DOI:
10.1039/D0QO01157F
(Review Article)
Org. Chem. Front., 2021,
8, 169-191
Chemistry of 3-hydroxy-2-aryl acrylate: syntheses, mechanisms, and applications
Received
22nd September 2020
, Accepted 3rd November 2020
First published on 9th November 2020
Abstract
The discovery of the 3-hydroxy-2-aryl acrylate was a significant breakthrough in the field of organic and medicinal chemistry. Due to the presence of both electrophilic and nucleophilic centers, this scaffold acts as a fascinating building block of many bioactive compounds. More importantly, 3-hydroxy-2-aryl acrylate is a vital precursor in the synthesis of natural products and in the development of essential drugs. In this first review, we outline different approaches for preparing 3-hydroxy-2-aryl acrylate with mechanisms and its application in a variety of chemical fields.
 Mizzanoor Rahaman | Mizzanoor Rahaman received his MS degree in Organic Chemistry from the University of Dhaka, Bangladesh. He completed his PhD in Organic Chemistry under the supervision of Professor M Mahmun Hossain from the University of Wisconsin-Milwaukee. During graduate studies, his research was focused on total synthesis of natural products and synthesis of histone deacetylase (HDAC) inhibitors. After completing his PhD, Mizzanoor joined as a Postdoctoral Fellow in the Silverman group at the Northwestern University. Currently, he is working on design and synthesis of mutant superoxide dismutase 1 (SOD1) dependent protein aggregation inhibitors for the treatment of amyotrophic lateral sclerosis (ALS). |
 M. Shahnawaz Ali | M. Shahnawaz Ali received his MS in Applied Chemistry & Chemical Technology from the University of Dhaka, Bangladesh. He then moved to the University of Wisconsin-Milwaukee in 2015 to pursue his Doctoral Degree in Organic Chemistry under the supervision of Professor M Mahmun Hossain. His research interest focuses on the catalytic development of all carbon quaternary stereocenter. |
 Khorshada Jahan | Khorshada Jahan completed her MS in Organic Chemistry at the University of Dhaka, Bangladesh. Currently, she is a PhD student under the supervision of Professor M Mahmun Hossain at the University of Wisconsin-Milwaukee. Her research involves mainly in the area of total synthesis of novel natural products. |
 Jawad Bin Belayet | Jawad Bin Belayet received his MS degree from the University of Dhaka, Bangladesh in 2014. Since 2016, he has been conducting PhD research in the area of Medicinal Chemistry under the supervision of Professor M. Mahmun Hossain at the Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, USA. During his PhD studies, he received multiple Chancellor's Fellowship Awards. His research area is focused on the design and synthesis of drug molecules as well as investigating their diverse chemistry associated with the biological system using mass spectroscopy. |
 A. F. M. Towheedur Rahman | A. F. M. Towheedur Rahman obtained his Bachelor of Pharmacy degree from the University of Rajshahi, Bangladesh in 2010, and MSc in Drug Design and Discovery with Court Medal Award from the University of the West of Scotland, United Kingdom in 2013. Currently, he is conducting PhD research in the area of Medicinal Chemistry under the supervision of Professor M. Mahmun Hossain at the Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, USA. During his graduate studies, he received multiple Chancellor's Fellowship Awards. |
 M. Mahmun Hossain | M. Mahmun Hossain received his MSc degree in chemistry from the University of Dhaka, Bangladesh. In 1985, he received his PhD from the University of South Carolina. After about three years of postdoctoral study with Professor Jack Halpern at the University of Chicago, he joined the Department of Chemistry & Biochemistry at the University of Wisconsin-Milwaukee as an Assistant Professor. Later, he was promoted to Professor, and received a research award from the UWM Foundation for his outstanding research and creativity. |
1. Introduction
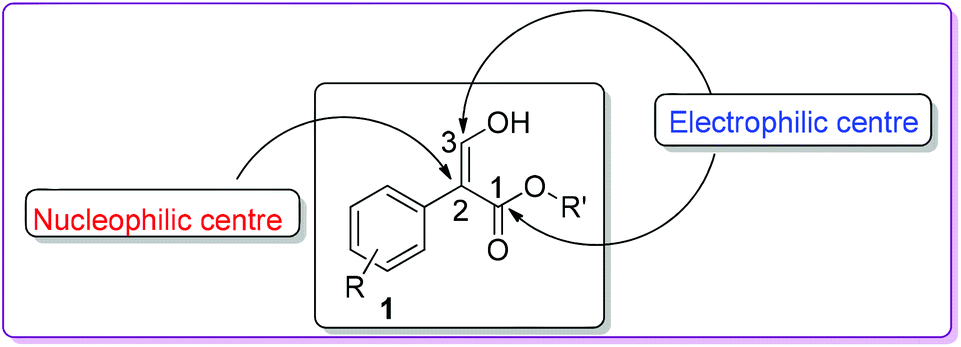
In organic synthesis, small molecules containing multiple functional groups are important since they are pivotal for the efficient construction of complex structures. One such molecule is 3-hydroxy-2-aryl acrylate 1 (Fig. 1), which has proven to be a valuable precursor in the preparation of various biologically active natural and synthetic compounds.1 Compound 1 mainly exists as the Z-enol form due to the hydrogen bonding between the carbonyl of the ester functional group and the enolic hydroxy group.2
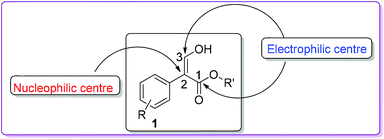 |
| | Fig. 1 Structure of 3-hydroxy-2-aryl acrylate. | |
Due to the presence of one nucleophilic and two electrophilic carbon centres (Fig. 1), compound 1 has proven to be a useful substrate in the synthesis of aromatic organic molecules, including many heterocycles such as phenanthrolines,3 benzofurans,4 indoles,5 oxazoles,6 imidazoles,7 and quinolines.8 Herein, we undertake a systematic approach to describe different synthetic methods, mechanisms, and applications of compound 1 in organic synthesis. To the best of our knowledge, so far, no comprehensive review of chemistry involving compound 1 has been reported.
2. Syntheses
2.1 Na/NaH catalysed reaction
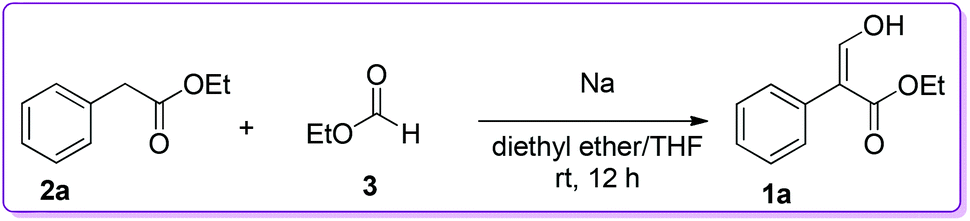
The condensation of alkyl arylacetate with alkyl formate was one of the most direct approaches to prepare 3-hydroxy-2-aryl acrylate 1. Such an example was reported by Wislicenus et al. for the synthesis of 3-hydroxy-2-phenyl acrylate 1a by condensing ethyl phenylacetate 2a with ethyl formate 3 in the presence of sodium metal in diethyl ether (Scheme 1).9–11
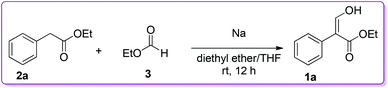 |
| | Scheme 1 Sodium catalysed synthesis of 1. | |
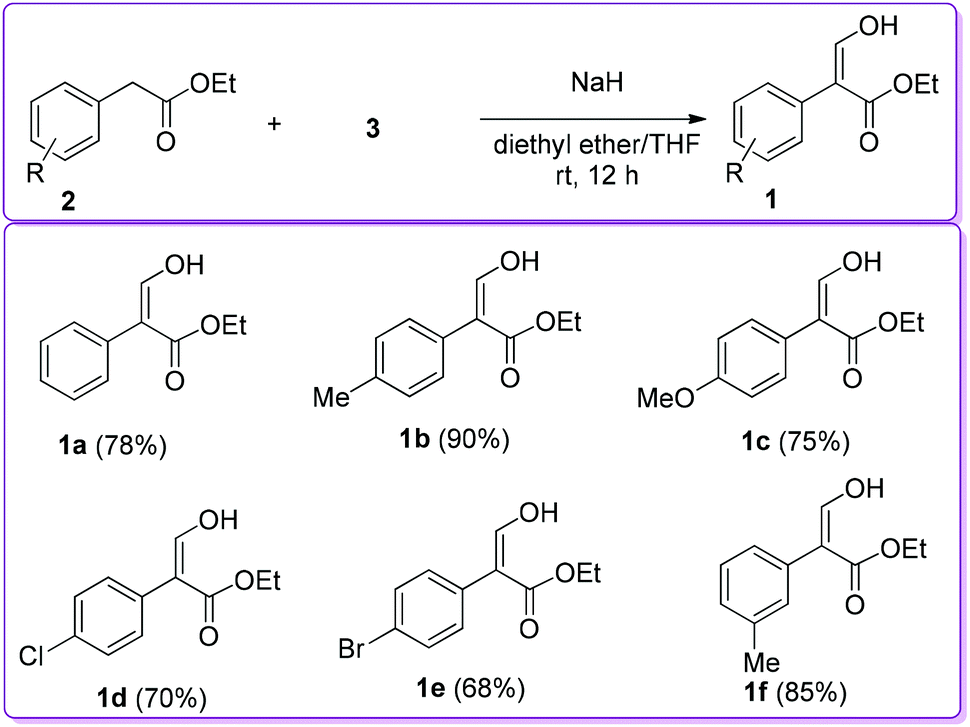
Later, other researchers applied the same protocol to synthesize 1 with substituted ethyl phenylacetate 2 by using sodium hydride as a base instead of sodium metal to obtain better yield (Scheme 2).12–14
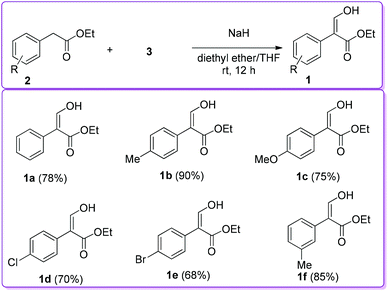 |
| | Scheme 2 Sodium hydride catalysed synthesis of 1. | |
2.2 Lewis acid catalysed reaction
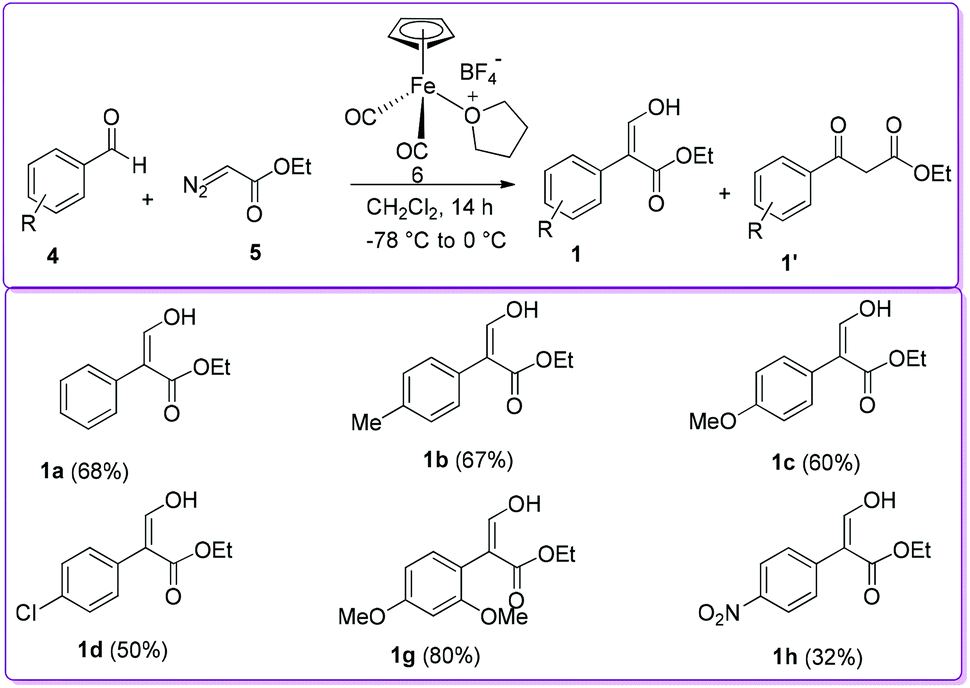
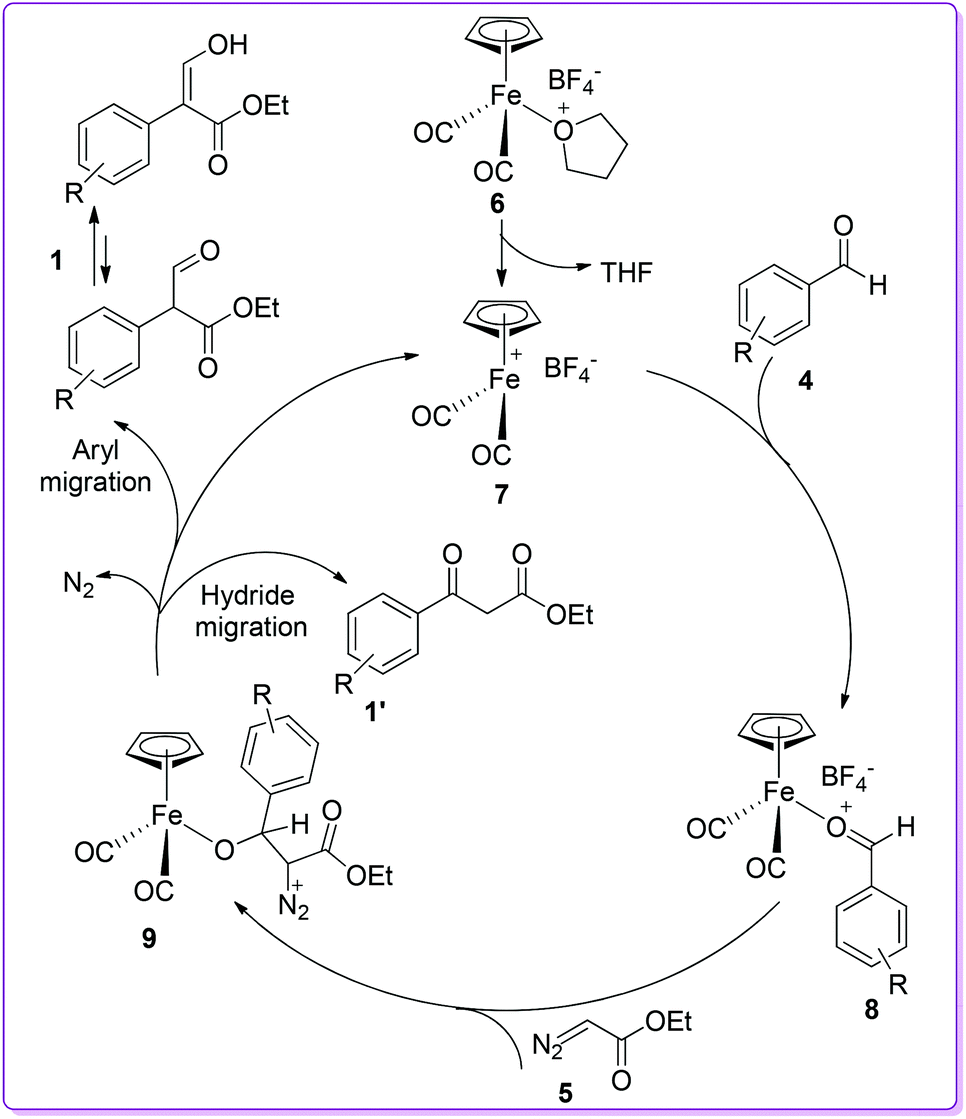
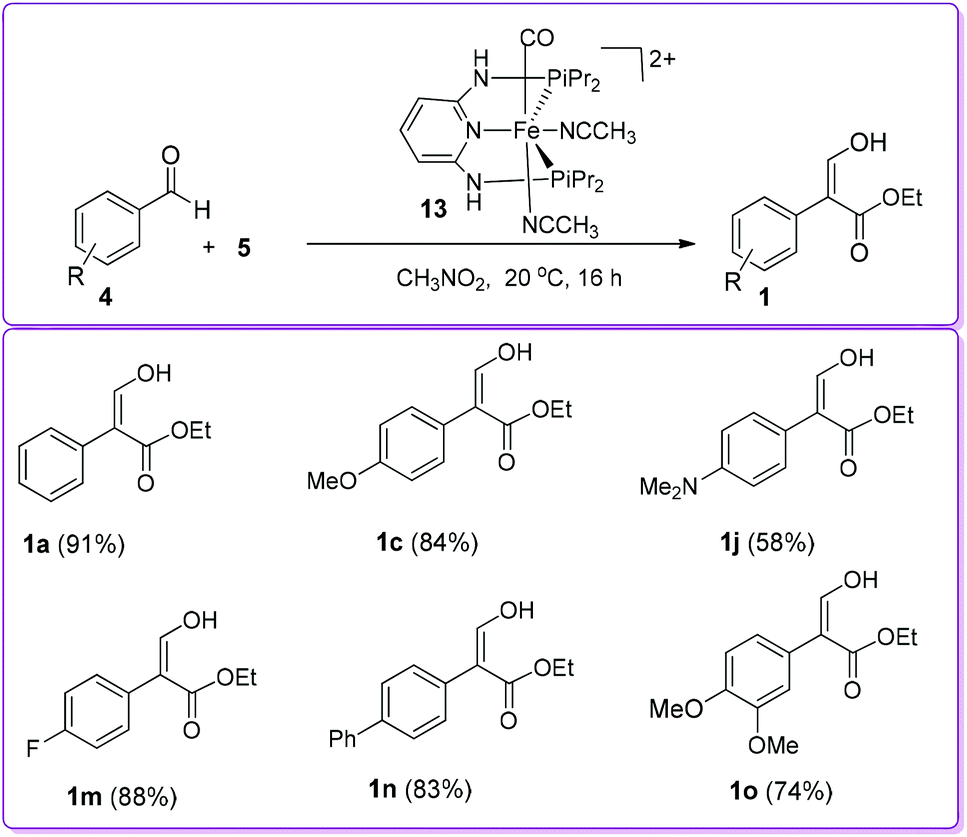
In 1998, the Hossain group reported the formation of 1 from the reaction of aromatic aldehyde 4 with ethyl diazoacetate (EDA) 5 in the presence of iron Lewis acid catalyst, [η5-(C5H5)Fe+(CO)2(THF)]BF4−6, by 1,2-aryl migration (Scheme 3).15 Previously, similar reactions using different Lewis acid catalysts such as BF3, ZnCl2, AlCl3, and SnCl4 produced 3-oxo-3-arylpropanoic acid ethyl ester (β-ketoester) 1′ as the major product.16 In contrast, iron Lewis acid 6 catalysed reaction produced desired compound 1 along with β-ketoester 1′ from moderate to excellent yields. The authors reported that benzaldehyde having electron donating groups provided higher yield of product 1 compared to having electron withdrawing groups.
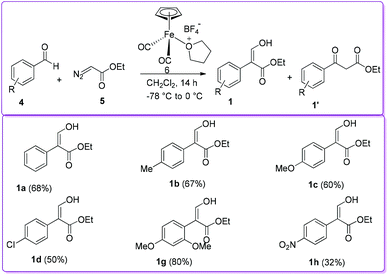 |
| | Scheme 3 Synthesis of 1 using iron Lewis acid catalyst. | |
To explain the formation of products, a catalytic cycle was proposed by the Hossain group. First, the removal of the THF ligand from catalyst 6 formed the highly reactive 16e− complex 7 which subsequently reacted with aromatic aldehyde 4 to form the σ-bonded aldehyde iron complex 8. The reaction of EDA 5 with aldehyde-iron complex 8 gave intermediate 9. Finally, the 1,2-aryl migration over hydride migration in complex 9 provided compound 1 as the major product (Scheme 4).15
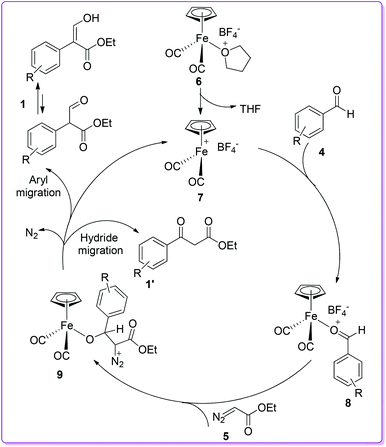 |
| | Scheme 4 Mechanism of iron Lewis acid catalysed reaction. | |
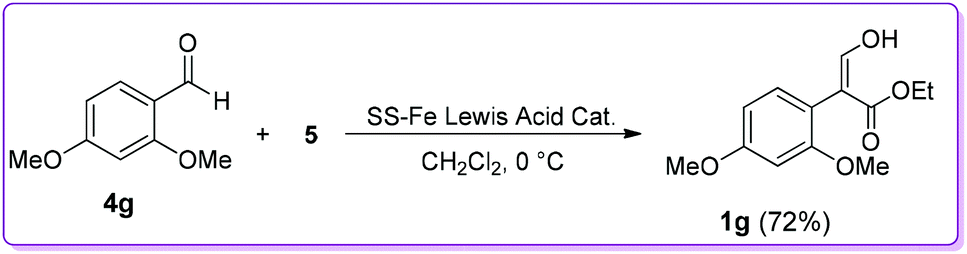
The Hossain group also reported the reaction of 2,4-dimethoxybenzaldehyde 4g with EDA 5 in the presence of a silica-supported iron (SS-Fe) Lewis acid catalyst to observe the efficacy and durability of catalyst 6 (Scheme 5).17 The group found that the efficiency of SS-Fe Lewis acid catalyst was comparable to the unsupported iron Lewis acid catalyst 6 (72% vs. 80% yield, 1g of Scheme 5vs.Scheme 3). It should be mentioned that the SS-Fe was reusable for several times.
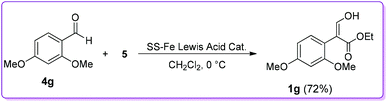 |
| | Scheme 5 Silica-supported iron Lewis acid catalysed synthesis of 1. | |
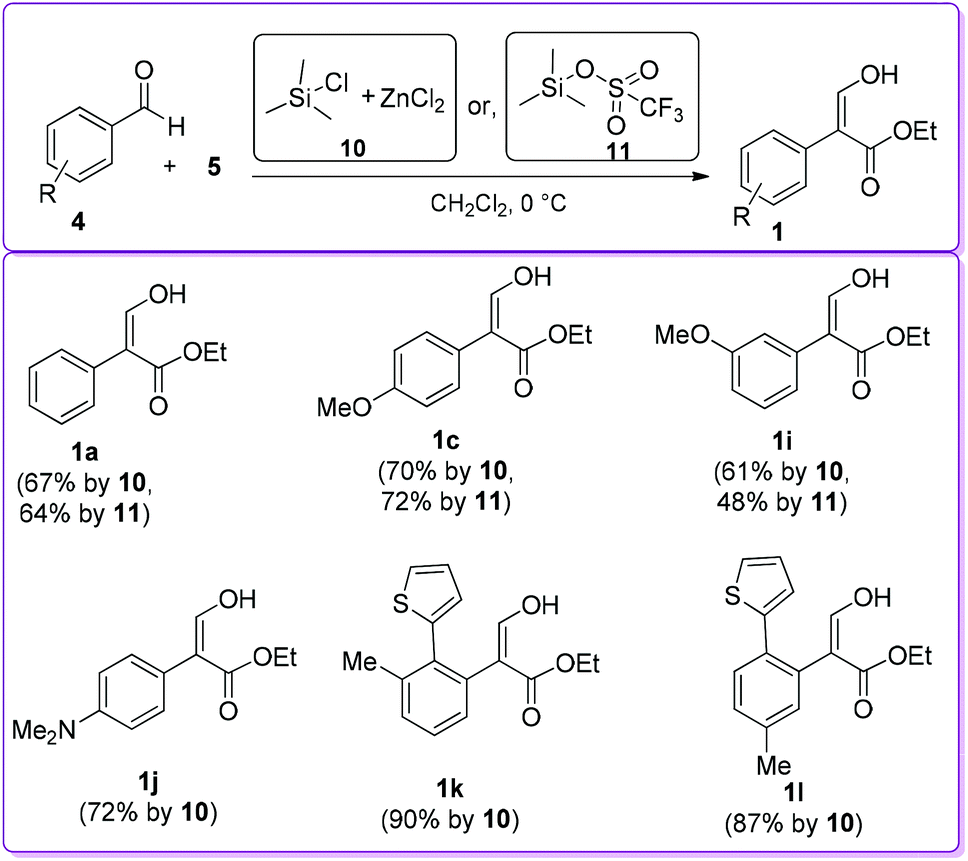
In 1999, Kanemasa et al. described the synthesis of 1 by the condensation of 4 and 5 utilizing two different catalysts, namely ZnCl2 in the presence of chlorotrimethylsilane 10 and trimethylsilyl trifluoro-methanesulphonate 11 (Scheme 6).18 Both catalysts 10 and 11 produced corresponding acrylate 1 with moderate to high yields; in several cases they obtained a mixture of products 1 and 1′.
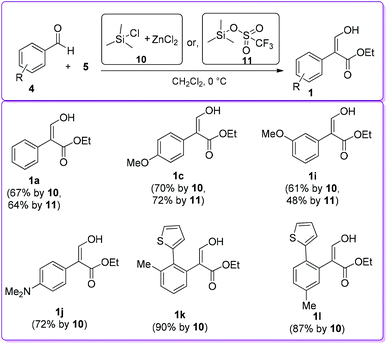 |
| | Scheme 6 Synthesis of 1 by silane-based Lewis acid catalyst. | |
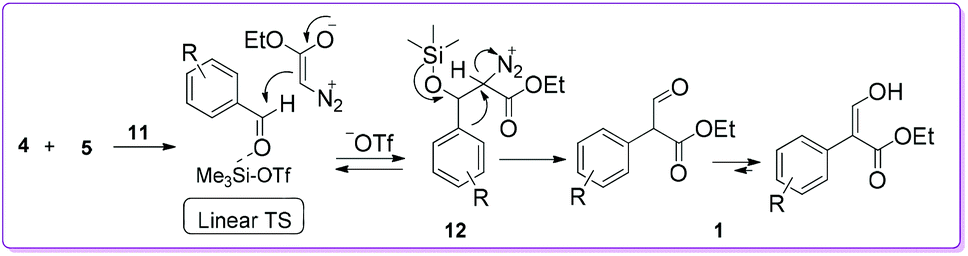
Kanemasa group proposed a mechanism to explain the formation of 1 by considering catalyst 11 (Scheme 7).18 Initially, aromatic aldehyde 4 was activated by coordinating with catalyst 11, so that EDA 5 could easily react to the carbonyl carbon of the aldehyde 4 to form diazonium silyloxy intermediate 12 through a linear transition structure (TS). Later, the aryl group migrates from behind of the diazonium group to produce compound 1 by the loss of N2.
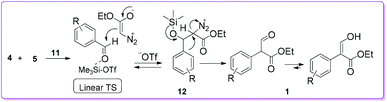 |
| | Scheme 7 Mechanism of trimethylsilyl trifluoromethanesulphonate catalysed formation of 1. | |
Kirchner and co-workers reported the selective formation of 1 (and less than 3% β-ketoester 1′) from aromatic aldehyde 4 and EDA 5 using cationic iron pincer complexes such as [Fe(PNP)(CH3CN)3](BF4)2 and [Fe(PNP)(CO)(CH3CN)2](BF4)2, where PNP are various tridentate pincer-type ligands based on 2,6-diaminopyridine, and R = Ph, iPr, biphenol, PhHex. The most effective catalyst was found to be cis-[Fe(PNP-iPr)(CO)(CH3CN)2]BF413, which provided the acrylate 1 in higher yield (Scheme 8).19
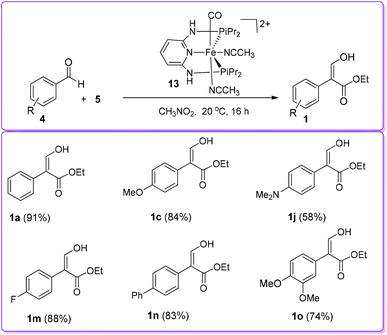 |
| | Scheme 8 Formation of 1 by cis-iron(II) pincer complex catalyst. | |
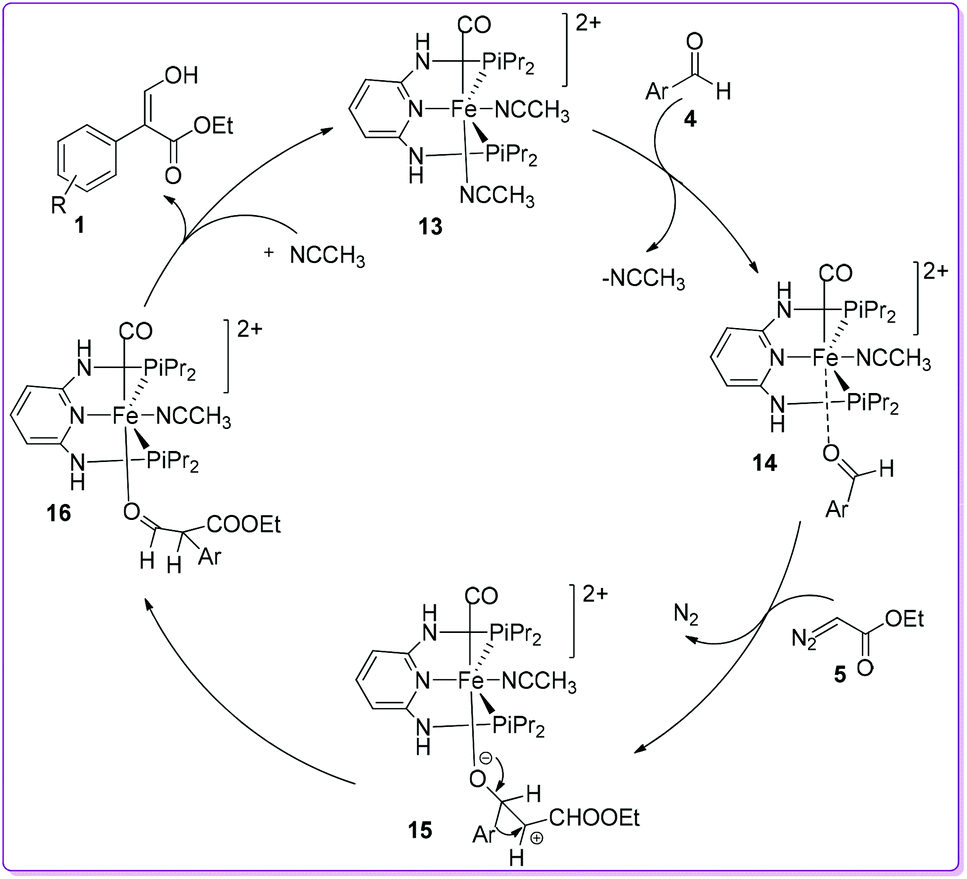
Kirchner et al. proposed a similar mechanistic pathway for the formation of 1 as suggested by Hossain et al.15 In the catalyst 13, the presence of CO ligand makes CH3CN more labile due to its stronger trans-effect and easily substituted by incoming aromatic aldehyde 4 to form [Fe (PNP-iPr)(CO)(CH3CN)(κ1(O)-aldehyde)]2+ complex 14. The resulted complex 14 was attacked by EDA 5 and formed the complex 15 by subsequent liberation of N2. Preferential migration of the aryl group over hydride led to form a κ1O-coordinated aldehyde ester ligand 16. The aldehyde ester ligand was easily removed from 16 by an incoming CH3CN, and rapidly tautomerized to yield the thermodynamically more stable compound 1 (Scheme 9).19
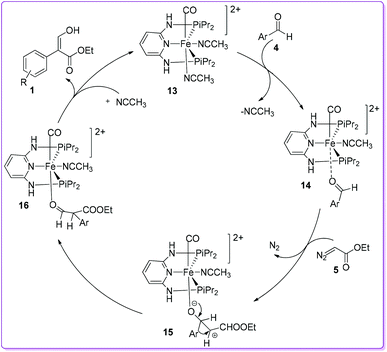 |
| | Scheme 9 Mechanism of cis-iron(II) pincer complex catalysis. | |
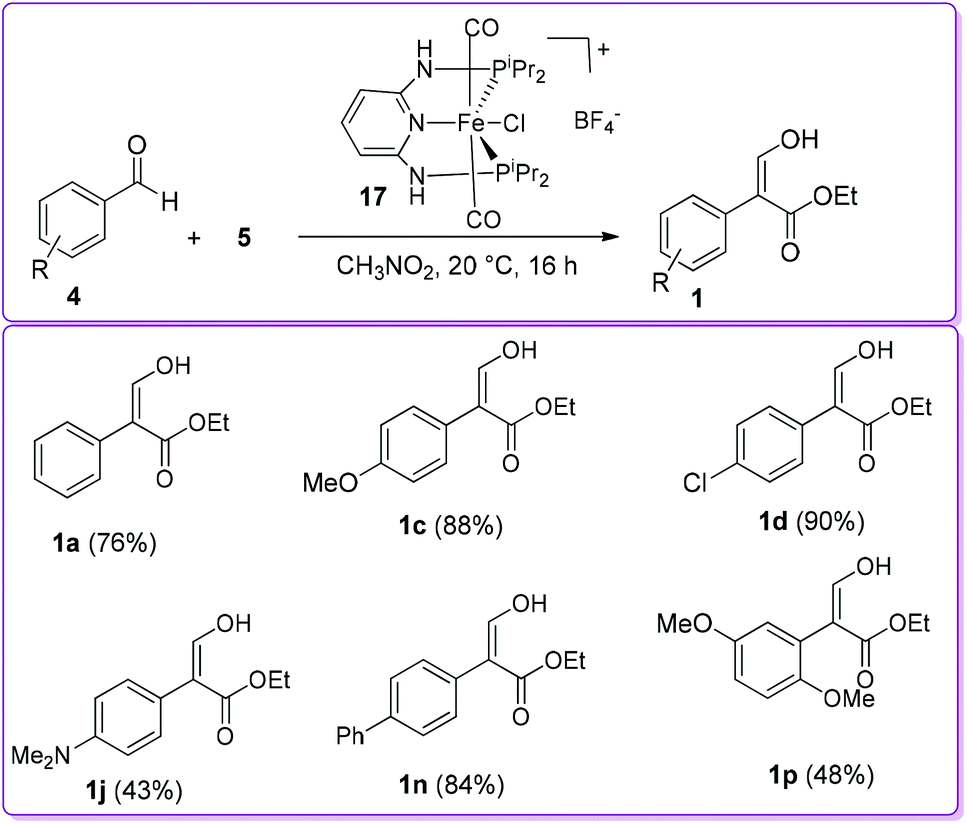
Later, the Kirchner group studied the effect of the counterion on selective formation of compound 1 by utilizing mono cationic dicarbonyl trans-[Fe(PNP-iPr)(CO)2Cl](X) complex, where X = NO3−, CF3COO−, CF3SO3−, BF4−, PF6−, SbF6− and B[3,5-(CF3)2C6H3]4−. With BF4− as a counterion, the reaction was proceeded with conversions up to 90% but other counterions such as NO3−, CF3COO−, CF3SO3−, SbF6− and [3,5-(CF3)2C6H3]4− did not show any activity (Scheme 10).20
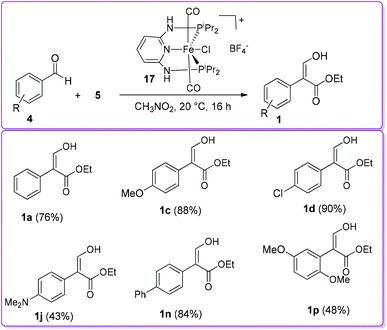 |
| | Scheme 10 Formation of 1 by trans-iron(II) pincer complex catalyst. | |
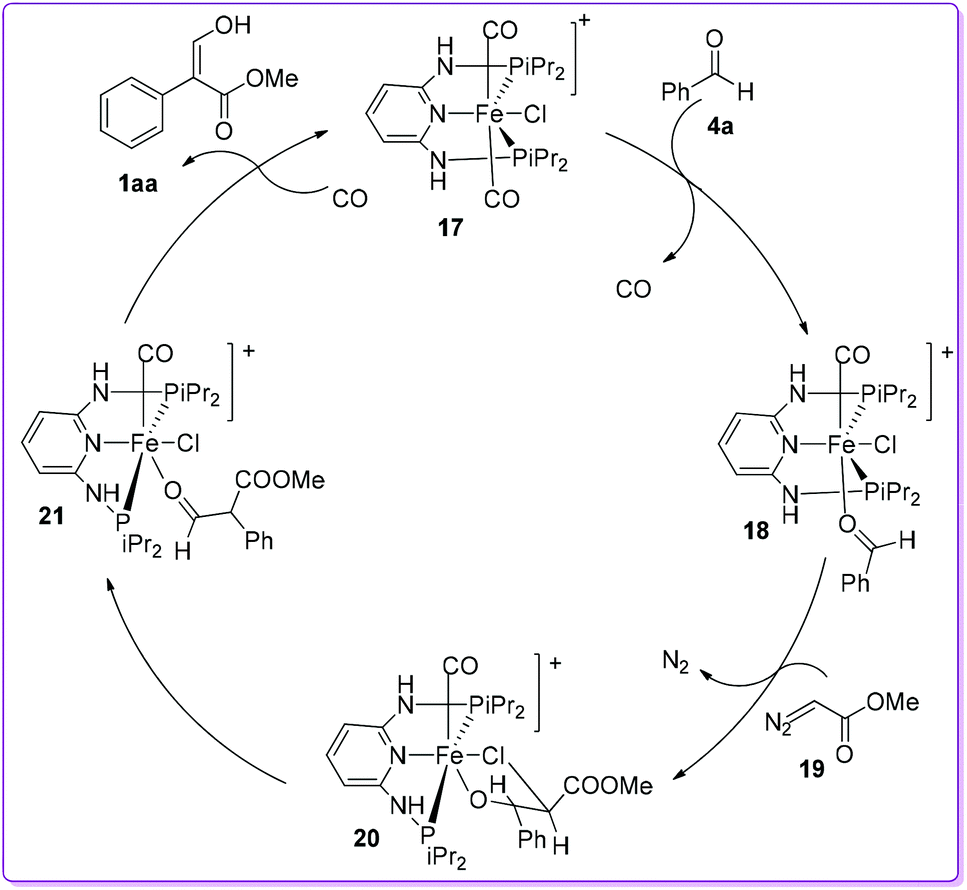
Based on density functional theory (DFT)/B3LYP calculation, a conceivable mechanism for the formation of acrylate 1 was proposed by Kirchner et al. using trans-iron(II) pincer complex 17.20 Initially, the CO ligand of the complex 17 was substituted by benzaldehyde 4a to form [Fe(PNP-iPr)(CO)(Cl)(κ1(O)-aldehyde)]+ complex 18. The reaction of 18 with methyl diazoacetate (MDA) 19 formed the complex 20 by the extrusion of N2. The preferential migration of the phenyl group from 20 produced the complex 21. Finally, substitution of κ1O-coordinated aldehyde ester by a CO ligand provided the acrylate 1aa after tautomerization and regenerated the catalyst 17 (Scheme 11).
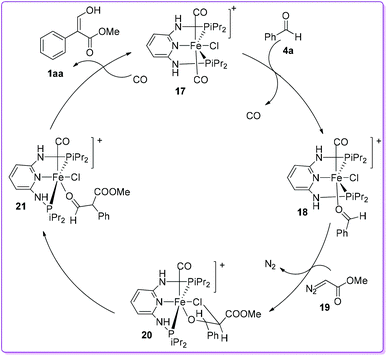 |
| | Scheme 11 Mechanism of trans-iron(II) pincer complex catalysis. | |
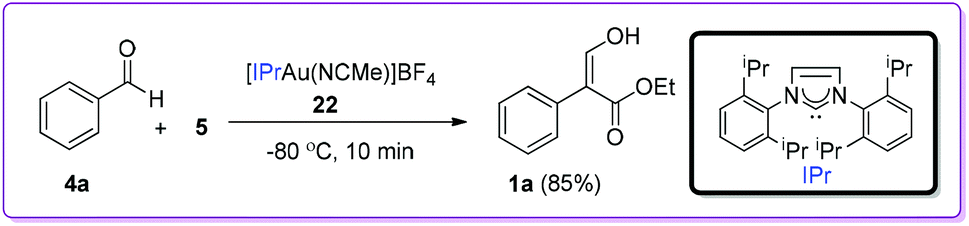
In 2009, Pérez and co-workers reported the first example of Group 11 metal based catalysts for the coupling reaction between benzaldehyde 4a and EDA 5 (Scheme 12).21 Among different N-heterocyclic carbene based gold complexes, [IPrAu(NCMe)]BF422 was found to be the most effective catalyst with excellent turnover frequency (TOF) for the formation of 1a.
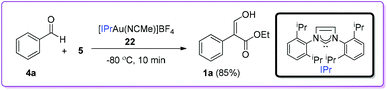 |
| | Scheme 12 Gold metal complex catalysed formation of 1. | |
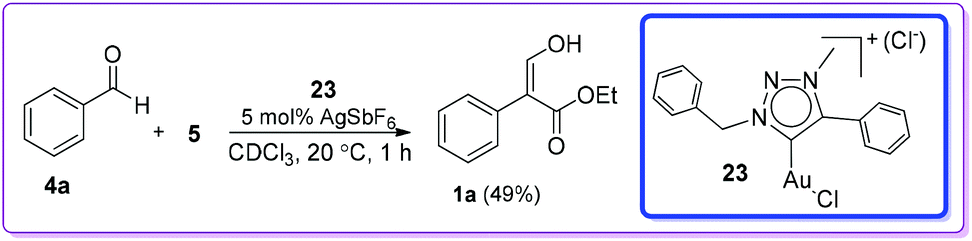
Later, Crowley and co-workers described a gold(I) carbene(1,2,3-triazolylidene) complex 23/AgSbF6 catalysed formation of 1a from the reaction of 4a and EDA 5 (Scheme 13).22
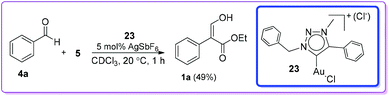 |
| | Scheme 13 AgSbF6 with gold complex catalysed formation of 1. | |
2.3 Brønsted type acid catalysed reaction
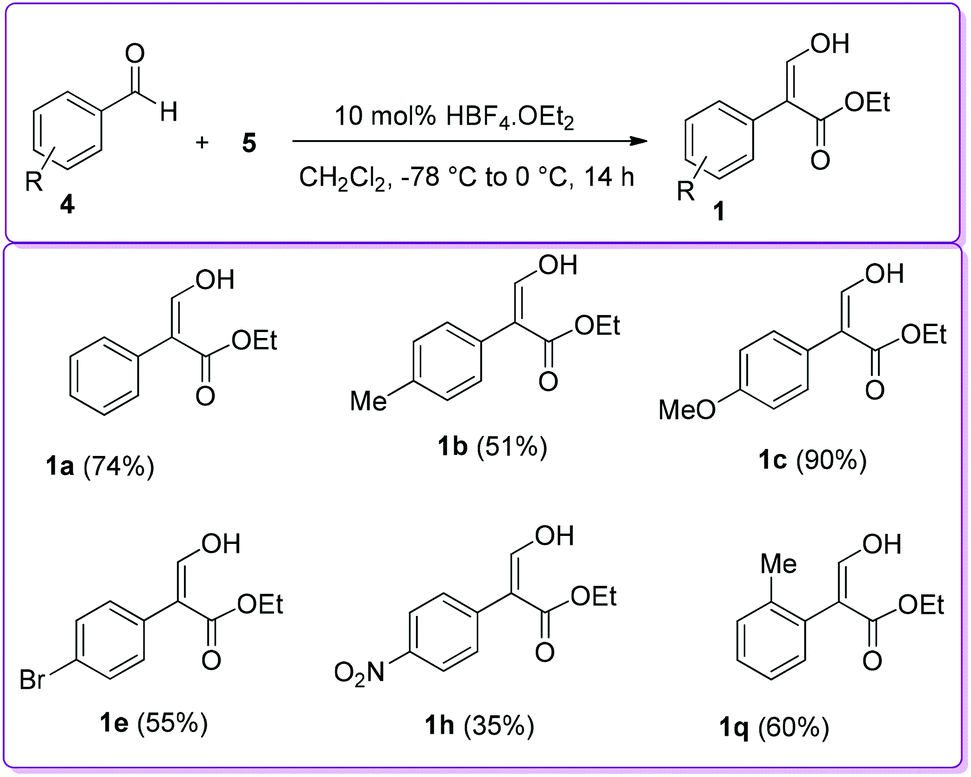
In 2004, Hossain and co-workers explored several Brønsted acid catalysts, including HBF4·OEt2, to prepare 1 from the coupling reaction of aromatic aldehydes 4 and EDA 5 (Scheme 14).23 The catalytic activity of HBF4·OEt2 was screened with a series of aromatic aldehyde at different temperatures. It was found that electron donating substituents on the aromatic ring and reaction at lower temperature significantly enhanced the formation of the desired product 1.
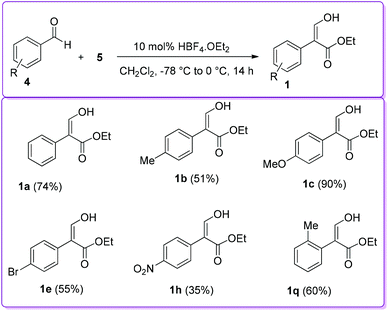 |
| | Scheme 14 First Brønsted type acid catalysed formation of 1. | |
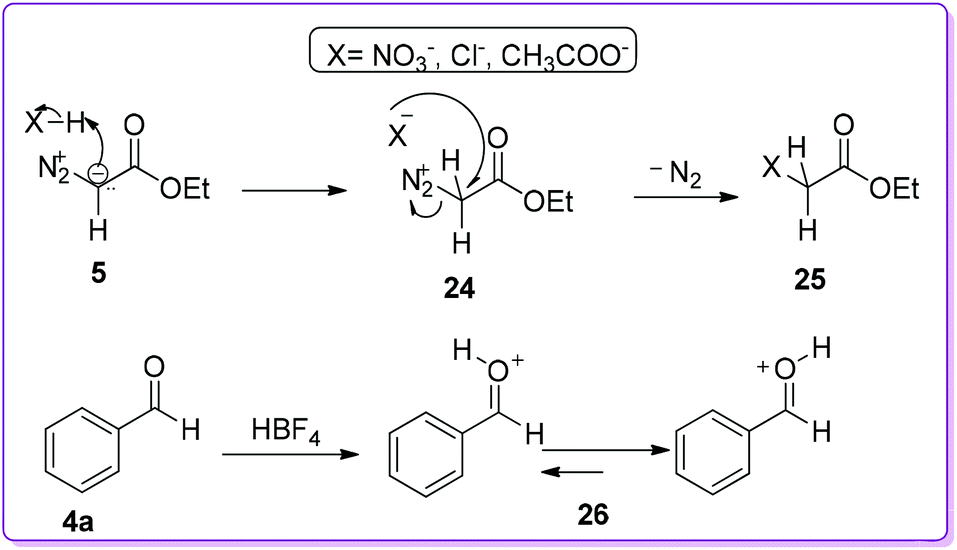
The group also screened several commercially available Brønsted acids, such as HNO3, H2SO4, HClO4, HCl besides HBF4, to see the counterion effect on selective product formation. They found that the acids with nucleophilic counterions, such as Cl−, AcO−, and NO3−, provided a small amount of product 1 since these acids readily quenched by EDA 5 and formed byproduct 25 (Scheme 15). On the other hand, HBF4 did not quench by EDA due to the presence of non-nucleophilic counterion (BF4−); it rather bound to the carbonyl oxygen and generated O-protonated benzaldehyde 26 which initiated the reaction (Scheme 15).23
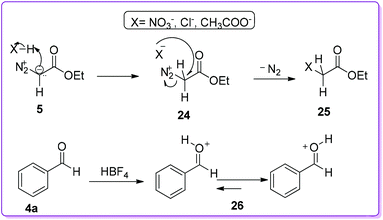 |
| | Scheme 15 Reaction of Brønsted type acids with EDA and aldehyde. | |
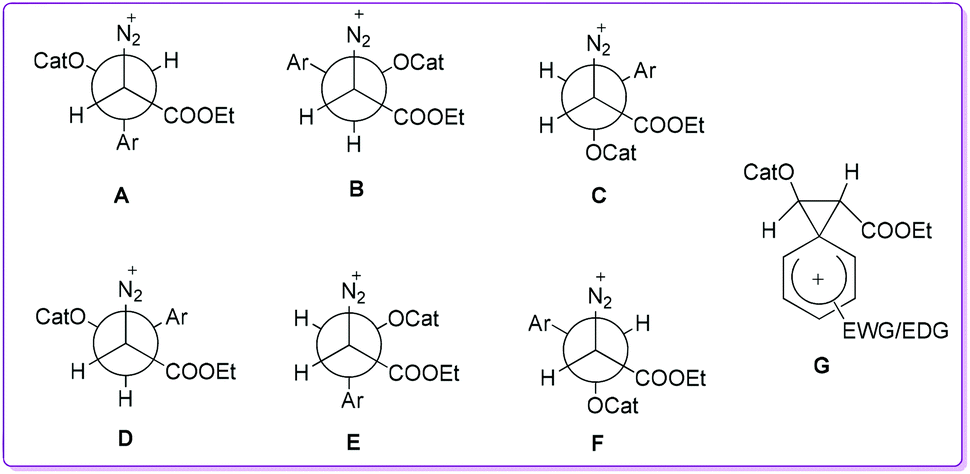
The Hossain group proposed that the catalyst HBF4 first attached to the aldehyde and then the nucleophilic methine carbanion of EDA 5 attacked the re-face of the protonated aldehyde, thus three rotamers (Newman projections) A, B, and C (or their enantiomers) could form (Fig. 2). Rotamers A–C were interconvertible, and A had the lowest energy due to less steric interactions. In the rotamer A, migrating aryl group and leaving diazonium group were anti to each other, thus the formation of hydroxy acrylate 1 was favourable. Similarly, if the EDA attacked to the si-face of the protonated aldehyde, three more interconvertible rotamers D, E, and F (or their enantiomers) could form. The rotamer D had the lowest energy due to less steric interactions of bulky groups, where hydride migrating group was anti to the leaving group which favoured the formation of β-keto ester 1′. However, the aryl migration was more favourable over hydride (Fig. 2, rotamer Avs. rotamer D) due to its ability to stabilize an intermediate carbocation by the formation of phenonium ion (Fig. 2, G). Since electron-donating groups (EDG) could enhance the stability of the resultant phenonium ion which favoured selective formation of 1. Likewise, electron-withdrawing groups (EWG) destabilized the phenonium ion resulting in decreased formation of 1.
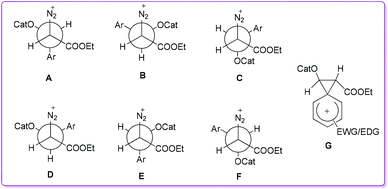 |
| | Fig. 2 Newman projections of six possible rotamers (A–F) and an intermediate (G) phenonium ion resulting from aryl migration. | |
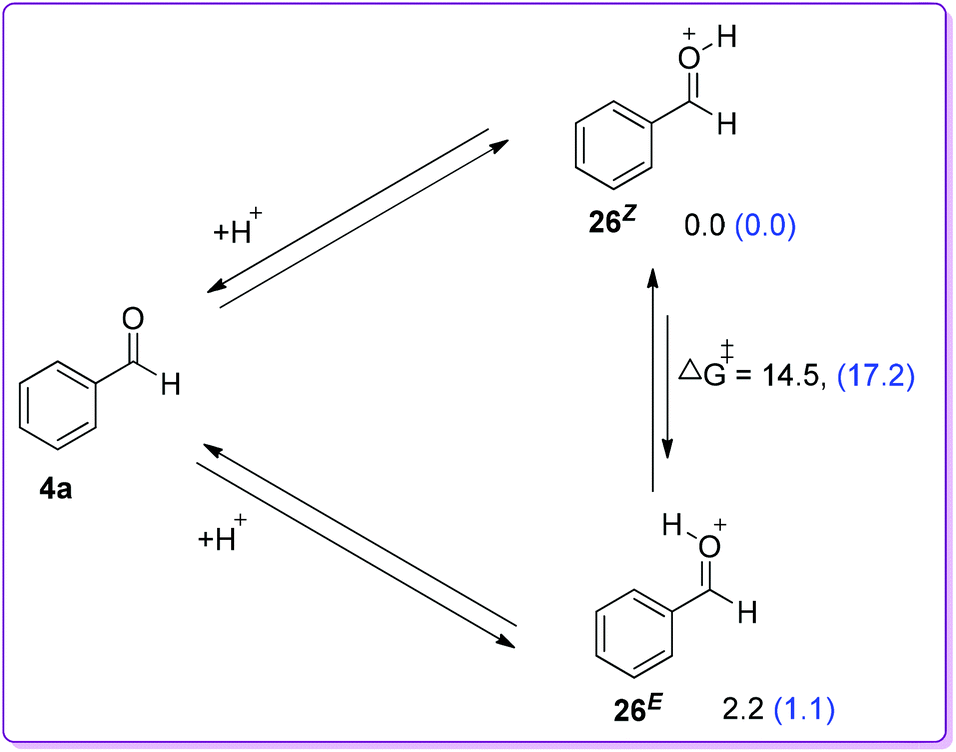
The favorable migratory aptitude of phenyl over hydride was later verified by Kirchner group using (GIAO method at the B3LYP/6-31G** basis set) density functional theory (DFT).24 They have studied the coupling between MDA and benzaldehyde in DCM using HBF4·OEt2 as a catalyst. Results of stepwise analysis of the probable protonation of benzaldehyde in DFT study agreed with the experimental findings that the Z-isomer of 26 was slightly favourable by 2.2 kcal mol−1 than the E-isomer (Scheme 16).23 They also stated the facile intramolecular E/Z isomerization at room temperature with an energy barrier of 14.5 kcal mol−1.
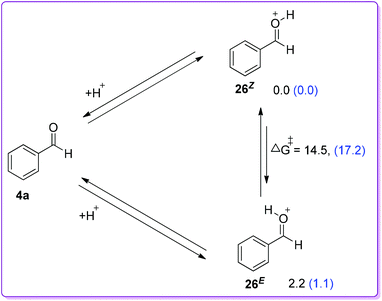 |
| | Scheme 16 Energy difference between E-and Z-isomer of O-protonated benzaldehyde by computational study [parentheses values (blue) refer to solvent DCM corrected energies]. | |
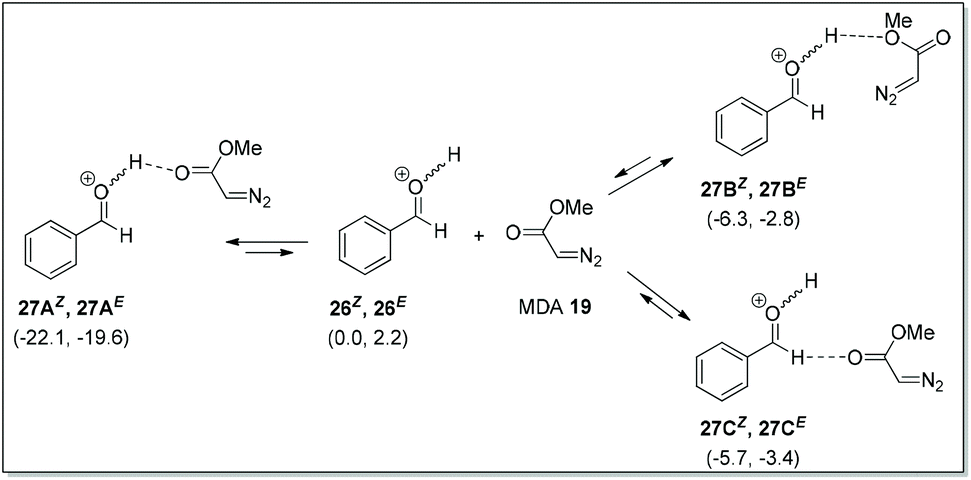
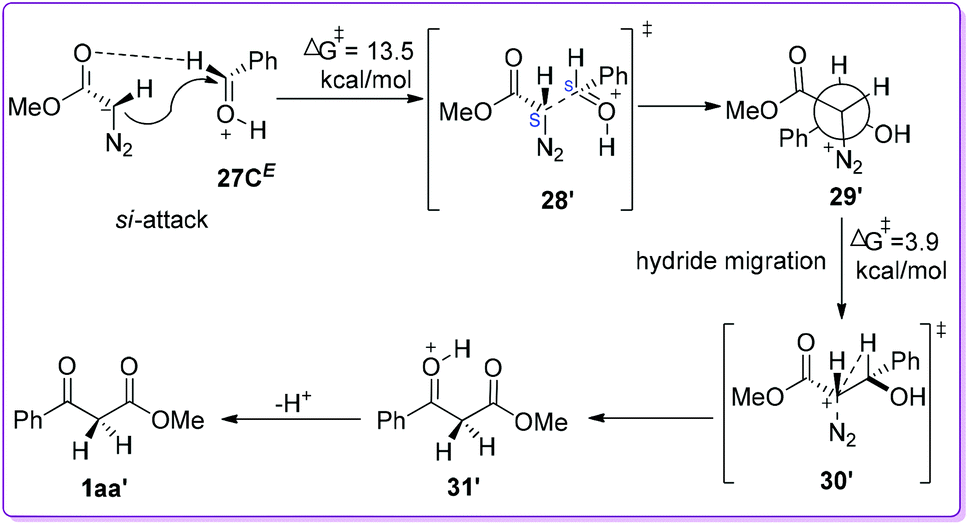
Kirchner et al. predicted that when O-protonated benzaldehyde isomers (26E/26Z) were dissolved in DCM along with MDA, three associated species 27A–27C could be formed via hydrogen bonding (Scheme 17). These species were rapidly interconvertible (fast pre-equilibria) with respect to the subsequent carbon–carbon bond forming reactions. Detailed calculations for all possible nucleophilic attack (si-/re-face) of MDA on 27A–27C were reported up to the product formation.
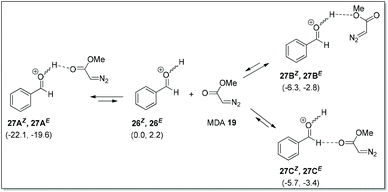 |
| | Scheme 17 Possible hydrogen bonding between O-protonated benzaldehyde and MDA. | |
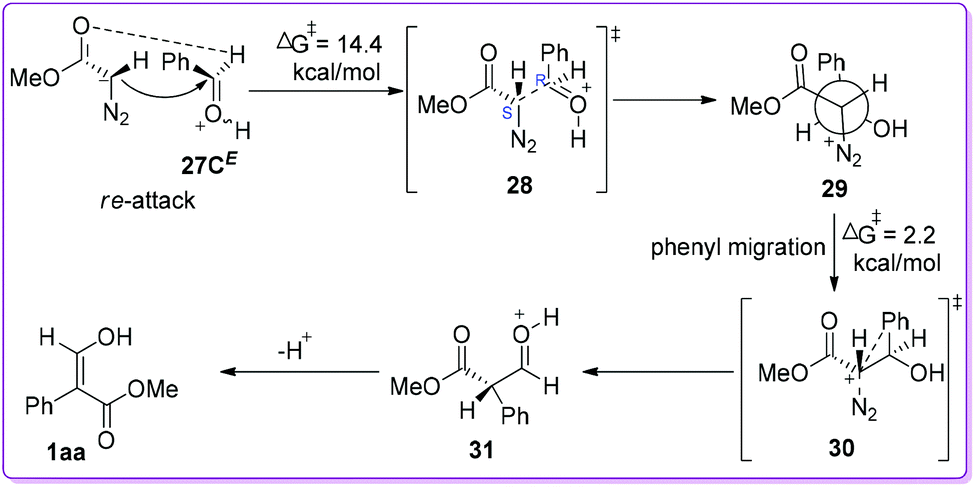
Initially, the authors presented the data involving re-face attack of MDA 19 on 27CE with which provided the diastereomeric intermediate 29 (Scheme 18) adopting 1S, 2R-configuration (or the other enantiomer, i.e. 1R, 2S). This reaction, proceeding through transition state 28, is endergonic (7.5 kcal mol−1) with a free energy of activation of 14.4 kcal mol−1. Due to the antiperiplanar orientation of phenyl to N2 (as a prerequisite), the rotamer 29 easily undergoes phenyl migration to form protonated aldehydic ester 31via transition state 30 with a free energy of activation of 2.2 kcal mol−1. The formation of 31 from 29 is strongly exergonic by 40.5 kcal mol−1. Finally, the phenyl migration product (thermodynamically more stable) 3-hydroxyacrylate 1aa was obtained by the deprotonation of 31 (Scheme 18).24
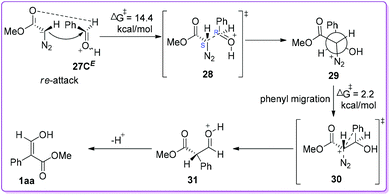 |
| | Scheme 18 Computational mechanistic studies of O-protonated benzaldehyde and MDA via re-face attack. | |
On the other hand, when MDA 19 attacks on the other enantioface (si-face) of 27CE to form the diastereomeric intermediate 29′ (Scheme 19) adopting 1S, 2S-configuration (or the other enantiomer, i.e., 1R, 2R). The reaction is shown to be endergonic (7.4 kcal mol−1) and proceeds through transition state 28′ with 13.5 kcal mol−1 free energy of activation. The rotamer 29′ is favourable for subsequent hydride-migration due to the antiperiplanar orientation of hydride to N2 (as a prerequisite). This migration occurs through the transition state 30′ with an activation energy of 3.9 kcal mol−1 to form the O-protonated β-ketoester 31′. The formation of 31′ from 29′ is strongly exergonic with energy 55.0 kcal mol−1. Eventually upon loss of the proton from the 31′ formed β-keto ester 1aa′ (Scheme 19). The DFT calculation of these two approaches clearly indicated that the formation of phenyl migrated product 1aa is slightly more favourable than hydride migrated product 1aa′ (Scheme 18 and 19).
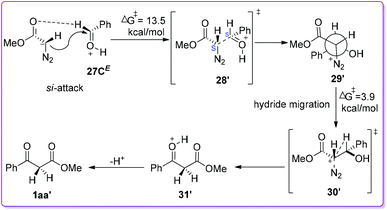 |
| | Scheme 19 Computational mechanistic studies of O-protonated benzaldehyde and MDA via si-face attack. | |
Similarly, detailed energy calculation involving other species of 27 where the phenyl migration from corresponding rotamers were also found to be slightly favourable compare to the hydride migration. Hence, the theoretical data clearly substantiate the experimental findings of HBF4·Et2O catalysed selective formation of 3-hydroxy-2-aryl acrylate by the Hossain group.
2.4 Organic base catalysed reaction
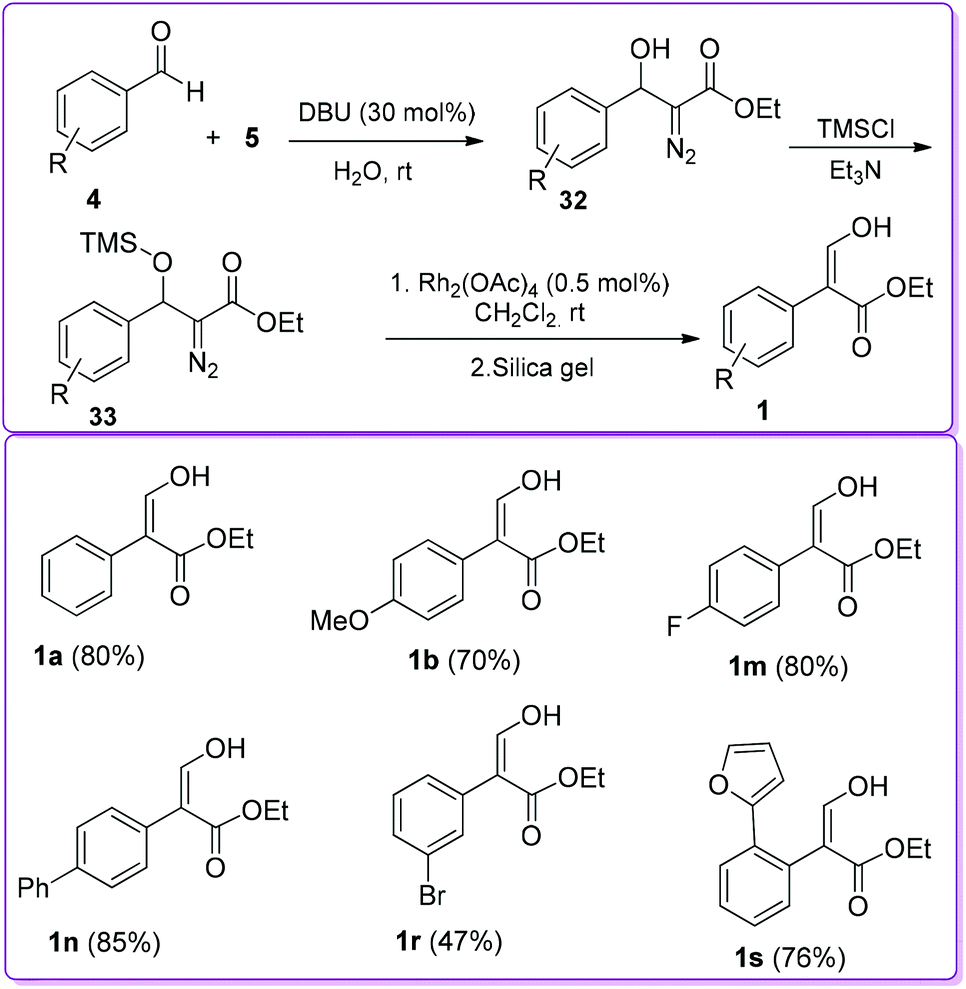
In 2007, Wang et al. reported 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) catalysed reaction between 4 and EDA 5 to synthesize β-hydroxy diazo ester 32. The β-hydroxy group of 32 was protected by trimethylsilyl chloride (TMSCl) to form compound 33. In the presence of Rh(II)-catalyst, compound 33 underwent 1,2-aryl migration to provide the final product 1 (Scheme 20).25 The steric factors play an important role in the 1,2-migratory aptitude of Rh(II)-catalysed reaction. When the substituent was bulkier at the β-position, such as a siloxy group, aryl migration was favoured over hydride migration.26
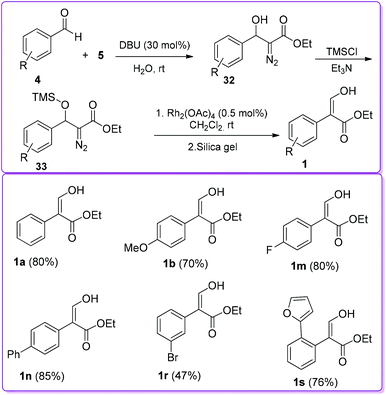 |
| | Scheme 20 DBU and Rh(II)-catalysed formation of 1. | |
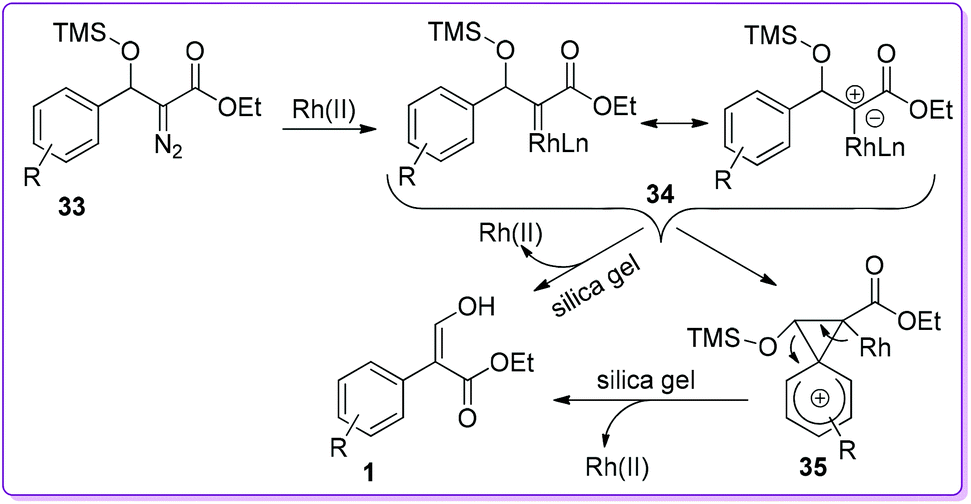
Based on the reaction, Wang et al. proposed two ways mechanism involving Rh(II)-catalyst for the formation of acrylate 1 (Scheme 21).26–28 In both ways, the initial step was the formation of Rh(II)-carbene intermediate 34 which could provide compound 1 by 1,2-aryl migration via a bridged phenonium ion intermediate 35. On the other hand, product 1 could be formed directly from carbene 34 by a concerted pathway where aryl group could migrate simultaneously with the dissociation of the catalyst.
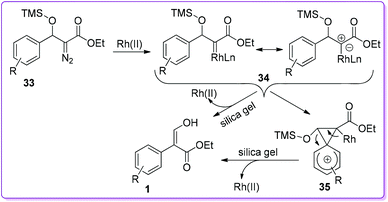 |
| | Scheme 21 Proposed mechanism of Rh(II)-catalysed 1,2-migration. | |
3. Synthetic applications
3.1 Quinolone/quinolinone type compounds
4-Quinolones and 2-quinolones captured great interest by medicinal chemists due to their effective biological properties.29–31 Specifically, 3-aryl-4-quinolones showed antitumor32a and antimalarial activity,32b and acted as a platelet aggregation inhibitor,32c while 3-aryl-2-quinolones reported to be acting as an integrin antagonist.33
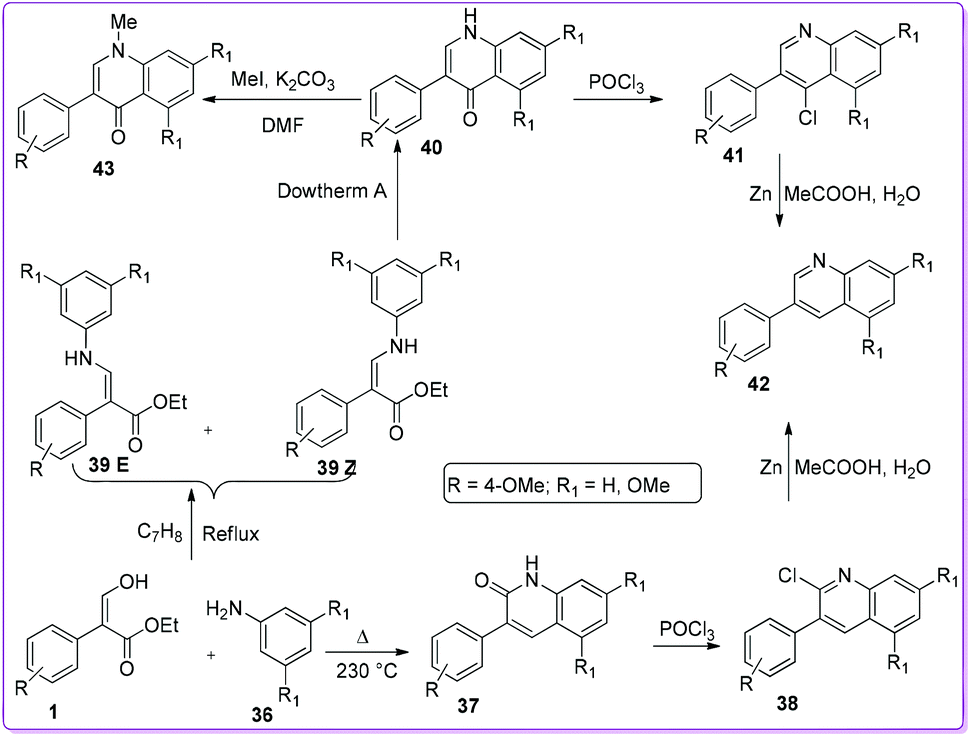
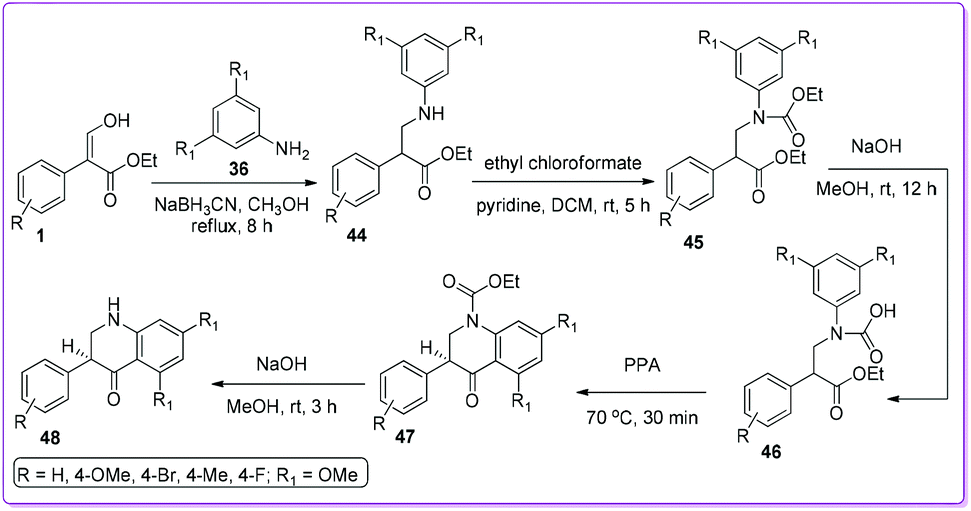
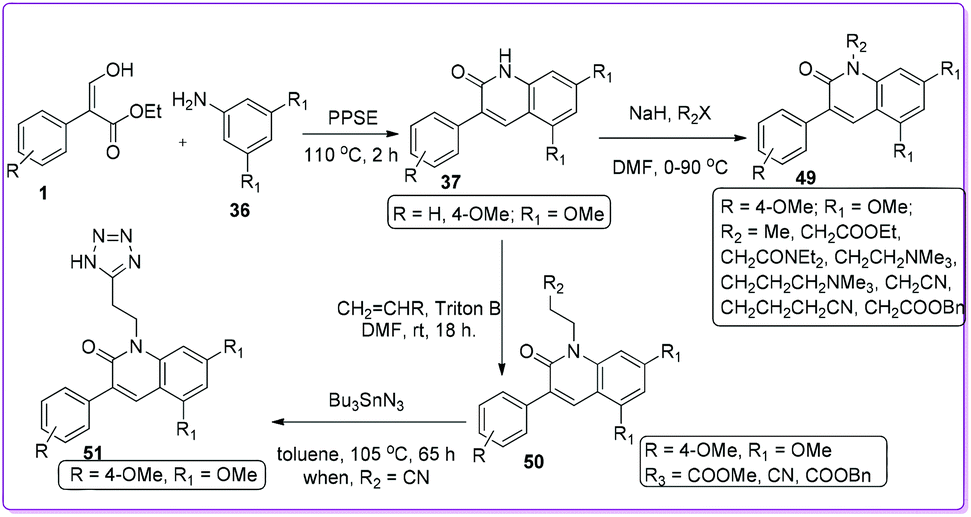
The first approach to synthesize 3-aryl-2-quinolones 37 and 3-aryl-4-quinolones 40 from 3-hydroxy-2-aryl acrylate 1 was reported in 1997 by Bisagni et al.34 Condensation reactions between 1 and 3,5-dimethoxy aniline 36 formed a E/Z mixture of intermediate 39 which underwent cyclization by refluxing in Dowtherm A (biphenyl/diphenyl ether mixture 26.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :73.5) to prepare 40. On the other hand, by heating 1 and 36 at 230 °C they obtained 3-phenyl-2-quinolone 37. They also prepared other quinolone derivatives such as 38, 41, 42, and 43 from quinolones 37 and 40 (Scheme 22).
:73.5) to prepare 40. On the other hand, by heating 1 and 36 at 230 °C they obtained 3-phenyl-2-quinolone 37. They also prepared other quinolone derivatives such as 38, 41, 42, and 43 from quinolones 37 and 40 (Scheme 22).
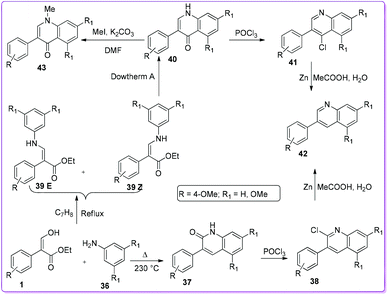 |
| | Scheme 22 Synthesis of 2-and 4-quinolinones from 1. | |
Kumar et al. followed the similar protocol of Bisagni34 to synthesize 40 and attempted to prepare dihydroquinolone 48 by direct reduction of compound 40.35 However, the reduction reaction was unsuccessful, subsequently they developed an alternate route to synthesize 48 from 1 (Scheme 23). The authors screened the anticancer activity of 40 as well as 48 against a range of cancer cell lines, including H460 lung carcinoma, DU145 prostate carcinoma, A-431 skin (epidermoid) carcinoma, HT-29 colon adenocarcinoma and MCF7 breast adenocarcinoma. They found that 4-quinolone derivatives 40 was more potent than 2,3-dihydroquinolin-4-one derivatives 48.
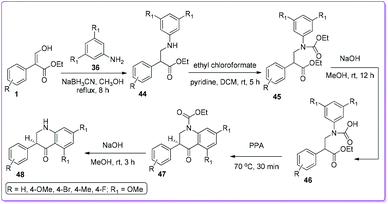 |
| | Scheme 23 Synthesis of 2,3-dihydroquinolinones from 1. | |
In 2002, Joseph et al. prepared numbers of N-substituted 3-aryl-2-quinolone derivatives 49 starting from 1 to develop nontoxic antimigratory compounds.36 They proposed that such 2-quinolone derivatives 49 could also complement the anticancer action of conventional drugs. After synthesizing compound 37 from 1, alkylation led to the preparation of targeted N-alkylated derivative 49 and 50. Further transformation of 50 formed the lead compound 51 by using tributyltin azide (Scheme 24). Their study indicated that the efficacy of conventional drugs was improved when nontoxic antimigratory drug 51 was combined with conventional cytotoxic drugs either ETO (etoposide inhibitor) or DOX (doxorubicin) (which are topoisomerase II inhibitors) in treatment for MXT mouse mammary tumour.36
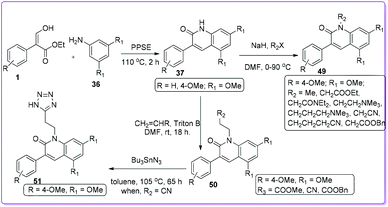 |
| | Scheme 24 Synthesis of N-substituted 2-quinolinones from 1. | |
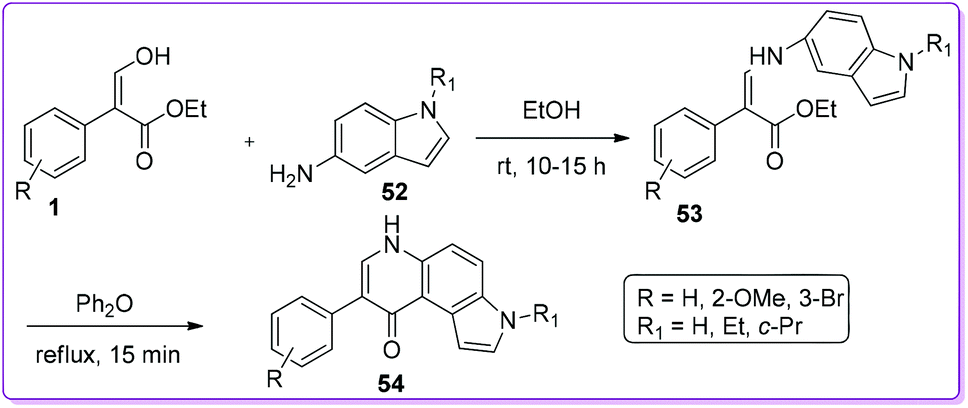
Viola et al. synthesized a series of 7-phenyl-3H-pyrrolo[3,2-f]quinolin-9-ones 54 and evaluated their anti-proliferative activity (Scheme 25).8a During synthesizing their target molecules, compound 1 was condensed with 5-aminoindoles 52 to prepare enamine 53. The mixture of E-and Z-isomer of 53 used for thermal cyclization to form 54. The final derivatives were screened against solid tumour and leukemic cell lines for observing in vitro anti-proliferative activity. Some of the synthetic compounds executed very promising micromolar potency compare to well-known colchicine class of compounds.8a
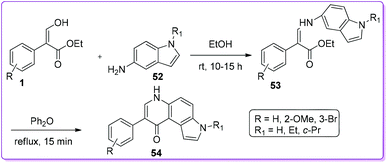 |
| | Scheme 25 Synthesis of pyrroloquinolinones from 1. | |
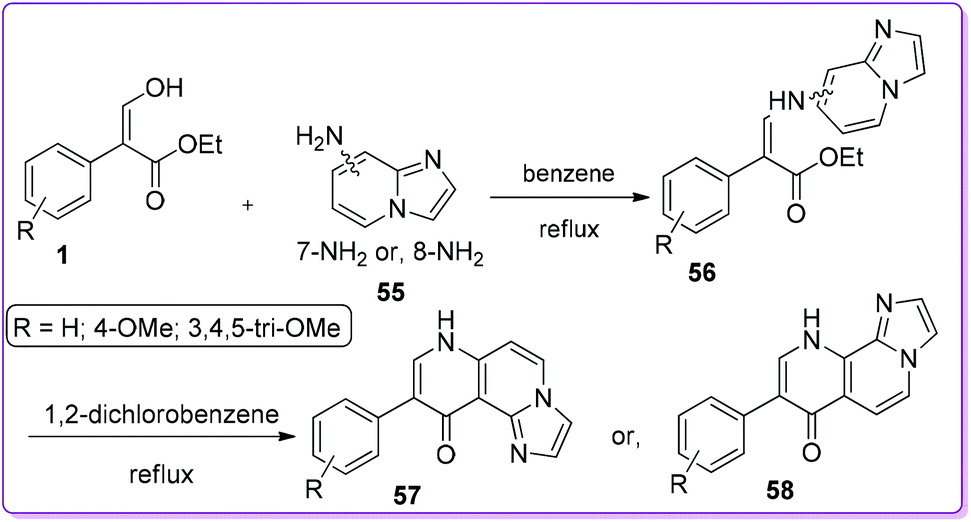
Moreau and co-workers synthesized a series of β-arylimidazonaphthyridine-7-ones 57 and β-arylimidazonaphthyridi-ne-10-ones 58 from 1 by Conrade–Limpach reaction (Scheme 26).7 They screened antiproliferative activity of compounds 57 and 58 against four tumour cancer cell lines; HT-1080 (fibrosarcoma), HT-29 (colon carcinoma), M21 (melanoma) and MCF7 (breast carcinoma) and found that 58 showed low to moderate cytotoxic activity, where 57 was inactive.
 |
| | Scheme 26 Synthesis of β-imidazonaphthyridinones from 1. | |
3.2 Pyrimidine/pyrimidinone type compounds
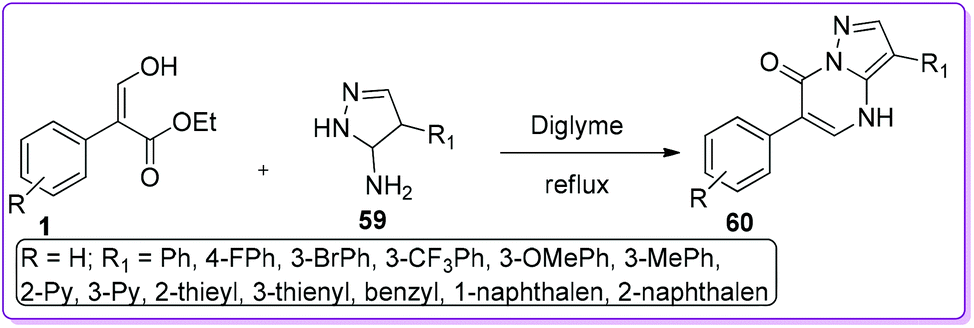
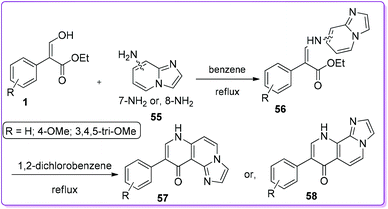
Various neurological disorders related to the GABAergic system can be treated by targeting the GABAA-Receptors (GABAA-RS).37 Compounds of different structural classes can modulate, directly activate or inhibit GABAA-R subtypes. Therefore, different types of fused nitrogen-containing heterocyclic systems have been extensively investigated to synthesize GABAA-Receptors sub-type ligands.38 Selleri et al. described the synthesis of second-generation pyrazole-pyrimidine compound 60, bearing an aryl ring at 6-position as an isostere of the thiophene ring to evaluate in vitro GABAA-receptors affinity.39 Compound 60 was prepared by a one-step reaction between a suitable pyrazole 59 and 1 in diglyme at reflux condition (Scheme 27).
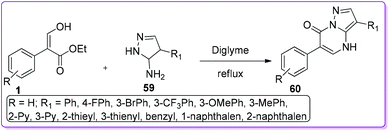 |
| | Scheme 27 Synthesis of pyrazolopyrimidinone from 1. | |
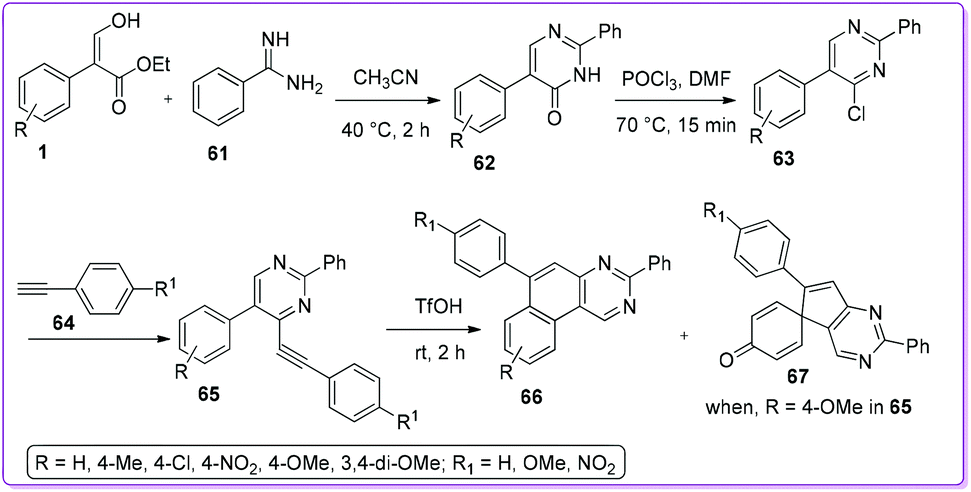
Kuznetsov et al. reported one of the simplest synthetic protocols for deriving 5-aryl-4-(arylethynyl)pyrimidines 63 from compound 1. First, acrylate 1 and benzamidine 61 reacted to form 2,5-diphenylpyrimidin-4(3H)-one 62 which was then converted to 4-chloropyrimidines 63 by using POCl3 (Scheme 28).40 The resulting pyrimidine 63 underwent the Sonogashira coupling reaction with ethynylbenzene 64 and yielded 2,5-diphenyl-4-(phenylethynyl) pyrimidine 65. Acidic cyclization of 65 produced benzo[f]quinazoline 66. With para-methoxy aryl acrylate, spiro[cyclohexa-2,5-diene-1,5′-cyclopenta[d]pyrimidin]-4-one 67 was obtained along with product 66.
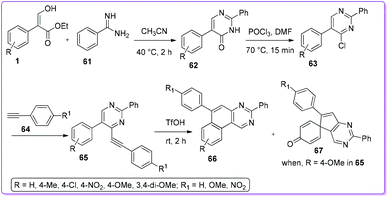 |
| | Scheme 28 Synthesis of benzoquinazolines and pyrimidinone from 1. | |
3.3 Uracil type compounds
Uracil is one of the three pyrimidines widely spread in naturally occurring nucleic acids.41 Due to structural similarities with nucleosides and nucleotides, uracil plays a pivotal role in the treatment for human immunodeficiency virus (HIV),42 hepatitis B virus (HBV),43 hepatitis C virus (HCV),44 herpes simplex virus (HSV),45 and various types of cancer.46
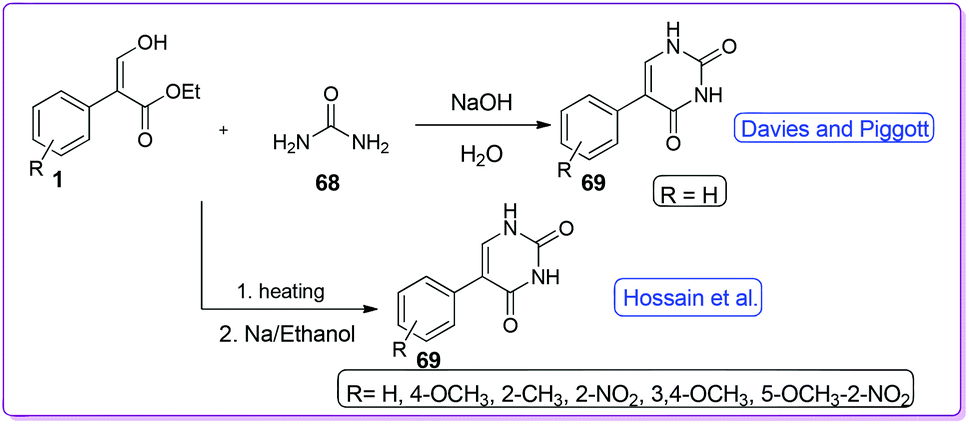
Davies and Piggott introduced an approach for the synthesis of uracil type compounds, 5-phenylpyrimidine-2,4(1H,3H)-dione 69 from the reaction of 1 and 68 in the presence of NaOH (Scheme 29).47 Interestingly, whenever other amides were attempted to be used in this reaction, no product was yielded. In 2007, Hossain et al. reported NaOEt catalysed synthesis of a series of derivatives 69 using a variety of acrylates including both electron donating and electron withdrawing groups (Scheme 29).48
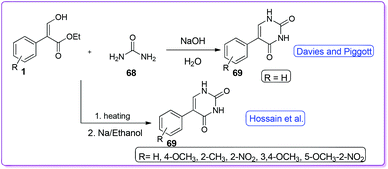 |
| | Scheme 29 Synthesis of 5-phenyl uracil from 1. | |
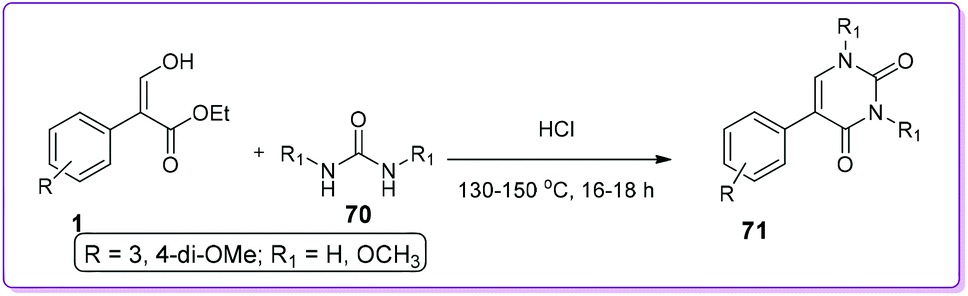
Takaya et al. reported an acid catalysed condensation between 1 and 70 to synthesize 5-aryl uracil derivatives 71 at 130–150 °C under reduced pressure (Scheme 30).49 This condensation reaction was only limited with 3,4-dimethoxyphenyl acrylate.
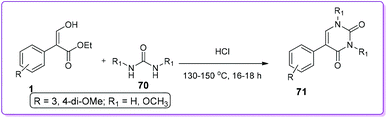 |
| | Scheme 30 Synthesis of N-substituted 5-aryl uracil from 1. | |
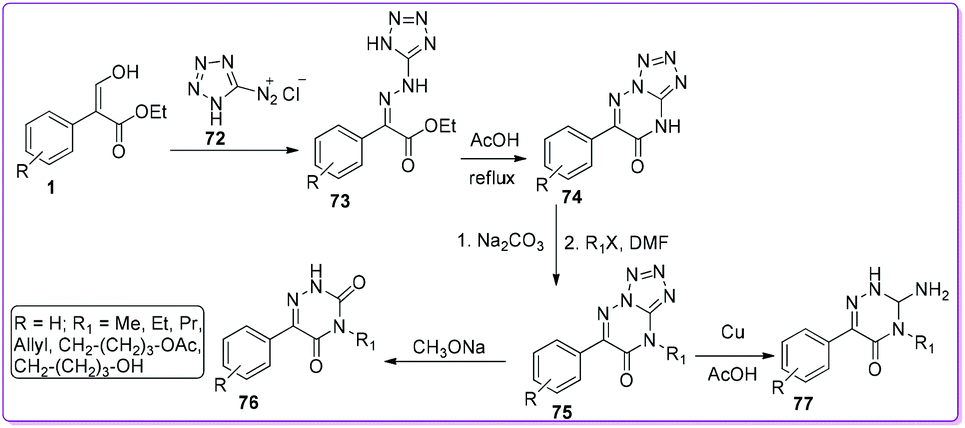
Shestakova et al. reported the synthesis of azauracils 76 and azaisocytosines 77 by the azo-coupling reaction of diazotetrazole 72 with 1 (Scheme 31).50 At first, the compound 72 reacted with 1 to form hydrazine 73 which transformed into 6-phenyltetrazolo[1,5-b]triazine-7-one 75via tetrazole 74 by refluxing with acetic acid. Depending on the reaction condition, 75 produced either azauracils 76 or azaisocytosines 77. These analogues were demonstrated as effective in the biosynthesis of anomalous nucleosides.50
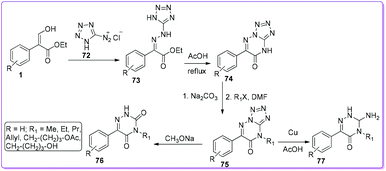 |
| | Scheme 31 Synthesis of azauracils and azaisocytosines from 1. | |
3.4 Pyrone type compounds
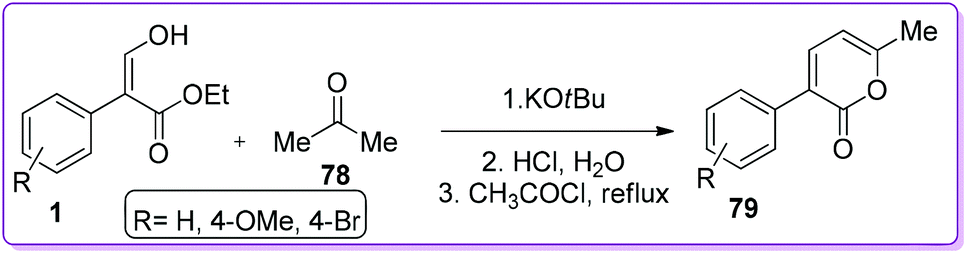
2-Pyrone is a conjugated diene found in pheromonal, antifungal, antitumor and neurologically effective compounds. It is widely used in Diels–Alder cycloaddition reactions.51,52 In 1985, Kovshev et al. first reported the synthesis of 3-aryl-6-methyl-2-pyrones 79 by condensation of 1 with acetone 78 in the presence of potassium tert-butoxide (Scheme 32).53
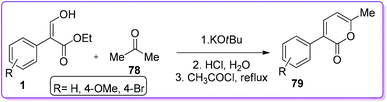 |
| | Scheme 32 Synthesis of 2-pyrone from 1. | |
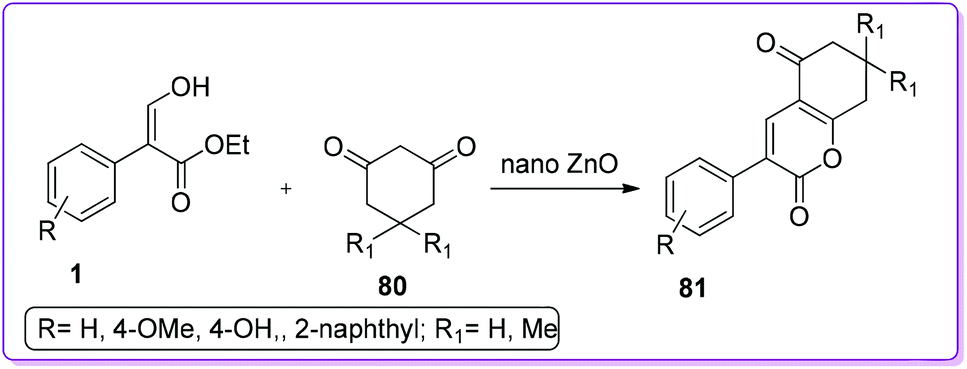
Moghaddam and co-workers reported the synthesis of highly substituted 2-pyrone derivatives 81 by tandem Knoevenagel condensation followed by lactonization reaction of 1,3-cyclohexadiones 80 with 1 in the presence of nanocatalyst, ZnO (Scheme 33).54
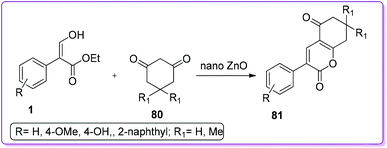 |
| | Scheme 33 Synthesis of bicyclic 2-pyrone from 1. | |
3.5 Isoxazoline type compounds
Isoxazolinone derivatives are promising molecules for bacterial (Gram-positive) and inflammatory lung diseases (H1N1 and SARS virus infection).6,55 They are the versatile building blocks in organic synthesis mainly because of the weak N–O bond (BDE, D0298 = 151 kcal mol−1) cleavage and CO2 extrusion.56,57 Several approaches to synthesize isoxazolinone derivatives were reported from 1 (Scheme 34).
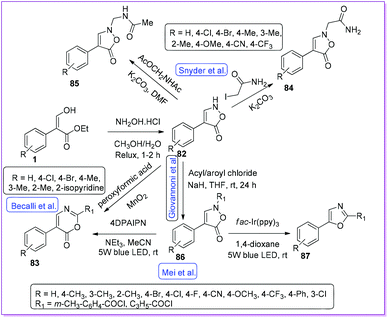 |
| | Scheme 34 Synthesis of isoxazolone derivatives from 1. | |
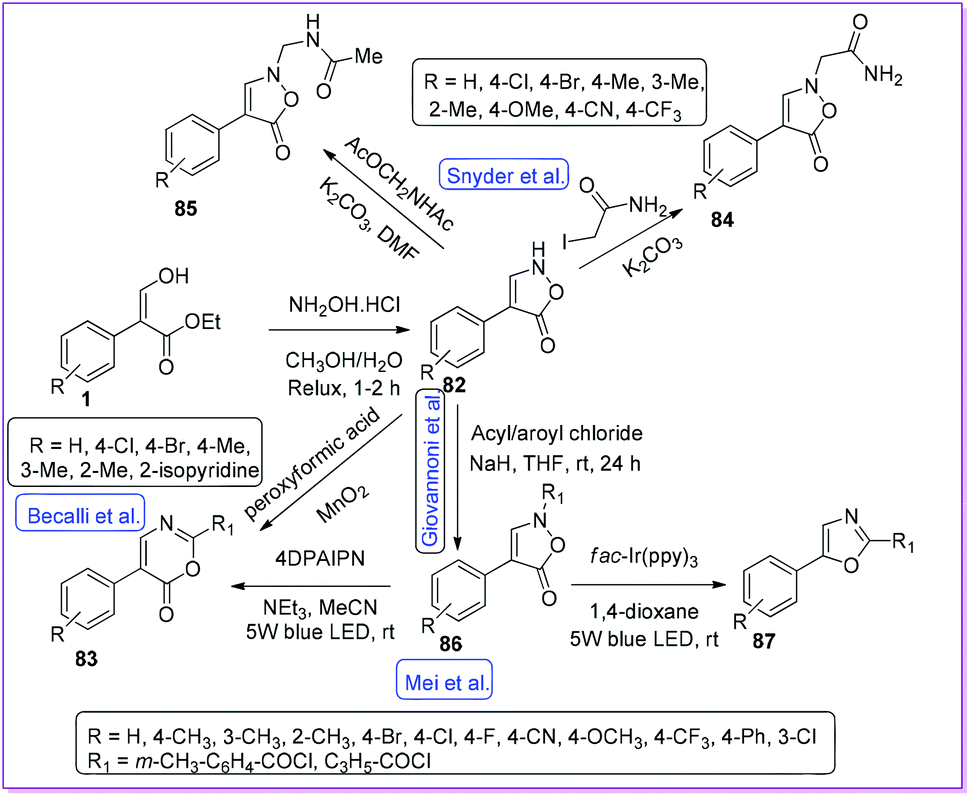
In 1984, Beccalli and co-workers reported a convenient way for the synthesis of 4-arylisoxazolone derivatives 82 through a cyclocondensation reaction between 1 and hydroxylamine hydrochloride.12 The authors also published that the compound 82 undergoes oxidative rearrangement to form 1,3-oxazin-6-one derivatives 83 (Scheme 34). Later, Snyder and co-workers further extended the scope of 82 moiety to prepare 84 and 85 by alkylation with iodoacetamide and acetamido methyl acetate respectively.58 After in vitro antibacterial activity screening, compound 84 was found active, whereas retroamide 85 was inactive (Scheme 34). In 2019, Giovannoni et al. modified N-alkylation reaction by using appropriate acyl/aroyl chloride with 82 to synthesize N-acyl-4-aryl-isoxazol-5-one 86 (Scheme 34).6 Mei et al. reported the visible light promoted selective decarboxylative rearrangement of 86 to oxazole 87 by photocatalytic reaction.59 Switching the photocatalyst from fac- tris[2-phenylpyridinato-C2,N]iridium(III)(Ir(ppy)3) to donor–acceptor fluorophore 1,3-dicyano-2,4,5,6-tetrakis(N,N-diphenyl amino)-benzene (4DPAIPN), selective formation of compound 1,3-oxazin-6-one 83 was obtained. It should be mentioned that both 83 and 87 are vital heterocyclic cores in a wide range of bioactive compounds, as exemplified by antidiabetic agent AD-5061,60 antimycobacterial texaline61 and salinazinone A.62
3.6 Phenanthrene/phenanthroline type compounds
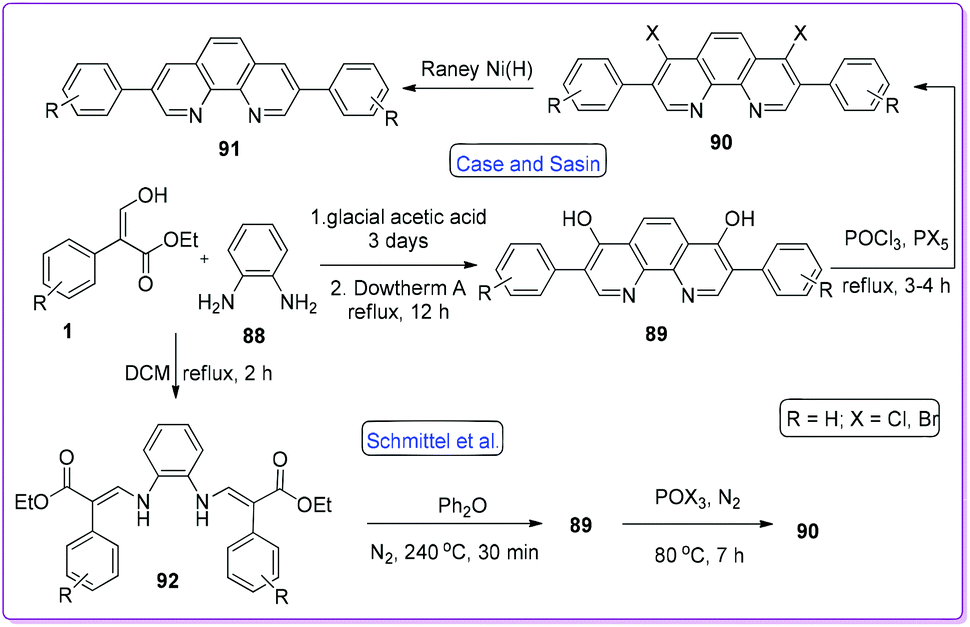
Phenanthroline plays a significant role in supramolecular coordination chemistry63,64 as well as in colorimetric assay of multiple metal ions.65,66 It can bind to metal through N and O-donor atoms and this interesting feature attracted chemists to explore different synthetic strategies. In 1955, Case and Sasin reported the synthesis of 4,7-dihydroxy-3,8-diphenyl-1,10-phenanthrolines 89 by the cyclization reaction of 1 and o-phenylenediamine 88.67 Successive halogenation of the hydroxy group and reduction by RANEY®-Ni formed the phenanthroline compound 91via compound 90 (Scheme 35).
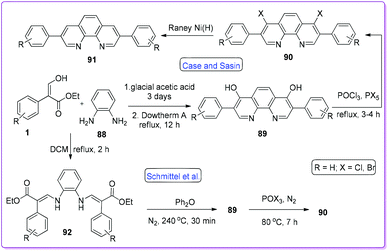 |
| | Scheme 35 Synthesis of phenanthrolines from 1. | |
Later, Schmittel and co-workers reported the method of preparing 92 by refluxing 1 along with 88 in dichloromethane.3 Cyclization of 92 at 240 °C yielded compound 89 which was then converted to 90 by similar conditions as reported earlier by Case and Sasin (Scheme 35).67
3.7 Synthesis of vinyl derivatives
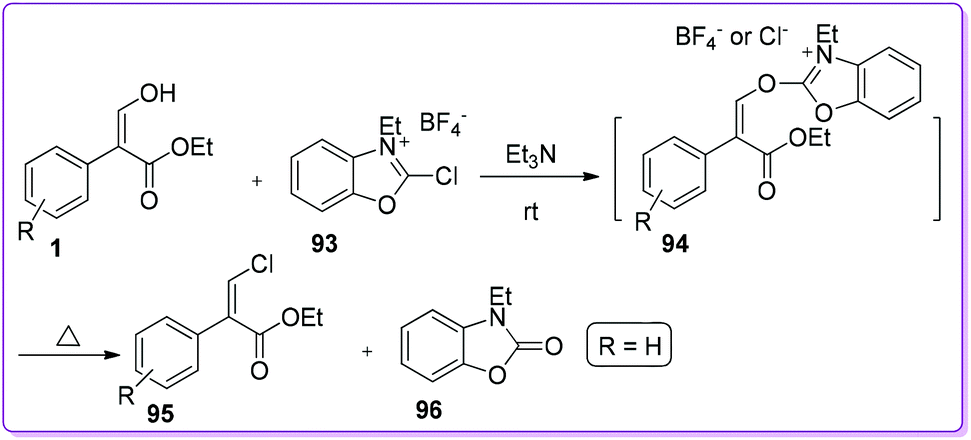
First selective method for the preparation of vinyl chloride 95 from 1 was reported by Mukaiyama et al.68 Treatment of 1 with 2-chloro-3-ethylbenzoxazolium tetrafluoroborate 93 in the presence of triethylamine formed an active intermediate 94. Successive nucleophilic attack on the intermediate by chloride ion afforded the corresponding vinyl chloride 95 (Scheme 36).
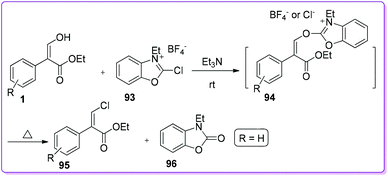 |
| | Scheme 36 Synthesis of chloro vinyl derivatives from 1. | |
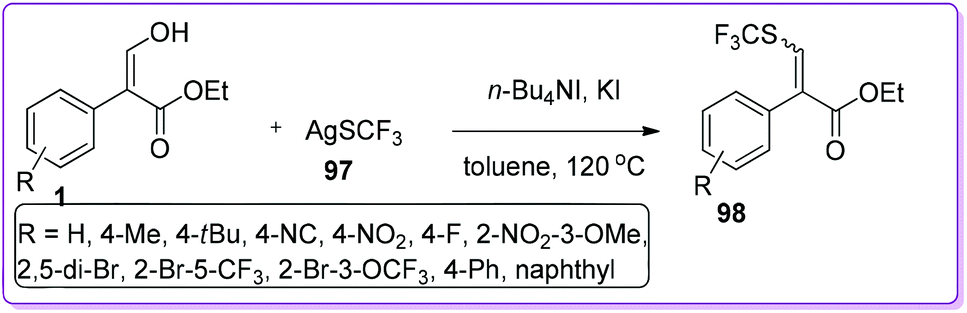
Qing and co-workers synthesized a variety of trifluoromethyl sulfides 98 by direct trifluoromethylthiolation of CSP2–OH bond activation of 1 using silvertrifluoromethanethiol 97.69 The employment of both of n-Bu4NI and KI activated 97 to afford the desired product 98 (Scheme 37). This convenient approach of incorporating such an extremely high lipophilic70 trifluoromethylthio group (SCF3) might simplify the synthesis of many pharmaceutical and agrochemical compounds such as toltrazuril,71 fipronil72 and tiflorex.73
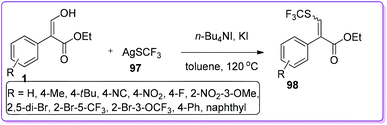 |
| | Scheme 37 Synthesis of thio vinyl derivatives from 1. | |
3.8 Non-steroidal anti-inflammatory drugs (NSAIDs)
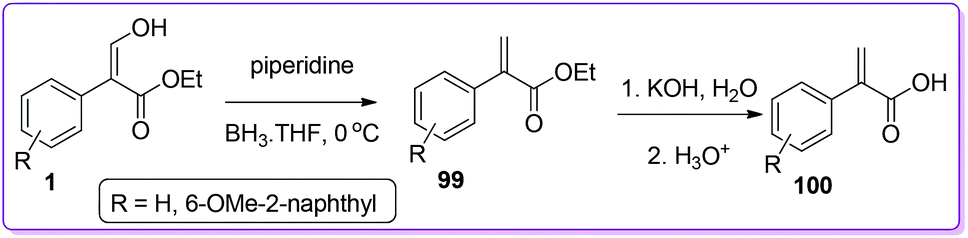
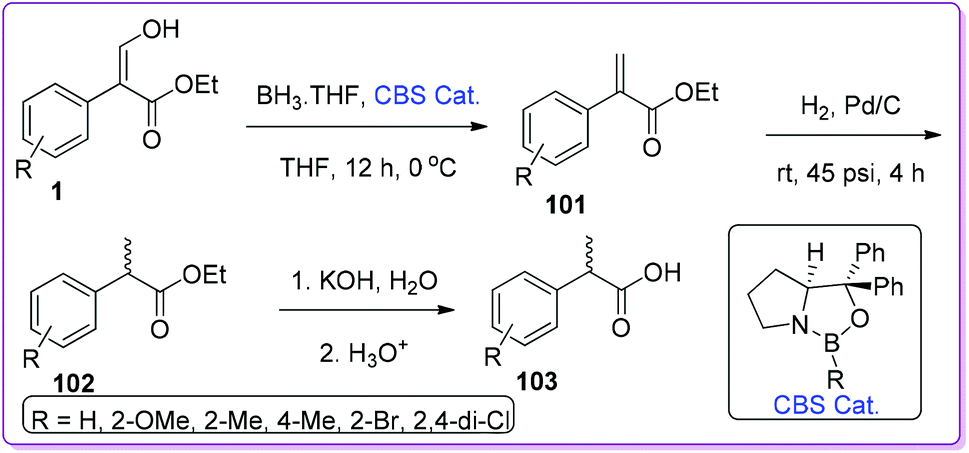
α-Arylpropanoic acids containing non-steroidal anti-inflammatory agents (NSAIDs) such as naproxen, profens etc. are globally used as potential candidates for pain relief and the treatment of chronic rheumatoid arthritis.74–76 Approaches using multistep synthetic route or CO gas at high pressure have limited their use in high scale production of α-arylpropanoic acids.77 In 2002, Hossain et al. described the synthesis of naproxen precursor 100 from 1 in only two steps.1a The synthesis includes a unique reduction of 1 by using BH3·THF to form 99 followed by base catalysed preparation of 100 (Scheme 38).
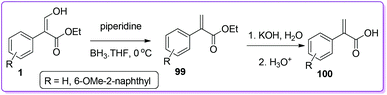 |
| | Scheme 38 Synthesis of naproxen precursor from 1. | |
Recently, Hossain group advanced the catalytic system for generalizing the reduction of 1 with Corey–Bakshi–Shibata (CBS) catalyst. They started with the prepared α-arylpropenoic ester 101 to further reduce it to saturated α-arylpropanoic ester 102. Finally, after hydrolysis, they separated the desired racemic α-arylpropanoic acid 103 (Scheme 39).78
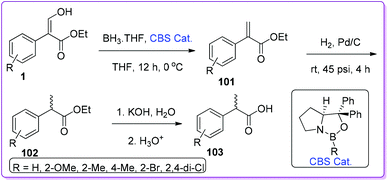 |
| | Scheme 39 Synthesis of 2-aryl propanoic acid from 1. | |
3.9 All-carbon quaternary centre containing compounds
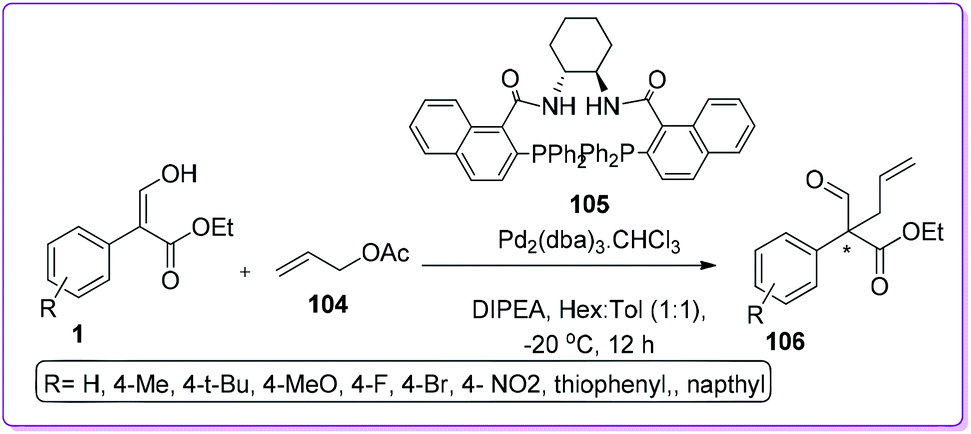
Quaternary carbon stereo centres are prevalent scaffold in many bioactive natural products and pharmaceuticals such as taxol,79 gelsemine,80 morphoine,81 and vinblastine.82 Lots of attention had been devoted to the development of efficient methods for the stereo-controlled construction of quaternary carbon centers.83 The formation of acyclic quaternary carbon stereocenters is more challenging due to enhanced conformational mobility compare to ring structure.84 A couple of successful methods (both in intra and intermolecular way) were reported by Hossain group to prepare such achiral quaternary centre from 1.
A unique route of intermolecular Pd-catalysed asymmetric allylic alkylation (Pd-AAA) of 1 to synthesize all-carbon quaternary centre containing compound 106 was first introduced by Hossain and co-workers (Scheme 40).85 They explained that base induced deprotonation of 1 resulted in enolate, which acted as a nucleophilic counterpart against electrophilic Pd-allyl complex in the presence of (R,R)-DACH naphthyl Trost ligand 105. Optimized reaction conditions provided good to excellent yields (75–99%) and enantioselectivities (75–94%) of 106.
 |
| | Scheme 40 Intermolecular synthesis of all-carbon quaternary centres from 1. | |
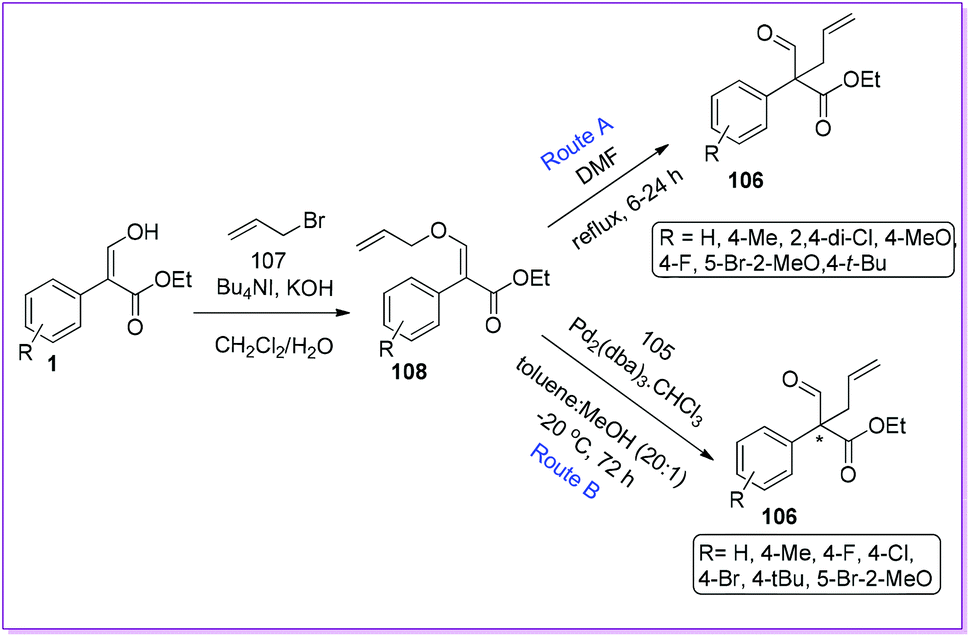
The first initiative of an intramolecular Claisen rearrangement of O-allyl enol-ether 108 to prepare racemic 106 was reported by Hossain et al. in 2010 (Route A, Scheme 41).86 Compound 108 was made from 1 by O-allylation reaction using n-Bu4NI as a phase transfer catalyst (PTC). Later, the same group published an asymmetric synthesis of chiral 106 by Pd-AAA using Trost chiral diphosphine ligand.87 The authors observed that enol ethers 108 was used as precursors for pi-allylpalladium intermediates to form the all-carbon quaternary aldehyde 106 (Route B, Scheme 41).
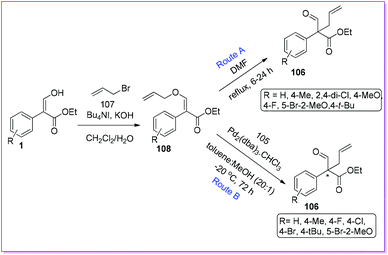 |
| | Scheme 41 Intramolecular synthesis of all-carbon quaternary centres from 1via enol-ether. | |
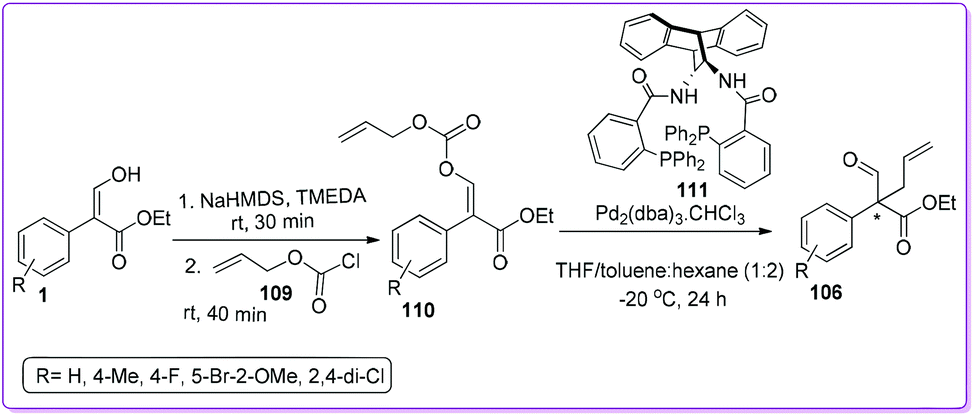
The group also reported a Pd-catalysed decarboxylative allylic alkylation (DAAA) reaction using DACH phenyl ANDEN Trost ligand 111 for the formation of 106 (Scheme 42).88 They accomplished the stereoselective preparation of Z- or E-isomer of the carboxy allyl acrylate 110 in very high Z/E ratios (50:1 or 1:99 respectively) from 1 by screening different bases and chelating agents. Due to the better reactivity and enantioselectivity, the E-isomer was further explored for the DAAA reaction to make 106 with high yields (96–99%) and moderate to good enantioselectivities (53–73% ee).
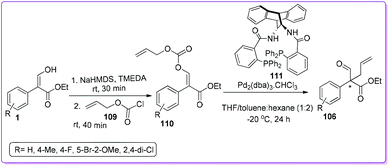 |
| | Scheme 42 Intramolecular synthesis of all-carbon quaternary centres from 1via carboxylate. | |
3.10 Benzofuran and related compounds
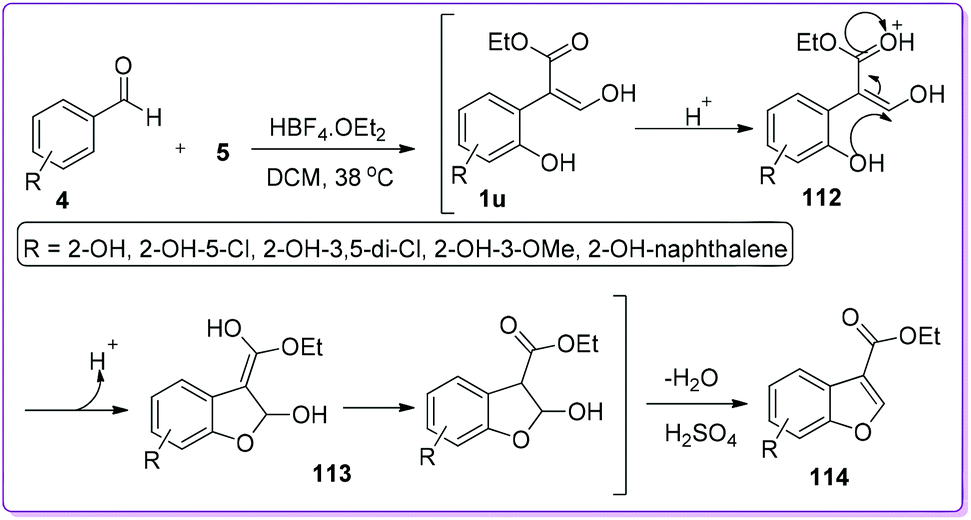
Benzofurans are pivotal precursors for the synthesis of many pharmaceutical and biologically active compounds.89,90 Several methods were reported for the synthesis of 2-substituted or 2,3-disubstituted benzofurans but the preparation of 3-substituted benzofurans was limited. In 2006, Hossain et al. reported an unexpected formation of 3-ethoxycarbonylbenzofurans while studying the substrate scope of benzaldehyde 4 to prepare substituted 3-hydroxy-2-arylacrylates 1.91 To explain the results, they proposed a mechanism which indicates that ortho-hydroxybenzaldehyde 4 first reacts with EDA 5 to form 1u which subsequently undergoes cyclization in the presence of acid to form the hemiacetal 113. Finally, the resulted hemiacetal 113 in the presence of concentrated H2SO4 undergoes dehydration to form 3-ethoxycarbonylbenzofuran 114 (Scheme 43).92 This protocol for the synthesis of 114 was further explored by other groups to synthesize medicinal and natural products which are shown in Schemes 44–50.
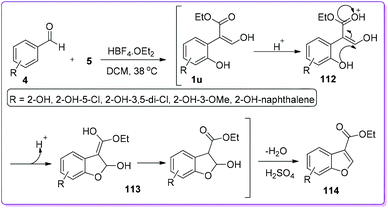 |
| | Scheme 43 Synthesis of 3-ethoxycarbonylbenzofurans from 1. | |
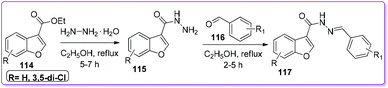 |
| | Scheme 44 Synthesis of benzofuran-3-carbohydrazide. | |
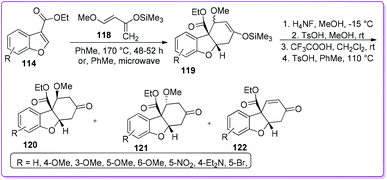 |
| | Scheme 45 Synthesis of building block of natural products from 1. | |
 |
| | Scheme 46 Synthesis of 2,3-disubstituted benzofuran from 1. | |
 |
| | Scheme 47 Synthesis of pterocarpenes and coumestans from 1. | |
 |
| | Scheme 48 Synthesis of (+)-paeoveitol and (−)-paeoveitol from 1. | |
 |
| | Scheme 49 Synthesis of enantiopure (R)-BRL-37959 from 1. | |
 |
| | Scheme 50 Synthesis of BRL-37959 and its analogues from 1. | |
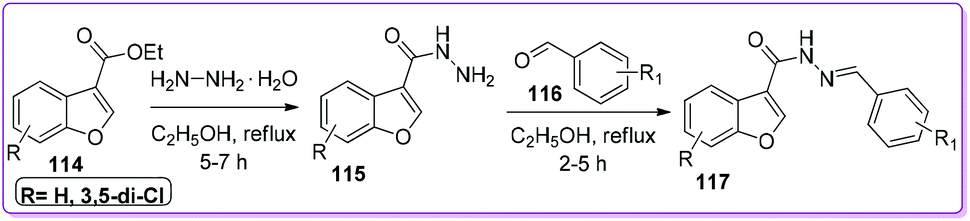
Telvekar and co-workers used 114 as a precursor to synthesize a novel series of N′-benzylidene benzofuran-3-carbohydrazide compounds 117 (Scheme 44).93 The authors mentioned that the synthesized compound 117 and its analogues showed promising activity as an anti-mycobacterial agent against Mycobacterium tuberculosis (Mtb) H37Rv strain.
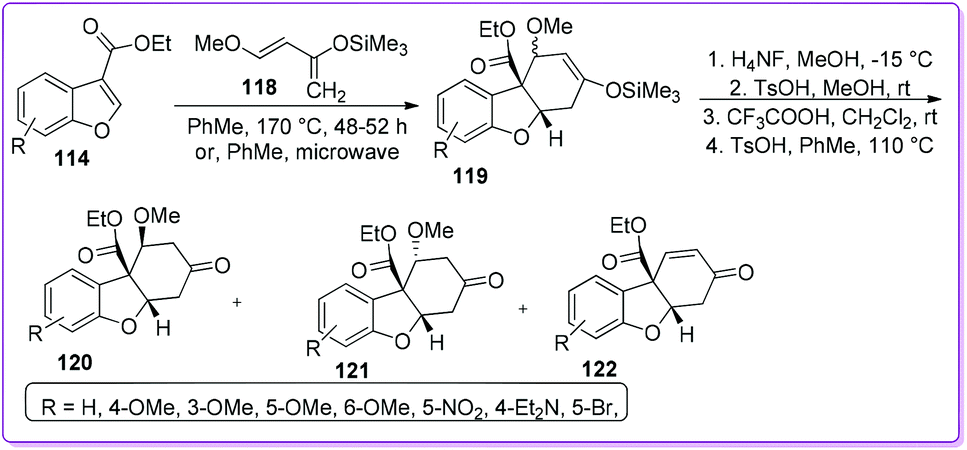
In 2013, Tolstikov et al. reported regioselective Diels–Alder reactions of Danishefsky's diene 118 with substituted 3-ethoxycarbonyl benzofurans 114 (Scheme 45).94 These reactions provided an effective method for the construction of heterocyclic skeletons of hexahydrodibenzofuran-7-ones 120 and 121 as well as tetrahydrodibenzofuran-7-one 122. These tricyclic fragments are the structural components of many pharmacologically vital substances, such as plant alkaloids morphine, galanthamine, lycoramine, lunarine, linderol A, and several selective estrogen receptor β agonists.95–97
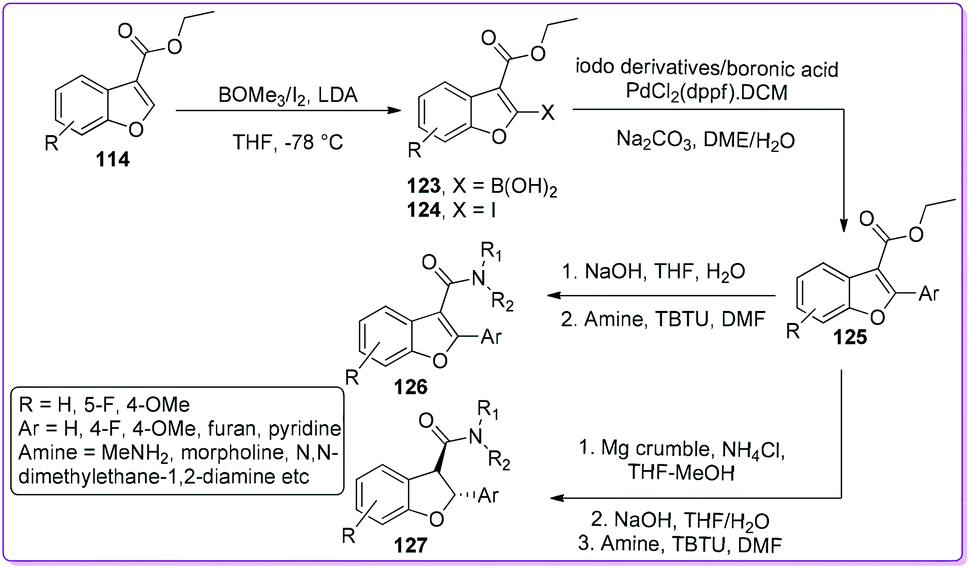
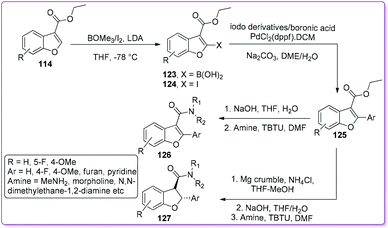
In 2017, Elofsson et al. synthesized a series of 2-aryl benzofuran 3-carboxamides 126 and trans-2,3-dihydrobenzofuran-2-aryl benzofuran 3-carboxamides 127 from 3-ethoxycarbonyl benzofuran 114.98 The authors reported that 114 first converted to 3-ethoxycarbonyl-2-aryl benzofuran 125 by Suzuki coupling reaction as shown in Scheme 46. Then, the compound 126 was synthesized from 125 by amidation whereas the compound 127 was obtained by reduction of 125 followed by amide formation. The resulted scaffolds have been considered as a core component in many biologically active natural and synthetic compounds including antiviral, antibacterial, anti-inflammatory, antiangiogenic, and antimitotic activities.99
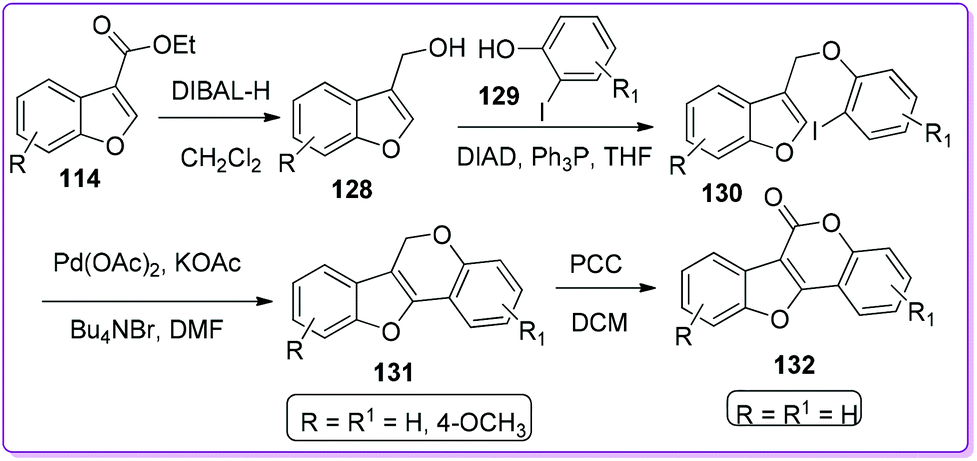
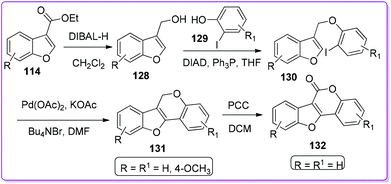
Morrow et al. synthesized pterocarpenes 131 and coumestans 132 type heterocycles from 3-ethoxycarbonylbenzofuran 114via a potential Heck cyclization reaction. At first, the compound 114 reduced in the presence of DIBAL-H to form 3-hydroxymethylbenzofuran 128 which undergoes Mitsunobu coupling with ortho-iodophenols 129 to form aryloxymethylbenzofurans 130. Pterocarpans 131 was obtained from 130 by Heck cyclization under Jeffery's conditions. Unsubstituted pterocarpans converted to coumestan 132 by oxidizing in the presence of pyridinium chlorochromate (PCC) (Scheme 47).100 Pterocarpans show a broad-spectrum activity against Gram-positive bacteria and vancomycin-resistant strains of enterococci; on the other hand, coumestans have antibacterial, antifungal, and antimyotoxic effects.101
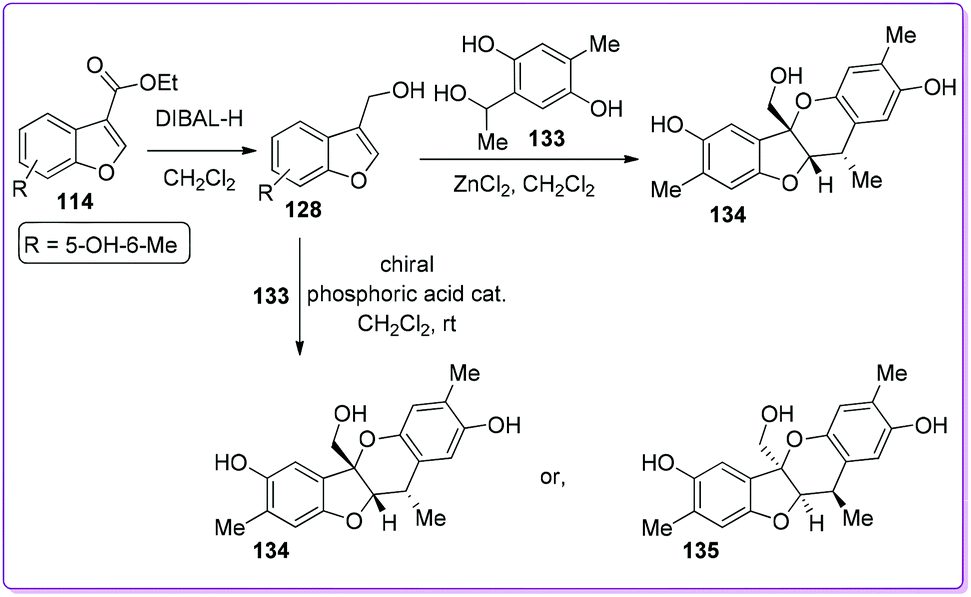
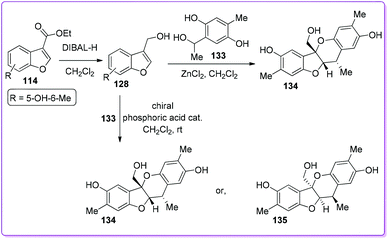
Paeoveitol is one of the norditerpene natural products, used as an antidepressant agent for many years.102 Zhao et al. first reported a highly efficient total synthesis of paeoveitol 134 from 114 (Scheme 48).103 The compound 114 was reduced to paeoveitol D 128 and underwent an unusual intermolecular cycloaddition with ortho-quinone methide 133 to synthesize 134. In this method, the final product retained an excellent regio- and diastereoselectivity.
Later on, Chen and co-workers reported the first catalytic asymmetric total synthesis of (+)-paeoveitol 134 and (−)-paeoveitol 135 from 3-ethoxycarbonyl benzofuran 114via a biomimetic hetero-Diels–Alder reaction in the presence of chiral phosphoric acid catalysts (Scheme 48).104
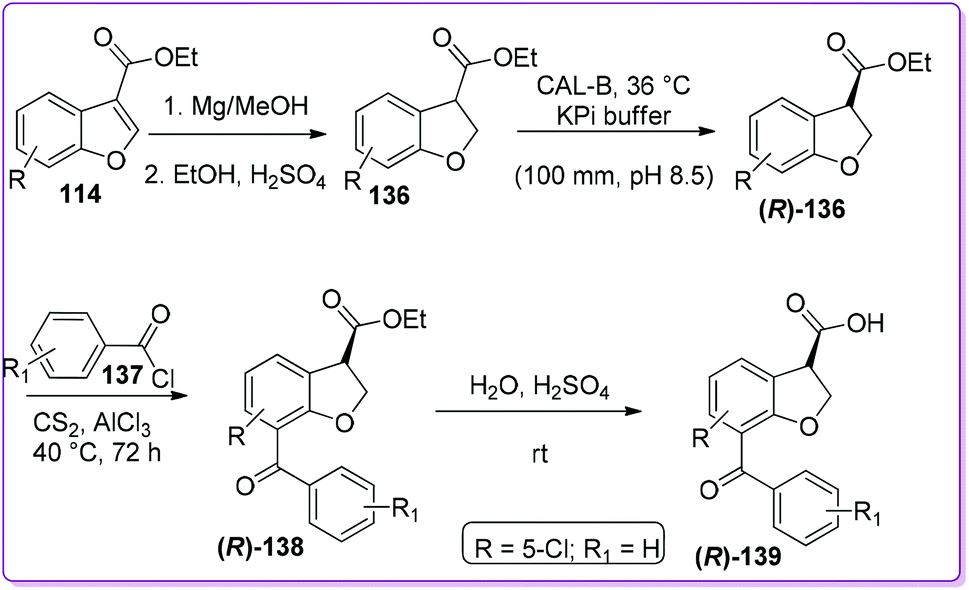
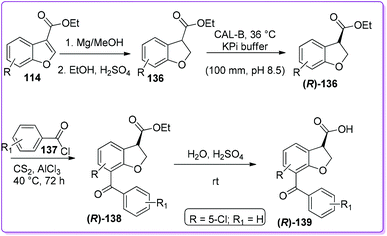
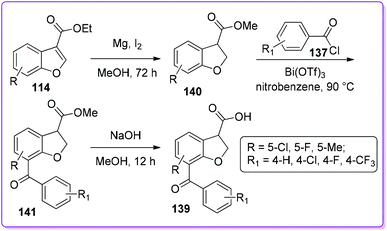
Bongen et al. reported an efficient asymmetric synthesis of 7-benzoyl-2,3-dihydro-1-benzofuran-3-carboxylic acid, BRL-37959 139 (Scheme 49),105 which acts as a very potent analgesic agent with low gastric irritancy.106 At first, 114 was reduced by magnesium turnings in methanol to form racemic 2,3-dihydrobenzofuran-3-carboxylic acid ethyl ester 136. This racemic product 136 was then resolved by dynamic kinetic resolution (DKR) followed by the Friedel–Crafts acylation producing enantiopure product (R)-136. Finally, the acylated intermediate (R)-138 undergoes acidic hydrolysis to produce the desired (R)-BRL-37959 139. In the synthesis, the DKR was found to be a useful method to prepare (R)-136 although the acylation to make (R)-138 was low yielding.
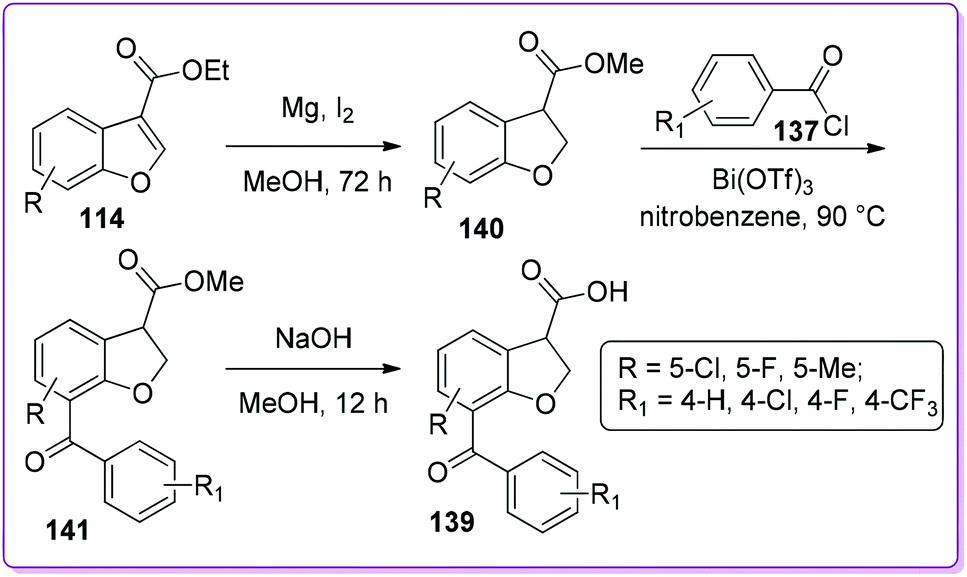
Recently, the Hossain group reported an efficient method to synthesize BRL-37959 and its analogue 139 from 114 (Scheme 50).107 The main challenge associated with acylation encountered by Bongen et al.105 was overcome by using bismuth(III) trifluoromethanesulfonate as a catalyst. This three-step synthesis has been proven to be a highly concise method to produce 139 as well as its analogue in good yields.
3.11 Other applications
3.11.1 Indole/gramine type compounds.
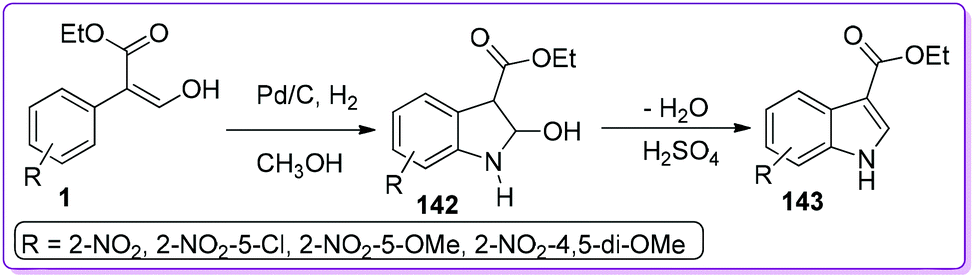
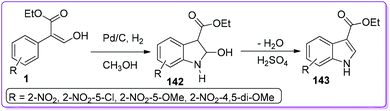
Indole is considered as an important building block of many pharmaceutical and biologically active compounds such as indocin (anti-inflammatory),108 lescol (cholesterol-lowering),109 vinblastine (anti-cancer),110 oncovin (anti-cancer),111 and tryprostatins (microtubule inhibitor).112 Out of many different approaches of mono- and di-substituted indole syntheses, there are only a few methods for preparing 3-substituted indole.113 Hossain et al. developed a convenient two-step procedure for the synthesis of 3-ethoxycarbonylindole 143 from commercially available 3-hydroxy-2-nitrophenyl acrylate.5a Selective reduction of the nitro group provided 2-hydroxyindoline-3-carboxylate 142, and subsequent acid-induced dehydration resulted in the functionalized indole ester 143 (Scheme 51).
 |
| | Scheme 51 Synthesis of 3-ethoxycarbonylindole from 1. | |
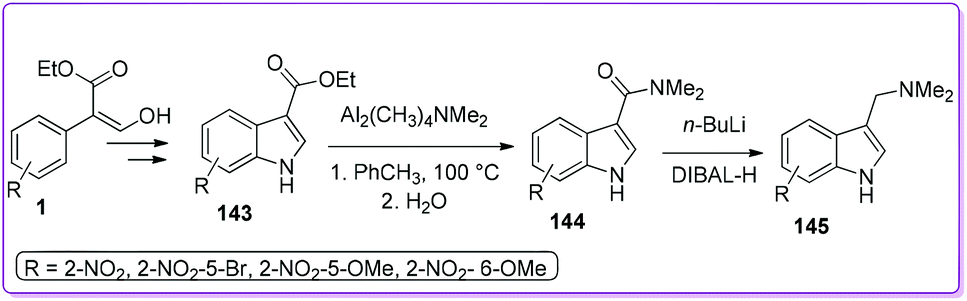
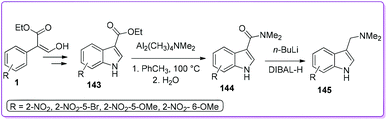
Later, the same group developed an efficient method for the preparation of a versatile synthetic intermediate, 3-dimethylaminomethylindole (gramine) 145 by adding two more steps to 143 (Scheme 52).5b The simplistic amidation of 143 using dimeric amidoaluminium complex is the key step for this synthetic approach. An extended scope of preparing various substituted indoles and gramines have been offered by using these efficient methods.
 |
| | Scheme 52 Synthesis of gramine from 1. | |
3.11.2 Isoquinoline type compounds.
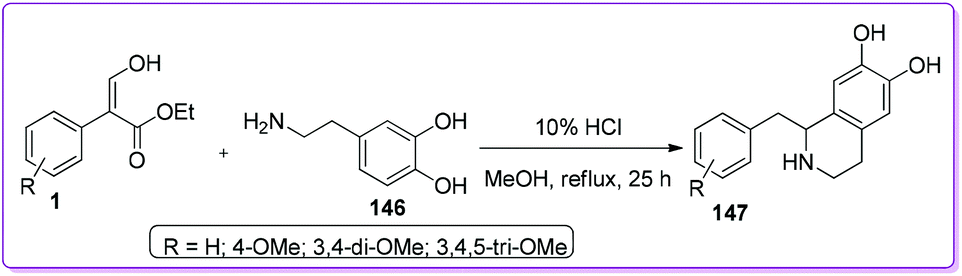
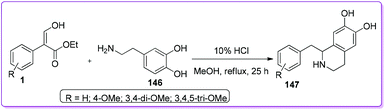
1-Substituted isoquinoline hydrochloride such as 1-(3,4,5-trimethoxybenzyl)-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline hydrochloride 147 is already used as a potent bronchodilator in practical clinics.114 Yamato et al. reported a convenient protocol of preparing such isoquinolines 147 by reacting compound 1 with amine 146 in acidic methanol under reflux condition.8c The resulting crude was solidified by trituration and further purified to form colourless crystals 147 (Scheme 53).
 |
| | Scheme 53 Synthesis of isoquinoline derivatives from 1. | |
3.11.3 Enamine type compounds.
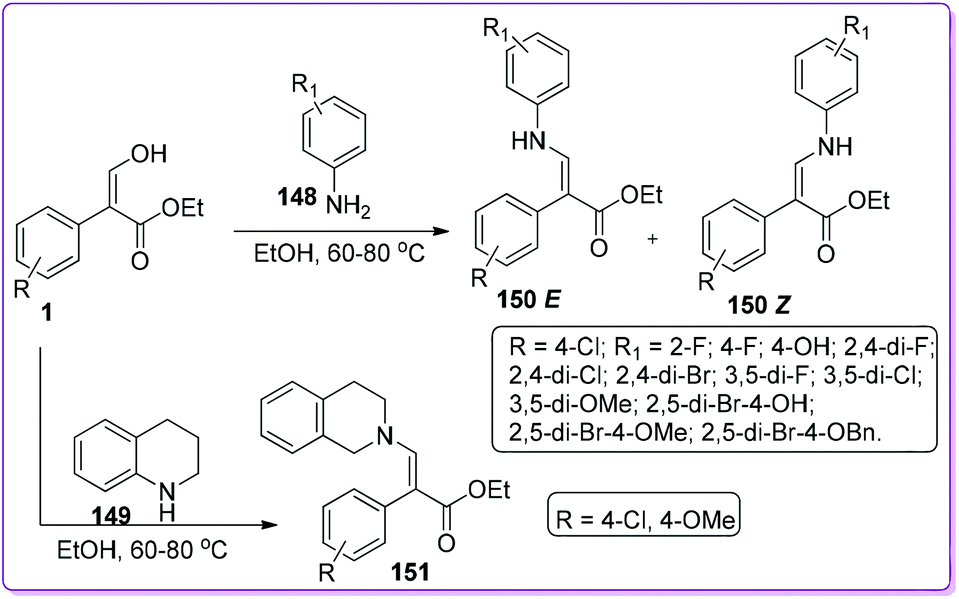
Stable enamines such as 150 and 151 possess antibacterial activity reported by Xiao et al.115 The group published structure–activity relationships (SARs) of their twenty six synthesized enamine compounds based upon the bioassay using Staphylococcus aureus ATCC 6538 and Pseudomonas fluorescens ATCC 13525 strains.116,117 They synthesized the enamines 150 and 151 by a dehydration reaction of 1 with different arylamines 148 and 149 respectively (Scheme 54).
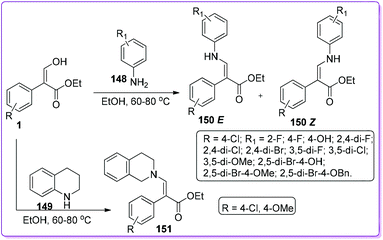 |
| | Scheme 54 Synthesis of enamine derivatives from 1. | |
3.11.4 Tropic acid type compounds.
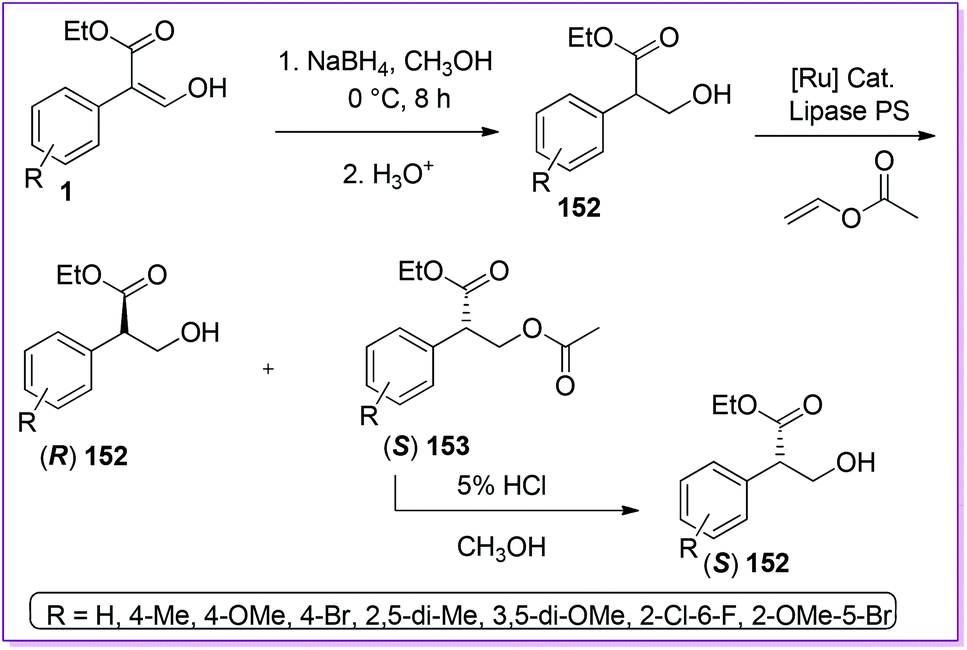
Hossain et al. reported the preparation of racemic mixtures of tropic acid ethyl ester derivatives 152 from 1 using sodium borohydride in methanol.118 To obtain chiral tropic acid, they developed an asymmetric kinetic resolution by utilizing lipase PS enzyme. Moreover, to improve the yield of one enantiomer, they explored dynamic kinetic resolution (DKR) of 152 and its derivatives using ruthenium catalyst to initiate the racemization (Scheme 55).119 Tropic acid is an integral part of tropen alkaloids such as l-scopolamine, l-hyoscyamine, l-atrovent etc. are considered to be prospective drug targets for treating mental illness,120 cardiacarytmia121 and chronic bronchitis122 respectively.
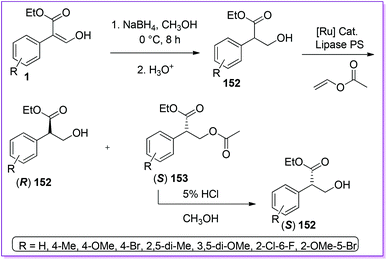 |
| | Scheme 55 Synthesis of TAEE from 1via dynamic kinetic resolution. | |
3.11.5 Thiazolidine type compounds.
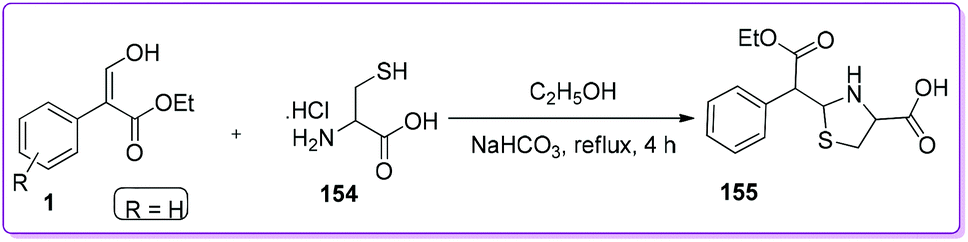
Strukov et al. discovered the method for the synthesis of thiazolidine core containing compound such as leucogen 155 by the reaction of L-cysteine hydrochloride 154 and 1 (Scheme 56).123,124 Leucogen, (L-2(α-phenyl-α-ethoxycarbonylmethyl)-thizolidine-4-carboxylic acid) is one of the active ingredients of veterinary vaccine for the treatment of leucosis and preclude blood systemic disease.125
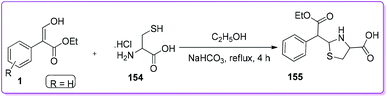 |
| | Scheme 56 Synthesis of leucogen from 1. | |
3.11.6 Thiolethione type compounds.
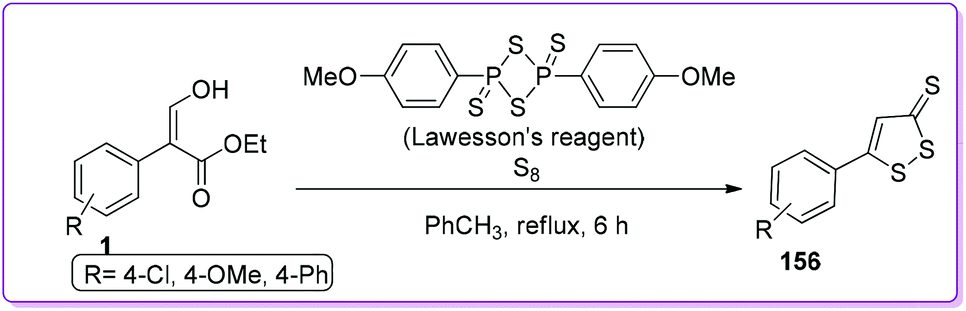
Brown group reported the synthesis of dithiolethiones 156 by reacting 1 with elemental sulfur and Lawesson's thionating reagent (Scheme 57).126 Dithiolethiones work as exogenous molecules to increase the cellular antioxidant, glutathione by controlling its biosynthesis and have the potential to treat the Parkinson's disease.127
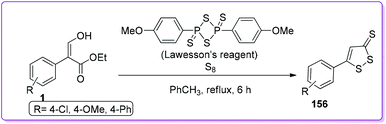 |
| | Scheme 57 Synthesis of dithiolethione from 1. | |
3.11.7 Non-β-lactam inhibitors.
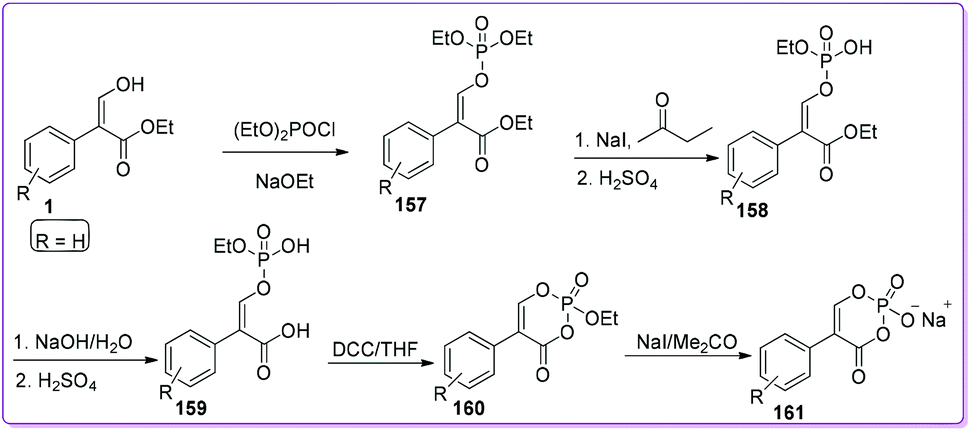
Pratt group developed phosphonate type compounds 161 which showed promising activities, specifically against class C β-lactamases as non-β-lactam inhibitors.128 Surprisingly, they also observed an unusual irreversible inhibitory activity of this class of compounds against class A β-lactamases. Compound 1 was converted into a mixture of E- and Z-diethyl 2-carbethoxystyryl-1-phosphate 157 by the general procedure reported by Craig and Moyle.129 Selective hydrolysis of 157 to 158 was achieved by the method developed by Marecek and Griffith.130 Alkaline hydrolysis of the ester 158 yielded the carboxylic acid 159 which underwent cyclization to form 160. Finally, treatment of 160 with sodium iodide yielded the compound 1-hydroxy-4-phenyl-2,6-dioxaphosphorinanone(3)-1-oxide sodium salt 161, which was purified by Sephadex G-10 chromatography (Scheme 58).
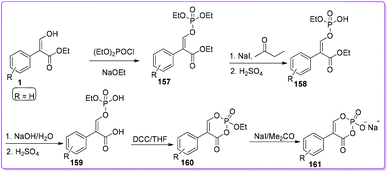 |
| | Scheme 58 Synthesis of β-lactam from 1. | |
3.11.8 α-Fluoroester compounds.
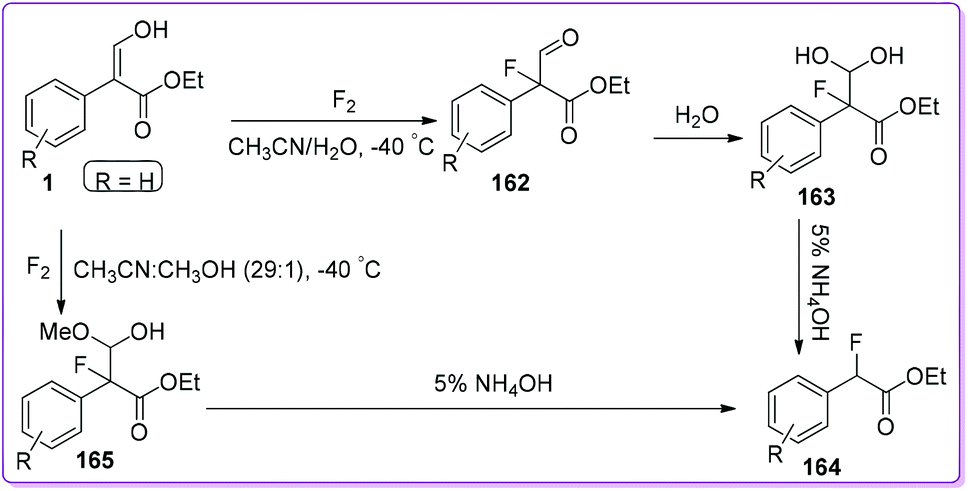
α-Fluoroketones and α-fluoroesters have significant impact from both synthetic and biological point of view.131 Kamaya et al. introduced a site-specific fluorination technique for carbonyl compounds that may be a valuable tool for preparing fluorine containing building blocks.132 Molecular fluorine efficiently reacts with 1 to give α-fluoro aldehyde 162 by releasing HF as a by-product. Resulting aldehyde 162 readily deformylated to form 164 through compound 163 which was produced in the presence of water. The yield of the compound 164 was significantly improved by an alternative route through the synthesis of 165 from 1 using fluorine in methanol (Scheme 59). The economic benefit of the synthesis is the use of molecular fluorine (as fluorinating agent) to expand the scope of large-scale synthesis.133,134
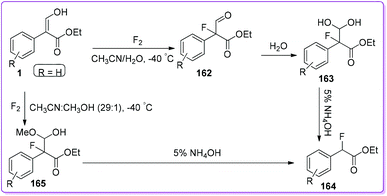 |
| | Scheme 59 Synthesis of α-fluoroester from 1. | |
3.11.9 Chromenone-crown ethers.
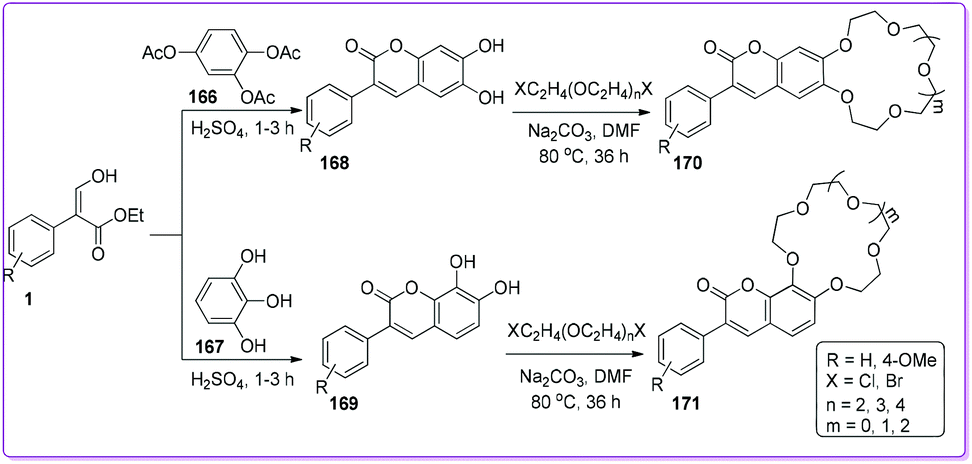
Macrocyclic ethers have been used to estimate cationic recognition and selective cation binding using different physical approaches such as filtration, absorption etc.135–137 Erk et al. reported the synthesis of various 3-phenyl chromenone-crown ether moieties 170 and 171 which was used for the detection of cations such as Li+, Na+, K+, and Rb+ as well as perchlorates with fluorescence spectroscopy.138,139 Compound 170 and 171 were synthesized by the addition of crown ether (12-Crown-4, 15-Crown-4, and 18-Crown-6) to chromenones 168 and 169; which was obtained from the cyclization of triacetate 166 or triol 167 with compound 1 (Scheme 60).
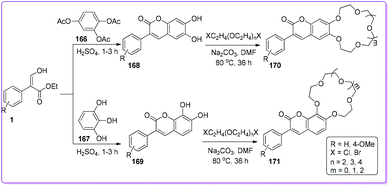 |
| | Scheme 60 Synthesis of chromenone-crown ethers from 1. | |
4. Conclusion
Herein, we summarized the main developments in synthesizing 3-hydroxy-2-aryl acrylate 1 from aromatic aldehydes and diazo compounds. Earlier syntheses mainly studied base catalysed reactions, but recent research has instead focused mostly on employing different Lewis acid and Brønsted acid catalysts. This review has compiled several mechanisms for the formation of 1, all of which involve 1,2-migration of the aryl group. This valuable scaffold contains multifunctional centres, which can be transformed into indole, gramine, benzofuran, oxazole, imidazole, and quinoline type building blocks. Based on such transformations, scientists may be able to obviate the conventional hit-to-lead stage of medicinal and chemical biology programs. Furthermore, we believe that there remain extensive opportunities for expanding the selective formation of 1 and its applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are thankful to the Graduate School of University of Wisconsin-Milwaukee for the support through grant RGI-101X351.
References
-
(a) S. J. Mahmood, C. Brennan and M. M. Hossain, A Convenient New Synthesis of A Naproxen Precursor, Synthesis, 2002, 1807–1809 CAS;
(b) G. Khodarahmi, P. Asadi, F. Hassanzadeh and E. Khodarahmi, Benzofuran as a promising scaffold for the synthesis of antimicrobial and antibreast cancer agents: A review, J. Res. Med. Sci., 2015, 20, 1094–1104 CrossRef CAS.
- S. T. Yoffe, P. V. Petrovakii, E. I. Fedin, K. V. Vatsuro, P. S. Burenko and M. I. Kabachnik, The kinetic investigation of trans-aldo-cis-enol transformation of alkyl 2-formyl-2-phenylacetates, Tetrahedron Lett., 1967, 46, 4525–4529 CrossRef.
- M. Schmittel and H. Ammon, A Short Synthetic Route to 4,7−Dihalogenated 1,10−Phenanthrolines with Additional Groups in 3,8−Position: Soluble Precursors for Macrocyclic Oligophenanthrolines, Eur. J. Org. Chem., 1998, 785–792 CrossRef CAS.
- M. Rahaman and M. M. Hossain, Discussion Addendum for: Convenient Preparation of 3-Ethoxycarbonyl Benzofurans from Salicylaldehydes and Ethyl Diazoacetate, Org. Synth., 2019, 96, 98–109 CrossRef CAS.
-
(a) M. S. Islam, C. Brennan, Q. Wang and M. M. Hossain, Convenient Method of Synthesizing 3-Ethoxycarbonyl Indoles, J. Org. Chem., 2006, 71, 4675–4677 CrossRef CAS;
(b) R. Todd and M. M. Hossain, A Practical Synthesis of Indole-Based Heterocycles Using an Amidoaluminum-Mediated Strategy, Synthesis, 2009, 1846–1850 CAS.
- M. P. Giovannoni, L. Crocetti, N. Cantini, G. Guerrini, C. Vergeli, A. Lacovone, E. Teodori, I. A. Schepetkin, M. T. Quinn, S. Ciattini, P. Rossi and P. Paoli, New 3-unsubstituted Isoxazolones as Potent Human Neutrophil Elastase Inhibitors: Synthesis and Molecular Dynamic Simulation, Drug Dev. Res., 2020, 81, 338–349 CrossRef CAS.
- N. Masurier, E. Debiton, A. Jacquemet, A. Bussière, J.-M. Chezal, A. Ollivier, D. Tétégan, M. Andaloussi, M.-J. Galmier, J. Lacroix, D. Canitrot, J.-C. Teulade, R. C. Gaudreault, O. Chavignon and E. Moreau, Imidazonaphthyridine Systems (Part 2): Functionalization of the Phenyl Ring Linked to the Pyridine Pharmacophore and Its Replacement by a Pyridinone Ring Produces Intriguing Differences in Cytocidal Activity, Eur. J. Med. Chem., 2012, 52, 137–150 CrossRef CAS.
-
(a) M. G. Ferlin, R. Bortolozzi, P. Brun, I. Castagliuolo, E. Hamel, G. Basso and G. Viola, Synthesis and in vitro evaluation of 3h-pyrrolo[3,2-f]-quinolin-9-one derivatives that show potent and selective anti-leukemic activity, ChemMedChem, 2010, 5, 1373–1385 CrossRef CAS;
(b) V. Gasparotto, I. Castagliuolo and M. G. Ferlin, 3-Substituted 7-Phenyl-Pyrroloquinolinones Show Potent Cytotoxic Activity in Human Cancer Cell Lines, J. Med. Chem., 2007, 50, 5509–5513 CrossRef CAS;
(c) E. Yamato, New Synthesis of 1-Benzyl-6, 7-dihydroxy-1, 2, 3, 4-tetrahydroisoquinoline Derivatives, Chem. Pharm. Bull., 1970, 18, 2038–2042 CrossRef CAS.
-
(a) W. Wislicenus, Ueber einige Aldehydeaureeeter, Chem. Ber., 1887, 20, 2930–2934 CrossRef;
(b) W. Wislicenus, Ueber die Isomerie der-Formylphenylessigester, Chem. Ber., 1895, 28, 767–774 CrossRef CAS;
(c) W. Wislicenus, Ueber die Isoinerie der l'ormylphenylessigester, Justus Liebigs Ann. Chem., 1896, 291, 147–216 CrossRef CAS.
- H. Watanabe, A. Ide, N. Sugimoto, Y. Noguchi, R. Ishida and Y. Kowa, Studies on the Thiazolidinecarboxylic Acid Derivatives as the Protective and Restorative Agents against Radiation Injury, Yakugaku Zasshi, 1963, 83, 1118–1124 CrossRef CAS.
- J. N. Marx, J. C. Argyle and L. R. Norman, Migration of electronegative substituents. I. Relative migratory aptitude and migration tendency of the carbethoxy group in the dienone-phenol rearrangement, J. Am. Chem. Soc., 1974, 96, 2121–2129 CrossRef CAS.
- E. M. Beccalli, C. L. Rosa and A. Marchesini, Oxidation of 4-aryl-substituted isoxazolin-5-ones. A new synthesis of 2,5-diaryl-1,3-oxazin-6-ones, J. Org. Chem., 1984, 49, 4287–4290 CrossRef CAS.
- I. Pevet, C. Brulé, A. Tizot, A. Gohier, F. Cruzalegui, J. A. Boutin and S. Goldstein, Synthesis and Pharmacological Evaluation of thieno[2,3-b]pyridine Derivatives as Novel c-Src Inhibitors, Bioorg. Med. Chem., 2011, 19, 2517–2528 CrossRef CAS.
- B. Latli, M. Stiasni, M. Hrapchak, Z. Li, N. Grinberg, H. Lee, C. A. Busacca and C. H. Senanayake, Buscopan labeled with carbon–14 and deuterium, J. Labelled Compd. Radiopharm., 2016, 59, 557–564 CrossRef CAS.
- S. J. Mahmood and M. M. Hossain, Iron Lewis Acid Catalysed Reactions of Aromatic Aldehydes with Ethyl Diazoacetate: Unprecedented Formation of 3-Hydroxy-2-arylacrylic Acid Ethyl Esters by a Unique 1,2-Aryl Shift, J. Org. Chem., 1998, 63, 3333–3336 CrossRef CAS.
-
(a) H. J. Liu and S. P. Majumdar, On the regioselectivity of boron trifluoride catalysed ring expansion of cycloalkanones with ethyl diazoacetate, Synth. Commun., 1975, 5, 125–130 CrossRef CAS;
(b) W. L. Mock and M. Hartman, Synthetic scope of the triethyloxonium ion catalysed homologation of ketones with diazoacetic esters, J. Org. Chem., 1977, 42, 459–465 CrossRef;
(c) V. Dave and E. W. Warnhoff, Regiospecific homologation of unsymmetrical ketones, J. Org. Chem., 1983, 48, 2590–2598 CrossRef CAS;
(d) C. R. Holmquist and E. J. Roskamp, A Selective Method for Diazoacetate Catalysed the Direct Conversion of Aldehydes into β-Keto Esters with Ethyl by Tin(II) Chloride, J. Org. Chem., 1989, 54, 3258–3260 CrossRef CAS;
(e) C. R. Holmquist and E. J. Roskamp, Tin(II) chloride catalysed addition of diazo sulfones, diazo phosphine oxides, and diazo phosphonates to aldehydes, Tetrahedron Lett., 1992, 33, 1131–1134 CrossRef CAS;
(f) A. Padwa, S. F. Hornbuckle, Z. Zhang and L. Zhi, Synthesis of 1,3-Diketones Using α-Diazo Ketones and Aldehydes in the Presence of Tin(II) Chloride, J. Org. Chem., 1990, 55, 5297–5299 CrossRef CAS;
(g) S. G. Sudrik, B. S. Balaji, A. P. Singh, R. B. Mitra and H. R. Sonawane, Zeolite-mediated Synthesis of β-Keto Esters: Condensation of Ethyl Diazoacetate with Aldehydes, Synlett, 1996, 369–370 CrossRef CAS;
(h) K. Nomura, T. Iida, K. Hori and E. Yoshii, Synthesis of γ-unsubstituted α-acyl-β-tetronic acids from aldehydes, J. Org. Chem., 1994, 59, 488–490 CrossRef CAS.
- M. Redlich, S. J. Mahmood, M. F. Mayer and M. M. Hossain, Silica Supported Catalysis: A Practical use of an Iron Lewis Acid, Synth. Commun., 2000, 30, 1401–1411 CrossRef CAS.
- S. Kanemasa, T. Kanai, T. Araki and E. Wada, Lewis acid-catalysed reactions of ethyl diazoacetate with aldehydes. Synthesis of α-formyl esters by a sequence of aldol reaction and 1,2-nucleophilic rearrangement, Tetrahedron Lett., 1999, 40, 5055–5058 CrossRef CAS.
- D. Benito-Garagorri, J. Wiedermann, M. Pollak, K. Mereiter and K. Kirchner, Iron(II) Complexes Bearing Tridentate PNP Pincer-Type Ligands as Catalysts for the Selective Formation of 3-Hydroxyacrylates from Aromatic Aldehydes and Ethyldiazoacetate, Organometallics, 2007, 26, 217–222 CrossRef CAS.
- L. G. Alves, G. Dazinger, L. F. Veiros and K. Kirchner, Unusual Anion Effects in the Iron–Catalysed Formation of 3−Hydroxyacrylates from Aromatic Aldehydes and Ethyl Diazoacetate, Eur. J. Inorg. Chem., 2010, 3160–3166 CrossRef CAS.
- M. R. Fructos, M. M. Díaz-Requejo and P. J. Pérez, Highly active gold-based catalyst for the reaction of benzaldehyde with ethyl diazoacetate, Chem. Commun., 2009, 5153–5155 RSC.
- K. J. Kilpin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Gold(I) “click” 1,2,3-triazolylidenes: Synthesis, self-assembly and catalysis, Chem. Commun., 2011, 47, 328–330 RSC.
- M. E. Dudley, M. M. Morshed, C. L. Brennan, M. S. Islam, M. S. Ahmad, M.-R. Atuu, B. Branstetter and M. M. Hossain, Acid-Catalysed Reactions of Aromatic Aldehydes with Ethyl Diazoacetate: An Investigation on the Synthesis of 3-Hydroxy-2-arylacrylic Acid Ethyl Esters, J. Org. Chem., 2004, 69, 7599–7508 CrossRef CAS.
- G. Dazinger and K. A. Kirchner, Acid catalysed coupling of aromatic aldehydes and methyldiazoacetate-a theoretical mechanistic study, New J. Chem., 2012, 36, 781–788 RSC.
- F. Xiao, Y. Liu and J. Wang, DBU-catalysed condensation of acyldiazomethanes to aldehydes in water and a new approach to ethyl β-hydroxy α-arylacrylates, Tetrahedron Lett., 2007, 48, 1147–1149 CrossRef CAS.
- F. Xiao and J. Wang, 1,2-Migration in Rhodium(II) Carbene Transfer Reaction: Remarkable Steric Effect on Migratory Aptitude, J. Org. Chem., 2006, 71, 5789–5791 CrossRef CAS.
- N. Jiang, Z. Ma, Z. Qu, X. Xing, L. Xie and J. Wang, Investigation of the Transition-Metal- And Acid-Catalysed Reactions of beta-(N-tosyl)amino Diazo Carbonyl Compounds, J. Org. Chem., 2003, 68, 893–900 CrossRef CAS.
- W. Shi, F. Xiao and J. Wang, 1,2-Vinyl and 1,2-Acetylenyl Migration in Rh(II) Carbene Reaction: Remarkable Bystander Effect, J. Org. Chem., 2005, 70, 4318–4322 CrossRef CAS.
-
(a) T. Akiyama, J. Ishida, S. Nakagawa, H. Ogawara, S. Watanabe, N. Itoh, M. Shibuya and Y. Fukami, Genistein, a Specific Inhibitor of Tyrosine-Specific Protein Kinases, J. Biol. Chem., 1987, 262, 5592–5595 CAS;
(b) J. Markovits, C. Linassier, P. Fossé, J. Couprie, J. Pierre, A. Jacquemin-Sablon, J.-M. Saucier, J.-B. L. Pecq and A. K. Larsen, Inhibitory Effects of the Tyrosine Kinase Inhibitor Genistein on Mammalian DNA Topoisomerase II, Cancer Res., 1989, 49, 5111–5117 CAS.
- M. H. Cynamon and G. H. Wilcox, The Role of 4-quinolones in the Treatment of Infections, Compr. Ther., 1987, 13, 37–43 CAS.
- S. Heeb, M. P. Fletcher, S. R. Chhabra, S. P. Diggle, P. Williams and M. Cámara, Quinolones: from antibiotics to autoinducers, FEMS Microbiol. Rev., 2011, 35, 247–274 CrossRef CAS.
-
(a) Z.-P. Xiao, P.-C. Lv, S.-P. Xu, T.-T. Zhu and H.-L. Zhu, Synthesis, Antiproliferative Evaluation, and Structure–Activity Relationships of 3−Arylquinolines, ChemMedChem, 2008, 3, 1516–1519 CrossRef CAS;
(b) A. Monastyrskyi, D. E. Kyle and R. Manetsch, 4(1H)-Pyridone and 4(1H)-Quinolone Derivatives as Antimalarials with Erythrocytic, Exoerythrocytic, and Transmission Blocking Activities, Curr. Top. Med. Chem., 2014, 14, 1693–1705 CrossRef CAS;
(c) L. Huang, M.-C. Hsieh, C.-M. Teng, K.-H. Lee and S.-C. Kuo, Synthesis and antiplatelet activity of phenyl quinolones, Bioorg. Med. Chem., 1998, 6, 1657–1662 CrossRef CAS.
- S. Fiorucci, X. Lin, K. Sadoul, G. Fournet, D. Bouvard, O. Vinogradova, B. Joseph and M. R. Block, Targeting Integrin-Dependent Adhesion and Signaling with 3-Arylquinoline and 3-Aryl-2-Quinolone Derivatives: A new Class of Integrin Antagonists, PLoS One, 2015, 10, e0141205, DOI:10.1371/journal.pone.0141205.
- M. Croisy, C. Huel and E. Bisagni, Synthesis of 3-(4-methoxyphenyl)-5,7-dimethoxy-(1H)-quinolin-2- or 4-ones and derivatives, Heterocycles, 1997, 45, 683–690 CrossRef CAS.
- S. Rajput, C. R. Gardner, T. W. Failes, G. M. Arndt, D. S. Black and N. Kumar, Synthesis and anticancer evaluation of 3-substituted quinolin-4-ones and 2,3-dihydroquinolin-4-ones, Bioorg. Med. Chem., 2014, 22, 105–115 CrossRef CAS.
- B. Joseph, F. Darro, A. Béhard, B. Lesur, F. Collignon, C. Decaestecker, A. Frydman, G. Guilaumet and R. Kiss, 3-Aryl-2-Quinolone Derivatives: Synthesis and Characterization of In Vitro and In Vivo Antitumor Effects with Emphasis on a New Therapeutical Target Connected with Cell Migration, J. Med. Chem., 2002, 45, 2543–2555 CrossRef CAS.
-
(a) J. R. Atack, GABAA Receptor Subtype-Selective Modulators. II. α5-Selective Inverse Agonists for Cognition Enhancement, Curr. Top. Med. Chem., 2011, 11, 1203–1214 CrossRef CAS;
(b) R. W. Olsen, Chapter Seven - Allosteric Ligands and Their Binding Sites Define γ-Aminobutyric Acid (GABA) Type A Receptor Subtypes, Adv. Pharmacol., 2015, 73, 167–202 CAS.
- U. Rudolph and F. Knoflach, Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes, Nat. Rev. Drug Discovery, 2011, 10, 685–697 CrossRef CAS.
- G. Guerrini, G. Ciciani, S. Daniele, L. D. C. Mannelli, C. Ghelardini, C. Martini and S. Selleri, Synthesis and pharmacological evaluation of pyrazolo[1,5-a]pyrimidin-7(4H)-one derivatives as potential GABAA-R ligands, Bioorg. Med. Chem., 2017, 25, 1901–1906 CrossRef CAS.
- A. N. Shestakov, A. S. Pankova, P. Golubev, A. F. Khlebnikov and M. A. Kuznetsov, Brønsted acid mediated cyclizations of ortho-aryl(ethynyl)pyrimidines, Tetrahedron, 2017, 73, 3939–3948 CrossRef CAS.
- R. Olinski, M. Jurgowiak and T. Zaremba, Uracil in DNA—Its biological significance, Mutat. Res., 2010, 705, 239–245 CAS.
-
(a) H. Tanaka, H. Takashima, M. Ubasawa, K. Sekiya, N. Inouye, M. Baba, S. Shigeta, R. T. Walker, E. De Clercq and T. Miyasaka, Synthesis and Antiviral Activity of 6-Benzyl Analogs of 1-[(2-Hydroxyethoxy)methyl]-5-(phenylthio) thymine (HEPT) as Potent and Selective Anti-HIV-1 Agents, J. Med. Chem., 1995, 38, 2860–2865 CrossRef CAS;
(b) T. Miyasaka, H. Tanaka, M. Baba, H. Hayakawa, R. T. Walker, J. Balzarini and E. De Clercq, A novel lead for specific anti-HIV-1 agents: 1-[(2-hydroxyethoxy)methyl]-6-(phenylthio)thymine, J. Med. Chem., 1989, 32, 2507–2509 CrossRef CAS.
- C. K. Chu, F. D. Boudinot, S. F. Peek, J. H. Hong, Y. Choi, B. E. Korba, J. L. Gerin, P. J. Cote, B. C. Tennant and Y. C. Cheng, Preclinical Investigation of L-FMAU as an Anti-Hepatitis B Virus Agent, Antiviral Ther., 1998, 3, 113–121 CAS.
- A. C. Krueger, J. T. Randolph, D. A. DeGoey, P. L. Donner, C. A. Flentge, D. K. Hutchinson, D. Liu, C. E. Motter, T. W. Rockway, R. Wagner, D. W. A. Beno, G. Koev, H. B. Lim, J. M. Beyer, R. Mondal, Y. Liu, W. M. Kati, K. L. Longenecker, A. Molla, K. D. Stewart and C. J. Maring, Aryl uracil inhibitors of hepatitis C virus NS5B polymerase: Synthesis and characterization of analogs with a fused 5,6-bicyclic ring motif, Bioorg. Med. Chem. Lett., 2013, 23, 3487–3490 CrossRef CAS.
- Y. Sekino, S. D. Bruner and G. L. Verdine, Selective Inhibition of Herpes Simplex Virus Type-1 Uracil-DNA Glycosylase by Designed Substrate Analogs, J. Biol. Chem., 2000, 275, 36506–36508 CrossRef CAS.
-
(a) C. H. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, R. J. Schnitzer, E. Pleven and J. Scheiner, Fluorinated Pyrimidines, A New Class of Tumour-Inhibitory Compounds, Nature, 1957, 179, 663–666 CrossRef CAS;
(b) E. De Clercq, Potential of bromovinyldeoxyuridine in anticancer chemotherapy, Anticancer Res., 1986, 6, 549–556 CAS.
- W. H. Davies and H. A. Piggott, A novel pyrimidine synthesis. Part I. 4-Amino-5-phenylpyrimidine, J. Chem. Soc., 1945, 347–351 RSC.
- M. M. Morshed, Q. Wang, S. Islam and M. M. Hossain, Convenient Synthesis of 5−Aryl Uracils, Synth. Commun., 2007, 37, 4173–4181 CrossRef CAS.
-
T. Takaya, M. Murata and K. Ito, Substituted-3,4-dihydro-4-(2,4,6-trimethoxyphenylimino)-2(1H)-pyrimidones useful as cardiotonic, antihypertensive, cerebrovascular vasodilator and anti-platelet agent, US Pat, 4612376A, Fujisawa Pharmaceutical Co. Ltd, 1986 Search PubMed.
- T. S. Shestakova, S. L. Deev, E. N. Ulomsky, V. L. Rusinov, O. N. Chupakhin, O. A. D′yachenko, O. N. Kazheva, A. N. Chekhlov, P. A. Slepukhin and M. I. Kodess, New approach to the synthesis of azauracils and azaisocytosines, Russ. Chem. Bull. Int. Ed., 2006, 55, 2071–2080 CrossRef CAS.
-
(a) T. F. Tam and P. Coles, 4-Alkoxy-6-alkyl-2-pyrones as precursors for 4-alkoxy-6-alkyl-2-aminobenzoic acid derivatives, Synthesis, 1988, 383–386 CrossRef CAS;
(b) K. Afarinkia, V. Vinader, T. D. Nelson and G. H. Posner, Diels-Alder cycloadditions of 2-pyrones and 2-pyridones, Tetrahedron, 1992, 48, 9111–9171 CrossRef CAS;
(c) K. Afarinkia and J. Berna-Canovas, Diels–Alder cycloaddition of 5-aryl-2-pyrones, Tetrahedron Lett., 2000, 41, 4955–4958 CrossRef CAS;
(d) C. H. Chen and C. C. Liao, One-Pot Stereoselective Synthesis of Tricyclic Bislactones from 2-Pyrones and 2-Methoxyfuran, Org. Lett., 2000, 2, 2049–2052 CrossRef CAS.
-
(a) J. S. Lee, Recent Advances in the Synthesis of 2-Pyrones, Mar. Drugs, 2015, 13, 1581–1620 CrossRef CAS;
(b) G. P. McGlacken and I. J. S. Fairlamb, 2-Pyrone natural products and mimetics: isolation, characterisation and biological activity, Nat. Prod. Rep., 2005, 22, 369–385 RSC.
- N. Lyukmanov and E. Kovshev, Synthesis, halogenation, and nitration of 6-methyl-3-phenyl-2-pyrone derivatives, Chem. Heterocycl. Compd., 1985, 21, 1215–1218 CrossRef.
- F. M. Moghaddam, Z. Mirjafary, M. J. Javan, S. Motamen and H. Saeidian, Facile synthesis of highly substituted 2-pyrone derivatives via a tandem Knoevenagel condensation/lactonization reaction of β-formyl-esters and 1,3-cyclohexadiones, Tetrahedron Lett., 2014, 55, 2908–2911 CrossRef CAS.
- C. A. Quesnelle, P. Gill, S. Roy, M. Dodier, A. Marinier, A. Martel, L. B. Snyder, S. V. D'Andrea, J. J. Bronson, M. Frosco, D. Beaulieu, G. A. Warr, K. L. DenBleyker, T. M. Stickle, H. Yang, S. E. Chaniewski, C. A. Ferraro, D. Tylor, J. W. Rusell, K. S. Santone, J. Clarke, R. L. Drain, J. O. Knipe, K. Mosure and J. F. Barrett, Biaryl isoxazolinone antibacterial agents, Bioorg. Med. Chem. Lett., 2005, 15, 2728–2733 CrossRef CAS.
-
(a) S. Z. Zard, New syntheses of alkynes: a tale of serendipity and design, Chem. Commun., 2002, 1555–1563 RSC;
(b) R. H. Prager and Y. Singh, The chemistry of 5-oxodihydroisoxazoles VII1 conversion of heterocyclylisoxazol-5(2H)-ones to imidazoles by flash vacuum pyrolysis, Tetrahedron, 1993, 49, 8147–8158 CrossRef CAS;
(c) K. H. Ang, R. H. Prager, J. A. Smith, B. Weber and C. M. Williams, The synthesis of oxazoles by thermolysis or photolysis of 2-acylisoxazol-5-ones, Tetrahedron Lett., 1996, 37, 675–678 CrossRef CAS;
(d) S. Huppé, H. Rezaei and S. Z. Zard, A new synthesis of 1-chloroalkynes, Chem. Commun., 2001, 1894–1895 RSC.
-
(a) K. Okamoto, T. Oda, S. Kohigashi and K. Ohe, Palladium–catalysed decarboxylative intramolecular aziridination from 4H–Isoxazol–5−ones leading to 1−Azabicyclo[3.1.0]hex–2−enes, Angew. Chem., Int. Ed., 2011, 50, 11470–11473 CrossRef CAS;
(b) K. Okamoto, T. Shimbayashi, E. Tamura and K. Ohe, Palladium–catalysed
Aza–Wittig–type condensation of Isoxazol–5(4H)–ones with aldehydes, Chem. – Eur. J., 2014, 20, 1490–1494 CrossRef CAS;
(c) P. C. Too, Y.-F. Wang and S. Chiba, Rhodium(III)-catalysed synthesis of isoquinolines from aryl ketone O-Acyloxime derivatives and internal alkynes, Org. Lett., 2010, 12, 5688–5691 CrossRef CAS;
(d) K. Okamoto, T. Shimbayashi, M. Yoshida, A. Nanya and K. Ohe, Synthesis of 2H–Azirines by Iridium–catalysed decarboxylative ring contraction of Isoxazol–5(4H)–ones, Angew. Chem., Int. Ed., 2016, 55, 7199–7202 CrossRef CAS.
- L. B. Snyder, Z. Meng, R. Mate, S. V. D'Andrea, A. Marinier, C. A. Quesnelle, P. Gill, K. L. Denbleyker, J. C. Fung-tomc, M. B. Frosco, A. Martel, J. F. Barrett and J. J. Bronson, Discovery of isoxazolinone antibacterial agents. Nitrogen as a replacement for the stereogenic center found in oxazolidinone antibacterials, Bioorg. Med. Chem. Lett., 2004, 14, 4735–4739 CrossRef CAS.
- M. Mei, D. Anand and L. Zhou, Divergent conversion of N-Acyl-isoxazol-5(2H)-ones to Oxazoles and 1,3-Oxazin-6-ones using photoredox catalysis, Org. Lett., 2019, 21, 3548–3553 CrossRef CAS.
- Y. Momosa, T. Maekawa, T. Yamano, M. Kawada, H. Odaka, H. Ikeda and T. Sohda, Novel 5-substituted 2, 4-thiazolidinedione and 2, 4-oxazolidinedione derivatives as insulin sensitizers with antidiabetic activities, J. Med. Chem., 2002, 45, 1518–1534 CrossRef.
- A. C. Giddens, H. I. M. Boshoff, S. G. Franzblau, C. E. Barry III and B. R. Copp, Antimycobacterial natural products: synthesis and preliminary biological evaluation of the oxazole-containing alkaloid texaline, Tetrahedron Lett., 2005, 46, 7355–7357 CrossRef CAS.
-
(a) M. C. Kim, J. H. Lee, B. Shin, L. Subedi, J. W. Cha, J.-S. Park, D.-C. Oh, S. Y. Kim and H. C. Kwon, Salinazinones A and B: Pyrrolidinyl-Oxazinones from solar saltern-derived Streptomyces sp. KMF-004, Org. Lett., 2015, 17, 5024–5027 CrossRef CAS;
(b) P. Fu, S. La and J. B. MacMillan, 1,3-Oxazin-6-one derivatives and Bohemamine-Type pyrrolizidine alkaloids from a marine-derived Streptomyces spinoverrucosus, J. Nat. Prod., 2016, 79, 455–462 CrossRef CAS.
-
(a) G. Accorsi, A. Listorti, K. Yoosaf and N. Armaroli, 1, 10-Phenanthrolines: versatile building blocks for luminescent molecules, materials and metal complexes, Chem. Soc. Rev., 2009, 38, 1690–1700 RSC;
(b) A. Bencini and V. Lippolis, 1,10-Phenanthroline: A versatile building block for the construction of ligands for various purposes, Coord. Chem. Rev., 2010, 254, 2096–2180 CrossRef CAS.
-
(a) U. Velten and M. Rehahn, First synthesis of soluble, well defined coordination polymers from kinetically unstable copper(I) complexes, Chem. Commun., 1996, 2639–2640 RSC;
(b) J.-M. Kern, J.-P. Sauvage and J.-L. Weidmann, Multiring interlocked systems via transition metal-templated strategy: The single-cyclization synthesis of [3]-catenates, Tetrahedron, 1996, 52, 10921–10934 CrossRef CAS.
-
(a) P. G. Sammes and G. Yahioglu, 1, 10-Phenanthroline: a versatile ligand, Chem. Soc. Rev., 1994, 23, 327–334 RSC;
(b) D. S. Sigman, Nuclease activity of 1, 10-phenanthroline-copper ion, Acc. Chem. Res., 1986, 19, 180–186 CrossRef CAS.
- M. Schmittel, H. Ammon and C. Wöhrle, Tris(1,10−phenanthroline)iron(II) Complexes—broad variation of the redox potential by 4,7−Substitution at the phenanthroline ligands, Chem. Ber., 1995, 128, 845–850 CrossRef CAS.
- F. H. Case and R. Sasin, Substituted 1, 10-phenanthrolines. VIII. 2- and 3-phenyl derivatives, J. Org. Chem., 1955, 20, 1330–1336 CrossRef CAS.
- Y. Echigo and T. Mukaiyama, Preparation of vinyl chlorides from enolizable aldehydes, Chem. Lett., 1978, 465–466 CrossRef CAS.
- Y.-L. Liu, X.-H. Xu and F.-L. Qing, Synthesis of vinyl and electron-deficient aryl trifluoromethyl sulfides via Csp2− OH bond activation with AgSCF3 and n-Bu4NI/KI, Tetrahedron, 2018, 74, 5827–5832 CrossRef CAS.
-
(a) C. Hansch, A. Leo, S. H. Unger, K. H. Kim, D. Nikaitani and E. J. Lien, Aromatic substituent constants for structure-activity correlations, J. Med. Chem., 1973, 16, 1207–1216 CrossRef CAS;
(b) C. Hansch, A. Leo and R. W. Taft, A survey of Hammett substituent constants and resonance and field parameters, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- M. Diaferia, F. Veronesi, G. Morganti, L. Nisoli and D. P. Fioretti, Efficacy of toltrazuril 5% suspension (Baycox®, Bayer) and diclazuril (Vecoxan®, Janssen-Cilag) in the control of Eimeria spp. in lambs, Parasitol. Res., 2013, 112, 163–168 CrossRef.
- D. B. Gant, A. E. Chalmers, M. A. Wolff, H. B. Hoffman and D. F. Bushey, Fipronil: action at the GABA receptor, Rev. Toxicol., 1998, 2, 147–156 CAS.
- J. F. Giudicelli, C. Richer and A. Berdeaux, Preliminary assessment of flutiorex, a new anorectic drug, in man, Br. J. Clin. Pharmacol., 1976, 3, 113–121 CrossRef CAS.
-
(a) L. Laine, Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient, Gastroenterology, 2001, 120, 594–606 CrossRef CAS;
(b) R. A. Moore, M. R. Tramer, D. Carroll, P. J. Wiffen and H. J. McQuay, Quantitive systematic review of topically applied non-steroidal anti-inflammatory drugs, Br. Med. J., 1998, 316, 333–338 CrossRef CAS.
- W. F. Kean and W. W. Buchanan, The use of NSAIDs in rheumatic disorders 2005: a global perspective, Inflammopharmacology, 2005, 13, 343–370 CrossRef CAS.
- T. J. Schnitzer, Update of ACR guidelines for osteoarthritis: role of the coxibs, J. Pain Symptom Manage., 2002, 23, 24–30 CrossRef.
-
(a) J. P. Rieu, A. Boucherle, H. Cousse and G. Mouzin, Tetrahedron Report Number 205: Methods for the synthesis of antiinflammatory 2-Aryl propionic acids, Tetrahedron, 1986, 42, 4095–4131 CrossRef CAS;
(b) T. Hiyama, N. Wakasa, T. Ueda and T. Kusumoto, A facile, practical synthesis of 2-(6-methoxy-2-naphthyl) propenoic acid, Bull. Chem. Soc. Jpn., 1990, 63, 640–642 CrossRef CAS;
(c) V. Sunjic, I. Habus, G. Comisso and F. Moimas, Asymmetric Hydrogenation of α–Arylpropenoic Acids Catalysed by Rhodium(I) Complexes of Chiral Ligands Derived from Some Monosaccharides, Gazz. Chim. Ital., 1989, 119, 229–234 CAS;
(d) A. Scrivanti and U. Matteoli, A convenient synthesis of 2-(6-methoxy-2-naphthyl) propenoic acid (a naproxen precursor), Tetrahedron Lett., 1995, 36, 9015–9018 CrossRef CAS.
- M. S. Islam, S. Ahmad, M.-R. Attu, F. H. Forsterling and M. M. Hossain, Concise synthesis of 2−Arylpropanoic acids and study of unprecedented reduction of 3−Hydroxy–2−arylpropenoic acid ethyl ester to 2−Arylpropenoic Acid ethyl ester by BH3·THF, Helv. Chim. Acta, 2015, 98, 1273–1286 CrossRef.
-
(a) T. Mukaiyama, I. Shiina, H. Iwadare, M. Saitoh, T. Nishimura, N. Ohkawa, H. Sakoh, K. Nishimura, Y.-I. Tani, M. Hasegawa, K. Yamada and K. Saitoh, Asymmetric Total Synthesis of Taxol, Chem. – Eur. J., 1999, 5, 121–161 CrossRef CAS;
(b) D. J. Newman and G. M. Cragg, Natural products as sources of new drugs from 1981 to 2014, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS.
-
(a) Y. Schun and G. A. Cordell, Studies on the NMR Spectroscopic properties of Gelsemine–revisions and refinements, J. Nat. Prod., 1985, 48, 969–971 CrossRef CAS;
(b) X. Zhou, T. Xiao, Y. Iwama and Y. Qin, Biomimetic total synthesis of (+)–Gelsemine, Angew. Chem., Int. Ed., 2012, 51, 4909–4912 CrossRef CAS;
(c) X. Chen, S. Duan, C. Tao, H. Zhai and F. G. Qiu, Total synthesis of (+)-gelsemine via an organocatalytic Diels–Alder approach, Nat. Commun., 2015, 6, 1–7 Search PubMed.
-
(a) P. R. Blakemore and J. D. White, Morphine: the Proteus of organic molecules, Chem. Commun., 2002, 1159–1168 RSC;
(b) J. Zezula and T. Hudlicky, Recent progress in the synthesis of morphine alkaloids, Synlett, 2005, 388–405 CAS;
(c) N. Chida, Recent advances in the synthesis of morphine and related alkaloids, Top. Curr. Chem., 2010, 299, 1–28 CrossRef.
-
(a) J. W. Moncrief and W. N. Lipscomb, Structure of leurocristine methiodide dihydrate by anomalous scattering methods: relation to leurocristine (vincristine) and vincaleukoblastine (vinblastine), Acta Crystallogr., 1966, 21, 322–331 CrossRef CAS;
(b) H. Ishikawa, D. A. Colby, S. Seto, P. Va, A. Tam, H. Kakei, T. J. Rayl, I. Hwang and D. L. Boger, Total synthesis of vinblastine, vincristine, related natural products, and key structural analogues, J. Am. Chem. Soc., 2009, 131, 4904–4916 CrossRef CAS.
-
(a) B. M. Trost and C. Jiang, Catalytic enantioselective construction of all-carbon quaternary stereocenters, Synthesis, 2006, 369–396 CrossRef CAS;
(b) A. Y. Hong and B. M. Stoltz, The construction of all-carbon quaternary stereocenters by use of Pd-catalysed asymmetric allylic alkylation reactions in total synthesis, Eur. J. Org. Chem., 2013, 2745–2759 CrossRef CAS.
-
(a) J. P. Das and I. Marek, Enantioselective synthesis of all-carbon quaternary stereogenic centers in acyclic systems, Chem. Commun., 2011, 47, 4593–4623 RSC;
(b) I. Marek and G. Sklute, Creation of quaternary stereocenters in carbonyl allylation reactions, Chem. Commun., 2007, 43, 1683–1691 RSC;
(c) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, All-carbon quaternary stereogenic centers in acyclic systems through the creation of several C–C bonds per chemical step, J. Am. Chem. Soc., 2014, 136, 2682–2694 CrossRef CAS.
- S. A. Asad, J. Ulicki, M. Shevyrev, N. Uddin, E. Alberch and M. M. Hossain, First Example of the Intermolecular Palladium–Catalysed Asymmetric Allylic Alkylation of Hydroxyacrylates: Synthesis of All–Carbon α–Aryl Quaternary Aldehydes, Eur. J. Org. Chem., 2014, 5695–5699 CrossRef CAS.
- E. Alberch, N. Uddin, M. Shevyrev and M. M. Hossain, Synthesis of compounds containing α-aryl quaternary carbon centers, ARKIVOC, 2010, 139–146 Search PubMed.
- N. Uddin, M. Rahaman, E. Alberch, S. A. Asad and M. M. Hossain, Palladium (0)-catalysed rearrangement of allyl enol ethers to form chiral quaternary carbon centers via asymmetric allylic alkylation, Tetrahedron Lett., 2018, 59, 3401–3404 CrossRef CAS.
- E. Alberch, C. Brook, S. A. Asad, M. Shevyrev, J. Ulicki and M. M. Hossain, Stereoselective Allyl Enol Carbonates for the Synthesis of Chiral Aldehydes Bearing All Carbon Quaternary Stereocenters via the Decarboxylative Asymmetric Allylic Alkylation (DAAA), Synlett, 2015, 26, 388–392 CrossRef CAS.
- Y.-H. Miao, Y.-H. Hu, J. Yang, T. Liu, J. Sun and X.-J. Wang, Natural source, bioactivity and synthesis of benzofuran derivatives, RSC Adv., 2019, 9, 27510–27540 RSC.
-
(a) E. A. Boyle, F. R. Morgan, R. E. Markwell, S. A. Smith, M. J. Thomson, R. W. Ward and P. A. Wyman, 7-Aroyl-2,3-dihydrobenzo[b]furan-3-carboxylic acids and 7-benzoyl-2,3-dihydrobenzo[b]thiophene-3-carboxylic acids as analgesic agents, J. Med. Chem., 1986, 29, 894–898 CrossRef CAS;
(b) J. Suh, K. Y. Yi, Y.-S. Lee, E. Kim, E. K. Yum and S.-E. Yoo, Synthesis and biological evaluation of 3-substituted-benzofuran-2-carboxylic esters as a novel class of ischemic cell death inhibitors, Bioorg. Med. Chem. Lett., 2010, 20, 6362–6365 CrossRef CAS.
- M. E. Dudley, M. M. Morshed and M. M. Hossain, A Convenient Method of Synthesizing 3-Ethoxycarbonylbenzofurans from Salicylaldehydes and Ethyl Diazoacetate, Synthesis, 2006, 1711–1714 CAS.
- M. E. Dudley, M. M. Morshed and M. M. Hossain, Convenient Preparation of 3−Ethoxycarbonyl Benzofurans from Salicylaldehydes and Ethyl Diazoacetate, Org. Synth., 2009, 86, 172–180 CrossRef CAS.
- V. N. Telvekar, A. Belubbi, V. K. Bairwa and K. Satardekar, Novel N’-benzylidene benzofuran-3-carbohydrazide Derivatives as Antitubercular and Antifungal Agents, Bioorg. Med. Chem., 2012, 22, 2343–2346 CrossRef CAS.
- A. S. Kil'met'ev, E. E. Shul'ts, M. M. Shakirov, T. V. Rybalova and G. A. Tolstikov, Diels-alder reactions with ethyl 1-benzofuran-3-carboxylates, Russ. J. Org. Chem., 2013, 49, 872–885 CrossRef.
- J.-Q. Chen, J.-H. Xie, D.-H. Bao, S. Liu and Q.-L. Zhou, Total Synthesis of (−)-Galanthamine and (−)-Lycoramine via Catalytic Asymmetric Hydrogenation and Intramolecular Reductive Heck Cyclization, Org. Lett., 2012, 14, 2714–2717 CrossRef CAS.
- M. Yamashita, N. D. Yadav, T. Sawaki, I. Takao, I. Kawasaki, Y. Sugimoto, A. Miyatake, K. Murai, A. Takahara, A. Kurume and S. Ohta, Asymmetric Total Synthesis of (−)-Linderol A, J. Org. Chem., 2007, 72, 5697–5703 CrossRef CAS.
- H. Sundén and R. Olsson, Asymmetric Synthesis of a Tricyclic Benzofuran Motif: A Privileged Core Structure in Biologically Active Molecules, Org. Biomol. Chem., 2010, 8, 4831–4833 RSC.
- L. Qin, D.-D. Vo, A. Nakhai, C. D. Andersson and M. Elofsson, Diversity-Oriented Synthesis of Libraries Based on Benzofuran and 2,3-Dihydrobenzofuran Scaffolds, ACS Comb. Sci., 2017, 19, 370–376 CrossRef CAS.
- R. J. Nevagi, S. N. Dighe and S. N. Dighe, Biological and Medicinal Significance of Benzofuran, Eur. J. Med. Chem., 2015, 97, 561–581 CrossRef CAS.
- K. J. Fowler, J. L. Ellis and G. W. Morrow, 6-Endo Heck Cyclization of 3-(2-Iodophenoxy)methylbenzofurans: A Useful Approach to Pterocarpenes, Synth. Commun., 2013, 43, 1676–1682 CrossRef CAS.
-
(a) H. Tanaka, M. Sato, T. Oh-Uchi, R. Yamaguchi, H. Etoh, H. Shimizu, M. Sako and H. Takeuchi, Antibacterial Properties of a New Isoflavonoid From Erythrina Poeppigiana Against Methicillin-Resistant Staphylococcus Aureus, Phytomedicine, 2004, 11, 331–337 CrossRef CAS;
(b) C. C. Li, Z. X. Xie, Y. D. Zhang, J. H. Chen and Z. Yang, Total Synthesis of Wedelolactone, J. Org. Chem., 2003, 68, 8500–8504 CrossRef CAS;
(c) T. Nehybová, J. Šmarda and P. Beneš, Plant Coumestans: Recent Advances and Future Perspectives in Cancer Therapy, Anti-Cancer Agents Med. Chem., 2014, 14, 1351–1362 CrossRef.
- Q.-Q. Mao, S.-P. Ip, S.-H. Tsai and C.-T. Che, Antidepressant-like effect of peony glycosides in mice, J. Ethnopharmacol., 2008, 119, 272–275 CrossRef CAS.
- L. Xu, F. Liu, L.-W. Xu, Z. Gao and Y.-M. Zhao, A Total Synthesis of Paeoveitol, Org. Lett., 2016, 18, 3698–3701 CrossRef CAS.
- T.-Z. Li, C.-A. Geng, X.-J. Yin, T.-H. Yang, X.-L. Chen, X.-Y. Huang, Y.-B. Ma, X.-M. Zhang and J.-J. Chen, Catalytic Asymmetric Total Synthesis of (+)- and (−)-Paeoveitol via a Hetero-Diels–Alder Reaction, Org. Lett., 2017, 19, 429–431 CrossRef CAS.
- P. Bongen, J. Pietruszka and R. C. Simon, Dynamic Kinetic Resolution of 2,3-dihydrobenzo[b]furans: Chemoenzymatic Synthesis of Analgesic Agent BRL 37959, Chem. – Eur. J., 2012, 18, 11063–11070 CrossRef CAS.
- E. A. Boyle, F. R. Mangan, R. E. Markwell, S. A. Smith, M. J. Thomson, R. W. Ward and P. A. Wyman, 7-Aroyl-2,3-dihydrobenzo[b]furan-3-carboxylic acids and 7-benzoyl-2,3-dihydrobenzo[b]thiophene-3-carboxylic acids as analgesic agents, J. Med. Chem., 1986, 29, 894–898 CrossRef CAS.
- S. A. Ahmed, D. J. Hinz, M. J. Jellen and M. M. Hossain, A Concise Synthesis of Potential COX Inhibitor BRL–37959 and Analogs Involving Bismuth(III) Catalysed Friedel−Crafts Acylation, Chem. Biodiversity, 2018, 15, e1800334, DOI:10.1002/cbdv.201800334.
- C. A. Winter, E. D. Risley and R. H. Silber, Antiinflammatory Activity of Indomethacin and Plasma Corticosterone in Rats, J. Pharmacol. Exp. Ther., 1968, 162, 196–201 CAS.
- G. L. Plosker and A. J. Wagstaff, Fluvastatin: A Review of its Pharmacology and Use in the Management of Hypercholesterolemia, Drugs, 1996, 51, 433–459 CrossRef CAS.
-
(a) P. Magnus, M. Ladlow and J. Elliott, Models for a hypothetical mechanism of action of the anticancer agent vinblastine, J. Am. Chem. Soc., 1987, 109, 7929–7930 CrossRef CAS;
(b) R. Silvestri, New Prospects for Vinblastine Analogues as Anticancer Agents, J. Med. Chem., 2013, 56, 625–627 CrossRef CAS;
(c) W. N. Dahl, R. Oftebro, E. O. Pettersen and T. Brustad, Inhibitory and Cytotoxic Effects of Oncovin (Vincristine Sulfate) on Cells of Human Line NHIK 3025, Cancer Res., 1976, 36, 3101–3105 CAS.
-
(a) H. Woehlecke, H. Osada, A. Herrmann and H. Lage, Reversal of Breast Cancer Resistance Protein-Mediated Drug Resistance by Tryprostatin A, Int. J. Cancer, 2003, 107, 721–728 CrossRef CAS;
(b) H. Wang, T. Usui, H. Osada and A. Ganesan, Synthesis and Evaluation of Tryprostatin B and Demethoxyfumitremorgin C Analogues, J. Med. Chem., 2000, 43, 1577–1585 CrossRef CAS.
- M. Huisman, M. Rahaman, S. Asad, S. Oehm, S. Novin, A. L. Rheingold and M. M. Hossain, Total Synthesis of Tryprostatin B: Synthesis and Asymmetric Phase-Transfer-Catalysed Reaction of Prenylated Gramine Salt, Org. Lett., 2019, 21, 134–137 CrossRef CAS.
-
(a) G. W. Gribble, Recent developments in indole ring synthesis—methodology and applications, J. Chem. Soc., Perkin Trans. 1, 2000, 1045–1075 RSC;
(b) T. L. Gilchrist, Synthesis of aromatic heterocycles, J. Chem. Soc., Perkin Trans. 1, 1999, 2849–2866 RSC;
(c)
R. J. Sundberg, Indoles, Academic Press, San Diego, CA, 1996 Search PubMed;
(d) C. J. Moody, Synthesis of Carbazole Alkaloids, Synlett, 1994, 9, 681–688 CrossRef;
(e) G. W. Gribble, Recent developments in indole ring synthesis—methodology and applications, Contemp. Org. Synth., 1994, 1, 145–172 RSC;
(f) U. Pindur and R. Adam, Synthetically attractive indolization processes and newer methods for the preparation of selectively substituted indoles, J. Heterocycl. Chem., 1988, 25, 1–8 CrossRef CAS.
- E. Yamato, M. Hirakura and S. Sugasawa, Synthesis of 6, 7-dihydrox-1, 2, 3, 4-tetrahydroisoquinoline derivatives, Tetrahedron, 1966, 22, 129–134 CrossRef.
-
(a) Z.-P. Xiao, J.-Y. Xue, S.-H. Tan, H.-Q. Li and H.-L. Zhu, Synthesis, structure, and structure–activity relationship analysis of enamines as potential antibacterials, Bioorg. Med. Chem., 2007, 15, 4212–4219 CrossRef CAS;
(b) Z.-P. Xiao, R.-Q. Fang, H.-Q. Li, J.-Y. Xue, Y. Zhang and H.-L. Zhu, Enamines as novel antibacterials and their structure–activity relationships, Eur. J. Med. Chem., 2008, 43, 1828–1836 CrossRef CAS.
- G. Reybrouck, A theoretical approach of disinfectant testing, Zentralbl. Bakteriol. Orig. B, 1975, 160, 342–367 CAS.
- M. J. Meier, R. M. Subasinghe and L. A. Beaudette, Draft Genome Sequence of the Industrially Significant Bacterium Pseudomonas fluorescens ATCC 13525, Microbiol. Resour. Announce., 2018, 7, e01368-18 CrossRef.
- M. R. Atuu, S. J. Mahmood, F. Laib and M. M. Hossain, Kinetic resolution of tropic acid ethyl ester and its derivatives by lipase PS, Tetrahedron: Asymmetry, 2004, 15, 3091–3101 CrossRef CAS.
- M. R. Attu and M. M. Hossain, Dynamic kinetic resolution of racemic tropic acid ethyl ester and its derivatives, Tetrahedron Lett., 2007, 48, 3875–3878 CrossRef.
-
(a) E. Leete, The Biogenesis of Tropic Acid and Related Studies on the Alkaloids of Datura stramonium, J. Am. Chem. Soc., 1960, 82, 612–614 CrossRef CAS;
(b) N. W. Hamon and J. L. Eyolfson, Biosynthesis of Tropic Acid in Datura innoxia Root Tissue, J. Pharm. Sci., 1972, 61, 2006–2008 CrossRef CAS;
(c) E. Leete, N. Kowanko and R. A. Newmark, Use of carbon-13 nuclear magnetic resonance to establish that the biosynthesis of tropic acid involves an intramolecular rearrangement of phenylalanine, J. Am. Chem. Soc., 1975, 97, 6826–6830 CrossRef CAS.
-
(a) M. E. Conklin, Genetic and biochemical aspects of the development of Datura, Monogr. Dev. Biol., 1975, 12, 1–170 Search PubMed;
(b) P. W. Le Quesne, Alkaloids: biochemistry, ecology, and medicinal applications, J. Nat. Prod., 1999, 62, 664, DOI:10.1021/np980259j.
- P. Montuschi, Pharmacological treatment of chronic obstructive pulmonary disease, Int. J. Chronic Obstruct. Pulm. Dis., 2006, 1, 409–423 CAS.
- I. T. Strukov, Tiazolidin-4-karbonovaya kislota I EE proizvodnye .2. S-zameshchennye tsisteina I ikh prevrashcheniya V proizvodnye tiazolidin-4-karbonovoi kisloty, Zh. Obshch. Khim., 1952, 22, 1028 Search PubMed.
- V. G. Foshkin, N. S. Kovaleva, F. B. Naidis and A. S. Vitvitskaya, Improvement of the technological method for the synthesis of leucogen, Pharm. Chem. J., 1976, 10, 1090–1091 CrossRef.
- L. Redding and D. B. Weiner, DNA vaccines in veterinary use, Expert Rev. Vaccines, 2009, 8, 1251–1276 CrossRef CAS.
- D. A. Brown, S. Betharia, J.-H. Yen, Q. Tran, H. Mistry and K. Smith, Synthesis and structure–activity relationships study of dithiolethiones as inducers of glutathione in the SH-SY5Y neuroblastoma cell line, Bioorg. Med. Chem. Lett., 2014, 24, 5829–5831 CrossRef CAS.
- D. A. Brown, S. Betharia, J.-H. Yen, P.-C. Kuo and H. Mistry, Further structure–activity relationships study of substituted dithiolethiones as glutathione-inducing neuroprotective agents, Chem. Cent. J., 2016, 10, 1–11 CrossRef.
- K. Kaur and R. F. Pratt, Mechanism of reaction of acyl phosph(on)ates with the β-lactamase of Enterobacter cloacae P99, Biochemistry, 2001, 40, 4610–4621 CrossRef CAS.
- J. C. Craig and M. Moyle, The Synthesis of Acetylenes and Allenes from Enol Phosphates, J. Chem. Soc., 1963, 3712–3718 RSC.
- J. F. Marecek and D. L. Griffith, Enol phosphates of. beta.-keto acids. Synthesis and hydrolysis of ethyl. beta.-hydroxy-trans-cinnamic acid cyclic phosphate, J. Am. Chem. Soc., 1970, 92, 917–921 CrossRef CAS.
-
(a) J. Mann, Modern methods for the introduction of fluorine into organic molecules: An Approach to Compounds with Altered Chemical and Biological Activities, Chem. Soc. Rev., 1987, 16, 381–436 RSC;
(b) J. T. Welch, Advances in the preparation of biologically active organofluorine compounds, Tetrahedron, 1987, 43, 3123–3197 CrossRef CAS;
(c) G. Resnati, Synthesis of chiral and bioactive fluoroorganic compounds, Tetrahedron, 1993, 49, 9385–9445 CrossRef CAS.
- H. Kamaya, M. Sato and C. Kaneko, An efficient method for α-monofluorination of carbonyl compounds with molecular fluorine: Use of α-hydroxymethylene substituent as directing and activating groups, Tetrahedron Lett., 1997, 38, 587–590 CrossRef CAS.
-
(a) J. A. Wilkinson, Recent advances in the selective formation of the C-F bond, Chem. Rev., 1992, 92, 505–519 CrossRef CAS;
(b) S. Rozen, Selective fluorinations by reagents containing the OF group, Chem. Rev., 1996, 96, 1717–1736 CrossRef CAS;
(c) G. S. Lal, G. P. Pez and R. G. Syvret, Electrophilic NF fluorinating agents, Chem. Rev., 1996, 96, 1737–1755 CrossRef CAS.
-
(a) S. T. Purrington, B. S. Kagan and T. M. Patrick, The application of elemental fluorine in organic synthesis, Chem. Rev., 1986, 86, 997–1018 CrossRef CAS;
(b) S. Rozen, Elemental fluorine as a “Legitimate” reagent for selective fluorination of organic compounds, Acc. Chem. Res., 1988, 21, 307–312 CrossRef CAS.
- R. M. Izatt, K. Pawlak, J. S. Bradshaw and R. L. Bruening, Thermodynamic and kinetic data for macrocycle interaction with cations, anions, and neutral molecules, Chem. Rev., 1995, 95, 2529–2586 CrossRef CAS.
- J. S. Bradshaw and R. M. Izatt, Crown ethers: the search for selective ion ligating agents, Acc. Chem. Res., 1997, 30, 338–345 CrossRef CAS.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Crown Ethers: Sensors for Ions and Molecular Scaffolds for Materials and Biological Models, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS.
- Ç. Erk, Cation Recognition with fluorophore crown ethers, Ind. Eng. Chem. Res., 2000, 39, 3582–3588 CrossRef.
- M. Bulut and Ç. L. Erk, The synthesis of novel crown ethers, part IX, 3-phenyl chromenone-crown ethers, J. Heterocycl. Chem., 2001, 38, 1291–1295 CrossRef CAS.
|
| This journal is © the Partner Organisations 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
M. Shahnawaz
Ali
,
Khorshada
Jahan
,
Jawad Bin
Belayet
,
A. F. M. Towheedur
Rahman
and
M. Mahmun
Hossain
,
M. Shahnawaz
Ali
,
Khorshada
Jahan
,
Jawad Bin
Belayet
,
A. F. M. Towheedur
Rahman
and
M. Mahmun
Hossain