
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImplementation of the emulsification-diffusion method by solvent displacement for polystyrene nanoparticles prepared from recycled material
Ana María Pineda-Reyes a,
Mauricio Hernández Delgadoa,
María de la Luz Zambrano-Zaragozab,
Gerardo Leyva-Gómezc,
Nestor Mendoza-Muñozd and
David Quintanar-Guerrero*a
a,
Mauricio Hernández Delgadoa,
María de la Luz Zambrano-Zaragozab,
Gerardo Leyva-Gómezc,
Nestor Mendoza-Muñozd and
David Quintanar-Guerrero*a
aLaboratorio de Investigación y Posgrado en Tecnología Farmacéutica, Facultad de Estudios Superiores Cuautitlán, Universidad Nacional Autónoma de México, Av. 1o. de Mayo s/n, Cuautitlán Izcalli, Estado de México 54740, Mexico. E-mail: quintana@unam.mx
bLaboratorio de Procesos de Transformación y Tecnologías Emergentes de Alimentos, Departamento de Ingeniería y Tecnología, Facultad de Estudios Superiores Cuautitlán, Universidad Nacional Autónoma de México, Km 2.5 Carretera Cuautitlán–Teoloyucan, San Sebastián Xhala, Cuautitlán Izcalli, Estado de México 54714, Mexico
cDepartamento de Farmacia, Facultad de Química, Universidad Nacional Autónoma de México, Ciudad de México, 04510, Mexico
dFacultad de Ciencias Quimicas, Universidad de Ciencias Quimicas, Colimas, 28400, Mexico
First published on 8th January 2021
Abstract
From an integral perspective, nanotechnology can be used to care for the environment by improving current preparation methods and facilitating industrial scale-up. This article discusses the implementation of techniques for obtaining polystyrene nanoparticles (PSN), as an added value, using an emulsification and solvent displacement method (EDSD); the solvent displacement is a novel modification to the emulsion-diffusion methodology, where the diffusion is done directly on the emulsion, which allows concentrated dispersions that facilitate direct use to be obtained, eliminating the dilution with water and the recovery of water by additional processes. The solvent was recovered by reduced pressure, which is class 3, conforming to ICH, and making this method sustainable. The optimization of this process has not been reported elsewhere. This approach made it possible to obtain highly-concentrated nanoparticles while allowing the reuse of the solvent. A scaling proposal is presented that integrates the conditioning of the solid urban waste material called expanded polystyrene (EPS) foam that constitutes a serious environmental problem both nationally and globally. Hence, the article presents an alternative to the recycling of EPS, and a methodology in the context of green chemistry, because solvent is recovered to prepare other batches. The PSN obtained from this waste material had a minimum particle size of 225.8 nm, with a polydispersion index of 0.158. Process performance was 97.1%, and the solvent was recovered at a maximum rate of 85%. The morphology of the PSN was spherical and uniform, with a smooth surface.
1. Introduction
One of the most important challenges of nanotechnology consists of preparing nanoparticles by simple, non-polluting processes. In this regard, the use of preformed polymers is preferable to those that require polymerization reactions, due to the residues that may be generated.1 Other objectives of preparing preformed polymer particles are: (i) obtaining dispersions with a high concentration of solids; (ii) using the minimum amount of solvents or, preferably, “green” solvents; (iii) devising more environmentally friendly processes (e.g., by reusing solvents or using waste polymers); and (iv) achieving technological feasibility such as easy implementation and susceptibility to industrial scale-up.1–6 One recent proposal that has drawn attention consists of recycling polymers for industrial use by forming nanoparticles for later applications.7,8 One polymer of interest is EPS, a waste material. In 2016, discarded plastics accounted for 12% of all solid waste worldwide. In Mexico, 10.9% of solid urban waste consisted of plastics, with EPS being one of the most common thermoplastics, after polyolefins and polyvinyl chloride. However, in Mexico in 2012, only around 9.63% of the total volume of urban waste generated was recycled (INECC, 2012).9 EPS can be used to prepare polystyrene nanoparticles that could have various uses at a low cost, with scale-up potential, and environment-friendly processes. The task of scaling up nanoparticle production demands evaluating process design, optimizing preparation variables, and an adaptation process from small- to large-scale operating conditions.10,11 Polystyrene nanoparticles obtained of EPS can be used for a huge variety of different purposes which can include: in the form of a coating on Kraft paper,12 treatment of phenol contaminated industrial wastewater,13 a paint binder,14 wood plastic composites,15 concrete impregnated,16 DVD cases and office equipment,17 materials for use in filtration applications,18 among others.Rajeev et al. reported the preparation of polystyrene particles from annealed expanded polystyrene by the nanoprecipitation or solvent displacement method using polar solvents. The final diameter was less than 500 nm.19 Mangalara and Varughese obtained polystyrene nanoparticles using D-limonene as polystyrene solvent and PVAL as stabilizer by the emulsification-diffusion obtaining a maximum of 86% PE in the final dispersion. These authors stood out the sustainability of the method.20
The literature contains several reports on preparation methods that incorporate preformed polymers into emulsified systems, developed as a first step towards increasing the exposed surface area and permitting processes of polymeric aggregation in non-solvent microphases. A second step involves forming nanoparticles by controlled aggregation and “diffusion-stranding” stabilization of the polymer.2,21–23 The best-known processes are emulsification–evaporation,1,24–28 salting-out,5,22,29–31 solvent displacement (or nanoprecipitation)4,21,32–34 and emulsification-diffusion.24,27,35–38 Modalities of these processes include dialysis,21 emulsification with solvent displacement,1,3,39 and spray-drying.21,40–42 In this regard, Campardelli et al. evaluated adding a continuous supercritical fluid processing step to improve the emulsification-diffusion method by eliminating the organic solvent from the nanosuspension obtained.43 Naranjos et al. evaporated the solvent using reduced pressure.24 Kwon et al. removed the nanoparticles by dialysis to separate the added water from the nanoparticles,37 while Quintanar et al. used distillation, or cross-flow, filtration to eliminate the organic solvent, according to its boiling point,5 and recovering the solvent by a distillation process.20 Staff et al. prepared nanoparticles of polystyrene by the emulsion–solvent evaporation process, using chloroform, toluene and other solvents.44
The emulsification-diffusion method by solvent displacement is a modality of the emulsification-diffusion method that consists in carrying out the diffusion phase via direct extraction of the solvent under moderate temperature and reduced pressure. This process was developed to obtain concentrated pseudolatexes,1,3,39 but has been proposed, as well, for applications in biotechnology and nanomedicine.1 It has important advantages over other methods; for example, using acceptable organic solvents, allowing the preparation of nanospheres and nanocapsules with high yield and excellent reproducibility, and facilitating particle size control, among others.45 The emulsion-diffusion method consists of two steps. First, an emulsion is formed from a mixture of a partially-miscible solvent in water (previously saturated with water) that contains the polymer, with an aqueous phase (previously saturated with the solvent) containing the stabilizer. Once the emulsion is obtained, the second step consists in diffusing the solvent from the internal phase (globules) by diluting the emulsion, using considerable amounts of water. This change in composition causes the polymer to enter a “non-solvent” phase that produces polymer aggregates called “protonanoparticles” of colloidal size from which nanoparticles will form, provided the stabilizer impedes coalescence.3,5,21,36,46 This method has been adapted successfully and has produced important results, such as developing micro and nanospheres in which drugs can be either trapped in a matrix or dispersed into an oil core covered by a polymeric membrane that can be dissolved, chemically adsorbed, or bonded to the surface of the particles.2,47
The aims of this project are to develop and optimize the emulsion-diffusion method for preparing polystyrene nanoparticles as a recycling procedure, presenting a modification to the emulsion-diffusion method where one of the main advantages is that the diffusion is done directly on the emulsion (EDSD), which allows us to obtain concentrated dispersions that facilitate its direct use, evaluating the parameters for industrial scale-up. The use of nanoparticles preformed from waste material can have potential technological applications for the industry. This can minimize the useful life of large volumes of polymer waste, while decreasing production in synthesizing new materials with a favourable impact on the environment.
2. Experimental protocol
2.1. Materials
EPS waste from packaging materials used with household appliances was collected in signposted containers at the Faculty in a pilot waste recovery program. Ethyl acetate (EtAc) was supplied by Fermont, Mexico. The stabilizer, polyvinyl alcohol (PVA, Mowiol® 4-88 Mw ∼ 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), was purchased from Sigma-Aldrich, USA. Purified water was of Milli-Q quality (Millipore, USA-Bedford, MD). All other reagents were of at least analytical grade and were used without further purification.
000), was purchased from Sigma-Aldrich, USA. Purified water was of Milli-Q quality (Millipore, USA-Bedford, MD). All other reagents were of at least analytical grade and were used without further purification.
2.2. Methods
:2 EtAc/water.1 For this purpose, the EtAc and water were mutually saturated in a separating funnel under manual agitation for 1 minute before use to ensure the thermodynamic equilibrium of both liquids. The densified EPS was dissolved from 1 to 10% (w/v) in the saturated EtAc (organic phase) using magnetic agitation until dissolution was complete. The organic phase was emulsified in a saturated aqueous solution containing from 1 to 5% (w/v) of PVA (aqueous phase) under agitation at 733.04, 1152.92 and 1623.16 s−1 from 1 to 10 min in a rotor–stator device (Ultra-Turrax® T18 Basic, IKA-Werke, Staufen, Germany). Finally, the emulsion was subjected to distillation under reduced pressure to remove the solvent by evaporation (Laborota 4000 Heidolph Instruments, Schwabach, Germany) at 40 °C and 9.3 kPa with stirring at 6.28 s−1, until complete evaporation was achieved. As much as 85% of the EtAc was recovered and reused to prepare other batches.
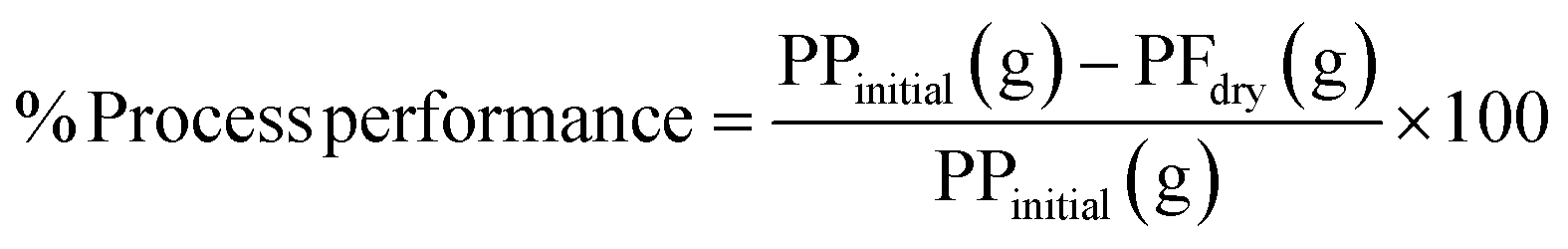 | (1) |
| Factor | Range analysed |
|---|---|
| Polymer | 1.0–10.0% |
| Stabilizer | 1.0–5.0% |
| Stirrer speed | 7000, 11000 and 15500 rpm |
| Agitation time | 1–10 minutes |
| Batch no. | Polymera (w/v%) | Stabilizerb (w/v%) | Batch no. | Polymera (w/v%) | Stabilizerb (w/v%) |
|---|---|---|---|---|---|
| a Organic phase.b Aqueous phase. | |||||
| 1 | 5.77 | 4.70 | 14 | 10.00 | 1.00 |
| 2 | 5.86 | 5.00 | 15 | 1.00 | 5.00 |
| 3 | 5.95 | 1.00 | 16 | 8.65 | 1.00 |
| 4 | 8.65 | 1.00 | 17 | 1.00 | 1.00 |
| 5 | 1.00 | 1.00 | 18 | 1.00 | 3.64 |
| 6 | 1.00 | 4.56 | 19 | 1.00 | 5.00 |
| 7 | 5.32 | 3.56 | 20 | 1.00 | 3.64 |
| 8 | 10.00 | 3.50 | 21 | 5.77 | 4.70 |
| 9 | 10.00 | 5.00 | 22 | 1.00 | 1.00 |
| 10 | 10.00 | 5.00 | 23 | 10.00 | 3.50 |
| 11 | 1.00 | 1.52 | 24 | 10.00 | 1.00 |
| 12 | 5.32 | 3.56 | 25 | 5.65 | 1.00 |
| 13 | 10.00 | 2.95 | — | — | — |
The information corresponding to the analysis by the 3-D response surface methodology proceeds from the results obtained from the characterization of the 25 experimental runs proposed by the I-optimal design. The variables analyzed to study the relation between the response and independent variables and so determine the optimal preparation conditions were particle size, the polydispersion index, and process performance. It is important to mention, however, that these results were determined by the mathematical model that best fitted the data obtained from the I-optimal design. Likewise, the equation from the model used in the diagnostic graphs was analysed using the quadratic polynomial model, after validation through a suitability verification analysis, according to the assumptions of normality, constant variance, and independence, to ensure the accuracy of the representation of the real system.49 According to Hernández this procedure generates results with a strong correlation between the model's predictions and real results, and so allows analyses of the values of the factors that optimized those of the variables.8
2.2.5.1. Particle size analysis. The average particle size and polydispersity index were determined using the dynamic light-scattering (DLS) technique on a Nano ZS Zetasizer (ZEN3600, Malvern Instruments Ltd., Enigma Business Park, Grovewood Road, UK) at 25 °C with a detection angle of 173°. All samples were diluted with an excess of purified water and analyzed in triplicate.
2.2.5.2. Determination of zeta potential. The electrical charge of the nanoparticles in dispersion was determined by electrophoretic mobility using Doppler Laser Electrophoresis equipment and a Nano ZS Zetasizer (ZEN3600, Malvern Instruments Limited, UK). Measurements were made in triplicate at 25 °C after diluting the samples in water.
2.2.5.3. Differential thermal analysis. The thermal stability of the undensified and densified EPS, and of the lyophilized PSN, was analyzed by differential scanning calorimetry (DSC) in a Q10 TA Instruments, DSC Discovery (New Castle, DE, USA), with a heating ramp of 10 °C min−1, from 0 to 200 °C in a nitrogen atmosphere at a flow of 50 mL min−1. The PSN were previously lyophilized in a Labconco freeze-dryer (77520-00 Labconco, Millrock Technology, Inc. Kinsgston, NY, USA).
2.2.5.4. Scanning electron microscopy. The morphology of the PSN was registered by scanning electron microscopy (SEM) in a microscope (model LV-SEM, JSM 5600 LV, JEOL, Tokyo, Japan) with a resolution of 5 nm at 7 kV, and a voltage chamber pressure of 12–20 Pa. The samples were prepared by covering the surface with a gold film (∼20 nm) in a sputter coater cathode evaporator (JFC-1100, JEOL, Tokyo, Japan) under exposure to the environment until removal of the excess water.
3. Results and discussion
3.1. Densification of EPS waste
This study was designed to evaluate the use of waste material with potential technological applications, such as preparing PSN to introduce them in useful materials giving them functionality or improvement of properties, such as construction materials, as in gypsum-boards mentioned in section 3.5. In this regard, Fig. 1 shows the material in its original volume (Fig. 1a), and after heat treatment (Fig. 1b). The thermal treatment sufficed to reduce the volume/weight ratio and facilitate the final disposition for easy handling with a low generation of electrostatic charge. Thermal densification was achieved by changing the foamy state to a compact solid state (rigid texture). This approach is consistent with the study by Kan and Demirboğa, who described a rigid texture with a morphology distinct from that of the original foams after EPS thermal treatment.48 This result justifies proposing the thermal treatment method to reduce the volume of EPS at the industrial level to approximately 90%, thus facilitating its handling, storage, and disposal. | ||
| Fig. 1 Waste EPS cut into pieces (a) and densified by heat treatment (b). | ||
3.2. Preparation of the PSN
Another objective was to prepare preformed PSN from recycled material, while evaluating the maximum quantities of EPS that can be transformed into nanoparticles by EDSD in a modification of the emulsification-diffusion method1 (Fig. 2). In addition, we proposed determining optimal fabrication conditions. It is important to emphasize that one of the main advantages of this method is that it induces diffusion from the emulsion by evaporation instead of water dilution. This allowed us to obtain dispersions with high solid content, as we report in detail in the following section. Introducing a water-soluble organic solvent to the continuous phase has favourable effects on the formation of the nanoparticles. He et al. observed the evolution of the internal structures of polystyrene microspheres went from porous to hollow, after introducing ethanol which allowed control of the internal structures of the microspheres as well as decreased the particle size of them, attributing this effect to the decrease in surface tension caused by solvent.50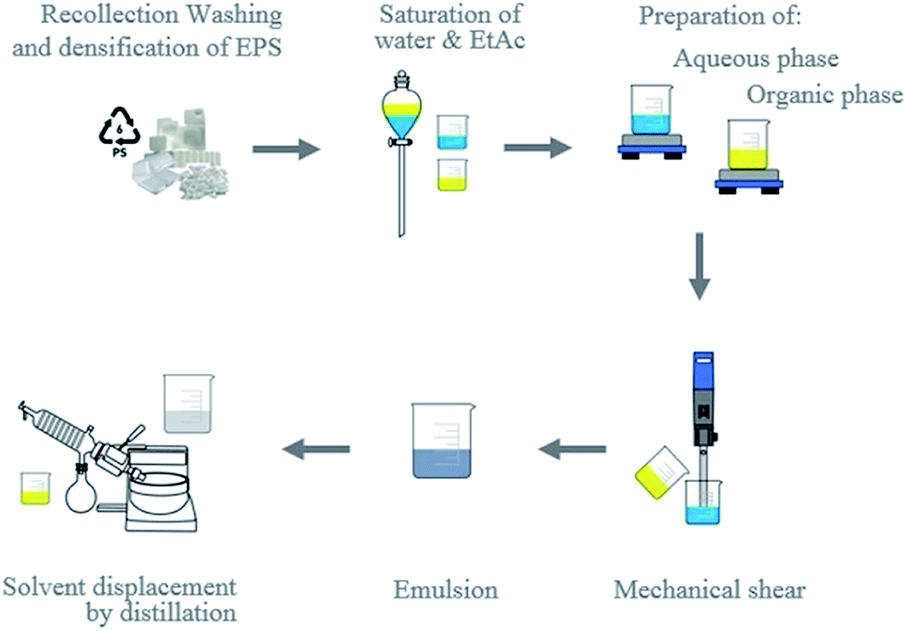 | ||
| Fig. 2 Emulsification-displacement process of the solvent at laboratory scale.36 | ||
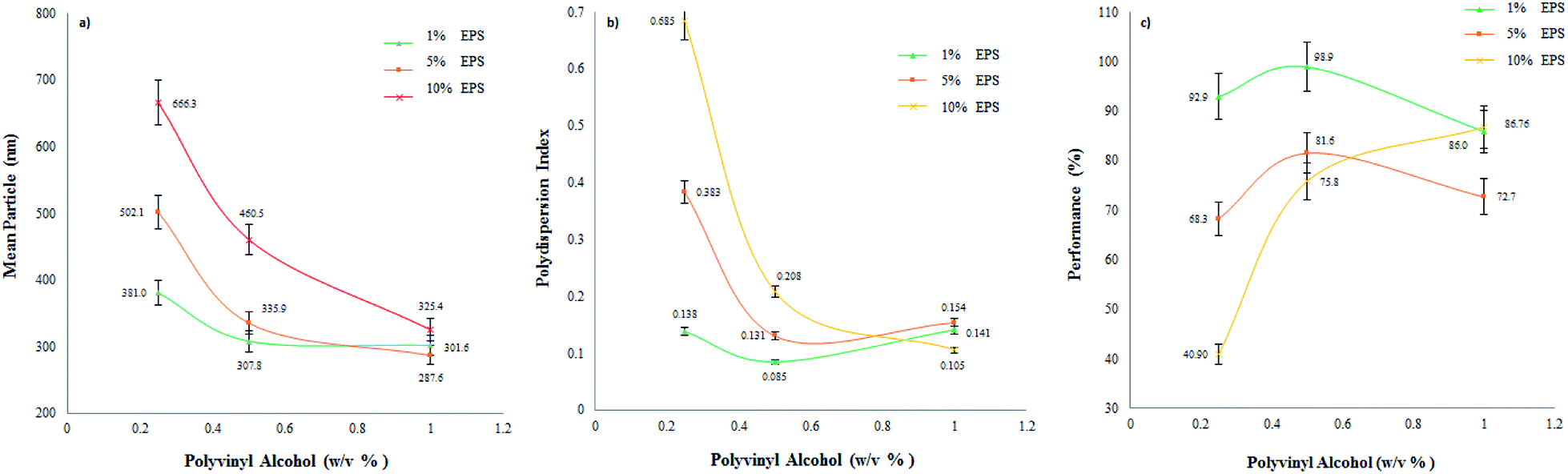 | ||
| Fig. 3 Effect of polymer and stabilizer concentrations on average particle size (a), the polydispersion index (b), and process performance (c) by the EDSD method. | ||
The manufacturing batches produced were characterized by a concentration of up to 10%, though particle size increased significantly as the amount of EPS increased, mainly at low stabilizer concentrations. This behaviour is related to the mechanism of the formation of the nanoparticles, since during the diffusion stage a higher polymer concentration contributes to forming a more saturated aggregation layer. Known as “diffusion–stranding”, this procedure generates the formation of nanoparticles,38,54 considering that the stabilizer concentration is involved in the complex interfacial phenomena formed by the emulsion-diffusion technique,55 but without taking into account the nucleation and growth of the nanoparticles.38 Thus, even though the stabilizer may function adequately, the probability of particle aggregates passing from a more concentrated region is very high, which would explain the particle size increments with the increase in EPS. Concordant results were observed for the polydispersion index (Fig. 3b), as the distribution curve becomes wider at higher EPS concentrations. These differences were significant at low PVA concentrations (0.25%) but not at high ones, suggesting that the stabilizer function is appropriate for these concentrations (1.0%). PVA can stabilize globules in the emulsion and impede aggregation during diffusion in the solid–liquid interface.54 This behaviour was also observed by Mora et al. where, regardless of the polymer, particle size increased at higher concentrations.36
Process performance in relation to the stabilizer content for different EPS concentrations is depicted in Fig. 3c. Low stabilizer concentrations (0.25%) make it possible to obtain dispersions with low yield. Differences among EPS concentrations are marked. PVA concentrations of 0.5% allowed yields above 70%, a level considered adequate. Quintanar et al. prepared nanoparticles of poly(D,L-lactic acid) with a yield >95%. In their study, diffusion was performed by adding water.51 High production yields of particles (>70%) have been reported using emulsion-diffusion during the preparation of nanocapsules; however, it is essential to note that water must be added during diffusion.43,54,56–59 Concentrations above 1.0% did not significantly increase the yield at any EPS concentration.
The response surface graph is shown in Fig. 4, illustrating the effect of polymer and stabilizer concentrations on process performance. The EPS concentration has the greatest influence on the process performance of the PSN, possibly due to the diffusion of the solvent, which allows the transport of polystyrene molecules into the “non-solvent” phase, and from the globules into the aqueous phase, leading to the formation of supersaturated areas of polymer, where the presence of the stabilizer prevents coalescence. With respect to the PVA concentration, this factor has a greater influence on process performance at higher concentrations. This mechanism allows the particles to form polymer aggregates in drops by interfacial phenomena, which are stabilized by the PVA, thus avoiding fusion and the formation of agglomerates, while remaining at the interfaces. PVA presents an adequate protection effect during the step of forming the PSN,5,60 which requires a few milliseconds during the diffusion stage of the solvent from the emulsion drops to the aqueous phase of the system.61
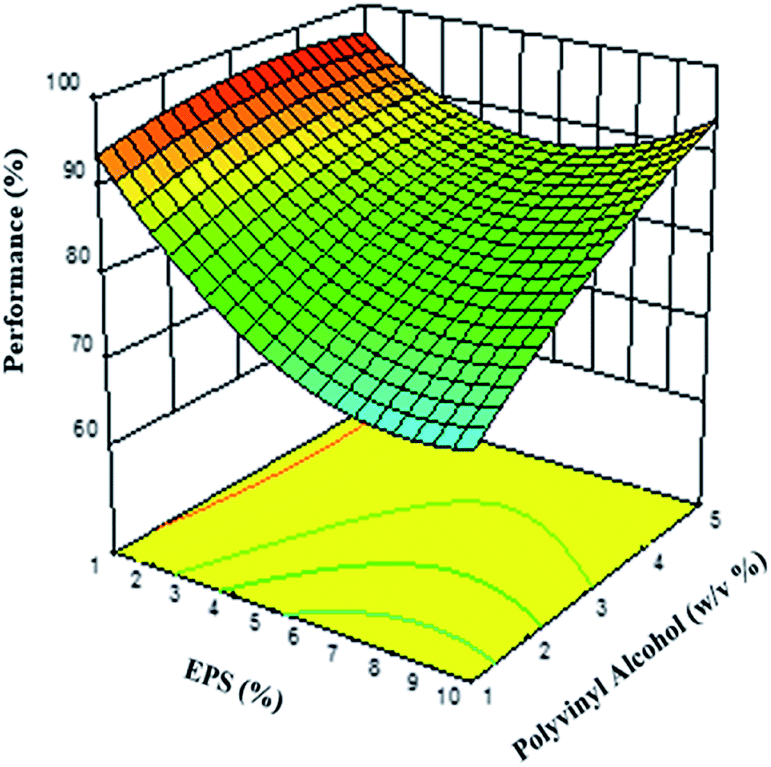 | ||
| Fig. 4 3-D response surface graph of the effect of polymer and stabilizer concentrations on process performance by the EDSD method. | ||
| Batch no. | Zeta potential (mV) | Batch no. | Zeta potential (mV) |
|---|---|---|---|
| 1 | −31.80 | 14 | −34.17 |
| 2 | −28.70 | 15 | −30.33 |
| 3 | −30.20 | 16 | −28.37 |
| 4 | −33.77 | 17 | −33.37 |
| 5 | −31.50 | 18 | −32.00 |
| 6 | −31.90 | 19 | −33.87 |
| 7 | −32.33 | 20 | −32.10 |
| 8 | −31.53 | 21 | −26.40 |
| 9 | −32.23 | 22 | −26.87 |
| 10 | −32.20 | 23 | −26.57 |
| 11 | −19.23 | 24 | −28.90 |
| 12 | −32.47 | 25 | −26.97 |
| 13 | −35.40 | — | — |
3.3. Differential scanning calorimetry
Thermal analysis showed that the thermal densification process at 130 °C does not affect EPS. It further revealed that the curves obtained by DSC were similar for densified EPS, virgin EPS, and the reference polystyrene (PS) (Fig. 5). Likewise, a change in the baseline at approximately 100 °C was observed, indicating the glass transition of the densified polystyrene, likely due to the evaporation of the gases contained in the cellular structure of this material, which presented a negative slope at lower temperatures. Similar results were reported by Samper et al. who observed glass transition temperatures around 105 °C for EPS, without observing degradation of the material up to 136 °C.60 Those authors also reported an important fact regarding the glass transition temperature of unprocessed polystyrene particles, which is lower than that for EPS, thus allowing a higher state of thermal resistance of EPS than of unprocessed polystyrene particles. The physical transitions of EPS have also been studied by Mehta et al. with results for the density temperature between approximately 110 and 120 °C. In comparison, the melting temperature was observed at 160 °C. The vaporization temperature was reported to be 275 °C, while total volatilization occurred between 460 and 500 °C. This allowed us to observe temperature ranges of EPS handling that did not affect the environment when the material decomposed due to the effect of temperature. This also facilitated proper handling.63 With respect to the undensified EPS, our analysis demonstrated a glass transition of the polymer from 100 to 110 °C, similar to that of densified polystyrene. This finding could be due to the effect just mentioned. Therefore, the DSC analysis confirmed that the densification process was adequate, since it verified that there was no degradation of the polymer.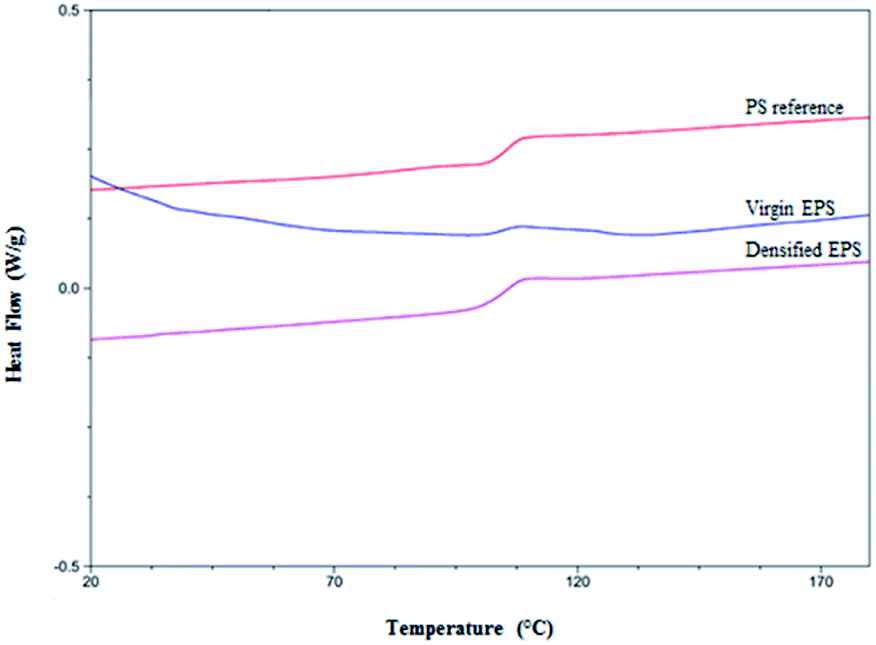 | ||
| Fig. 5 Results of differential scanning calorimetry analysis for the characterization of virgin EPS and densified EPS vs. reference PS. | ||
3.4. Scanning electron microscopy
SEM depicted a spherical, uniform, smooth surface morphology of the PSN (Fig. 6). This shows that the EDSD method is suitable for forming nanoparticles of uniform morphology using low amounts of stabilizer. The sample shown corresponded to the optimized formulation of the I-optimal design, which agreed in size with the measurements made in DLS; that is 230.6 nm. Magnification at 7500× was sufficient to observe a well-defined morphology in the samples.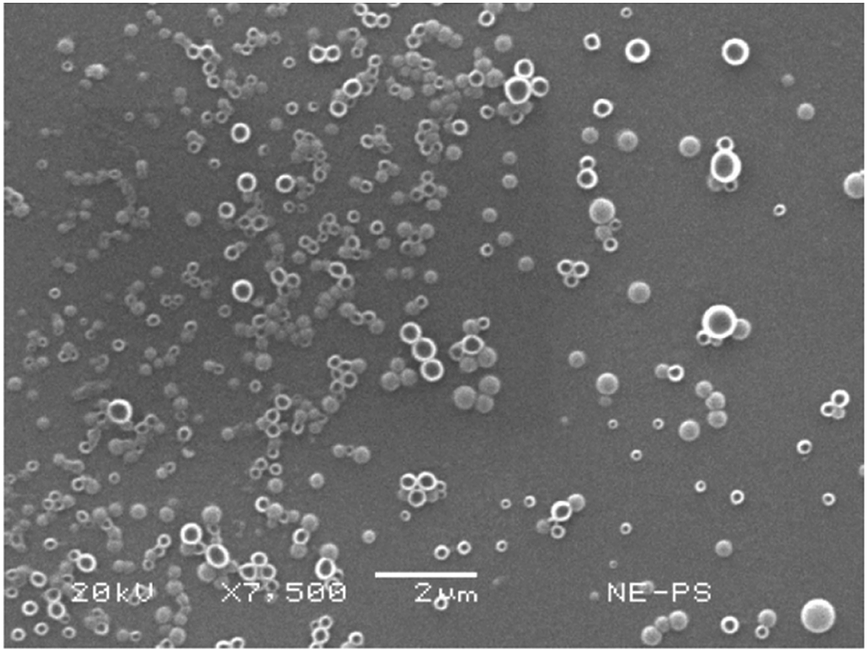 | ||
| Fig. 6 Micrographs of nanoparticles obtained by the EDSD method at 5000 and 7000×. | ||
3.5. Possible scale-up of PSN production
Considering the high production yields obtained, and the high concentrations achieved from PSN, it is possible to predict successful industrial-scale production that will give commercial value to the production of PSN, highlighting production based on recycling EPS waste and solvent recovery. PSN can be used in several applications with no risk for the environment; for example, in the construction industry. An application in gypsum-boards produced satisfactory results obtained by measuring the percentage of moisture absorption, which was lower than in gypsum-boards with no PSN incorporated.8Based on the emulsification-diffusion method – which has been widely-researched – and total compression in the formation of nanoparticles, which is essential for easy scale-up.53,54,58,64–68 All authors have predicted the factors and conditions that determine the optimization of the method. In this sense, Quintanar et al. mentioned as a key factor the relation between rapid solvent displacement and nanoparticle formation during diffusion.35 This facilitates scale-up because, as the present study proposes, the solvent can be recovered for future use and the process fosters polymer aggregation, resulting in high nanoparticle yields when compared to the evaporation or filtration techniques commonly used as a subsequent emulsion step to form nanoparticles.69 Mendoza et al. described the evaporation stage as a limitation in scale-up to produce large batches of nanoparticles because it must be efficient to obtain residual solvent.3 In contrast to using solvent displacement in this method, the approach proposed is easy, versatile, and useful. In addition, the displacement of the solvent from the internal to the external phase is fast and, ultimately, leads to polymer aggregation.35
Fig. 7 presents a proposal for both the laboratory scale and industrial scale-up for producing PSN, and indicates the critical variables involved. The main components at the industrial scale are a mill and heating tunnel for the densification process. The fabrication of nanoparticles comprises various phases: the saturation system, mixing with a propeller stirrer, solvent evaporation under reduced pressure, and storage in a tank. Laboratory studies show that the critical variables – the bulk size of EPS, stirring times, volumetric mixtures ratios, concentrations, stirring times and rates, and temperatures – must all be controlled to prepare small nanoparticles with a low polydispersity index. Thus, the operating equipment required does not have to be highly-sophisticated to enable efficient scale-up.3,35,54 Solvent evaporation under reduced pressure permits solvent recovery. As mentioned above, removing the organic solvent is the main aim in the solvent displacement step in this novel method, which has been used to resolve the formation of nanoparticles by rapid diffusion and allow the formation of supersaturated regions near the interface. In addition, environmental problems are avoided, so we can disregard this obstacle to the implementation of an efficient system for extracting the solvent and quickly achieving scale-up.3
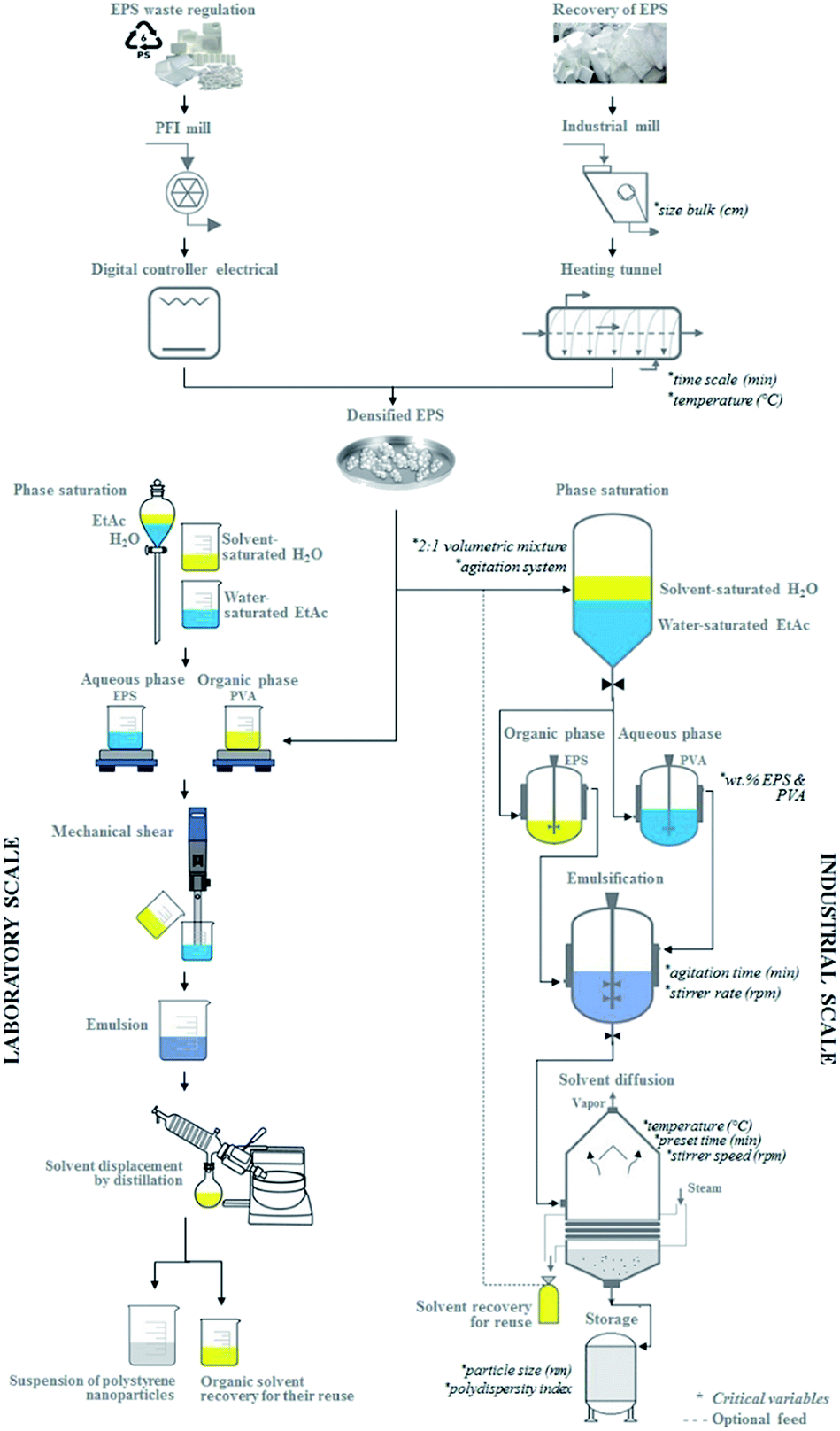 | ||
| Fig. 7 Schematic description of scaling-up to prepare polystyrene nanoparticles from urban waste using the EDSD method. | ||
Other authors have identified drawbacks of scale-up; for example, that the emulsification-diffusion method requires diffusing a large amount of water to dissolve the organic phase during diffusion and, as a consequence, the need to dispose of large volumes of water.54,64 In this case, the nanoprecipitation method must be controlled throughout the process, since the preparation of the emulsion requires large amounts of both phases, causing certain difficulties.34 The high-pressure homogenization method demands a substantial increase in the circulation time of continuous 10 kg batches; thus requiring a change of batch size or a more discontinuous system.70 Emulsification–reverse salting out methods, meanwhile, have potential problems of incompatibility between the salts and the drugs during the manufacture of nanocapsules, and due to the presence of electrolytes, which makes an additional stage of purification necessary.53
4. Conclusions
EPS foams can be treated by recycling to help reduce the problem of urban waste. The EDSD method allowed us to obtain satisfactory results when producing high-concentration polystyrene nanoparticles that meet the optimized formulation of the I-optimal design, with a 97.1% of process yield. This method not include the diffusion step with large amount water, and allowing the recovery of the solvent by up to 85%. The polystyrene nanoparticles obtained from this waste material presented a minimum particle size of 225.8 nm, and a polydispersion index of 0.158, with process values of 10% polystyrene and 4.8% stabilizer. The morphology of the polystyrene nanoparticles was spherical and uniform, with a smooth surface. The use of recycled material to produce polystyrene nanoparticles can aid in the treatment of solid urban waste, generate a value-added product with broad potential in the field of construction, and contribute to mitigating environmental pollution. In addition, solvent recovery for use in new batches can also benefit the environment, and it is a solvent class 3 considered not human health hazard conform to ICH. The experience in the handling of this technique allowed us to demonstrate the feasibility of industrial scale-up, due to such advantages as high reproducibility and versatility.Conflicts of interest
There are no conflicts to declare by any of the authors.Acknowledgements
Pineda-Reyes thanks the postdoctoral scholarship program, Dirección General de Asuntos del Personal Académico (DGAPA), for the funding received. Hernández-Delgado thanks the Consejo Nacional de Ciencia y Tecnología (CONACyT) for the grant received (number 597099). The authors acknowledge the financial support for this work provided by PAPIIT IN 222520 and IN 222420 (DGAPA), PIAPI 2040, Conacyt CB 221629 and Conacyt INFRA 251940.References
- D. Quintanar-Guerrero, E. Allémann, H. Fessi and E. Doelker, Int. J. Pharm., 1999, 188(2), 155–164 CrossRef CAS
.
- G. Subedi, A. K. Shrestha and S. Shakya, Open Pharmaceutical Sciences Journal, 2016, 3, 182–195 CrossRef
- N. Mendoza-Muñoz, S. Alcalá-Alcalá and D. Quintanar-Guerrero, in Polymer Nanoparticles for Nanomedicines, ed. C. Vauthier and G. Ponchel, Springer, Switzerland, 2016, ch. 4, pp. 87–121 Search PubMed
- K. Miladi, S. Sfar, H. Fessi and A. Elaissari, in Polymer Nanoparticles for Nanomedicines, ed. C. Vauthier and G. Ponchel, Springer, Switzerland, 2016, ch. 2, pp. 17–53 Search PubMed
- D. Quintanar-Guerrero, E. Allémann, H. Fessi and E. Doelker, Drug Dev. Ind. Pharm., 1998, 24(12), 1113–1128 CrossRef CAS
- E. K. Noriega-Peláez, N. Mendoza-Muñoz, A. Ganem-Quintanar and D. Quintanar-Guerrero, Drug Dev. Ind. Pharm., 2011, 37(2), 160–166 Search PubMed
- MX Pat., MX/a/2019/009694 Search PubMed
- M. Hernández, Master thesis, Universidad Nacional Autónoma de México, 2018
- Informe del Medio Ambiente en México 2015 [Internet], Informe del Medio Ambiente, 2016, available https://apps1.semarnat.gob.mx:8443/dgeia/informe15/index.html, accessed June 25, 2020 Search PubMed.
- A. A. Ruiz and H. Álvarez, Información Tecnológica, 2011, 22(6), 33–52 CrossRef CAS
- A. Anaya-Durand and P.-F. Humberto, Escalamiento, el arte de la ingeniería química: plantas piloto, el paso entre el huevo y la gallina, Tecnol., Cienc., Educ., 2008, 23(1):31–39, recuperado de: https://www.redalyc.org/articulo.oa?id=48223105 Search PubMed.
- M. Jimenez-Francisco, J. A. Caamal-Canche, J. G. Carrillo and R. H. Cruz-Estrada, J. Polym. Environ., 2017, 26(4), 1573–1580 CrossRef
- A. N. Siyal, S. Q. Memon, S. Parveen, A. Soomro, M. I. Khaskheli and M. Y. Khuhawar, J. Chem., 2013, 1–8, DOI:10.1155/2013/842435
- S. A. Osemeahon, J. T. Barminas and A. L. Jang, IOSR J. Environ. Sci., Toxicol. Food Technol., 2013, 5(4), 01–07 CrossRef
- M. Poletto, J. Dettenborn, M. Zeni and A. J. Zattera, Waste Manage., 2011, 31(4), 779–784 CrossRef CAS
- M. Amianti and V. R. Botaro, Cem. Concr. Compos., 2008, 30(1), 23–28 CrossRef CAS
- E. Aminudin, M. F. Md Din, Z. Mohamad, Z. Z. Noor and K. Iwao, presented in part at International Conference on Environment and Industrial Innovation IPCBEE, Singapore, vol. 12, 2011 Search PubMed
- C. Shin, Packag. Technol. Sci., 2005, 18(6), 331–335 CrossRef CAS
- A. Rajeev, V. Erapalapati, N. Madhavan and M. G. Basavaraj, J. Appl. Polym. Sci., 2016, 133(4) DOI:10.1002/app.42904
- S. C. H. Mangalara and S. Varughese, ACS Sustainable Chem. Eng., 2016, 4(11), 6095–6100 CrossRef CAS
- J. Allouche, in Nanomaterials: A Danger or a Promise? A Chemical and Biological Perspective, ed. R. Brayner, F. Fiévet and T. Coradin, Springer, Verlag London, 2013, ch. 2, pp. 34–42 Search PubMed
- J. P. Rao and K. E. Geckeler, Prog. Polym. Sci., 2011, 36(7), 887–913 CrossRef CAS
- P. A. Grabnar and J. Kristl, J. Microencapsul., 2011, 28(4), 323–335 CrossRef CAS
- N. Naranjos-Ramírez, D. I. Torres-Cantú, V. I. Castillo-Rodríguez, S. A. Galindo-Rodríguez and A. Chávez-Montes, Rev. Mex. Fis., 2011, 57(2), 41–43 Search PubMed
- C. Wischke, ýInt. J. Pharm., 2020, 584, 1–19 Search PubMed
- K. Kızılbey, ACS Omega, 2019, 4(1), 555–562 CrossRef
- M. G. Nava-Arzaluz, E. Pinon-Segundo, A. Ganem-Rondero and D. Lechuga-Ballesteros, Recent Pat. Drug Delivery Formulation, 2012, 6(3), 209–223 CrossRef CAS
- S. Ji, D. Srivastava, N. J. Parker and I. Lee, Polymer, 2012, 53(1), 205–212 CrossRef CAS
- E. B. Souto, P. Severino and M. H. A. Santana, Polímeros, 2012, 22(1), 101–106 CrossRef CAS
- N. Mendoza-Muñoz, D. Quintanar-Guerrero and E. Allemann, Recent Pat. Drug Delivery Formulation, 2012, 6(3), 236–249 CrossRef
- H. Ibrahim, C. Bindschaedler, E. Doelker, P. Buri and R. Gurny, Int. J. Pharm., 1992, 87(1–3), 239–246 CrossRef CAS
- T. Querettea, E. Fleury and N. Sintes-Zydowicza, Eur. Polym. J., 2019, 114, 434–445 CrossRef
- K. Krishnaswamy and V. Orsat, in Nano- and Microscale Drug Delivery Systems, ed. A. M. Grumezescu, Elsevier, Sainte-Anne-de-Bellevue, QC, Canada, 2017, ch. 2, pp. 17–32 Search PubMed
- C. J. Martínez, M. Tarhini, W. Badri, K. Miladi, H. Greige-Gerges, Q. A. Nazari and A. Elaissari, Int. J. Pharm., 2017, 532(1), 66–81 CrossRef
- D. Quintanar-Guerrero, M. L. Zambrano-Zaragoza, E. Gutierrez-Cortez and N. Mendoza-Munoz, Recent Pat. Drug Delivery Formulation, 2012, 6(3), 184–219 CrossRef CAS
- C. E. Mora-Huertas, H. Fessi and A. Elaissari, Adv. Colloid Interface Sci., 2011, 163(2), 90–122 CrossRef CAS
- H.-Y. Kwon, J.-Y. Lee, S.-W. Choi, Y. Jang and J.-H. Kim, Colloids Surf., A, 2001, 182(1–3), 123–130 CrossRef CAS
- D. Quintanar-Guerrero, E. Allémann and E. Doelker, Colloid Polym. Sci., 1997, 275, 640–647 CrossRef CAS
- C. A. Nguyen, Y. N. Konan-Kouakou, E. Allemann, E. Doelker, D. Quintanar-Guerrero, H. Fessi and R. Gurny, AAPS PharmSciTech, 2006, 7(3), E54–E60 CrossRef
- C. R. Hoffmeister, T. L. Durli, S. R. Schaffazick, R. P. Raffin, E. A. Bender, R. C. Beck and S. S. Guterres, Nanoscale Res. Lett., 2012, 7(251), 1–13 Search PubMed
- X. Li, N. Anton, C. Arpagaus, F. Belleteix and T. F. Vandamme, J. Controlled Release, 2010, 147(2), 304–310 CrossRef CAS
- S. S. Guterres, V. Weiss, L. de L. Freitas and A. R. Pohlmann, Drug Delivery, 2000, 7(4), 195–199 CrossRef CAS
- R. Campardelli, M. Cherain, C. Perfetti, C. Iorio, M. Scognamiglio, E. Reverchon and G. D. Porta, J. Supercrit. Fluids, 2013, 82, 34–40 CrossRef CAS
- R. H. Staff, D. Schaeffel, A. Turshatov, D. Donadio, H.-J. Butt, K. Landfester and D. Crespy, Small, 2013, 9(20), 3514–3522 CrossRef CAS
- M.-Y. Lee, S.-G. Min, S. Bourgeois and M.-J. Choi, J. Microencapsul., 2009, 26(2), 122–129 CrossRef CAS
- E. Cohen-Sela, M. Chorny, N. Koroukhov, H. D. Danenberg and G. Golomb, J. Controlled Release, 2009, 133(2), 90–95 CrossRef CAS
- J. Robles, R. Castro, A. González and A. Chávez, Implementación de la técnica de emulsión/difusión para la nanoencapsulación de furosemida, Monterrey, N.L. México, 2015 Search PubMed
- A. Kan and R. Demirboğa, J. Mater. Process. Technol., 2009, 209(6), 2994–3000 CrossRef CAS
- M. M. Ba-Abbad, A. A. H. Kadhum, A. B. Mohamad, M. S. Takriff and K. Sopian, J. Ind. Eng. Chem., 2013, 19(1), 99–105 CrossRef CAS
- Y. He, X. Li, T. Zhu, M. Shan, L. Zhu, T. Si and Y. Sun, Polymers, 2018, 10, 789, DOI:10.3390/polym10070789
- D. Quintanar-Guerrero, E. Allémann, E. Doelker and H. Fessi, Pharm. Res., 1998, 15(7), 1056–1062 CrossRef CAS
- D. Moinard-Chécot, Y. Chevalier, S. Briançon, L. Beney and H. Fessi, J. Colloid Interface Sci., 2008, 317(2), 458–468 CrossRef
- J. S. Hong, D. Srivastava and I. Lee, J. Appl. Polym. Sci., 2018, 135, 46199–46207 CrossRef
- E. Piñón-Segundo, V. G. Llera-Rojas, G. Leyva-Gómez, Z. Urbán-Morlán, N. Mendoza-Muñoz, and D. Quintanar-Guerrero, in Nanoscale Fabrication, Optimization, Scale-up and Biological Aspects of Pharmaceutical Nanotechnology, ed. A. M. Grumezescu, Elsevier Inc., Bucharest, Romania, 2018, ch. 2, pp. 51–83 Search PubMed
- D. Quintanar-Guerrero, H. Fessi, E. Allémann and E. Doelker, Int. J. Pharm., 1996, 143(2), 133–141 CrossRef CAS
- S. M. Habib, A. S. Amr and I. M. Hamadneh, J. Am. Oil Chem. Soc., 2011, 89(4), 695–703 CrossRef
- M.-J. Choi, A. Soottitantawat, O. Nuchuchua, S.-G. Min and U. Ruktanonchai, Food Res. Int., 2009, 42(1), 148–156 CrossRef CAS
- D. Moinard, Y. Chevalier, S. Briançon, H. Fessi and S. Guinebretière, J. Nanosci. Nanotechnol., 2006, 6, 2664–2681 CrossRef
- C. E. Mora-Huertas, O. Garrigues, H. Fessi and A. Elaissari, Eur. J. Pharm. Biopharm., 2012, 80(1), 235–239 CrossRef CAS
- S. Samper, D. Garcia-Sanoguera, F. Parres and F. López, Prog. Rubber, Plast. Recycl. Technol., 2010, 26(2), 51–60 CrossRef
- T. M. Riddick, Control of Colloid Stability through Zeta Potential: With a Closing Chapter on Its Relationship to Cardiovascular Disease, Zeta-Meter, Minnesota, 1968 Search PubMed
- K. E. Sapsford, K. M. Tyner, B. J. Dair, J. R. Deschamps and I. L. Medintz, Anal. Chem., 2011, 83(12), 4453–4488 CrossRef CAS
- S. Mehta, S. Biederman and S. Shivkumar, J. Mater. Sci., 1995, 30, 2944–2949 CrossRef CAS
- C. Vauthier and K. Bouchemal, Pharm. Res., 2009, 26(5), 1025–1058 CrossRef CAS
- H. Souguir, F. Salaün, P. Douillet, I. Vroman and S. Chatterjee, Chem. Eng. J., 2013, 221, 133–145 CrossRef CAS
- S. Surassmo, S.-G. Min, P. Bejrapha and M.-J. Choi, Food Res. Int., 201, 43(1), 8–17 CrossRef
- D. Quintanar-Guerrero, D. Tamayo-Esquivel, A. Ganem-Quintanar, E. Allémann and E. Doelker, Eur. J. Pharm. Sci., 2005, 26(2), 211–218 CrossRef CAS
- D. Quintanar-Guerrero, E. Allémann, H. Fessi and E. Doelker, Int. J. Pharm., 1999, 188(2), 155–164 CrossRef CAS
- K. Lim and Z. A. A. Hamid, in Applications of Nanocomposite Materials in Drug Delivery, ed. Inamuddin, A. M. Asiri and A. Mohammad, Woodhead Publishing, 2nd edn, 2018, ch. 10, pp. 217–237 Search PubMed
- A. Dingler and S. Gohla, J. Microencapsulation, 2002, 19(1), 11–16 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2021 |