
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of modified fly ash doped carbon paste electrodes and metal film electrodes on the determination of trace cadmium(II) by anodic stripping voltammetry†
Jinying Hou ,
Yuping Fan,
Xiaomin Ma,
Xianshu Dong* and
Suling Yao
,
Yuping Fan,
Xiaomin Ma,
Xianshu Dong* and
Suling Yao
College of Mining Engineering, Taiyuan University of Technology, Taiyuan, 030024, China. E-mail: dxshu520@163.com
First published on 10th May 2021
Abstract
Fly ash, as waste from coal combustion, has been effectively modified to improve its adsorption to remove toxic metals, and modified fly ash could be used for electrode modification to improve the sensitivity of the electrode. In this paper, a modified fly ash doped carbon paste electrode for the detection of trace cadmium was first and successfully developed. Several parameters affecting the anodic stripping voltammetric response of Cd(II) were optimized, such as the composition of the paste, pH of the measurement solution, the concentration of Sb(III) (or Sb(III) and Bi(III)), deposition potential and deposition time. Compared with Sb/MFA-CPE, the square wave anodic stripping voltammetry (SWASV) response of Cd(II) at Sb/MMFA-CPE had a higher linear range and lower sensitivity. Relative to MFA, MMFA-CPE, due to the introduction of CTAB, provided a larger effective area for interacting with analytes, more binding sites and further facilitating electron transfer at the electrode, and amplified the electrochemical signal. Compared with Sb/MMFA-CPE, the SWASV response of Cd(II) at Sb–Bi/MMFA-CPE had a higher linear range and similar sensitivity, since mechanisms at bismuth and antimony film electrodes were different. Besides, the electrode reactions of Cd(II) at bismuth film electrodes involved adsorption phenomena while they were free of adsorption at antimony film electrodes.
1. Introduction
Toxic metals are mainly encountered from industrial wastewater and are non-biodegradable and durable, posing a severe threat to organisms.1–3 Among the toxic metals, cadmium ions, even in trace amounts, accumulate in the body and adversely affect virtually every system of the body. Cadmium ions can damage the kidneys, the nervous system, the productive system, and cause high blood pressure4 and anemia.5,6 Several methods have been applied to the determination of trace cadmium ions, including Inductively Coupled Plasma Mass Spectrometry (ICP-MS),7 X-Ray Fluorescence (XRF),8 flame atomic absorption spectrometry,9 Solid Phase Spectrophotometry (SPS),10 etc. However, these methods rely on the collection of liquid discrete samples and even require a pre-concentration step to measure toxic metal ions. Based on this, electrochemical technology has been proposed and successfully applied to the determination of toxic metal ions, which has desirable characteristics including specificity for targeted metal ions, enhanced measurement frequency and precision, robustness, inexpensiveness, and infrequent regeneration.11Electrochemical methods are favorable techniques for the determination of metal ions because of their low cost, high sensitivity, easy operation and portability.12 Cyclic voltammetry (CV),13 differential pulse voltammetry (DPV),14–16 anodic stripping voltammetry (ASV)17–19 and other electrochemical methods have been applied to the detection of toxic metal ions. ASV has been recognized as a powerful technology for electrochemical measurement of trace toxic metal ions in various samples from environmental, clinical and industrial sources.20–22 ASV is used for the detection of trace toxic metal ions, first developed from the mercury film electrode (MFE) and hanging mercury electrode (HMDE).12 However, due to the high toxicity of mercury, alternative environmentally friendly electrode materials are needed. In 2000, Wang et al. successfully proposed the use of bismuth film electrodes (BiFEs) as an alternative to MFE.23 In 2007, Hocevar et al. successfully proposed the use of antimony film electrodes (SbFEs) for the detection of metal ions.24 Both of two electrodes have been extensively studied in the detection of trace toxic metal ions.25–27 Compared to the BiFEs, the SbFEs have significant properties in a more acidic solution (pH ≤ 2), which is advantageous in the electrochemical analysis of trace toxic metals.28
SbFEs were successfully introduced into electroanalytical applications using carbon substrate for the preparation of the film. Among the carbon substrates, carbon paste electrodes are widely used because of its additional advantage of an easy surface renewal.27 The performance of the stripping techniques is strongly influenced by the working electrode material. Stripping voltammetry using a chemically modified electrode (CME) has higher selectivity due to the ability of the modifier to capture the specific metal ions. Inorganic materials, such as zeolites, clays and nanocomposite,29–31 have been successfully introduced into the application in the electrochemical analysis as a modifier.
Fly ashes are waste products of coal combustion in electric and thermal power plants. To reduce the harm to the environment, fly ash has been widely used in building materials, soil amendment, production of glass-ceramics, extraction of alumina and so on. Besides, since fly ash contains high contents of silicates and alumina-silicates, fly ashes can be converted into zeolite-like crystalline materials as a result of chemical treatment, which are effective adsorbents for environmental pollutants.32,33 Zeolite has been used in electroanalysis to measure toxic metal ions.34–36 However, to the best of our knowledge, electrochemical determination of cadmium using the antimony film modified zeolite modified by fly ash doped carbon paste electrode has not been reported up to now. Thus, modification of fly ash and application to the electrochemical detection of cadmium ions not only provide a supplementary method for the detection of cadmium ions but also provide new possibilities for the utilization of fly ash.
In this work, fly ash was chemically modified to convert into zeolite-like crystalline materials or to load amino functional groups to increase the adsorption of metal ions. And modified fly ash doped carbon paste electrode used at metal film electrodes for the determination of trace Cd(II) was described. Upon the optimization of the parameters affecting electroanalytical response, the different sensing systems were studied to provide a wide detection range and a low detection limit.
2. Materials and methods
2.1. Apparatus
SWASV was performed with a VersaSTAT3 electrochemical station (Princeton, Advanced Measurement Technology, Inc., USA). A conventional three-electrode system consisted of a carbon paste working electrode with 3.0 mm diameter, a counter electrode made of platinum wire and an Ag|AgCl reference electrode. A 20.0 mL cell was used for the electrochemical measurements. All potentials were given with respect to the Ag|AgCl reference electrode. The pH meter (Shanghai INESA Scientific Instrument Co., Ltd, China) was used to measure pH in this experiment. An ultrasonic cleaner (Kunshan Ultrasonic Instrument Co., Ltd., Jiangsu, China) was used for the ultrasonication of fly ash. The chemical composition of fly ash used in this experiment was measured with an XRF (PANalytical B.V., Netherlands). The surface morphology and pore structure of fly ash before and after modification were measured with the scanning electron microscope (SEM) and automatic surface area and porosity analyzer, respectively.2.2. Reagent and solution
Fly ash was obtained from a power plant (Hebei, China). Graphite powder was spectrum pure and purchased from Tianjin Kemiou Chemical Reagent Co., Ltd. (Tianjin, China). Paraffin oil was purchased from Sangon Biotech (Shanghai) Co., Ltd. (China). Antimony standard solution, cadmium standard solution, cetyltrimethylammonium bromide (CTAB) and potassium ferricyanide were purchased from Shanghai Aladdin Bio-Chem Technology Co., LTD (Shanghai, China). Dilute NaOH solution and HNO3 solution served as the adjustment of pH. After the electrochemical test, the volumetric flask, electrolytic cell, three electrodes and rotor should be washed with 10% dilute HNO3 solution. All other chemicals were analytical grade, or better, and used as received. All solutions were prepared with ultrapure water (18.25 MΩ cm).2.3. Modification of fly ash
A mixture containing 30.0 g of fly ash and 200.0 mL of 3.0 mol L−1 NaOH solution was stirring for 24.0 h, in which temperature was 348.15 K and stirring rate was 300.0 rpm. Then the mixture was washed thoroughly with ultrapure water to remove the residual NaOH and dried. MFA was prepared. A mixture containing 5.0 g MFA and 25.0 mL of 0.1 mol L−1 CTAB was ultrasonicated for 4.0 h. The mixture was centrifuged and dried to get MMFA.2.4 Electrode preparation
The modified carbon paste electrode (CPE) was prepared by thoroughly homogenizing 4.0 mg of MFA (or MMFA) with 200.0 mg of graphite powder and 80.0 μL of paraffin oil using the agate mortar and pestle. The prepared electrodes were dried at room temperature for 48.0 h. Before every measurement, the surface of the CPE was polished on a clean weighing paper to get a smooth surface. It should be noted that the mass of graphite powder was 200.0 mg, which in MFA-CPE, MMFA-CPE and CPE was fixed.2.5. Measurement procedure
SWASV was a two-step process: antimony and cadmium deposited on the working electrode as cathode, and then stripped from the working electrode as anode. Take the determination of Cd(II) at Sb/MFA-CPE for example, the preconcentration was carried out at −1.2 V for 280.0 s under stirring. Then the stirring was stopped and after a 10.0 s equilibration period (in this period, the voltage was −1.2 V), the positive-going square wave anodic stripping voltammograms were recorded from −1.2 V to 0.2 V with a pulse amplitude of 50.0 mV, frequency of 25.0 Hz, and scan rate of 100.0 mV s−1. A 30.0 s conditioning step at 0.3 V (with solution stirring) was used to remove the cadmium and the antimony film, prior to the next cycle. Cyclic voltammetry (CV) was performed in [Fe(CN)6]3−/4− as an electrochemical probe from −0.5 V to 1.0 V to study the electrochemical behavior of modified and unmodified electrodes. Electrochemical impedance spectroscopy (EIS) was performed to determine the electron transfer resistance of modified and unmodified electrodes. The impedance spectra were recorded in the frequency range from 50 kHz to 0.05 Hz using an ac signal amplitude of 10 mV. All experiments were carried out at 298.15 K temperature.3. Results and discussion
3.1 Characterization of FA, MFA and MMFA
As waste from coal combustion, fly ash has been effectively modified to improve its adsorption of toxic metals. The crystalline structure of fly ash is mainly mullite and quartz (see in Fig. 1d), and there are well-developed particles in the shape of rather smooth balls in the sample of fly ash (see in Fig. 1a). One of the most common ways to improve the adsorbability of fly ash is that fly ash is treated by hydrothermal treatment to form a variety of zeolitic types.37 Fly ash is treated with NaOH solution (MFA), which leads to a partial conversion to zeolite A (Na96Al96Si96O384·284H2O), and some quartz and mullite are still present (see in Fig. 1b). That is because quartz and mullite in ash are inert and difficult to dissolve. Thus, MFA is co-crystallized zeolite with original crystalline phases. Amorphous materials (aluminosilicate glasses) in fly ash are dissolved readily in NaOH solution, and the sample has a transformation of majority of ball-shaped particles into agglomerations of undefined shape (see in Fig. 1b). These alterations in the fly ash are commensurate with increasing inter particle pore characterization (i.e., pore volume, pore size and surface area) (see in Fig. 1e). When continuing to treat the sample with CTAB (MMFA), the sample has no significant change in the crystalline structure and morphology (see in Fig. 1c and d). As a result of functionalization, a significant decrease in the surface area, pore volume and pore diameter is observed (see in Fig. 1e).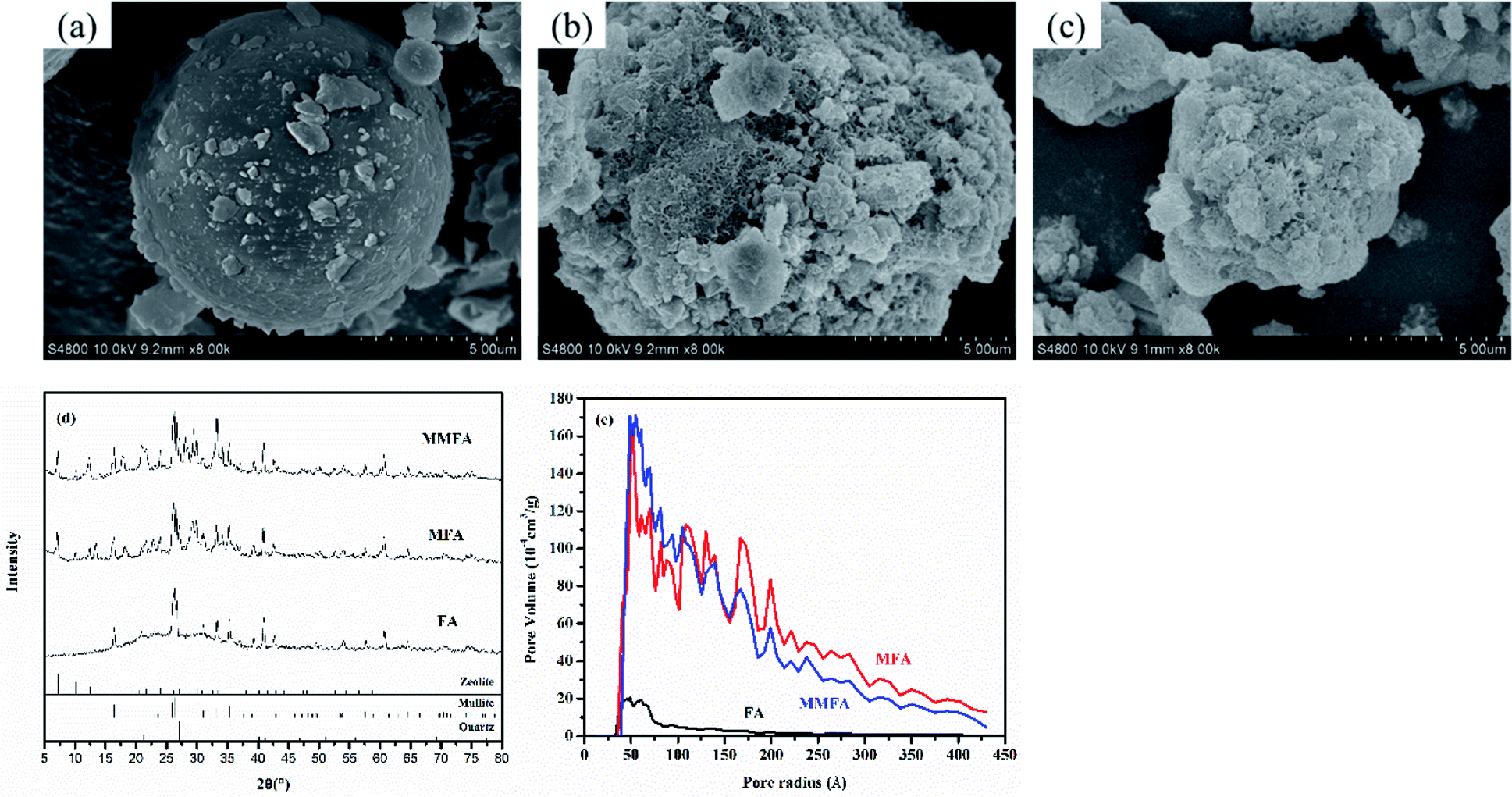 | ||
| Fig. 1 Scanning electron micrographs of (a) FA, (b) MFA and (c) MMFA. (d) XRD data and (e) pore volume distribution as a function of the pore radius for FA, MFA and MMFA. | ||
3.2 Electrochemical characterization of modified and unmodified electrodes
The electrochemical behavior of the electrodes before and after doping with MFA or MMFA was studied by using CV. [Fe(CN)6]3−/4− was selected as an electrochemical probe to understand the changes in the electrochemical behavior of electrodes after doping with MFA or MMFA. As shown in Fig. 2a, the peak-to-peak potential difference (ΔEP) of ferri/ferrocyanide redox couple at CPE was 122.38 mV. The larger ΔEP was attributed to result from low electrical conductivity. When CPE was doped with MFA, the ΔEP of ferri/ferrocyanide redox couple at MFA-CPE slightly decreased, from 122.38 mV to 121.46 mV. However, the ΔEP of ferri/ferrocyanide redox couple at MMFA-CPE decreased from 122.38 mV to 108.59 mV. On the contrary, when doping with MFA or MMFA, the peak currents of the electrodes increased from 94.21 to 106.07 μA for doping MFA and to 156.47 μA for doping MMFA. It both indicates that the doping of MFA does not show a significant improvement in the electron transfer property on the electrochemical behavior of [Fe(CN)6]3−/4− on the electrode, but the improvement of doping with MMFA is obvious. The electrochemical behavior of the electrodes was examined in [Fe(CN)6]3−/4− at different scan rates (from 50 to 800 mV s−1) to understand the dependence of the peak current of ferri/ferrocyanide on the scan rate. As shown in Fig. S1,† as the increase of the scan rate, the anodic peak potential of three electrodes all shifted positively. Besides, the anode peak current of all three electrodes had a good linear relationship with v1/2, which indicated the oxidation process was controlled by diffusion at all three electrodes. The values of active surface area could be calculated by Randles–Sevcik eqn (1).38–41| Ipa = 2.69 × 105n3/2AD1/2v1/2C | (1) |
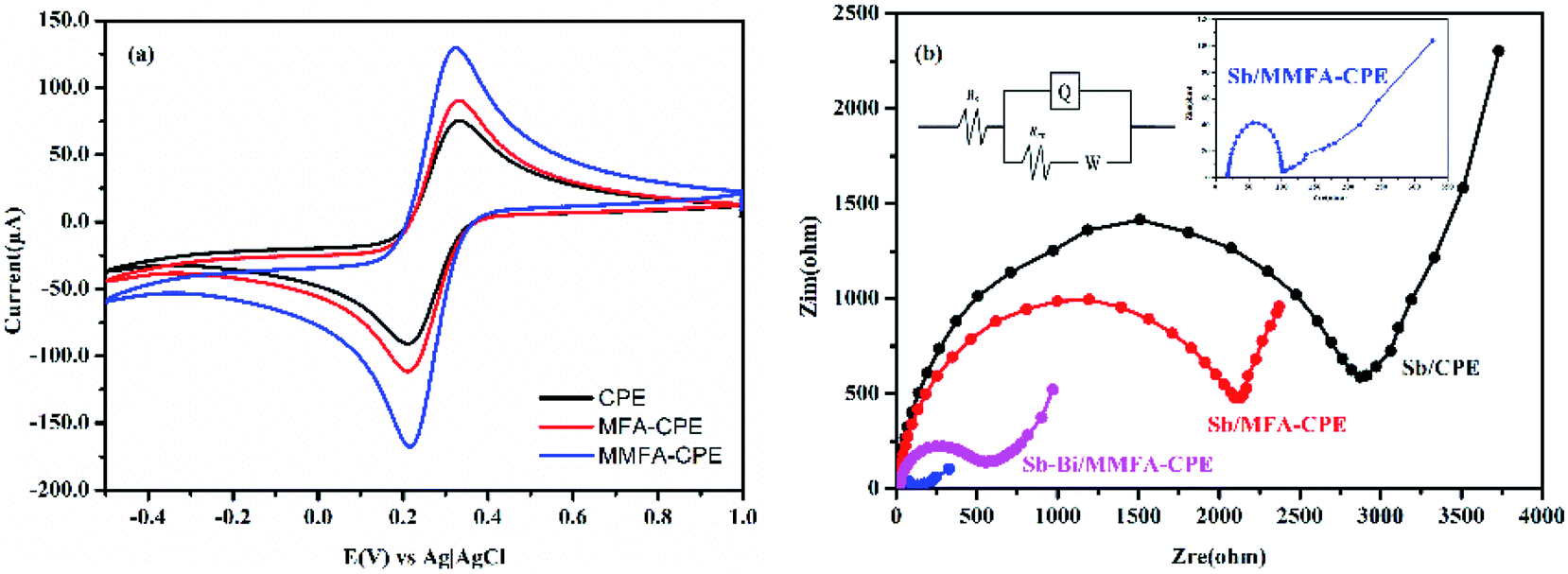 | ||
| Fig. 2 (a) CVs of CPE, MFA-CPE and MMFA-CPE in 5 mM [Fe(CN)6]3−/4− containing 1 M KCl at a scan rate of 150 mV s−1. (b) Nyquist plots of EIS of Sb/CPE, Sb/MFA-CPE, Sb/MMFA-CPE and Sb–Bi/MMFA-CPE in 80.0 μg L−1 Cd(II). Inset section are an equivalent circuit and the Nyquist plot of EIS of Sb/MMFA-CPE. | ||
EIS was performed in 80.0 μg L−1 Cd(II) at Sb/CPE, Sb/MFA-CPE, Sb/MMFA-CPE and Sb–Bi/MMFA-CPE, and the Nyquist plots of EIS were shown in Fig. 2b. The Nyquist plots were fitted considering the Randles electrical equivalent circuit, where Rct was the electron transfer resistance. Rct was equal to the diameter of the semicircle in the Nyquist plot, which controlled the electron transfer kinetics of metal ions at the surface of the electrodes. For Sb/CPE, Sb/MFA-CPE and Sb/MMFA-CPE, the Rct values were 2.56, 1.90 and 0.081 kΩ, respectively. It indicated that doping MFA and MMFA to CPE could decrease the electron transfer resistance, and the reduction effect of doping MMFA was obvious. Due to the introduction of CTAB, MMFA-CPE provides more binding sites which facilitated the adsorption of metal ions and promoted electron transfer. Compared to Sb/MMFA-CPE, the Rct value of Sb–Bi/MMFA-CPE (Rct = 0.47 kΩ) was larger indicating the higher electron resistance. However, the Rct value of Sb–Bi/MMFA-CPE was about 1.43 kΩ smaller than that of Sb/MFA-CPE.
According to the Rct obtained by EIS, the electron transfer rate constant (k) could be calculated by eqn (2).42
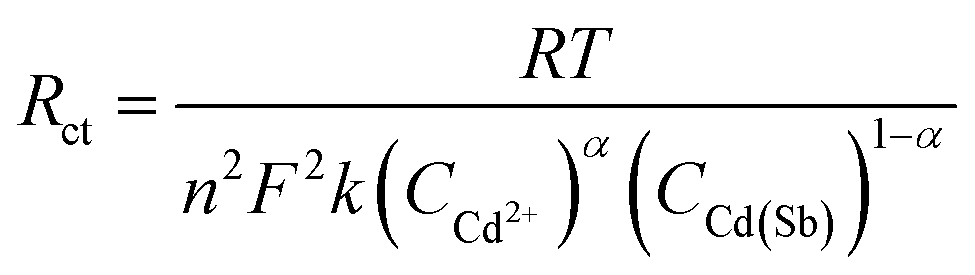 | (2) |
3.3 Optimization of experimental conditions for determination of Cd(II) at Sb/MFA-CPE, Sb/MMFA-CPE and Sb–Bi/MMFA-CPE
In the determination of Cd(II) at the Sb/MFA-CPE, several factors including the amounts of MFA, the amount of Sb(III), pH, deposition potential and time that affect the test results should be optimized firstly. The stripping responses of Cd(II) at the Sb/MFA-CPE with these five factors were studied by single-factor variable analysis, and the optimization of experimental conditions for determination of Cd(II) was identified. The peak current of Cd(II) increases with the increase of MFA content, which can be attributed to the ion exchange properties of MFA. Nevertheless, excessive MFA is not conducive to electron transfer, affecting the conductivity of the electrode, so the response of Cd(II) decreases when the amount of MFA exceeds a certain value. Fig. 3a shows the stripping responses of Cd(II) at the Sb/MFA-CPE with 2.0–6.0 mg, and it reaches a maximum peak current of Cd(II) at the amount of MFA of 4.0 mg which was selected in the subsequent experiments. In this paper, in situ antimony film electrode is selected to measure trace Cd(II), and the amount of Sb(III) is a key factor affecting the stripping response of Cd(II) at the Sb/MFA-CPE. The stripping responses of Cd(II) at the concentrations of Sb(III) of 2.0–6.0 mg L−1 were studied (Fig. 3b). The peak current of Cd(II) when the concentration of Sb(III) is 4.0 mg L−1 is higher than that when the concentration of Sb(III) is higher or lower. When there are few amounts of Sb(III), the available Sb surface area increases with the increase of amount, which is beneficial to metal ion codeposition/entrapment within the inner layers of Sb particles. When there is more than 4.0 mg L−1 of Sb(III), after metal ion codeposition/entrapment within the inner layers of Sb particles, the metal ion is difficult to diffuse out of the film and return to the solution during the stripping step43,44 because of the thick antimony film coated on the electrode surface. So 4.0 mg L−1 of Sb(III) was selected for the following experiments. As we all know, ions exist in different forms under different pH conditions. Sb(III) exists as neutral molecule (Sb(OH)3 or SbO(OH) or HSbO2) in a wide range of pH (about 2.0–10.0), as SbO+ or Sb(OH)2+ in strong acid and as SbO2− or Sb(OH)4− in strong alkali. Fig. 3c shows peak currents of Cd(II) at Sb/MFA-CPE in a solution with the pH range of 2.0–4.0, and the peak current of Cd(II) in SWASV is the highest in the pH of 3.0. The pH of 3.0 was chosen for the following experiments. Deposition potential and deposition time are crucial factors for stripping voltammetry method that influences the sensitivity and selectivity of the determination.45 The deposition potential at which the in situ antimony film is deposited together with the analytes was studied in the potential range of −1.1 to −1.4 V, and the results are shown in Fig. 3d. The optimum deposition potential was −1.2 V. So the deposition potential of −1.2 V was chosen for the following experiment. The deposition time was studied in the range of 50.0–350.0 s for a solution containing 80.0 μg L−1 Cd(II). As shown in Fig. 3e, the SWASV responses increased considerably along with the deposition time prolonged in the range of 50.0–280.0 s. As deposition time longer than 280.0 s, there was no significant change in the peak current. This is because further increasing the deposition time, the electrode surface saturation and the exhaustion of the accessible adsorption sites on the electrode lead to the plateau. Therefore, the deposition time of 280.0 s was set in our later studies.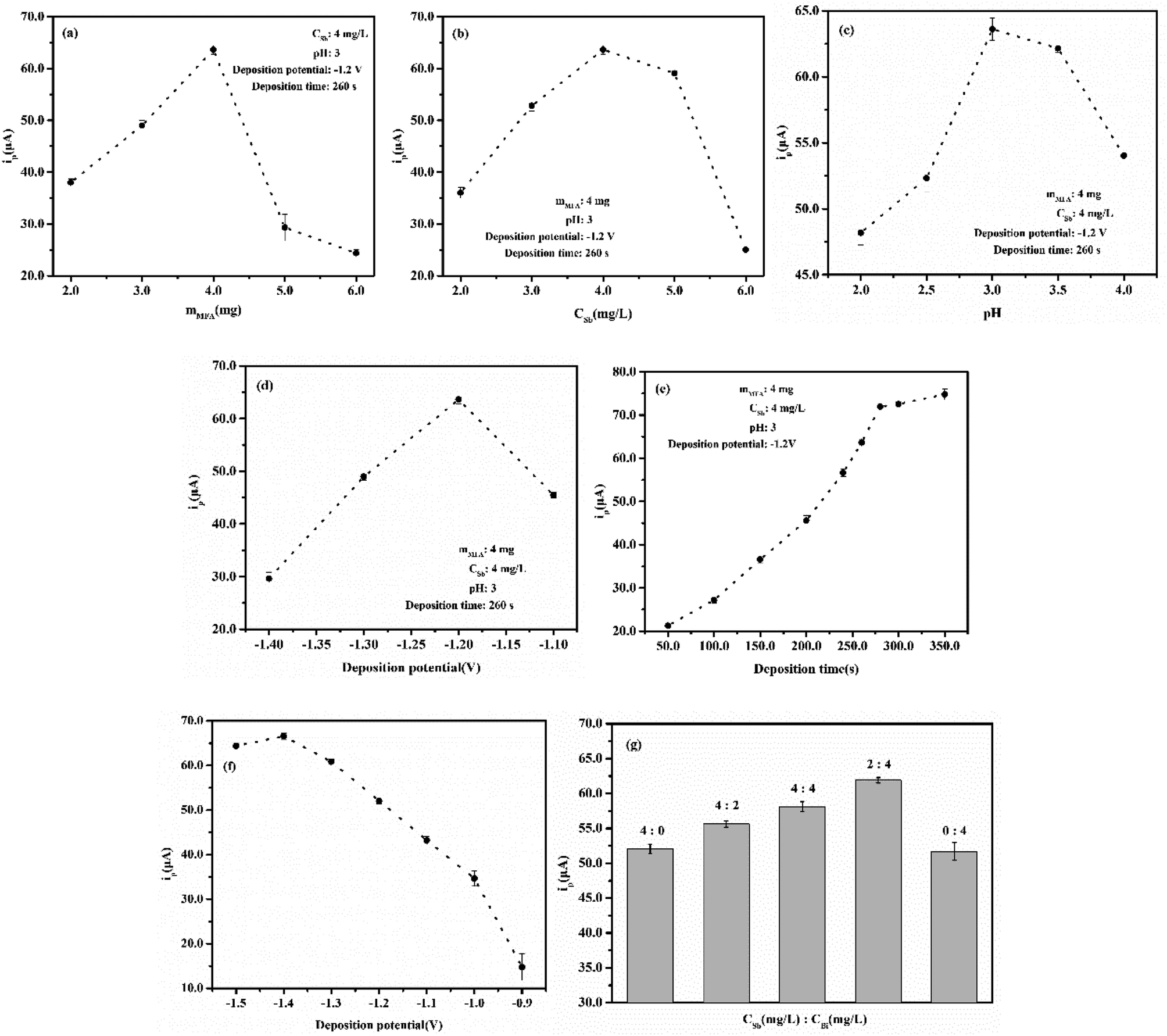 | ||
| Fig. 3 Effect of different (a) amounts of MFA, (b) amounts of Sb(III), (c) pH, (d) deposition potential and (e) deposition time on the SWASV response of 80.0 μg L−1 Cd(II) at Sb/MFA-CPE. Effect of different (f) deposition potential on the SWASV response of 80.0 μg L−1 Cd(II) at Sb/MMFA-CPE. Effect of different (g) amounts of Sb(III) and Bi(III) on the SWASV response of 80.0 μg L−1 Cd(II) at Sb–Bi/MMFA-CPE. | ||
The experimental conditions for the determination of Cd(II) at Sb/MMFA-CPE and Sb–Bi/MMFA-CPE were optimized. The optimal conditions were the same as the measuring conditions at Sb/MFA-CPE, except that the deposition potential at Sb/MMFA-CPE and the concentrations of Sb(III) and Bi(III) at Sb–Bi/MMFA-CPE. The deposition potential at Sb/MMFA-CPE was studied in the potential range of −0.9 to −1.5 V, and the optimum deposition potential was −1.4 V (as shown in Fig. 3f). The different concentrations of Sb(III) and Bi(III) (4.0 and 0, 4.0 and 2.0, 4.0 and 4.0, 2.0 and 4.0, 0 and 4.0 mg L−1) were studied, and Sb–Bi/MMFA-CPE in the concentrations of 2.0 mg L−1 of Sb(III) and 4.0 mg L−1 Bi(III) had better electrochemical response to cadmium ions (as shown in Fig. 3g).
The instrumental parameters counting pulse amplitude, the frequency and scan rate were studied, and they had the same trend of influence on SWASV responses at Sb/MFA-CPE, Sb/MMFA-CPE and Sb–Bi/MMFA-CPE, as shown in Fig. 4. The pulse amplitude, the frequency and scan rate of 50 mV, 25 Hz and 100 mV s−1 were set in our later studies.
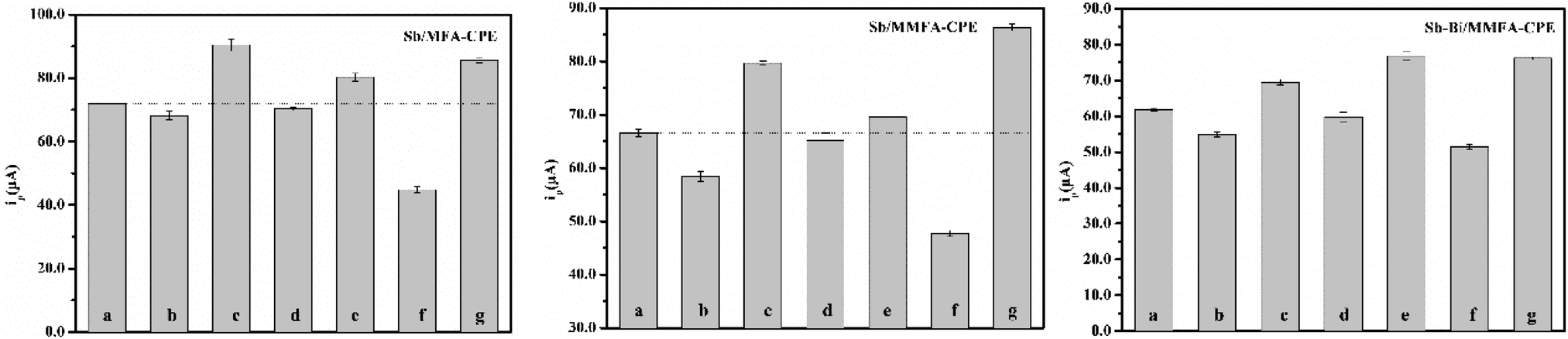 | ||
| Fig. 4 SWASV responses of 80 μg L−1 Cd(II) at Sb/MFA-CPE, Sb/MMFA-CPE and Sb–Bi/MMFA-CPE with pulse amplitude, the frequency and scan rate of (a) 50, 25 and 100, (b) 40, 25 and 100, (c) 60, 25 and 100, (d) 50, 20 and 100, (e) 50, 30 and 100, (f) 50, 25 and 50, (g) 50 mV, 25 Hz and 150 mV s−1, respectively. | ||
3.4 Square wave voltammetric determination of Cd(II)
Under the optimum conditions of each factor, the linear regression equation (peak current versus Cd(II) concentration), linear range and detection limit obtained for Sb/MFA-CPE, Sb/MMFA-CPE and Sb–Bi/MMFA-CPE are shown in Table 1 and Fig. 5. As shown in this table, the biggest linear range (2.0–150.0 μg L−1) and the lowest detection limit (1.36 μg L−1) for Sb–Bi/MMFA-CPE were obtained. Three electrodes were compared to other sensors for the detection of metal ions by anodic stripping voltammetry (Table S3†). Due to the introduction of CTAB to carbon paste electrodes and Bi to metal film electrodes, the detection of Cd(II) at Sb–Bi/MMFA-CPE could achieve wide detection range and lower detection limit.| Electrode | Linear regression equation (ip: μA, C: μg L−1) | Liner range (μg L−1) | Detection limit (μg L−1) |
|---|---|---|---|
| Sb/MFA-CPE | ip = 1.44C − 43.18 R2 = 0.999 | 40.0–80.0 | 2.63 |
| Sb/MMFA-CPE | ip = 0.69C + 13.15 R2 = 0.995 | 40.0–150.0 | 2.25 |
| Sb–Bi/MMFA-CPE | ip = 0.75C + 3.76 R2 = 0.992 | 2.0–150.0 | 1.36 |
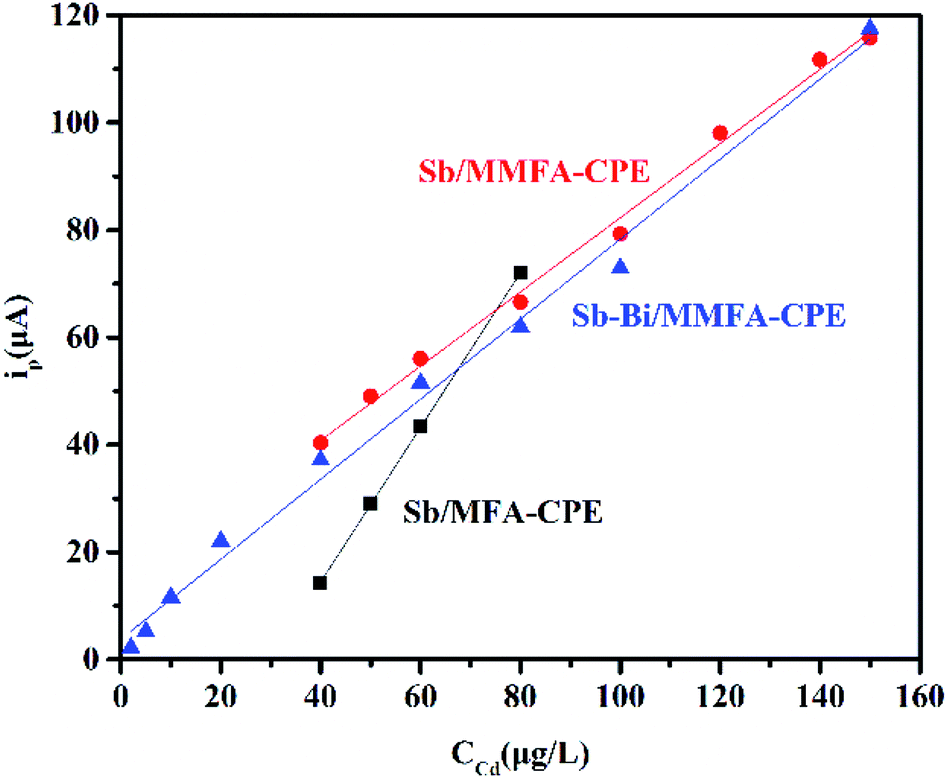 | ||
| Fig. 5 Calibration curves for the determination of Cd(II) of different concentrations. | ||
As shown in Table S2,† the proposed procedure is proved to be effective in overcoming small variations of deposition potential (Ed), pulse amplitude (Ea) and pH, and has good robustness. It demonstrates that the determination of trace cadmium(II) at three electrodes by anodic stripping voltammetry is robust.
The stability of the electrodes is an important consideration for the practical application. SWASV responses of 80 μg L−1 Cd(II) at Sb/MFA-CPE and Sb/MMFA-CPE were determined to study the long-term stability of MFA-CPE and MMFA-CPE. The peak currents of Cd(II) showed no significant change within three consecutive days (Table S4†). After 30 days of irregular measurement, the peak currents of Cd(II) at both two electrodes kept stable (Fig. S5†), demonstrating a good stability.
3.5 Interference studies
Various interferent ions, including Cl−, NO3−, SO42−, Na+, K+, Mg2+, Al3+, Fe3+, Pb2+, Ca2+, Zn2+ and Cu2+, were studied to check the selectivity of the proposed stripping voltametric method for the cadmium ions. The tolerance limit of interference is shown in Table 2.| Electrodes | Inorganic substance | Tolerated ratio | Inorganic substance | Tolerated ratio |
|---|---|---|---|---|
| a Even affected when adding 5.0 μg L−1 Cu2+. | ||||
| Sb/MFA-CPE | Na+ | 3000 | Al3+ | 80 |
| K+ | 3000 | Fe3+ | 4 | |
| Cl− | 100 | Pb2+ | 0.25 | |
| NO3− | 8000 | Ca2+ | 2 | |
| SO42− | 100 | Zn2+ | 2 | |
| Mg2+ | 60 | Cu2+ | —a | |
| Sb/MMFA-CPE | Na+ | 2000 | Al3+ | 120 |
| K+ | 3000 | Fe3+ | 2 | |
| Cl− | 600 | Pb2+ | 0.5 | |
| NO3− | 5000 | Ca2+ | 1 | |
| SO42− | 600 | Zn2+ | 1 | |
| Mg2+ | 400 | Cu2+ | 0.125 | |
| Sb–Bi/MMFA-CPE | Na+ | 3000 | Al3+ | 520 |
| K+ | 3000 | Fe3+ | 4 | |
| Cl− | 1500 | Pb2+ | 0.25 | |
| NO3− | 8000 | Ca2+ | 0.5 | |
| SO42− | 1500 | Zn2+ | 0.25 | |
| Mg2+ | 600 | Cu2+ | 0.25 | |
3.6 Analytical applications
In order to verify the practical feasibility of the electrodes, the Sb/MFA-CPE, Sb/MMFA-CPE and Sb–Bi/MMFA-CPE were applied to the analysis of Cd(II) in tap water sample which was collected from our laboratory. 0.1 milliliter of the water sample was pipetted into a 20.0 mL calibrated flask and determined under the optimum conditions, and the peak currents of Cd(II) in tap water which was added 50, 60, 70 μg L−1 at Sb/MFA-CPE, Sb/MMFA-CPE and Sb–Bi/MMFA-CPE were obtained (Table 3). It demonstrated that Cd(II) was not found in tap water samples.| Sample | Electrodes | Original (μg L−1) | Added (μg L−1) | Founda (μg L−1) | RSD (%) | Recovery (%) |
|---|---|---|---|---|---|---|
| a Mean of three determinations.b Not detected. | ||||||
| Ultrapure water with 0.1 mL tap water | Sb/MFA-CPE | NDb | 50.0 | 50.14 | 2.06 | 100.28 |
| NDb | 60.0 | 60.31 | 2.82 | 100.51 | ||
| NDb | 70.0 | 70.71 | 0.30 | 101.02 | ||
| Sb/MMFA-CPE | NDb | 50.0 | 49.82 | 3.61 | 99.65 | |
| NDb | 60.0 | 59.90 | 3.36 | 99.83 | ||
| NDb | 70.0 | 71.48 | 7.84 | 102.12 | ||
| Sb–Bi/MMFA-CPE | NDb | 50.0 | 49.07 | 4.74 | 98.14 | |
| NDb | 60.0 | 63.86 | 1.74 | 106.43 | ||
| NDb | 70.0 | 70.64 | 2.70 | 100.91 | ||
3.7 Discussion
Comparing Sb/MMFA-CPE with Sb/MFA-CPE, the calibration curves for the determination of Cd(II) show significant differences in the linear range (40.0–150.0 versus 40.0–80.0 μg L−1) and the slope (0.69 versus 1.44 μA μg−1 L−1) at the same preconcentration time (as shown in Fig. 5), which means that SWASV response of Cd(II) at Sb/MMFA-CPE has a higher linear range and lower sensitivity. Sánchez et al.46 has found that the larger pore size of the modifier of the electrode could increase the diffusion coefficient in the process of metal ions deposition and achieve a higher linear range. In this paper, compared with MFA, the characteristics of pore structure of MMFA have small changes, that is, the surface area (from 74.401 to 72.892 m2 g−1), pore volume (from 0.27 to 0.20 cm3 g−1) and pore size (from 14.6 to 11.1 nm) slightly reduce due to the channel blocking by amine molecules. However, due to the introduction of CTAB, MMFA-CPE provides a larger effective area for interacting with analytes and more binding sites and possesses more adsorption capacity. Besides, it further facilitates electron transfer at the electrode and amplifies the electrochemical signal. Thus, the SWSV responses of Cd(II) at Sb/MMFA-CPE are better than those at Sb/MFA-CPE within the detection range. It indicates that binding sites that can be reached by the Cd(II) play a crucial role in the determination process for the electrode modified with modified fly ash with similar pore structure. For these two electrodes, the detection limits of Sb/MMFA-CPE and Sb/MFA-CPE are 2.25 and 2.63 μg L−1, respectively.Under the same test conditions, the SWASV responses of Cd(II) at Sb–Bi/MMFA-CPE are all better than that at Sb/MMFA-CPE or Bi/MMFA-CPE (as shown in Fig. 3g). This fact points out that Sb–Bi thin film can give higher electrical conductivity than single Sb thin film or single Bi thin film, and the optimal concentrations of Sb(III) and Bi(III) are 2.0 mg L−1 and 4.0 mg L−1, respectively. Similar results were obtained by Yi et al.45 who found the optimal concentration proportion of Sb(III) and Bi(III) was 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5. Comparing Sb–Bi/MMFA-CPE with Sb/MMFA-CPE, the calibration curves for the determination of Cd(II) show significant difference in the linear range (2.0–150.0 versus 40.0–150.0 μg L−1) and no significant difference in slope (0.75 versus 0.69 μA μg−1 L−1) at the same preconcentration time (as shown in Fig. 5), which means that SWASV response of Cd(II) at Sb–Bi/MMFA-CPE has a higher linear range and similar sensitivity. The different SWASV responses at two electrodes are because of the different electrode mechanisms at bismuth compared to SbFEs. The electrode mechanisms of Cd(II) at BiFEs are the attractive forces between the accumulated metal particles and coupled by partial adsorption of the metal ions. Compared to BiFEs, the electrode mechanisms of Cd(II) at SbFEs are affected by attractive interactions, and the electrode mechanisms of Cd(II) at SbFEs are free of adsorption phenomena. Therefore, the calibration curves for the determination of Cd(II) at Sb–Bi/MMFA-CPE could achieve a wider linear range, and Sb–Bi/MMFA-CPE could be used for the determination of the lower concentration of cadmium than Sb/MMFA-CPE. The detection limits of Sb–Bi/MMFA-CPE and Sb/MFA-CPE are found to be 1.36 and 2.25 μg L−1, respectively.
:5. Comparing Sb–Bi/MMFA-CPE with Sb/MMFA-CPE, the calibration curves for the determination of Cd(II) show significant difference in the linear range (2.0–150.0 versus 40.0–150.0 μg L−1) and no significant difference in slope (0.75 versus 0.69 μA μg−1 L−1) at the same preconcentration time (as shown in Fig. 5), which means that SWASV response of Cd(II) at Sb–Bi/MMFA-CPE has a higher linear range and similar sensitivity. The different SWASV responses at two electrodes are because of the different electrode mechanisms at bismuth compared to SbFEs. The electrode mechanisms of Cd(II) at BiFEs are the attractive forces between the accumulated metal particles and coupled by partial adsorption of the metal ions. Compared to BiFEs, the electrode mechanisms of Cd(II) at SbFEs are affected by attractive interactions, and the electrode mechanisms of Cd(II) at SbFEs are free of adsorption phenomena. Therefore, the calibration curves for the determination of Cd(II) at Sb–Bi/MMFA-CPE could achieve a wider linear range, and Sb–Bi/MMFA-CPE could be used for the determination of the lower concentration of cadmium than Sb/MMFA-CPE. The detection limits of Sb–Bi/MMFA-CPE and Sb/MFA-CPE are found to be 1.36 and 2.25 μg L−1, respectively.
4. Conclusions
Fly ash was treated by hydrrothermal procedure with NaOH (MFA), which led to a partial conversion to zeolite A, the transformation of the morphology and great improvement of adsorption. When MFA was doped carbon paste electrode, SWASV of Cd(II) at Sb/MFA-CPE had better electrochemical response than that at Sb/CPE. Compared to Sb/MFA-CPE, the SWASV response of Cd(II) at Sb/MMFA-CPE had a higher linear range and lower sensitivity because the introduction of CTAB was beneficial for interacting with analytes and facilitating electron transfer at the electrode. The application of the Sb–Bi/MMFA-CPE for measuring trace Cd(II) revealed better electroanalytical performance than Sb/MMFA-CPE since Sb–Bi thin film could give higher electrical conductivity than single Sb thin film or single Bi thin film.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by International Cooperation and Exchange of the National Natural Science Foundation of China (No. 51820105006), National Natural Science Foundation of China (No. 51674174), International Cooperation in Science and Technology of Major Research Plan of Shanxi Province (No. 201803D421104) and Science and Technology Research Foundation for Young Scholars of Shanxi Province (No. 201801D221347). This paper was funded by China Scholarship Council.References
- L. Luo, X. Wang, Y. Ding, Q. Li, J. Jia and D. Deng, Appl. Clay Sci., 2010, 50, 154–157 CrossRef CAS.
- J. O. Nriagu and J. M. Pacyna, Nature, 1988, 333, 134–139 CrossRef CAS PubMed.
- A. Jang, Y. Seo and P. L. Bishop, Environ. Pollut., 2005, 133, 117–127 CrossRef CAS PubMed.
- M. El Mhammedi, M. Achak, M. Hbid, M. Bakasse, T. Hbid and A. Chtaini, J. Hazard. Mater., 2009, 170, 590–594 CrossRef CAS PubMed.
- E. Shams and R. Torabi, Sens. Actuators, B, 2006, 117, 86–92 CrossRef CAS.
- Z. Zou, A. Jang, E. Macknight, P.-M. Wu, J. Do, P. L. Bishop and C. H. Ahn, Sens. Actuators, B, 2008, 134, 18–24 CrossRef CAS.
- H. Zhang, Z. Gao, Y. Liu, C. Ran, X. Mao, Q. Kang, W. Ao, J. Fu, J. Li and G. Liu, et al., J. Hazard. Mater., 2018, 355, 128–135 CrossRef CAS PubMed.
- K. Nakano, K. Okubo and K. Tsuji, Powder Diffr., 2009, 24, 135 CrossRef CAS.
- H. Shahsavand and M. R. Nateghi, J. Water Chem. Technol., 2018, 40, 86–90 CrossRef.
- J. Vuković, S. Matsuoka, K. Yoshimura, V. Grdinić, R. J. Grubešić and O. Županić, Talanta, 2007, 71, 2085–2091 CrossRef PubMed.
- M. A. El Mhammedi, M. Achak and A. Chtaini, J. Hazard. Mater., 2009, 161, 55–61 CrossRef CAS PubMed.
- G. Chen, X. Hao, B. L. Li, H. Q. Luo and N. B. Li, Sens. Actuators, B, 2016, 237, 570–574 CrossRef CAS.
- Y. Gómez, L. Fernández, C. Borrás, J. Mostany and B. Scharifker, Talanta, 2011, 85, 1357–1363 CrossRef PubMed.
- M. F. Philips, A. I. Gopalan and K.-P. Lee, J. Hazard. Mater., 2012, 237, 46–54 CrossRef PubMed.
- B. Chen, L. Wang, X. Huang and P. Wu, Microchim. Acta, 2011, 172, 335–341 CrossRef CAS.
- L. Cao, J. Jia and Z. Wang, Electrochim. Acta, 2008, 53, 2177–2182 CrossRef CAS.
- B. Kong, B. Tang, X. Liu, X. Zeng, H. Duan, S. Luo and W. Wei, J. Hazard. Mater., 2009, 167, 455–460 CrossRef CAS PubMed.
- C. Zhang, Y. Zhou, L. Tang, G. Zeng, J. Zhang, B. Peng, X. Xie, C. Lai, B. Long and J. Zhu, Nanomaterials, 2016, 6, 7 CrossRef PubMed.
- X. Han, Z. Meng, H. Zhang and J. Zheng, Microchim. Acta, 2018, 185, 274 CrossRef PubMed.
- M. de la Gala Morales, M. R. P. Marín, L. C. Blázquez and E. P. Gil, Anal. Methods, 2014, 6, 8668–8674 RSC.
- D. Demetriades, A. Economou and A. Voulgaropoulos, Anal. Chim. Acta, 2004, 519, 167–172 CrossRef CAS.
- N. Spano, A. Panzanelli, P. C. Piu, M. I. Pilo, G. Sanna, R. Seeber and A. Tapparo, Anal. Chim. Acta, 2005, 553, 201–207 CrossRef CAS.
- J. Wang, J. Lu, S. B. Hocevar, P. A. Farias and B. Ogorevc, Anal. Chem., 2000, 72, 3218–3222 CrossRef CAS PubMed.
- S. B. Hocevar, I. Švancara, B. Ogorevc and K. Vytřas, Anal. Chem., 2007, 79, 8639–8643 CrossRef CAS PubMed.
- B. Petovar, K. Xhanari and M. Finšgar, Anal. Chim. Acta, 2018, 1004, 10–21 CrossRef CAS PubMed.
- G. Zhao, H. Wang, G. Liu and Z. Wang, Sens. Actuators, B, 2016, 235, 67–73 CrossRef CAS.
- N. Serrano, J. M. Díaz-Cruz, C. Arino and M. Esteban, TrAC, Trends Anal. Chem., 2016, 77, 203–213 CrossRef CAS.
- G. Kefala and A. Economou, Anal. Chim. Acta, 2006, 576, 283–289 CrossRef CAS PubMed.
- E. Asadian, S. Shahrokhian, A. I. Zad and F. Ghorbani-Bidkorbeh, Sens. Actuators, B, 2017, 239, 617–627 CrossRef CAS.
- J. C. Kemmegne-Mbouguen and L. Angnes, Sens. Actuators, B, 2015, 212, 464–471 CrossRef CAS.
- H. S. Hashemi, A. Nezamzadeh-Ejhieh and M. Karimi-Shamsabadi, Mater. Sci. Eng., C, 2016, 58, 286–293 CrossRef CAS PubMed.
- Z. Sarbak and M. Kramer-Wachowiak, Powder Technol., 2002, 123, 53–58 CrossRef CAS.
- T. C. Nguyen, P. Loganathan, T. V. Nguyen, J. Kandasamy and S. Vigneswaran, Environ. Sci. Pollut. Res., 2017, 25, 1–9 Search PubMed.
- M. Abrishamkar, S. N. Azizi and J. B. Raoof, Monatsh. Chem., 2012, 143, 409–412 CrossRef CAS.
- A. Ismail, A. Kawde, O. Muraza, M. A. Sanhoob and A. R. Al-Betar, Microporous Mesoporous Mater., 2016, 225, 164–173 CrossRef CAS.
- B. Kaur, R. Srivastava and B. Satpati, New J. Chem., 2015, 39, 5137–5149 RSC.
- J. Scott, J. Chem. Technol. Biotechnol., 2010, 77, 63–69 CrossRef.
- H. Ibrahim and Y. Temerk, Talanta, 2020, 208, 120362 CrossRef CAS PubMed.
- Y. M. Temerk, H. S. M. Ibrahim and W. Schuhmann, Electroanalysis, 2016, 28, 372–379 CrossRef CAS.
- M. Ibrahim, H. Ibrahim, N. Almandil and A. N. Kawde, Sens. Actuators, B, 2018, 274, 123–132 CrossRef CAS.
- H. Ibrahim and Y. Temerk, Sens. Actuators, B, 2015, 206, 744–752 CrossRef CAS.
- E. Barsoukov and R. J. Macdonald, Impedance Spectroscopy: Theory, Experiment, and Applications, Wiley-Interscience, 2005 Search PubMed.
- L. Cao, J. Jia and Z. Wang, Electrochim. Acta, 2008, 53, 2177–2182 CrossRef CAS.
- Z. Guo, F. Feng, Y. Hou and N. Jaffrezic-Renault, Talanta, 2005, 65, 1052–1055 CrossRef CAS PubMed.
- W. J. Yi, Y. Li, G. Ran, H. Q. Luo and N. B. Li, Sens. Actuators, B, 2012, 166–167, 544–548 CrossRef CAS.
- A. Sánchez, S. Morante-Zarcero, D. Pérez-Quintanilla, I. D. Hierro and I. Sierra, J. Electroanal. Chem., 2013, 689, 76–82 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07493d |
| This journal is © The Royal Society of Chemistry 2021 |