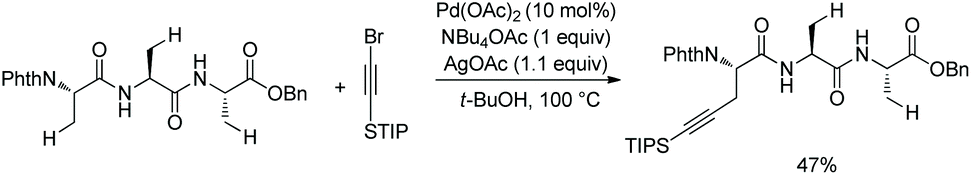DOI:
10.1039/D0QO00988A
(Review Article)
Org. Chem. Front., 2021,
8, 133-168
Site-selective and diastereoselective functionalization of α-amino acid and peptide derivatives via palladium-catalyzed sp3 C–H activation
Received
16th August 2020
, Accepted 24th October 2020
First published on 26th October 2020
Abstract
α-Amino acid and peptide derivatives are important in synthetic organic chemistry and medicinal chemistry, which attract many chemists to develop new methods for their synthesis. Palladium-catalyzed sp3 C–H activation/functionalization has been developed greatly for the modification of α-amino acids and peptides. This review introduces palladium-catalyzed sp3 C–H activation/functionalization of amino acids and peptides. The content includes intermolecular functionalization (arylation, alkylation, alkenylation, alkynylation, acetoxylation, fluorination, silylation, borylation, sulfonylation, chalcogenation and carbonylative cyclization) and intramolecular functionalization (intramolecular C–N bond-forming cyclization and peptide macrocyclization).
 Ming Zhang | Ming Zhang was born in Jiangxi, China, in 1968. He studied at East China Normal University from 1985 to 1992 for his bachelor's and master's degree. After he left ECNU, he carried out research at Dalian Institute of Chemical Physics of the Chinese Academy of Science and Zhejiang University. He synthesized a series of cyclopentadienyl titanium complexes, and applied them in hydrogenation of olefins. Now he is working at Jiangxi Normal University, and his current interest is development of organic reactions catalyzed by transition metals. |
 Yiyuan Peng | Yiyuan Peng received his B.S. degree from the Department of Chemistry at Jiangxi Normal University in 1985. He received his M.S. degree from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, under the supervision of Prof. Xikui Jiang and Prof. Chengxue Zhao in 1988. He obtained his Ph.D. degree from Nankai University under the supervision of Prof. Jinpei Cheng in 2003. Then he joined the College of Chemistry & Chemical Engineering, Jiangxi Normal University, as a professor. His research interests focus on organic synthesis and chemical biology. |
 Ai Qin Zhang | Ai Qin Zhang was born in Jiangxi, China, in 1971. She studied at Nanchang University from 2007 to 2010 for her Ph.D. She is currently an associate professor at Nanchang Hangkong University. Her research interest focuses on the development of organic reactions catalyzed by transition metals. |
1. Introduction
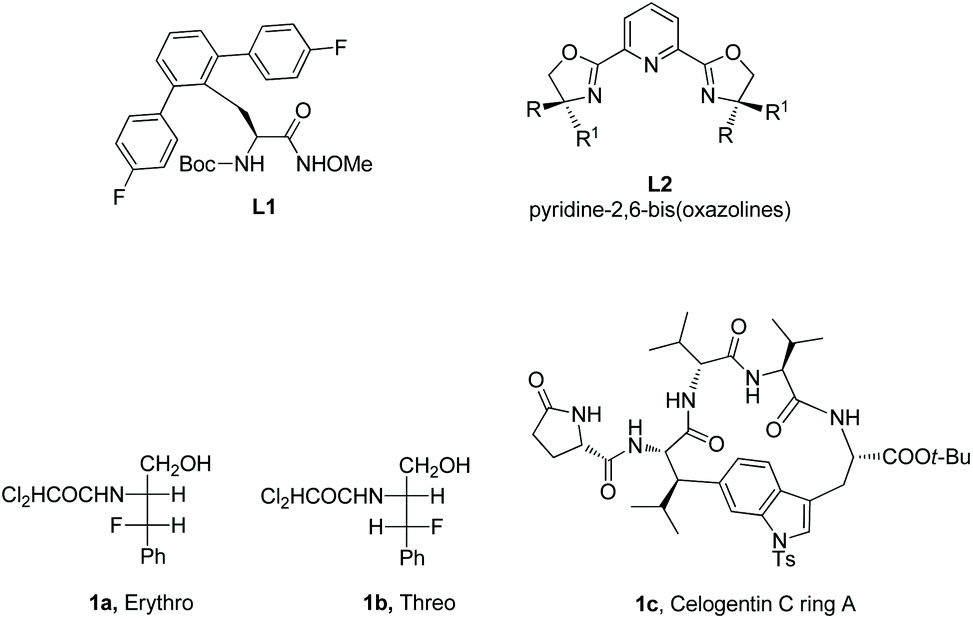
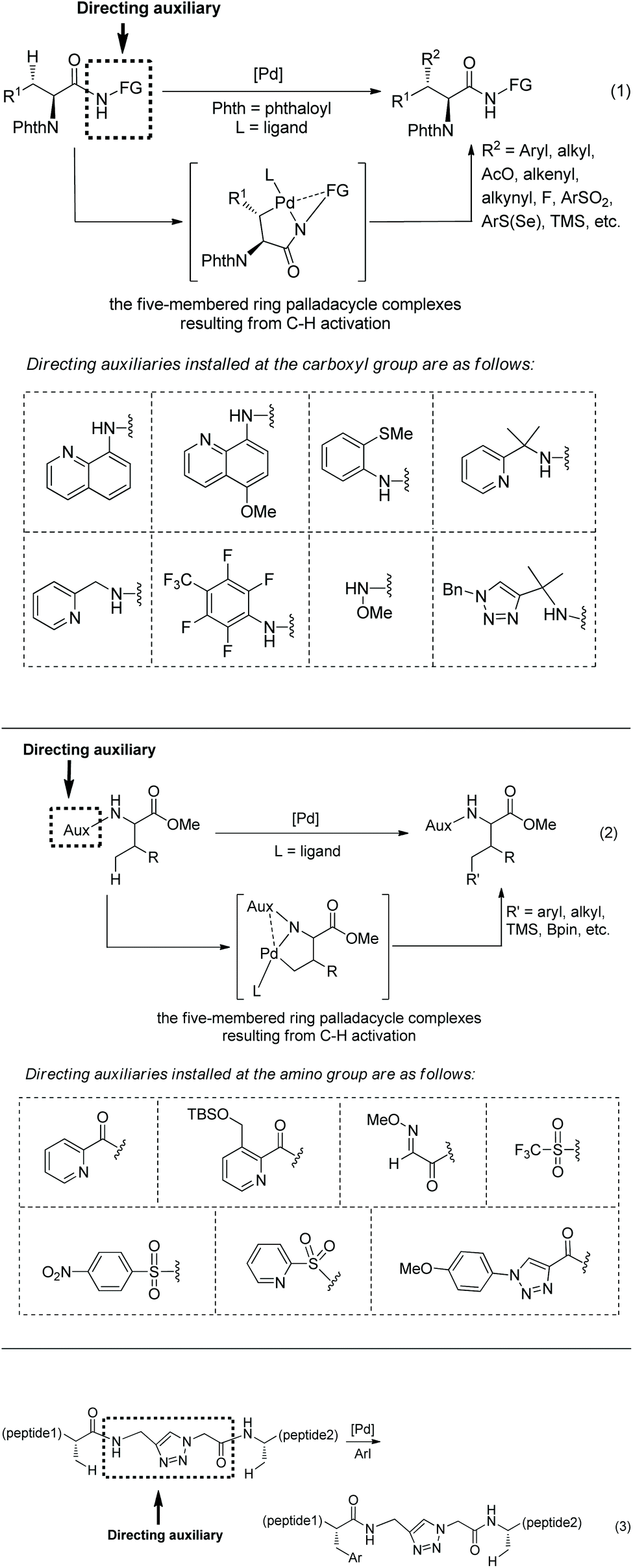
α-Amino acid and peptide derivatives play great roles in synthetic organic chemistry and medicinal chemistry, such as L1 can be used as a ligand in palladium-catalyzed enantioselective sp3 C–H activation,1aL2 can serve as ligands in many asymmetric reactions,1b1a and 1b have antifungal and antibacterial activities,2 Celogentin C existing in the seeds of Celosia argentea has strong inhibitory ability towards polymerization of tubulin, and the ring A of Celogentin C 1c is a cylcopeptide (Scheme 1).3 A recent review described the applications of fluorine-containing amino acids for drug design.4 Given the importance of fluorinated amino acid derivatives, the synthesis of fluorinated amino acid derivatives has been attracting much attention from organic chemists.5 The main method for the synthesis of fluorinated amino acid derivatives in the previous reviews is nucleophilic substitution. In recent years, the C–H activation strategy has turned out to be effective for site-selective and diastereoselective fluorination or other functionalizations of α-amino acid and peptide derivatives. Although many reviews involved C–H functionalization via sp3 C–H activation,6 the description of functionalization of α-amino acid and peptide derivatives via sp3 C–H activation is rare in the published reviews. This review introduces the recent developments of site-selective and diastereoselective functionalization of α-amino acid and peptide derivatives via sp3 C–H activation. The C–H activation strategy for the functionalization has four methods. (1) The directing auxiliaries are installed at the carboxyl group of amino acids (Scheme 2, eqn (1)); (2) the directing auxiliaries are installed at the amino group of amino acids (Scheme 2, eqn (2)); (3) an internal triazole moiety is installed in peptides (Scheme 2, eqn (3)); (4) endogenous groups such as native free carboxyl/amino groups in amino acids, and native N,N-/N,O-bidentate ligands in peptides act as directing groups, and exogenous auxiliary is not needed.
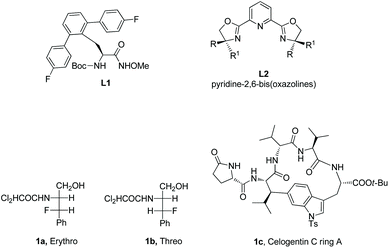 |
| | Scheme 1 Useful α-amino acid and peptide derivatives. | |
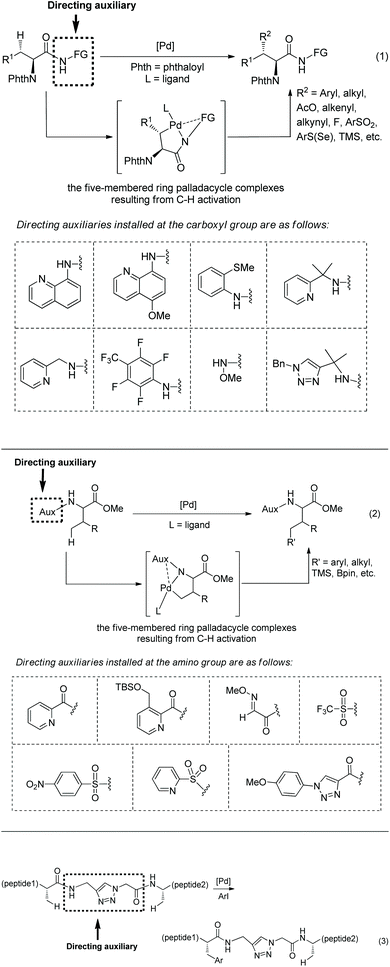 |
| | Scheme 2 Three ways to install directing auxiliaries. | |
2. Functionalization assisted by the directing groups derived from the carboxyl group of amino acids, an internal triazole moiety installed in peptides, or endogenous groups
2.1. Functionalization of α-amino acid derivatives
2.1.1. C–C, C–O and C–N bond formation.
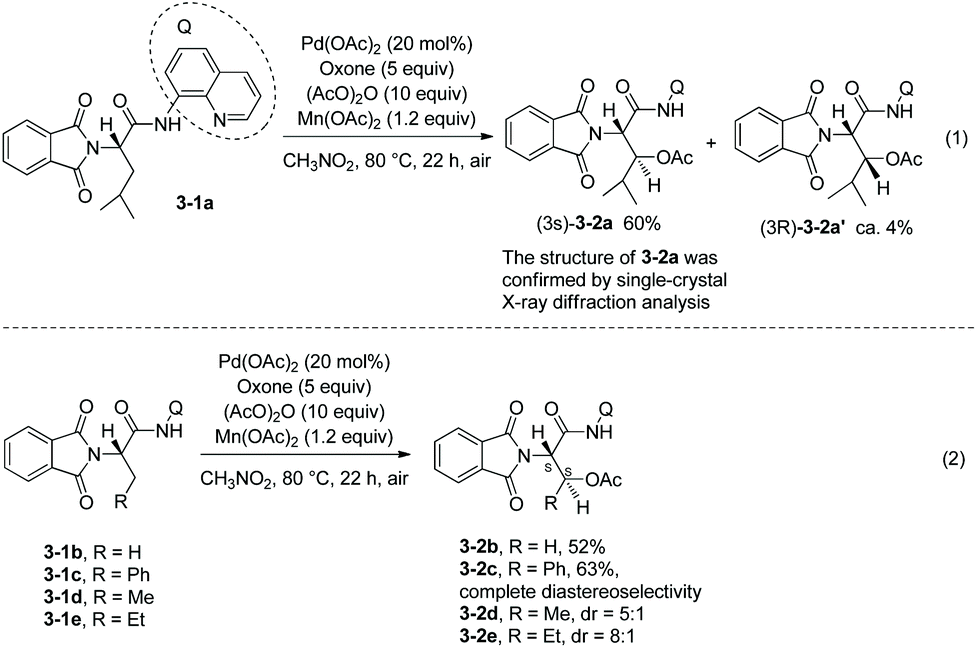
As early as in 2006, Corey and co-workers started the research on the functionalization of α-amino acid derivatives via sp3 C–H activation.7 The amino group of α-amino acids was protected by phthaloyl, 8-aminoquinoline was installed at the carboxyl group, and the formed amide directed the Pd-catalyzed functionalization of L-α-amino acid derivatives. Acetoxylation and arylation were investigated. N-Phthaloyleucine 8-aminoquinoline amide 3-1a was acetoxylated at the β position (Scheme 3, eqn (1)), palladium acetate was used as the catalyst, and the combination of two oxidants (oxone and manganous acetate) was used. The major product is (3S)-diastereomer 3-2a with 60% yield, and the (3R)-diastereomer 3-2a′ was isolated in ca. 4% yield. The alanine, β-phenylalanine, β-methylalanine and β-ethylalanine derivatives (3-1b)–(3-1e) could all be acetoxylated at the β position under the same reaction conditions (Scheme 3, eqn (2)). The alanine derivative 3-1b gave the serine derivative 3-2b in 52% yield. β-Phenylalanine derivative 3-1c gave the (3S)-diastereomer 3-2c exclusively in 63% yield. In the cases of 3-1d and 3-1e, the diastereomeric ratios were 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 8:1, respectively, and the (3S)-diastereomers 3-2d and 3-2e were preferentially formed. The regio- and diastereo-selectivities of the acetoxylation could be explained by the mechanism (Scheme 4). In the step of C–H activation from 4A to 4B, cyclopalladation favors to form a new five-membered ring over a six- or seven-membered ring, giving the Pd(II) complex 4B, which is the reason for regioselectivity that the acetoxylation occurs at the β position. The sterically favored trans-intermediate 4B could account for the diastereoselectivity, leading to the anti diastereomer product 3-2a.
:1 and 8:1, respectively, and the (3S)-diastereomers 3-2d and 3-2e were preferentially formed. The regio- and diastereo-selectivities of the acetoxylation could be explained by the mechanism (Scheme 4). In the step of C–H activation from 4A to 4B, cyclopalladation favors to form a new five-membered ring over a six- or seven-membered ring, giving the Pd(II) complex 4B, which is the reason for regioselectivity that the acetoxylation occurs at the β position. The sterically favored trans-intermediate 4B could account for the diastereoselectivity, leading to the anti diastereomer product 3-2a.
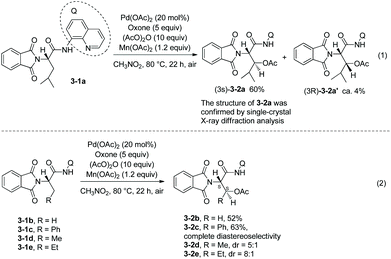 |
| | Scheme 3 Acetoxylation of L-α-amino acid derivatives. | |
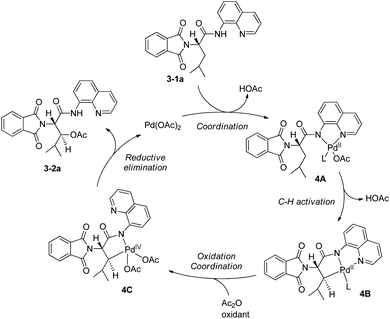 |
| | Scheme 4 Mechanism of acetoxylation. | |
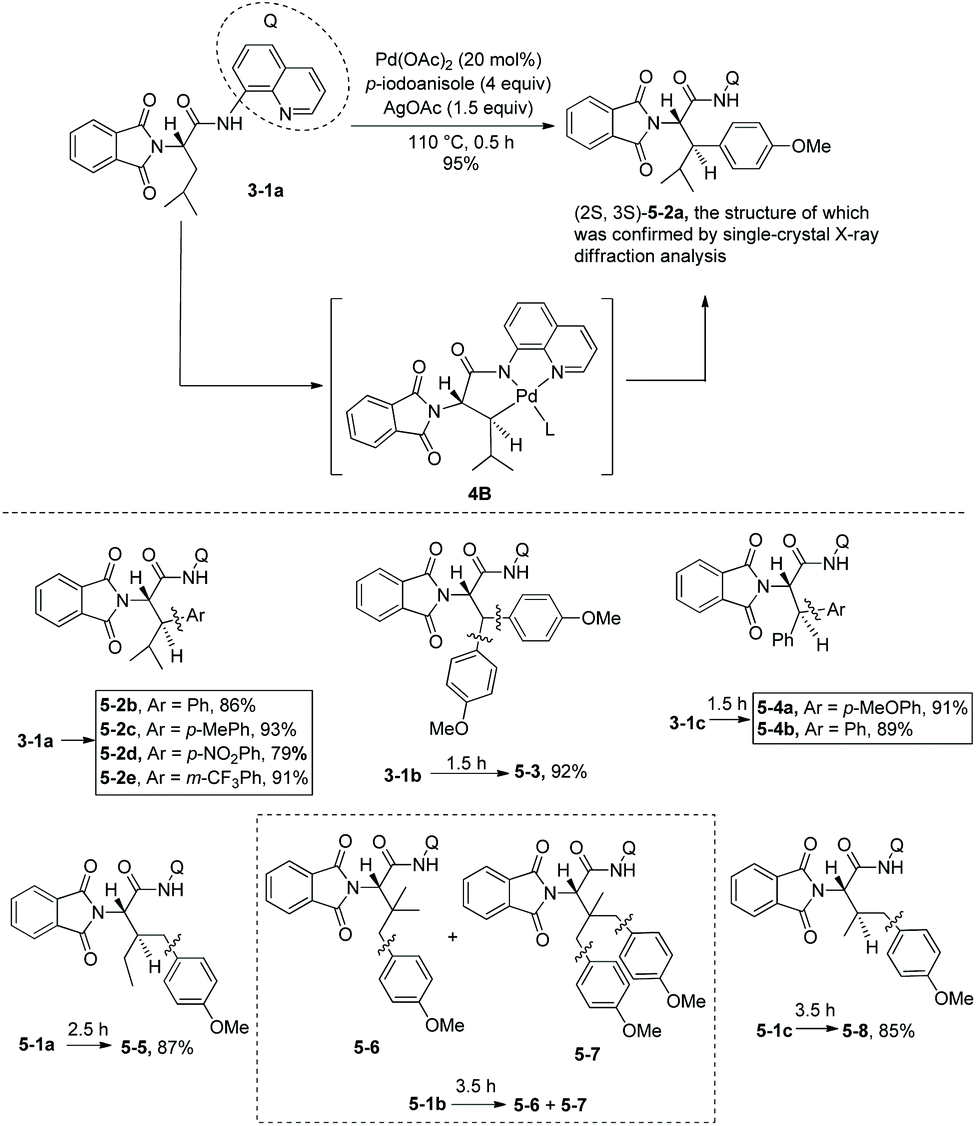
Arylation of N-phthaloyleucine 8-aminoquinoline amide 3-1a with p-idoanisole (4 equiv.) was affected under palladium catalysis, the product (2S,3S)-diastereomer 5-2a was obtained in 95% yield (Scheme 5, equation),7 and no solvent was used in the reaction. The diastereoselectivity of arylation of 3-1a confirmed further the structure of the trans-palladacycle 4B, and the trans-palladacycles could well account for the stereochemistry of acetoxylation and arylation, favoring the anti diastereomer products. In the arylation of the N-phthaloyleucine 8-aminoquinoline amide 3-1a, many electron-donating or -withdrawing groups (MeO, Me, NO2, and CF3 groups) could be tolerated, the β-arylated leucine derivatives (5-2b)–(5-2e) were obtained readily. Subsequently, arylation of a series of L-α-amino acid derivatives was investigated under the standard reaction conditions but with different reaction times. The arylation of N-phthaloyalanine 8-aminoquinoline amide 3-1b with p-idoanisole (4 equiv.) gave the diarylated alanine derivative 5-3 in 92% yield, which is also a β-arylated tyrosine derivative. Arylation of the N-phthaloylated phenylalanine amide 3-1c with p-iodoanisole or iodobenzene gave the β-arylated phenylalanine derivatives 5-4a and 5-4b smoothly. Chiral 5-4a is difficult to prepare in other ways. Arylation of the isoleucine derivative 5-1a with p-iodoanisole afforded the γ-arylated product 5-5 in 87% yield. It shows that the order of C–H activation reactivities is: γ-primary methyl C–H > β-tertiary C–H. Arylation of the t-leucine derivative 5-1b with p-iodoanisole also occurred at the γ position, affording a mixture of mono/diarylation products 5–6 and 5–7. N-Phthaloylvaline 8-aminoquinoline amide 5-1c was monoarylated at the γ position with 4 equivalents of p-iodoanisole, giving the products 5–8 in 85% yield.
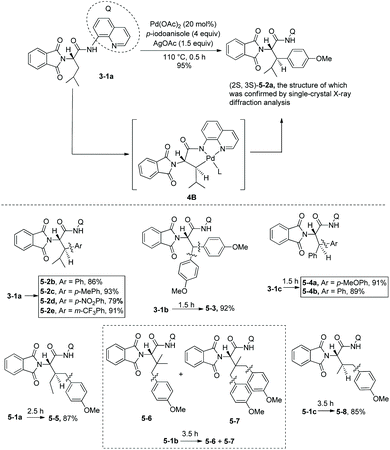 |
| | Scheme 5 Arylation of L-α-amino acid derivatives. | |
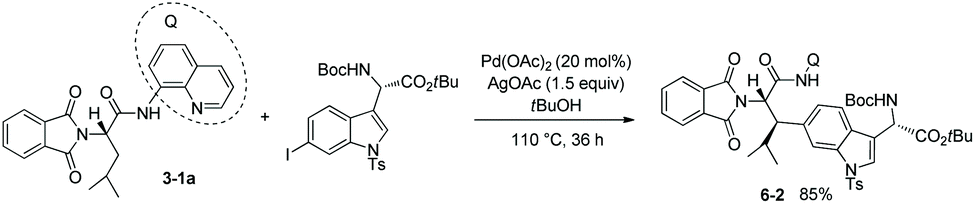
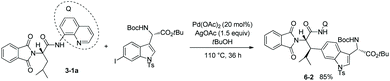
Based on the works of Corey and co-workers,7 the stereoselective β-indolylation of the N-phthaloyleucine 8-aminoquinoline amide 3-1a was developed in 2010 by Chen and co-workers as a key step for the concise total synthesis of celogentin C with biological activities existing in Celosia argentea (Scheme 6).8tert-Butanol was used as the reaction solvent. The product 6-2 was formed with complete diastereoselectivity and obtained in 85% yield on a 4 gram scale.
 |
| | Scheme 6 Indolylation of the leucine amide. | |
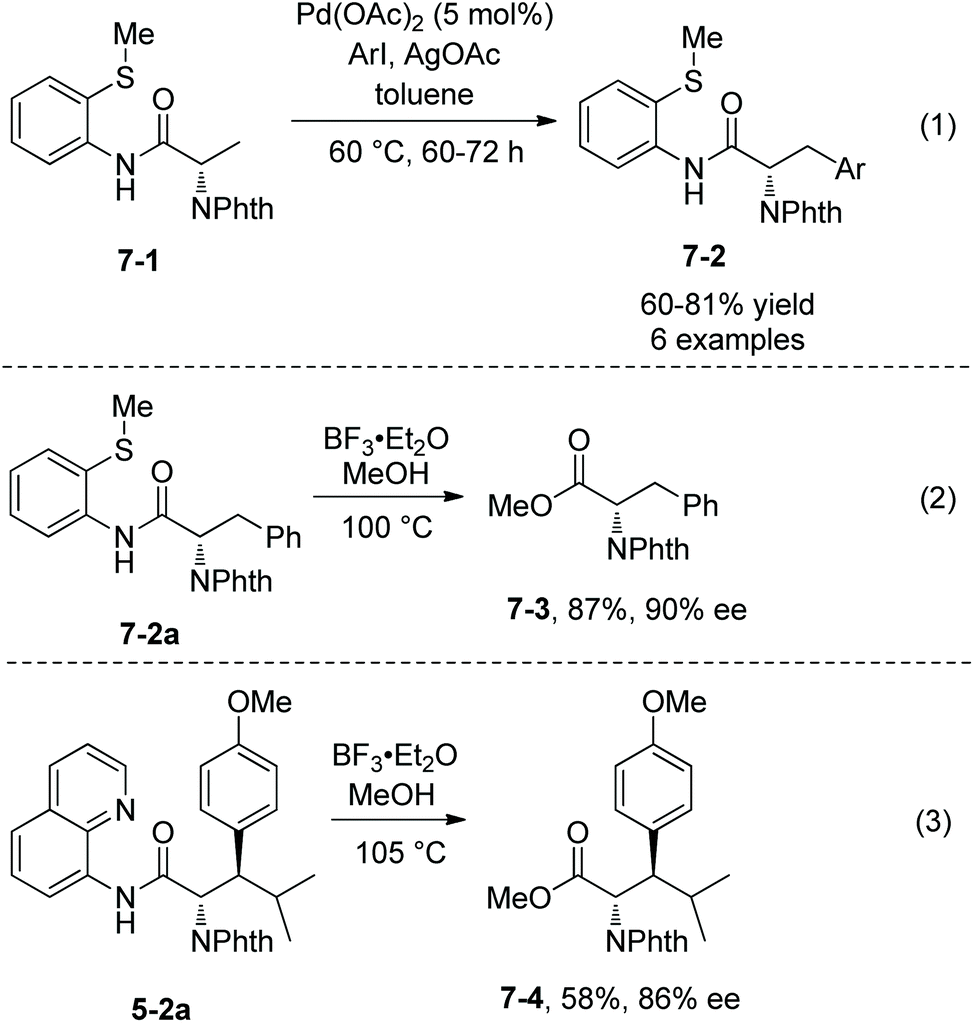
Palladium-catalyzed monoarylation of N-phthaloyalanine derivatives 7-1 was achieved by Daugulis et al. in 2012, using 2-(methylthio)aniline amides as the directing group (Scheme 7, eqn (1)), affording monoarylated products 7-2 in 60–81% yields.9 The directing auxiliary 2-(methylthio)aniline in arylation product 7-2a was removed readily with BF3·Et2O, giving the N-phthaloyalanine methyl ester 7-3 in 87% yield and with 90% ee (Scheme 7, eqn (2)).
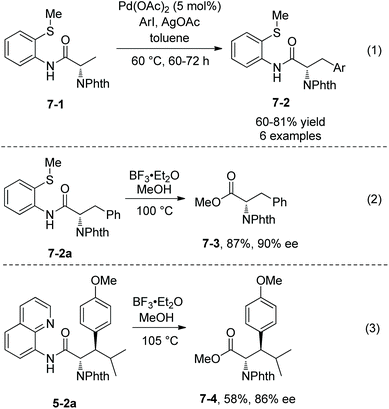 |
| | Scheme 7 Monoarylation of alanine derivatives and removal of auxiliaries. | |
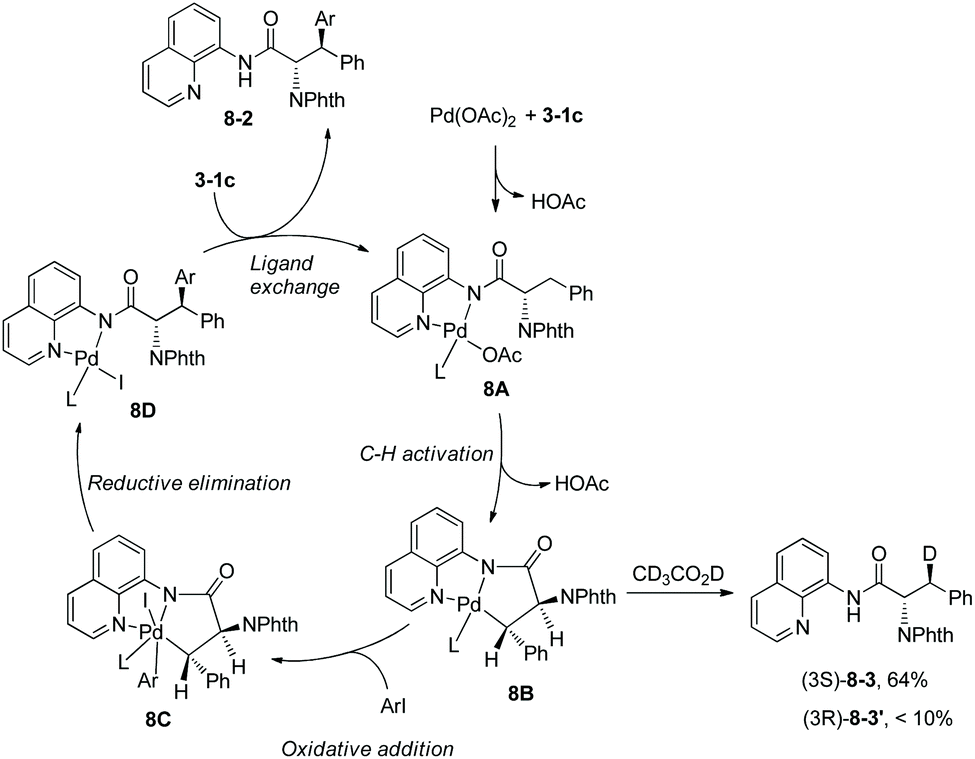
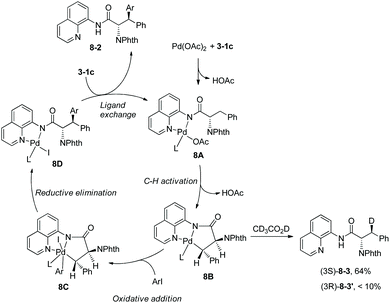
Arylation of N-phthaloylated L-α-amino acid 8-aminoquinoline amides was investigated by Daugulis et al. again.9 The phenylalanine derivative 3-1c, lysine derivative and leucine derivative 3-1a could be monoarylated at the β position smoothly with Pd(OAc)2 (5–11%), AgOAc, and 4-iodoanisole or 2-iodothiophene at 60 °C for 72–96 hours. The yield is up to 95%, the diastereoselectivity of the arylation is up to >50:1, and anti diastereomer products were favored. N-Phthaloyalanine 8-aminoquinoline amide 3-1b was diarylated at the β position with Pd(OAc)2 (5–6%), AgOAc, and 3,4-dimethyl-1-iodobenzene or 4-iodobenzoic acid ethyl ester at 60 °C for 62–70 hours, and the yield is 92% or 84%, respectively. The directing auxiliary 8-aminoquinoline in arylation products could be removed readily by treatment with BF3·Et2O in methanol at 105 °C (Scheme 7, eqn (3)), and the β-arylated N-phthaloyleucine methyl ester 7-4 was produced in 58% yield and 86% ee. To gain insight into the mechanism of arylation, the H/D exchange was conducted in 3-1c under palladium catalysis in a mixture of CD3COOD and [D8] toluene. 64% of the (3S)-deuterated product 8-3 was observed, and less than 10% of the (3R)-deuterated product 8-3′ was formed. According to this observation, the palladacycle 8B resulting from C–H activation was considered to have a trans configuration, which leads to the diastereoselectivity of the arylation (Scheme 8), and anti-8-2 is the major product in the arylation of 3-1c.
 |
| | Scheme 8 Mechanism of arylation. | |
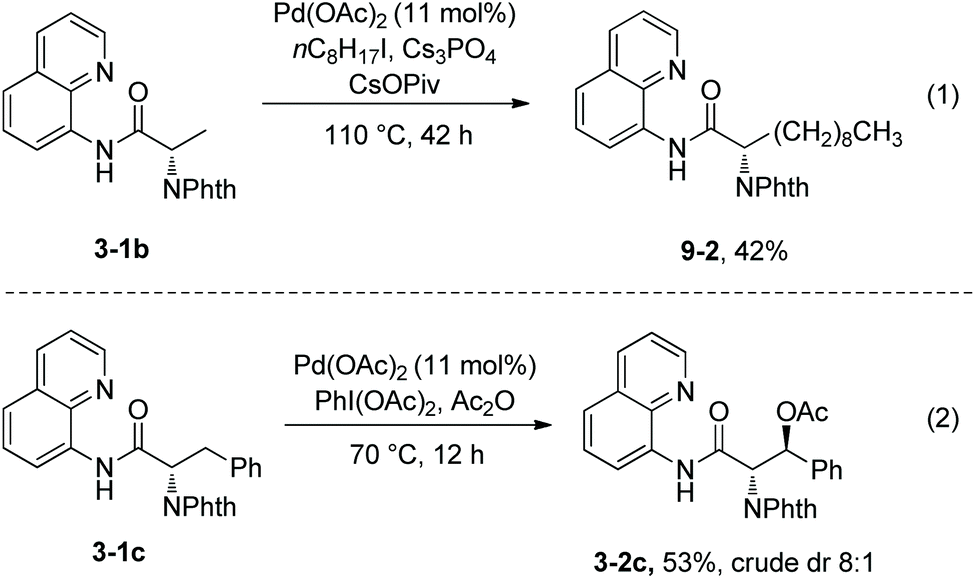
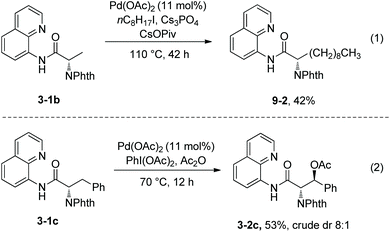
Alkylation of N-phthaloyalanine 8-aminoquinoline amide 3-1b and actoxylation of N-phthaloyl-β-phenylalanine 3-1c were also reported (Scheme 9).9 Alkylation of 3-1b with 1-iodooctane gave the lipidic amino acid derivative 9-2 in 42% yield (Scheme 9, eqn (1)). Actoxylation of 3-1c afforded (3S)-3-2c in 53% yield, using PhI(OAc)2 as an oxidant (Scheme 9, eqn (2)).
 |
| | Scheme 9 Alkylation and acetoxylation. | |
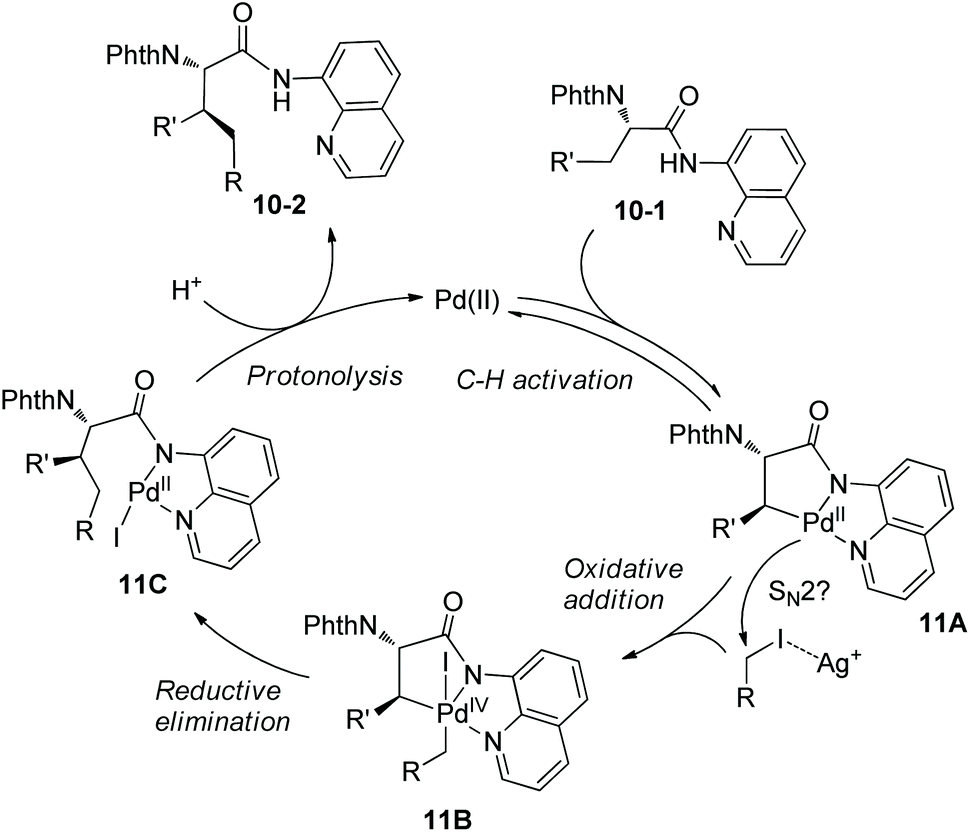
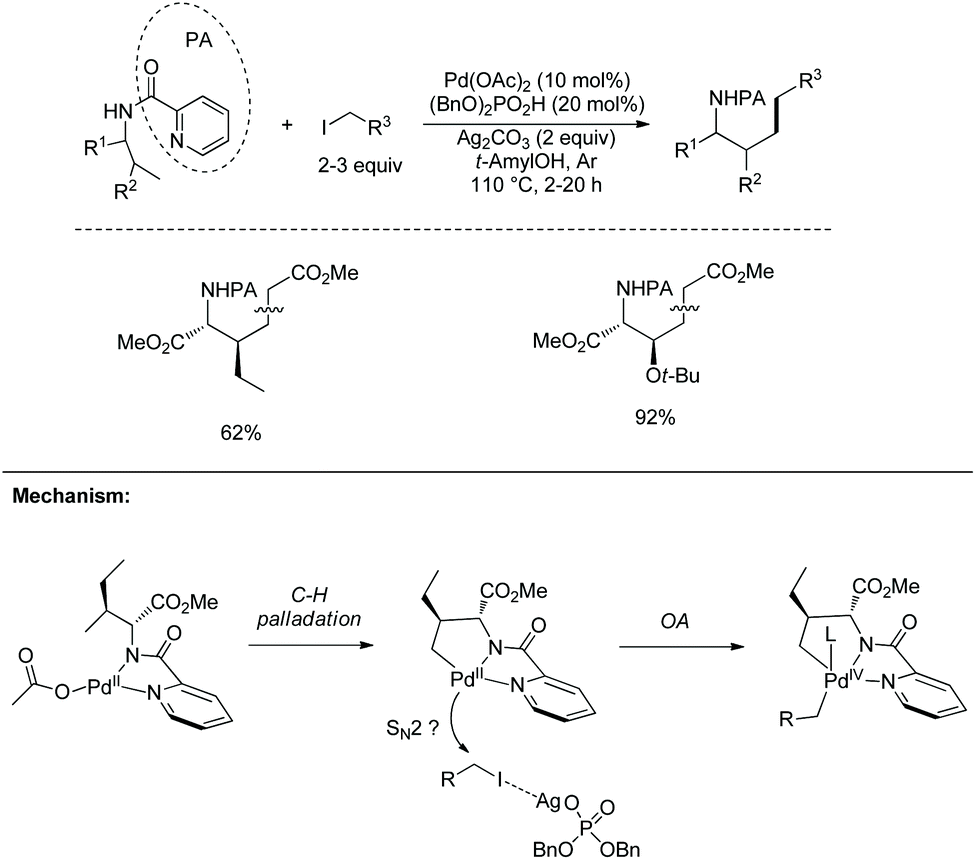
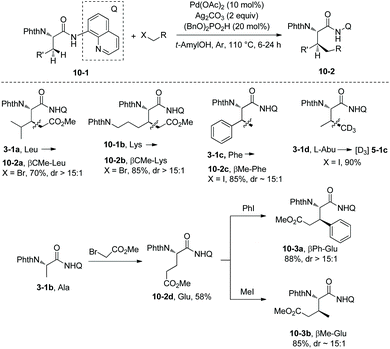
On the basis of alkylation of N-phthaloyalanine 8-aminoquinoline amide 3-1b at a β-primary methyl C–H bond by the Daugulis’ laboratory,9 Chen's group developed new reaction conditions in 2013 for the stereoselective alkylation of the β-methylene C(sp3)–H bonds in α-amino acid derivatives, also using palladium acetate as the catalyst and N-phthaloylated L-α-amino acid 8-aminoquinoline amides as substrates (Scheme 10).10 The method is also suitable for N-(8-quinolinyl)-aliphatic carboxamides without an α-amino group. The leucine derivative 3-1a (Leu), lysine derivative 10-1b (Lys), phenylalanine derivative 3-1c (Phe), L-α-aminobutyramide derivative 3-1d (Abu), and alanine derivative 3-1b (Ala) could all be β-alkylated smoothly, affording the alkylated products, for example, for (10-2a)–(10-2c), isotope-labeled [D3] 5-1c, and 10-2d, the yield is up to 90%, and the dr is up to >15:1. The alanine derivative 3-1b (Ala) undergoes preferentially monoalkylation with methyl 2-bromoacetate to give the glutamic acid derivative 10-2d, and 3-1b could be alkylated/arylated, alkylated/alkylated or arylated/alkylated sequentially twice at the β position with two hydrocarbonyl iodides or bromides to give β,β-disubstituted alanine derivatives (for example, 10-3a and 10-3b) in good yields and with high diastereoselectivities.
 |
| | Scheme 10 Alkylation by Chen. | |
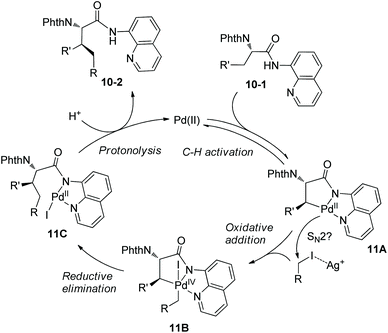
A Pd(II)/Pd(IV) catalytic cycle was proposed for the alkylation (Scheme 11). The intermediate 11A resulting from C–H activation has a sterically favored trans configuration (R′ ≠ H), which determines the stereoselectivity of the transformation, anti-10-2 is formed finally. Oxidative addition of alkyl iodide to 11A is promoted by Ag+, which proceeds probably through the SN2 pathway. Ag+ could also promote reductive elimination and regeneration of a more active Pd(II) catalyst by removal of the iodide ligand from 11B and 11C. (BnO)2PO2H could combine with Ag2CO3 to form a soluble complex, also serve as a ligand for palladium in the steps of oxidative addition and reductive elimination, and promote the protonolysis of 11C.
 |
| | Scheme 11 Mechanism of alkylation by Chen. | |
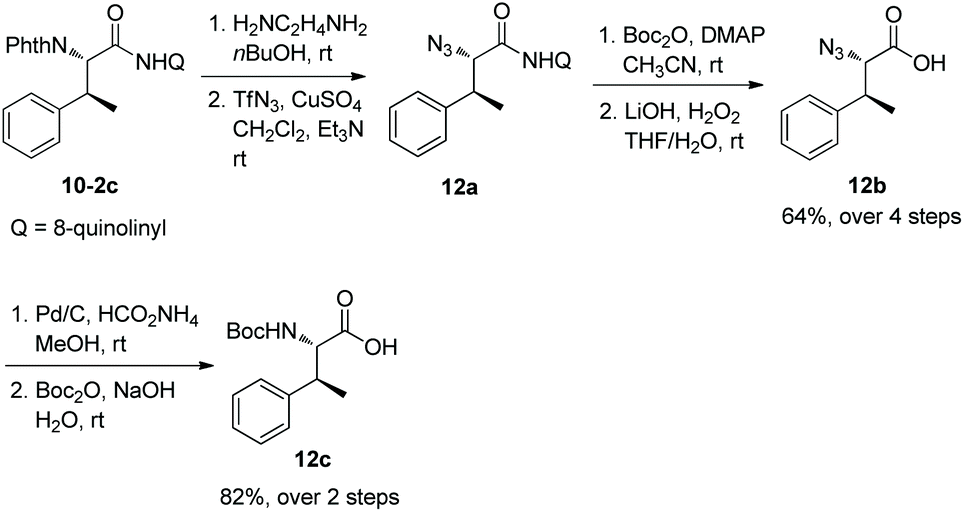
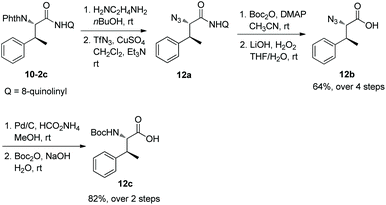
The auxiliary 8-aminoquinoline in the alkylated products can be removed under mild conditions at room temperature. 10-2c can be deprotected with ethylenediamine and subsequent transformation with TfN3 gives an azido derivative 12a, and treatment of Boc2O and LiOH/H2O2 produces an azido acid 12b. Reduction of 12b by hydrogen and protection of the obtained amino group with Boc2O afford the Boc-protected phenylalanine derivative 12c (Scheme 12).
 |
| | Scheme 12 Removal of 8-aminoquinoline. | |
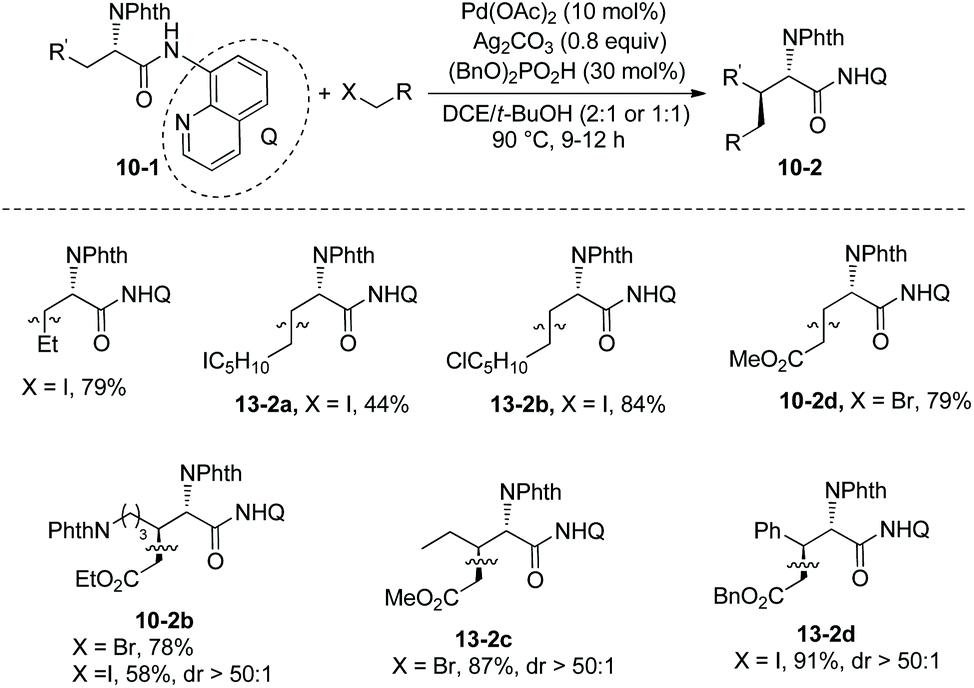
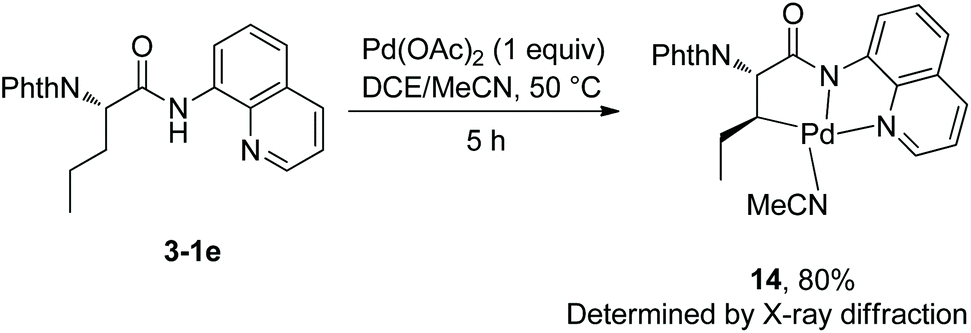
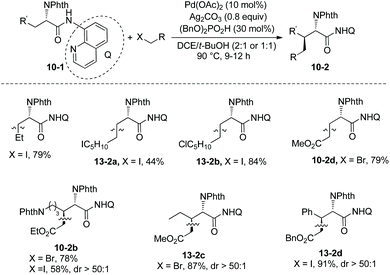
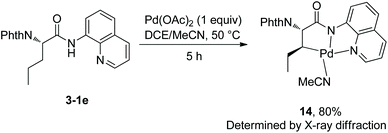
At the same time (in 2013), Shi and co-workers also reported the β-alkylation of N-phthaloylated L-α-amino acid 8-aminoquinoline amides with alkyl iodides or bromides, using the Pd(OAc)2/Ag2CO3/(BnO)2PO2H/DCE-tBuOH reaction system (Scheme 13).11 The reaction systems used by Shi et al. and Chen et al.10 are almost the same except for the solvents. The yield is up to 91% (13-2d), and the dr is up to >50:1 (10-2b, 13-2c and 13-2d). The Cl, I, CF3, COOR groups, or alkenyls and an acetal are compatible in the transformation. The trans-palladacycle 14 from the reaction of norvaline derivative 3-1e with Pd(OAc)2 was isolated and its structure was determined by X-ray diffraction (Scheme 14). The trans configuration of palladacycles creates the stereoselectivity of β-functionalization.
 |
| | Scheme 13 Alkylation by Shi. | |
 |
| | Scheme 14 The isolated palladacycle. | |
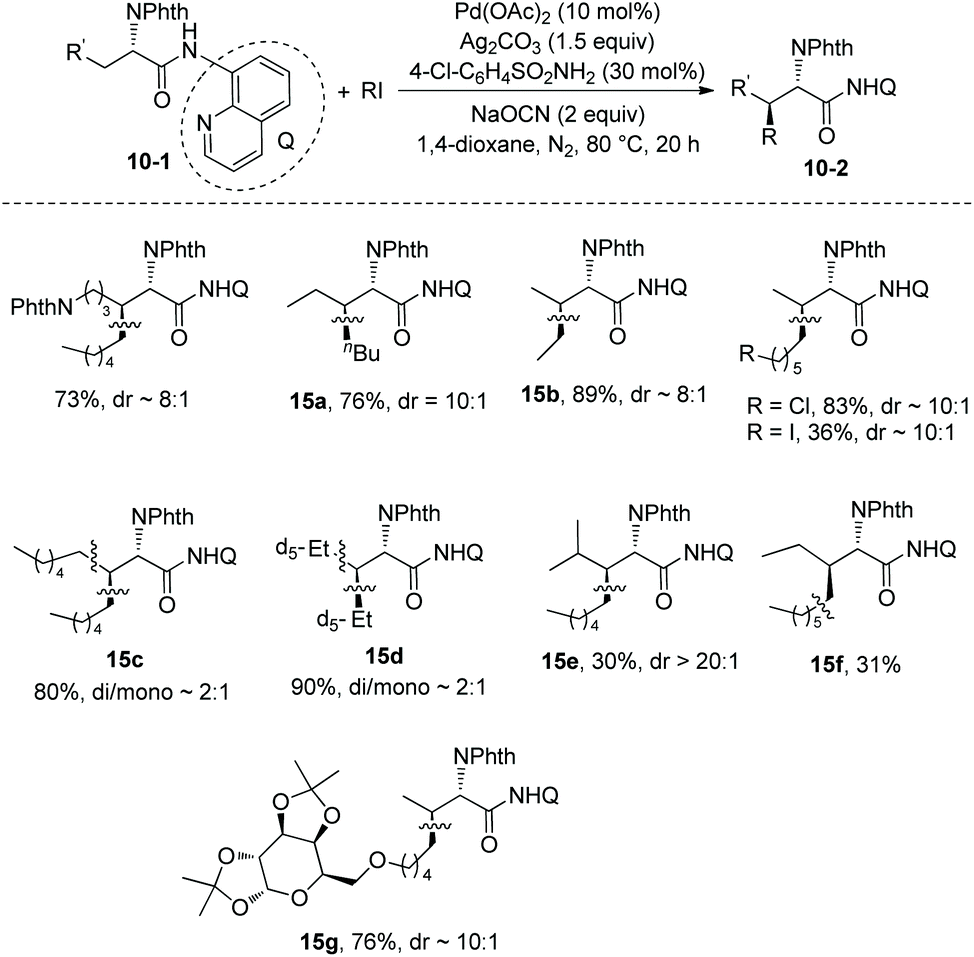
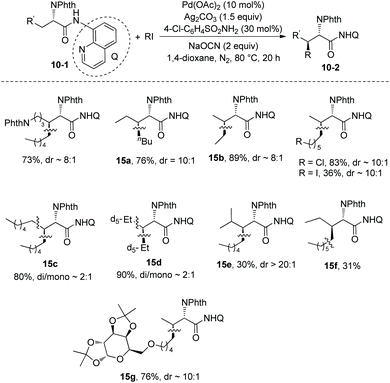
Then in 2014, Shi and co-workers developed another reaction system for the β-alkylation of N-phthaloylated L-α-amino acid 8-aminoquinoline amides with alkyl iodides, that is the Pd(OAc)2/Ag2CO3/NaOCN/4-Cl-C6H4SO2NH2/1,4-dioxane system (Scheme 15),12 the efficiency of the alkylation of L-norvaline derivative 3-1e with n-butyliodide using the new system (15a, 76% yield, dr 10:1) is much higher than that using the Pd(OAc)2/Ag2CO3/(BnO)2PO2H/DCE-tBuOH reaction system (13% yield, dr 9:1). The L-norvaline derivative 3-1e (Nva), L-α-aminobutyramide derivative 3-1d (Abu), phenylalanine derivative 3-1c (Phe), lysine derivative 10-1b (Lys), ornithine derivative (Orn), and leucine derivative 3-1a undergo monoalkylation at the β position, the yield is up to 89% (15b), and the dr is up to >20:1 (15e). Alkylation of 3-1a gives the product 15e in lower yield (30%) because of the bigger steric hindrance of 3-1a. The alanine derivative 3-1b (Ala) undergoes preferentially dialkylation, producing mixtures of mono/dialkylation products (15c, 80% and 15d, 90%), and this protocol can provide an isotope-labeled amino acid derivative (15d). Alkylation of 3-1d with a sugar-containing alkyl iodide gives a complex molecule 15g successfully. The isoleucine derivative 5-1a gives a γ-alkylated product 15f in 31% yield. The chloro, iodo, trifuoromethyl, methoxycarbonyl, 1,3-dioxolan-2-yl, cyano, silyl, Cbz-protected amino, alkynyl, and alkenyl groups are compatible in this transformation.
 |
| | Scheme 15 Alkylation with a sulfonamide ligand. | |
Combination of this protocol and the previously developed protocol for alkylation and arylation by themselves11 can modify structurally the alanine derivative 3-1b (Ala), and β,β-disubstituted alanine derivatives can be afforded in good yields and with high stereoselectivity (Scheme 16).
 |
| | Scheme 16 Synthesis of β,β-disubstituted alanine derivatives. | |
It is speculated that the dissociation of 4-Cl-C6H4SO2NH2 from Pd(II) of a palladacycle provides a coordination site for oxidative addition to form a Pd(IV) complex, and the coordination of 4-Cl-C6H4SO2NH2 to the resulting Pd(IV) complex can accelerate reductive elimination to form a new C–C bond.
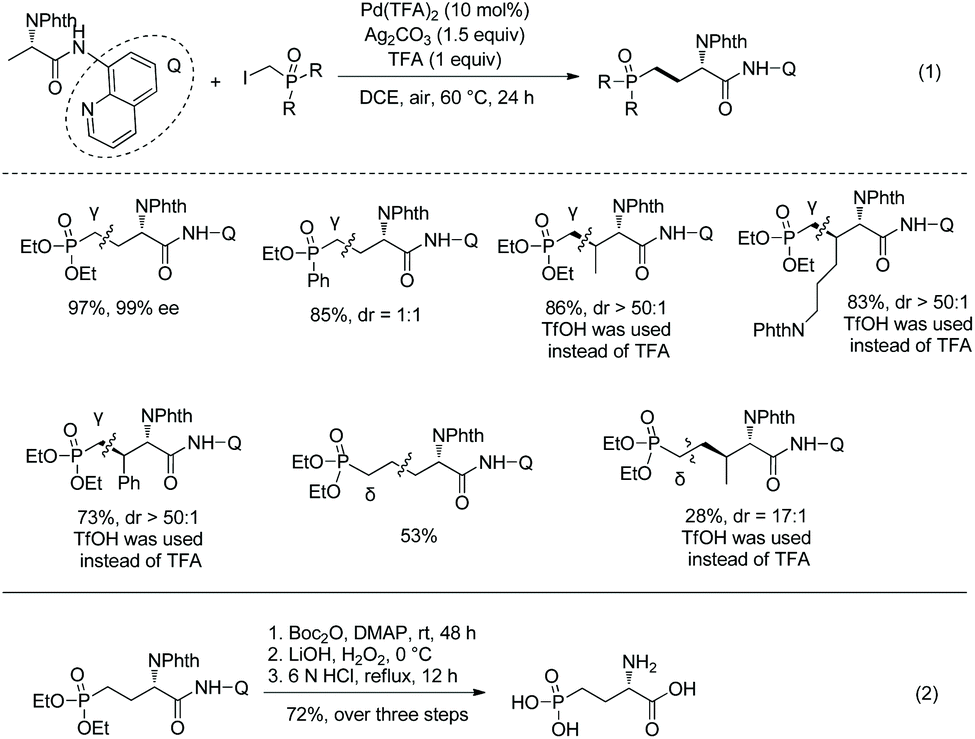
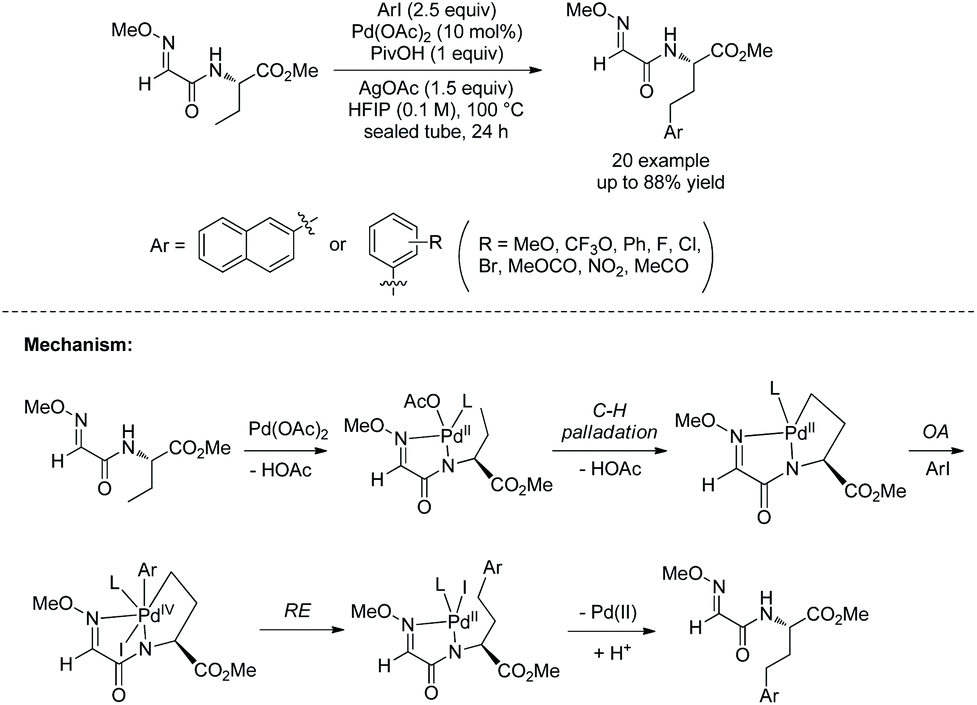
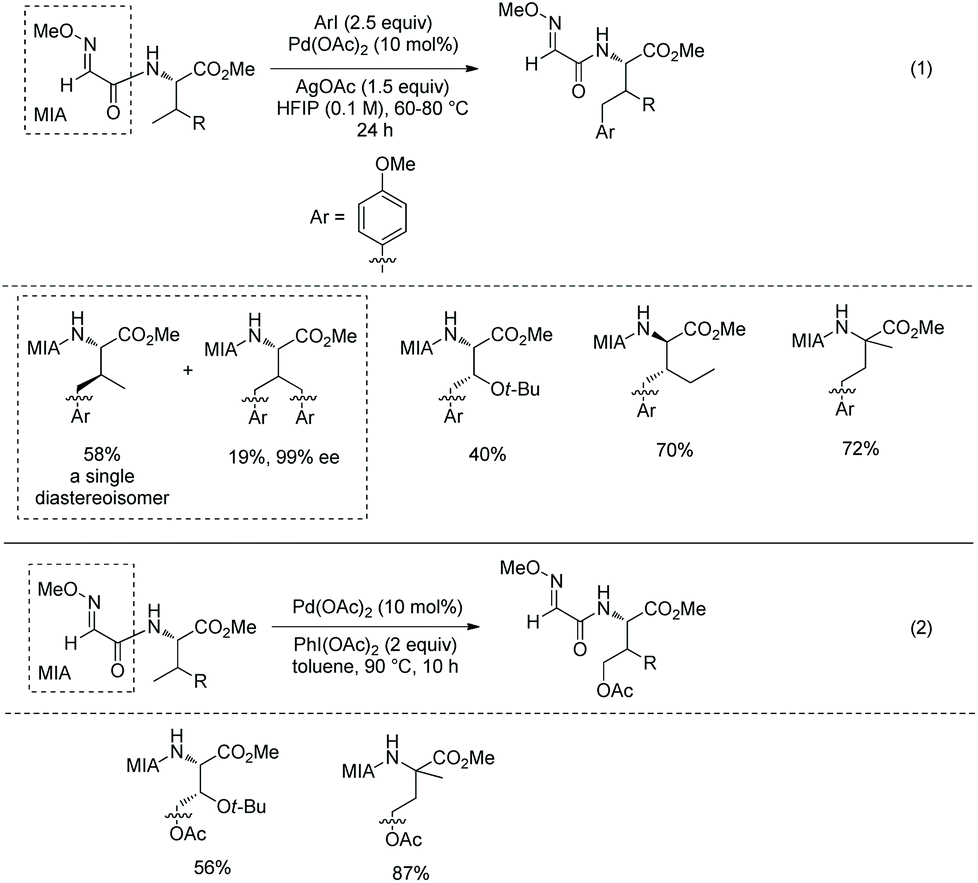
A Pd-catalyzed sp3 C–H alkylation was developed to construct γ/δ-phosphono-α-amino acids (Scheme 17, eqn (1)).13 β-Primary sp3 C–H (in alanine), γ-primary sp3 C–H (in valine) and β-secondary sp3 C–H (such as in Leu, Abu, Cha, Lys, Phe, etc.) can be alkylated smoothly. Diastereoselectivity of alkylation of β-secondary sp3 C–H is excellent (dr is up to >50:1). A phosphono-α-amino amide of 8-aminoquinoline can be converted to the corresponding carboxylic acid by treatment with Boc2O and LiOH, H2O2 (Scheme 17, eqn (2)). Then the Phth (phthaloyl) group is removed by treatment with 6 N HCl, and ester groups are also hydrolyzed. The obtained product is an important intermediate for the synthesis of bioactive molecules.
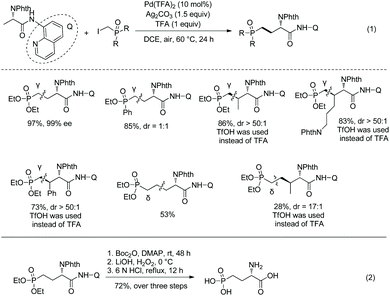 |
| | Scheme 17 Synthesis of phosphono-α-amino acids by alkylation and removal of auxiliary. | |
The mechanism is a Pd(II)/Pd(IV) catalytic cycle, undergoing cyclopalladation, oxidative addition and reductive elimination.
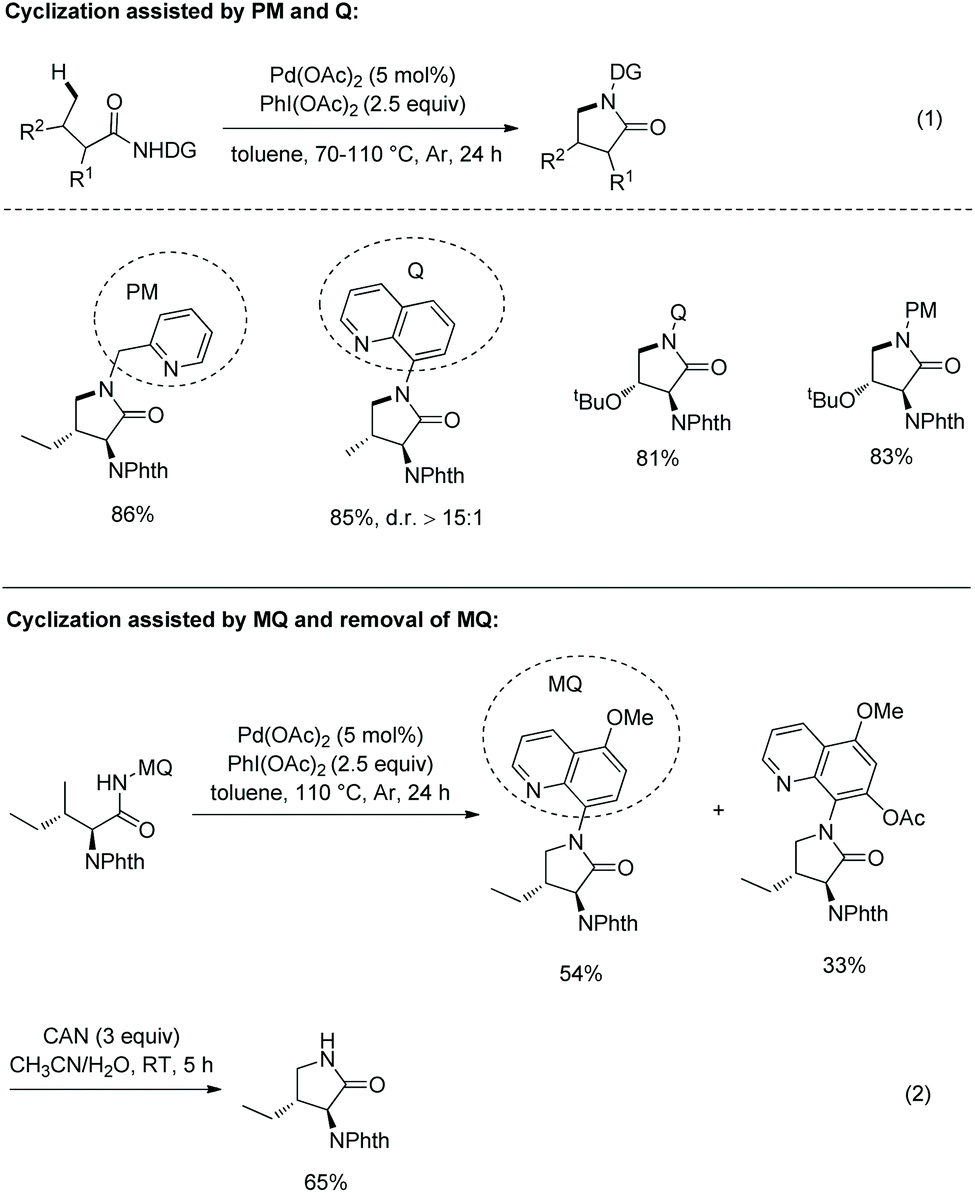
In 2013, 8-aminoquinoline, 5-methoxyquinolin-8-amine or 2-pyridylmethyl amine derived amides of N-phthaloyl protected amino acids were converted to pyrrolidones (γ-lactams) via Pd-catalyzed γ-sp3 C–H activation/intramolecular C–N bond formation (Scheme 18, eqn (1)).14 The auxiliary 5-methoxy-quinolin-8-yl group (MQ) and its derivative can be removed readily by treatment with ceric ammonium nitrate (CAN) at room temperature (Scheme 18, eqn (2)).
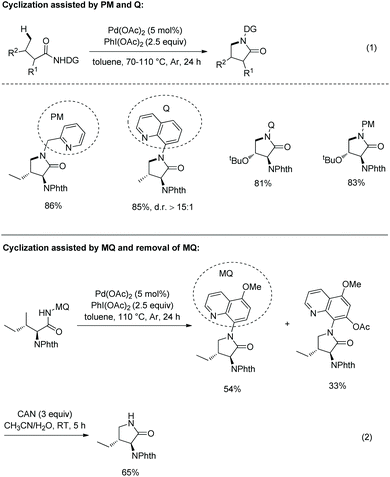 |
| | Scheme 18 Synthesis of pyrrolidones. | |
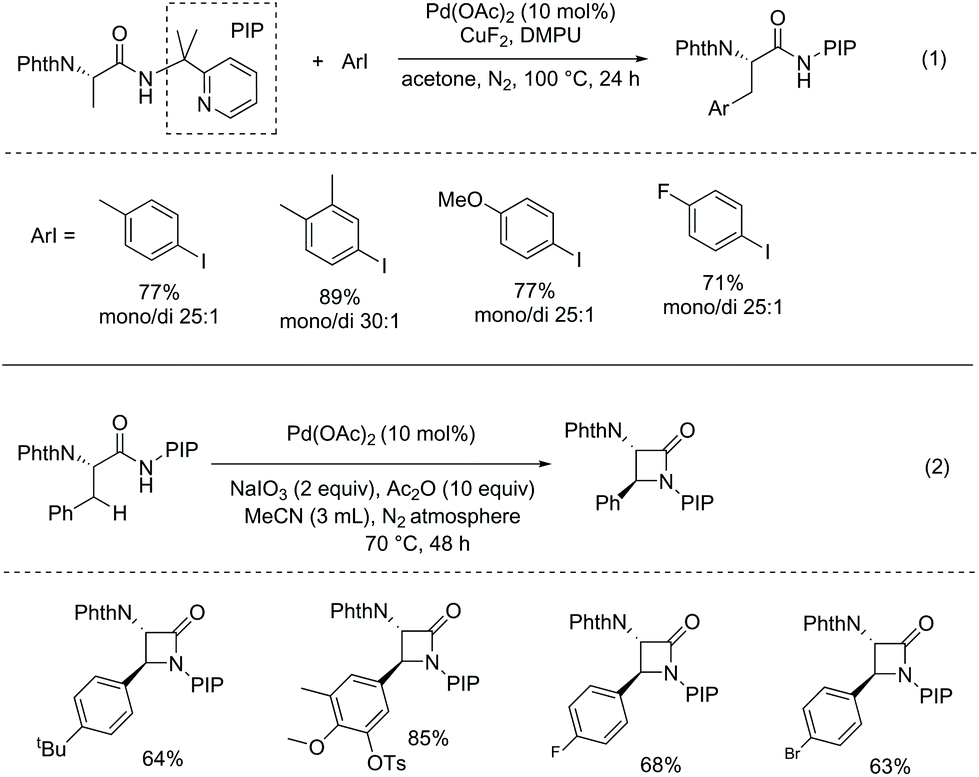
In 2013, Shi and co-workers developed Pd-catalyzed monoarylation of β-C(sp3)–H bonds using N-phthaloyl protected alanine 2-(pyridin-2-yl)propan-2-amine amide as a substrate (Scheme 19, eqn (1)),15 and subsequent C–N coupling cyclization can provide a series of α-amino-β-lactams in acceptable yields (Scheme 19, eqn (2)). The transformations can tolerate many functional groups; however, 2-(pyridin-2-yl)isopropyl (PIP) cannot be removed from the β-lactams.
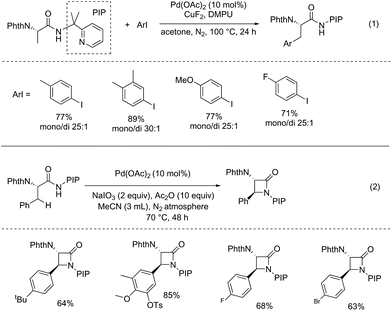 |
| | Scheme 19 Monoarylation and cyclization with PIP. | |
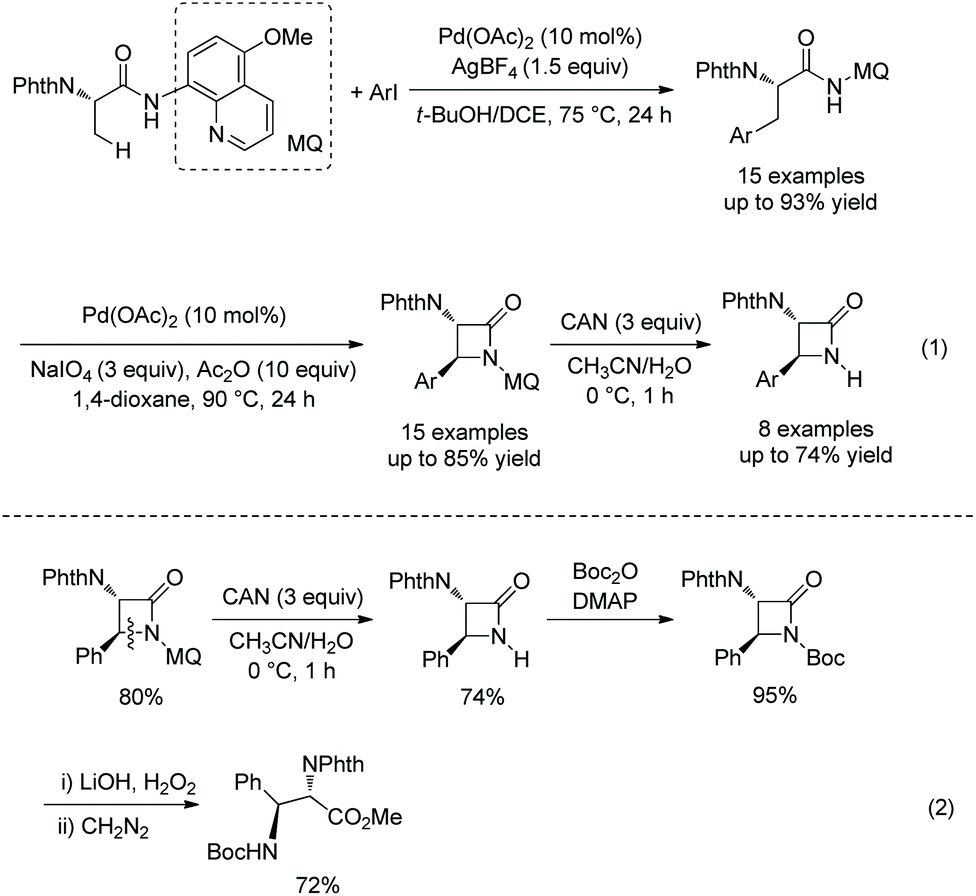
The same group developed another monoarylation of β-C(sp3)–H bonds under palladium catalysis using N-phthaloyl protected alanine 5-methoxyquinolin-8-amine amide as a substrate in 2017 (Scheme 20, eqn (1)),16 a series of α-amino-β-lactams can also be obtained by subsequent intramolecular C–N coupling, and the auxiliary 5-methoxyquinolin-8-yl (MQ) can be removed readily from the β-lactams, which broadens the application of this method in organic synthesis. Orthogonally protected anti-α,β-diamino acids can be prepared from N-unprotected β-lactams (Scheme 20, eqn (2)).
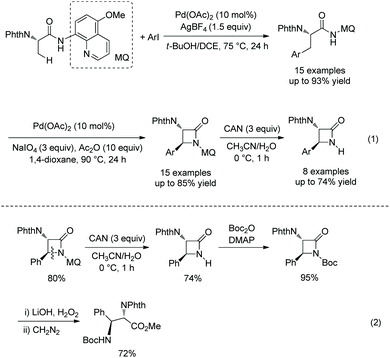 |
| | Scheme 20 Monoarylation and cyclization with MQ. | |
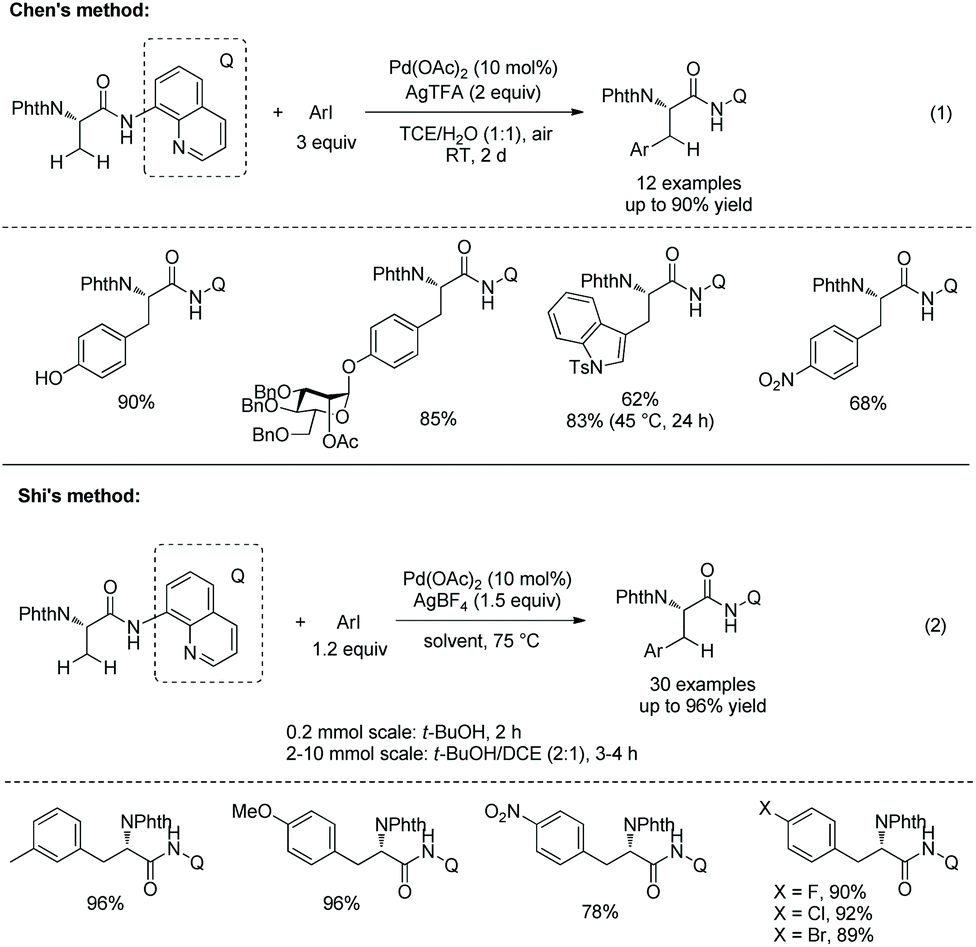
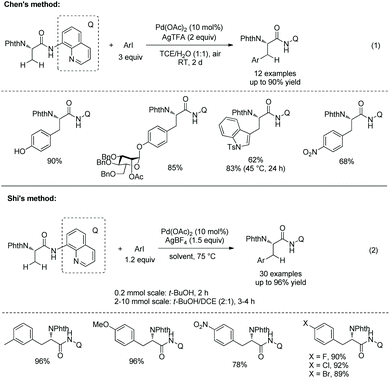
In 2014, Chen's group and Shi's group developed Pd-catalyzed β-monoarylation of N-phthaloyalanine 8-aminoquinoline amide independently (Scheme 21).17,18 The arylation reported by Chen's group proceeds at room temperature, and sensitive groups such as diethyl phosphate and Bn-protected O-mannose can be tolerated (Scheme 21, eqn (1)). Unprotected 4-iodophenol and 5-iodoindole work well. N-Ts-3-iodoindole gives the Trp derivative in good yield (62%) at room temperature, and a higher yield (83%) can be obtained at 45 °C for 24 h.
 |
| | Scheme 21 Monoarylation with Q. | |
Using sequential monoarylation (or monoalkylation)/arylation (or alkylation) by the methods for monoarylation of alanine and the known methods for C–H functionalization of phenylalanine, β,β-disubstituted alanine derivatives can be obtained.17,18 Two diastereomers can be obtained by using different orders of C–H functionalizations.
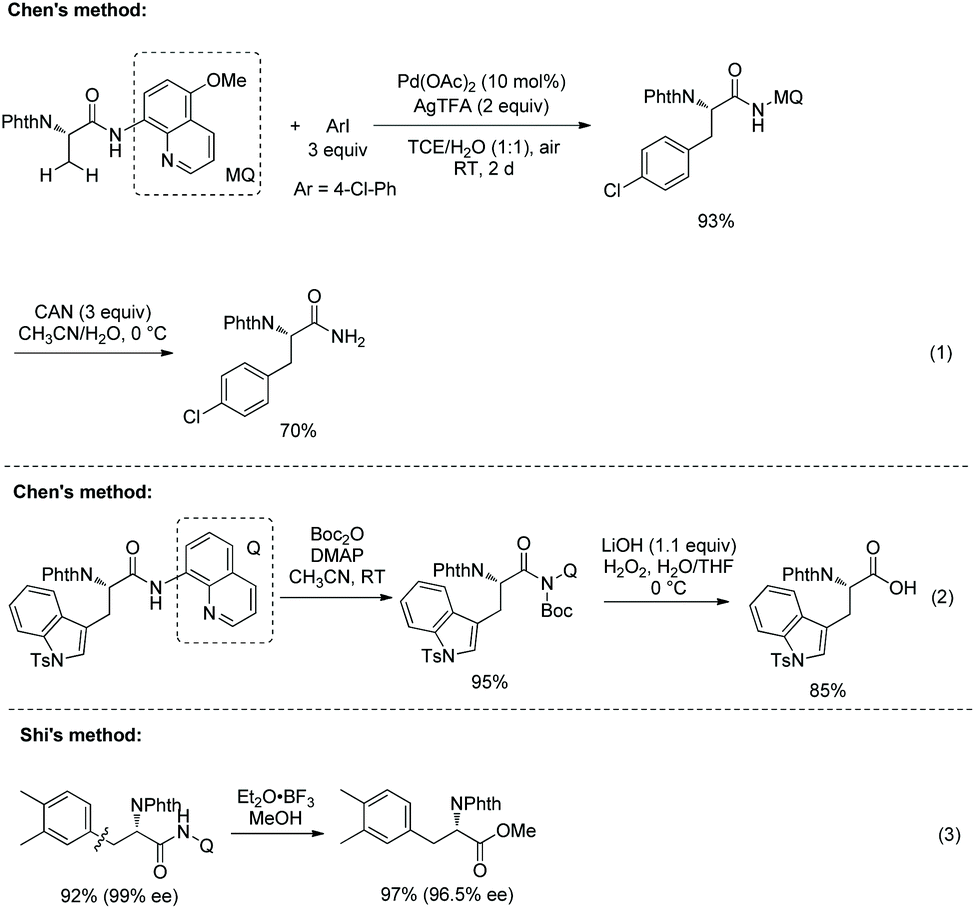
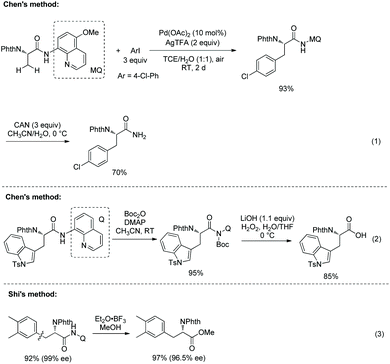
Chen's group investigated the removal of the auxiliaries. C–N cleavage occurs when an arylated N-phthaloyalanine 5-methoxyquinolin-8-amine amide is treated with CAN (cerium ammonium nitrate) (Scheme 22, eqn (1)),17 and a primary amide is obtained. An arylated N-phthaloyalanine 8-aminoquinoline amide is activated at the N atom with Boc and then C–N cleavage gives a free carboxylic acid (Scheme 22, eqn (2)). Shi's group treats an arylated N-phthaloyalanine 8-aminoquinolin amide with Et2O·BF3 in methanol to give the corresponding methyl ester in excellent yield (97%) without obvious change of optical purity (Scheme 22, eqn (3)).18
 |
| | Scheme 22 Removal of auxiliaries by Chen and Shi. | |
The mechanism for monoarylation of N-phthaloyalanine 8-aminoquinoline amide was considered as a Pd(II)/Pd(IV) cycle by Chen's group.17 Cyclopalladation occurs with the assistance of N,N-bidentate coordination, oxidative addition of aryl iodide forms a Pd(IV) complex, and then reductive elimination gives the arylated product and releases the Pd(II) species. Water can promote protonolysis of the arylation product coordinating Pd(II), Ag+ can clean up I−, and TFA (trifluoroacetate) may stabilize the Pd(II) palladacycle as a ligand and promote the oxidative addition with aryl iodide.
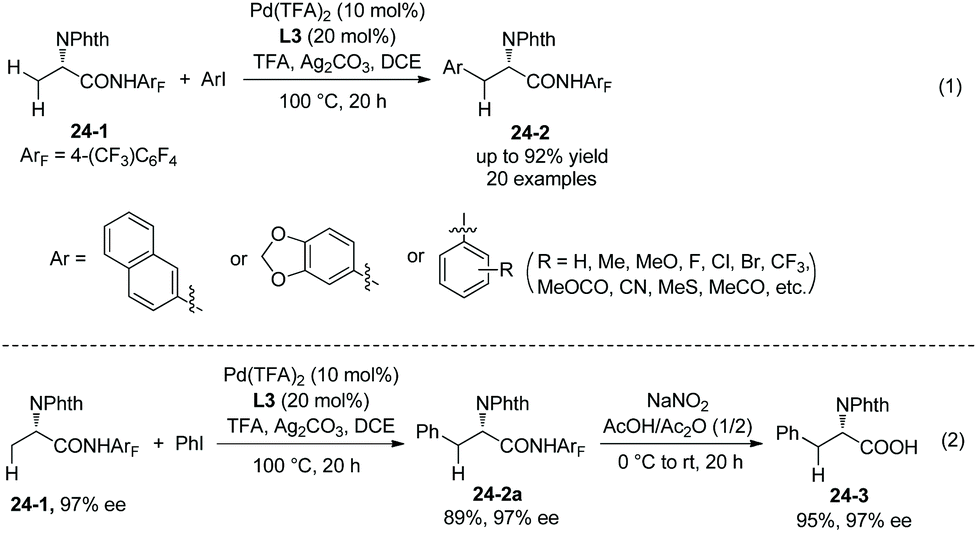
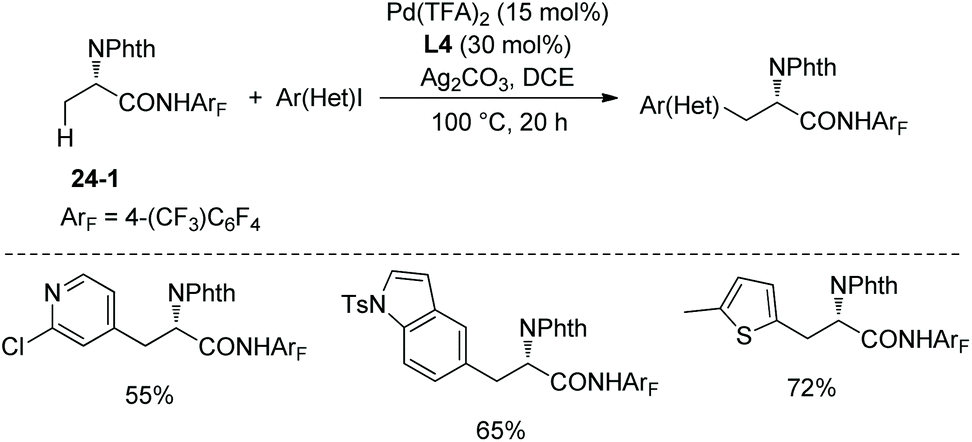
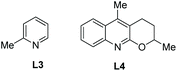
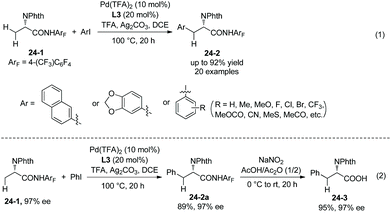
The N,N-bidentate directing group in 8-aminoquinoline amides is employed for C–H functionalization in the above examples, and the N-monodentate directing group in perfluorinated p-toluidine amides was found to be effective for the arylation and olefination of alanine derivative 24-1, as reported by Yu et al. in 2014.19 The mono/diarylation can be controlled by ligands, the monoarylation is enabled by 2-picoline L3 (Scheme 23), and the transformation occurs at β-primary methyl C–H bonds and remaining at unreacted β-secondary methylene C–H bonds (Scheme 24, eqn (1), 24-2). The steric and electronic properties of L3 are just suitable for the first arylation of alanine derivative 24-1. A broad range of electron-donating or -withdrawing functional groups are compatible in the transformation. The monophenylation product 24-2a is obtained in 89% yield and with 97% ee, the ee value remains unchanged during the arylation showing that no racemization occurred. The auxiliary is removed by treatment with sodium nitrite under mild conditions, affording the N-phthaloyalanine 24-3 in 95% yield and with 97% ee, also no racemization occurs.
 |
| | Scheme 23 Ligands by Yu. | |
 |
| | Scheme 24 Monoarylation and removal of auxiliary by Yu. | |
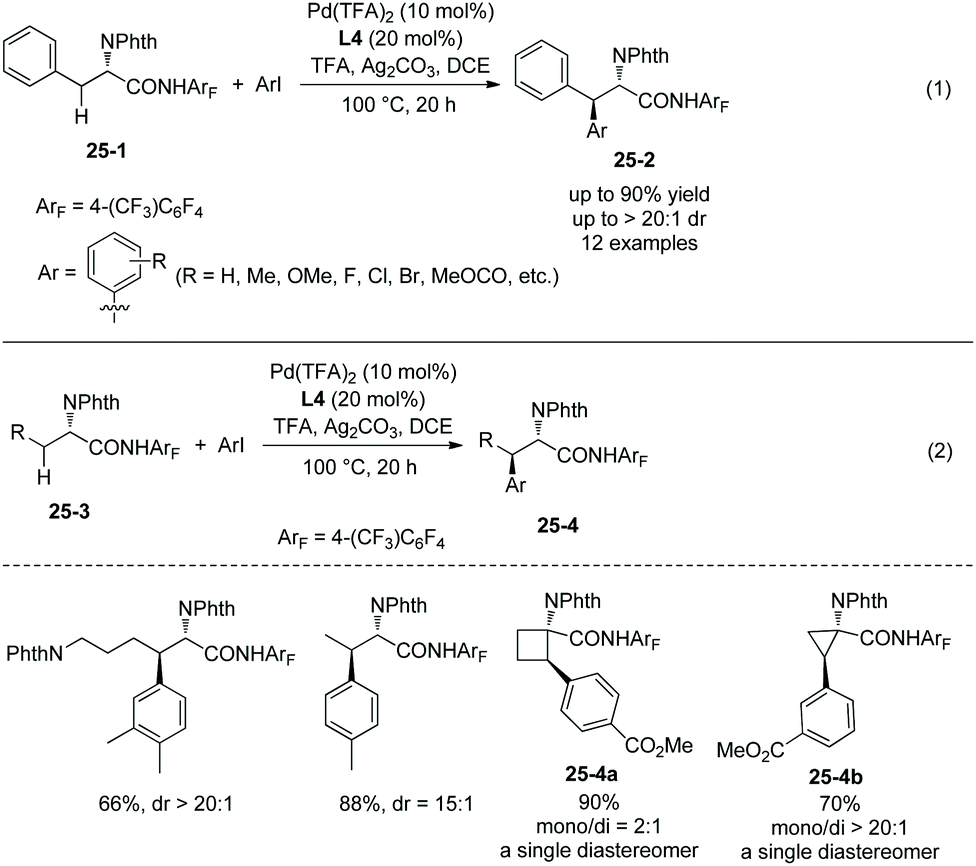
The tricyclic ligand L4 (Scheme 23) can enable the second arylation of the alanine derivative 24-1 (i.e. β-methylene secondary C(sp3)–H arylation of phenylalanine derivative 25-1, Scheme 25, eqn (1))19 or arylation of other amino acid derivatives 25-3 at β-secondary methylene C–H bonds (Scheme 25, eqn (2)), affording diarylation products 25-2 of the alanine derivative 24-1, or arylation products 25-4 of other amino acid derivatives 25-3 in good yields and with high diastereoselectivities. In two cases, the products (25-4a and 25-4b) are single diastereomers. The rigid oxygen of L4 undergoes more readily p–π conjugation with pyridine and enhances the coordinating ability to palladium, and larger catalytic activity of palladium can be produced with the assistance of ligand L4.
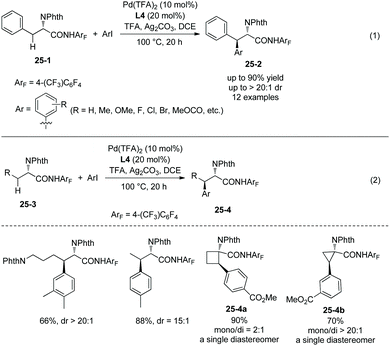 |
| | Scheme 25 Arylation at methylene. | |
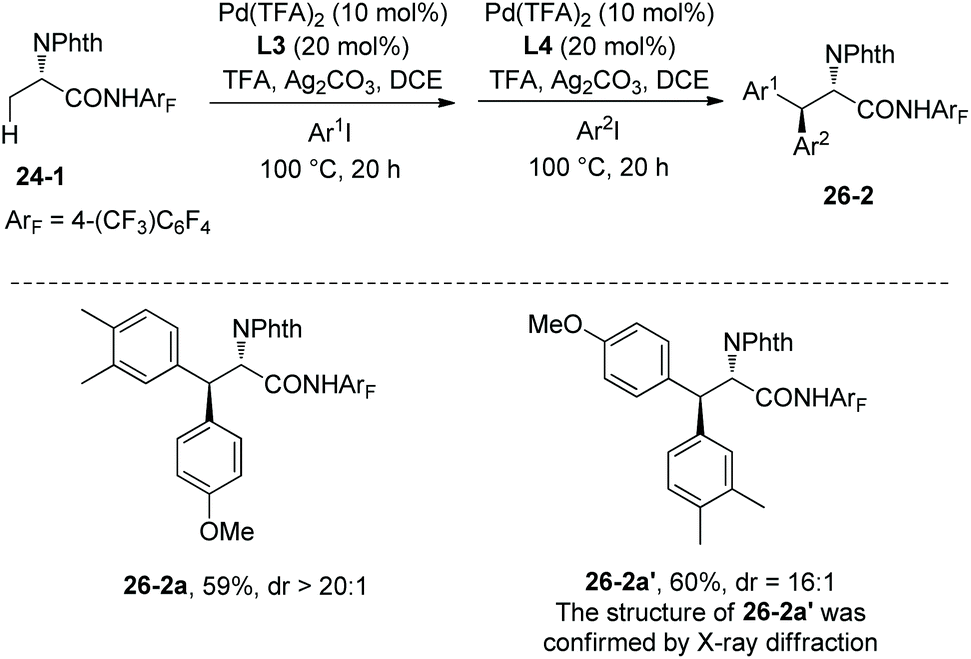
One-pot synthesis of β-Ar1–β-Ar2-alanine derivatives 26-2 was successfully achieved by employing the ligands L3 and L4 one after another. Two diastereomers (26-2a and 26-2a′) can be obtained by different orders of addition of aryl iodides (Scheme 26).19
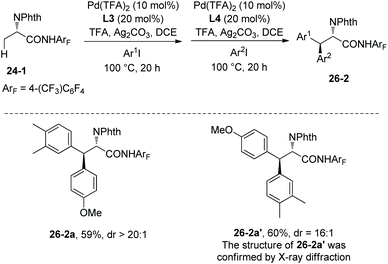 |
| | Scheme 26 One-pot synthesis. | |
Pyridyl, indolyl, and thiophenyl iodides cannot react as a coupling partner in secondary C(sp3)–H arylation of phenylalanine derivative 25-1 with ligand L4, whereas they can act as a coupling partner for primary C(sp3)–H monoarylation of alanine derivative 24-1 with ligand L4, affording arylation products in moderate to high yields (Scheme 27).19
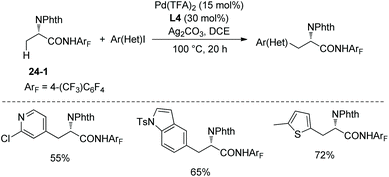 |
| | Scheme 27 Arylation with heteroaryl iodides. | |
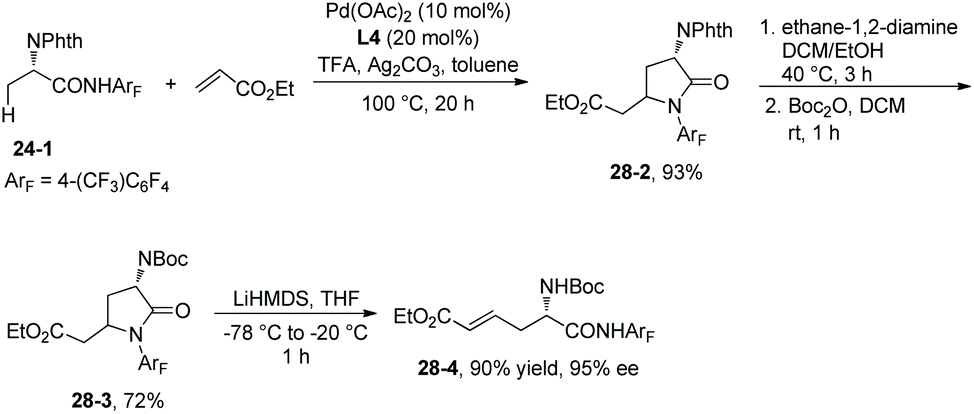
Ligand L4 can also enable the olefination of alanine derivative 24-1, and a lactam 28-2 is produced in 93% yield via olefination and Michael addition in situ. 28-2 is converted to N-Boc-protected lactam 28-3 in 72% yield, which is then converted to a versatile intermediate β-olefinated α-amino acid 28-4 in 90% yield and with 95% ee (Scheme 28).19
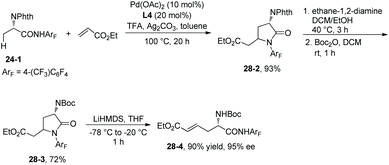 |
| | Scheme 28 Olefination with L4. | |
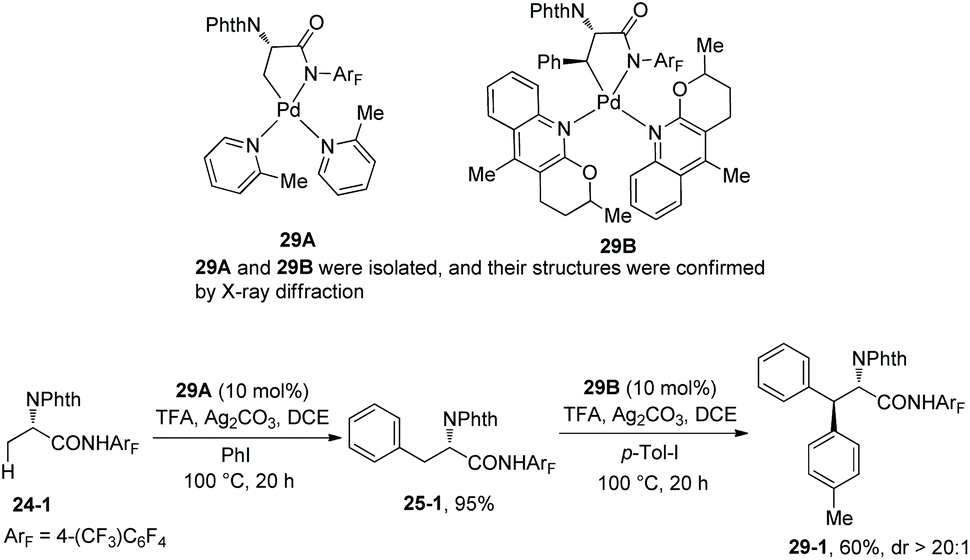
To gain insight into the mechanism, intermediates 29A and 29B were isolated from the reaction of 24-1 with L3 (2 equiv.) and palladium trifluoroacetate (1 equiv.) or the reaction of 25-1 with L4 (2 equiv.) and palladium trifluoroacetate (1 equiv.) respectively (Scheme 29).19 Then 29A and 29B were successfully used as a catalyst for arylation of 24-1 and 25-1, respectively, the diarylation product 29-1 was obtained, which indicates that palladacycles are formed during the arylations and ligands L3 and L4 play crucial coordinating roles in primary C(sp3)–H or secondary C(sp3)–H functionalization, respectively. It also shows that both the intermediates 29A and 29B undergo further oxidative addition and reductive elimination, and the two arylations proceed via Pd(II)/Pd(IV) catalytic cycles.
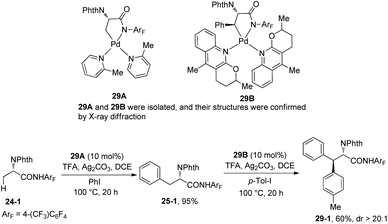 |
| | Scheme 29 Mechanistic studies. | |
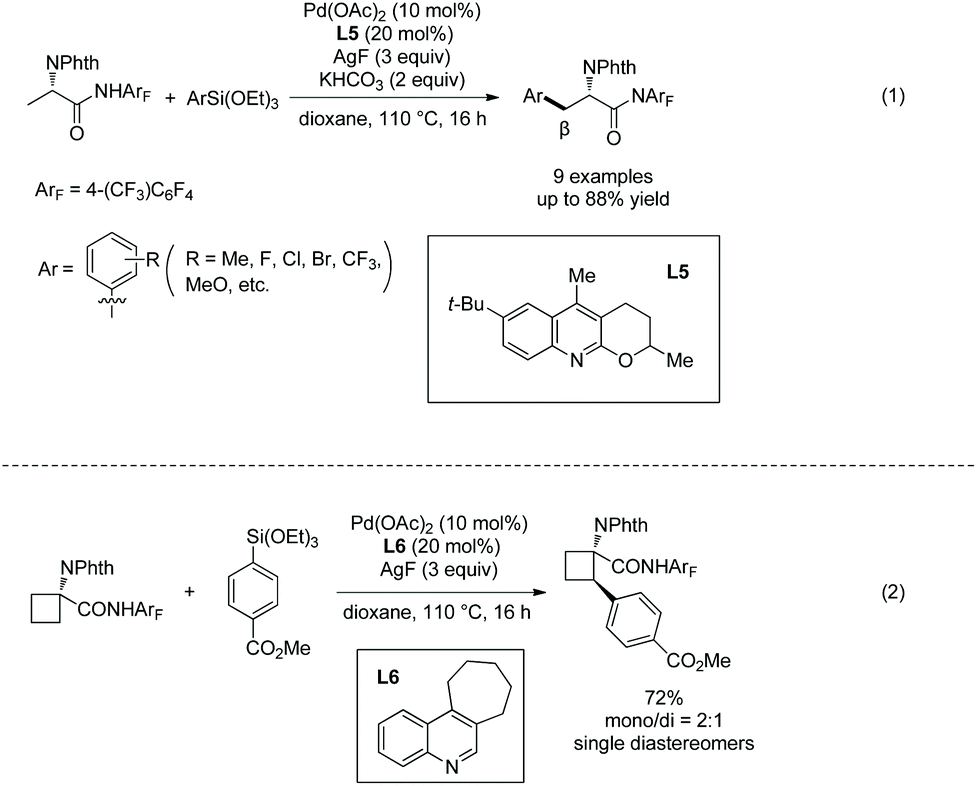
The arylating reagents arylsilanes can also be coupled with alanine derivatives under palladium catalysis successfully, and perfluorinated p-toluidine amides are also employed as an N-monodentate directing group (Scheme 30).20 β-Methyl of the alanine derivative is arylated in up to 88% yield (Scheme 30, eqn (1)). β-Methylene of 1-aminocyclobutane-1-carboxylic acid can also be arylated, affording the arylated product in 72% yield (Scheme 30, eqn (2)).
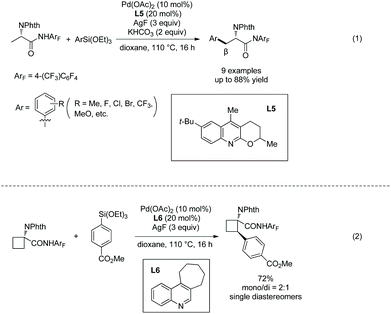 |
| | Scheme 30 Arylation with arylsilane. | |
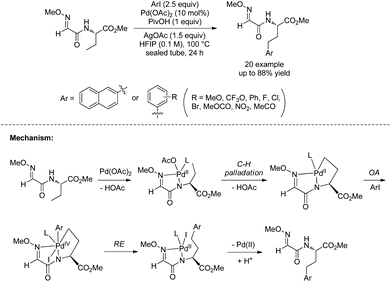
The same group used the above N-monodentate directing group (perfluorinated p-toluidine amides) to process γ-sp3 C–H arylation of valine, isoleucine and tert-leucine (Scheme 31, eqn (1)).21 The arylated products are produced in high to excellent yields and with high diastereoselectivity. The groups of F, Cl, Br, Me, Ph, OMe, OAc, NHBoc, MeO2C, MeCO, CF3, etc. are tolerated in this arylation. Phenyl iodides, 2-iodonaphthalene, 4-iodo-2-(trifluoromethyl)pyridine and ethyl 6-iodo-4-oxo-4H-chromene-2-carboxylate all work well in the transformation. The amide auxiliary can be removed and a thioester or free carboxylic acid can be obtained in high yield (Scheme 31, eqn (2) and (3)). Phth can also be removed and Boc-protected amine can be provided (Scheme 31, eqn (3)).
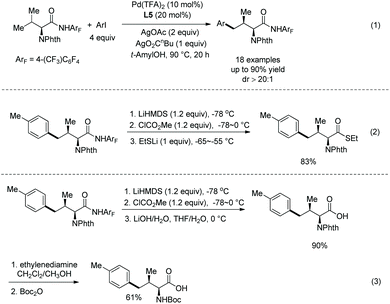 |
| | Scheme 31 Arylation of valine and removal of auxiliary. | |
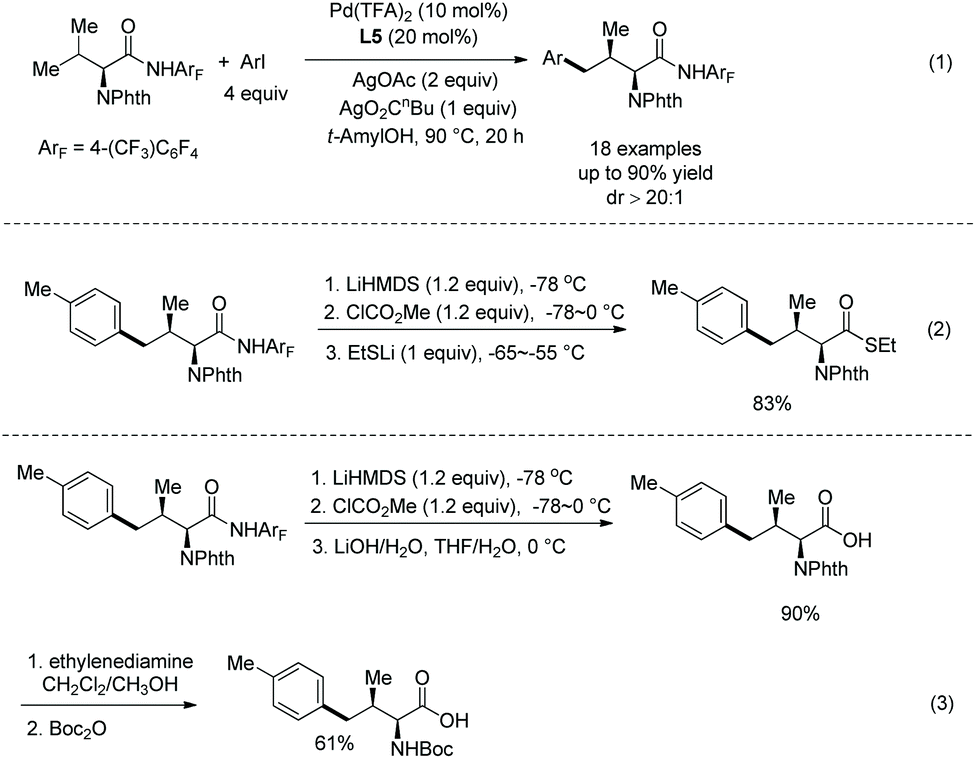
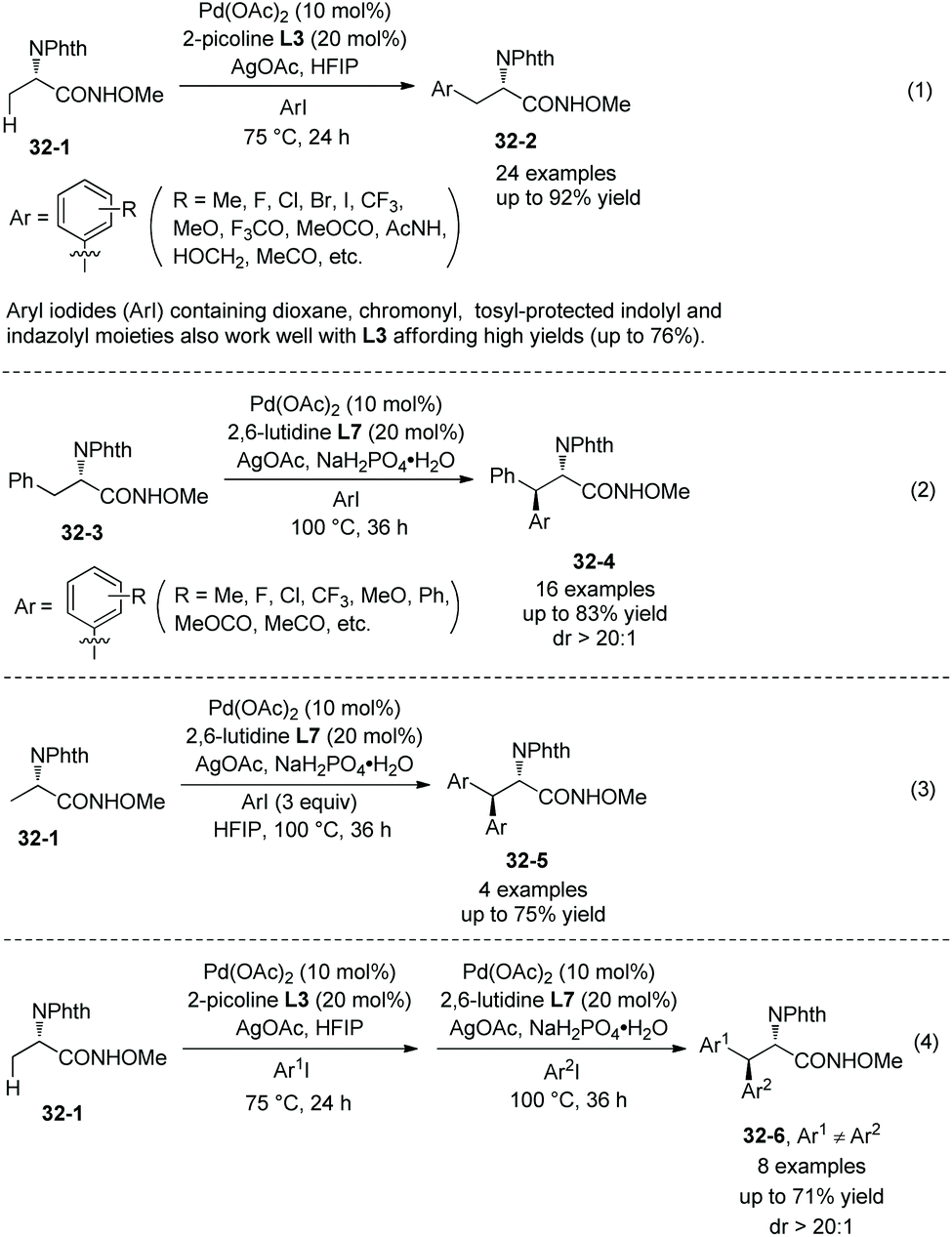
Another N-monodentate directing group, which is in N-methoxyamides, was also found to be effective for the arylation of alanine derivative 32-1 and phenylalanine derivative 32-3 (Scheme 32, eqn (1) and (2)).22 Monoarylation of 32-1 and β-methylene secondary C(sp3)–H arylation of 24-3 can be enabled by ligand L3 (2-picoline) and ligand L5 (2,6-lutidine), respectively, affording β-arylalanine derivative 32-2 and β-arylphenylalanine derivative 32-4 in good to excellent yields, and the diastereoselectivities of the arylation of 32-3 are high. Aryl iodides (ArI) containing dioxane, chromonyl, tosyl-protected indolyl and indazolyl moieties also work well with L3 affording 32-2 in high yields (up to 76%). Homodiarylation of alanine derivative 32-1 can be accomplished with the assistance of ligand L5 (2,6-lutidine), affording the β,β-diarylalanine derivatives 32-5 in high yields (Scheme 32, eqn (3)). Heterodiarylation of alanine derivative 32-1 can also be accomplished by two successive arylations with two different aryl iodides in a one-pot manner, and β-aryl1–β-aryl2 alanine derivatives 32-6 can be obtained in good yields and with high diastereoselectivities (Scheme 32, eqn (4)). Two diastereomers can be prepared respectively by switching the arylation order of Ar1I and Ar2I.
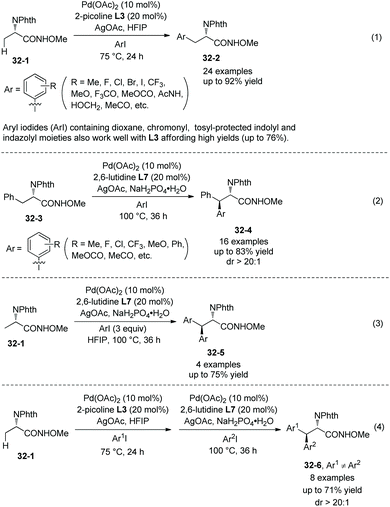 |
| | Scheme 32 Arylation by Yu. | |
Strong coordinative abilities of heteroatoms in heterocycles for palladium cause catalyst poisoning. When pyridyl iodides are coupled with 32-1 under the standard conditions (Scheme 33, eqn (1)), the arylation is not effective, the yields are not more than 20%. But switching the ligand L3 (2-picoline) to ligand L5 (2,6-lutidine), the arylation with pyridyl, quinolinyl and quinoxalinyl iodides can work well except for 4-pyridyl iodide, affording the products 32-7 in synthetically useful yields (Scheme 33).22
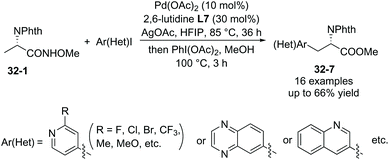 |
| | Scheme 33 Arylation with pyridyl iodides. | |
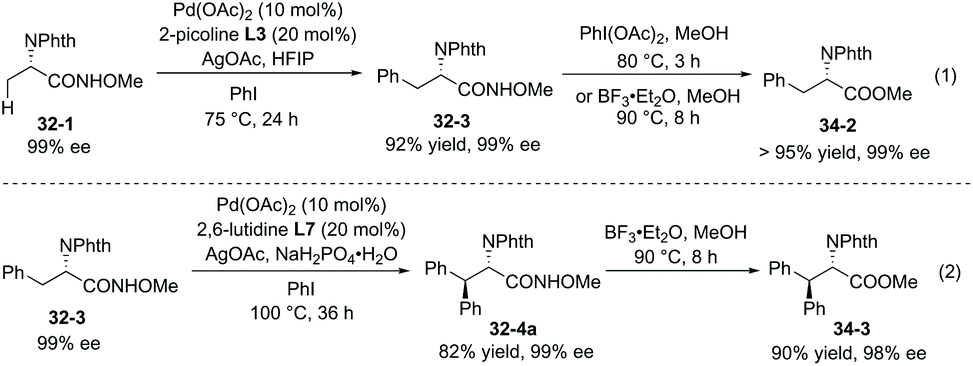
The enantiopurity of the α-carbon is retained in arylation and removal of the auxiliary (Scheme 34).22 Removal of the auxiliary in monoarylation products (32-3) was achieved by treatment of PhI(OAc)2 or BF3·Et2O, and indole-containing amides cannot work efficiently using PhI(OAc)2. The ester 34-2 is afforded in excellent yield. Removal of the auxiliary in diarylation products 32-4a can be achieved by treatment of BF3·Et2O, affording the ester 34-3 in excellent yield.
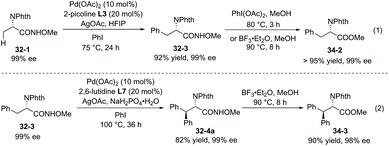 |
| | Scheme 34 Removal of the auxiliary NHOMe. | |
In 2014, Chen's group developed Pd-catalyzed olefination of alanine using 8-aminoquinoline as a directing group (Scheme 35, eqn (1)).23 The transformation proceeds at room temperature and can avoid cyclization via Michael addition of the olefinated products. Iodinated terminal and internal olefins all can be coupled with alanine, and the steric configurations of olefins are retained in products. TIPS protected acetylene bromide also works well but at a higher temperature 65 °C. The olefinated products (β-vinyl alanines) can be further converted to γ-lactams by intramolecular Michael addition at room temperature (Scheme 35, eqn (2)). By contrast, the olefination reported by Yu employs terminal olefins, and only cyclization products are produced.19 The directing auxiliary in olefinated and alkynylated products can be removed under mild conditions by Boc activation and C–N cleavage (Scheme 36).
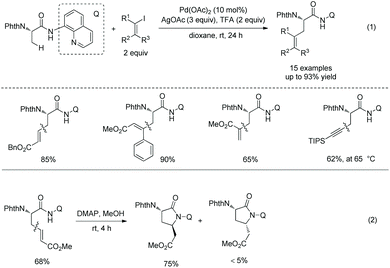 |
| | Scheme 35 Olefination by Chen. | |
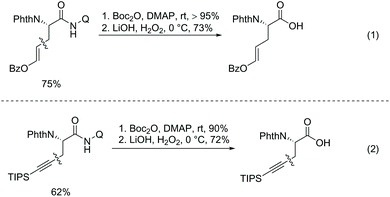 |
| | Scheme 36 Removal of auxiliary by Chen. | |
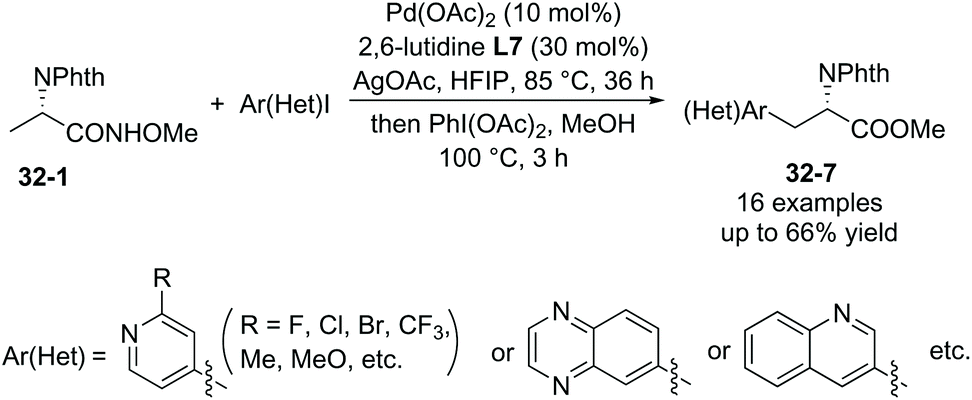
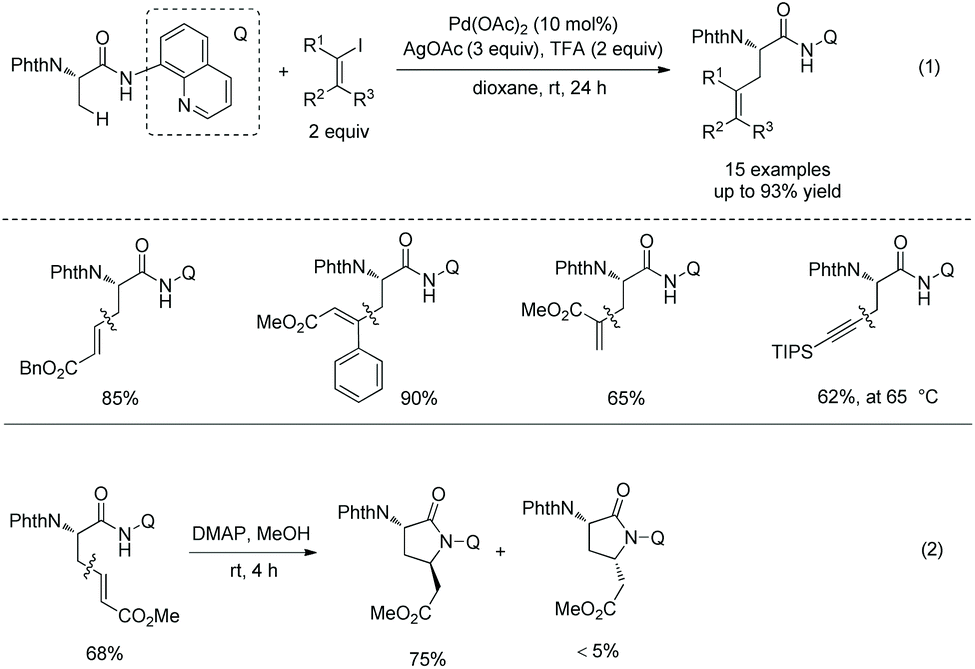
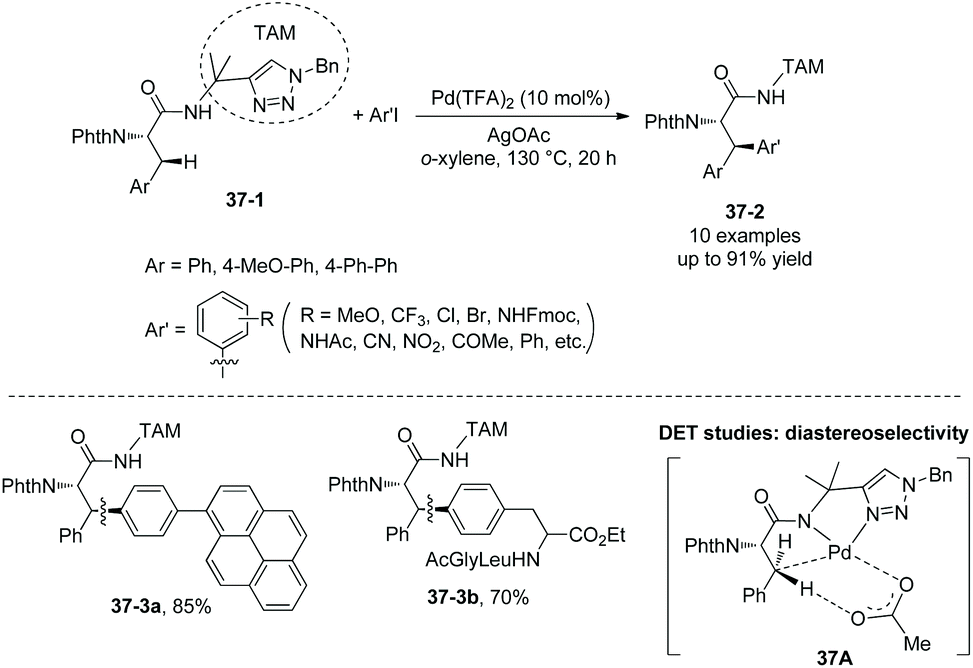
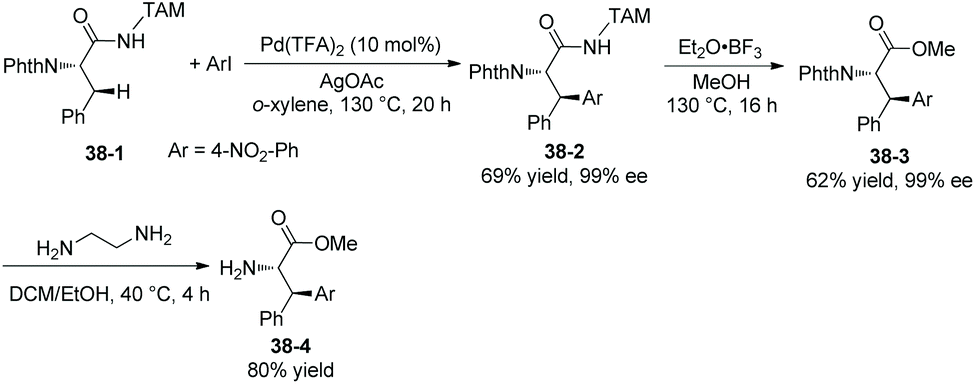
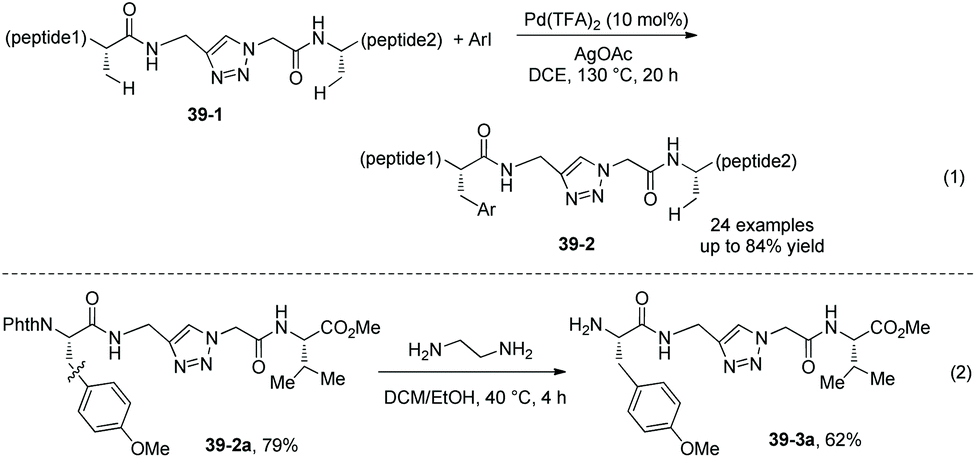
In 2018, a triazole moiety incorporated into amino acids (e.g.37-1) or peptides enables β-sp3 C–H arylation with high chemo/site-selectivity and diastereoselectivity (dr >20:1) (Schemes 37 and 39).24 A wide range of electrophiles with either electron-withdrawing or electron-donating groups were proved to be compatible with the arylation. A fluorescent moiety and iodophenylalanine derivatives are successfully coupled with Phe (e.g.37-3a and 37-3b). A trans palladacycle will be formed diastereoselectively from 37A according to density functional theory (DFT) studies. No racemization at the chiral centers is observed. The TAM auxiliary and N-protecting group Phth can be removed conveniently in two steps (Scheme 38); finally the N-unprotected β-arylphenylalanine methyl ester 38-4 is obtained.
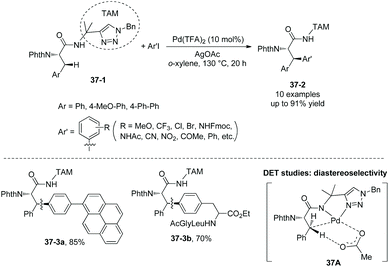 |
| | Scheme 37 Arylation with TAM. | |
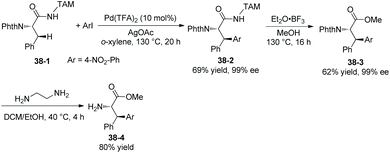 |
| | Scheme 38 Removal of the TAM auxiliary and Phth. | |
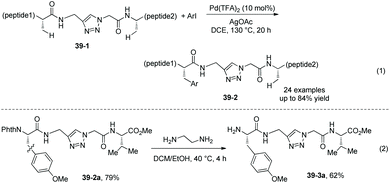 |
| | Scheme 39 Arylation of peptides and removal of Phth. | |
Arylation of the peptides containing the internal triazole 39-1 is also developed, affording the arylated peptides 39-2 in moderate to high yields (Scheme 39, eqn (1)).24 The N-protecting group Phth in an arylated peptide (39-2a) is removed under mild conditions, affording the N-unprotected arylated peptide 39-3a in 62% yield (Scheme 39, eqn (2)).
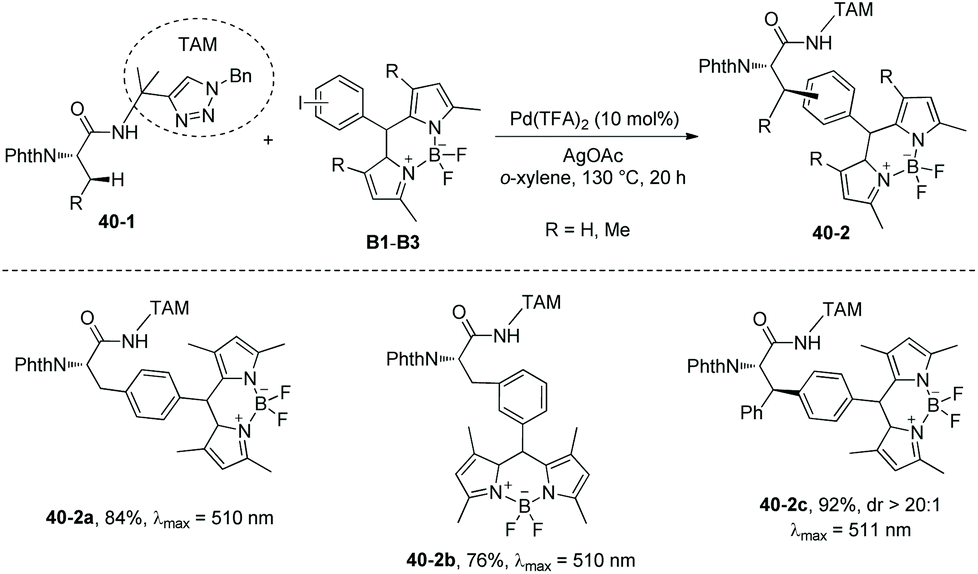
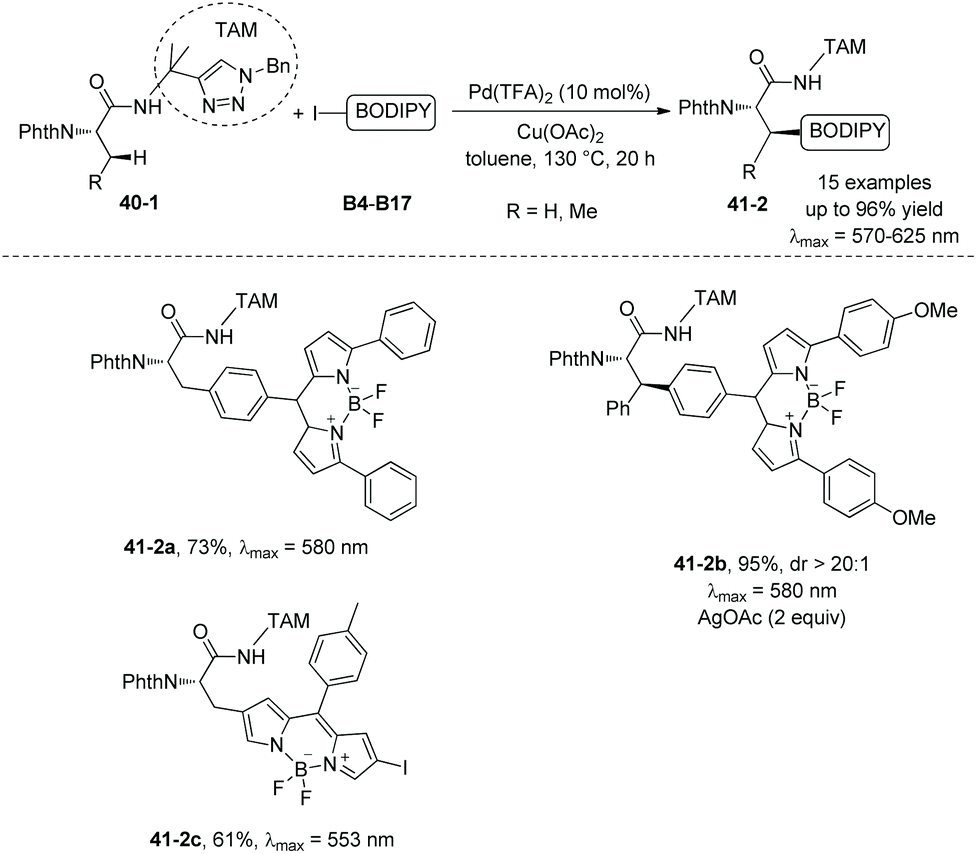
In 2018, the same group used the triazole moiety for BODIPY amino acid/peptide labeling via sp3 C–H activation,25 affording peptidomimetic BODIPY sensors (Schemes 40 and 41). The BODIPY fluorescent dye units are incorporated onto Ala and Phe successfully in good to excellent yields, and β-primary methyl sp3 C–H and secondary methylene sp3 C–H bonds are functionalized. Phe is labeled with excellent diastereoselectivity (e.g.40-2c and 41-2b). The BODIPY labeling proceeds without racemization of the chiral centers of amino acids. Seventeen electron-poor or -rich BODIPYs are labeled successfully. Iodine is tolerated in the labeling (e.g.41-2c, Scheme 41).
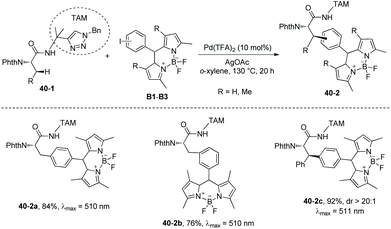 |
| | Scheme 40 BODIPY labeling with B1–B3. | |
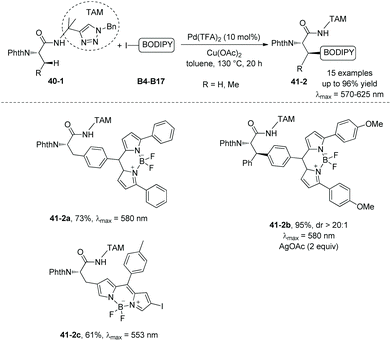 |
| | Scheme 41 BODIPY labeling with B4–B17. | |
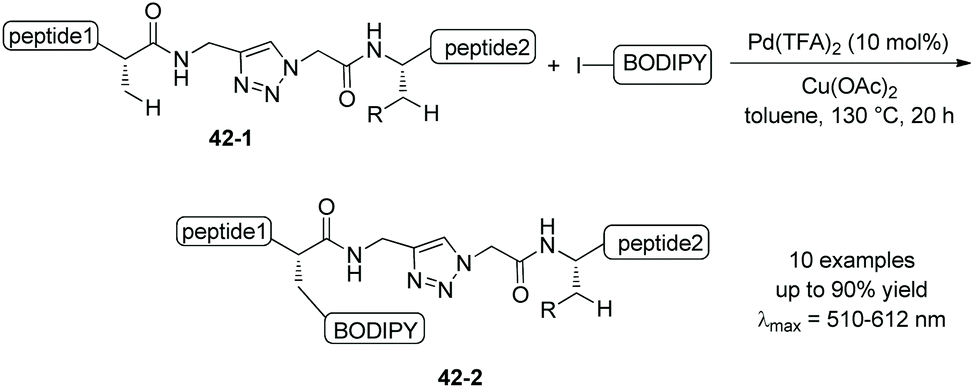
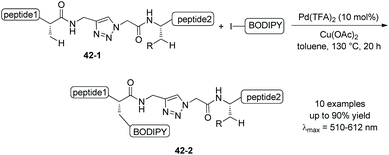
Fluorescently labeled peptides 42-2 also can be prepared via triazole-assisted β-sp3 C–H activation from the substrates with triazole embedded 42-1 (Scheme 42).25
 |
| | Scheme 42 Fluorescently labeled peptides. | |
The BODIPY-labeled parent amino acids and peptides can be obtained by treatment of 40-2, 41-2 or 42-2 with BF3·Et2O or ethylenediamine.25
In 2017, Yu's group developed unprotected carboxylate-directed Pd-catalyzed sp3 C–H arylation of α-amino acids, and ligands are crucial to these transformations (Scheme 43).26 β-sp3 C–H bonds of alanine derivatives are arylated smoothly, and a wide range of functional groups are compatible with the transformation (Scheme 43, eqn (1)). The tert-Leucine derivative can be arylated at the γ-position (Scheme 43, eqn (2)). The carboxyl group acts as a directing group, and no exogenous auxiliary needs to be installed, which makes the synthetic route of arylation of amino acids shorter. The transformation undergoes carboxylate-directed cyclopalladation (when the substrate is Ala, a five-membered ring palladacycle is formed), oxidative addition of the palladacycle with aryl iodides, and reductive elimination forming a new Ar–C(sp3) bond.
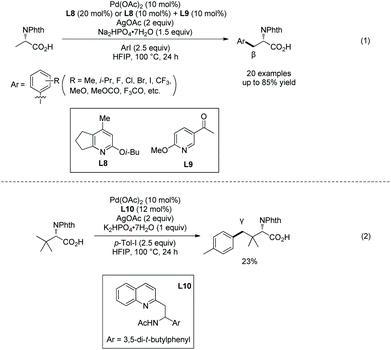 |
| | Scheme 43 Carboxylate-directed arylation by Yu. | |
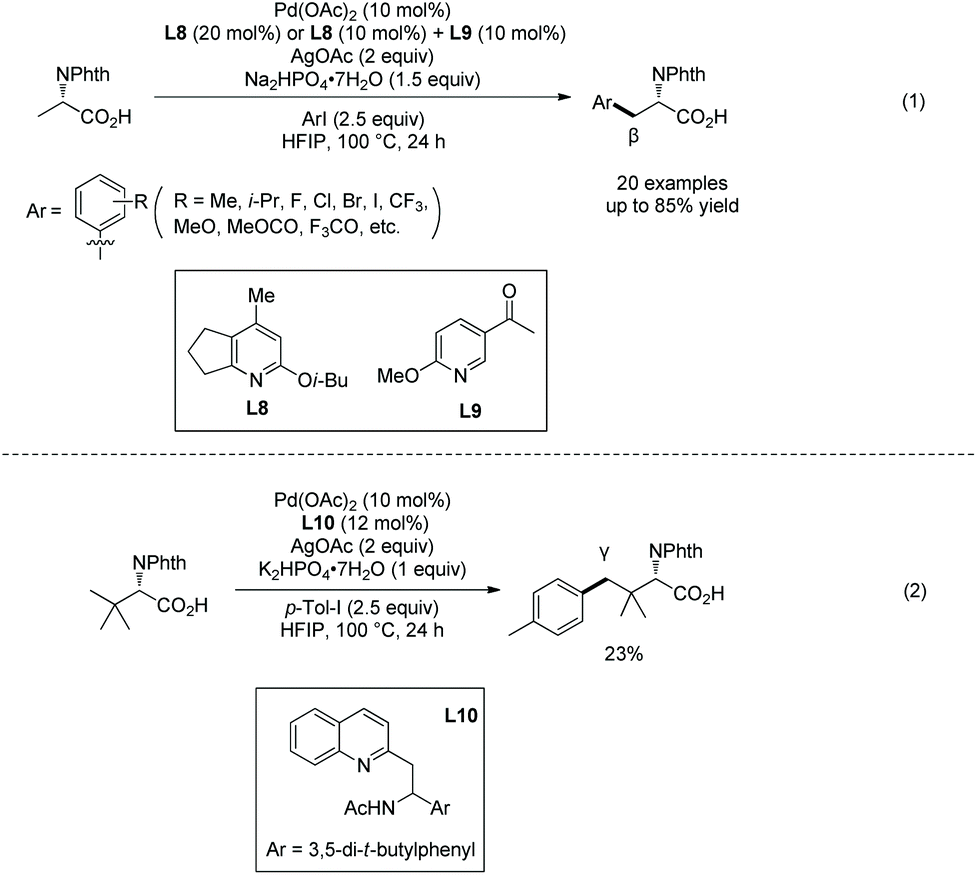
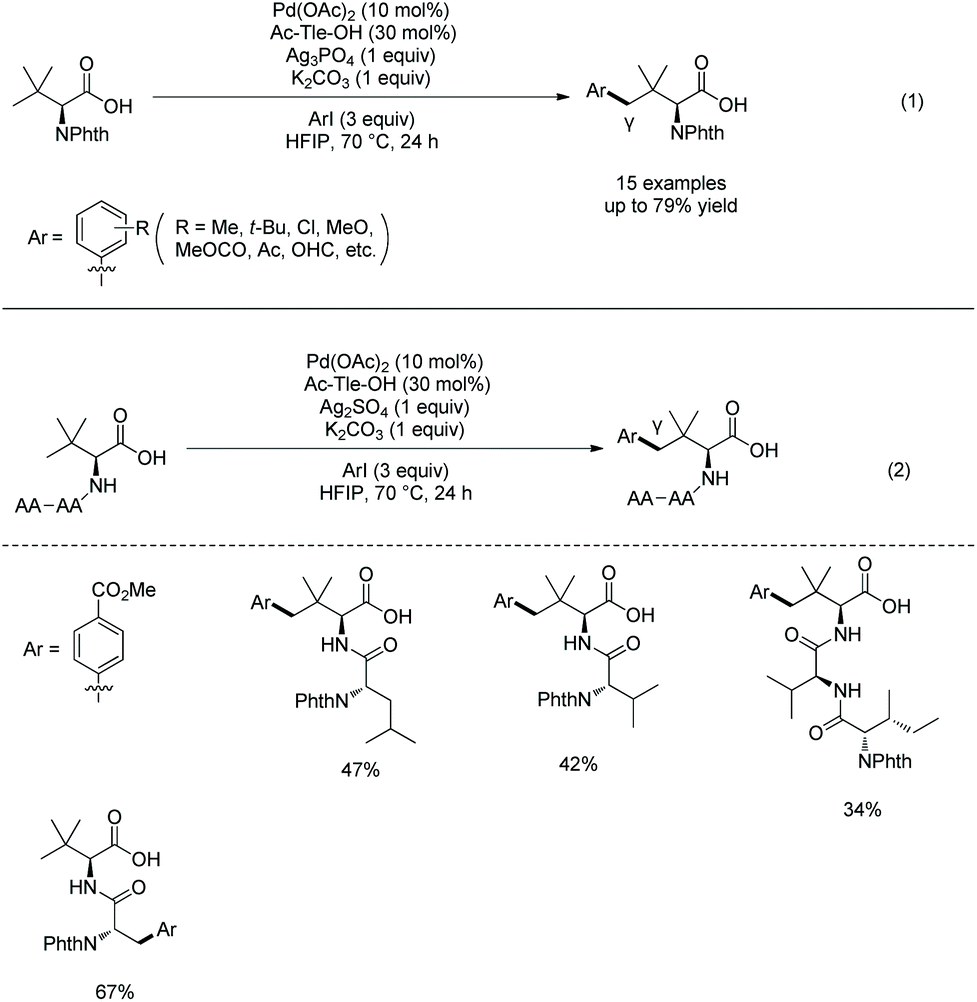
In 2020, Shi's group found that N-acetyl-tert-leucine as a ligand could promote significantly carboxylate-directed γ-C(sp3)–H arylation of tert-leucine and its peptide derivatives under palladium catalysis (Scheme 44).27 A six-membered ring palladacycle is formed during the arylation. The arylated tert-leucine derivatives can serve as a catalyst for Co-catalyzed enantioselective sp3 C–H amidation. When the N-termini are Val, Ile and Tle residues, the C-terminal Tle residue of di- or tripeptides can be arylated at the γ-position (Scheme 44, eqn (2)), and backbone-directed γ-C(sp3)–H arylation does not occur at N-termini. When N-Phth-Ala-Tle-OH is used as the substrate, arylation occurs at the β-position of the N-terminal Ala residue, which results from N,O-bidentate ligand-directed sp3 C–H palladation, forming a five-membered ring palladacycle.
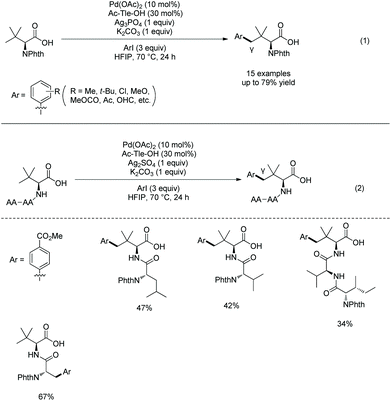 |
| | Scheme 44 Carboxylate-directed arylation by Shi. | |
2.1.2. C–F bond formation.
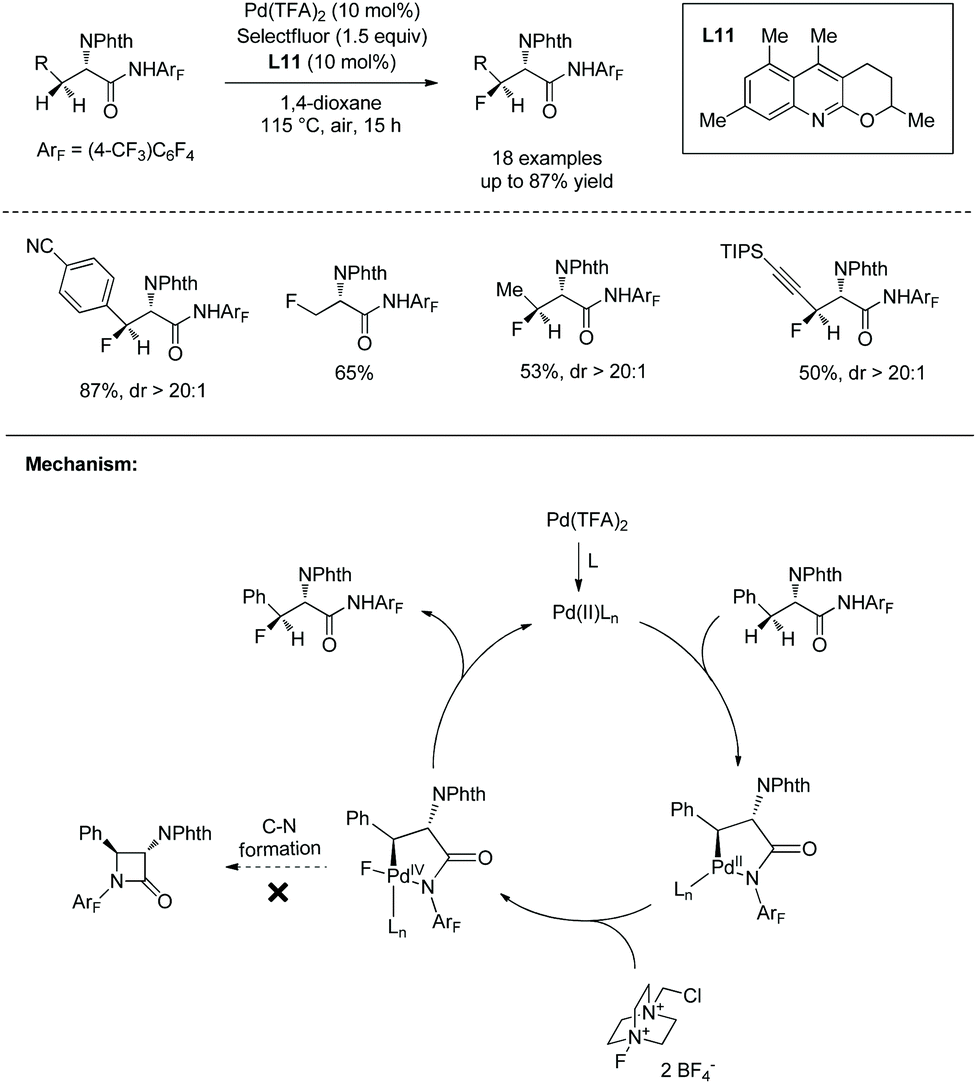
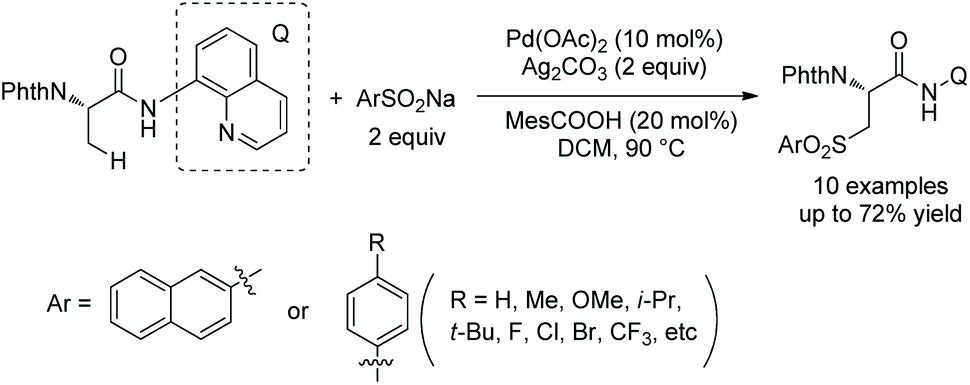
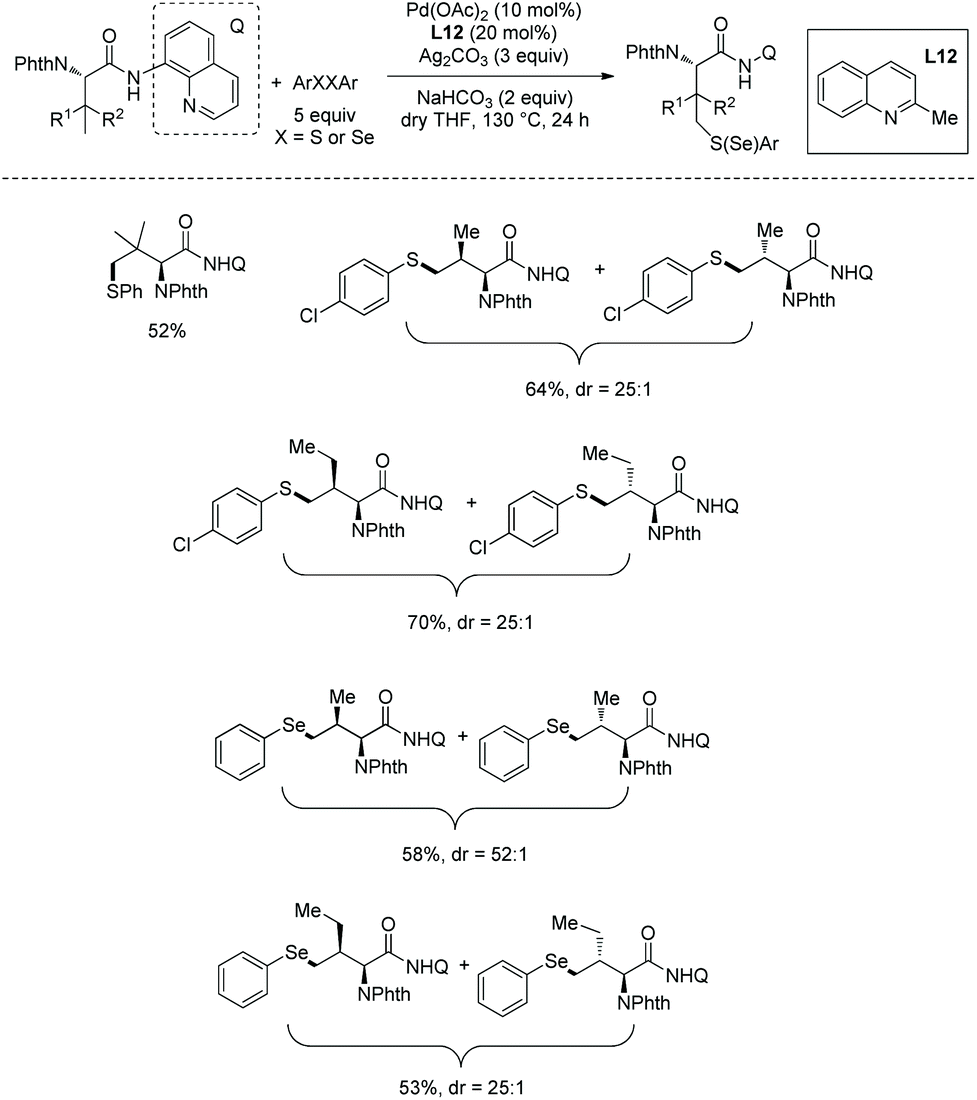
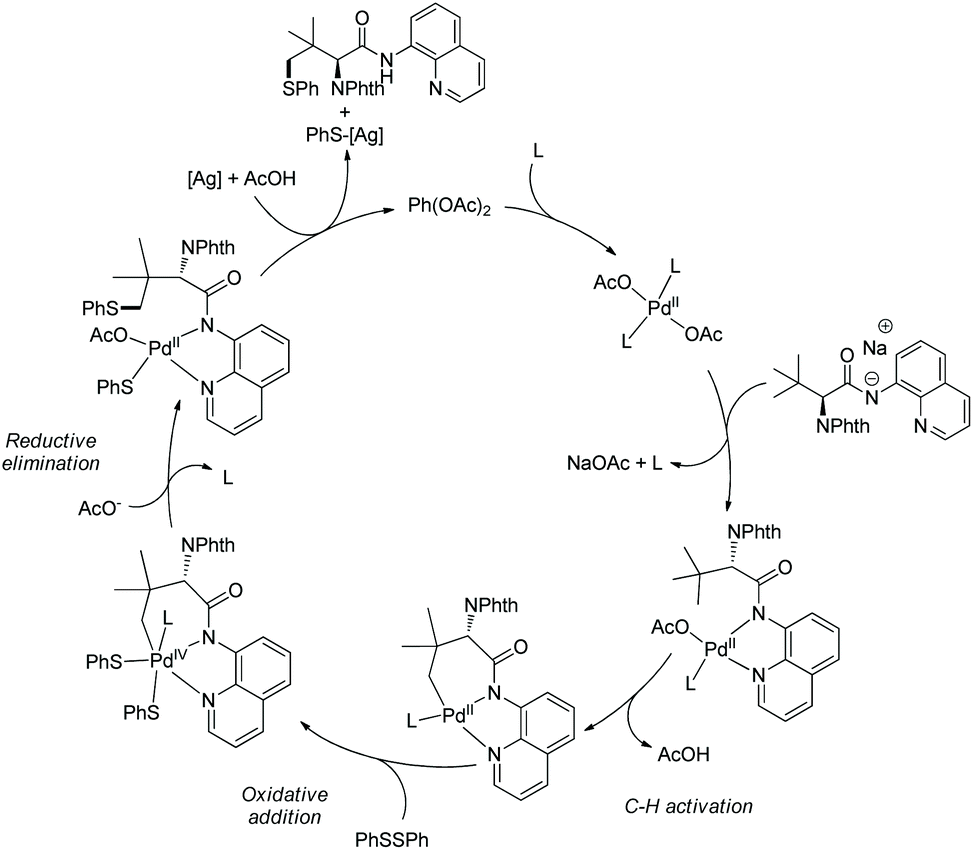
In May 2015, Yu's group made use of perfluorinated p-toluidine amides to achieve β-C–H fluorination of amino acids (Scheme 45).28 β-Methyl of the alanine derivative is fluorinated in 65% yield. Benzylic, aliphatic, and propargylic methylenes all work well, affording the fluorinated product in up to 87% yields and with high diastereoselectivities (dr >20:1). Many functional groups are compatible with this transformation, such as F, Cl, Br, MeOCO, Ac, NC, NO2, CF3, pyridyl, etc. The mechanism includes cyclopalladation forming a five-membered ring palladacycle, oxidative addition to form a F-ligated Pd(IV) complex, and reductive elimination affording the product. The ligand is considered to promote the C–F bond forming reductive elimination, and intramolecular C–N bond forming reductive elimination does not occur.
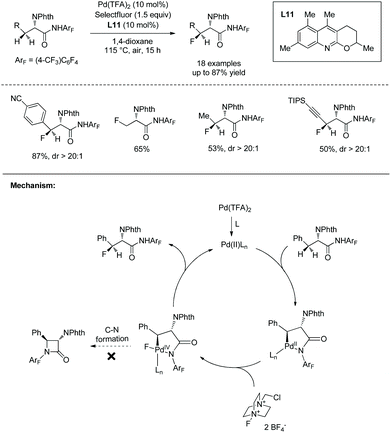 |
| | Scheme 45 Fluorination by Yu. | |
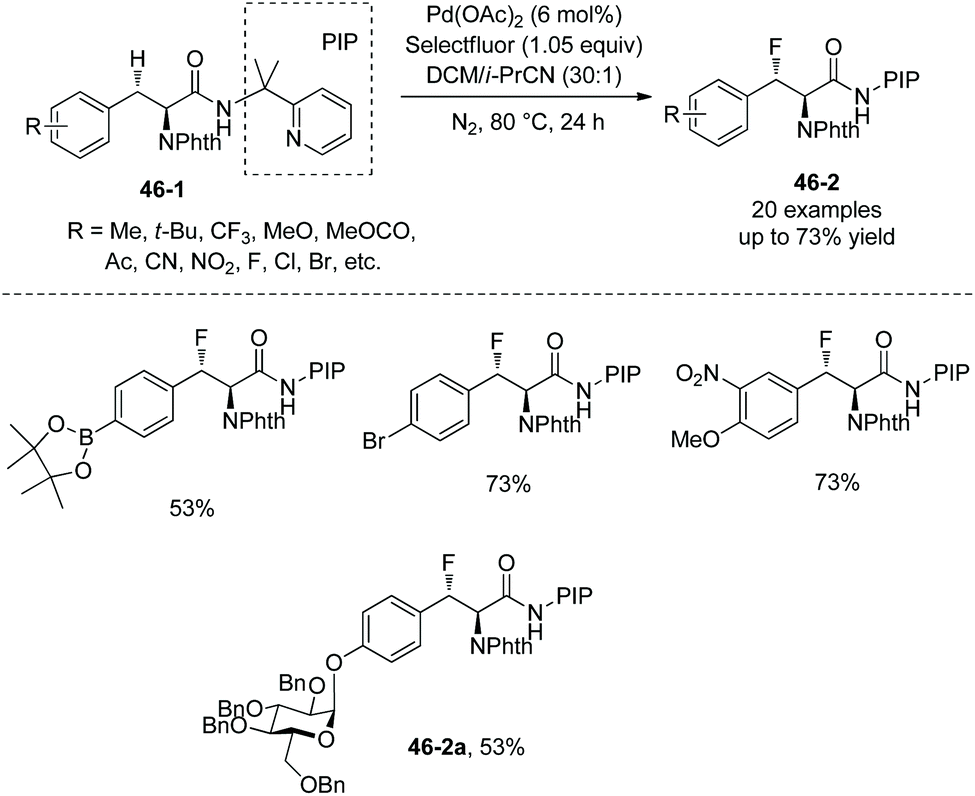
In June 2015, Shi and co-workers reported palladium-catalyzed fluorination of β-methylene C(sp3)–H bonds of L-α-amino acids.29 The N,N-bidentate directing group in N-phthaloylated L-α-amino acid 2-(pyridin-2-yl)propan-2-amine (PIP-amine) amides (46-1 and 47-1) is used for the fluorination (Schemes 46 and 47). Steric bulk of PIP is considered to be able to promote reductive elimination. Fluorination of benzylic methylene C(sp3)–H bonds proceeds smoothly with moderate to good yields (Scheme 46), affording products 46-2 as a single diastereoisomer, and the structures of four products are confirmed by X-ray diffraction. Many groups, such as acetyl, methoxycarbonyl, cyano, nitro, pinacolborane moiety, fluoride, chloride, and bromide, are tolerated in the transformation.
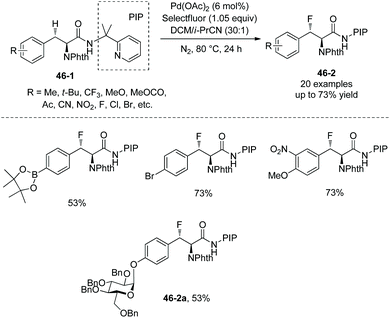 |
| | Scheme 46 Fluorination of benzylic methylene C–H bonds. | |
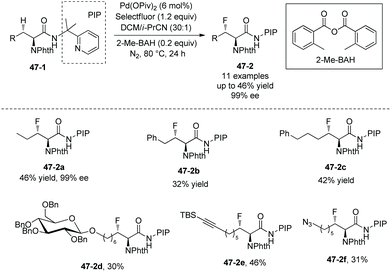 |
| | Scheme 47 Fluorination of aliphatic methylene C–H bonds. | |
Fluorination of aliphatic methylene C(sp3)–H of α-amino acid derivatives 47-1 also proceeds smoothly (Scheme 47),29 giving products 47-2 in moderate yields, using Pd(OPiv)2 as the catalyst, and the role of the additive 2-Me-BAH is not clear. A cyclic acetal, silyl-protected alcohols, and an alkene are compatible with this transformation. No epimerization occurs, and the products 47-2 have only a single diastereomer with a trans arrangement of F and phthaloyamino groups.
The fluorinated complex molecules containing glucose (46-2a and 47-2d) and “clickable” functional groups (a terminal alkyne 47-2e and an azide 47-2f) have been synthesized successfully, which are important intermediates for the synthesis of bioactive peptides and proteins.
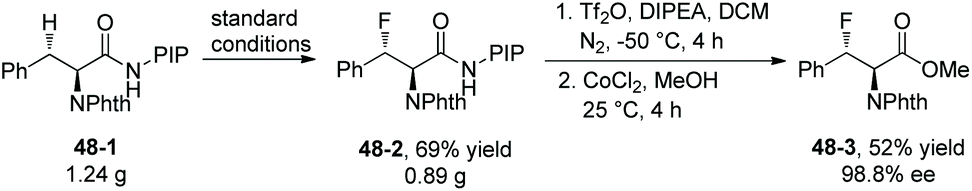
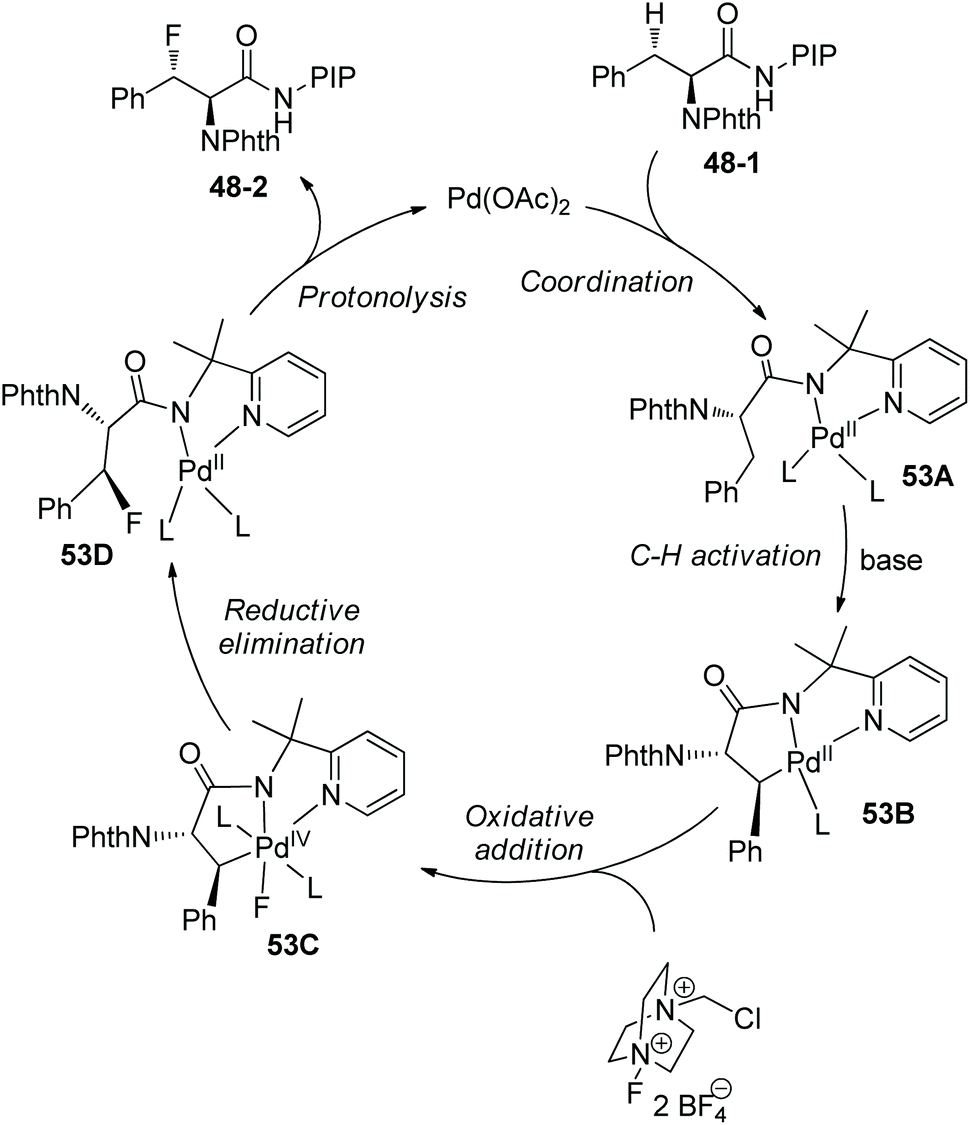
Fluorination of phenylalanine derivative 48-1 on a gram scale affords the product 48-2 in 69% yield (Scheme 48).29 Removal of PIP in product 48-2 gives the ester 48-3 in 52% yield and with 98.8% ee.
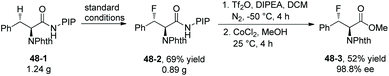 |
| | Scheme 48 Removal of PIP by Shi. | |
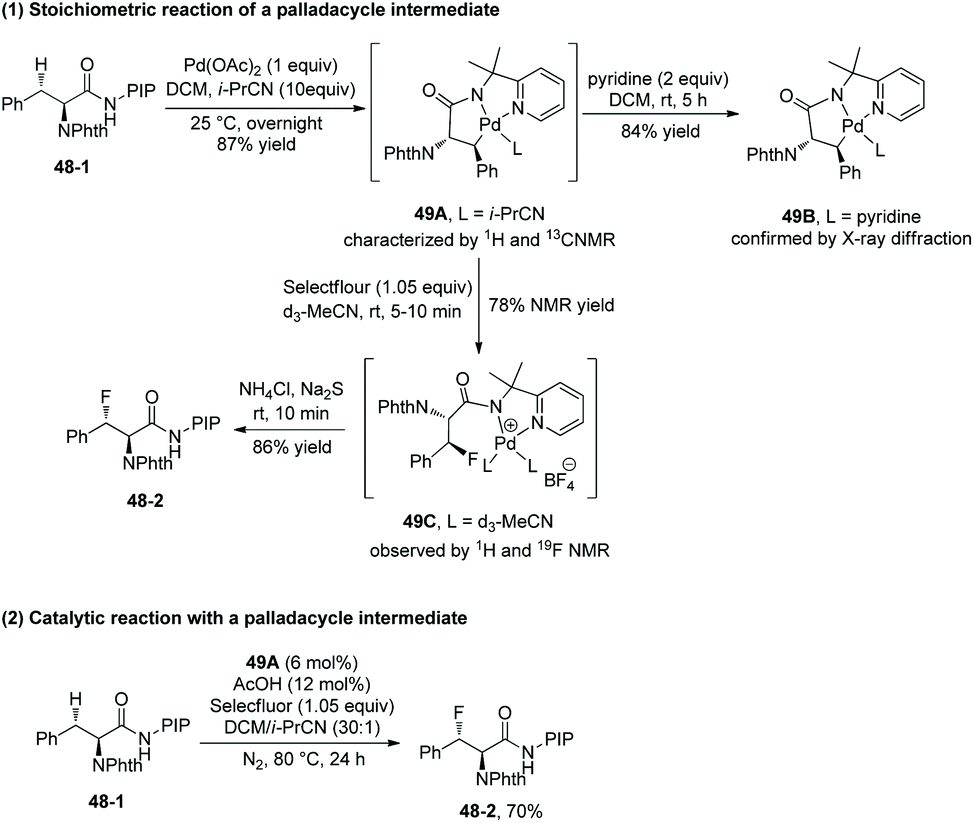
A palladacycle intermediate 49A is prepared, whose trans configuration is confirmed by X-ray diffraction of analogous complex 49B (Scheme 49-1).29 The stoichiometric reaction of 49A with Selectfluor gives 49C in 78% yield, and then 49C gives the fluorinated product 48-2 diastereospecifically in 86% yield; it is evidence for direct C–F reductive elimination. 48-2 can be obtained in 70% yield with catalytic 49A, comparable with the yield under the standard conditions (Scheme 49-2). This implies that 49A is an intermediate during the fluorination and can act as a catalyst for the transformation.
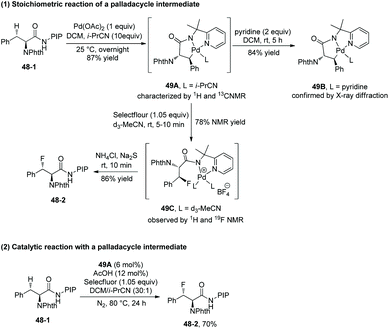 |
| | Scheme 49 Mechanistic studies for fluorination by Shi. | |
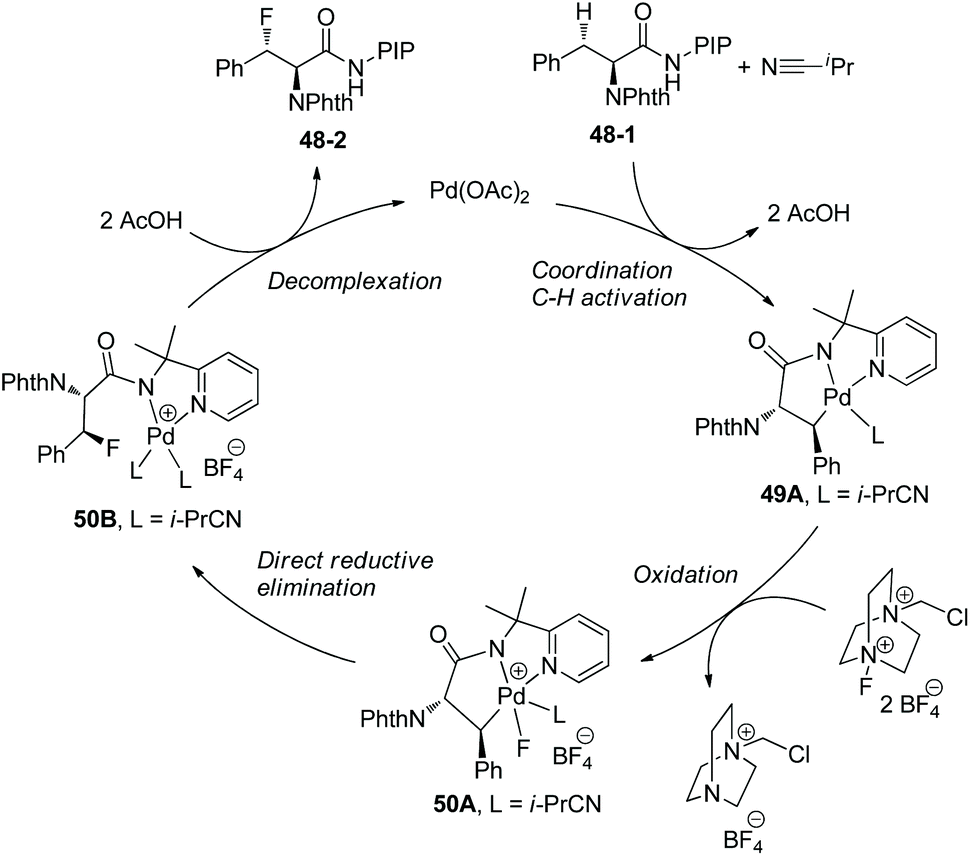
Based on mechanistic studies, a Pd(II)/Pd(IV) catalytic cycle is proposed for the fluorination (Scheme 50).29 i-PrCN plays two roles beneficial for the formation of C–F bonds. One is promoting the formation of palladacycle 49A as a neutral ligand, and another is promoting reductive elimination of 50A during steric crowding between the PIP auxiliary and i-PrCN.
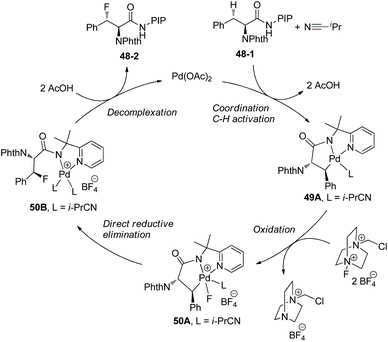 |
| | Scheme 50 Mechanism for fluorination by Shi. | |
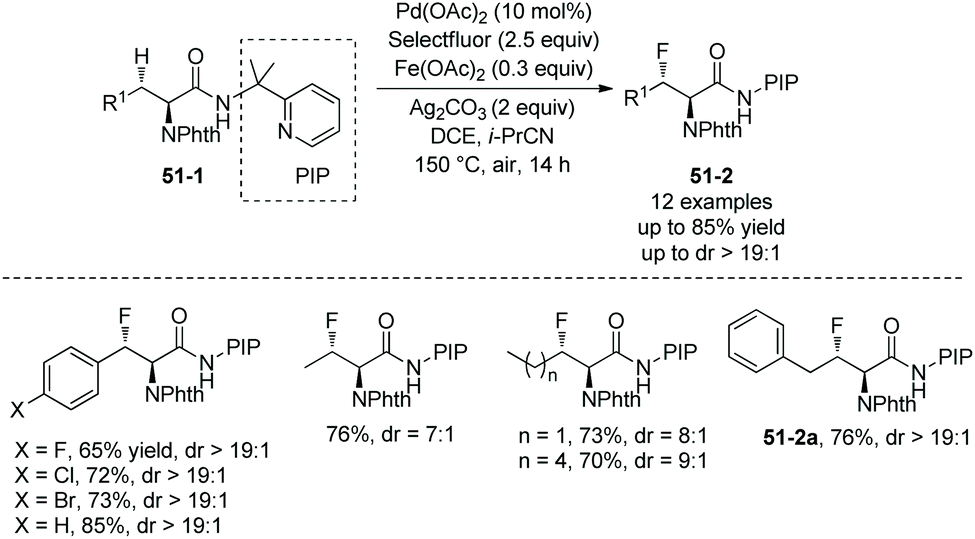
In July 2015, the N,N-bidentate directing group in N-phthaloylated L-α-amino acids 2-(pyridin-2-yl)propan-2-amine (PIP-amine) amides 51-1 was used for fluorination again by Ge and co-workers (Scheme 51).30 It was discovered that Fe(OAc)2 could improve the yields significantly. The fluorination proceeds smoothly in air, affording the products 51-2 with yields up to 85%, and no obvious racemization of α-carbon occurs. β-Benzylic and aliphatic methylene C(sp3)–H can all be fluorinated in high yields. β-Methylene C(sp3)–H bonds are preferentially fluorinated over γ-benzylic C(sp3)–H bonds (51-2a).
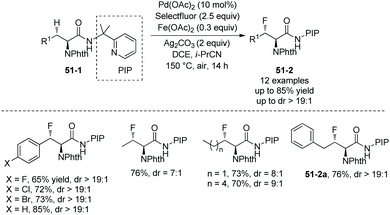 |
| | Scheme 51 Fluorination by Ge. | |
The protecting and directing groups (Phth and PIP) can be removed, affording the corresponding products (52-2 and 52-3) in good yields (Scheme 52).
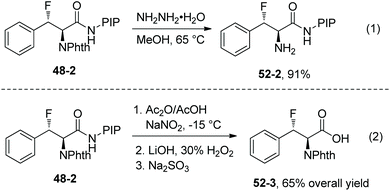 |
| | Scheme 52 Removal of PIP and Phth by Ge. | |
A Pd(II)/Pd(IV) catalytic cycle was also considered as the mechanism for the fluorination (Scheme 53),30 which is similar to that proposed by Shi (Scheme 50).29 Ag2CO3 probably promotes the ligand exchange and C–H bond activation as a base, and also promotes the oxidative addition of the palladacycle intermediate 53B with Selectfluor. Fe(OAc)2 can promote the release of Pd(II) species from the complex 53D.
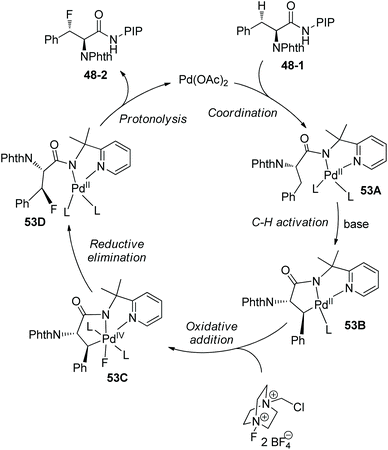 |
| | Scheme 53 Mechanism for fluorination by Ge. | |
2.1.4. C–Si bond formation.
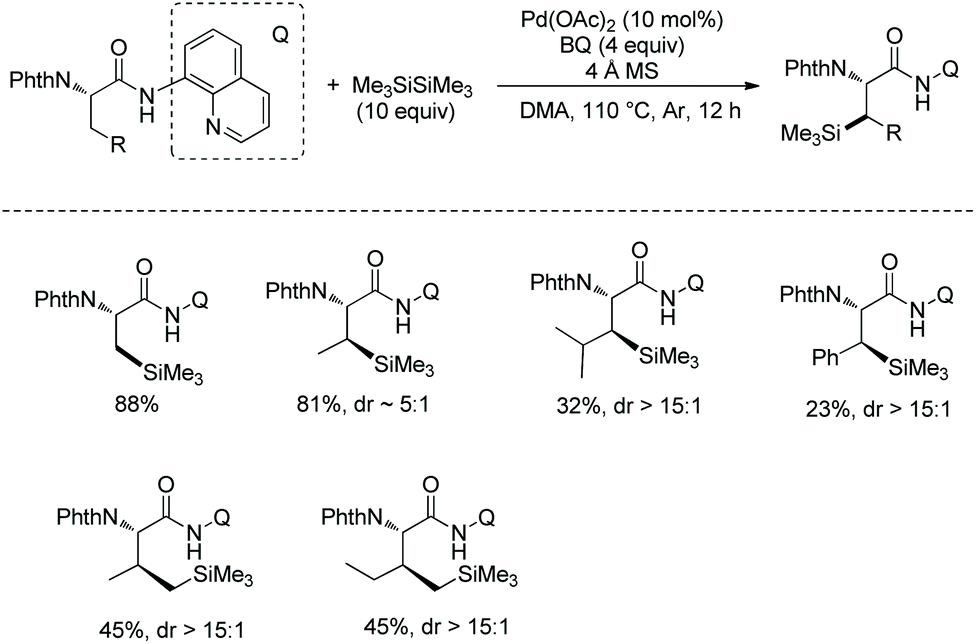
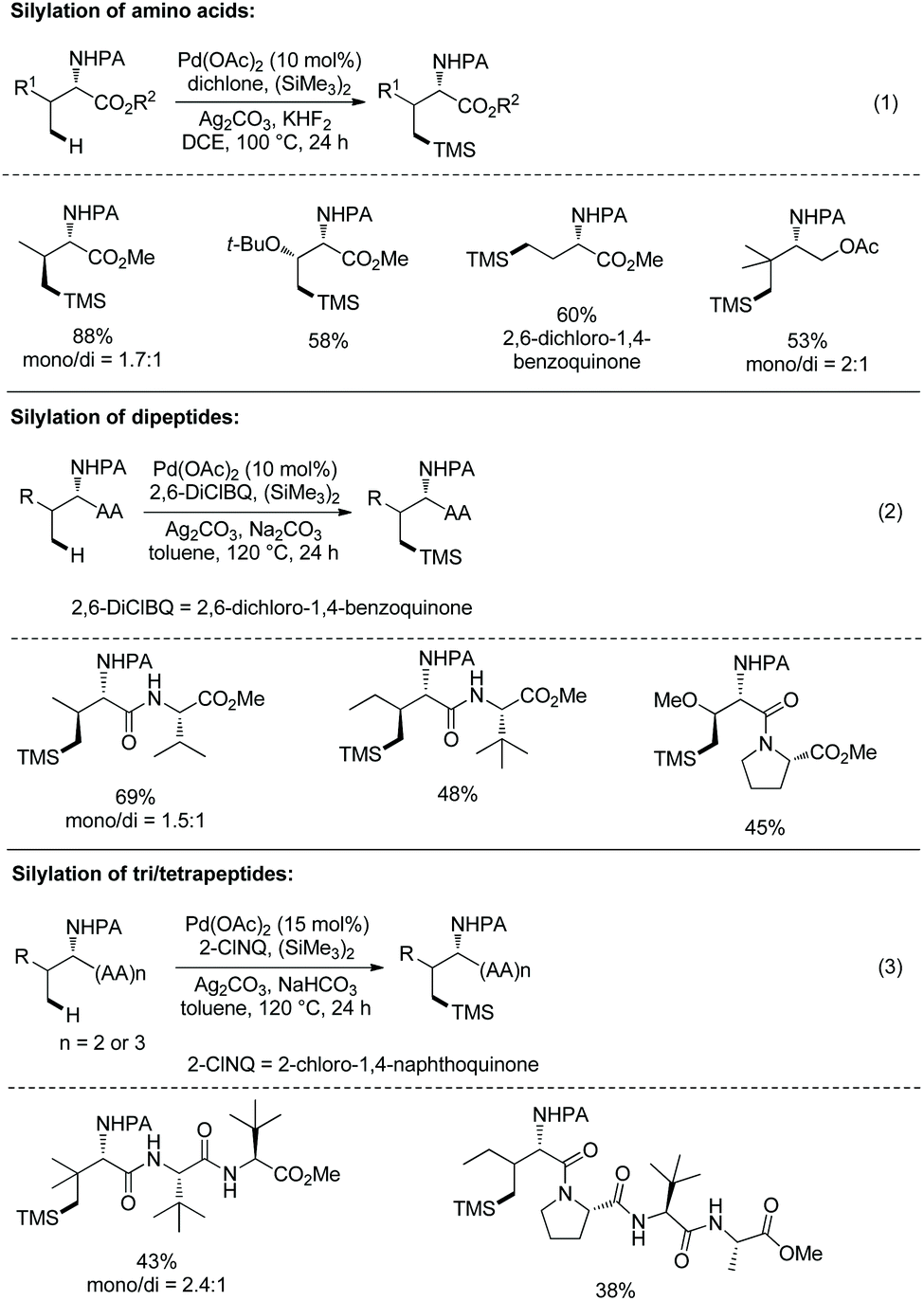
In September 2016, Shi and co-workers reported palladium-catalyzed silylation of amino acids, and the silylating reagent is hexamethyldisilane (Scheme 57).33aN-Boc-Val-OH is crucial to the silylation of the alanine derivative (Scheme 57, eqn (1)), and the directing auxiliary can be removed readily. No racemization occurs during the silylation and removal of the auxiliary. β-Benzylic or aliphatic secondary C–H bonds all can be silylated successfully with high diastereoselectivities (dr >20:1) (Scheme 57, eqn (2)).
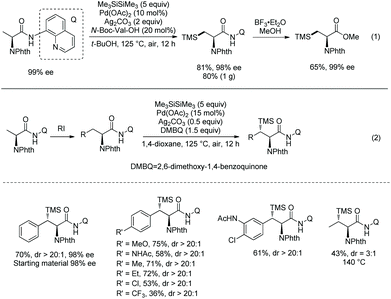 |
| | Scheme 57 Silylation by Shi. | |
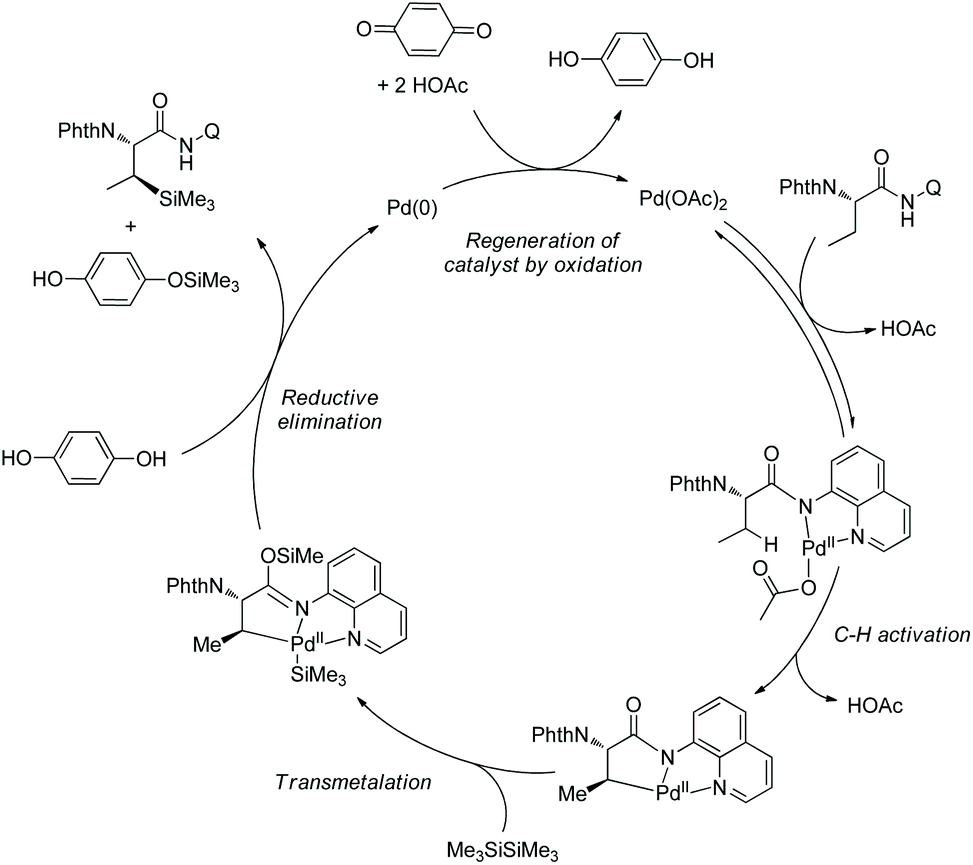
In October 2016, palladium-catalyzed silylation of amino acids was reported by another group, Zhang and co-workers (Scheme 58),33b the silylating reagent is also hexamethyldisilane, but the oxidant is benzoquinone (BQ). The following three kinds of C–H bonds are silylated successfully, (1) β-methyl of alanine derivative, (2) β-methylene of α-aminobutyric acid, leucine, and phenylalanine derivatives, and (3) γ-methyl of valine and isoleucine derivatives. The diastereoselectivity is up to >15:1. The mechanism is proposed as a Pd(II)/Pd(0) catalytic cycle (Scheme 59). The silylation undergoes C–H activation forming a fused two five-membered ring palladium(II) complex, transmetalation to give a silyl-ligated palladacycle, and reductive elimination affording the silylated product and releasing the Pd(0). Pd(0) is reoxidized to Pd(II), completing the catalytic cycle. In the step of C–H activation, a new five- or six-membered ring palladacycle with a sterically favored trans configuration is formed, which is the origin of diastereoselectivity.
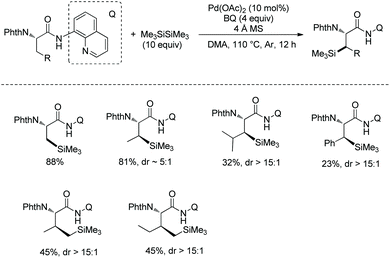 |
| | Scheme 58 Silylation. | |
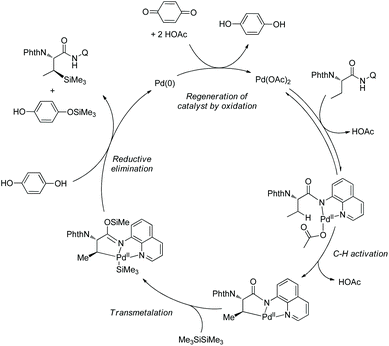 |
| | Scheme 59 Mechanism for silylation. | |
2.2. Functionalization of peptide derivatives and formation of cyclic peptides
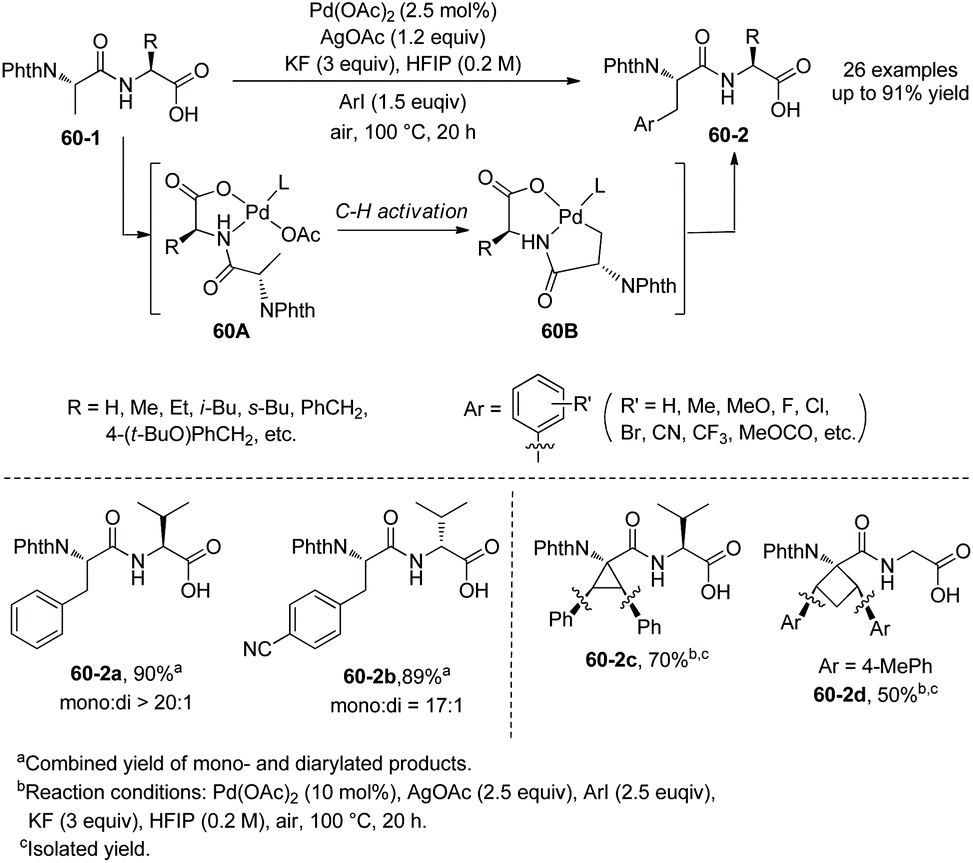
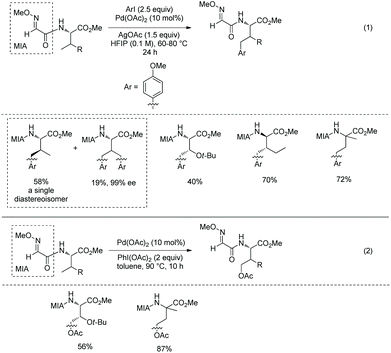
In 2014, Yu's group reported palladium-catalyzed backbone-directed sp3 C–H intermolecular functionalization of peptides.34 The functionalization proceeds site-selectively at the β-C(sp3)–H bonds of N-terminal amino acid residues in peptides without an extra directing group. Di-, tri-, and tetrapeptides are surveyed for the functionalization. The C-terminal amino acid in dipeptides serves as a directing group with an N,O-bidentate ligand (60A and 60B), enabling arylation to proceed smoothly (Scheme 60). When the C-terminal carboxyl group is protected, the C-terminus cannot serve as a directing group and promote the functionalization. The phenylation of N-phthaloyl protected dipeptide 60-1a with phenyl iodide proceeds smoothly in HFIP, giving 60-2a as a single diastereomer in high yield, and also can proceed in HFIP containing 20% water (v/v) with 10% Pd(OAc)2, affording the phenylation product 60-2a with 55% NMR yield. It shows that water can be used as a cosolvent in cases where solubility of peptides in organic solvents is poor. Arylation of 60-1a tolerates a wide range of functional groups at aryl iodides, including both electron-donating and -withdrawing groups. Heteroaryl iodides (4-iodopyridine, 3-iodopyridine, 2-iodo-5-methyl-thiophene, 3-iodothiophene, and 5-iodo-1H-indole) cannot undergo arylation with 60-1a, and the reaction of 5-iodo-1-tosyl-1H-indole with 60-1a gives a poor yield (∼30% NMR yield) of the arylated product. Besides L-valine, other amino acids (L-alanine, L-α-aminobutyric acid, L-leucine, L-isoleucine, L-phenylalanine, glycine, L-tyrosine and 2-aminoisobutyric acid) at C-termini in dipeptides also give satisfactory results. D-Valine at the C-terminus also works well (60-2b). When the N-terminal amino acid in dipeptides is 2-aminoisobutyric acid, 1-aminocyclopropanecarboxylic acid or 1-aminocyclobutanecarboxylic acid, the diarylation products are obtained in moderate to good yields (e.g.60-2c and 60-2d).
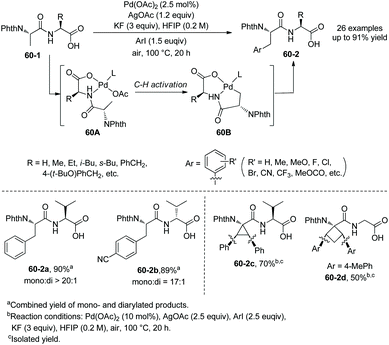 |
| | Scheme 60 Arylation of dipeptides. | |
Subsequently, tripeptides are investigated for site-selective arylation (Scheme 61, eqn (1)).34 The C-terminal free carboxyl group of N-phthaloyl tripeptides is not beneficial to arylation, leading to too much lower yield. Arylation of N-phthaloyl tripeptides proceeds smoothly when the C-terminal carboxylic acid is converted to a benzyl ester or methyl ester (61-1), affording products 61-2 in moderate to good yields. The N,N-bidentate ligand at backbones can promote the β-C(sp3)–H activation/arylation at the N-terminal amino acid residues by chelating palladium (61A and 61B). Tripeptides containing alanine, leucine, valine, isoleucine, proline, glycine, threonine, phenylalanine, tryptophan, aspartic acid, or 2-aminoisobutyric acid can be arylated smoothly. A broad range of functional groups at aryl iodides are compatible with this transformation.
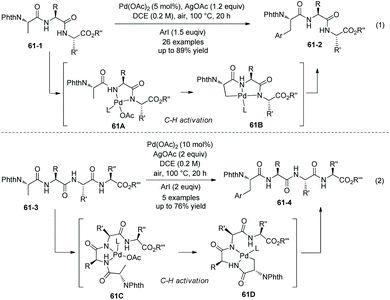 |
| | Scheme 61 Arylation of tri- and tetrapeptides. | |
Site-selective arylation of tetrapeptides 61-3 also occurs at β-C(sp3)–H bonds of N-terminal amino acid residues, affording the arylated products 61-4 as major products with 60–76% yields (Scheme 61, eqn (2)). It implies that site-selective arylation of longer peptides can be achieved. The native N,N-bidentate ligand (61C and 61D) also promotes the site-selective C–H functionalization at N-termini as in the case of tripeptides. The arylation of a tetrapeptide can proceed with water as a cosolvent (20% v/v) in lower yield. The palladium residue in an arylation product is 280 ppm.
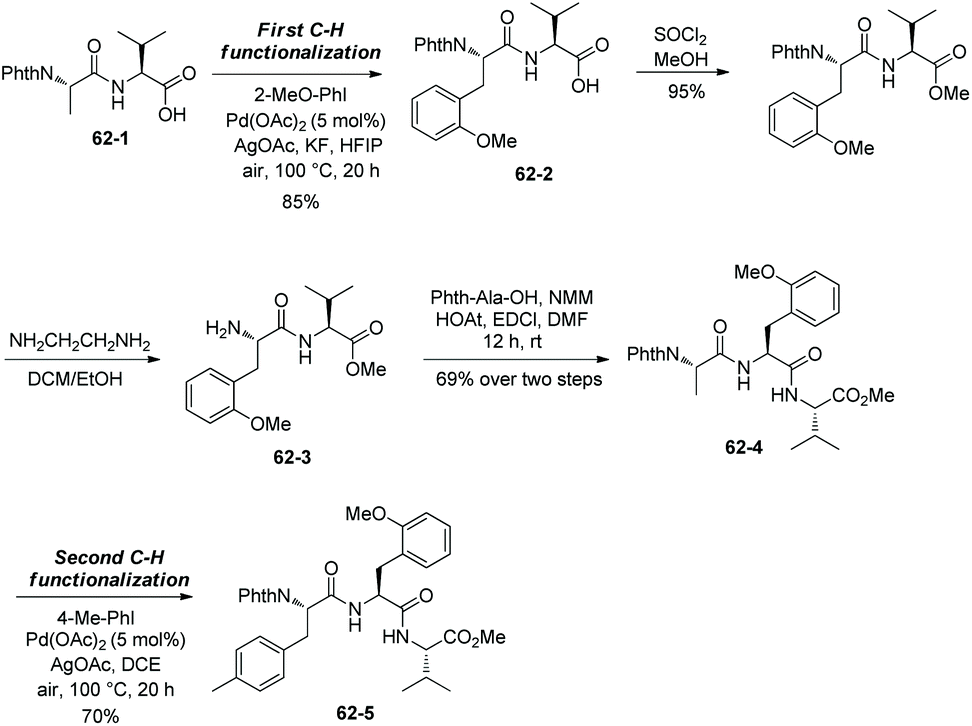
While the arylation occurs at N-termini site-selectively, the amino acid residue next to a N-terminus can also be arylated by a sequential method (Scheme 62).34 Dipeptide 62-1 is arylated at the N-terminus first, giving the arylated product 62-2. Esterification of the carboxylic acid at the C-terminus and deprotection of PhthN give the dipeptide 62-3, which is converted to the tripeptide 62-4. The second arylation occurs at the N-terminus end, affording the diarylated product 62-5.
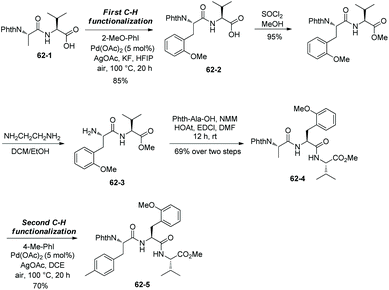 |
| | Scheme 62 Sequential arylations. | |
Acetoxylation of tripeptides 62-1 also proceeds site-selectively, affording the acetoxylated products 63-2 in moderate yields (45–66%, Scheme 63).34 Many tripeptides containing proline, leucine, isoleucine, valine, glycine, or alanine residues can undergo site-selective acetoxylation at the N-termini.
 |
| | Scheme 63 Acetoxylation of tripeptides. | |
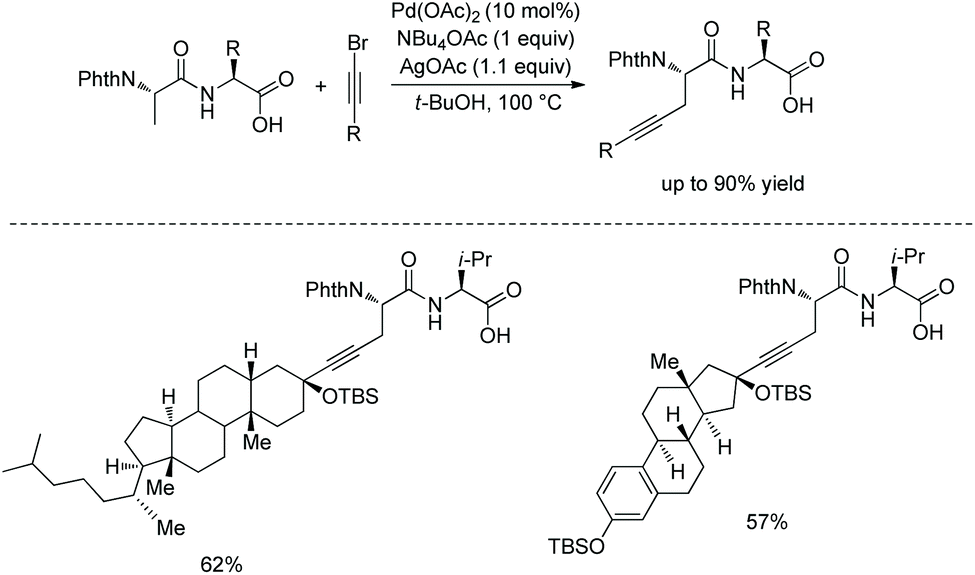
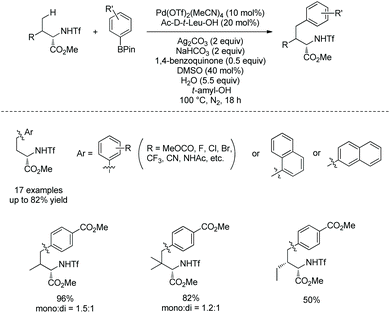
In 2017, the same group reported alkylnylation of oligopeptides via site-selective sp3 C–H activation under palladium catalysis.35 The dipeptides with a free carboxyl group at the C-termini can be alkynylated smoothly at β-sp3 C–H bonds of the N-terminal Ala (Scheme 64), coprostanol and estradiol (pharmacophores) can be linked to a dipeptide, which is useful for drug studies.
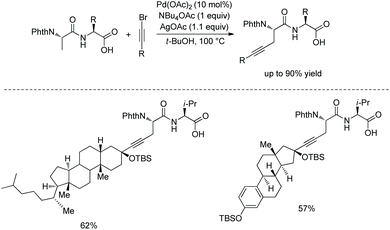 |
| | Scheme 64 Alkynylation of dipeptides. | |
A tripeptide with an esterified carboxyl group at C-terminus also can be alkynylated under the same conditions (Scheme 65).35 The N,N-bidentate ligand in the backbone of the tripeptide promotes the C–H activation/coupling. Although the tripeptide possesses three Ala residues, the coupling occurs site-selectively at β-sp3 C–H of the N-terminus.
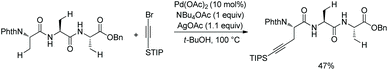 |
| | Scheme 65 Alkynylation of a tripeptide. | |
Inspired by the works of Yu et al. and Ackermann et al. about peptide functionalization using native N,O-/N,N-bidentate ligands34,35 and triazole-containing internal directing groups,24,25 respectively, the groups of Weng and Ackermann developed native asparagine residue-directed arylation of di-, tri-, and tetrapeptides containing Asn next to N-termini (Scheme 66).36 The arylation occurs at the β-position of N-terminal alanine residues with aryl iodides. A wide range of functional groups at aryl iodides are tolerated in this arylation. The mechanism was considered as a Pd(II)/Pd(IV) catalytic cycle. The asparagine residue with a primary amide moiety acts as a N,N-bidentate ligand, coordination of which to palladium forms a six-membered ring palladium complex, and the NH of the internal amide is deprotonated, which is supported by computational studies. β-sp3 C–H activation at N-terminal Ala forms a 5,6-fused bicyclic palladacycle intermediate, which undergoes oxidative addition, reductive elimination and protonolysis. Finally, the arylated product is afforded. The computational studies show that the C–H activation is the rate-determining step, the activation energy of which is 19.6 kcal mol−1.
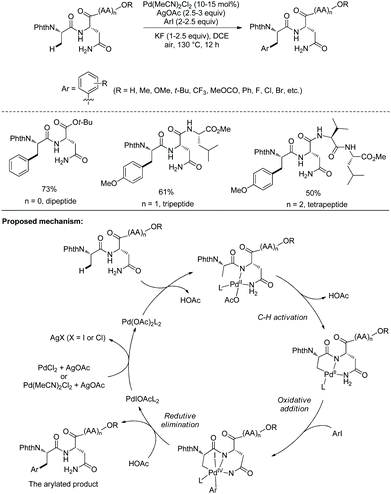 |
| | Scheme 66 Asparagine-directed arylation. | |
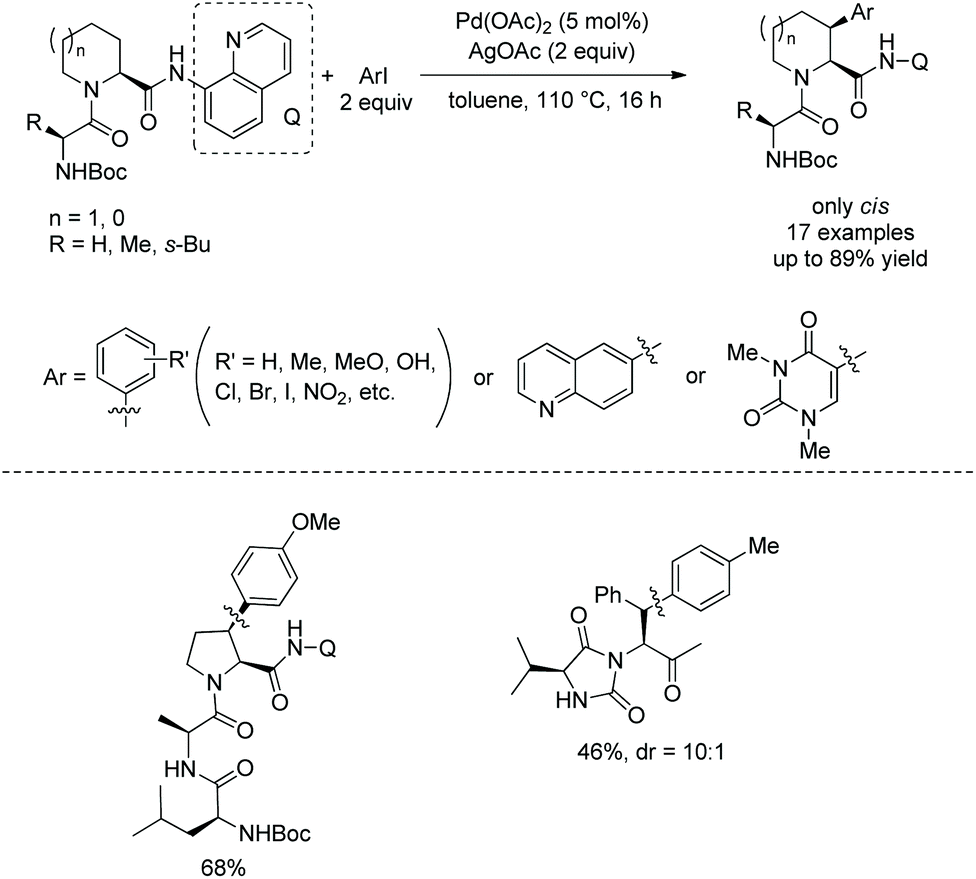
In 2016, Pd-catalyzed arylation of di- and tripeptides was achieved using C-terminal 8-aminoquinoline amides as directing groups (Scheme 67).37 Arylation occurs at aliphatic or benzylic β-positions of C-termini. The arylation has excellent stereoselectivity. When an aryl group is introduced at a ring, only a cis product is obtained. When an aryl group is introduced at a benzylic position, the dr is up to 10:1. 8-Aminoquinoline was removed readily in an arylated dipeptide, and the resulting peptide with a carboxyl group was prolonged, forming a tripeptide (Scheme 68).
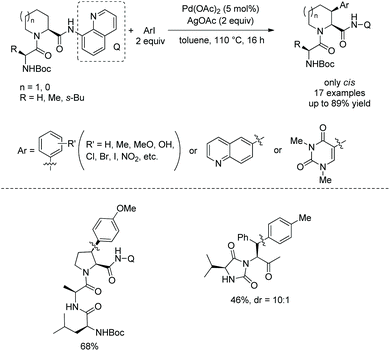 |
| | Scheme 67 Arylation of peptides with Q. | |
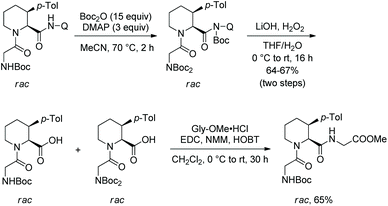 |
| | Scheme 68 Removal of auxiliary in arylated peptides. | |
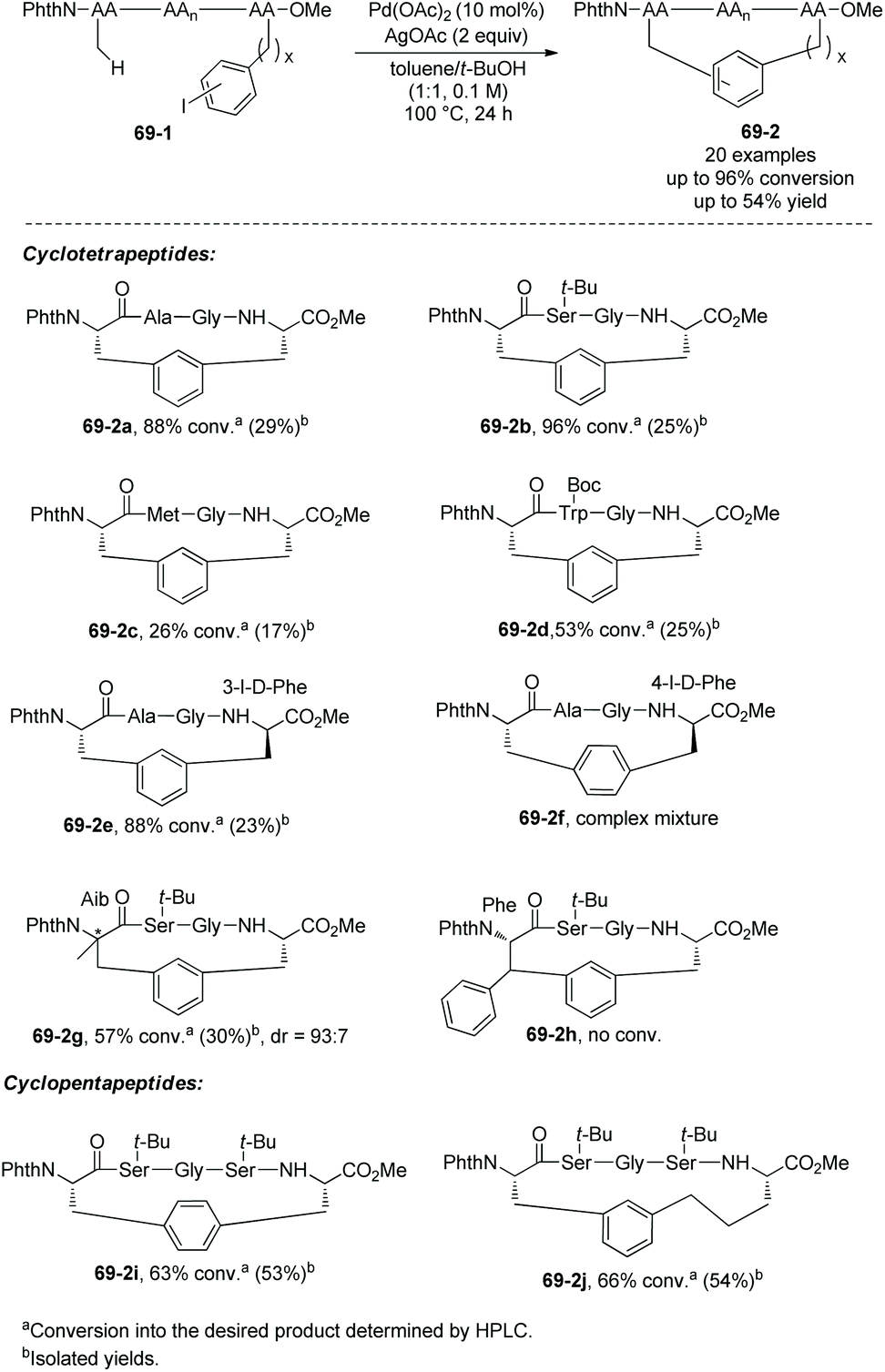
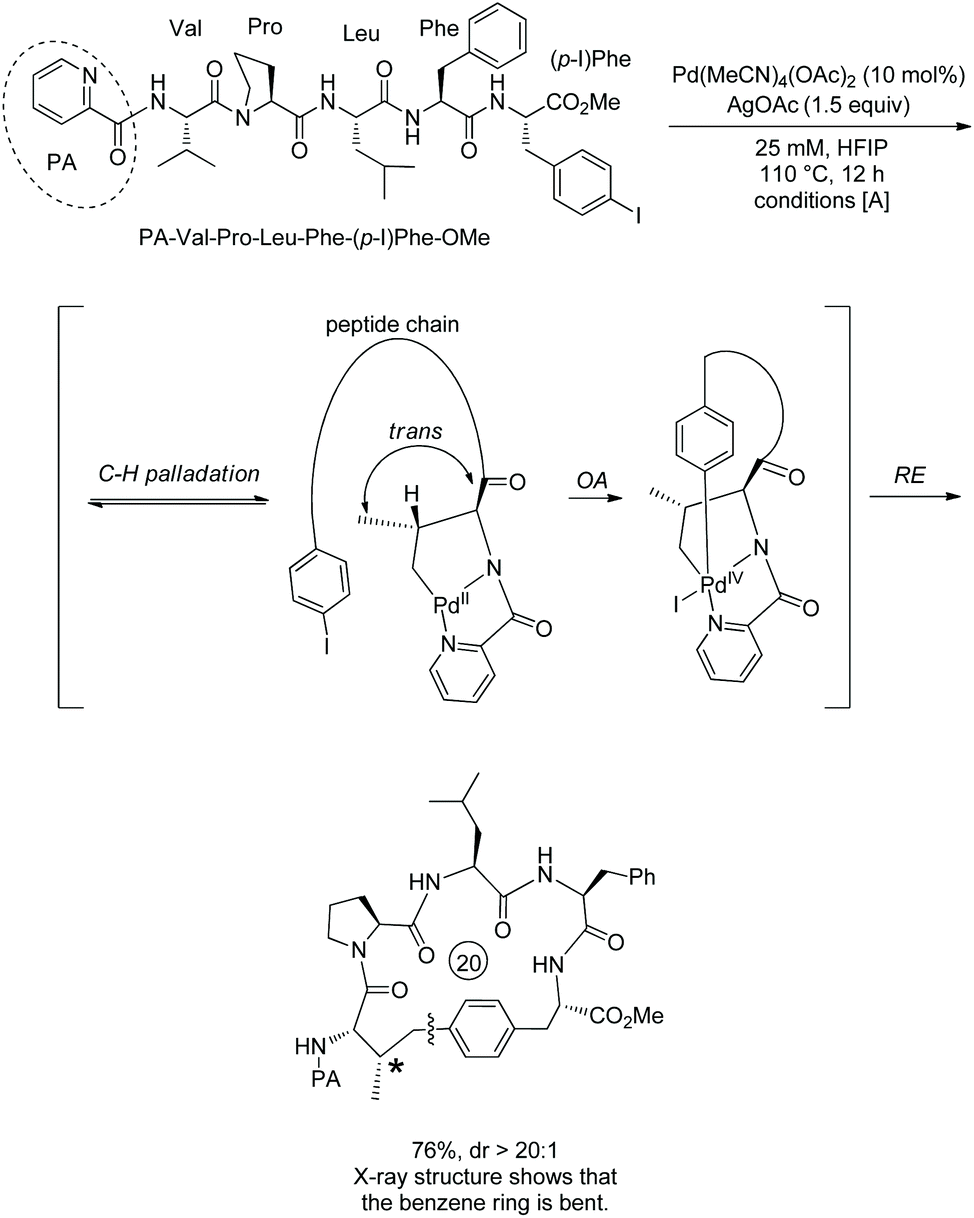
Based on the backbone-enabled C(sp3)–H activation/intermolecular arylation of peptides by Yu et al.,34,35 Noisier and co-workers synthesized cyclic tetrapeptides and pentapeptides by palladium-catalyzed late-stage C(sp3)–H activation/intramolecular arylation, i.e. peptide macrocyclization (Scheme 69).38 The β-C(sp3)–H bonds at N-termini of linear peptides 69-1 are activated under palladium catalysis and with the assistance of a native N,N-bidentate ligand, then coupled with intramolecular m-(p-)iodophenyl of another amino acid residue, forming cyclic tetrapeptides or pentapeptides (69-2). No epimerization of amino acid residues is found in the transformation, and 69-2a is produced as a single diastereoisomer. Linear peptides containing O-tert-butyl-L-serine (Ser(tBu)), Nε-tert-butyloxycarbonyl-L-lysine (Lys(Boc)), Nβ-(2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl)-L-arginine (Arg(Pbf)), Nγ-trityl-L-asparagine (Asn-(Trt)), and L-glutamic acid 5-tert-butyl ester (Glu(OtBu)) can be stapled with good conversion. Tetrapeptides with branched O-tert-butyl-L-threonine Thr(tBu) or L-leucine (Leu) in position i + 2 can undergo the late-stage macrocyclization, affording Si,i+3S(5) peptides with good conversion. Conformational bias from L-proline (Pro) in position i + 2 of a tetrapeptide prevents the coupling from forming a Si,i+3S(5) peptide. Whereas L-methionine (Met) or L-Trp-(Boc) has sulfur atoms deactivating palladium and more reactive C(sp2)–H bonds, peptides containing L-methionine (Met) or L-Trp-(Boc) also undergo the macrocyclization via C(sp3)–H activation (69-2c and 69-2d). When the C-terminus is 3-I-D-Phe (69-1e) with different stereo configuration from 69-1a, the Cβ–Ph coupling also proceeds smoothly with good conversion (69-2e). When the C-terminus is 4-I-D-Phe, the conversion results in a complex mixture 69-2f. When the N-terminus is 2-aminoisobutyric acid, a β-sp3 C–H bond of prochiral Aib residues is coupled with phenyl of the C-terminus highly diastereoselectively (dr = 93:7) in 30% yield (69-2g). When the N-terminus is Phe, the Cβ–Ph coupling failed (69-2h), and the β-sp3 C–H coupling is probably prevented by steric hindrance. Si,i+3S(5), Si,i+3S(7), Si,i+4S(5), Si,i+4S(6), and Si,i+4S(7) peptides all have successful examples resulting from stapling linear peptides, and Si,i+3S(6), Si,i+3S(8) and Si,i+2S(5) peptides were not obtained. These indicate that the lengths of the cross-links have influences on the macrocyclization for a certain stapling (i,i + 2; i,i + 3 or i,i + 4).
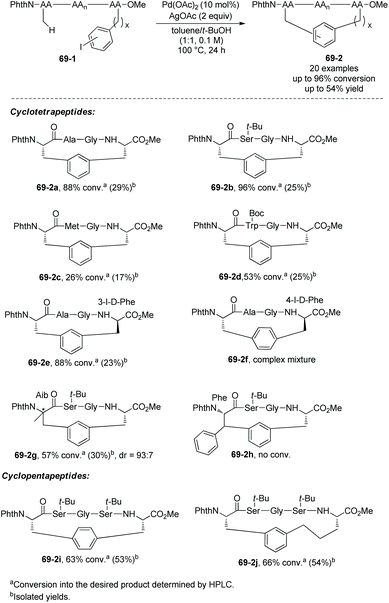 |
| | Scheme 69 Stapling peptides. | |
The method for stapling peptides was extended successfully to solid phase synthesis (Scheme 70).38 The peptides were bound to an amide resin linker (the methylbenzhydrylamine (MBHA) Rink) 70-1, macrocyclization gives cyclic peptides 70-2, deprotection and elongation of peptides 70-2 give 70-4. Si,i+3S(5) peptide 70-4a, Si,i+3S(7) peptide 70-4b, and Si,i+4S(7) peptide 70-4c were obtained in 11–15% yield.
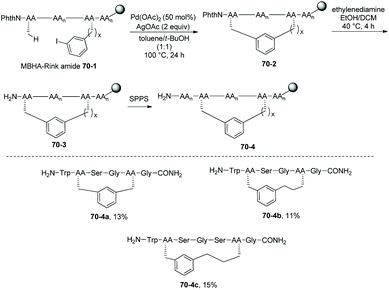 |
| | Scheme 70 Solid phase synthesis. | |
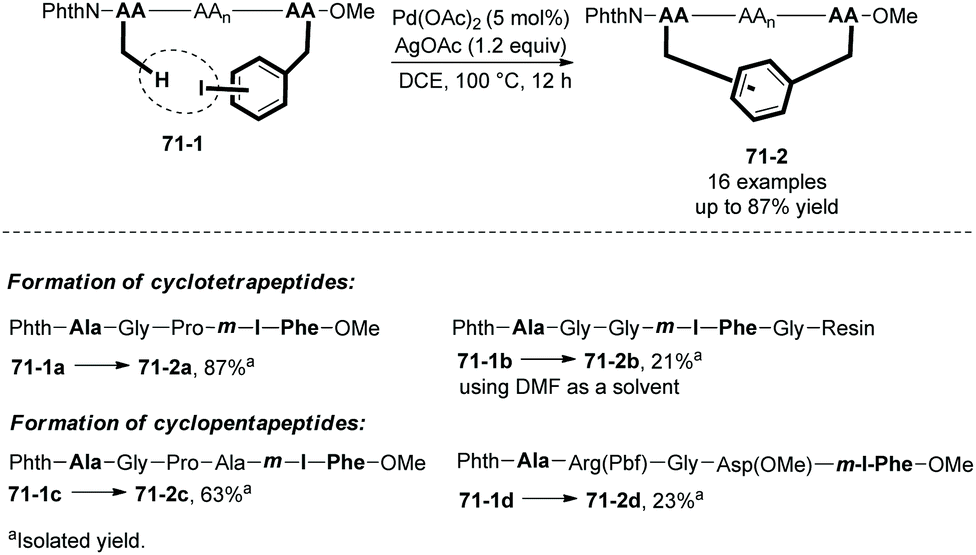
In the same year (2017), Wang and co-workers also reported peptide 71-1 macrocyclization via palladium-catalyzed sp3 C–H activation/intramolecular arylation, forming cyclotetrapeptides and cylcopentapeptides in 50–87% isolated yields (71-2, Scheme 71).39 The intramolecular arylation occurs between the β-sp3-C–H bonds of the N-terminal amino acid residue and the m(p)-I-Phe residue. A cyclotetrapeptide 71-2b was obtained by a solid phase synthesis method.
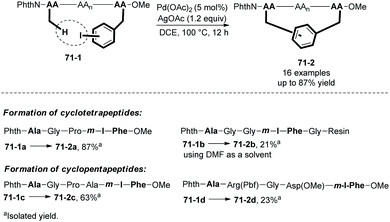 |
| | Scheme 71 Peptide macrocyclization. | |
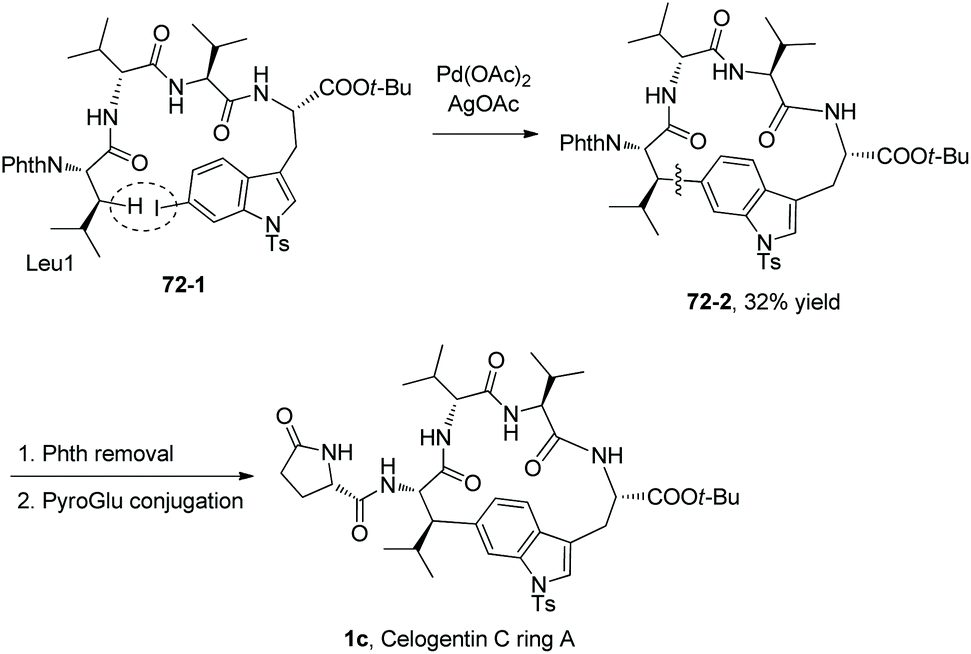
The macrocyclization method is employed for the synthesis of a natural product (Scheme 72).39 The tetrapeptide 72-1 with Leu at the N-terminus and 6′-I-Trp at the C-terminus produce the cyclotetrapeptide 72-2via intramolecular sp3 C–H arylation. The sp3 C–H activation occurs at the β-position of N-terminal Leu, the transformation is stereospecific and only one diastereomer is formed. 72-2 can be converted to 1c by deprotection and conjugation of a PyroGlu residue, which is ring A of the natural product celogentin C.
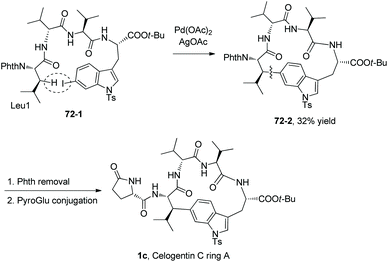 |
| | Scheme 72 Application of macrocyclization. | |
A RGD cyclopentapeptide 71-2d obtained by this method proves to have higher proteolytic stability and binding affinity to integrin than a commercially available cyclo-(RGDfK).39 This method will provide structurally multifarious cyclopeptides and will be helpful for biological studies.
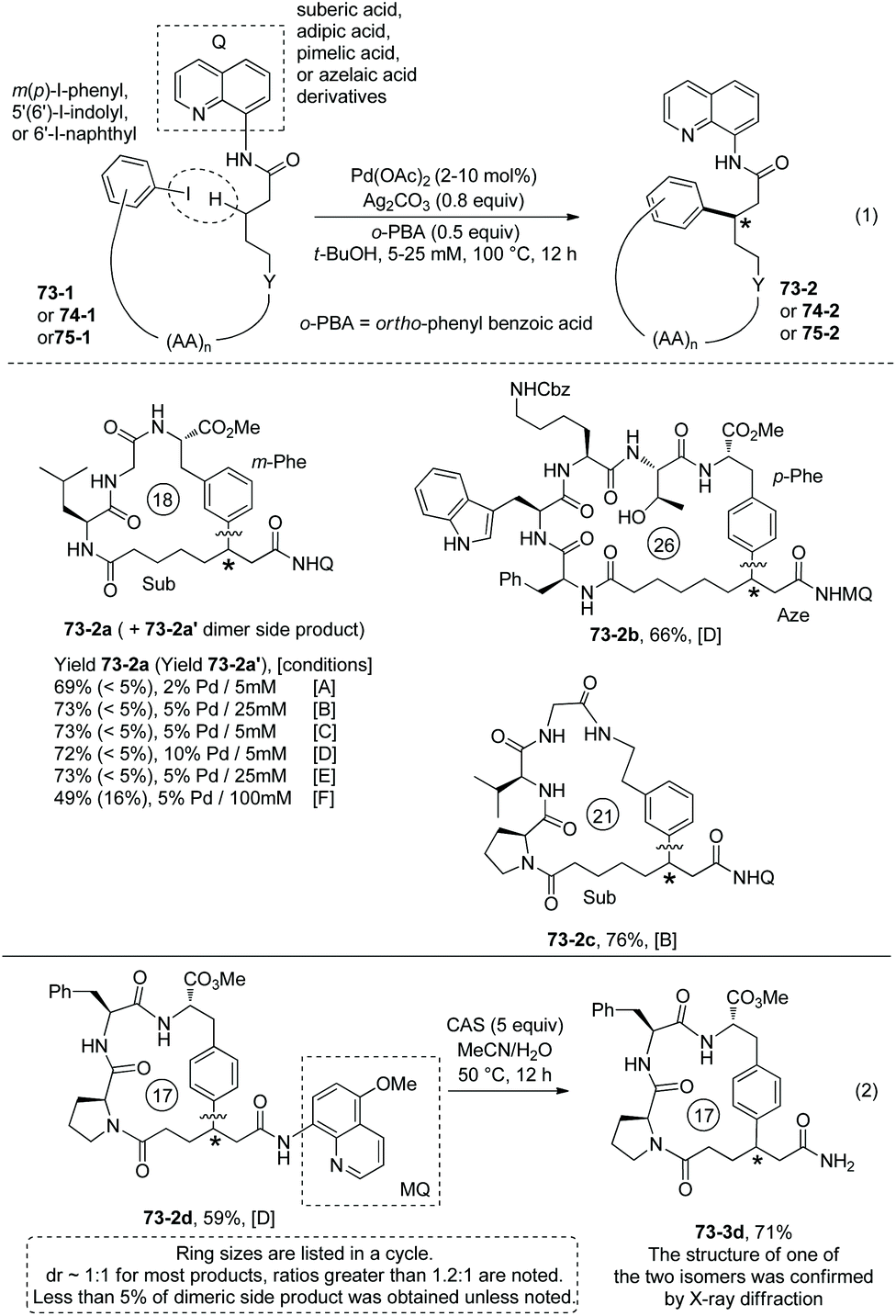
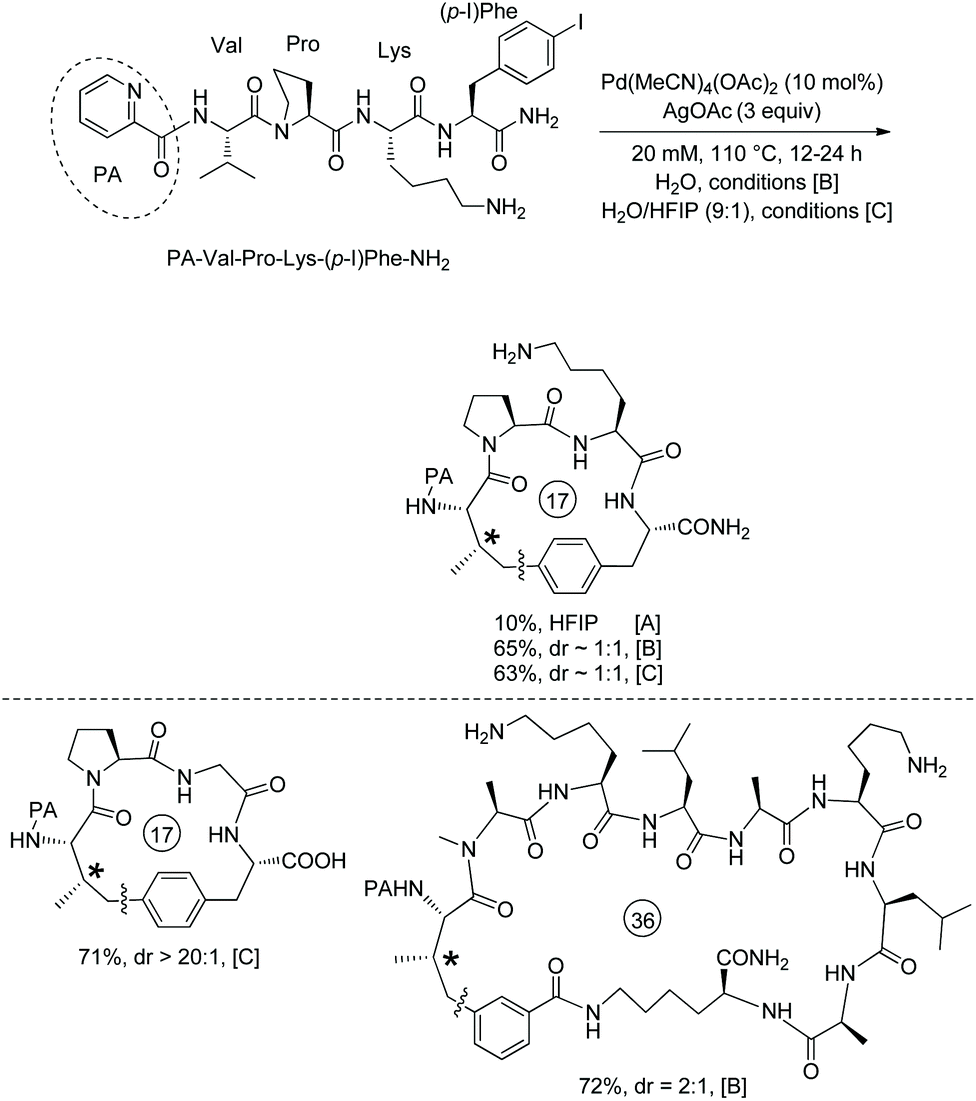
In the above two examples, a native N,N-bidentate ligand is employed for peptide macrocyclization.38,39 In 2018, 8-aminoquinoline was used as a directing group for the formation of cyclophane-braced peptide macrocycles via intramolecular sp3 C–H arylation (Scheme 73, eqn (1)).40
 |
| | Scheme 73 Formation of peptide macrocycles and removal of MQ. | |
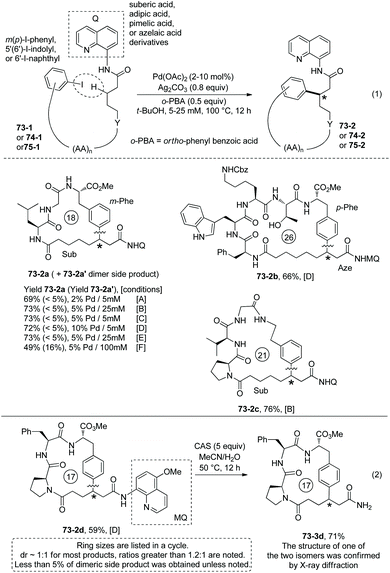
Tripeptide QNH-Sub-Leu-Gly-m-I-Phe-OMe (73-1a) with m-I-Phe residue and an alkyl tail of QNH2 (8-aminoquinoline)-installed suberic acid gives a good yield for the synthesis of 18-membered meta-cyclophane product 73-2a. While HOAc and PivOH are usually helpful for C–H activation, ortho-phenyl benzoic acid (oPBA) was found to be the best acid for the cyclization. The intramolecular C–H arylation can be achieved at m(p)-I-Phe, m-I-Tyr, or 7′-I-Trp residues. Unprotected amino acid units (e.g. Thr 73-2b, Trp 51-2b and Ser) and protected amino acid units (e.g. Lys 73-2b, Arg and Met) can be well tolerated. Besides suberic acid (Sub, C8), other alkyl diacids (e.g. adipic acid (Adi, C6), pimelic acid (Pim, C7), and azelaic acid (Aze, C9)) and 6-aminohexanoic acid (Ahx) can also be used for an alkyl tail. However, succinic acid (C4) or glutaric acid (C5) is not suitable for an alkyl tail, probably due to the interference of the proximate and competitive C–H activation assisted by the peptide's terminal amino groups. The auxiliary Q (quinolin-8-yl group) is difficult to remove, and MQ (5-methoxy-quinolin-8-yl group) can also promote the C–H activation/cyclization efficiently. Treatment of MQ containing 73-2d with cerium ammonium sulfate (CAS) gives 73-3d in 71% yield under mild conditions (Scheme 73, eqn (2)).40
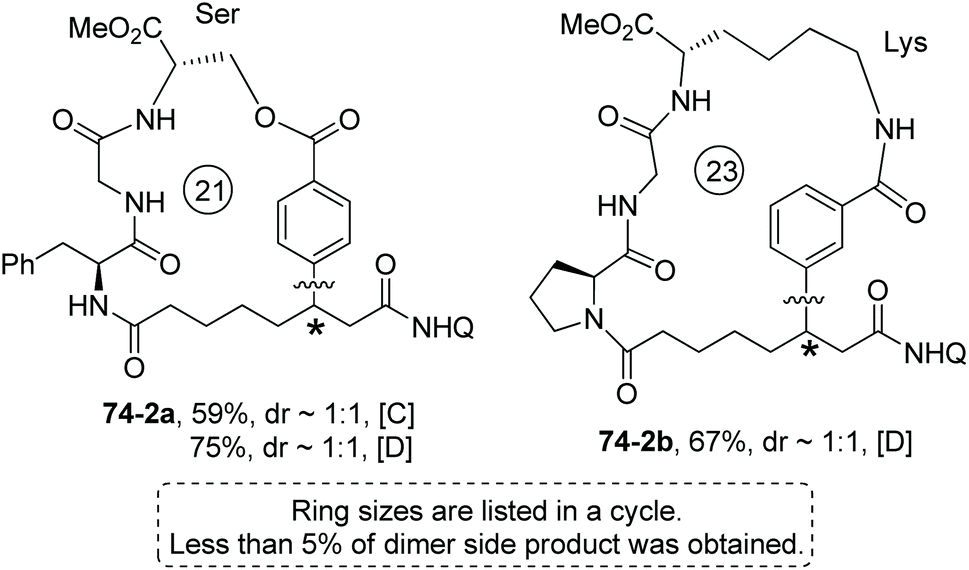
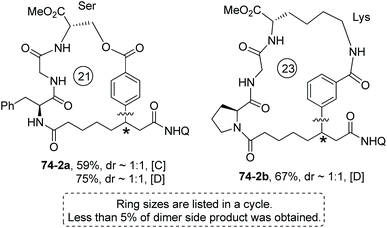
Modified Ser, Glu and Lys containing m(p)-I-phenyl or 6′-I-naphthyl groups can provide aryl components for intramolecular βC-Ar coupling, affording cyclophane smoothly (e.g.74-2a, 74-2b, Scheme 74).40
 |
| | Scheme 74 Macrocyclization at modified nonaromatic amino acid residues. | |
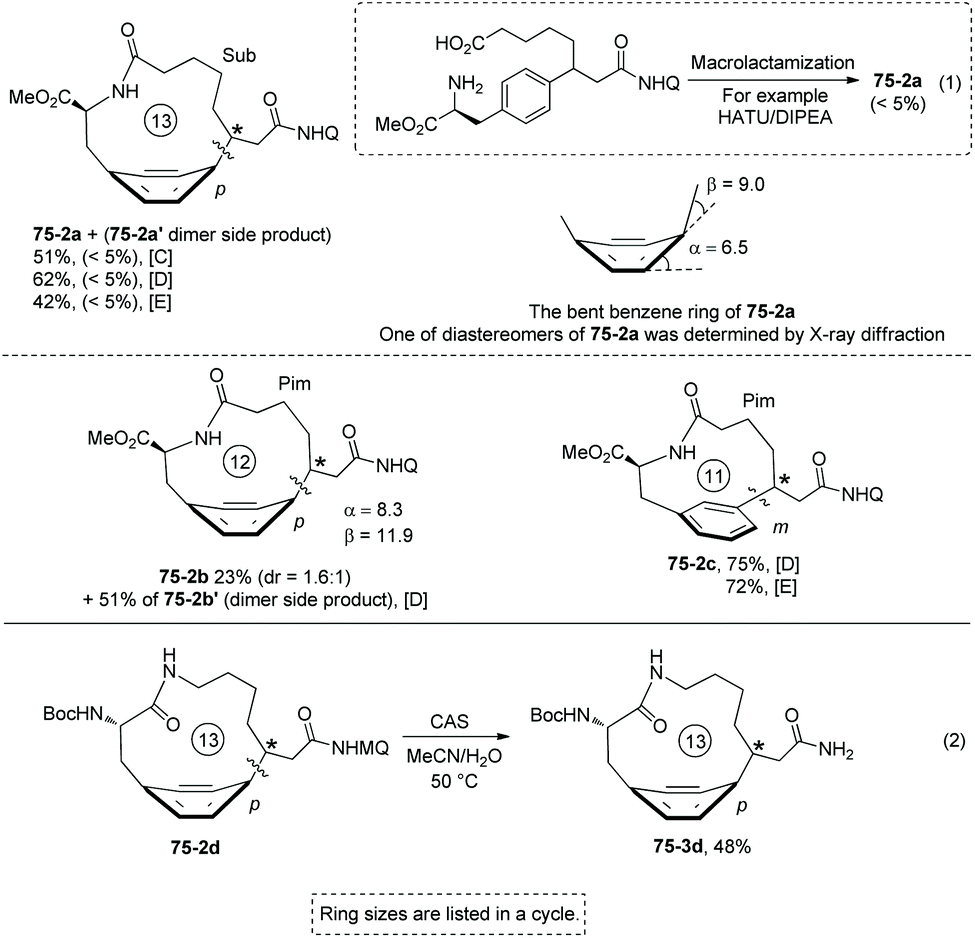
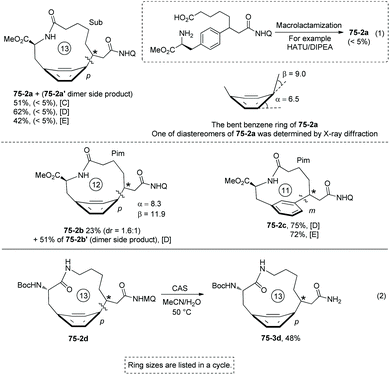
Some small-sized cyclophanes possessing a bent benzene ring and significant ring strain are also provided by the intramolecular sp3 C–H arylation (Scheme 74).40 13-Membered p-cyclophane 75-2a with a calculated ring strain of 7.7 kcal mol−1 was prepared in 62% yield under conditions D, and the conventional lactamization gives only less than 5% yield of product 75-2a (Scheme 75, eqn (1)). 12-Membered p-cyclophane 75-2b with a more bent benzene ring and bigger ring strain was also prepared albeit with a lower yield (23%); meanwhile a more undesired dimer was formed in 51% yield. 11-Membered m-cyclophane 75-2c with smaller ring strain was prepared in good yield (75%) under conditions D. MQ (5-methoxy-quinolin-8-yl group) of 13-membered p-cyclophane 75-2d was also removed by treatment of cerium ammonium sulfate (CAS), affording product 75-3d in 48% yield (Scheme 75, eqn (2)).
 |
| | Scheme 75 Synthesis of small-sized cyclophanes. | |
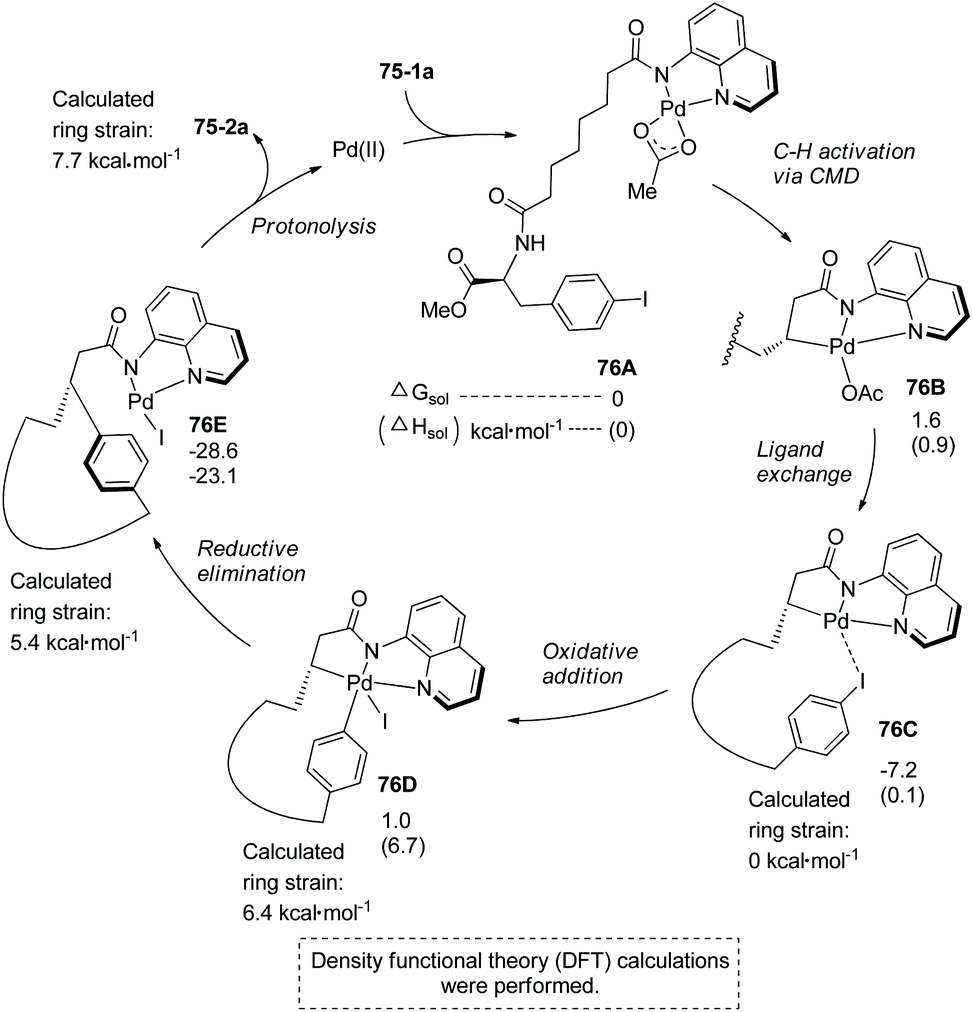
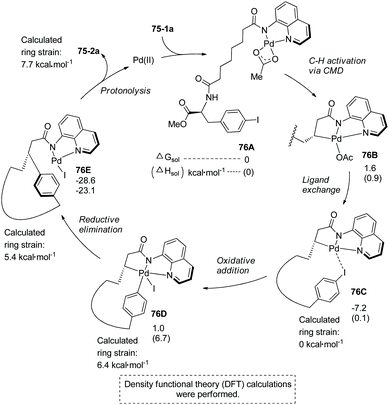
The mechanism was proposed and understood with the assistance of density functional theory (DFT) calculations for the synthesis of 75-2a (Scheme 76).4075-1a first coordinates to palladium forming the complex 76A, subsequent C–H activation via a concerted metalation–deprotonation pathway (CMD) gives the palladacycle 76B, ligand exchange produces complex 76C, intramolecular oxidative addition with an energy barrier of 17.4 kcal·mol−1 affords Pd(IV) complex 76D with a calculated ring strain of 6.4 kcal mol−1, reductive elimination with a barrier of 6.7 kcal mol−1 produces the cyclized complex 76E, and protonolysis gives the cyclized product 75-2a with a calculated ring strain of 7.7 kcal mol−1, and releases the palladium(II) catalyst. A catalytic cycle is completed. Whereas the ring strain increases from 76C to 76E, the process releases 21.4 kcal mol−1 of energy, and the exothermic process drives the transformation. Intramolecular coordination of the iodo group to palladium forming stable complex 76C is entropically preferred over the intermolecular coordination. Low energy barriers of OA and RE are probably due to stabilizing the Pd(IV) complex 76D by the formation of a chelate palladium complex with 8-aminoquinoline. The intramolecular arylation is entropically favored over the intermolecular arylation, leading to the success of the intramolecular transformation.
 |
| | Scheme 76 Mechanism for synthesis of 75-2a. | |
A 21-membered cyclophane 73-2c prepared by this method has selective cytotoxicity toward proliferative Myc-dependent cancer cell lines, and thus is a potent lead compound.40 This method can provide various cyclophane-braced peptide macrocycles and has significant potential in drug discovery.
On the basis of the above work,40 Chen and co-workers developed another palladium-catalyzed transformation for peptide macrocyclization (Scheme 77).41 8-Aminoquinoline was installed on the C-terminal N-methyl L(D)-Ala of peptides rather than the alkyl tail via amide conjugation, β-sp3 C–H activation occurs at C-termini, and then intramolecular arylation gives cyclophane-braced peptide macrocycles as a single diastereomer. The tetrapeptide 77-1ap-I-Phe-Ile-Gly-N(Me)-Ala-NHQ gives a 16-membered cyclic peptide 77-2a in 68% yield under the standard conditions, along with 8% yield of a side product 77-2a′. If the solvent is toluene, an undesired compound 77-3 is produced as a major product with 33% yield. The increase of reactant concentration leads to more dimeric cyclization. If there is not an N-protecting group on Ala in the molecule of 77-1a, the cyclization cannot proceed. This is probably due to the coordination of the amino group on Ala to palladium interfering the sp3 C–H activation. If N(Me)-Phe is replaced with N(Me)-Nva or N(Me)-Phe units in the molecule of 77-1a, the cyclization also cannot occur. This is probably due to the increase of steric hindrance.
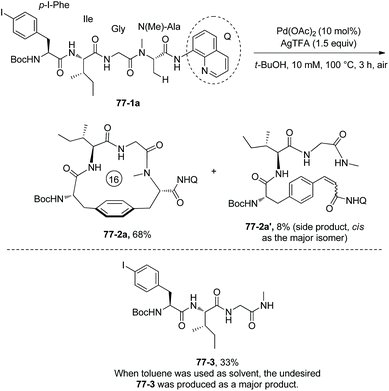 |
| | Scheme 77 Macrocyclization at N-methyl-Ala. | |
Many linear tripeptides, tetrapeptides or pentapeptides give cyclopeptides with medium-sized rings in good yields, and less than 10% yields of alkene byproducts are produced via cyclization and elimination in these cases (e.g.Scheme 78, (77-2c)–(77-2f)).41 However, small (or large)-sized ring products are obtained in lower yields (e.g.77-2b with two AA residues and 77-2g with six AA residues and a prosthetic group). No obvious epimerization of AA residues occurs. The cyclization tolerates the following AA residues: methionine, unprotected threonine (77-2e), tryptophan, asparagine with the protected primary carboxamide, aspartic acid with the protected carboxyl group (77-2g), argnine with the protected guanidine (77-2g), and nonproteinogenic AA (e.g. tert-Leucine, 2-aminobutyric acid, and β-alanine) residues. Nonaromatic AA residues installed with prosthetic groups contain m-I-phenyl groups and can serve as a coupling partner for the intramolecular C–H arylation (e.g.77-2g).
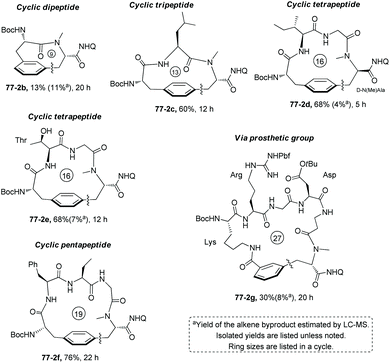 |
| | Scheme 78 Substrate scope. | |
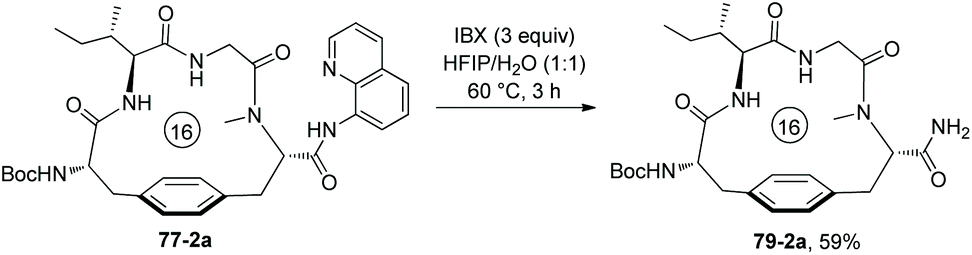
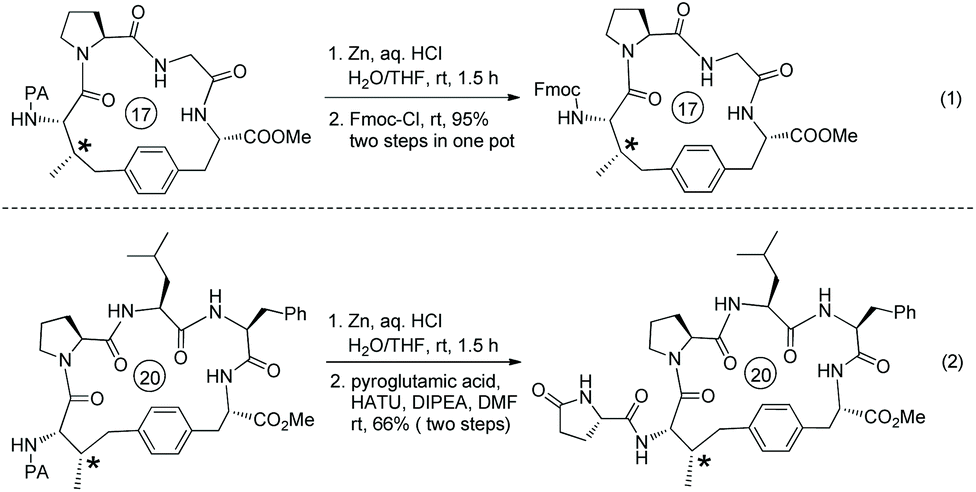
The auxiliary Q (quinolin-8-yl group) of 79-2a can be removed by treatment of 2-iodoxybenzoic acid (IBX), affording the primary amide 57-2a in 59% yield (Scheme 79).41
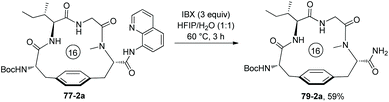 |
| | Scheme 79 Removal of quinolin-8-yl group (Q). | |
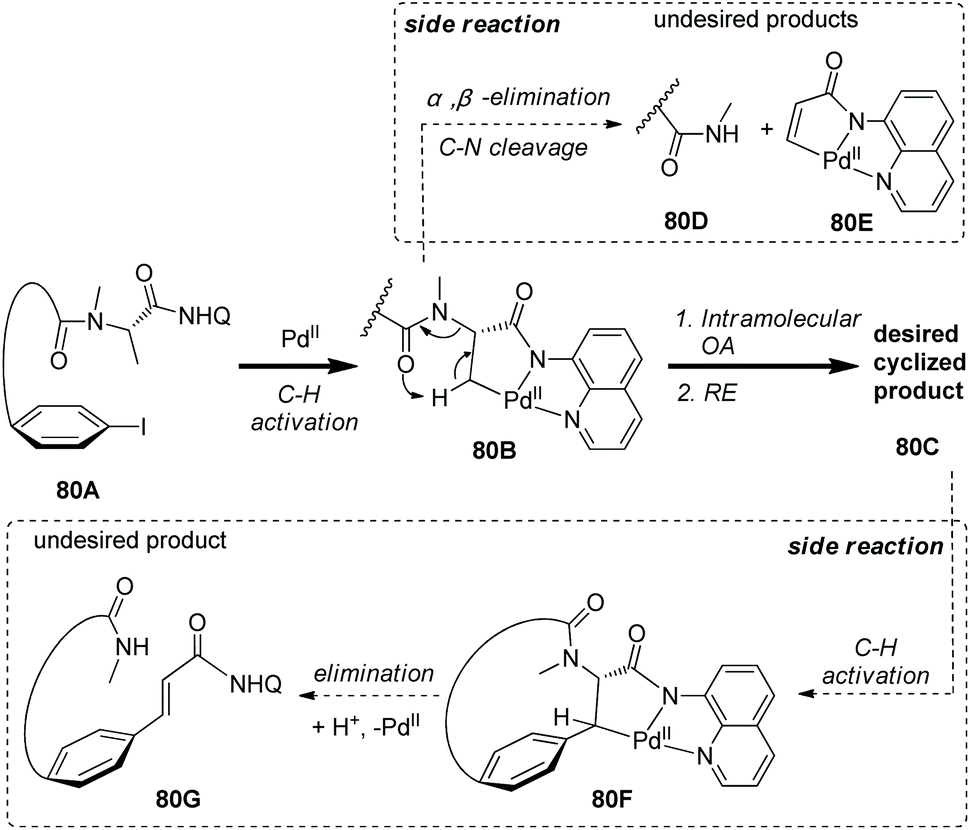
A mechanism was proposed to understand the macrocyclization and the formation of side products (Scheme 80).41 Aminoquinoline-assisted C–H activation gives the palladacycle 80B, and the desired product 80C is formed via intramolecular oxidative addition and reductive elimination. 80B also can give the side products 80D and 80Evia α,β-elimination leading to C–N cleavage, and this process may be initiated by the amide O atom promoted deprotonation via a six-membered transition state. The desired product 80C gives the palladacycle 80Fvia further C–H activation, and the alkene byproduct 80D can be produced via α,β-elimination and subsequent protodepalladation.
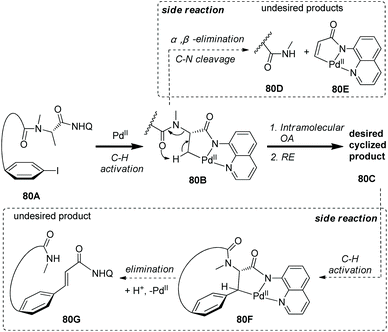 |
| | Scheme 80 Mechanism for macrocyclization by Chen. | |
3. Functionalization assisted by the directing groups derived from the amino group of amino acids, or endogenous unprotected amino groups
3.1. Modification of amino acids
3.1.1. C–C, C–O, and C–N bond formation.
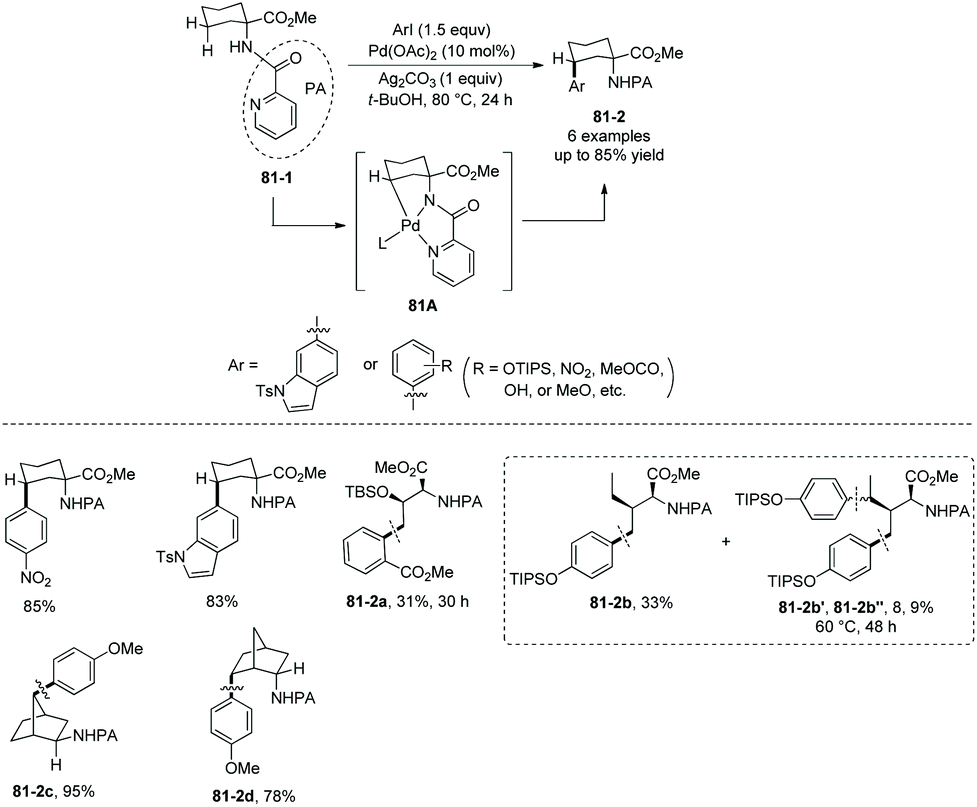
The directing groups also can be installed at the amino group of amino acids. In 2011, a palladium-catalyzed picolinamide-directed γ-sp3 C–H functionalization of amino acid, bornylamine, and 2-methylcyclohexylamine derivatives was developed (Scheme 81) by Chen's group.42 Methyl 1-(picolinamido)cyclohexanecarboxylate 81-1 is monoarylated stereoselectively at the γ-position with aryl iodides; the site-/stereoselectivities are possibly due to the formation of five-membered ring palladacycle 81A in the C–H activation step. Threonine and isoleucine derivatives also work well, affording the monoarylated products as major products (81-2a and 81-2b). The structure of major product 81-2b shows that the C–H activation reactivities of the γ-primary methyl sp3 C–H bonds are bigger than that of γ-secondary methylene sp3 C–H bonds in this transformation. Bisarylated products (81-2b′ and 81-2b′′) are afforded as side products, but less than 3% of gem-diarylated products are found in all cases due to the steric effect. Bornylamine derivatives give the monoarylated products in high to excellent yields (81-2c and 81-2d).
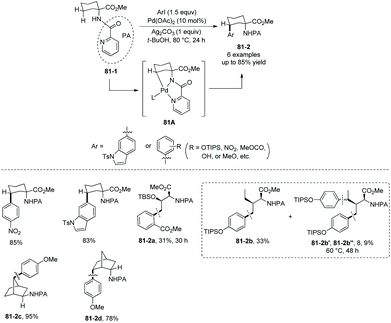 |
| | Scheme 81 Picolinamide-directed arylation. | |
A Pd(II)/Pd(IV) pathway was considered as the mechanism for the picolinamide-directed arylation. Through mechanistic studies including γ-H/D exchange of a substrate under palladium catalysis, it was found that the C–H activation occurs before oxidation addition of Pd(II) with an aryl iodide, and C–H activation is reversible in this transformation.
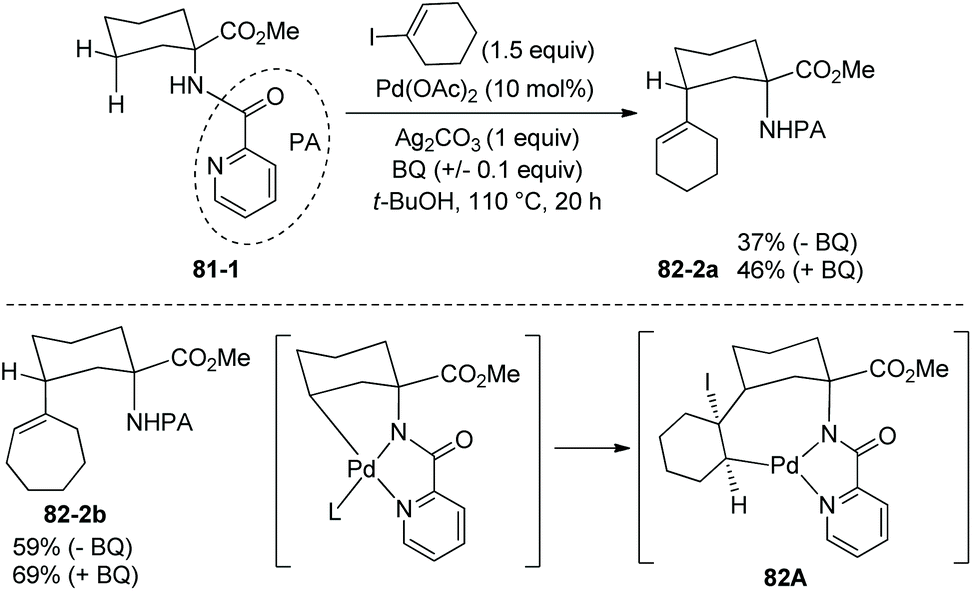
The picolinamide group also can enable alkenylation of 81-1 with cyclic vinyl iodides, and alkenylated products can be obtained successfully (e.g.82-2a and 82-2b, Scheme 82).42 The mechanism for alkenylation is possibly a Pd(II)/Pd(IV) pathway as that for arylation. Migratory insertion of a vinyl iodide across the Pd–C bond is also possible, forming the intermediate 82A, and a trans β-iodine elimination gives the alkenylated products 82-2.
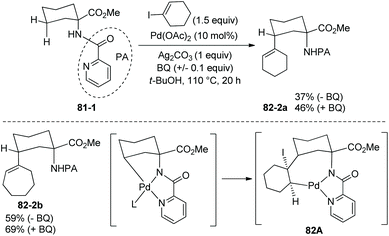 |
| | Scheme 82 Picolinamide-directed alkenylation. | |
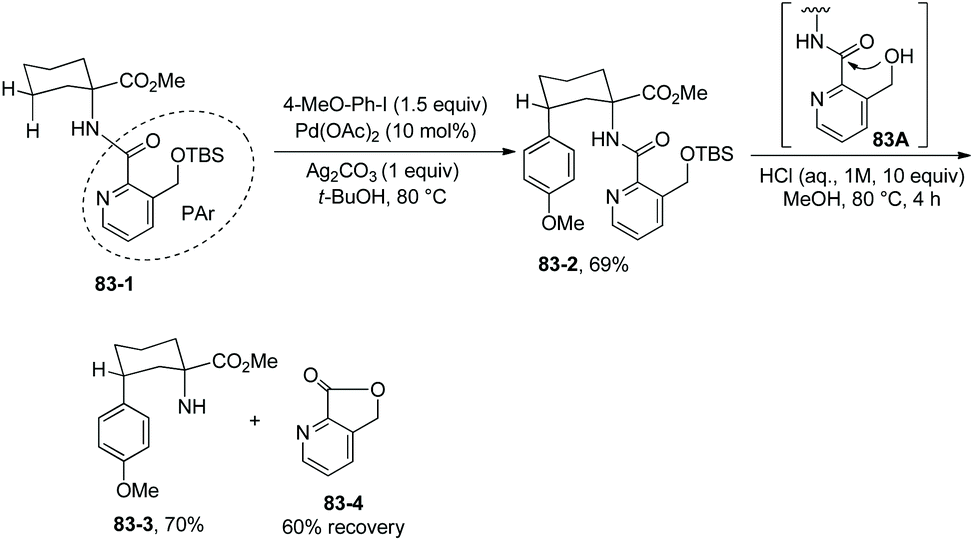
To remove the auxiliary easily, a modified picolinic acid was prepared from a commercially available material and installed on the amino substrate (Scheme 83, 83-1).4283-1 can give the arylated product 83-2 in good yield under the standard conditions. Removal of the new auxiliary PAr is easily accomplished upon treatment of 83-2 with aqueous HCl, and the N-unprotected product 83-3 was obtained in 70% yield; compound 83-4 (a material for preparation of the modified picolinic acid PAr-OH) was recovered in 60% yield. Easy removal of the PAr is probably due to the intramolecular nucleophilic attack of the ortho methylene hydroxy group at the carbonyl group (83A).
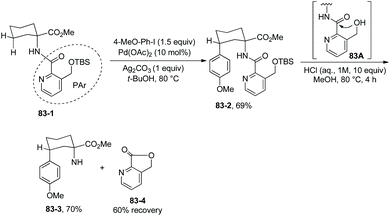 |
| | Scheme 83 A more readily removable auxiliary. | |
In 2013, Chen's group also used picolinamide to achieve alkylation of amino acids (Scheme 84).43 Allo-isoleucine and protected threonine are alkylated at γ-methyl in 62% and 92% yield, respectively. The mechanism involves C–H palladation via a concerted deprotonation/palladation process, oxidative addition probably via a SN2 process and reductive elimination. Dibenzyl phosphate probably acts as a phase transfer catalyst (PTC), bringing silver cations into the liquid phase to polarize the alkyl iodide.
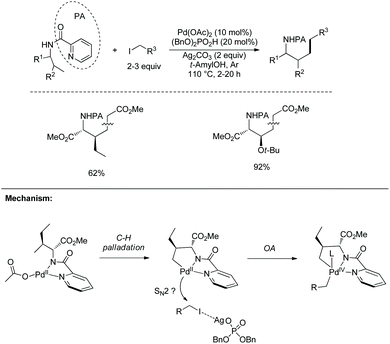 |
| | Scheme 84 Alkylation with PA by Chen. | |
In 2013, Ma's group made use of 2-methoxyiminoacetyl as a directing auxiliary to accomplish the arylation of N-protected methyl (2S)-aminobutanate (Scheme 85).44 The arylation occurs at γ-methyl in up to 88% yield. The mechanism includes C–H palladation forming a fused two five-membered ring Pd(II) complex, oxidative addition of palladium with aryl iodide forming an aryl ligated Pd(IV) complex, and reductive elimination affording the product and releasing Pd(II) (Scheme 85).
 |
| | Scheme 85 Arylation of aminobutanate by Ma. | |
Arylation of the L-valine derivative affords a mixture of mono/diarylated products in a combined yield of 77%; the monoarylated product is produced as a single diastereoisomer with (3S) configuration in 58% yield. L-Threonine, D-allo-isoleucine, and α-quaternary amino acid derivatives all can be monoarylated at the γ-position (Scheme 86, eqn (1)).44L-Threonine derivatives and α-quaternary amino acid derivatives also can be acetoxylated, affording the products in 56% and 87% yield, respectively (Scheme 86, eqn (2)). γ-Methylene in N-protected methyl 2-aminopentanoate, and δ-methyl in N-protected methyl 2-amino-4-methylpentanoate cannot undergo arylation under the arylating reaction conditions.
 |
| | Scheme 86 Arylation and acetoxylation by Ma. | |
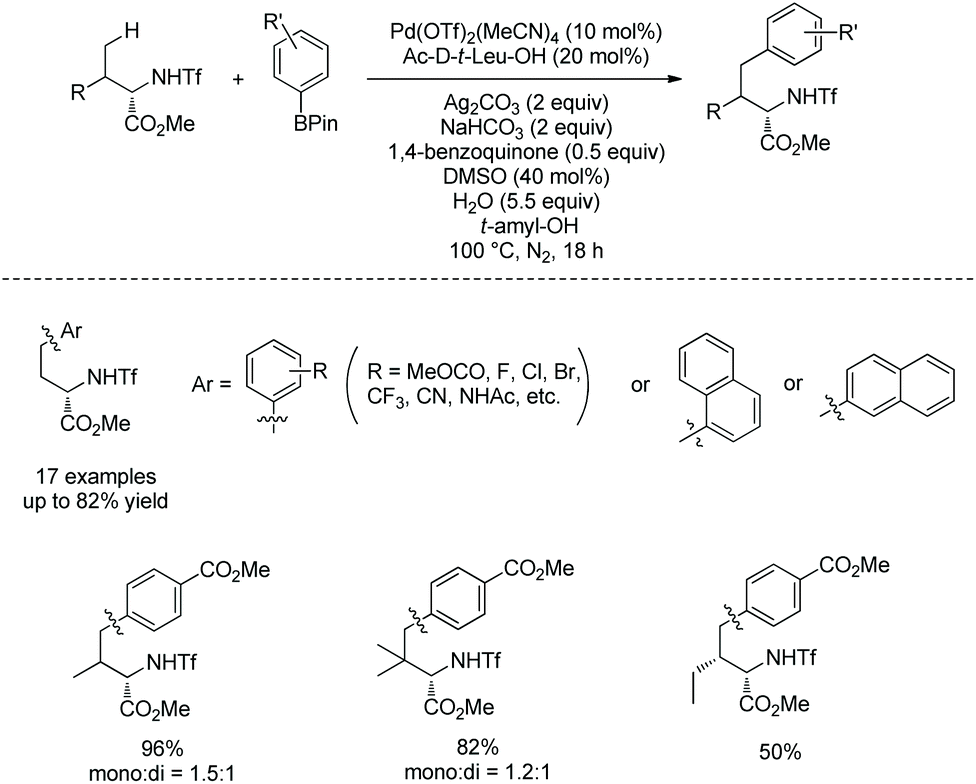
Besides aryl iodides, arylboron reagents were also used as arylating reagents to modify amino acids. Yu's group developed a Pd-catalyzed γ-C(sp3)–H arylation of triflyl-protected α-amino acids employing phenylboronic acid pinacol esters (Scheme 87).45 Methyl N-triflyl-(2S)-aminobutanate can be arylated successfully with a wide range of arylboron reagents. Arylation of valine or t-leucine derivatives affords a mixture of mono/diarylation products in 96% and 82% yield, respectively. Isoleucine derivatives also work well. The arylation with arylboron reagents proceeds via the Pd(II)/Pd(0) catalytic cycle. By contrast, the arylation with aryl iodide proceeds via the Pd(II)/Pd(IV) catalytic cycle.
 |
| | Scheme 87 Arylation with arylboron by Yu. | |
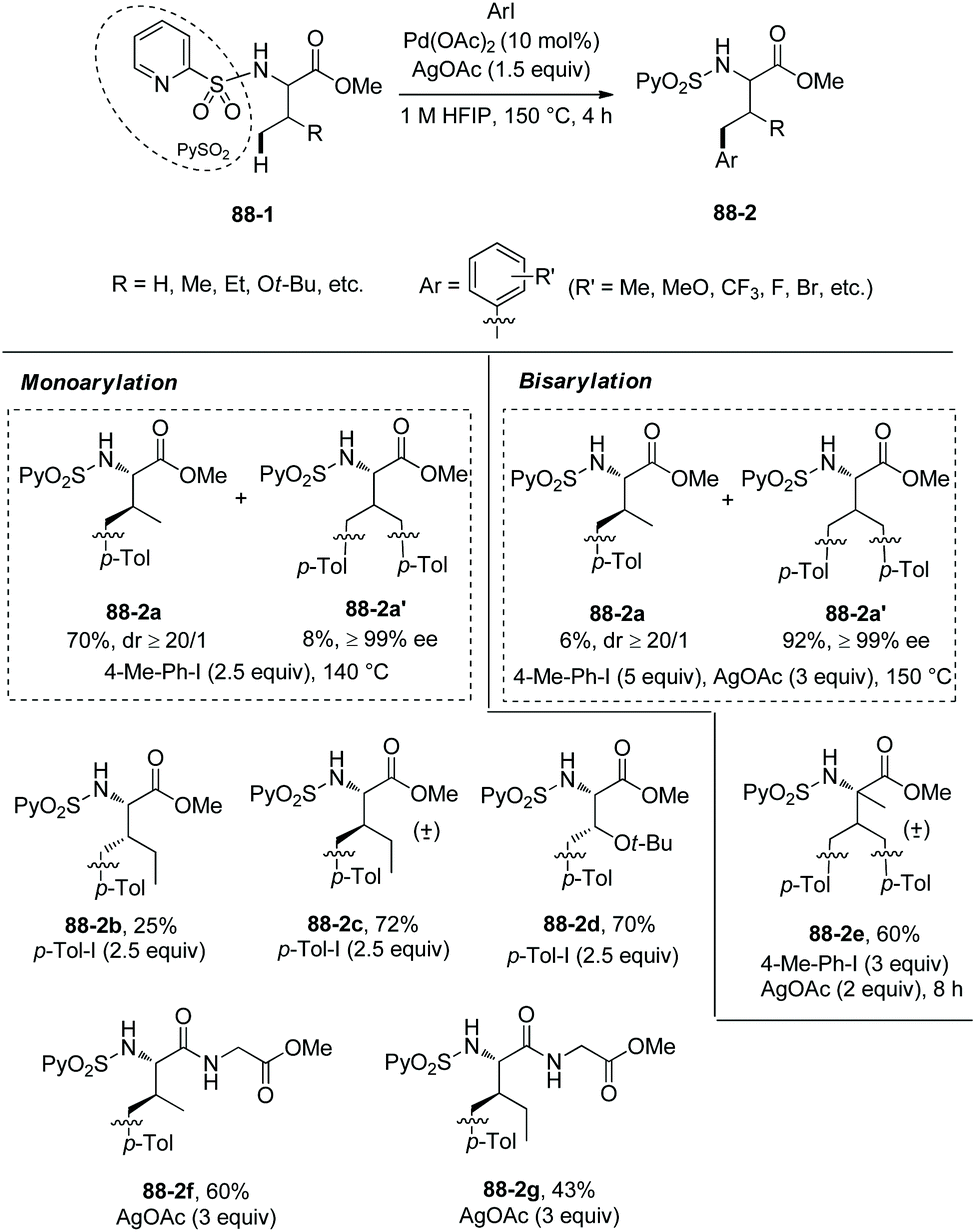
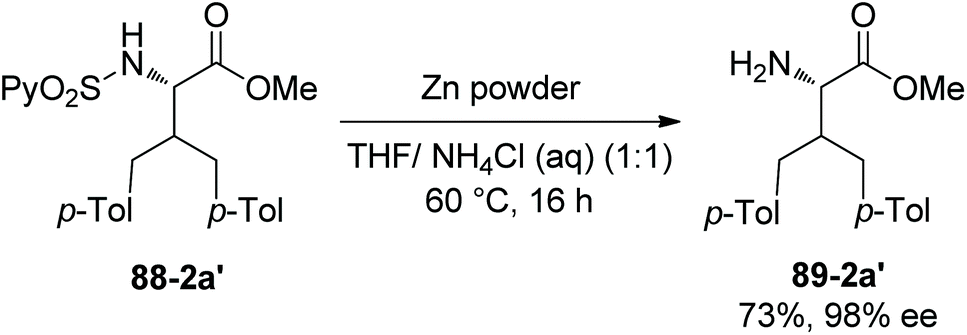
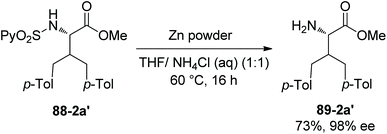
In 2013, N-(2-pyridyl)sulfonyl was used as a directing group for γ-sp3 C–H arylation of amino acid derivatives under palladium catalysis, as reported by the groups of Fernandez-Ibanez and Carretero (Scheme 88).46 The arylation occurs at the γ-position site-selectively, monoarylated product 88-2a is afforded with high diastereoselectivity (dr ≥20:1), the configuration of β-carbon of 88-2a is S, and this is due to forming the more sterically favored five-membered ring palladacycle with trans configuration in the step of C–H activation. The bisarylated product 88-2a′ is obtained with high enantiopurity (99% ee), indicating that no racemization occurs at α-carbon. Gem-diarylation does not occur owing to steric hindrance. Monoarylation product 88-2a is obtained in 70% yield with 2.5 equivalents of 4-Me-Ph-I, and bisarylation product 88-2a′ is afforded in 92% yield with 5 equivalents of 4-Me-Ph-I and 3 equivalents of AgOAc. Besides the N-(2-pyridyl)sulfonamide of L-valine methyl ester (88-2a), the derivatives of isoleucine, allo-isoleucine, protected threonine, homoalanine, α-methylvaline, and a β-amino acid work well, giving mono/bisarylation products smoothly (e.g.(88-2a)–(88-2e), 88-2a′). The derivatives of two dipeptides (valine-glycine and allo-isoleucine-glycine) also can undergo the arylation, affording the products with a γ-aryl group at the N-terminus (88-2f and 88-2g). The groups of Me, MeO, CF3, F and Br at aryl iodides are tolerated in the arylation. The auxiliary PySO2 can be easily removed by treatment of zinc powder (Scheme 89), and the N-unprotected bisarylation product 89-2a′ is afforded in 73% yield and with 98% ee.
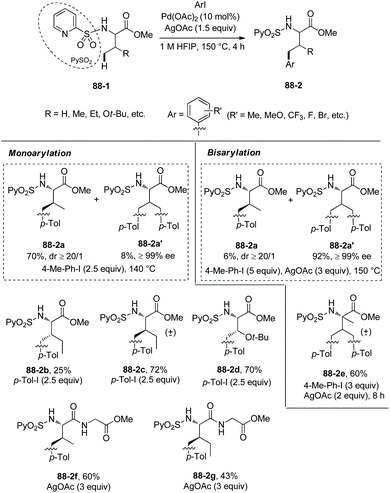 |
| | Scheme 88
N-(2-Pyridyl)sulfonyl directed arylation. | |
 |
| | Scheme 89 Removal of PySO2. | |
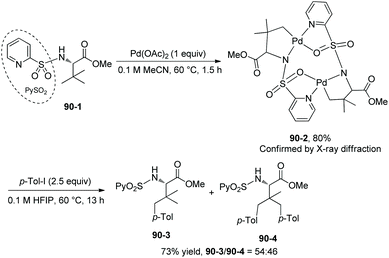
To obtain insights into the mechanism for N-(2-pyridyl)sulfonyl directed arylation, a bimetallic palladacycle 90-2 is obtained by the reaction of N-(2-pyridyl)sulfonyl tert-leucine methyl ester 90-1 and 1 equivalent of palladium acetate (Scheme 90).46 The C–H activation occurs at the γ-position of tert-leucine, forming a five-membered ring palladacycle. The reaction of 90-2 with 2.5 equivalents of p-Tol-I gives mono/bisarylated products (90-3 and 90-4) in high yield (73%).
 |
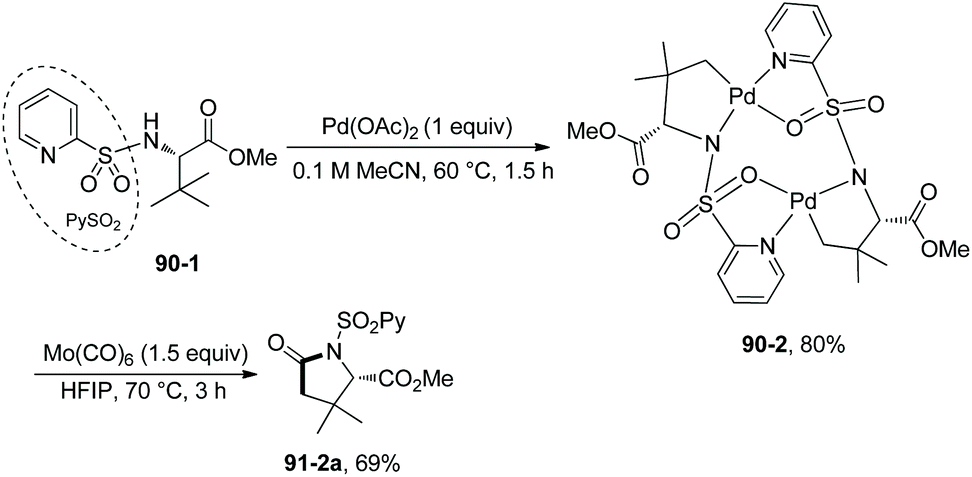
| | Scheme 90 Mechanistic studies. | |
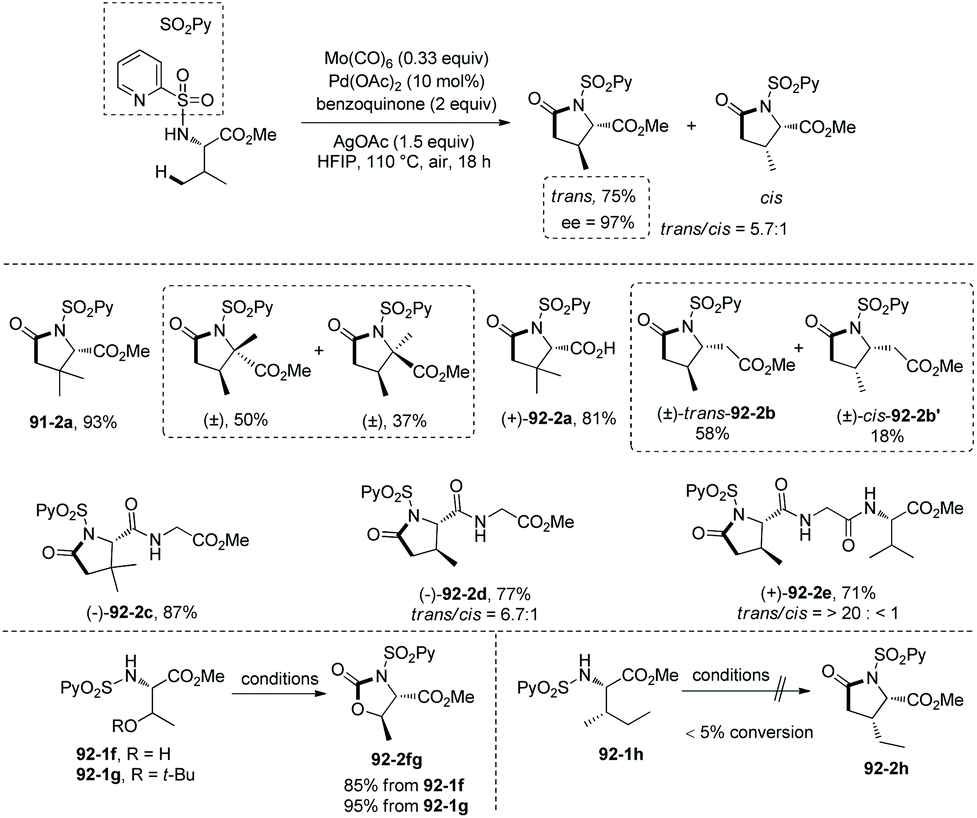
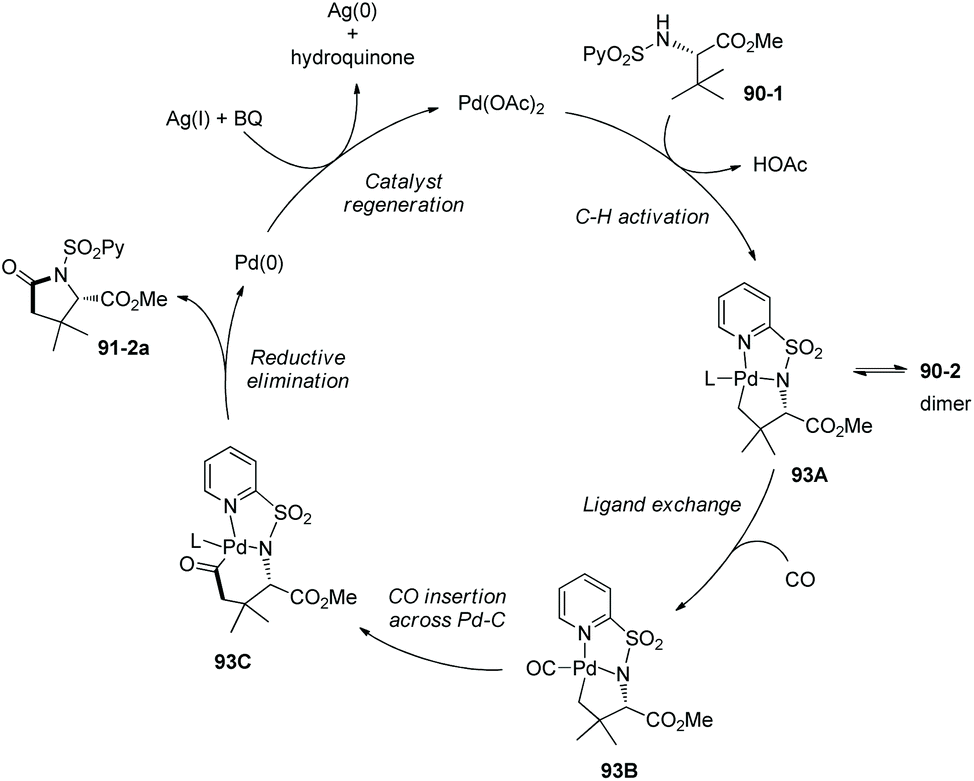
Based on N-(2-pyridyl)sulfonyl-directed arylation of amino acids and peptides, an N-(2-pyridyl)sulfonyl-directed carbonylative cyclization of AAs and peptides was developed in 2016.47 The bimetallic palladacycle 90-2 was used to react with Mo(CO)6, and a lactam 91-2a was obtained in 69% yield (Scheme 91). Then catalytic carbonylative cyclization of AAs and peptides was developed (Scheme 92). The (+)-L-valine, (±)-allo-isoleucine, (±)-2-methylvaline and (+)-L-tert-leucine derivatives give γ-lactams in good yields via γ-sp3 C–H activation/carbonylative cyclization (e.g.91-2a, 92-2a and 92-2c). 92-2a is prepared from the tert-leucine derivative with a free COOH group. A β-amino acid derivative also works well, affording the corresponding γ-lactam successfully (92-2b and 92-2b′). Both the threonine derivatives with a free hydroxyl group (92-1f) or a protecting group (O-t-Bu, 92-1g) give a cyclic carbamate (92-2fg) in good to excellent yields under the same conditions, and γ-methyl C–H activation/carbonylative cyclization does not occur. The (+)-L-isoleucine diastereomer 92-1h cannot give the desired product 92-2h; this possibly results from the less sterically favored palladacycle formed in the step of γ-C–H activation where the ethyl and methyloxycarbonyl groups are orientated in a cis configuration. Two dipeptide derivatives (tert-leucine-glycine and valine-glycine) and a tripeptide derivative (valine-glycine-valine) also can undergo carbonylative cyclization at γ-positions of N-termini, affording the modified peptides ((92-2c)–(92-2e)) in good yields. The formation of tripeptide derivative 92-2e shows that the γ-C(sp3)–H activation/carbonylative cyclization preferentially occurs at the N-terminus over the C-terminus, which indicates that the directing capability of N-(2-pyridyl)sulfonyl overrides that of the native N,N-bidentate ligand in the backbone of the tripeptide. Carbonylative cyclization of aliphatic amine derivatives including an estrone derivative also can be achieved, providing richly functionalized γ-lactams successfully. This method is also effective for γ-, δ-, and ε-sp2 C–H carbonylative cyclization, affording the cyclized products (isoindolinone, 3,4-dihydroisoquinolin-1(2H)-one, and benzo[c]azepine-1-one derivatives) in moderate to excellent yields. The order of sp3 C–H versus sp2 C–H reactivities towards carbonylative cyclization is as follows: γ-sp2 C–H bonds > γ-primary methyl sp3 C–H bonds; δ-sp2 C–H bonds > γ-primary methyl sp3 C–H bonds; and γ-primary methyl sp3 C–H bonds > ε-sp2 C–H bonds.
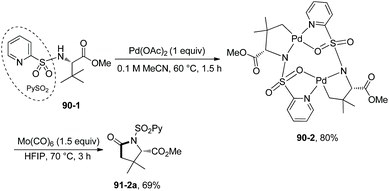 |
| | Scheme 91 Carbonylative cyclization with a palladacycle. | |
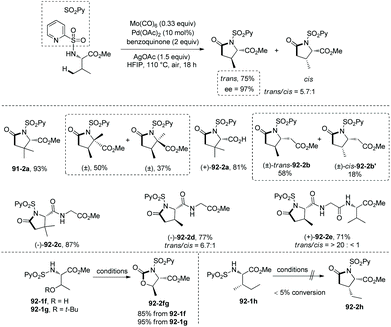 |
| | Scheme 92 Catalytic carbonylative cyclization. | |
A Pd(II)/Pd(0) catalytic cycle was proposed for the carbonylative cyclization (Scheme 93).47 C–H activation with the assistance of the bidentate N-(2-pyridyl)sulfonyl group gives the Pd(II) complex 93A, subsequent ligand exchange affords the intermediate 93B, CO insertion across Pd–C produces the intermediate 67C, reductive elimination forms the product 91-2a, and finally regeneration of the catalyst is achieved with oxidants (AgOAc and benzoquinone).
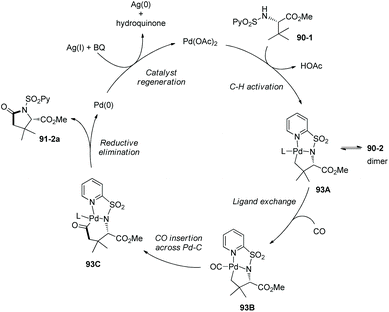 |
| | Scheme 93 Mechanism for carbonylative cyclization. | |
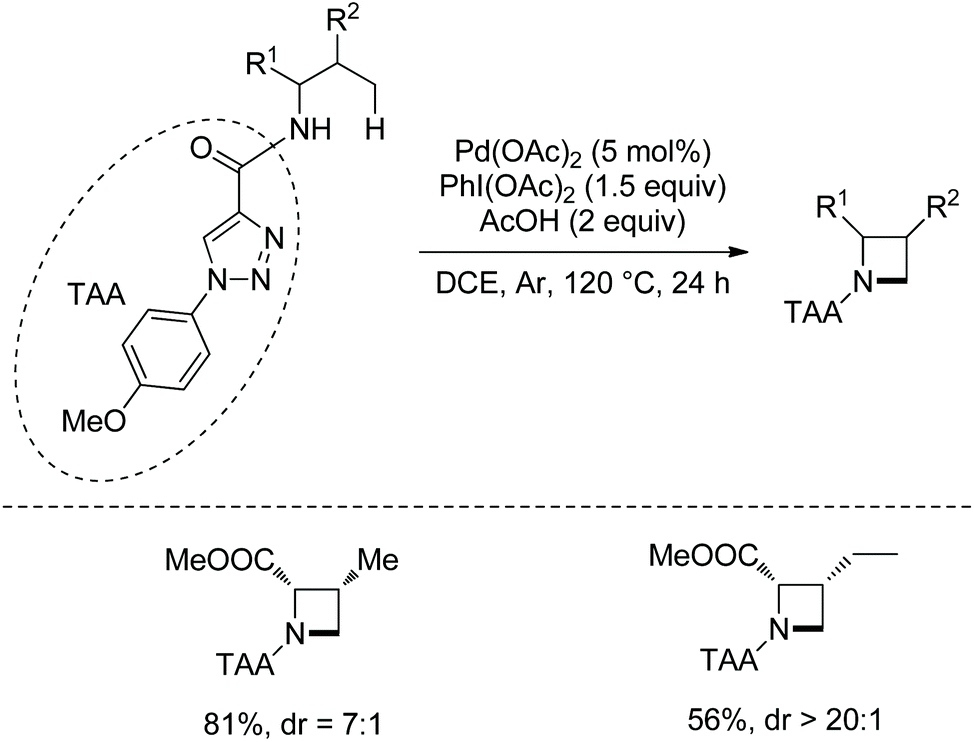
When N1-p-methyoxyphenyl-1,2,3-triazole-4-formyl is installed on the amino group of amino acids, azetidines are formed under palladium catalysis and with PhI(OAc)2 as an oxidant (Scheme 94).48 Valine and isoleucine derivatives cyclized in 81% and 56% yields, respectively. The cyclization occurs selectively at γ-methyl in the isoleucine derivative. The transformation undergoes directed γ-C(sp3)–H activation and intramolecular C–N coupling.
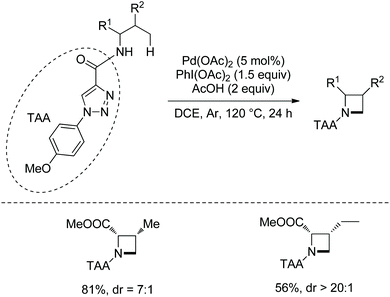 |
| | Scheme 94 Synthesis of azetidines with TAA. | |
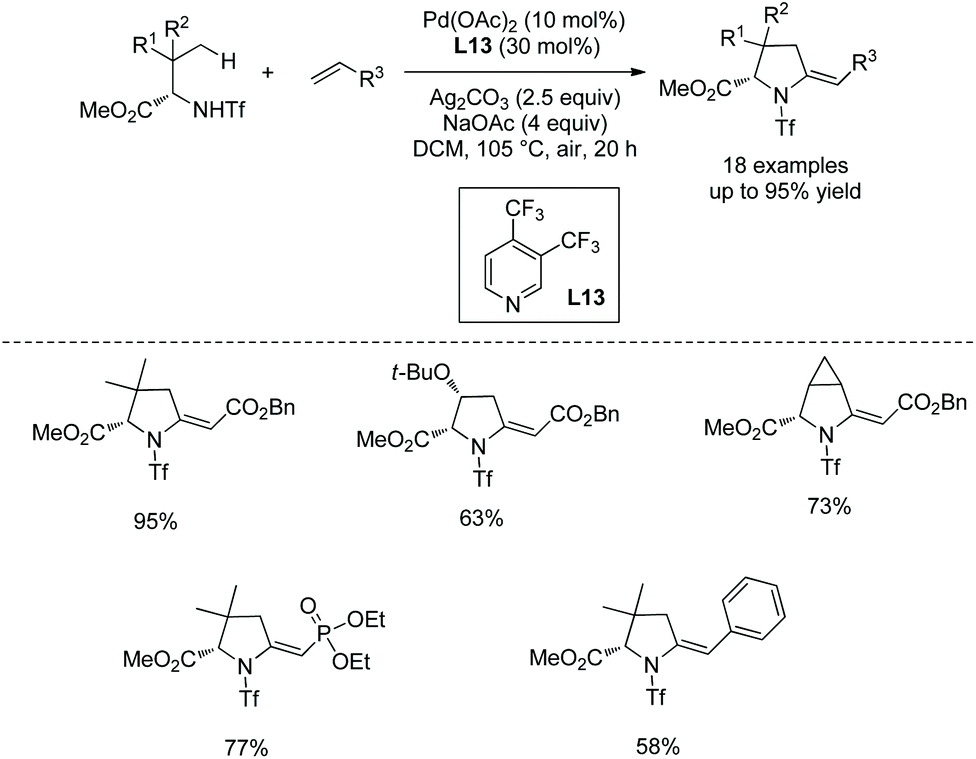
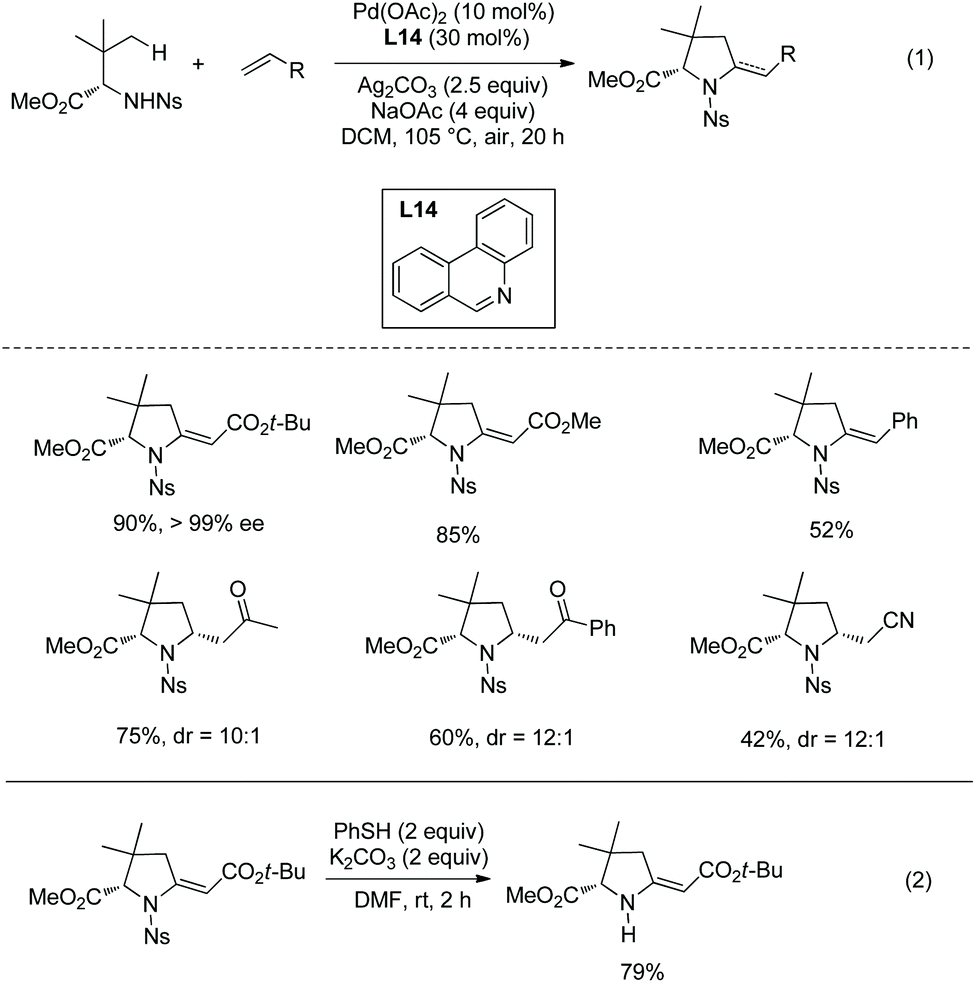
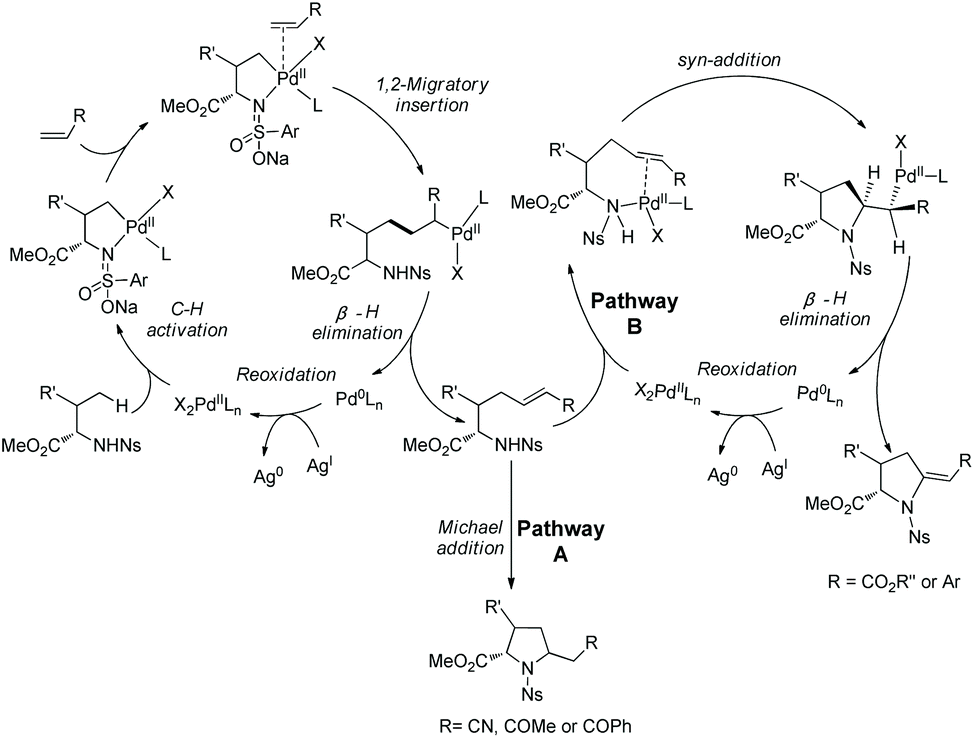
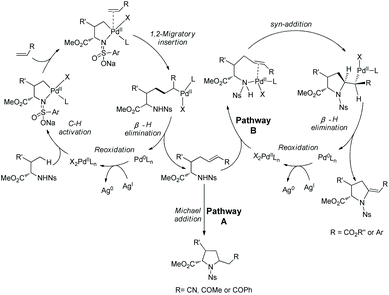
Cyclization following olefination of amino acids affords a variety of 5-substituted-pyrrolidine-2-carboxylates. N-Triflyl (Tf) and 4-nitrobenzenesulfonyl (Ns) protected amino acids can be olefinated with acrylates, diethyl vinylphosphonate, and styrenes on γ-C(sp3)–H bonds.49 The resultant olefins undergo further transformation (aza-Wacker oxidative cyclization or Michael addition) in situ, affording pyrrolidine-2-carboxylates. N-Triflyl (Tf) protected amino acids provide 5-methylene-pyrrolidine-2-carboxylates in up to 95% yields (Scheme 95). L-Valine, L-tert-leucine, L-isoleucine, L-allo-isoleucine, TBSO- and t-Bu-protected L-threonine derivatives etc. all work well. The N-(4-Nitro)benzenesulfonyl (Ns) protected L-tert-leucine derivative provides 5-methylene-pyrrolidine-2-carboxylates when acrylates and styrene are used as olefinating reagents, and gives 5-alkyl-pyrrolidine-2-carboxylates when methyl vinyl ketone, 1-phenylprop-2-en-1-one, and acrylonitrile are used as olefinating reagents (Scheme 96, eqn (1)). The Ns group in a pyrrolidine is removed readily, giving the N-unprotected pyrrolidine with two unchanged ester groups (Scheme 96, eqn (2)). The possible mechanism was proposed (Scheme 97). γ-Olefinated amino acids are produced firstly. Subsequent intramolecular Michael addition gives 5-alkyl-pyrrolidine-2-carboxylates (pathway A). Pathway B affords 5-methylene-pyrrolidine-2-carboxylates via syn-addition and β-H elimination.
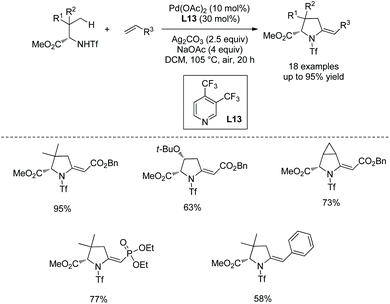 |
| | Scheme 95 Synthesis of pyrrolidines with Tf-protected AAs. | |
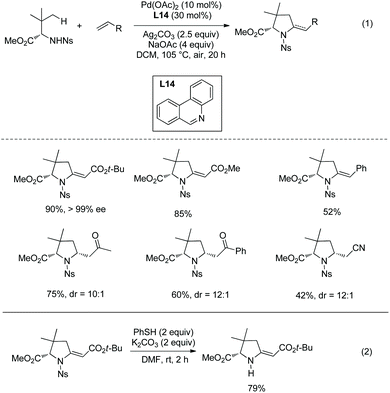 |
| | Scheme 96 Synthesis of pyrrolidines with Ns-protected AAs and removal of Ns. | |
 |
| | Scheme 97 Mechanism for olefination by Yu. | |
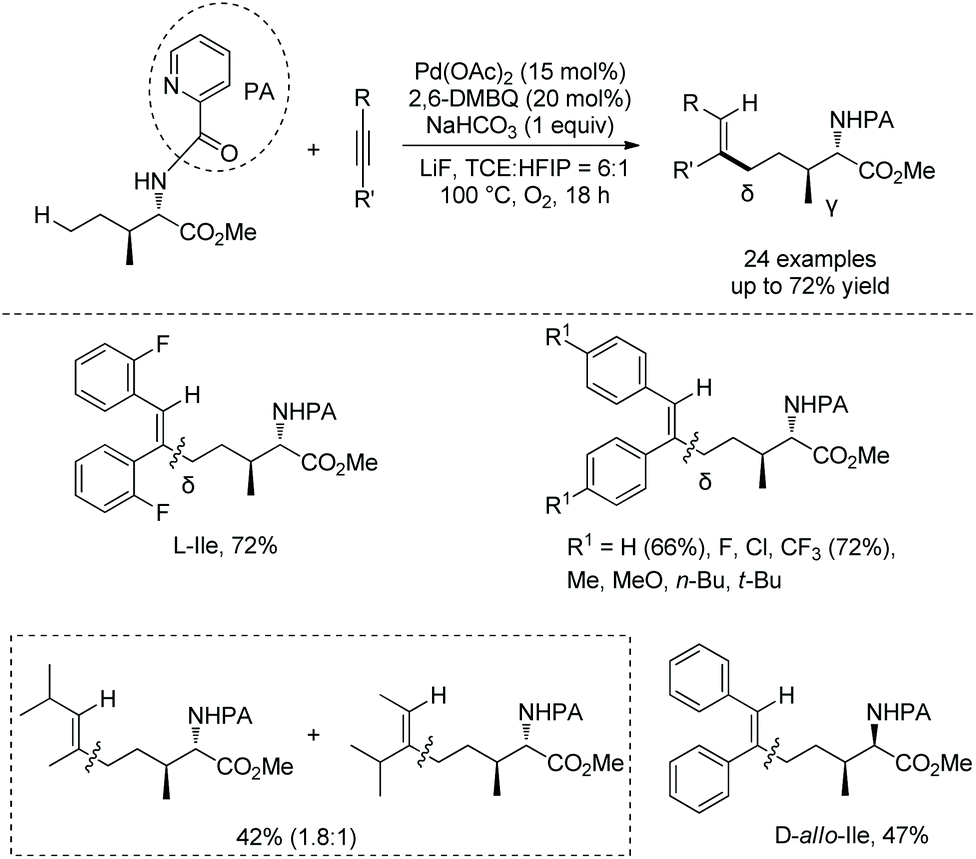
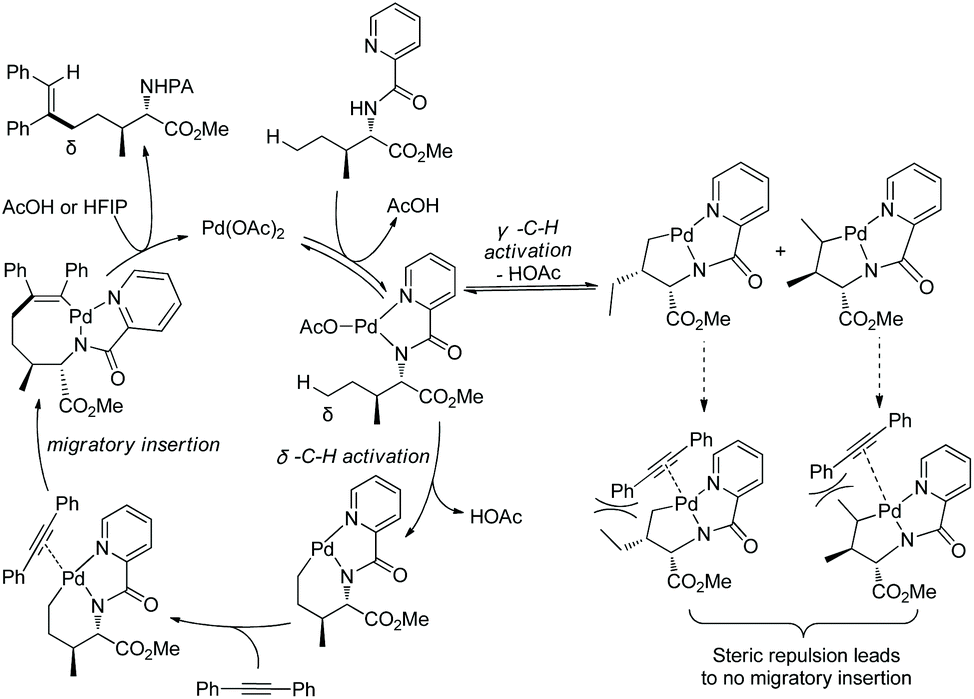
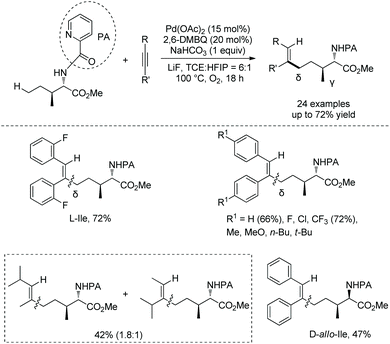
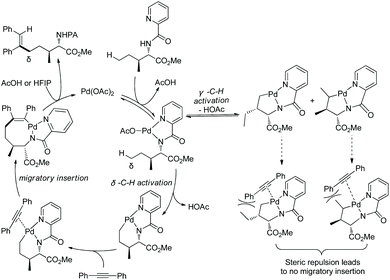
Generally, γ-methyl sp3 C–H bonds of an amine are more favorably activated and functionalized over δ-methyl sp3 C–H bonds under palladium catalysis, which results from the preferential formation of a five-membered ring palladacycle over a six-membered ring palladacycle. By contrast, in 2016, Shi and co-workers reported a δ-sp3 C–H alkenylation of amino acids with alkynes under palladium catalysis (Scheme 98).50 Alkenylation of L-Ile, D-allo-Ile and DL-Ile derivatives all can be achieved. Diarylacetylenes bearing electron-donating or -withdrawing groups including F and Cl are compatible with this reaction. Alkenylation with dialkylacetylenes is also accomplished in moderate yields. The resulting alkenes can be applied to the synthesis of bioactive molecules. The selective δ-C–H alkenylation can be explained by the proposed mechanism (Scheme 99). Big steric repulsion exists when alkynes coordinate to palladium in the palladacycles resulting from γ-C–H activation, and further migratory insertion cannot occur. In the case of δ-C–H activation, there is no such steric repulsion, and the palladacycle resulting from δ-C–H activation can undergo migratory insertion further, leading to the final formation of a δ-alkenylation product.
 |
| | Scheme 98 δ-C–H alkenylation by Shi. | |
 |
| | Scheme 99 Mechanism for alkenylation with alkynes by Shi. | |
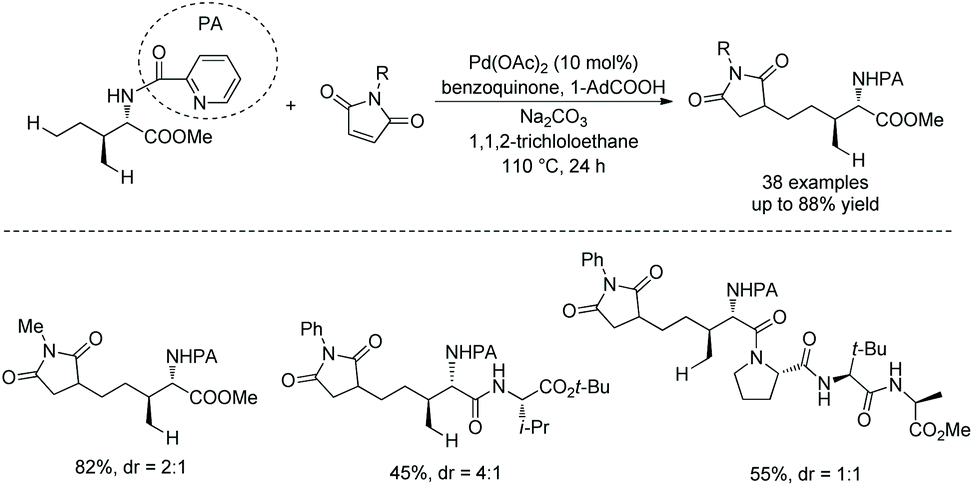
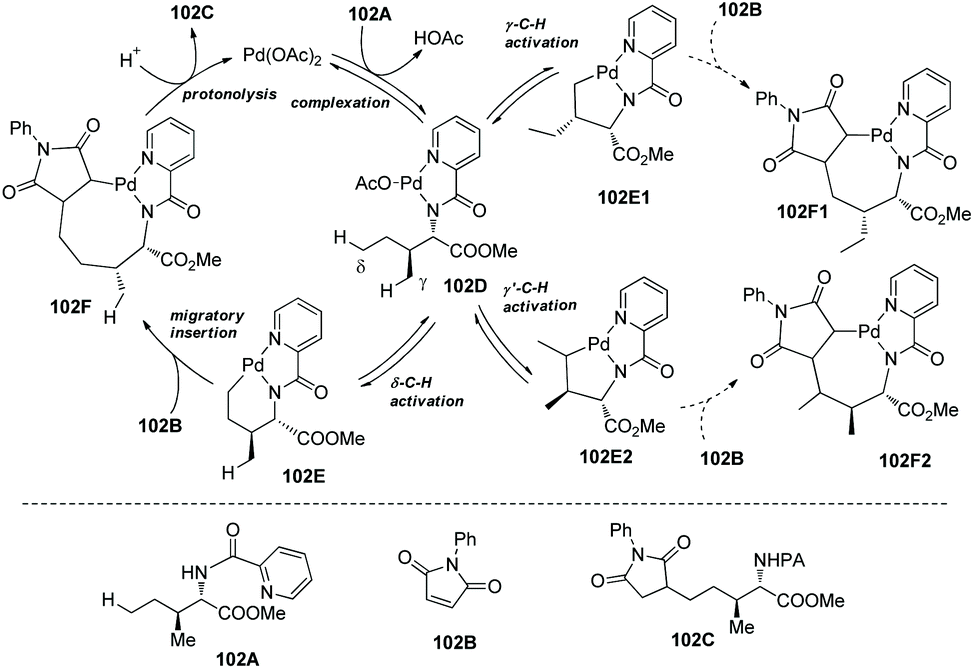
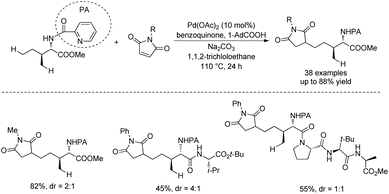
In 2018, Shi and co-workers reported another example about unusual reactivity of δ-sp3 C–H in amino acids. δ-sp3 C–H alkylation of amino acids and peptides with maleimides is achieved, which results from the formation of a six-membered ring palladacycle (Scheme 100).51N-Picolinoyl L-isoleucine methyl ester, and peptides (tri-, tetra-, or pentapeptides) containing N-picolinoyl Ile at N-termini and an ester group at C-termini can be alkylated smoothly with a variety of maleimides.
 |
| | Scheme 100 δ-Alkylation of amino acids and peptides. | |
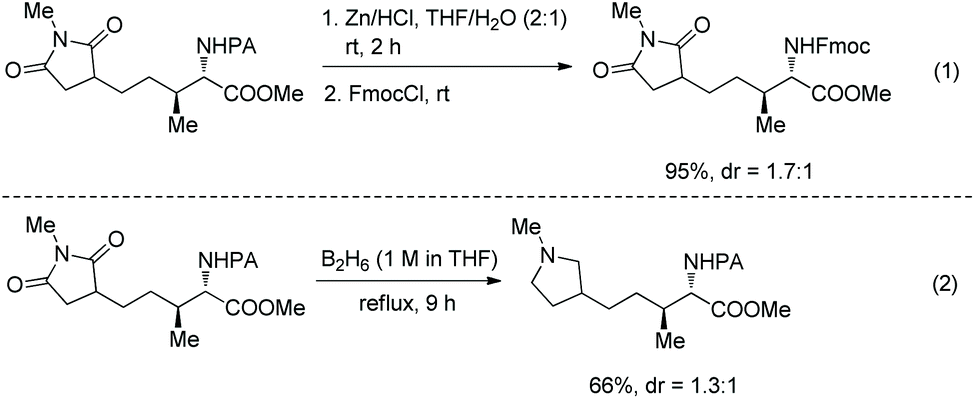
The PA group is removed easily upon treatment of an alkylated product with zinc powder, and useful protecting group Fmoc (fluorenylmethyloxycarbonyl) is installed on the resulting amino group in excellent yield (95% over two steps) (Scheme 101, eqn (1)).51 Reduction of an alkylated product gives tetrahydropyrrole in 66% yield (Scheme 101, eqn (2)).
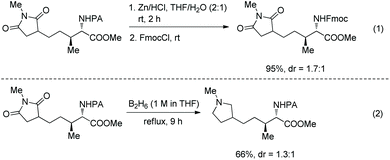 |
| | Scheme 101 Removal of PA and reduction of a product. | |
The H/D exchange experiment shows that the γ- and δ-C–H activation is reversible, and γ-C–H activation is kinetically favored.51 Although the five-membered ring palladacycles 102E1 and 102E2 are preferentially formed over the six-membered ring palladacycle 102E, 102E undergoes migratory insertion of a maleimide much faster than 102E1 and 102E2, then an eight-membered ring palladacylce 102F is produced, and protodepalladation gives the alkylated product 102C (Scheme 102).
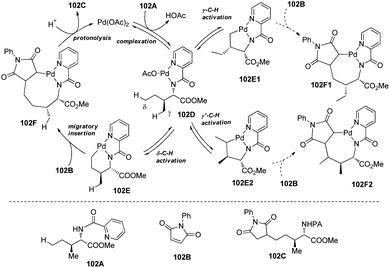 |
| | Scheme 102 Mechanism for δ-C–H alkylation. | |
In 2018, a catalytic transient directing group (2-hydroxynicotinaldehyde) was used for γ-C(sp3)–H and γ-C(sp2)–H arylation of amino acid derivatives under palladium catalysis (Scheme 103).52 Ethyl 2-aminolbutanoate, ethyl 2-amino-2-methylbutanoate, and ethyl 2-amino-3-phenylbutanoate can undergo site-selective γ-C(sp3)–H arylation in moderate to good yields (Scheme 103, eqn (1)). A variety of (hetero) aryl iodides are compatible with the arylation. β-amino acid esters and amino acid bis-esters also work well. However, complete racemization occurs in the arylation of ethyl (S)-2-aminobutanoate. Phenylglycine ethyl esters can undergo γ-C(sp2)–H arylation using slightly modified reaction conditions, giving γ-arylated products in up to 94% yield (Scheme 103, eqn (2)). The transformation tolerates many electron-donating or -withdrawing groups on aryl iodides or phenylglycine ethyl esters. However, the reaction of ethyl (S)-2-amino-2-phenylacetate with ethyl 4-iodobenzoate gives a completely racemic product.
 |
| | Scheme 103 Arylation with TDG. | |
Arylation of ethyl 2-amino-2-methylbutanoate with ethyl 4-iodobenzoate on a 2 mmol scale gives the product in 51% yield (Scheme 104, eqn (1)),52 which can be further arylated with a different aryl iodide (1-iodo-4-methylbenzene), affording a γ-diarylated product with 5:1 dr (Scheme 104, eqn (2)). A 1,2,3,4-tetrahydro-1,8-naphthyridine-2-carboxylate can be synthesized by the arylation of ethyl 2-amino-2-methylbutanoate with 2-fluoro-3-iodopyridine and nucleophilic aromatic substitution in one pot (Scheme 104, eqn (3)). 1,2,3,4-Tetrahydro-1,8-naphthyridine-2-carboxylates are important intermediates for the synthesis of complex organic molecules and pharmaceuticals. Fingolimod (Gilenya) can be afforded by reduction of an arylated product (Scheme 104, eqn (4)).
 |
| | Scheme 104 Synthetic applications. | |
In 2019, unprotected amino groups of amino acids were used as directing groups to modify amino acids, di- and tripeptides under palladium catalysis, as reported by Yao and co-workers (Scheme 105).53 Arylation of ethyl L-tert-leucinate with an aryl iodide gives a mixture of γ-C–H mono-, di- and triarylated products. MeOCO, MeO, F, Me, and NO2 groups are tolerated with this transformation. A little racemization occurs during monoarylation, and the enantiomeric excess (ee) of the corresponding acetamides of monoarylation products is up to 94%. Racemization gradually increases during di- and triarylation. The monoarylation product of ethyl (S)-2-aminobutanoate is almost racemic (5% ee of the corresponding acetamides). Ethyl L-valinate and ethyl L-isoleucinate afford the monoarylated product with moderate dr (2.4:1 and 1.4:1 respectively). Ten dipeptides and a tripeptide with an N-unprotected Tle residue at N-termini all can be arylated at γ-C(sp3)–H, and the steric configuration of all α-carbons is retained. Val, Ala, Phe, Ile, Leu, Tle, and some unnatural amino acids are tolerated at C-termini.
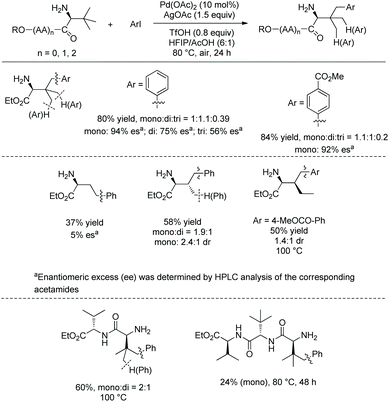 |
| | Scheme 105 Amino-directed arylation. | |
3.1.2. C–Si bond formation.
In 2019, Shi's group made use of picolinamide to synthesize silicon-containing amino acids and peptides with hexamethyldisilane via palladium-catalyzed γ-C(sp3)–H silylation (Scheme 106).54N-Picolinoyl (PA) amino acid derivatives such as L-valine methyl ester, L-valine tert-butyl ester, L-isoleucine esters (methyl, ethyl, tert-butyl, and benzyl esters), t-Bu protected L-threonine methyl ester, L-tert-leucine methyl ester, and L-2-aminobutyric acid methyl ester can give a γ-C(sp3)–H monosilylated product or a mixture of mono/disilylated products (Scheme 106, eqn (1)). The silylation proceeds smoothly with moderate to high yields (up to 88%) and with excellent diastereoselectivities (up to 99% ee). No racemization was observed. The stereoselectivity is probably due to the formation of a palladacycle with a sterically favored trans configuration. N-Picolinoyl L-norvaline methyl ester gives solely the δ-methyl C–H silylation product in 11% yield. Amino alcohol derivatives such as N-picolinoyl (PA)-protected O-acetyl-protected L-tert-leucinol and L-valinol also can afford γ-C(sp3)–H silylation products in acceptable yields (53%, mono:di = 2:1; 69%, mono:di = 3.3:1, respectively). Other disilanes and hexamethyldigermane also can be coupled with amino acids albeit with lower yields. Simple aliphatic picolinamides without an ester or alcohol group give low yields. Di-, tri-, and tetrapeptides also can be silylated with hexamethyldisilane under slightly changed reaction conditions (Scheme 106, eqn (2) and (3)). The silylation occurs at the γ-positions of N-termini. Many amino acid residues such as L-valine, L-tert-leucine, L-2-aminobutyric acid, methyl protected L-threonine, and L-isoleucine residues at N-termini are tolerated in the reaction, giving the silylated products in reasonable yields.
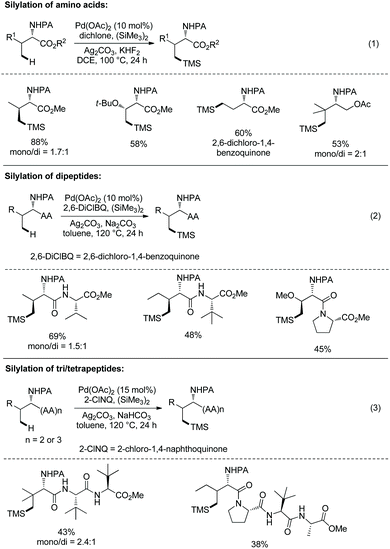 |
| | Scheme 106 Silylation of amino acids and peptides. | |
The auxiliary PA in silylation products can be removed by treatment with zinc powder, and the resulting amino group can be protected with a fluorenylmethyloxycarbonyl group (Fmoc) (Scheme 107, eqn (1)).54 A silylated product is further methoxylated at the γ-position where TMS has been coupled (Scheme 107, eqn (2)). The unusual chemoselectivity is probably due to the α-silicon effect.
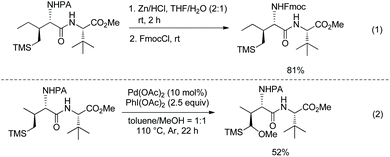 |
| | Scheme 107 Removal of PA and further C–H methoxylation. | |
3.1.3. C–B bond formation.
Organoboron compounds are widely used in coupling reactions such as Suzuki coupling. In 2014, Shi and co-workers developed Pd-catalyzed picolinamide-directed γ-C(sp3)–H borylation with bis(pinacolato)diboron to modify amino acids (Scheme 108).55 Cheap and harmless oxygen is employed as the oxidant, so it is relatively an economic and environmentally friendly transformation. Borylation of L-valine gives a γ-borylated product in 68% yield with high diastereoselectivity (dr = 83:17). L-Isoleucine, DL-isoleucine, L-O-t-Bu-threonine and L-tert-leucine also work well, affording the γ-borylated product in high to good yields.
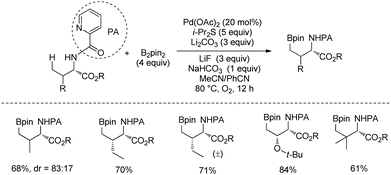 |
| | Scheme 108 Borylation. | |
3.2. Formation of cyclic peptides
At the front of the article, the synthesis of cyclophane-braced peptide macrocycles has been introduced, and the synthetic methods include the palladium-catalyzed backbone-assisted N-terminal sp3 C–H activation/macrocyclization reported by the groups of Noisier, Albericio and Wang,38,39 and the palladium-catalyzed 8-aminoquinoline-assisted C-terminal sp3 C–H activation/macrocyclization by the group of Chen.40,41 In 2019, Chen and co-workers developed another method for the synthesis of cyclophane-braced peptide macrocycles, and the palladium-catalyzed picolinamide-directed N-terminal γ-sp3 C–H activation/macrocyclization (Scheme 109).56 The cyclization undergoes directed C–H palladation forming a five-membered ring palladacycle, oxidative addition (OA) of the palladacycle with aryl iodide forming a Pd(IV) complex, and reductive elimination (RE) of the Pd(IV) complex to form the intramolecularly arylated product. L-Abu, L-Ile, D-allo-Ile, L-Val, L-Tle, L-CPrg (cyclopropylglycine), L-Chg (cyclohexylglycine), and L-Cpg (cyclopentylglycine), and L-Nva at the N-termini of peptides work well to give macrocyclization products. Both γ-methyl sp3 C–H bonds and γ-methylene sp3 C–H bonds can undergo the cyclization. (m-I)Phe, (p-I)Phe and (m-I)Pyr at the C-termini are used for macrocyclization, in which aryl groups are coupled with the sp3 C–H bonds at the N-termini. Nonaromatic residues (Ser, Lys, and Glu) with prosthetic iodinated aromatic groups installed also can serve as an intramolecular coupling partner. An organic solvent (HFIP) is suitable for some organic solvent-soluble substrates (Scheme 76). The product with a bent benzene ring is formed via reaction in HFIP, showing that the transformation is powerful. Protected polar side chains such as Lys(Cbz), Arg(Pbf), and Glu(tBu) can be well tolerated in reactions in HFIP. Unprotected carboxyl and amino groups inhibited the macrocyclization in HFIP due to the strong coordinative ability to palladium. Water or a mixed solvent of water and organic solvents is the choice when substrates are water soluble or less water soluble (Scheme 110). Unprotected groups of COOH (Asp), CO2NH2 (Gln), NH2 (Lys), OH (Thr and Ser), and guanidine (Arg) are tolerated in aqueous medium, which is probably due to their weakened coordinating abilities in the slightly acidic aqueous solution. However, the groups of SH (Cys), imidazole (His), and indole (Trp) cannot be tolerated. Large-sized rings such as a 10-mer with a 36-membered ring can be synthesized in good yields.
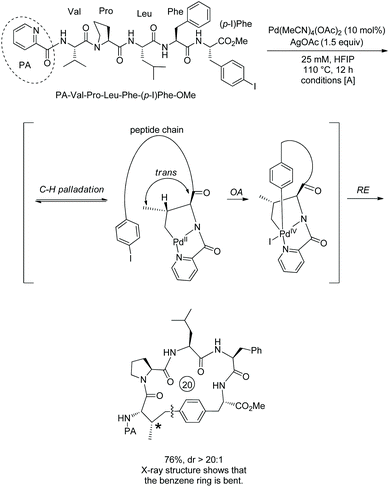 |
| | Scheme 109 Peptide macrocyclization in organic solvents. | |
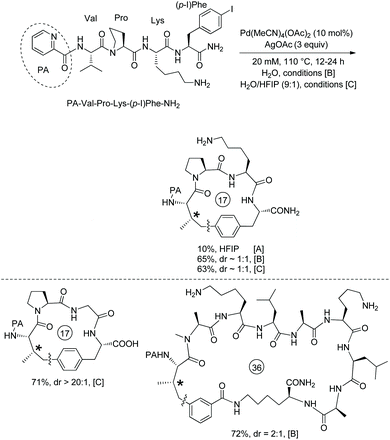 |
| | Scheme 110 Peptide macrocyclization in aqueous medium. | |
The directing auxiliary PA can be removed easily by treatment of the cyclopeptides with zinc powder at room temperature, and further modification provides a Fmoc-protected cyclopeptide (Scheme 111, eqn (1)), or a cyclopeptide with pyroglutamic acid coupled (a close mimic of hibispeptin, Scheme 111, eqn (2)). The methyl ester group and peptide bonds remain untouched in these conversions.
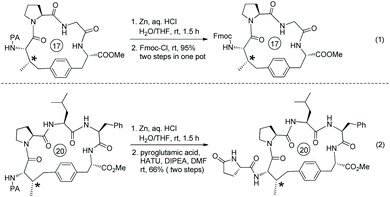 |
| | Scheme 111 Removal of PA and further modification. | |
4. Conclusions
The palladium-catalyzed C–H activation/functionalization of amino acids and peptides occurs at the β-, γ-, or δ-sp3 C–H bonds of amino acids or residues respectively with the assistance of different directing groups. In some cases, the site-/diastereoselectivities are excellent. Sixteen directing auxiliaries for the sp3 C–H activation/functionalization of amino acid and peptide derivatives are introduced, which are installed on the substrates, forming N-monodentate, or N,N-/N,O-/N,S-bidentate ligands. These directing auxiliaries enable sp3 C–H activation/functionalization with different site- (or chemo, diastereo) selectivities or reactivities. The functionalization assisted with these directing auxiliaries includes arylation, alkylation, fluorination, acetoxylation, alkenylation, silylation, borylation, sulfonylation, chalcogenation, intramolecular C–N bond-forming cyclization, and carbonylative cyclization. The native free carboxyl or amino groups also can promote arylation of amino acids or peptides. The native N,O- or N,N-bidentate ligands of peptide backbones or asparagine residue are also introduced, promoting sp3 C–H arylation, acetoxylation, and alkynylation. Intramolecular arylation of peptides forms cyclophane-braced peptide macrocycles. A transient directing group (2-hydroxynicotinaldehyde) is introduced as well, enabling arylation of amino acid derivatives; however, complete racemization occurs at the α-carbon of some substrates. The main trends are as follows. (1) The sp3 C–H activation/functionalization of amino acid and peptide derivatives with a transient directing group (TDG) remains to be developed further, and the stereoselectivity of the transformation with TDG remains to be improved. It is expected that in the future transient directing groups with unique structures will be designed and employed for these purposes. (2) The alkylating reagents used are only limited to alkyl halides, and other reagents such as alkylating boron or silicon derivatives will be used. (3) Stoichiometric chemical oxidants are used to form carbon–oxygen bonds, green oxidation methods involving electrochemistry will be introduced in the functionalization of amino acids and peptides to form new bonds including carbon–oxygen bonds. (4) Palladium catalysis is mainly used to modify amino acids and peptides, and cheap metal catalysts such as copper, cobalt and nickel catalysts will be used for the C–H functionalization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21262017 and 21366023).
Notes and references
-
(a) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, Palladium(II)-Catalyzed Enantioselective C(sp3)-H Activation Using a Chiral Hydroxamic Acid Ligand, J. Am. Chem. Soc., 2014, 136, 8138–8142 CrossRef CAS;
(b) G. Desimoni, G. Faita and P. Quadrelli, Pyridine-2,6-bis(oxazolines), Helpful Ligands for Asymmetric Catalysts, Chem. Rev., 2003, 103, 3119–3154 CrossRef CAS.
-
(a) T. Tsushima, K. Kawada, T. Tsuji and K. Tawara, Fluorine-containing amino acids and their derivatives. 4. Synthesis and antibacterial activity of threo and erythro 1-fluorodehydroxylated chloramphenicol analogs, J. Med. Chem., 1985, 28, 253–256 CrossRef CAS;
(b) A. A. Kaspar and J. M. Reichert, Future directions for peptide therapeutics development, Drug Discovery Today, 2013, 18, 807–817 CrossRef CAS;
(c) F. Albericio and H. G. Kruger, Therapeutic peptides, Future Med. Chem., 2012, 4, 1527–1531 CrossRef CAS.
- J. Kobayashi, H. Suzuki, K. Shimbo, K. Takeya and H. Morita, Celogentins A–C, New Antimitotic Bicyclic Peptides from the Seeds of Celosia argentea, J. Org. Chem., 2001, 66, 6626–6633 CrossRef CAS.
- H. Mei, J. Han, K. D. Klika, K. Izawa, T. Sato, N. A. Meanwell and V. A. Soloshonok, Applications of fluorine-containing amino acids for drug design, Eur. J. Med. Chem., 2020, 186, 111826 CrossRef CAS.
-
(a) X.-L. Qiu and F.-L. Qing, Recent Advances in the Synthesis of Fluorinated Amino Acids, Eur. J. Org. Chem., 2011, 3261–3278 CrossRef CAS;
(b) X.-L. Qiu, W.-D. Meng and F.-L. Qing, Synthesis of fluorinated amino acids, Tetrahedron, 2004, 60, 6711–6745 CrossRef CAS;
(c) A. Sutherland and C. L. Willis, Synthesis of fluorinated amino acids, Nat. Prod. Rep., 2000, 17, 621–631 RSC;
(d) A. M. Remete, M. Nonn, S. Fustero, F. Fulop and L. Kiss, Synthesis of fluorinated amino acid derivatives through late-stage deoxyfluorinations, Tetrahedron, 2018, 74, 6367–6418 CrossRef CAS.
-
(a) J. Diesel and N. Cramer, Generation of Heteroatom Stereocenters by Enantioselective C-H Functionalization, ACS Catal., 2019, 9, 9164–9177 CrossRef CAS;
(b) J. Thongpaen, R. Manguin and O. Balse, Chiral N-Heterocyclic Carbene Ligands Enable Asymmetric C-H Bond Functionalization, Angew. Chem., 2020, 59, 10242–10251 CrossRef CAS;
(c) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak,
N-Heterocyclic Carbene Complexes in C-H Activation Reactions, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS;
(d) S. Rej, Y. Ano and N. Chatani, Bidentate Directing Groups: An Efficient Tool in C-H Bond Functionalization Chemistry for the Expedient Construction of C-C Bonds, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS;
(e) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, From Pd(OAc)2 to Chiral Catalysts: The Discovery and Development of Bifunctional Mono-N-Protected Amino Acid Ligands for Diverse C-H Functionalization Reactions, Acc. Chem. Res., 2020, 53, 833–851 CrossRef CAS;
(f) L. Ackermann, Metalla-electrocatalyzed C-H Activation by Earth-Abundant 3d Metals and Beyond, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS;
(g) K. Yang, M. Song, H. Liu and H. Ge, Palladium-catalyzed direct asymmetric C-H bond functionalization enabled by the directing group strategy, Chem. Sci., 2020 10.1039/d0sc03052j;
(h) M. Zhang, Q. Wang, Y. Peng, Z. Chen, C. Wan, J. Chen, Y. Zhao, R. Zhang and A. Q. Zhang, Transition metal-catalyzed sp3 C-H activation and intramolecular C-N coupling to construct nitrogen heterocyclic scaffolds, Chem. Commun., 2019, 55, 13048–13065 RSC;
(i) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Transition Metal-Catalyzed Enantioselective C-H Functionalization via Chiral Transient Directing Group Strategies, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS;
(j) Z. Chen, M.-Y. Rong, J. Nie, X.-F. Zhu, B.-F. Shi and J.-A. Ma, Catalytic alkylation of unactivated C(sp3)–H bonds for C(sp3)-C(sp3) bond formation, Chem. Soc. Rev., 2019, 48, 4921–4942 RSC;
(k) Q. Zhang and B.-F. Shi, From Reactivity and Regioselectivity to Stereoselectivity: An Odyssey of Designing PIP Amine and Related Directing Groups for C-H Activation, Chin. J. Chem., 2019, 37, 647–656 CrossRef CAS;
(l) H.-R. Tong, B. Li, G. Li, G. He and G. Chen, Postassembly Modifications of Peptides via Metal-Catalyzed C-H Functionalization, CCS Chem., 2020, 2, 1797–1820 CrossRef.
- B. V. S. Reddy, L. R. Reddy and E. J. Corey, Novel Acetoxylation and C-C Coupling Reactions at Unactivated Positions in α-Amino Acid Derivatives, Org. Lett., 2006, 8, 3391–3394 CrossRef CAS.
- Y. Feng and G. Chen, Total Synthesis of Celogentin C by Stereoselective C-H Activation, Angew. Chem., Int. Ed., 2010, 49, 958–961 CrossRef CAS.
- L. D. Tran and O. Daugulis, Nonnatural Amino Acid Synthesis by Using Carbon-Hydrogen Bond Functionalization Methodology, Angew. Chem., Int. Ed., 2012, 51, 5188–5191 CrossRef CAS.
- S.-Y. Zhang, Q. Li, G. He, W. A. Nack and G. Chen, Stereoselective Synthesis of β-Alkylated α-Amino Acids via Palladium-Catalyzed Alkylation of Unactivated Methylene C(sp3)-H Bonds with Primary Alkyl Halides, J. Am. Chem. Soc., 2013, 135, 12135–12141 CrossRef CAS.
- K. Chen, F. Hu, S.-Q. Zhang and B.-F. Shi, Pd(II)-catalyzed alkylation of unactivated C(sp3)-H bonds: efficient synthesis of optically active unnatural α-amino acids, Chem. Sci., 2013, 4, 3906–3911 RSC.
- K. Chen and B.-F. Shi, Sulfonamide-Promoted Palladium(II)-Catalyzed Alkylation of Unactivated Methylene C(sp3)-H Bonds with Alkyl Iodides, Angew. Chem., Int. Ed., 2014, 53, 11950–11954 CrossRef CAS.
- Q. Yang and S.-D. Yang, Highly Efficient and Divergent Construction of Chiral γ-Phosphono-α-Amino Acids via Palladium-Catalyzed Alkylation of Unactivated C(sp3)-H Bonds, ACS Catal., 2017, 7, 5220–5224 CrossRef CAS.
- G. He, S.-Y. Zhang, W. A. Nack, Q. Li and G. Chen, Use of a Readily Removable Auxiliary Group for the Synthesis of Pyrrolidones by the Palladium-Catalyzed Intramolecular Amination of Unactivated γ C(sp3)-H Bonds, Angew. Chem., Int. Ed., 2013, 52, 11124–11128 CrossRef CAS.
- Q. Zhang, K. Chen, W. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Stereoselective Synthesis of Chiral α-Amino-β-Lactams through Palladium(II)-Catalyzed Sequential Monoarylation/Amidation of C(sp3)-H Bonds, Angew. Chem., Int. Ed., 2013, 52, 13588–13592 CrossRef CAS.
- P.-X. Ling, S.-L. Fang, X.-S. Yin, Q. Zhang, K. Chen and B.-F. Shi, Palladium-catalyzed sequential monoarylation/amidation of C(sp3)-H bonds: stereoselective synthesis of α-amino-β-lactams and anti-α,β-diamino acid, Chem. Commun., 2017, 53, 6351–6354 RSC.
- B. Wang, W. A. Nack, G. He, S.-Y. Zhang and G. Chen, Palladium-catalyzed trifluoroacetate-promoted mono-arylation of the β-methyl group of alanine at room temperature: synthesis of β-arylated α-amino acids through sequential C-H functionalization, Chem. Sci., 2014, 5, 3952–3957 RSC.
- K. Chen, S.-Q. Zhang, J.-W. Xu, F. Hu and B.-F. Shi, A general and practical palladium-catalyzed monoarylation of β-methyl C(sp3)-H of alanine, Chem. Commun., 2014, 50, 13924–13927 RSC.
- J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Ligand-Controlled C(sp3)-H Arylation and Olefination in Synthesis of Unnatural Chiral α-Amino Acids, Science, 2014, 343, 1216–1220 CrossRef CAS.
- J. He, R. Takise, H. Fu and J.-Q. Yu, Ligand-Enabled Cross-Coupling of C(sp3)-H Bonds with Arylsilanes, J. Am. Chem. Soc., 2015, 137, 4618–4621 CrossRef CAS.
- S. Li, R.-Y. Zhu, K.-J. Xiao and J.-Q. Yu, Ligand-Enabled Arylation of γ-C-H Bonds, Angew. Chem., Int. Ed., 2016, 55, 4317–4321 CrossRef CAS.
- G. Chen, T. Shigenari, P. Jain, Z. Zhang, Z. Jin, J. He, S. Li, C. Mapelli, M. M. Miller, M. A. Poss, P. M. Scola, K.-S. Yeung and J.-Q. Yu, Ligand-Enabled β-C-H Arylation of α-Amino Acids Using a Simple and Practical Auxiliary, J. Am. Chem. Soc., 2015, 137, 3338–3351 CrossRef CAS.
- B. Wang, C. Lu, S.-Y. Zhang, G. He, W. A. Nack and G. Chen, Palladium-Catalyzed Stereoretentive Olefination of Unactivated C(sp3)-H Bonds with Vinyl Iodides at Room Temperature: Synthesis of β-Vinyl α-Amino Acids, Org. Lett., 2014, 16, 6260–6263 CrossRef CAS.
- M. Bauer, W. Wang, M. M. Lorion, C. Dong and L. Ackermann, Internal Peptide Late-Stage Diversification: Peptide-Isosteric Triazoles for Primary and Secondary C(sp3)-H Activation, Angew. Chem., Int. Ed., 2018, 57, 203–207 CrossRef CAS.
- W. Wang, M. M. Lorion, O. Martinazzoli and L. Ackermann, BODIPY Peptide Labeling by Late-Stage C(sp3)-H Activation, Angew. Chem., Int. Ed., 2018, 57, 10554–10558 CrossRef CAS.
- G. Chen, Z. Zhuang, G.-C. Li, T. G. Saint-Denis, Y. Hsiao, C. L. Joe and J.-Q. Yu, Ligand-Enabled β-C-H Arylation of α-Amino Acids Without Installing Exogenous Directing Groups, Angew. Chem., Int. Ed., 2017, 56, 1506–1509 CrossRef CAS.
- L. Liu, Y.-H. Liu and B.-F. Shi, Synthesis of amino acids and peptides with bulky side chains via ligand-enabled carboxylate-directed γ-C(sp3)-H arylation, Chem. Sci., 2020, 11, 290–294 RSC.
- R.-Y. Zhu, K. Tanaka, G.-C. Li, J. He, H.-Y. Fu, S.-H. Li and J.-Q. Yu, Ligand-Enabled Stereoselective β-C(sp3)-H Fluorination: Synthesis of Unnatural Enantiopure anti-β-Fluoro-α-amino Acids, J. Am. Chem. Soc., 2015, 137, 7067–7070 CrossRef CAS.
- Q. Zhang, X.-S. Yin, K. Chen, S.-Q. Zhang and B.-F. Shi, Stereoselective Synthesis of Chiral β-Fluoro α-Amino Acids via Pd(II)-Catalyzed Fluorination of Unactivated Methylene C(sp3)-H Bonds: Scope and Mechanistic Studies, J. Am. Chem. Soc., 2015, 137, 8219–8226 CrossRef CAS.
- J. Miao, K. Yang, M. Kurek and H. Ge, Palladium-Catalyzed Site-Selective Fluorination of Unactivated C(sp3)-H Bonds, Org. Lett., 2015, 17, 3738–3741 CrossRef CAS.
- W.-H. Rao, B.-B. Zhan, K. Chen, P.-X. Ling, Z.-Z. Zhang and B.-F. Shi, Pd(II)-Catalyzed Direct Sulfonylation of Unactivated C(sp3)-H Bonds with Sodium Sulfinates, Org. Lett., 2015, 17, 3552–3555 CrossRef CAS.
- S. Guin, A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, Promoting Highly Diastereoselective γ-C-H Chalcogenation of α-Amino Acids and Aliphatic Carboxylic Acids, ACS Catal., 2018, 8, 2664–2669 CrossRef CAS.
-
(a) Y.-J. Liu, Y.-H. Liu, Z.-Z. Zhang, S.-Y. Yan, K. Chen and B.-F. Shi, Divergent and Stereoselective Synthesis of β-Silyl-α-Amino Acids through Palladium-Catalyzed Intermolecular Silylation of Unactivated Primary and Secondary C-H Bonds, Angew. Chem., Int. Ed., 2016, 55, 13859–13862 CrossRef CAS;
(b) J.-L. Pan, Q.-Z. Li, T.-Y. Zhang, S.-H. Hou, J.-C. Kang and S.-Y. Zhang, Palladium-catalyzed direct intermolecular silylation of remote unactivated C(sp3)-H bonds, Chem. Commun., 2016, 52, 13151–13154 RSC.
- W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, Site-Selective C(sp3)-H Functionalization of Di-, Tri-, and Tetrapeptides at the N-Terminus, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef CAS.
- T. Liu, J. X. Qiao, M. A. Poss and J.-Q. Yu, Palladium(II)-Catalyzed Site-Selective C(sp3)-H Alkynylation of Oligopeptides: A Linchpin Approach for Oligopeptide-Drug Conjugation, Angew. Chem., Int. Ed., 2017, 56, 10924–10927 CrossRef CAS.
- Y. Weng, X. Ding, J. C. A. Oliveira, X. Xu, N. Kaplaneris, M. Zhu, H. Chen, Z. Chen and L. Ackermann, Peptide late-stage C(sp3)-H arylation by native asparagine assistance without exogenous directing groups, Chem. Sci., 2020, 11, 9290–9295 RSC.
- B. Mondal, B. Roy and U. Kazmaier, Stereoselective Peptide Modifications via β-C(sp3)-H Arylations, J. Org. Chem., 2016, 81, 11646–11655 CrossRef CAS.
- A. F. M. Noisier, J. Garcia, I. A. Ionut and F. Albericio, Stapled Peptides by Late-Stage C(sp3)-H Activation, Angew. Chem., Int. Ed., 2017, 56, 314–318 CrossRef CAS.
- J. Tang, Y. He, H. Chen, W. Sheng and H. Wang, Synthesis of bioactive and stabilized cyclic peptides by macrocyclization using C(sp3)-H activation, Chem. Sci., 2017, 8, 4565–4570 RSC.
- X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen, G. He, X. Qi, W. Shen, P. Liu and G. Chen, A general strategy for synthesis of cyclophanebraced peptide macrocycles via palladium-catalysed intramolecular sp3 C-H arylation, Nat. Chem., 2018, 10, 540 CrossRef CAS.
- X. Li, L. Qi, B. Li, Z. Zhao, G. He and G. Chen, Synthesis of Cyclophane-Braced Peptide Macrocycles via Palladium-Catalyzed Intramolecular C(sp3)-H Arylation of N-Methyl Alanine at C-Termini, Org. Lett., 2020, 22, 6209–6213 CrossRef CAS.
- G. He and G. Chen, A Practical Strategy for the Structural Diversification of Aliphatic Scaffolds through the Palladium-Catalyzed Picolinamide-Directed Remote Functionalization of Unactivated C(sp3)-H Bonds, Angew. Chem., Int. Ed., 2011, 50, 5192–5196 CrossRef CAS.
- S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, Palladium-Catalyzed Picolinamide-Directed Alkylation of Unactivated C(sp3)-H Bonds with Alkyl Iodides, J. Am. Chem. Soc., 2013, 135, 2124–2127 CrossRef CAS.
- M. Fan and D. Ma, Palladium-Catalyzed Direct Functionalization of 2-Aminobutanoic Acid Derivatives: Application of a Convenient and Versatile Auxiliary, Angew. Chem. Int. Ed., 2013, 52, 12152–12155 CrossRef CAS.
- K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Ligand-enabled cross-coupling of C(sp3)-H bonds with arylboron reagents via Pd(II)/Pd(0) catalysis, Nat. Chem., 2014, 6, 146–150 CrossRef CAS.
- N. Rodriguez, J. A. Romero-Revilla, M. A. Fernandez-Ibanez and J. C. Carretero, Palladium-catalyzed N-(2-pyridyl)sulfonyl-directed C(sp3)-H γ-arylation of amino acid derivatives, Chem. Sci., 2013, 4, 175–179 RSC.
- E. Hernando, J. Villalva, A. M. Martinez, I. Alonso, N. Rodriguez, R. G. Arrayas and J. C. Carretero, Palladium-Catalyzed Carbonylative Cyclization of Amines via γ-C(sp3)-H Activation: Late-Stage Diversification of Amino Acids and Peptides, ACS Catal., 2016, 6, 6868–6882 CrossRef CAS.
- X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Petersena and X. Shi, 1,2,3-Triazoles as versatile directing group for selective sp2 and sp3 C-H activation: cyclization vs substitution, Chem. Sci., 2013, 4, 3712–3716 RSC.
- H. Jiang, J. He, T. Liu and J.-Q. Yu, Ligand-Enabled γ-C(sp3)-H Olefination of Amines: En Route to Pyrrolidines, J. Am. Chem. Soc., 2016, 138, 2055–2059 CrossRef CAS.
- J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, Site-Selective Alkenylation of δ-C(sp3)-H Bonds with Alkynes via a Six-Membered Palladacycle, J. Am. Chem. Soc., 2016, 138, 10750–10753 CrossRef CAS.
- B.-B. Zhan, Y. Li, J.-W. Xu, X.-L. Nie, J. Fan, L. Jin and B.-F. Shi, Site-Selective δ-C(sp3)-H Alkylation of Amino Acids and Peptides with Maleimides via a Six-Membered Palladacycle, Angew. Chem., Int. Ed., 2018, 57, 5858–5862 CrossRef CAS.
- H. Lin, C. Wang, T. D. Bannister and T. M. Kamenecka, Site-Selective γ-C(sp3)-H and γ-C(sp2)-H Arylation of Free Amino Esters Promoted by a Catalytic Transient Directing Group, Chem. – Eur. J., 2018, 24, 9535–9541 CrossRef CAS.
- F. Yuan, Z.-L. Hou, P. K. Pramanick and B. Yao, Site-Selective Modification of α-Amino Acids and Oligopeptides via Native Amine-Directed γ-C(sp3)-H Arylation, Org. Lett., 2019, 21, 9381–9385 CrossRef CAS.
- B.-B. Zhan, J. Fan, L. Jin and B.-F. Shi, Divergent Synthesis of Silicon-Containing Peptides via Pd-Catalyzed Post-Assembly γ-C(sp3)-H Silylation, ACS Catal., 2019, 9, 3298–3303 CrossRef CAS.
- L.-S. Zhang, G. Chen, X. Wang, Q.-Y. Guo, X.-S. Zhang, F. Pan, K. Chen and Z.-J. Shi, Direct Borylation of Primary C-H Bonds in Functionalized Molecules by Palladium Catalysis, Angew. Chem., Int. Ed., 2014, 53, 3899–3903 CrossRef CAS.
- B. Li, X. Li, B. Han, Z. Chen, X. Zhang, G. He and G. Chen, Construction of Natural-Product-Like Cyclophane-Braced Peptide Macrocycles via sp3 C–H Arylation, J. Am. Chem. Soc., 2019, 141, 9401–9407 CrossRef.
|
| This journal is © the Partner Organisations 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Shengliang
Zhong
a,
Yiyuan
Peng
*a,
Shengliang
Zhong
a,
Yiyuan
Peng