
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient microwave-assisted Suzuki–Miyaura cross-coupling reaction of 3-bromo pyrazolo[1,5-a]pyrimidin-5(4H)-one: towards a new access to 3,5-diarylated 7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidine derivatives†
Badr Jismya,
Gérald Guillaumetb,
Mohamed Akssirac,
Abdellatif Tikadd and
Mohamed Abarbri *a
*a
aLaboratoire de Physico-Chimie des Matériaux et des Electrolytes pour l'Energie (PCM2E). EA 6299, Faculté des Sciences, Avenue Monge Parc de Grandmont, 37200 Tours, France. E-mail: mohamed.abarbri@univ-tours.fr; Fax: +33 2 47 36 70 73; Tel: +33 2 47 36 73 59 Tel: +33 6 70 22 15 77
bInstitut de Chimie Organique et Analytique (ICOA), Université d'Orléans, UMR CNRS 7311, BP 6759, rue de Chartres, 45067 Orléans Cedex2, France. E-mail: gerald.guillaumet@univ-orleans.fr; Tel: +33 2 38 41 70 73
cLaboratoire de Chimie Physique et de Chimie Bioorganique, URAC 22, BP 146, 28800 Mohammedia, Morocco. E-mail: akssira.m@gmail.com; Fax: +212 5 23315353; Tel: +212 5 23 314705 Tel: +212 5 23315352
dLaboratoire de Chimie Moléculaire et Substances Naturelles, Faculté des Sciences, Département de Chimie, Université Moulay Ismail, B. P. 11201, Zitoune, Meknès 50050, Morocco. E-mail: a.tikad@umi.ac.ma
First published on 4th January 2021
Abstract
A convenient and efficient synthetic route to C3-arylated 7-trifluoromethylpyrazolo[1,5-a]pyrimidin-5-one derivatives has been reported starting from 3-bromo-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-5-one through a Suzuki–Miyaura cross-coupling reaction. The arylation (heteroarylation) strategy can be performed using a wide variety of aryl and heteroaryl boronic acids and requiring a tandem catalyst XPhosPdG2/XPhos to avoid the debromination reaction. These optimized conditions were successfully extended to the synthesis of 7-, 8- and 9-arylated pyrimido[1,2-b]indazol-2-ones from their corresponding brominated starting materials. Furthermore, the second C-5 arylation of C3-arylated pyrazolo[1,5-a]pyrimidin-5-ones was achieved under standard Suzuki–Miyaura cross-coupling conditions, after activating the C–O bond of the lactam function with PyBroP, giving access to a small library of 3,5-diarylated 7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidines in good to excellent yields. The interest of this approach has been highlighted by the synthesis of a known anti-inflammatory agent. Additionally, a preliminary biological evaluation has revealed that a number of derivatives display micromolar IC50 values against monoamine oxidase B, an important target in the field of neurodegenerative disorders.
Introduction
Transition metal-catalyzed cross-coupling reactions are widely recognized to be powerful synthetic tools for carbon–carbon and carbon-heteroatom bond formation, giving access to more complex molecules.1 The Suzuki–Miyaura cross-coupling reaction is one of the most useful methods to create new carbon–carbon (C sp2–C sp2) bonds, thanks to the stability and the low toxicity of organoboron reagents.2 Moreover, organoboron compounds provide high selectivity in cross-coupling reactions, proving that the Suzuki–Miyaura reaction is particularly suitable for the elegant synthesis of biologically active molecules and natural products.3,4 The use of this coupling reaction in medicinal chemistry represents a considerable asset for inducing modification of the biochemical properties by introducing a functional diversification such as a vinyl, aryl or heteroaryl moiety.5 Despite the wide scope of this coupling reaction in various fields of chemistry, some difficulties and limitations may be encountered in the case of some heteroaromatic substrates. This is observed mainly for heterocycles having a free amino group, probably because of the complexation of the metal with NH function, leading to the inhibition or deactivation of the catalyst.6 For these reasons, the Pd-catalyzed Suzuki–Miyaura cross-coupling involving heteroaromatic substrates is therefore a major challenge for organic chemists7 and in the pharmaceutical discovery chemistry.8 On the other hand, pyrazolo[1,5-a]pyrimidine is a basic heterocycle of a variety of complex chemical compounds having large spectrum of biological properties,9 viz: COX-2 inhibitors,10 hepatitis C virus inhibitors,11 kinase inhibitors,12 anticancer agents,13 PET tumor imaging agents,14 anxiolytic agents,15 antimicrobial agents,16 and serotonin 5-HT6 receptor antagonists.17 In addition, pyrazolo[1,5-a]pyrimidine constitute the central core of several pharmaceutical drugs, such as Indiplon and Zaleplon, which are sedative-hypnotic, used to treat insomnia, and Ocinaplon which is an anxiolytic drug. Synthetically, a wide number of approaches giving access to pyrazolo[1,5-a]pyrimidine derivatives have been reported.18 The condensation of 3(5)-aminopyrazoles with 1,3-dicarbonyl compound appears to be the most general approach for the synthesis of these heterocycles.19 Other 1,3-bis electrophilic compounds, such as alkoxymethylene β-dicarbonyl derivatives, enaminonitriles and β-enaminones, have been used to generate this class of fused heterocycles.20Additionally, only few syntheses have been reported for pyrazolo[1,5-a]pyrimidin-5-one skeleton, involving essentially the condensation of aminopyrazoles with 1,3-dimethyluracil,20,21 3-ethoxyacrylate22 or ethyl 4,4,4-trifluoroacetoacetate.23
To date, despite significant progress in the development of palladium-catalyzed reactions of aminopyrazoles,24 only one example of a Suzuki–Miyaura coupling reaction has been disclosed by Ellermann and co-workers, starting from 3-halogeno pyrazolo[1,5-a]pyrimidin-5-ones.25 In this case, the cross-coupling was achieved using XPhosPdG3 (10 mol%) and XPhos (10 mol%) in THF at 80 °C for 16 h, providing the C3-arylated products in low yields ranging from 3 to 41%. Furthermore, no systematic study including reaction optimization conditions was reported. Consequently, the biological potential of the target arylated pyrazolo[1,5-a]pyrimidine derivatives and the lack of cross coupling exploration in this heterocycle led us to further develop the C3-arylation of fluorinated pyrazolo[1,5-a]pyrimidin-5-one 4a. Recently, Schmitt et al. have reported a two-pot synthesis of 7-(hetero)aryl pyrazolo[1,5-a]pyrimidin-5-ones from aryl (heteroaryl)halides and aminopyrazoles.26 Nevertheless, these strategies have various drawbacks: the scope of the substrate is restricted and pyrazolo[1,5-a]pyrimidin-5-ones were obtained in low to moderate yields.
To the best of our knowledge, the Suzuki–Miyaura cross-coupling of fluorinated 3-bromo pyrazolo[1,5-a]pyrimidin-5-ones has never been described in the literature. This is somewhat surprising since it has been shown that the incorporation of fluorine atoms into compounds has a significant impact on their physical, chemical and biological properties.27 For example, the presence of a CF3 moiety contributes to increase the lipophilicity of biologically active molecules.28 A versatile and flexible methodology for constructing 3-arylated pyrazolo[1,5-a]pyrimidin-5-ones, bearing a CF3 group is thus of major importance. However, there have been few reports describing the synthesis of 3,5-disubstituted pyrazolo[1,5-a]pyrimidines bearing a trifluoromethyl group at C7 position.29 Furthermore, selective and/or sequential arylation has never been studied on a pyrazolo[1,5-a]pyrimidin-5-one for the synthesis of substituted pyrazolo[1,5-a]pyrimidines.
New strategies are therefore necessary for easy and effective access to new variously substituted heterocycles targets, whose biological potential seems certain.
Recently, we reported the one-pot efficient regioselective synthesis of new fluorinated heterocycles via an addition/heterocyclization sequence using a fluorinated alkyne and 1,3-bisnucleophilic reagents.30 Continuing our efforts to develop the synthesis of new potentially active heterocycles,31 we report herein the first examples of a general and efficient Suzuki–Miyaura C3-arylation and heteroarylation of 3-bromo-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-5-one 4a using various aryl and heteroaryl boronic acids (Scheme 1). Some of 3-arylated pyrazolo[1,5-a]pyrimidin-5-ones were subsequently used as building blocks to synthesize novel fluorinated 3,5-disubstituted pyrazolo[1,5-a]pyrimidines. Indeed and based on our recently published work (Scheme 1),32 an efficient and divergent method was developed to synthesize 3,5-diarylated pyrazolo[1,5-a]pyrimidines bearing a trifluoromethyl group at C7 position through C–O activation of the amide function.33
 | ||
| Scheme 1 Convergent approach to arylation of fluorinated pyrazolo[1,5-a]pyrimidine and fluorinated pyrazolo[1,5-a]pyrimidin-5-one derivatives. | ||
Results and discussion
The synthesis of 7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-5-one 3 was accomplished according to our previously reported procedure through the condensation of commercially available 3-aminopyrazole 1 with ethyl 4,4,4-trifluorobutynoate 2 (Scheme 2).32 The reaction proceeded smoothly to provide regioselectively 7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-5-one 3 in 63% yield, without any trace of the regioisomer pyrazolo[1,5-a]pyrimidin-7-one (Scheme 2). It is noteworthy that for several years in our laboratory, alkyne 2 has been the subject of investigations as a basic element to synthesize new fluorinated compounds.34 Compound 3 was subsequently converted selectively into 3-bromo and 3-iodo pyrazolo[1,5-a]pyrimidin-5-ones 4a and 4b via NBS or NIS-mediated bromination or iodination in CH2Cl2 at room temperature in excellent yields [X = Br, 4a (94%), X = I, 4b (90%)] (Scheme 2). | ||
| Scheme 2 Synthesis of 3-halo pyrazolo[1,5-a]pyrimidin-5-ones 4a and 4b. | ||
The resulting halides 4a and 4b could be a key building blocks to modify the C3 position through the Suzuki–Miyaura coupling reaction.2a–c,35 This strategy is challenging because metal-catalyzed reactions present some problems and limitations especially when the used substrates contain an unprotected amino group.36 First, we examined the coupling reaction between 3-bromo pyrazolo[1,5-a]pyrimidin-5-one 4a and p-methoxyphenylboronic acid, using PdCl2(PPh3)2 (5 mol%) as a catalyst in the presence of Na2CO3 (2 equiv.) in dioxane at 110 °C (Table 1, entry 1). The analysis of the crude 1H and 19F NMR spectrums showed 9% of the expected product 5a along with 91% of the undesired debrominated product 3 (Table 1, entry 1). To optimize the reaction conditions, the solvent, catalyst, ligand, base and temperature were carefully screened and all obtained results are summarized in Table 1. Using PdCl2dppf instead of PdCl2(PPh3)2 in the presence of Na2CO3 or K2CO3, increased slightly the yield of 5a to 17% (Table 1, entries 2 and 3).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Organic solvent | Base | Catalyst (5 mol%) | Ligand (10 mol%) | T (°C) | Ratioa | |
| 5a | 3 | ||||||
| a The ratio of mixture (5a/3) was determined from crude 1H and 19F NMR spectrums.b 3 equiv. of K2CO3 were used.c The reaction was carried out under microwave irradiation for 40 minutes.d Yield of isolated product 5a.e Reaction was performed with XPhosPdG2 (2.5 mol%) and XPhos (5 mol%). | |||||||
| 1 | Dioxane | Na2CO3 | PdCl2(PPh3)2 | — | 110 | 9 | 91 |
| 2 | Dioxane | Na2CO3 | PdCl2dppf | — | 110 | 11 | 89 |
| 3 | Dioxane | K2CO3 | PdCl2dppf | — | 110 | 17 | 83 |
| 4 | Dioxane | K2CO3 | XPhosPdG2 | XPhos | 110 | 18 | 82 |
| 5 | Dioxane | K2CO3 | Pd2(dba)3 | XPhos | 110 | 4 | 96 |
| 6 | EtOH | K2CO3 | Pd2(dba)3 | XPhos | 110 | 45 | 55 |
| 7 | EtOH | K2CO3 | Pd(OAc)2 | XPhos | 110 | 52 | 48 |
| 8 | EtOH | K2CO3 | Pd(OAc)2 | SPhos | 110 | 41 | 59 |
| 9 | EtOH | K2CO3 | Pd(OAc)2 | CPhos | 110 | 24 | 76 |
| 10 | EtOH | K2CO3 | Pd(OAc)2 | DavePhos | 110 | 21 | 79 |
| 11 | EtOH | K2CO3 | Pd(OAc)2 | Dppf | 110 | 14 | 86 |
| 12 | EtOH | K2CO3 | Pd(OAc)2 | BINAP | 110 | 19 | 81 |
| 13 | EtOH | K2CO3 | Pd(OAc)2 | XantPhos | 110 | 20 | 80 |
| 14 | EtOH | K2CO3 | Pd(OAc)2 | PPh3 | 110 | 18 | 82 |
| 15 (ref. 24) | THF | K3PO4 | XPhosPdG3 | Xphos | 80 | 31 | 69 |
| 16 | EtOH | K2CO3 | XPhosPdG2 | XPhos | 110 | 92 | 8 |
| 17 | EtOH | K2CO3 | XPhosPdG2 | XPhos | 135 | 94 | 6 |
| 18b | EtOH | K2CO3 | XPhosPdG2 | XPhos | 110 | 78 | 22 |
| 19c | EtOH | K2CO3 | XPhosPdG2 | XPhos | 135 | 100(91)d | 0 |
| 20c | EtOH | K2CO3 | XPhosPdG2 | — | 135 | 45 | 55 |
| 21c,e | EtOH | K2CO3 | XPhosPdG2 | XPhos | 135 | 100(89)d | 0 |
| 22c | H2O | K2CO3 | XPhosPdG2 | XPhos | 135 | 100(86)d | 0 |

Under the same conditions, replacing PdCl2dppf by the tandem XPhosPdG2 (5 mol%) and XPhos (10 mol%) resulted in a similar 5a/3 ratio (Table 1, entry 4). No improvement in the yield of the coupling product 5a was observed when the reaction was carried out using Pd2(dba)3 (5 mol%) as a catalyst and XPhos (10 mol%) as a ligand (Table 1, entry 5). Using a polar protic solvent such as ethanol/H2O instead of dioxane/H2O increased the yield of the desired product 5a to 45%, decreasing the formation of by-product 3 to 55% (Table 1, entry 6). Encouraged by this result, the mixture of EtOH/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was chosen as solvents for further experiments. A similar result was recorded when the reaction was performed using Pd(OAC)2/XPhos tandem as catalyst (Table 1, entry 7). Additional optimizations were undertaken, using the same catalyst [Pd(OAc)2] and varying the ligand. In all cases, whatever the nature of the ligand employed in these reactions (Sphos, CPhos, DavePhos, dppf, BINAP, XantPhos and PPh3), the side product 3 was obtained mainly at the expense of the target heterocycle 5a (Table 1, entries 8–14). Surprisingly, under Ellermann conditions (XPhosPdG3/XPhos, K3PO4, THF, 80 °C),25 the yield of arylated adduct 5a did not exceed 31%, since the debrominated product 3 was also formed in 69% yield (Table 1, entry 15). Interestingly, when the coupling reaction was performed using the XPhos derived precatalyst XPhosPdG2 in EtOH/H2O (4:1) at 110 °C, the starting material was completely consumed after 12 hours, and the yield of arylated product 5a was improved considerably to 92% (Table 1, entry 16). Nevertheless, the crude 1H and 19F NMR spectrums showed the presence of 8% of by-product 3. Under the same reaction conditions and increasing the temperature to 135 °C, no significant difference in the result was observed (Table 1, entry 17). Using an excess of the base K2CO3 (3 equiv.) promoted the formation of the undesired product 3 (Table 1, entry 18). Gratifyingly, switching from conventional thermal heating to microwave irradiation at 135 °C, only the coupled product 5a was produced in 91% isolated yield (Table 1, entry 19). Most remarkably, the reaction time was reduced from 12 h to 40 min and no trace of the debrominated product 3 was detected in crude 1H and 19F NMR spectrums. When the reaction was conducted without ligand, both heterocycles 5a and 3 were formed equally (Table 1, entry 20), indicating the crucial role of the ligand in avoiding the side debromination reaction. Finally, reducing both XPhosPdG2 (2.5 mol%) and XPhos (5 mol%) loading did not affect significantly the reaction yield, providing compound 5a in 89% yield (Table 1, entry 21). Very interestingly, when the reaction was carried out in water as the sole solvent, the arylation product 5a was the only isolated product in 86% yield (Table 1, entry 22). This result was very promising since water was the solvent of choice for green chemistry. Moreover, it is known that working with water remains tricky because of its limited chemical compatibility as well as the low aqueous solubility of a large number of reagents. It is noteworthy that the nature of the halo group present on the pyrazolo[1,5-a]pyrimidin-5-one 3 plays an important role in this arylation reaction. In fact, whatever the conditions used, including optimized ones, the deiodination reaction cannot be avoided when the substrate 4b was used as a starting material, presumably because of the easier reduction ability of the C–I bond to a C–H bond compared to C–Br bond.
:1) was chosen as solvents for further experiments. A similar result was recorded when the reaction was performed using Pd(OAC)2/XPhos tandem as catalyst (Table 1, entry 7). Additional optimizations were undertaken, using the same catalyst [Pd(OAc)2] and varying the ligand. In all cases, whatever the nature of the ligand employed in these reactions (Sphos, CPhos, DavePhos, dppf, BINAP, XantPhos and PPh3), the side product 3 was obtained mainly at the expense of the target heterocycle 5a (Table 1, entries 8–14). Surprisingly, under Ellermann conditions (XPhosPdG3/XPhos, K3PO4, THF, 80 °C),25 the yield of arylated adduct 5a did not exceed 31%, since the debrominated product 3 was also formed in 69% yield (Table 1, entry 15). Interestingly, when the coupling reaction was performed using the XPhos derived precatalyst XPhosPdG2 in EtOH/H2O (4:1) at 110 °C, the starting material was completely consumed after 12 hours, and the yield of arylated product 5a was improved considerably to 92% (Table 1, entry 16). Nevertheless, the crude 1H and 19F NMR spectrums showed the presence of 8% of by-product 3. Under the same reaction conditions and increasing the temperature to 135 °C, no significant difference in the result was observed (Table 1, entry 17). Using an excess of the base K2CO3 (3 equiv.) promoted the formation of the undesired product 3 (Table 1, entry 18). Gratifyingly, switching from conventional thermal heating to microwave irradiation at 135 °C, only the coupled product 5a was produced in 91% isolated yield (Table 1, entry 19). Most remarkably, the reaction time was reduced from 12 h to 40 min and no trace of the debrominated product 3 was detected in crude 1H and 19F NMR spectrums. When the reaction was conducted without ligand, both heterocycles 5a and 3 were formed equally (Table 1, entry 20), indicating the crucial role of the ligand in avoiding the side debromination reaction. Finally, reducing both XPhosPdG2 (2.5 mol%) and XPhos (5 mol%) loading did not affect significantly the reaction yield, providing compound 5a in 89% yield (Table 1, entry 21). Very interestingly, when the reaction was carried out in water as the sole solvent, the arylation product 5a was the only isolated product in 86% yield (Table 1, entry 22). This result was very promising since water was the solvent of choice for green chemistry. Moreover, it is known that working with water remains tricky because of its limited chemical compatibility as well as the low aqueous solubility of a large number of reagents. It is noteworthy that the nature of the halo group present on the pyrazolo[1,5-a]pyrimidin-5-one 3 plays an important role in this arylation reaction. In fact, whatever the conditions used, including optimized ones, the deiodination reaction cannot be avoided when the substrate 4b was used as a starting material, presumably because of the easier reduction ability of the C–I bond to a C–H bond compared to C–Br bond.
Having established the optimized reaction conditions (Table 1, entry 22), the scope and limitations of the Suzuki–Miyaura cross-coupling reaction were examined with various aryl and heteroaryl boronic acids as coupling partners. The results are summarized in Scheme 3.
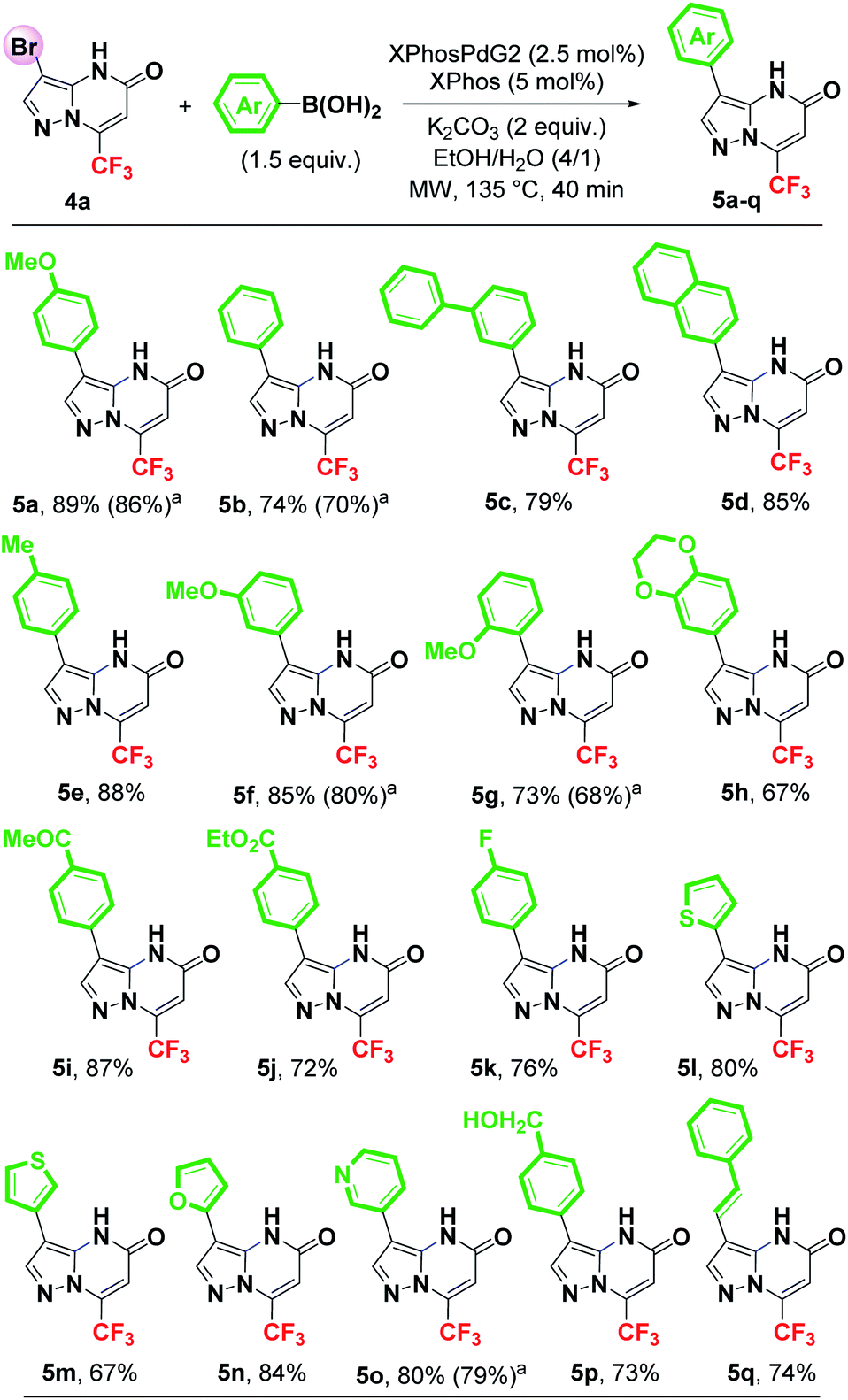 | ||
| Scheme 3 Synthesis of 3-arylated(heteroarylated) pyrazolo[1,5-a]pyrimidin-5-ones by Suzuki–Miyaura cross-coupling reaction of 4a. a Isolated yield using water as solvent. | ||
As shown in Scheme 3, in all cases the optimized conditions proved to be suitable for the coupling of 4a with a series of boronic acids, giving the expected C3-arylated products (5a–q) in good to excellent yields (67–89%). Unsubstituted aryls such as phenyl, biphenyl and naphthyl groups were efficiently introduced at C3 position of the pyrazolo[1,5-a]pyrimidin-5-one 4a in 74%, 79% and 85% yields, respectively (Scheme 3). A number of substituents (including functionalized ones) were tolerated on the boronic acids and very efficiently provided the desired products (5e–q). Arylboronic acid bearing an electron-donating group such as a methoxy group at para, meta or ortho position, reacted with 4a leading to the corresponding products 5a (89%), 5f (85%) and 5g (73%). As observed, it appears that the steric hindrance of the ortho-position has a significant effect on the reaction efficiency. The cross-coupling between 4a and 3,4-(ethylenedioxy)phenylboronic acid afforded the 3-arylated pyrazolopyrimidinone 5h in 67% yield. Boronic acids with electron-withdrawing substituent such as 4-acetyl and ethoxycarbonyl were tolerated, yielding the desired products 5i (87%) and 5j (72%), respectively. Boronic acid with a fluorine atom on the aromatic ring was also found to be compatible with this coupling reaction (5k, 76%). Moreover, the use of heteroarylboronic acids such as 2-thienyl, 3-thienyl, 2-furyl, 3-pyridyl was effective, providing the expected heterocycles in good yields [5l (80%), 5m (67%), 5n (84%) and 5o (80%)]. The introduction of a styryl group at C3 position was successfully achieved, affording the vinylation product 5q in 74% yield with retention of the double bond configuration. The presence of a free alcohol function on the boronic acid was tolerated and gave the expected product 5p in 73% yield. The success encountered during this arylation reaction allows easy diversification into biologically important biheterocyclic frameworks.
Encouraged by the efficiency of this Suzuki–Miyaura coupling reaction enabling the introduction of aryl, heteroaryl and vinyl groups at C3 position of pyrazol[1,5-a]pyrimidin-5-one 4a, we decided to extend our methodology to other electron-rich heterocycles such as pyrimido[1,2-b]indazol-2-ones. For this purpose, the tricyclic compounds 7a, 7b and 7c were first prepared by condensation of the corresponding brominated pyrimido[1,2-b]indazol-2-ones 6a, 6b and 6c with the fluorinated alkyne 2 in 64%, 58% and 60% yields, respectively (Scheme 4).
 | ||
| Scheme 4 Synthesis of 9, 8 and 7-brominated pyrimido[1,2-b]indazol-2-ones 7a–c. | ||
A variety of substituted arylboronic acids (with either electron-donating or electron-withdrawing groups) were used to provide the expected coupling products 8a–d, 9a–d and 10a,b in good to excellent yields (61–89%) (Scheme 5). Unsubstituted phenyl boronic acid was successfully coupled with 7a and 7b giving the expected pyrimido[1,2-b]indazol-2-ones 8a and 9a in 70% and 72% isolated yields, respectively. This result demonstrated that the position of bromine on the aromatic ring has no significant effect on the effeciency of this Suzuki–Miyaura coupling. The coupling reaction of 7a and 7b with an heteroarylboronic acid such as 2-furylboronic acid produced the heteroarylated compounds 9d and 8d in 73% and 83% yields, respectively. Moreover, 3-pyridylboronic acid was effectively coupled with 7c, providing the C-7 heteroarylated heterocycle 10b in 68% yield (Scheme 5).
 | ||
| Scheme 5 Suzuki–Miyaura coupling reaction of brominated pyrimido[1,2-b]indazol-2-ones 7a–c. a Isolated yield using water as solvent. | ||
The synthesized C3 heteroarylated pyrazolo[1,5-a]pyrimidin-5-ones 5a–q are attractive candidates for access to new 3,5-diarylated pyrazolo[1,5-a]pyrimidines because of their ability to undergo a transition-metal based coupling reaction.
The C3-arylated pyrazolo[1,5-a]pyrimidin-5-ones 5a, 5b, 5e, 5j and 5l were coupled with a variety of available boronic acids to give the desired 3,5-diarylated compounds using a combination of two conditions (Scheme 6), i.e., the phosphonium coupling condition [PyBrop (1.3 equiv.), Et3N (3 equiv.), 1,4-dioxane, rt, 2 h] then the Suzuki–Miyaura cross-coupling conditions [PdCl2(PPh3)2, Na2CO3, 110 °C, 12 h]. Direct arylation proceeded successfully providing the 3,5-diarylated-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidines 11a–h in good to excellent isolated yields (Scheme 6).
 | ||
| Scheme 6 Synthesis of 3,5-diarylated pyrazolo[1,5-a]pyrimidines 11a–h. | ||
Arylboronic acid bearing an electron-donating group such as a methoxy group at para position was easily coupled with 5b, 5e, 5j and 5l, producing the corresponding diarylated products 11a (88%), 11e (90%), 11f (74%) and 11g (87%), respectively. Phenyl boronic acid substituted with a methoxy group at ortho position gave the expected coupling product 11b in only 66% yield. Steric hindrance could explain this result. Boronic acid with an electron-withdrawing fluorine atom was also tolerated, yielding the desired product 11d in 78% yield. Furthermore, heteroarylboronic acids such as 2-thienyl was successfully coupled with 5l to yield the C5-heteroarylation product 11c in 87% yield. It seems that the nature of the boronic acids used in this coupling reaction had a significant effect on the obtained results.
To demonstrate the versatility and the synthetic potential of our method, we focused our attention on the synthesis of compound having an interesting biological activity via the introduction of suitable aryls at C3 and C5 positions of the pyrazolo[1,5-a]pyrimidine core. For our delight, when the compound 5b was subjected to the conditions of C5-arylation with p-bromophenylboronic acid, the known heterocyclic compound 11h was isolated in excellent yield (91%) (Scheme 6). It is noteworthy that the 3,5-diarylated pyrazolo[1,5-a]pyrimidin-5-one 11h exhibited the comparable anti-inflammatory activity (83.4%) to the standard drug Indomethacin (84.2%).29b,37
After successfully achieving stepwise sequential diarylation, we attempted to achieve this reaction sequence in a one-pot process to synthesize 3,5-diarylated pyrazolo[1,5-a]pyrimidines. The one-pot sequential arylation of 3-bromo pyrazolo[1,5-a]pyrimidin-5-one 4a was first performed with XPhosPdG2 (2.5 mol%)/XPhos (5 mol%) as catalyst in the presence of K2CO3 as base (2.0 equiv.) and p-methoxyphenylboronic acid (2.5 equiv.) in ethanol and water (4:1) at 135 °C under microwave irradiation for 40 min. Upon completion of the coupling (monitored by TLC), the solvents were evaporated before using a combination of two conditions to perform the second arylation: [PyBrop (1.3 equiv.), Et3N (3 equiv.), 1,4-dioxane, rt, 2 h], then the Suzuki–Miyaura cross-coupling conditions [PdCl2(PPh3)2, Na2CO3, 110 °C, 12 h] without isolating the intermediate product. Unfortunately, the sequential one-pot coupling reaction afforded the diarylated product 12 in low 25% overall yield (Scheme 7). Note that reversing the order of the arylation reactions using the same reaction conditions was unsuccessful, since only the debrominated product was formed and no trace of the expected diarylation product 12 was observed. This result shows that the stepwise sequential arylation of 4a, applying the earlier optimized conditions, was the best way to access 3,5-diarylated pyrazolo[1,5-a]pyrimidines in good yields.
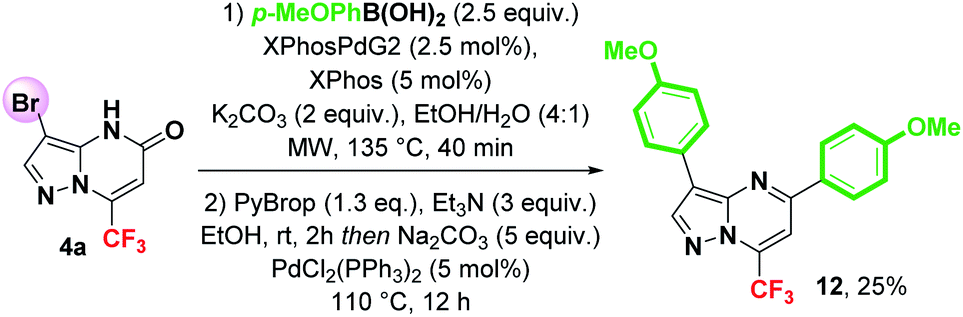 | ||
| Scheme 7 Sequential one-pot synthesis of fluorinated 3,5-diaryl pyrazolo[1,5-a]pyrimidine 12. | ||
After successfully performing the C5-arylation providing 3,5-diarylated pyrazolo[1,5-a]pyrimidines, we decided to introduce greater diversity using this strategy. A C5-alkynylation using the Sonogashira coupling reaction was conducted,38 wherein alkyne (1.5 equiv.), PdCl2(PPh3)2 (5 mol%), CuI (10 mol%), Et3N (5 equiv.) and dioxane were added after the prior addition of PyBroP and Et3N at room temperature for 2 h. The mixture was subsequently heated at 80 °C for 12 h, providing 5-alkynyl-3-aryl(heteroaryl) pyrazolo[1,5-a]pyrimidines 13a–c in good to excellent yields (Scheme 8). In particular, unsubstituted phenylacetylene was coupled with 5b to afford 13a in 90% isolated yield. Phenylacetylene with an electron-donating group such as a methoxy group at para position was easily coupled with 5b, yielding the desired product 13b in 88% yield. Phenylacetylene bearing an electron-withdrawing group such as a chlorine atom at para position provided the desired product 13c in 78% yield. The electronic effect of the substituents on the aryl group seems to have a small effect on the yield of Sonogashira coupling reaction.
 | ||
| Scheme 8 Synthesis of 5-alkynyl-3-phenyl pyrazolo[1,5-a]pyrimidines 13a–c. | ||
These successive C3-arylation/C5-arylation and C3-arylation/C5-alkynylation were carried out efficiently and open up interesting perspectives for the synthesis of diversely substituted and new potentially important biological bi (or tri)-heterocyclic frameworks.
In order to obtain further insight into the reaction mechanism of debromination, a series of control experiments were conducted to elucidate the role of each component in this reaction. The results obtained are presented in Table 2.

After examination of these results, we note that neither the presence of the boronic acid alone nor that of the ligand XPhos did not induce the formation of debrominated product 3, since only the starting material 4a was recovered (Table 2, entries 1 and 2). The use of XPhosPdG2 led to a low partial debromination (Table 2, entry 3). On the other hand, the treatment of compound 4a with K2CO3 (2 equiv.) at 135 °C resulted in the exclusive conversion of starting material 4a into the debrominated product 3 (Table 2, entry 4). The same result was obtained when K2CO3 was replaced by a base having a comparable basicity such as Na2CO3 or a stronger one such as KOH (Table 2, entries 5and 6). When the bromide 4a was heated to 135 °C without any additive, no trace of the debrominated product 3 has been detected (Table 2, entry 7), confirming without a doubt the involvement of the base in the promotion of this debromination reaction. These results are completely in agreement with those reported by Cankař et al. in the case of the Suzuki–Miyaura coupling reaction of brominated amino pyrazoles.39
Basing on these experimental results and the previous literature report,39 a possible mechanism is proposed as shown below in Scheme 9.
 | ||
| Scheme 9 Proposed mechanism for the debromination reaction. | ||
The reaction proceeds first by a deprotonation leading to the formation of anion 14a, which can be transformed by delocalization of electrons into the anionic species 14b. The presence of a polar protic solvent allows a protonation of the compound 14b giving access to the intermediate 14c. A subsequent aromatization process occurred along with bromium (Br+) release, affording the anionic intermediate 14d, which after protonation, provides the debrominated product 3.
Pyrazolopyrimidinone derivatives are also known for their activities against AChE and BuChE cholinesterases.40 It is worth mentioning that they showed moderate selectivity for BuChE over acetylcholinesterase (AChE). They are described as potential inhibitors of BuChE with micromolar IC50 values. Their inhibitory potencies against BuChE were even higher than the anti-AD drug rivastigmine.
The prepared pyrazolopyrimidinones 5a–q were thus screened against both cholinesterases (AChE and BChE), MAO-B and MAO-A using established protocols as described in the experimental section. The results of the assays are summarized in Table 3.
| hAChEa | hBChEb | hMAO-Ac | hMAO-Bd | |
|---|---|---|---|---|
| RAe at 100/10 μM | RAe at 100/10 μM | RAe at 100/10 μM | RAe at 100/10 μM | |
| a hAChE, human AChE.b hBChE, human BChE.c hMAO-A, human MAO-A.d hMAO-B, human MAO-B.e RA, residual activity at 100/10 μM compound concentration.f n.d., not determined. | ||||
| 5a | 86/93 | 103/90 | 99/95 | 26/73 |
| IC50 = 35.0 ± 11.2 μM | ||||
| 5f | 88/91 | 95/97 | 97/92 | 40/80 |
| IC50 = 101.1 ± 25.7 μM | ||||
| 5h | 86/91 | 95/94 | 95/110 | 8/36 |
| IC50 = 5.36 ± 0.25 μM | ||||
| 5i | 91/90 | 99/91 | 98/100 | 21/75 |
| IC50 = 26.2 ± 3.6 μM | ||||
| Tacrine | IC50 = 0.115 ± 0.009 μM | IC50 = 0.023 ± 0.003 μM | n.d.f | n.d.f |
| Pargyline | n.d.f | n.d.f | IC50 = 3.97 ± 0.28 μM | IC50 = 0.20 ± 0.02 μM |
Interestingly, some of the tested amides showed interesting biological activities. The products which inhibit the studied targets are 7-(trifluoromethyl) pyrazolo[1,5-a]pyrimidin-5(4H)-ones derivatives 5a, 5f, 5h and 5i. They selectively inhibit hMAO-B in the micromolar range (Table 3). This suggests that phenyl substituted at the C-3 and/or C-4 positions by an OMe or COMe group is required for inhibition of hMAO-B.41 Compound 5h was the most potent hMAO-B inhibitor with an IC50 value of 5.36 μM.
Conclusions
In conclusion, we have developed a catalytic system to synthesize a wide variety of new C3-arylated pyrazolo[1,5-a]pyrimidin-5-one derivatives via the Suzuki–Miyaura cross-coupling reaction of brominated pyrazolo[1,5-a]pyrimidin-5-one derivatives bearing a trifluoromethyl group, without the formation of any trace of the side debrominated product. The use of a catalytic amount of XPhosPdG2/XPhos tandem in the presence of K2CO3 in aqueous ethanol as green solvent allowed the efficient cross-coupling reaction of 3-bromo-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-5(4H)-one, 7-, 8- and 9-bromo 4-(trifluoromethyl)pyrimido[1,2-b]indazol-2(1H)-ones with a range of aryl, heteroaryl and styrylboronic acids, providing a rapid access to arylated and heteroarylated pyrazolo[1,5-a]pyrimidin-5-one derivatives in good to excellent yields. Some of synthesized C3-arylated pyrazolo[1,5-a]pyrimidin-5-ones were used as a building blocks for generating new 3,5-diarylated or 3-arylated/5-alkynylated pyrazolo[1,5-a]pyrimidines containing a trifluoromethyl group. The potential application of this environmentally protocol was also demonstrated by the synthesis of the known anti-inflammatory agent 11h, opening access for the design of new functionalized pyrazolo[1,5-a]pyrimidines, having other potential biological activities.Experimental
General information
Melting points of samples were measured using open capillary tubes. 1H, 13C and 19F NMR spectra were recorded at 300, 75 and 282 MHz respectively with a 300 MHz Bruker Avance FT-NMR spectrometer using CDCl3, acetone-d6, THF-d8 or DMSO-d6 as the solvents. All chemical shifts are given in ppm and they are referenced to tetramethylsilane (TMS) as an internal standard. Electrospray ionization high-resolution mass spectrometry experiments were performed with a hybrid tandem quadrupole/time-of flight (Q-TOF) instrument, equipped with a pneumatically assisted electrospray (Zspray) ion source (Micromass, Manchester, U.K.) operated in positive mode. Reactions were monitored by thin-layer chromatography (TLC) analysis using silica gel (60 F254) plates. All compounds were visualized by UV irradiation (longwave at 365 nm or shortwave at 254 nm). All column chromatography was performed using silica gel 60 (230–400.13 mesh, 0.040–0.063 mm). All chemicals reagents purchased from commercial suppliers were used as received. Triethylamine was distilled over calcium hydride and the 1,4-dioxane over sodium and benzophenone.The microwave-assisted reactions were performed using a Monowave 300 (microwave synthesis reactor: Anton Paar, 300 W maximum power). Microwave irradiation was carried out in sealed 10–30 mL vessels (borosilicate glass) with a PTFE-coated silicon septum and closed with a snap cap made of PEEK. The temperatures were measured on the surface of the vial with an IR sensor (measuring range: 30 to 300 °C; uncertainty: ±5 °C) and were measured with high precision in the reaction mixture with a ruby thermometer (measuring range: 30 to 300 °C; uncertainty: ±2 °C) that could be read directly from the instrument screen. The reaction time was measured from when the reaction mixture reached the stated temperature for temperature controlled experiments. The pressure was measured with a non-invasive pressure sensor located in the swiveling cover of the Monowave 300 (measuring range: 0 to 30 bar; uncertainty: ±1.5 bar).
Preparation and analytical data for 3-halo-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-5-one (4a–b)
NBS or NIS (1.05 equiv.) was added to a solution of pyrazolo[1,5-a]pyrimdin-5-one 3 (100 mg, 1 equiv.) in 8 mL of CH2Cl2 and the reaction was stirred under an argon atmosphere at room temperature for 12 h. The progress of the reaction was monitored by TLC (PE/EtOAc, 7/3). After completion of the reaction, evaporation of the solvent under reduced pressure provided the crude product 4a or 4b, which was purified by column chromatography.General procedure and analytical data for products 7a–c
In a microwave vial, 4(5 or 6)-bromo-1H-indazol-3-amine (0.47 mmol, 100 mg) was dissolved in 1,4-dioxane (4 mL). Ethyl 4,4,4-trifluorobut-2-ynoate (1.2 equiv.) was then added, and the mixture was degassed by argon bubbling for 10 min. The sealed tube was heated at 110 °C for 2 h. After cooling, MeONa (51.2 mg, 2 equiv.) was added and the reaction was stirred for 12 h at room temperature. The reaction mixture was neutralized with a solution of hydrochloric ether (2 M) and the solvent was removed under reduced pressure. The crude product was purified by silica-gel column chromatography.Pd-catalyzed arylation of 3-bromo 7-(trifluoromethyl)pyrazolo[1,5-a]pyrimdin-5-one 4a and C8(9 or 10)-bromo 4-(trifluoromethyl)pyrimido[1,2-b]indazol-2-one 7a–c with (hetero)aryl boronic acids.
A mixture of 4-bromo-7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidin-5-one 4a or 8(9 or 10)-bromo-4-(trifluoromethyl)benzo[4,5]imidazo[1,2-a]pyrimidin-2-one 7a–c (100 mg, 1 equiv.), boronic acid (1.5 equiv.), and K2CO3 (2 equiv.) in EtOH (4 mL) and water (1 mL) was thoroughly degassed with a stream of argon. Then, XPhos (5 mol%) and XPhosPdG2 (2.5 mol%) were added and the microwave vial containing the mixture was capped and inserted into microwave reactor. The reaction mixture was irradiated at 135 °C for 40 min. After that, the mixture was concentrated under reduced pressure. Purification of the crude product via column chromatography afforded desired 3-disubstituted pyrazolo[1,5-a]pyrimidin-5-one 5a–q and 8(9 or 10)- substituted benzo[4,5]imidazo[1,2-a]pyrimidin-2-ones 8a–d, 9a–d and 10a–b as solid, which were characterized by 1H, 13C NMR, 19F NMR and HRMS. In all cases, products were recrystallized from diethyl ether or CH2Cl2.
General procedure for the direct C5 arylation of 5b, 5e, 5a, 5j and 5l with (hetero)aryl boronic acids via C–OH bond activation
To a stirred solution of aromatic lactam 5a, 5b, 5e, 5j or 5l (100 mg, 1.0 equiv.) in 1,4-dioxane (3 mL) at room temperature was added PyBroP (1.3 equiv.) and Et3N (3 equiv.). The reaction mixture was stirred at room temperature for 2 h. The progress of the reaction was monitored by TLC. After completion of the first step, boronic acid (1.5 equiv.), Na2CO3 (5 equiv.), and PdCl2(PPh3)2 (5 mol%) were added successively at room temperature. After the mixture had been stirred at 110 °C for 12 h, it was diluted with CH2Cl2, washed with saturated NH4Cl solution, and dried with MgSO4. The crude product was purified by column chromatography to give pure 3,5-diarylated 7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidines 11a–h.General procedure for the direct alkynylation of 5b with alkynes via C–OH bond activation
To a stirred solution of aromatic lactam 5b (1.0 equiv.) in 1,4-dioxane (3 mL) at room temperature was added PyBroP (1.3 equiv.) and Et3N (3 equiv.). The reaction mixture was stirred at room temperature for 2 h. The progress of the reaction was monitored by TLC. After completion of the first step, alkyne (1.5 equiv.), PdCl2(PPh3)2 (5 mol%) and CuI (10 mol%) were added successively at room temperature. After the mixture had been stirred at 110 °C for 12 h, it was diluted with CH2Cl2, washed with saturated NH4Cl solution, and dried with MgSO4. The crude product was purified by column chromatography to give pure 3,5-disubstituted 7-(trifluoromethyl)pyrazolo[1,5-a]pyrimidines 13a–c.Protocols of inhibition
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the “Département d'analyses Chimiques et Médicales” (Tours, France) for chemical analyses. We would also like to thank Dr Damijan Knez from Ljubljana University for his help in carrying out the first biological tests.Notes and references
- (a) C. C. C. J. Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef; (b) F. S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC; (c) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS.
- (a) N. Miyaura and A. Suzuki, J. Chem. Soc., Chem. Commun., 1979, 866–867 RSC; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (c) A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef CAS; (d) J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC; (e) J. P. G. Rygus and C. M. Crudden, J. Am. Chem. Soc., 2017, 139, 18124–18137 CrossRef CAS.
- Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, ed M. L. Crawley and B. M. Trost, John Wiley & Sons, 2012 Search PubMed.
- (a) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS; (b) V. Chandrasekhar, R. S. Narayanan and P. Thilagar, Organometallics, 2009, 28, 5883–5888 CrossRef CAS; (c) M. Arthuis, R. Pontikis and J.-C. Florent, Org. Lett., 2009, 11, 4608–4611 CrossRef CAS; (d) B. Bhayana, B. P. Fors and S. L. Buchwald, Org. Lett., 2009, 11, 3954–3957 CrossRef CAS; (e) T. Martin, C. Laguerre, C. Hoarau and F. Marsais, Org. Lett., 2009, 11, 3690–3693 CrossRef CAS; (f) C. A. James, A. L. Coelho, M. Gevaert, P. Forgione and V. Snieckus, J. Org. Chem., 2009, 74, 4094–4103 CrossRef CAS; (g) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- (a) F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 2419–2440 CAS; (b) C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS; (c) S. Handa, M. Andersson, F. Gallou, J. Reilly and B. H. Lipshutz, Angew. Chem., Int. Ed., 2016, 55, 4914–4918 CrossRef CAS.
- (a) M. A. Dufert, K. L. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 12877–12885 CrossRef CAS; (b) J. Almond-Thynne, D. C. Blakemore, D. C. Pryde and A. C. Spivey, Chem. Sci., 2017, 8, 40–62 RSC; (c) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41(11), 1461–1473 CrossRef CAS.
- (a) M. Prieto, E. Zurita, E. Rosa, L. Munoz, P. Lloyd-Williams and E. Giralt, J. Org. Chem., 2004, 69, 6812–6820 CrossRef CAS; (b) T. Itoh and T. Mase, Tetrahedron Lett., 2005, 46, 3573–3577 CrossRef CAS; (c) P. Gunda, L. M. Russon and M. K. Lakshman, Angew. Chem., Int. Ed., 2004, 43, 6372–6377 CrossRef CAS.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- (a) S. Vichier-Guerre, L. Dugué and S. Pochet, Tetrahedron Lett., 2014, 55, 6347–6350 CrossRef CAS; (b) J. Tan, Y. Chen, H. Li and N. Yasuda, J. Org. Chem., 2014, 79, 8871–8876 CrossRef CAS; (c) E. Bratt, O. Verho, M. J. Johansson and J.-E. Bäckvall, J. Org. Chem., 2014, 79, 3946–3954 CrossRef CAS; (d) Y. Qian, M. Hamilton, A. Sidduri, S. Gabriel, Y. Ren, R. Peng, R. Kondru, A. Narayanan, T. Truitt, R. Hamid, Y. Chen, L. Zhang, A. J. Fretland, R. A. Sanchez, K.-C. Chang, M. Lucas, R. C. Schoenfeld, D. Laine, M. E. Fuentes, C. S. Stevenson and D. C. Budd, J. Med. Chem., 2012, 55, 7920–7939 CrossRef CAS.
- C. Almansa, A. F. de Arriba, F. L. Cavalcanti, L. A. Gomez, A. Miralles, M. Merlos, J. Garcia-Rafanell and J. Forn, J. Med. Chem., 2001, 44, 350–361 CrossRef CAS.
- J. Y. Hwang, M. P. Windisch, S. Jo, H. C. Kim, S. Kim, H. Kim, M. E. Lee, D.-S. Park, E. Park, S. Ahn, J. Cechetto, J. Kim, M. Liuzzi, Z. No and J. Lee, Bioorg. Med. Chem. Lett., 2012, 22, 7297–7301 CrossRef CAS.
- (a) T. Asano, H. Yamazaki, C. Kasahara, H. Kubota, T. Kontani, Y. Harayama, K. Ohno, H. Mizuhara, M. Yokomoto, K. Misumi, T. Kinoshita, M. Ohta and M. Takeuchi, J. Med. Chem., 2012, 55, 7772–7785 CrossRef CAS; (b) T. Kosugi, D. R. Mitchell, A. Fujino, M. Imai, M. Kambe, S. Kobayashi, H. Makino, Y. Matsueda, Y. Oue, K. Komatsu, K. Imaizumi, Y. Sakai, S. Sugiura, O. Takenouchi, G. Unoki, Y. Yamakoshi, V. Cunliffe, J. Frearson, R. Gordon, C. J. Harris, H. Kalloo-Hosein, J. Le, G. Patel, D. J. Simpson, B. Sherborne, P. S. Thomas, N. Suzuki, M. Takimoto-Kamimura and K. Kataoka, J. Med. Chem., 2012, 55, 6700–6715 CrossRef CAS.
- (a) A. Kamal, J. R. Tamboli, V. L. Nayak, S. F. Adil, M. V. P. S. Vishnuvardhan and S. Ramakrishna, Bioorg. Med. Chem. Lett., 2013, 23, 3208–3215 CrossRef CAS; (b) D. S. Williamson, M. J. Parratt, J. F. Bower, J. D Moore, C. M. Richardson, P. Dokurno, A. D. Cansfield, G. L. Francis, R. J. Hebdon, R. Howes, P. S. Jackson, A. M. Lockie, J. B. Murray, C. L. Nunns, J. Powles, A. Robertson, A. E. Surgenor and C. J. Torrance, Bioorg. Med. Chem. Lett., 2005, 15, 863–867 CrossRef CAS; (c) K. Paruch, M. P. Dwyer, C. Alvarez, C. Brown, T. Y. Chan, R. J. Doll, K. Keertikar, C. Knutson, B. McKittrick, J. Rivera, R. Rossman, G. Tucker, T. O. Fischmann, A. Hruza, V. Madison, A. A. Nomeir, Y. Wang, E. Lees, D. Parry, N. Sgambellone, W. Seghezzi, L. Schultz, F. Shanahan, D. Wiswell, X. Xu, Q. Zhou, R. A. James, V. M. Paradkar, H. Park, L. R. Rokosz, T. M. Stauffer and T. J. Guzi, Bioorg. Med. Chem. Lett., 2007, 17, 6220–6223 CrossRef CAS; (d) S. Ali, D. A. Heathcote, S. H. B. Kroll, S. Jogalekar, B. Scheiper, H. Patel, J. Brackow, A. Siwicka, M. J. Fuchter, M. Periyasamy, R. S. Tolhurst, S. K. Kanneganti, J. P. Snyder, D. C. Liotta, E. O. Aboagye, A. G. Barrett and R. C. Coombes, Cancer Res., 2009, 69, 6208–6215 CrossRef CAS.
- J. Xu, H. Liu, G. Li, Y. He, R. Ding, X. Wang, M. Feng, S. Zhang, Y. Chen, S. Li, M. Zhao, Y. Li and C. Qi, Naturforsch, 2012, 67, 827–834 CAS.
- (a) S. Selleri, F. Bruni, C. Costagli, A. Costanzo, G. Guerrini, G. Ciciani, P. Gratteri, F. Besnard, B. Costa, M. Montali, C. Martini, J. Fohlin, G. D. Siena and P. M. Aiello, J. Med. Chem., 2005, 48, 6756–6760 CrossRef CAS; (b) Y. L. Chen, WO1998008847A1, 1998; (c) J. P. Dusza, J. D. Albright A. S. Tomcufcik, U.S. Pat.5538977, 1996 Search PubMed.
- S. Bondock, W. Fadaly and M. A. Metwally, Eur. J. Med. Chem., 2010, 45, 3692–3701 CrossRef CAS.
- A. V. Ivashchenko, E. S. Golovina, M. G. Kadieva, V. M. Kysil, O. D. Mitkin and I. M. Okun, Pharm. Chem. J., 2012, 46, 406–410 CrossRef CAS.
- (a) I. L. Dalinger, I. A. Vatsadse, S. A. Shevelev and A. V. Ivachtchenko, J. Comb. Chem., 2005, 7, 236–245 CrossRef CAS; (b) M. Drev, U. Groselj, S. Mevec, E. Pusavec, J. Strekelj, A. Golobic, G. Dahmann, B. Stanovnik and J. Svete, Tetrahedron, 2014, 70, 8267–8279 CrossRef CAS; (c) M. H. Elnagdi and A. W. Erian, Bull. Chem. Soc. Jpn., 1990, 63, 1854–1856 CrossRef CAS; (d) S. M. Hussain, A. M. El-Reedy and S. A. El-Sharabasy, Tetrahedron, 1988, 44, 241–246 CrossRef CAS; (e) W. Ried and S. Aboul-Fetouh, Tetrahedron, 1988, 44, 7155–7162 CrossRef CAS.
- (a) D. R. Compton, S. Sheng, K. E. Carlson, N. A. Rebacz, I. Y. Lee, B. S. Katzenellenbogen and J. A. Katzenellenbogen, J. Med. Chem., 2004, 47, 5872–5893 CrossRef CAS; (b) P. J. Gilligan, C. Baldauf, A. Cocuzza, D. Chidester, R. Zaczek, L. W. Fitzgerald, J. McElroy, M. A. Smith, H. S. L. Shen, J. A. Saye, D. Christ, G. Trainor, D. W. Robertson and P. Hartig, Bioorg. Med. Chem., 2000, 8, 181–189 CrossRef CAS; (c) M. E. Fraley, W. F. Hoffman, R. S. Rubino, R. W. Hungate, A. J. Tebben, R. Z. Rutledge, R. C. McFall, W. R. Huckle, R. L. Kendall, K. E. Coll and K. A. Thomas, Bioorg. Med. Chem. Lett., 2002, 12, 2767–2770 CrossRef CAS; (d) B. T. Gregg, D. O. Tymoshenko, D. A. Razzano and M. R. Johnson, J. Comb. Chem., 2007, 9, 507–512 CrossRef CAS; (e) J. Quiroga, J. Portilla, R. Abonía, B. Insuasty, M. Nogueras and J. Cobo, Tetrahedron Lett., 2007, 48, 6352–6355 CrossRef CAS; (f) H. B. Abed, O. Mammoliti, G. V. Lommen and P. Herdewijn, Tetrahedron Lett., 2013, 54, 2612–2614 CrossRef; (g) J. Blaney, P. A. Gibbons, E. Hanan, J. P. Lyssikatos, S. R. Magnuson, R. Pastor, T. E. Rawson, A. Zhou, B.-Y. Zhu, WO2010051549 A1; 2010US Pat.063014, 2009; (h) S. Hölder, M. Vennemann, G. Beneke, A. Zülch, V. Gekeler, T. Beckers, A. Zimmermann, H. Joshi, WO2009021992 A2 EP Pat. 060690, 2008; 2009; (i) B. E. Evans, K. E. Rittle, M. G. Bock, R. M. DiPardo, R. M. Freidinger, W. L. Whitter, G. F. Lundell, D. F. Veber, P. S. Anderson, R. S. L. Chang, V. J. Lotti, D. J. Cerino, T. B Chen, P. J. Kling, K. A. Kunkel, J. P. Springer and J. Hirshfield, J. Med. Chem., 1988, 31, 2235–2246 CrossRef CAS; (j) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS; (k) D. A. Griffith, D. M. Hargrove, T. S. Maurer, C. A. Blum, S. De Lombaert, J. K. Inthavongsay, L. E. Klade, C. M. Mack, C. R. Rose, M. J. Sanders and P. A. Carpino, Bioorg. Med. Chem. Lett., 2011, 21, 2641–2645 CrossRef CAS; (l) H. Mukaiyama, T. Nishimura, S. Kobayashi, Y. Komatsu, S. Kikuchi, T. Ozawa, N. Kamada and H. Ohnota, Bioorg. Med. Chem., 2008, 16, 909–921 CrossRef CAS; (m) H. Mukaiyama, T. Nishimura, S. Kobayashi, T. Ozawa, N. Kamada, Y. Komatsu, S. Kikuchi, H. Oonota and H. Kusama, Bioorg. Med. Chem., 2007, 15, 868–885 CrossRef CAS; (n) H. Tye, S. G. Mueller, J. Prestle, S. Scheuerer, M. Schindler, B. Nosse, N. Prevost, C. J. Brown, A. Heifetz, C. Moeller, A. Pedret-Dunn and M. Whittaker, ACS Med. Chem. Lett., 2011, 21, 34–37 CrossRef CAS.
- (a) M. H. Elnagdi, M. R. H. Elmoghayar and G. H. Elgemeie, Adv. Heterocycl. Chem., 1987, 41, 319–376 CrossRef CAS; (b) J. Quiroga, A. Hormaza, B. Insuasty, C. Saitz, C. Jullian and A. Cannete, J. Heterocycl. Chem., 1998, 35, 61–64 CrossRef CAS; (c) F. M. A. El-Taweel and T. M. Abu Elmaati, J. Chin. Chem. Soc., 2002, 49, 1051–1055 CrossRef CAS; (d) R. Daniels, K. Kim, E. Lebois, H. Muchalski, M. Hughes and C. Lindsley, Tetrahedron Lett., 2008, 49, 305–310 CrossRef CAS; (e) A. V. Ivachtchenko, E. S. Golovina, M. G. Kadieva, V. M. Kysil, O. D. Mitkin, S. E. Tkachenko and I. M. Okun, J. Med. Chem., 2011, 54, 8161–8173 CrossRef CAS; (f) S. Ahmetaj, N. Velikanje, U. Grošelj, I. Šterbal, B. Prek, A. Golobič, D. Kočar, G. Dahmann, B. Stanovnik and J. Svete, Mol. Diversity, 2013, 17, 731–743 CrossRef CAS; (g) K. D. Khalil, H. M. Al-Matar, D. M. Al-Dorri and M. H. Elnagdi, Tetrahedron, 2009, 65, 9421–9427 CrossRef CAS; (h) J. Quiroga, D. Mejia, B. Insuasty, R. Abonia, M. Nogueras, A. Sanchez and J. Cobo, J. Heterocycl. Chem., 2002, 39, 51–54 CrossRef CAS.
- L. K. Gavrin, A. Lee, B. A. Provencher, W. W. Massefski, S. D. Huhn, G. M. Ciszewski, D. C. Cole and J. C. McKew, J. Org. Chem., 2007, 72, 1043–1046 CrossRef CAS.
- T. Haga, H. Kimura, M. Morita, T. Ueda, T. Ueki, K. Kiriyama, K. Yoshida, T. Hamamoto and T. Kodama, WO2010018853A1, February 18, 2010.
- (a) R. Balicki, Pol. J. Chem., 1983, 57, 789–797 CAS; (b) M. Yoshida, A. Mori, A. Inaba, M. Oka, H. Makino, M. Yamaguchi, H. Fujita, T. Kawamoto, M. Goto, H. Kimura, A. Baba and T. Yasuma, Bioorg. Med. Chem., 2010, 18, 8501–8511 CrossRef CAS.
- (a) L. Jedinák and P. Cankař, Eur. J. Org. Chem., 2016, 2013–2023 CrossRef; (b) H. A. Malik, B. L. H. Taylor, J. R. Kerrigan, J. E. Grob, K. N. Houk, J. Du Bois, L. G. Hamann and A. W. Pattersona, Chem. Sci., 2014, 5, 2352–2361 RSC.
- M. Ellermann, G. Valot, Y. Cancho Grande, J. Haßfeld, T. Kinzel, J. Köbberling, K. Beyer, S. Röhrig, WO2016071216A1, May 12, 2016.
- D. C. Schmitt, N. Niljianskul, N. W. Sach and J. I. Trujillo, ACS Comb. Sci., 2018, 20, 256–260 CrossRef CAS.
- (a) E. P. Gillis, J. K. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS; (b) P. Shah and A. D. Westwell, J. Enzyme Inhib. Med. Chem., 2007, 22, 527–540 CrossRef CAS; (c) T. Yamazaki, T. Taguchi, I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, 2009, pp. 3–46 Search PubMed; (d) R. Filler, Y. Kobayashi, L. M. Yugapolskii, Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications; Elsevier: Amsterdam, 1993 Search PubMed.
- (a) G. B. Elion, S. Gallawan, H. Nathan, S. Bicher, R. W. Randler and G. H. Hitchings, Biochem. Pharmacol., 1963, 12, 85–93 CrossRef CAS; (b) F. Ismail, J. Fluorine Chem., 2002, 118, 27–33 CrossRef CAS; (c) H.-J. Bohm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Muller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef; (d) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS; (e) J.-P. Bégué and D. Bonnet-Delpon, J. Fluorine Chem., 2006, 127, 992–1012 CrossRef; (f) K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef; (g) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (h) W. K. Hagmann, J. Med. Chem., 2008, 51, 2108–2114 CrossRef.
- (a) H. Wenyong, W. Jianshu, H. Kaishun, P. Li, Z. Yujue, C. Yongzheng, C. N. Faming Zhuanli Shenqing, Patent CN110627725A, December 31, 2019; (b) W. Jian-Shu, H. Kai-Shun, H. Wen-Yong, C. Bao-Dong, W. Nan-Wei and C. Yong-Zheng, Org. Lett., 2019, 21, 8751–8755 CrossRef; (c) E. E. Emelina, A. A. Petrov and A. V. Firsov, Russ. J. Org. Chem., 2001, 37, 852–858 CrossRef CAS.
- (a) J. Petrignet, E. Thiery, L. Silpa and M. Abarbri, Synthesis, 2014, 46, 947–954 CrossRef; (b) L. Silpa, J. Petrignet and M. Abarbri, Synlett, 2014, 25, 1827–1830 CrossRef CAS; (c) B. Jismy, A. Tikad, M. Akssira, G. Guillaumet and M. Abarbri, Molecules, 2020, 25, 2062 CrossRef CAS.
- (a) B. Jismy, G. Guillaumet, H. Allouchi, M. Akssira and M. Abarbri, Eur. J. Org. Chem., 2017, 6168–6178 CrossRef CAS; (b) L. Silpa, A. Niepceron, F. Laurent, F. Brossier, M. Pénichon, C. Enguehart-Gueiffier, M. Abarbri, A. Silvestre and J. Petrignet, Bioorg. Med. Chem. Lett., 2016, 26, 114–120 CrossRef CAS.
- B. Jismy, H. Allouchi, G. Guillaumet, M. Akssira and M. Abarbri, Synthesis, 2018, 50, 1675–1686 CrossRef CAS.
- (a) F. A. Kang, Z. Sui and W. V. Murray, J. Am. Chem. Soc., 2008, 130, 11300–11302 CrossRef CAS; (b) F. A. Kang, Z. Sui and W. V. Murray, Eur. J. Org. Chem., 2009, 461–479 CrossRef CAS; (c) F.-A. Kang, J. C. Lanter, C. Cai, Z. Sui and W. V. Murray, Chem. Commun., 2010, 46, 1347–1349 RSC; (d) C. Shi and C. C. Aldrich, Org. Lett., 2010, 12, 2286–2289 CrossRef CAS; (e) V. P. Mehta, S. G. Modha and E. Van der Eycken, J. Org. Chem., 2010, 75, 976–979 CrossRef CAS; (f) S.-M. Li, J. Huang, G.-J. Chena and F.-S. Han, Chem. Commun., 2011, 47, 12840–12842 RSC; (g) R. Belaroussi, A. El Hakmaoui, M. Akssira, G. Guillaumet and S. Routier, Eur. J. Org. Chem., 2016, 3550–3558 CrossRef CAS.
- (a) G. Prié, J. Thibonnet, M. Abarbri, J.-L. Parrain and A. Duchêne, Synlett, 1998, 839–840 CrossRef; (b) J. Thibonnet, G. Prié, M. Abarbri, J.-L. Parrain and A. Duchêne, Tetrahedron Lett., 1999, 40, 3151–3154 CrossRef CAS; (c) Y. Carcenac, K. Zine, J. C. Kizirian, J. Thibonnet, A. Duchêne, J.-L. Parrain and M. Abarbri, Adv. Synth. Catal., 2010, 352, 949–954 CrossRef CAS; (d) K. Zine, J. Petrignet, J. Thibonnet and M. Abarbri, Synlett, 2012, 23, 755–759 CrossRef CAS.
- (a) S. Kotha, K. Lahiri and D. Kashinath, Tetrahedron, 2002, 58, 9633–9695 CrossRef CAS; (b) F. Bellina, A. Carpita and R. Rossi, Synthesis, 2004, 2419–2440 CAS; (c) N. T. S. Phan, N. M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- (a) L. Jedinak and P. Cankař, Eur. J. Org. Chem., 2016, 2013–2023 CrossRef CAS; (b) S. Vichier-Guerre, L. Dugue and S. Pochet, Tetrahedron Lett., 2014, 55, 6347–6350 CrossRef CAS; (c) J. Tan, Y. Chen, H. Li and N. Yasuda, J. Org. Chem., 2014, 79, 8871–8876 CrossRef CAS; (d) E. Bratt, O. Verho, M. J. Johansson and J.-E. Backvall, J. Org. Chem., 2014, 79, 3946–3954 CrossRef CAS; (e) Y. Qian, M. Hamilton, A. Sidduri, S. Gabriel, Y. Ren, R. Peng, R. Kondru, A. Narayanan, T. Truitt, R. Hamid, Y. Chen, L. Zhang, A. J. Fretland, R. A. Sanchez, K.-C. Chang, M. Lucas, R. C. Schoenfeld, D. Laine, M. E. Fuentes, C. S. Stevenson and D. C. Budd, J. Med. Chem., 2012, 55, 7920–7939 CrossRef CAS; (f) S. D. Walker, T. E. Barder, J. R. Martinelli and S. L. Buchwald, Angew. Chem., Int. Ed., 2004, 43, 1871–1876 (Angew. Chem., 2004, 116, 1907–1912) CrossRef CAS.
- R. Aggarwala, E. Masana, P. Kaushik, D. Kaushik, C. Sharma and K. R. Aneja, J. Fluorine Chem., 2014, 168, 16–24 CrossRef.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 50, 4467–4470 CrossRef.
- L. Jedinák, R. Zatopkova, H. Zemankova, A. Sustkova and P. Cankař, J. Org. Chem., 2017, 82, 157–169 CrossRef.
- Y. F. Yu, Y. D. Huang, C. Zhang, X. N. Wu, Q. Zhou, D. Wu, Y. Wu and H. B. Luo, Neurosci, 2017, 8, 2522–2534 CAS.
- (a) C. Herrera-Arozamena, M. Estrada-Valencia, C. L. Pérez, L. Lagartera, J. A. Morales-García, A. Pérez-Castillo, J. F. Franco-Gonzalez, P. Michalska, P. Duarte, R. León, M. G. López, A. Mills, F. Gago, Á. J. García-Yagüe, R. Fernández-Ginés, A. Cuadrado and M. I. Rodríguez-Franco, Eur. J. Med. Chem., 2020, 190, 112090–112114 CrossRef CAS; (b) Z. Sang, K. Wang, P. Bai, A. Wu, J. Shi, W. Liu, G. Zhu, Y. Wang, Y. Lan, Z. Chen, Y. Zhao, Z. Qiao, C. Wang and Z. Tan, Eur. J. Med. Chem., 2020, 194, 112265 CrossRef CAS; (c) G. Zhu, K. Wang, J. Shi, P. Zhang, D. Yang, X. Fan, Z. Zhang, W. Liu and Z. Sang, Bioorg. Med. Chem. Lett., 2019, 29, 126625–126631 CrossRef CAS.
- (a) M. B. Youdim, D. Edmondson and K. F. Tipton, Nat. Rev. Neurosci., 2006, 7, 295–309 CrossRef CAS; (b) S. F. Carter, K. Herholz, P. Rosa-Neto, L. Pellerin, A. Nordberg and E. R. Zimmer, Trends Mol. Med., 2019, 25, 77–95 CrossRef CAS.
- U. Kosak, D. Knez, N. Coquelle, B. Brus, A. Pislar, F. Nachon, X. Brazzolotto, J. Kos, J. P. Colletier and S. Gobec, Bioorg. Med. Chem., 2017, 25, 633–645 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07959f |
| This journal is © The Royal Society of Chemistry 2021 |