
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mild one-pot synthesis of 2-iminothiazolines from thioureas and 1-bromo-1-nitroalkenes†
Yuan Xuab,
Xin Geb,
Yuhan Zhangb,
Hongbin Zhang a and
Xue-Wei Liu*b
a and
Xue-Wei Liu*b
aKey Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education and Yunnan Province, School of Chemical Science and Technology, Yunnan University, Kunming 650091, China
bDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 637371, Singapore. E-mail: xuewei@ntu.edu.sg
First published on 8th January 2021
Abstract
A mild method to access functionalized 2-iminothiazolines in a facile and efficient manner has been developed. The reaction started from 1,3-disubstituted thioureas and 1-bromo-1-nitroalkenes in the presence of triethylamine in THF and proceeded smoothly in air to afford 2-iminothiazoline derivatives in moderate to good yields.
Functionalized 2-iminothiazoline has been an important building block in organic chemistry.1 Its derivatives show significant pharmacological activities such as bactericidal and fungicidal activity.2 In addition, they are found in drug applications for treatment of allergies, hypertension, inflammations, bacterial and HIV infections.3 Pifithrin (Pft-α) (Fig. 1), isolated by screening of chemical libraries using 2-iminothiazoline skeleton, is a lead compound for p53 inactivation and has received increasing attention due to its possible applications in therapy of Alzheimer's disease, Parkinson's disease, stroke and other pathologies related to various signalling pathways.4 Hence, its utility and applicability are widely recognized in organic and biological areas.
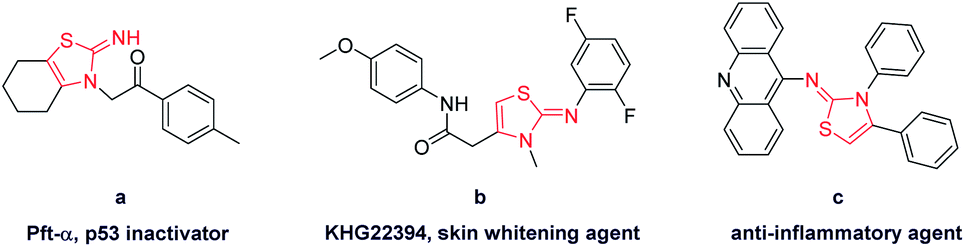 | ||
| Fig. 1 Pharmacologically important molecules consisting of 2-iminothiazoline core structure. (a) p53 inactivator; (b) skin whitening agent; (c) anti-inflammatory agent. | ||
The classical synthesis of 2-aminothiazole moiety involves the Hantzsch condensation reaction of thioureas and α-haloketones.5 Birkinshaw et al. reported the synthesis of N-alkylated imino-thiazolines by replacing thioureas with mono-N-substituted thioureas.6 Also, several alternative strategies have been devised, which include synthesis of highly functionalized thiazoles and 2-iminothiazolines by replacing α-haloketone with 2,2-dicyano-3,3-bis(trifluoromethyl)oxirane7 and 2-chlorooxirane,8 treatment of α-bromoketimines with potassium thiocyanate,9 reaction of N-monoalkylated thioureas with 3-bromomethyl-2-cyanocinnamonitrile,10 cycloadditions followed by elimination of 5-imino-1,2,4-thiazolidin-3-ones with enamines and ester enolate,11 ring transformation of 1-arylmethyl-2-(thiocyanomethyl)aziridines in the presence of TiCl4 and acyl chloride,12 reaction of N-propargylaniline with acyl isothiocyanates.13 Less general approaches towards the synthesis of these compounds involve the reaction of ketone either with N-alkyl rhodanamine or bisbenzyl formamidine disulfide14 or the reaction of α-chloroketones with thiosemicarbazide in an acidic medium,15 condensation of α-haloketones with N-benzoyl-N′-arylthioureas or N,N′-disubstituted thioureas.16
Although some of the methods used for preparing 2-iminothiazolines are convenient and effective, most procedures reported in literatures require arduous preparation of precursor substrates or harsh reaction conditions. Till now, only a few procedures on the one-pot synthesis of 2-iminothiazoline from N,N′-dialkylthiourea and in situ generated α-bromoketones have been reported.17 Herein, we reported a novel and efficient methodology for the synthesis of 2-imino-5-nitrothiazolines using 1,3-diarylthioureas and 1-bromo-1-nitroalkenes as starting materials.
The β-bromo-β-nitrostyrenes, with β-disubstituted styrene structure, showed versatile reactivity as a trifunctional synthon. Previous literatures showed their activity as Michael acceptors and [3 + 2] and [4 + 2] cycloaddition partners.18 In our preliminary experiments, the reaction of 1,3-diphenylthiourea and β-bromo-β-nitrostyrene 1a was studied, while the latter one could easily be prepared according to the reported procedure.18e We found that when these two reactants were treated with base such as K2CO3 in THF at room temperature under atmospheric air, a red crystalline product was obtained (Table 1, entry 1).
|
|||||
|---|---|---|---|---|---|
| Entrya | Base | Solvent | Temperature (°C) | Time (h) | Yield (%) |
| a Reactions were performed with β-bromo-β-nitrostyrene 1a (0.10 mmol) and 1,3-diphenylthiourea 2a (0.11 mmol) with base (0.02 mmol) in the indicated solvent (2.0 mL) under atmospheric air.b β-Bromo-β-nitrostyrene was completely consumed.c Reaction was carried out with 0.04 mmol of base.d Reaction was carried out with 0.01 mmol of base.e Reaction was carried out with 0.1 mmol of base. | |||||
| 1 | K2CO3 | THF | rt | 24 | 62 |
| 2b | K2CO3 | THF | 70 | 10 | 60 |
| 3 | Et3N | THF | rt | 24 | 72 |
| 4b | Et3N | THF | 70 | 10 | 65 |
| 5 | DBU | THF | rt | 24 | 63 |
| 6b | DBU | THF | 70 | 10 | 58 |
| 7 | KHCO3 | THF | rt | 24 | 55 |
| 8b | KHCO3 | THF | 70 | 10 | 60 |
| 9 | DIPEA | THF | rt | 24 | 68 |
| 10 | None | THF | rt | 24 | 29 |
| 11c | Et3N | THF | rt | 24 | 64 |
| 12d | Et3N | THF | rt | 24 | 58 |
| 13e | Et3N | THF | rt | 24 | 17 |
| 14 | Et3N | CH2Cl2 | rt | 24 | 45 |
| 15 | Et3N | Toluene | rt | 24 | 42 |
| 16b | Et3N | Toluene | 110 | 5 | 40 |
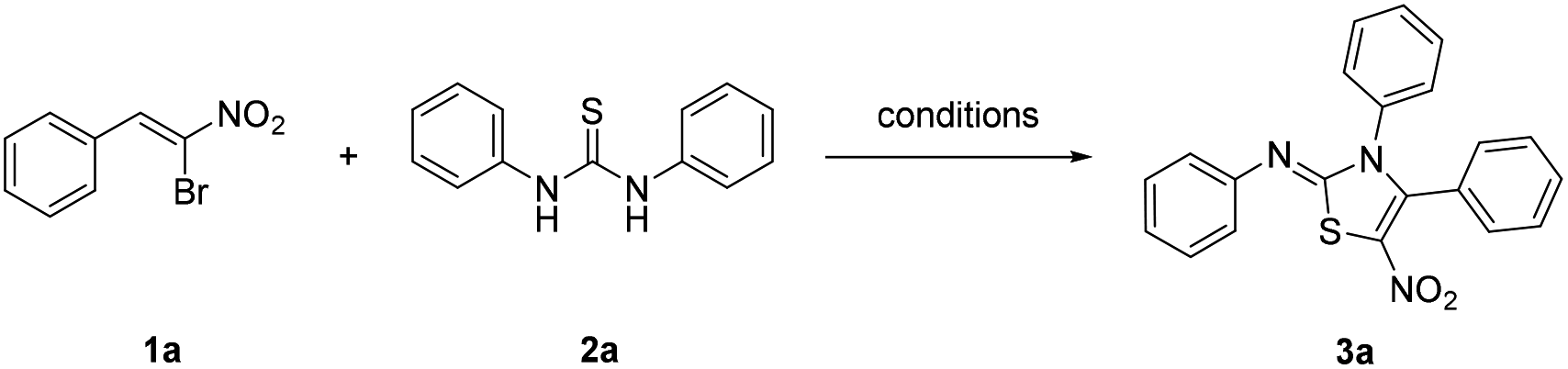
This structure was later confirmed by single crystal X-ray analysis as shown in Fig. 2. When the reaction temperature was increased to 70 °C, the reaction was completed in shorter time. However, the yield was lower due to an increase of side products (Table 1, entry 2).
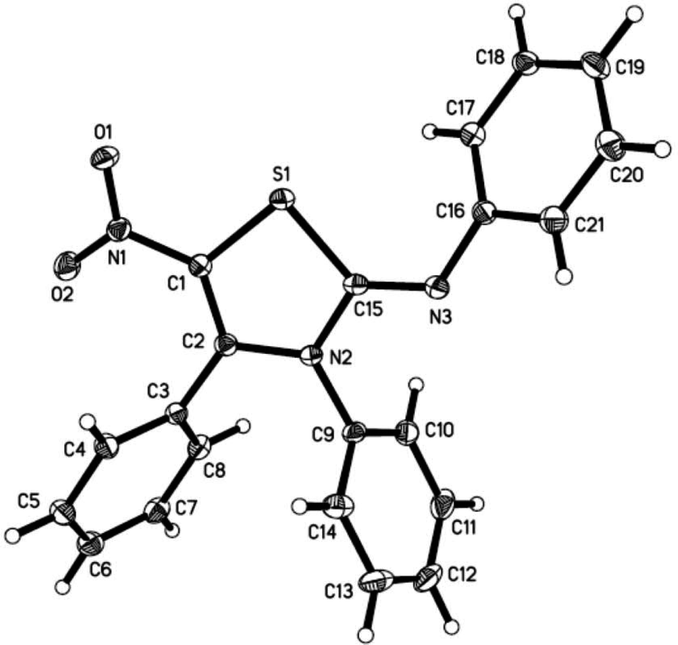 | ||
| Fig. 2 X-ray crystallography of compound 3a. | ||
Several other bases were tested such as K2CO3, Et3N, DBU, KHCO3, DIPEA, and Et3N was found to give the best results (Table 1, entries 1–9). When the reaction was conducted without base, most starting material remained, and very less desired product was obtained (Table 1, entry 10). Further examination was focussed on base loadings, providing the best results at 0.2 equiv. of base (Table 1, entries 11–13). Higher loading of base resulted in more side products rather than desired product while less or no loading of base were not enough to accelerate the reaction. These results suggest that suitable basicity and loading of employed base are critical for the competition between desired and side reactions. Different solvents were screened, and THF was identified to be the optimal solvent (Table 1, entries 14–16). When the reaction was conducted in an inert atmosphere, almost no desired products were formed. From the optimization results, we concluded that 0.2 equiv. of Et3N in THF at room temperature in the air presented the best set of condition for this reaction.
With the optimized conditions in hand, we investigated the scope of this reaction, and the results were shown in Table 2. In general, β-bromo-β-nitrostyrenes bearing halogen substituents on the phenyl ring give moderate to good yields (3b–3f) while stronger electron-donating and electron-withdrawing groups afford the products in relatively lower yields (3h–3j). While different types of thioureas including 1,3-diaryl, 1-aryl-3-alkyl and 1,3-dialkyl substituted ones were employed, only symmetrical 1,3-diarylthiourea could afford desired products. As an example, 1,3-ditolylthiourea could undergo the transformation smoothly under the optimal reaction condition, affording the desired products in moderate to good yields (3k–3n).
| a Reactions were performed with 1-bromo-1-nitroalkenes 1a–1n (0.10 mmol) and 1,3-diarylthioureas 2 (0.11 mmol) with Et3N (0.02 mmol) in THF (2.0 mL) at room temperature in the air for 24 hours. |
|---|
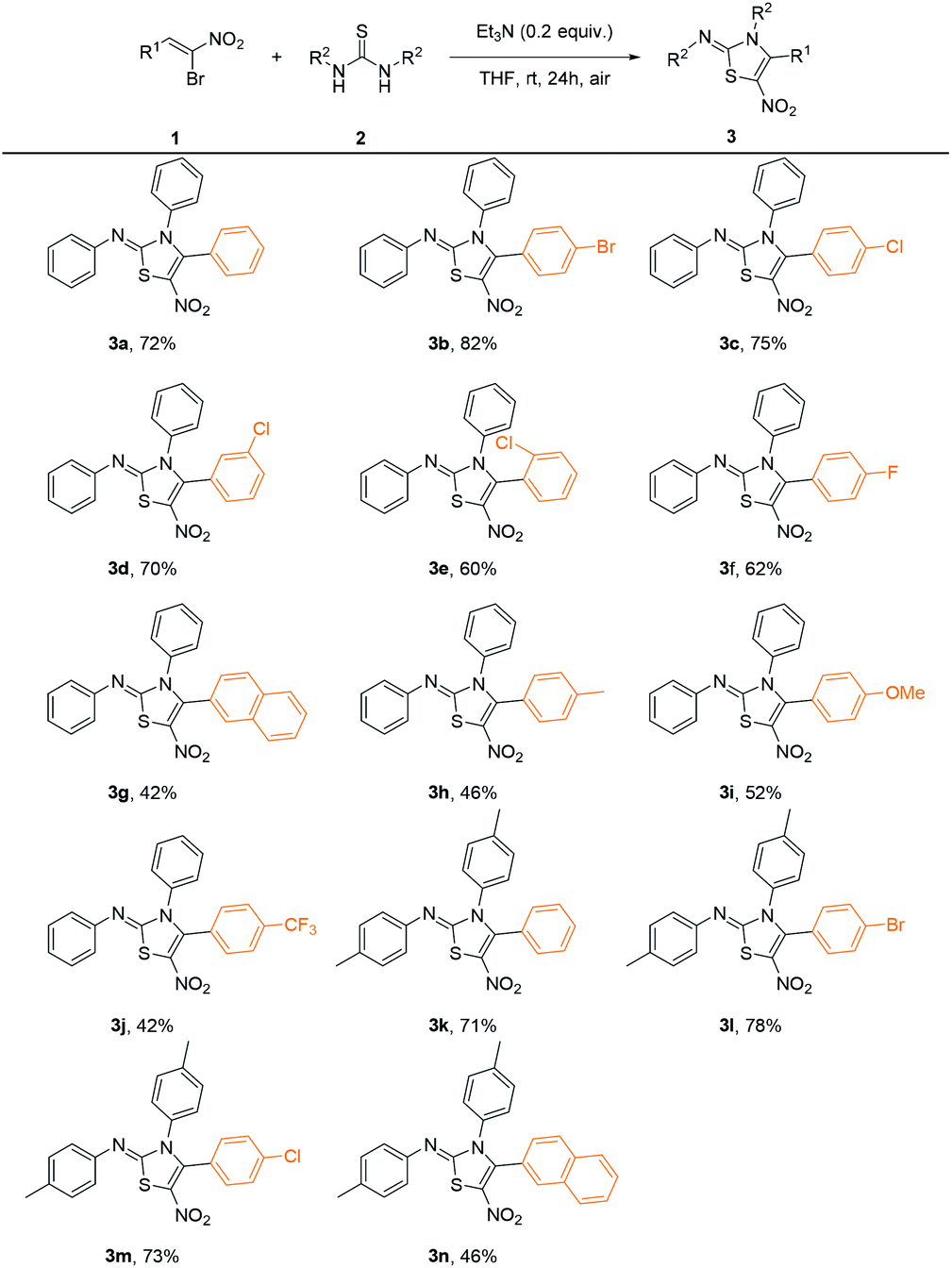 |
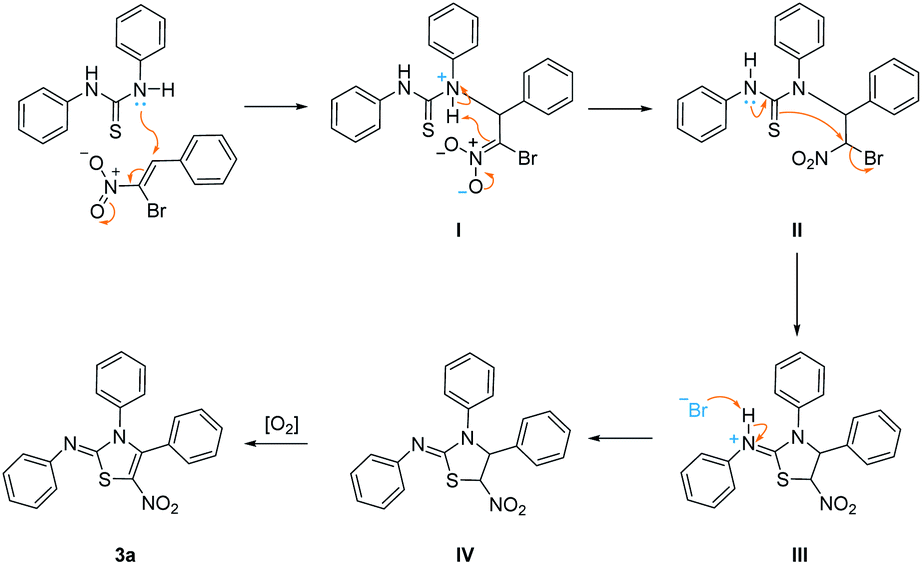
A plausible mechanism for this reaction has been proposed as shown in Scheme 1. Initially, a typical Michael addition happens with the β-bromo-β-nitrostyrene 1a,19 which is initiated by the attack of lone pair of nitrogen atom in 1,3-diphenylthiourea 2a, affording intermediate II. The successive tautomerism of thiourea structure and nucleophilic substitution in intermediate II generates five-membered ring intermediate III. Deprotonation of intermediate III by preceding bromide ion affords intermediate IV which subsequently undergoes aromatization with aid of atmospheric oxygen,20 yielding final product 3a.
 | ||
| Scheme 1 Plausible reaction pathway. | ||
Conclusions
In summary, we have developed a facile and mild synthesis to functionalized 2-imino-5-nitrothiazoline by reacting 1-bromo-1-nitroalkenes with 1,3-diarylthioureas in a mild basic condition. Although this method is limited to symmetrical thioureas, it provides a new approach for the synthesis of diverse 2-iminothiazolines, in which 1-bromo-1-nitroalkenes were used as a trifunctional synthon.Experimental
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on Bruker AV400 (400 MHz) or AV500 (500 MHz) NMR spectrometer. 1H and 13C NMR spectra are reported in parts per million (ppm) downfield from an internal standard, tetramethylsilane (0 ppm for 1H) and CHCl3 (7.26 ppm for 1H and 77.0 ppm for 13C). High-resolution mass spectra (HRMS) were obtained on a Finnigan/MAT LCQ quadrupole ion trap mass spectrometer, coupled with the TSP4000 HPLC system and the Crystal 310 CE system. Accurate masses are reported for the molecular ion [M + H]+ or a suitable fragment ion. X-ray crystallographic data was collected by using a Bruker X8 Apex diffractometer with Mo Kα radiation (graphite monochromator). All reagents were purchased from commercial suppliers and used without further purification except where noted otherwise. All reactions were conducted under an atmosphere of air, unless otherwise indicated. Analytical thin-layer chromatography (TLC) was performed on Merck 60 F254 silica gel plates. Flash chromatography was performed on silica gel 60 (0.010–0.063 mm, Merck). 1-Bromo-1-nitroalkenes 1a–1m were prepared from the standard literature procedures.18e 1,3-Diarylthioureas were obtained from commercial suppliers and used without further purification.General procedure for synthesis of compound 3
To a well-stirred solution of β-bromo-β-nitrostyrene 1 (0.1 mmol, 1.0 equiv., prepared as a mixture of Z- and E-isomers), 1,3-diarylthiourea 2 (0.11 mmol, 1.1 equiv.) in THF (2.0 mL) was added triethylamine (2.0 mg, 0.02 mmol, 0.2 equiv.). The reaction mixture was stirred at room temperature in the air for 24 h (TLC monitored). The resulting mixture was concentrated under reduced pressure to give the crude residue. The crude residue was then purified by flash column chromatography on silica gel (10% EtOAc in hexanes) to afford final product.Characterization of compounds 3a–3n
3a: (red crystal, 72%). 1H NMR (400 MHz, CDCl3): δ 7.36–7.27 (m, 8H), 7.24–7.17 (m, 4H), 7.13 (t, J = 7.2 Hz, 1H), 7.00 (dd, J = 8.0, 1.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 153.9, 149.6, 146.5, 135.5, 130.2, 129.7, 129.4, 129.2, 129.02, 128.99, 128.3, 127.5, 124.7, 120.6. HRMS (ESI) m/z calcd for C21H16N3O2S [M + H]+: 374.0963, found 374.0952.3b: (red crystal, 82%). 1H NMR (400 MHz, CDCl3): δ 7.43 (d, J = 8.4 Hz, 2H), 7.38–7.30 (m, 5H), 7.19–7.09 (m, 5H), 6.99 (d, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 153.8, 149.7, 145.3, 135.4, 131.9, 131.2, 129.9, 129.6, 129.4, 129.1, 126.4, 125.1, 125.0, 120.7. HRMS (ESI) m/z calcd for C21H1579BrN3O2S [M + H]+: 452.0068, found 452.0070; C21H1581BrN3O2S [M + H]+: 454.0048, found 454.0052.
3c: (yellow crystal, 75%). 1H NMR (400 MHz, CDCl3): δ 7.38–7.26 (m, 7H), 7.19–7.11 (m, 5H), 6.99 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 153.8, 149.7, 145.3, 136.8, 135.5, 131.0, 129.9, 129.6, 129.4, 129.1, 128.9, 126.0, 125.0, 120.7. HRMS (ESI) m/z calcd for C21H1535ClN3O2S [M + H]+: 408.0574, found 408.0571; C21H1537ClN3O2S [M + H]+: 410.0544, found 410.0540.
3d: (yellow crystal, 70%). 1H NMR (400 MHz, CDCl3): δ 7.38–7.32 (m, 6H), 7.30–7.24 (m, 2H), 7.22 (d, J = 10.8 Hz, 2H), 7.18–7.10 (m, 2H), 6.99 (d, J = 7.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 153.7, 149.6, 144.7, 135.3, 134.5, 130.6, 129.9, 129.8, 129.6, 129.53, 129.47, 129.3, 129.1, 127.7, 125.0, 120.7. HRMS (ESI) m/z calcd for C21H1535ClN3O2S [M + H]+: 408.0574, found 408.0576; C21H1537ClN3O2S [M + H]+: 410.0544, found 410.0541.
3e: (yellow solid, 60%). 1H NMR (400 MHz, CDCl3): δ 7.39–7.29 (m, 9H), 7.21–7.13 (m, 3H), 7.02 (d, J = 7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 153.8, 149.7, 143.4, 135.2, 133.7, 131.7, 130.5, 129.9, 129.8, 129.6, 129.4, 127.7, 127.0, 125.0, 120.8. HRMS (ESI) m/z calcd for C21H1535ClN3O2S [M + H]+: 408.0574, found 408.0571; C21H1537ClN3O2S [M + H]+: 410.0544, found 410.0540.
3f: (yellow solid, 62%). 1H NMR (400 MHz, CDCl3): δ 7.34–7.27 (m, 5H), 7.25–7.11 (m, 5H), 7.02–6.97 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 164.9, 162.4, 153.8, 149.7, 145.6, 135.6, 132.0, 131.9, 129.9, 129.5, 129.4, 129.1, 125.0, 123.50, 123.46, 120.8, 116.1, 115.8. HRMS (ESI) m/z calcd for C21H15FN3O2S [M + H]+: 392.0869, found 392.0852.
3g: (yellow solid, 42%). 1H NMR (400 MHz, CDCl3): δ 7.77–7.73 (m, 4H), 7.54–7.48 (m, 2H), 7.39–7.35 (m, 2H), 7.28–7.22 (m, 5H), 7.21–7.12 (m, 2H), 7.02 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 154.1, 149.8, 146.7, 135.7, 133.7, 132.5, 130.3, 129.9, 129.4, 129.2, 129.1, 128.6, 128.3, 128.0, 127.9, 126.9, 125.7, 124.9, 124.8, 120.8. HRMS (ESI) m/z calcd for C25H18N3O2S [M + H]+: 424.1120, found 424.1125.
3h: (yellow solid, 46%). 1H NMR (400 MHz, CDCl3) δ 7.40–7.23 (m, 5H), 7.22–7.16 (m, 2H), 7.15–7.06 (m, 5H), 6.99 (dd, J = 7.8, 1.2 Hz, 2H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.2, 149.9, 147.1, 140.8, 135.8, 129.9, 129.5, 129.3, 129.21, 129.18, 129.1, 124.9, 124.5, 120.8, 21.6. HRMS (ESI) m/z calcd for C22H18N3O2S [M + H]+: 388.1120, found 388.1119.
3i: (yellow solid, 56%). 1H NMR (400 MHz, CDCl3): δ 7.36–6.99 (m, 8H), 6.85–6.78 (m, 6H), 3.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.1, 154.1, 149.8, 147.0, 135.9, 131.4, 129.8, 129.4, 129.2, 129.1, 124.8, 120.8, 119.1, 113.9, 55.4. HRMS (ESI) m/z calcd for C22H18N3O3S [M + H]+: 404.1069, found 404.1054.
3j: (yellow solid, 42%). 1H NMR (500 MHz, CDCl3) δ 7.56 (d, J = 8.1 Hz, 2H), 7.41–7.27 (m, 7H), 7.21–7.11 (m, 3H), 6.99 (dd, J = 8.6, 1.1 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 153.7, 149.6, 144.7, 135.2, 132.4, 132.1, 131.4, 130.2, 129.9, 129.60, 129.61, 129.0, 125.6, 125.53, 125.50, 125.47, 125.1, 124.6, 122.5, 120.7. HRMS (ESI) m/z calcd for C22H15F3N3O2S [M + H]+: 442.0837, found 442.0839.
3k: (red solid, 71%). 1H NMR (500 MHz, CDCl3) δ 7.37–7.25 (m, 3H), 7.25–7.20 (m, 2H), 7.16 (d, J = 7.9 Hz, 2H), 7.11–7.04 (m, 4H), 6.90 (d, J = 7.9 Hz, 2H), 2.34 (s, 3H), 2.27 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 154.2, 147.4, 147.0, 139.2, 134.4, 133.0, 130.4, 130.3, 130.0, 129.5, 128.8, 128.4, 127.8, 120.6, 21.3, 21.1. HRMS (ESI) m/z calcd for C23H20N3O2S [M + H]+: 402.1276, found 402.1280.
3l: (red solid, 78%). 1H NMR (400 MHz, CDCl3) δ 7.45–7.39 (m, 2H), 7.17–7.06 (m, 6H), 7.06–7.00 (m, 2H), 6.90–6.84 (m, 2H), 2.32 (s, 3H), 2.29 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.8, 147.3, 145.6, 139.5, 134.5, 131.8, 131.1, 130.4, 130.2, 128.7, 126.6, 125.0, 120.5, 21.4, 21.1. HRMS (ESI) m/z calcd for C23H1979BrN3O2S [M + H]+: 480.0381, found 480.0378; HRMS (ESI) m/z calcd for C23H1981BrN3O2S [M + H]+: 482.0361, found 482.0359.
3m: (yellow solid, 73%). 1H NMR (400 MHz, CDCl3) δ 7.28–7.23 (m, 2H), 7.18–7.12 (m, 4H), 7.09 (d, J = 8.3 Hz, 2H), 7.05–7.00 (m, 2H), 6.89–6.84 (m, 2H), 2.32 (s, 3H), 2.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.8, 147.3, 145.6, 139.5, 136.6, 134.5, 132.8, 131.0, 130.4, 130.2, 128.9, 128.7, 126.1, 120.5, 21.3, 21.1. HRMS (ESI) m/z calcd for C23H1935ClN3O2S [M + H]+: 436.0887, found 436.0885; HRMS (ESI) m/z calcd for C23H1937ClN3O2S [M + H]+: 438.0857, found 438.0859.
3n: (red crystal, 46%). 1H NMR (400 MHz, CDCl3) δ 7.87–7.68 (m, 4H), 7.57–7.42 (m, 2H), 7.29–7.22 (m, 1H), 7.20–7.13 (m, 2H), 7.09 (d, J = 8.4 Hz, 2H), 7.04–6.98 (m, 2H), 6.94–6.85 (m, 2H), 2.33 (s, 3H), 2.17 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.2, 147.5, 147.0, 139.2, 134.4, 133.6, 133.0, 132.5, 130.4, 130.2, 130.1, 128.8, 128.6, 128.3, 128.0, 127.8, 126.8, 125.8, 125.0, 120.6, 21.3, 21.1. HRMS (ESI) m/z calcd for C27H22N3O2S [M + H]+: 452.1433, found 452.1435.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support by funding from the Natural Science Foundation of China (21572197 and U1702286) and Nanyang Technological University (RG120/18).Notes and references
- (a) Y. Wang, W.-X. Zhang, Z. Wang and Z. Xi, Angew. Chem., Int. Ed., 2011, 50, 8122–8126 CrossRef CAS; (b) G. Meng, M. Zheng, M. Dong, M. Wang, A. Zheng and Z. Guo, J. Heterocycl. Chem., 2016, 53, 588–594 CrossRef CAS; (c) G. S. Kumar, A. S. Kumar and H. M. Meshram, Synlett, 2016, 399–403 CAS.
- (a) J. P. Brincat, E. Carosati, S. Sabatini, G. Manfroni, A. Fravolini, J. L. Raygada, D. Patel, G. W. Kaatz and G. Cruciani, J. Med. Chem., 2011, 54, 354–365 CrossRef CAS; (b) S. Bae, H.-G. Hahn, K. D. Nam and H. Mah, J. Comb. Chem., 2005, 7, 7–9 CrossRef CAS.
- (a) K. D. Hargrave, F. K. Hess and J. T. Oliver, J. Med. Chem., 1983, 26, 1158–1163 CrossRef CAS; (b) W. C. Patt, H. W. Hamilton, M. D. Taylor, M. J. Ryan, D. G. Taylor, C. J. C. Connolly, A. M. Doherty, S. R. Klutchko and I. Sircar, J. Med. Chem., 1992, 35, 2562–2572 CrossRef CAS; (c) F. Haviv, J. D. Ratajczyk, R. W. DeNet, F. A. Kerdesky, R. L. Walters, S. P. Schmidt, J. H. Holms, P. R. Young and G. W. Carter, J. Med. Chem., 1988, 31, 1719–1728 CrossRef CAS; (d) K. Tsuji and H. Ishikawa, Bioorg. Med. Chem. Lett., 1994, 4, 1601–1606 CrossRef CAS; (e) F. W. Bell, A. S. Cantrell, M. Hoegberg, S. R. Jaskunas, N. G. Johansson, C. L. Jordan, M. D. Kinnick, P. Lind and J. M. Morin, J. Med. Chem., 1995, 38, 4929–4936 CrossRef CAS.
- (a) N. Pietrancosta, A. Moumen, R. Dono, P. Lingor, V. Planchamp, F. Lamballe, M. Bähr, J.-L. Kraus and F. Maina, J. Med. Chem., 2006, 49, 3645–3652 CrossRef CAS; (b) S. D. Barchéchath, R. I. Tawatao, M. Corr, D. A. Carson and H. B. Cottam, J. Med. Chem., 2005, 48, 6409–6422 CrossRef.
- (a) A. Hantzsch and J. H. Weber, Chem. Ber., 1887, 20, 3118–3132 CrossRef; (b) S. Kasmi, J. Hamelin and H. Benhaoua, Tetrahedron Lett., 1998, 39, 8093–8096 CrossRef CAS; (c) C. Bodhak and A. Pramanik, J. Org. Chem., 2019, 84, 7265–7278 CrossRef CAS.
- T. N. Birkinshaw, S. A. Harkin, P. T. Kaye, G. D. Meakins and A. K. Smith, J. Chem. Soc., Perkin Trans. 1, 1982, 939–943 RSC.
- W. J. Middleton, J. Org. Chem., 1966, 31, 3731–3734 CrossRef CAS.
- J. Gasteiger and C. Herzig, Tetrahedron, 1981, 37, 2607–2611 CrossRef CAS.
- (a) N. De Kimpe, M. Boelens and J.-P. Declercq, Tetrahedron, 1993, 49, 3411–3424 CrossRef CAS; (b) W. Zheng, A. Degterev, E. Hsu, J. Yuan and C. Yuan, Bioorg. Med. Chem. Lett., 2008, 18, 4932–4935 CrossRef CAS.
- (a) J. Svetlik, F. Turecek and I. Goljer, J. Org. Chem., 1990, 55, 4740–4744 CrossRef CAS; (b) J. Liebscher and E. Mitzner, Tetrahedron Lett., 1985, 26, 1835–1838 CrossRef CAS.
- F. Tittelbach, S. Vieth and M. Schneider, Eur. J. Org. Chem., 1998, 515–520 CrossRef CAS.
- M. D'Hooghe, A. Waterinckx and N. De Kimpe, J. Org. Chem., 2005, 70, 227–232 CrossRef.
- Y. Sanemitsu, S. Kawamura, J. Satoh, T. Katayama and S. Hashimoto, J. Pestic. Sci., 2006, 31, 305–310 CrossRef CAS.
- E. Schmitz and H. Striegler, J. Prakt. Chem., 1970, 312, 359–365 CrossRef CAS.
- (a) H. Beyer, W. lässig and E. Bulka, Chem. Ber., 1954, 87, 1385–1392 CrossRef CAS; (b) H. Beyer and G. Wolter, Chem. Ber., 1956, 89, 1652–1658 CrossRef CAS.
- (a) A. Bijev and P. Prodanova, Synth. Commun., 2006, 36, 3095–3101 CrossRef CAS; (b) X. C. Wang, F. Wang, Z. J. Quan, Z. Zhang and M. G. Wang, J. Heterocycl. Chem., 2006, 43, 1473–1477 CrossRef CAS; (c) A. Manaka, T. Ishii, K. Takahashi and M. Sato, Tetrahedron Lett., 2005, 46, 419–422 CrossRef CAS; (d) J. Schmeyers and G. Kaupp, Tetrahedron, 2002, 58, 7241–7250 CrossRef CAS.
- (a) S. Murru, C. B. Singh, V. Kavala and B. K. Patel, Tetrahedron, 2008, 64, 1931–1942 CrossRef CAS; (b) C. B. Singh, S. Murru, V. Kavala and B. K. Patel, Org. Lett., 2006, 8, 5397–5399 CrossRef CAS; (c) C.-Y. Chen, I. J. Barve and C.-M. Sun, ACS Comb. Sci., 2016, 18, 638–643 CrossRef CAS.
- (a) V. Mane, S. T. Sivanandan, R. G. Santana, A. Beatriz, E. N. da Silva Júnior and I. N. N. Namboothiri, J. Org. Chem., 2020, 85, 8825–8843 CrossRef CAS; (b) J.-W. Zhang, L.-S.-H. Yu, J.-L. Dong, Q.-C. Sun and J.-W. Xie, Synlett, 2018, 603–608 CAS; (c) J. Feng, X. Yuan, W. Luo, L. Lin, X. Liu and X. Feng, Chem.–Eur. J., 2016, 22, 15650–15653 CrossRef CAS; (d) V. Kumar, A. Awasthi, A. Metya and T. Khan, J. Org. Chem., 2019, 84, 11581–11595 CrossRef CAS; (e) W. E. Parham and J. L. Bleasdale, J. Am. Chem. Soc., 1951, 73, 4664–4666 CrossRef CAS; (f) J.-W. Xie, Z. Wang, W.-J. Yang, L.-C. Kong and D.-C. Xu, Org. Biomol. Chem., 2009, 7, 4352–4354 RSC; (g) S. E. Denmark, V. Guagnano, J. A. Dixon and A. Stolle, J. Org. Chem., 1997, 62, 4610–4628 CrossRef CAS.
- T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119–125 CrossRef CAS.
- A. C. Dawsey, V. Li, K. C. Hamilton, J. Wang and T. J. Williams, Dalton Trans., 2012, 41, 7994–8002 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of products. See DOI: 10.1039/d0ra00686f |
| This journal is © The Royal Society of Chemistry 2021 |