.
- S.-Y. Pan, E. E. Chang and P.-C. Chiang, CO2 Capture by accelerated carbonation of alkaline wastes: a review on its principles and applications, Aerosol Air Qual. Res., 2012, 12, 770–791 CrossRef CAS.
- K. C. Curry and USGS, Iron and steel slag, 2020 Search PubMed.
- J. K. Stolaroff, G. V. Lowry and D. W. Keith, Using CaO- and MgO-rich industrial waste streams for carbon sequestration, Energy Convers. Manage., 2005, 46, 687–699 CrossRef CAS.
- T. H. Adams, Coal Ash Recycling Rate Declines Amid Shifting Production and Use Patterns, ACAA, Washington, DC, USA, 2019 Search PubMed.
- M. Ahmaruzzaman, A review on the utilization of fly ash, Prog. Energy Combust. Sci., 2010, 36, 327–363 CrossRef CAS.
- G. Montes-Hernandez, R. Pérez-López, F. Renard, J. M. Nieto and L. Charlet, Mineral sequestration of CO2 by aqueous carbonation of coal combustion fly-ash, J. Hazard. Mater., 2009, 161, 1347–1354 CrossRef CAS.
- A. Ćwik, I. Casanova, K. Rausis, N. Koukouzas and K. Zarębska, Carbonation of high-calcium fly ashes and its potential for carbon dioxide removal in coal fired power plants, J. Cleaner Prod., 2018, 202, 1026–1034 CrossRef.
- Environmental Research & Education Foundation [EREF], Analysis of MSW landfill tipping fees, EREF, April 2019, retrieved from http://erefdn.org, https://nrcne.org/wp-content/uploads/2019/12/EREF-MSWLF-Tipping-Fees-2019-FINAL-revised.pdf Search PubMed.
- R. S. Iyer and J. A. Scott, Power station fly ash — a review of value-added utilization outside of the construction industry, Resour., Conserv. Recycl., 2001, 31, 217–228 CrossRef.
- C. L. Carlson and D. C. Adriano, Environmental impacts of coal combustion residues, J. Environ. Qual., 1993, 22, 227–247 CrossRef CAS.
- S. Chand, B. Paul and M. Kumar, Sustainable approaches for LD slag waste management in steel industries: A Review, Metallurgist, 2016, 60, 116–128 CrossRef.
- E. S. Rubin, J. R. Kalagnanam, H. C. Frey and M. B. Berkenpas, Integrated environmental control modeling of coal-fired power systems, J. Air Waste Manage. Assoc., 1997, 47, 1180–1188 CrossRef CAS.
- M. Loncnar, H. A. van der Sloot, A. Mladenovič, M. Zupančič, L. Kobal and P. Bukovec, Study of the leaching behaviour of ladle slags by means of leaching tests combined with geochemical modelling and mineralogical investigations, J. Hazard. Mater., 2016, 317, 147–157 CrossRef CAS.
- L. De Windt, P. Chaurand and J. Rose, Kinetics of steel slag leaching: Batch tests and modeling, J. Waste Manage., 2011, 31, 225–235 CrossRef CAS.
- Y.-H. Hsiao, X. Chen, E. C. La Plante, A. Kumar, M. Bauchy, D. Simonetti, D. Jassby, J. Israelachvili and G. Sant, Mineral dissolution under electric stimulation, J. Phys. Chem. C, 2020, 124, 16515–16523 CrossRef CAS.
- Z. Wei, Y.-H. Hsiao, X. Chen, E. C. La Plante, I. Mehdipour, D. Simonetti, N. Neithalath, L. Pilon, M. Bauchy, J. Israelachvili and G. Sant, Isothermal stimulation of mineral dissolution processes by acoustic perturbation, J. Phys. Chem. C, 2018, 122, 28665–28673 CrossRef CAS.
- D. Parkhurst and R. Webb, PHREEQC Version 3 [computer software], U.S. Geological Survey, Jan 7 2020, retrieved from https://www.usgs.gov/software/phreeqc-version-3 Search PubMed.
- N. M. Piatak, M. B. Parsons and R. R. Seal, Characteristics and environmental aspects of slag: A review, Appl. Geochem., 2015, 57, 236–266 CrossRef CAS.
- L. Malaeb and G. M. Ayoub, Reverse osmosis technology for water treatment: State of the art review, Desalination, 2011, 267, 1–8 CrossRef CAS.
- R. G. Bates, V. E. Bower and E. R. Smith, Calcium hydroxide as a highly alkaline pH standard, J. Res. Natl. Bur. Stand., 1956, 56, 305–312 CrossRef CAS.
- D. B. Gingerich and M. S. Mauter, Quantity, quality, and availability of waste heat from United States thermal power generation, Environ. Sci. Technol., 2015, 49, 8297–8306 CrossRef CAS.
- S.-J. Han, M. Yoo, D.-W. Kim and J.-H. Wee, Carbon dioxide capture using calcium hydroxide aqueous solution as the absorbent, Energy Fuels, 2011, 25, 3825–3834 CrossRef CAS.
- A. Pérez-González, A. M. Urtiaga, R. Ibáñez and I. Ortiz, State of the art and review on the treatment technologies of water reverse osmosis concentrates, Water Res., 2012, 46, 267–283 CrossRef.
- Y. Choi, H. Cho, Y. Shin, Y. Jang and S. Lee, Economic evaluation of a hybrid desalination system combining forward and reverse osmosis, Membranes, 2015, 6, 3 CrossRef.
- T. I. Yun, C. J. Gabelich, M. R. Cox, A. A. Mofidi and R. Lesan, Reducing costs for large-scale desalting plants using large-diameter, reverse osmosis membranes, Desalination, 2006, 189, 141–154 CrossRef CAS.
- M. Li, Reducing specific energy consumption in Reverse Osmosis (RO) water desalination: An analysis from first principles, Desalination, 2011, 276, 128–135 CrossRef CAS.
- A. Bódalo-Santoyo, Spiral-wound membrane reverse osmosis and the treatment of industrial effluents, Desalination, 2004, 160, 151–158 CrossRef.
- Y. Gendel, A. K. E. Rommerskirchen, O. David and M. Wessling, Batch mode and continuous desalination of water using flowing carbon deionization (FCDI) technology, Electrochem. Commun., 2014, 46, 152–156 CrossRef CAS.
- J. O. Bockris and A. K. N. Reddy, Modern electrochemistry, Plenum, New York, USA, 1970, vol. 1 Search PubMed.
- H. S. Harned and B. B. Owen, The Physical Chemistry of Electrolytic Solutions, Reinhold Publishing Corporation, New York, USA, 1939 Search PubMed.
- R. Gabrovšek, T. Vuk and V. Kaučič, Evaluation of the hydration of portland cement containing various carbonates by means of thermal analysis, Acta Chim. Slov., 2006, 7 Search PubMed.
- M. Khachani, A. E. Hamidi, M. Halim and S. Arsalane, Non-isothermal kinetic and thermodynamic studies of the dehydroxylation process of synthetic calcium hydroxide Ca(OH)2, 2014, p. 11 Search PubMed.
- M.-A. Popescu, R. Isopescu, C. Matei, G. Fagarasan and V. Plesu, Thermal decomposition of calcium carbonate polymorphs precipitated in the presence of ammonia and alkylamines, Adv. Powder Technol., 2014, 25, 500–507 CrossRef CAS.
- W. M. Deen, Analysis of Transport Phenomena, Oxford University Press, New York, 1998 Search PubMed.
- H. F. W. Taylor, Cement chemistry, T. Telford, London, 2nd edn, 1997 Search PubMed.
- W. J. J. Huijgen and R. N. J. Comans, Mineral CO2 sequestration by steel slag carbonation, Environ. Sci. Technol., 2005, 39, 9676–9682 CrossRef CAS.
- W. J. J. Huijgen and R. N. J. Comans, Carbonation of steel slag for CO2 sequestration: Leaching products and reaction mechanisms, Environ. Sci. Technol., 2006, 40, 2790–2796 CrossRef CAS.
- N. M. Piatak, in Environmental Geochemistry: Site Characterization, Data Analysis and Case Histories, Elsevier, 2018, pp. 487–519 Search PubMed.
- S. Yadav and A. Mehra, Dissolution of steel slags in aqueous media, Environ. Sci. Pollut. Res., 2017, 24, 16305–16315 CrossRef CAS.
- F. Zhang and H. Itoh, Extraction of metals from municipal solid waste incinerator fly ash by hydrothermal process, J. Hazard. Mater., 2006, 136, 663–670 CrossRef CAS.
- Y. Sun, V. Parikh and L. Zhang, Sequestration of carbon dioxide by indirect mineralization using Victorian brown coal fly ash, J. Hazard. Mater., 2012, 209–210, 458–466 CrossRef CAS.
- S. Eloneva, S. Teir, J. Salminen, C.-J. Fogelholm and R. Zevenhoven, Steel converter slag as a raw material for precipitation of pure calcium carbonate, Ind. Eng. Chem. Res., 2008, 47, 7104–7111 CrossRef CAS.
- S.-Y. Pan, P.-C. Chiang, Y.-H. Chen, C.-D. Chen, H.-Y. Lin and E.-E. Chang, Systematic approach to determination of maximum achievable capture capacity via leaching and carbonation processes for alkaline steelmaking wastes in a rotating packed bed, Environ. Sci. Technol., 2013, 47, 13677–13685 CrossRef CAS.
- M. Mulder, Basic Principles of Membrane Technology, Springer Netherlands, Amsterdam, 2nd edn, 1996 Search PubMed.
- B. Ladewig and B. Asquith, Desalination Concentrate Management, Springer, Berlin, Heidelberg, 2012, pp. 5–15 Search PubMed.
- J. Kucera, The essentials of reverse osmosis, Chem. Eng. Prog., 2019, 115(2), 30–37 CAS.
- J. Gilron and D. Hasson, Calcium sulphate fouling of reverse osmosis membrane - flux decline mechanism, Chem. Eng. Sci., 1987, 42, 2351–2360 CrossRef CAS.
- C. J. Lin, S. Shirazi, P. Rao and S. Agarwal, Effects of operational parameters on cake formation of CaSO4 in nanofiltration, Water Res., 2006, 40, 806–816 CrossRef CAS.
- M. Uchymiak, A. R. Bartman, N. Daltrophe, M. Weissman, J. Gilron, P. D. Christofides, W. J. Kaiser and Y. Cohen, Brackish water reverse osmosis (BWRO) operation in feed flow reversal mode using an ex situ scale observation detector (EXSOD), J. Membr. Sci., 2009, 341, 60–66 CrossRef CAS.
- E. M. Vrijenhoek, S. Hong and M. Elimelech, Influence of membrane surface properties on initial rate of colloidal fouling of reverse osmosis and nanofiltration membranes, J. Membr. Sci., 2001, 188, 115–128 CrossRef CAS.
- C. J. Gabelich, A. Rahardianto, C. R. Northrup, T. I. Yun and Y. Cohen, Process evaluation of intermediate chemical demineralization for water recovery enhancement in production-scale brackish water desalting, Desalination, 2011, 272, 36–45 CrossRef CAS.
- R. Sheikholeslami and J. Bright, Silica and metals removal by pretreatment to prevent fouling of reverse osmosis membranes, Desalination, 2002, 143, 255–267 CrossRef CAS.
- J. Gilron, M. Waisman, N. Daltrophe, N. Pomerantz, M. Milman, I. Ladizhansky and E. Korin, Prevention of precipitation fouling in NF/RO by reverse flow operation, Desalination, 2006, 199, 29–30 CrossRef CAS.
- D. Hou, C. Hu and Z. Li, Molecular simulation of the ions ultraconfined in the nanometer-channel of calcium silicate hydrate: Hydration mechanism, dynamic properties, and influence on the cohesive strength, Inorg. Chem., 2017, 56, 1881–1896 CrossRef CAS.
- F. P. Glasser, E. E. Lachowski and D. E. Macphee, Compositional model for calcium silicate hydrate (C-S-H) gels, their solubilities, and free energies of formation, J. Am. Ceram. Soc., 1987, 70, 481–485 CrossRef CAS.
- S. Goto, M. Daimon, G. Hosaka and R. Kondo, Composition and morphology of hydrated tricalcium silicate, J. Am. Ceram. Soc., 1976, 59, 281–284 CrossRef CAS.
- R. Gabrovšek, T. Vuk and V. Kaučič, Evaluation of the hydration of Portland cement containing various carbonates by means of thermal analysis, Acta Chim. Slov., 2006, 53, 159–165 Search PubMed.
- E. Ruiz-Agudo, K. Kudłacz, C. V. Putnis, A. Putnis and C. Rodriguez-Navarro, Dissolution and carbonation of portlandite [Ca(OH)2] single crystals, Environ. Sci. Technol., 2013, 47, 11342–11349 CrossRef CAS.
- C. Rodriguez-Navarro, E. Hansen and W. S. Ginell, Calcium hydroxide crystal evolution upon aging of lime putty, J. Am. Ceram. Soc., 2005, 81, 3032–3034 CrossRef.
- C. Speiser, T. Baumann and R. Niessner, Morphological and chemical characterization of calcium-hydrate phases formed in alteration processes of deposited municipal solid waste incinerator bottom ash, Environ. Sci. Technol., 2000, 34, 5030–5037 CrossRef CAS.
- X. Zhang, F. P. Glasser and K. L. Scrivener, Reaction kinetics of dolomite and portlandite, Cem. Concr. Res., 2014, 66, 11–18 CrossRef CAS.
- M. Galván-Ruiz, J. Hernández, L. Baños, J. Noriega-Montes and M. E. Rodríguez-García, Characterization of calcium carbonate, calcium oxide, and calcium hydroxide as starting point to the improvement of lime for their use in construction, J. Mater. Civ. Eng., 2009, 21, 694–698 CrossRef.
- G. Madras and B. J. McCoy, Growth and ripening kinetics of crystalline polymorphs, Cryst. Growth Des., 2003, 3, 981–990 CrossRef CAS.
- G. Madras and B. J. McCoy, Temperature effects on the transition from nucleation and growth to Ostwald ripening, Chem. Eng. Sci., 2004, 59, 2753–2765 CrossRef CAS.
- S.-J. Kim, S.-D. Park, Y. H. Jeong and S. Park, Homogeneous precipitation of TiO2 ultrafine powders from aqueous TiOCl2 solution, J. Am. Ceram. Soc., 1999, 82, 927–932 CrossRef CAS.
- F. Zeman, Energy and material balance of CO2 capture from ambient air, Environ. Sci. Technol., 2007, 41, 7558–7563 CrossRef CAS.
- A. Sagastume Gutiérrez, J. B. Cogollos Martínez and C. Vandecasteele, Energy and energy assessments of a lime shaft kiln, Appl. Therm. Eng., 2013, 51, 273–280 CrossRef.
- M. Stork, W. Meindertsma, M. Overgaag and M. Neelis, A Competitive and Efficient Lime Industry, Technical Report, European Lime Association EuLA, Brussels, Belgium, 2014 Search PubMed.
- H. Piringer, Lime shaft kilns, Energy Procedia, 2017, 120, 75–95 CrossRef.
- R. W. Fox, A. T. McDonald and P. J. Pritchard, Introduction to Fluid Mechanics, John Wiley & Sons, Hoboken, NJ, USA, 8th edn, 2011 Search PubMed.
- T. L. Bergman, A. S. Lavine, F. P. Incropera and D. P. Dewitt, Fundamentals of Heat and Mass Transfer, John Wiley & Sons, Hoboken, NJ, 7th edn, 2011 Search PubMed.
- J. K. Stolaroff, D. W. Keith and G. V. Lowry, Carbon dioxide capture from atmospheric air using sodium hydroxide spray, Environ. Sci. Technol., 2008, 42, 2728–2735 CrossRef CAS.
- Y. Peng, B. Zhao and L. Li, Advance in post-combustion CO2 capture with alkaline solution: a brief review, Energy Procedia, 2012, 14, 1515–1522 CrossRef.
- F. Kreith, P. Norton and D. Brown, A comparison of CO2 emissions from fossil and solar power plants in the united states, Energy, 1990, 15, 1181–1198 CrossRef CAS.
- J. J. Burkhardt, G. Heath and E. Cohen, Life cycle greenhouse gas emissions of trough and tower concentrating solar power electricity generation: Systematic review and harmonization, J. Ind. Ecol., 2012, 16, S93–S109 CrossRef CAS.
- J. A. de Chalendar, J. Taggart and S. M. Benson, Tracking emissions in the US electricity system, Proc. Natl. Acad. Sci., 2019, 116, 25497–25502 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08449b |
| ‡ Crystalline slags, unlike their amorphous counterparts (e.g., of which, the latter find use as cement replacement agents, that is, as supplementary cementitious materials: SCMs) – on account of their unreactive nature – are considered to be a low-value waste that is difficult to valorize, and that is often associated with a waste disposal (tipping) fee. For this reason, and in view of their Ca-based alkalinity which is equivalent to amorphous slags, we intentionally sought to use these less-reactive materials as a feedstock in our low-temperature process. |
| This journal is © The Royal Society of Chemistry 2021 |