DOI:
10.1039/D0QO01247E
(Research Article)
Org. Chem. Front., 2021,
8, 127-132
Synthesis of C3-alkylated benzofurans via palladium-catalyzed regiocontrolled hydro-furanization of unactivated alkenes†
Received
9th October 2020
, Accepted 16th November 2020
First published on 17th November 2020
Abstract
A new chelation-controlled hydrofuranization of unactivated olefins with α-alkynyl arylols is reported for the first time, and used to produce a wide range of C3-alkylated benzofurans with generally good yields under mild conditions. A bidentate directing group is used to suppress β-hydride elimination and in turn facilitate a protodepalladation event. In addition to 100% atom utilization, good functional group tolerance and mild conditions, the present catalysis did not demand any additional ligand, additives and bases/acids, making the transformation simple and facile and it thus opens new avenues for catalytic difunctionalization of alkynes and olefins.
Introduction
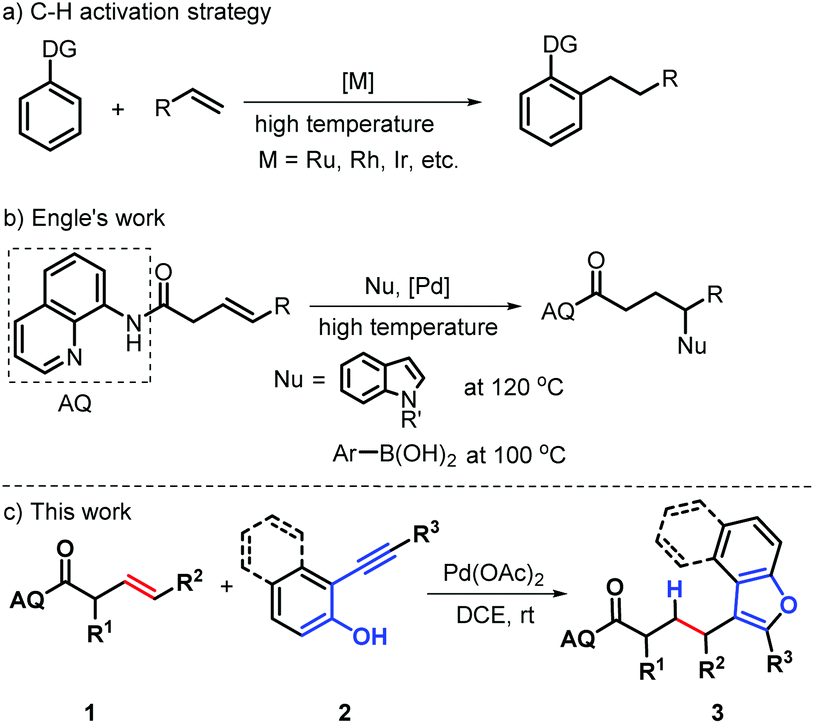
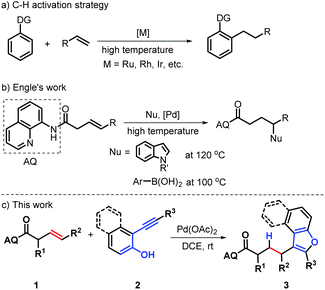
Transition-metal catalyzed hydroarylation of alkenes with arenes enabling direct formation of carbon–carbon bonds through the addition of arene C–H bonds across olefins is considered as one of the most straightforward approaches to access alkylated aromatics due to its characteristics of low-cost and sustainability.1 As such, great efforts have been contributed in the last decades towards establishing efficient and regioselective hydroarylation reactions of alkenes with various transition metal catalysts, including Ru, Rh, Ir, Pd and rare earth metal complexes.2 Of these methods, catalytic functional group directed activation of C–H bonds has become an important tool for synthesizing these molecules, generally depending on the activated alkenes and high temperatures (>100 °C) (Scheme 1a).2 Very recently, Engle's group reported an elegant Pd-catalyzed chelation-controlled hydroarylation of unactivated olefins with indoles or arylboronic acids as C–H nucleophiles through an amide-linked 8-aminoquinoline (AQ) assisted activation of double C–C bonds (Scheme 1b).3 During the formation of alkylated indoles, regioselective Wacker-type nucleopalladation of alkene occurs, giving palladacycle intermediate, which undergoes protodepalladation to give a γ-addition product. Notably, the presence of the AQ directing group suppress a competing β-H elimination process to control the regioselectivity of addition. Indeed, such a strategy can confer to the new double C–C activation mode and has recognized as one of the most attractive strategies to access alkylated aromatics with the controllable regioselectivity.4 Despite the remarkable advances gained in this field, further development of a new and general chelation-assisted strategy for achieving hydroarylation of alkenes is still highly desirable, especially for the direct assembly of privileged structural scaffolds.
 |
| | Scheme 1 Transition-metal-catalyzed hydroarylation of alkenes. | |
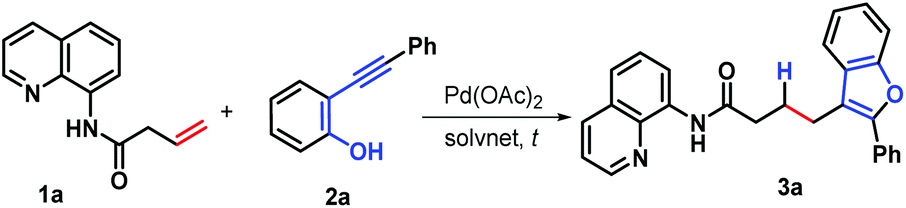
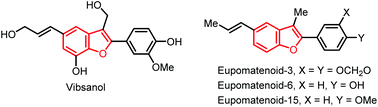
Benzofuran and its derivatives are widely present as scaffolds in a wide range of biologically potent naturally occurring products and synthetic substances.5 For example, prescribed agents incorporating this structural motif in the realms of pharmacologically active agents include the antidepressant (−)-BPAP6a and the antiarrythmic Amiodarone.6b Several representative natural products bearing benzofuran moiety include vibsanol7 and the eupomatenoid family (Fig. 1),8 which exhibit diverse biological activities.9 Demand for this important subunit have inspired numerous strategies and tactics for its construction. In general, the present synthetic methods can be divided into two pathways: (i) furanization of phenyl-containing building blocks, such as phenol,10 salicylaldehyde,11 and 2-methoxybenzoic acid,12 and (ii) benzannulation of furan derivatives, such as 2,5-dimethoxyltetrahydrofuran,13 2-alkoxy-3,4-dihydrofuran,14 and α-bromochalcones or α-bromocinnamates.15 Recently, we established a Pd-catalyzed decarboxylative allylation-furanization cascade of α-alkynyl arylols with vinylethylene carbonates, leading to 3-allylbenzofurans.16 While carrying out this project, we reasoned that under suitable palladium catalysis conditions, hydro-furanization of unactivated alkenes with furan precursors could be achieved by taking advantage of a methodology invented by Engle's group.3 Herein, we report the successful implementation of this concept with a new Pd-catalyzed hydrofuranization of unactivated alkenes with α-alkynyl arylols by using the amide-linked 8-aminoquinoline (AQ) as a bidentate chelating auxiliary under mild conditions, and used to produce a wide range of C3-alkylated benzofurans with good yields and 100% atom utilization (Scheme 1c). Notably, the current approach oriented an atom-economic and completely regioselective access to a wide range of structurally diverse amide-linked quinoline/benzofuran pairs.
 |
| | Fig. 1 Representative benzofuran-containing natural products. | |
For optimization of the hydro-furanization process, N-(quinolin-8-yl)but-3-enamide (1a) and 2-(phenylethynyl)phenol (2a) were selected as the model substrates, and the reaction was conducted under the Pd(II) catalysis. To our delight, with hexafluoroisopropanol (HFIP) being the solvent at 120 °C, the reaction proceeded readily to access C3-alkylated benzofuran 3a with 53% yield in the presence of Pd(OAc)2 (Table 1, entry 1).3a Very interestingly, decreasing the reaction temperature is beneficial for this transformation (entries 2–4). It is found that the reaction could worked more efficiently, providing a slightly higher yield of product 3a (56%) when the reaction temperature was adjusted to room temperature (entry 4). Moreover, further screening of various reaction parameters revealed that the solvent exerted a profound impact on the reaction outcome. With nonpolar solvents being the reaction medium, such as dichloromethane (DCM) and 1,2-dichloroethane (DCE), the higher yields could be obtained (entries 5 and 6) and of these two solvents, DCE proved to be the better choice for this process, resulting in 88% yield (entry 6). In contrast, the reaction efficiency was remarkably suppressed by switching the solvents to high-polarity methanol, acetonitrile (CH3CN), and tetrahydrofuran (THF) (entries 7–9). In another case of high polar N,N-dimethylformamide (DMF), the yield almost plateaued as compared with HFIP (entry 10 vs. entry 4). Without Pd(OAc)2, the reaction did not work (entry 11), indicating that the Pd(OAc)2 catalyst is crucial for this hydro-furanization reaction. Exchanging Pd(OAc)2 for Pd(TFA)2 or PdCl2 led to unsatisfactory results (entries 12 and 13). Significantly lower conversion was observed when the reaction was carried out under air conditions.
Table 1 Condition optimization for product 3aa
|

|
| Entry |
Cat. (10 mol%) |
Solvent |
T (°C) |
Yieldb (%) |
|
Reaction conditions: 1a (0.2 mmol), 2a (1.2 equiv.), Pd(OAc)2 (10 mol%), solvent (1 mL), Ar, 12 h.
Isolated yield based on substrate 1a.
Under air condition.
|
| 1 |
Pd(OAc)2 |
HFIP |
120 |
53 |
| 2 |
Pd(OAc)2 |
HFIP |
90 |
54 |
| 3 |
Pd(OAc)2 |
HFIP |
60 |
54 |
| 4 |
Pd(OAc)2 |
HFIP |
25 |
56 |
| 5 |
Pd(OAc)2 |
DCM |
25 |
76 |
| 6 |
Pd(OAc)2 |
DCE |
25 |
88 |
| 7 |
Pd(OAc)2 |
CH3OH |
25 |
Trace |
| 8 |
Pd(OAc)2 |
CH3CN |
25 |
Trace |
| 9 |
Pd(OAc)2 |
THF |
25 |
36 |
| 10 |
Pd(OAc)2 |
DMF |
25 |
58 |
| 11 |
— |
DCE |
25 |
N.R. |
| 12 |
Pd(TFA)2 |
DCE |
25 |
80 |
| 13 |
PdCl2 |
DCE |
25 |
38 |
| 14c |
Pd(OAc)2 |
DCE |
25 |
41 |
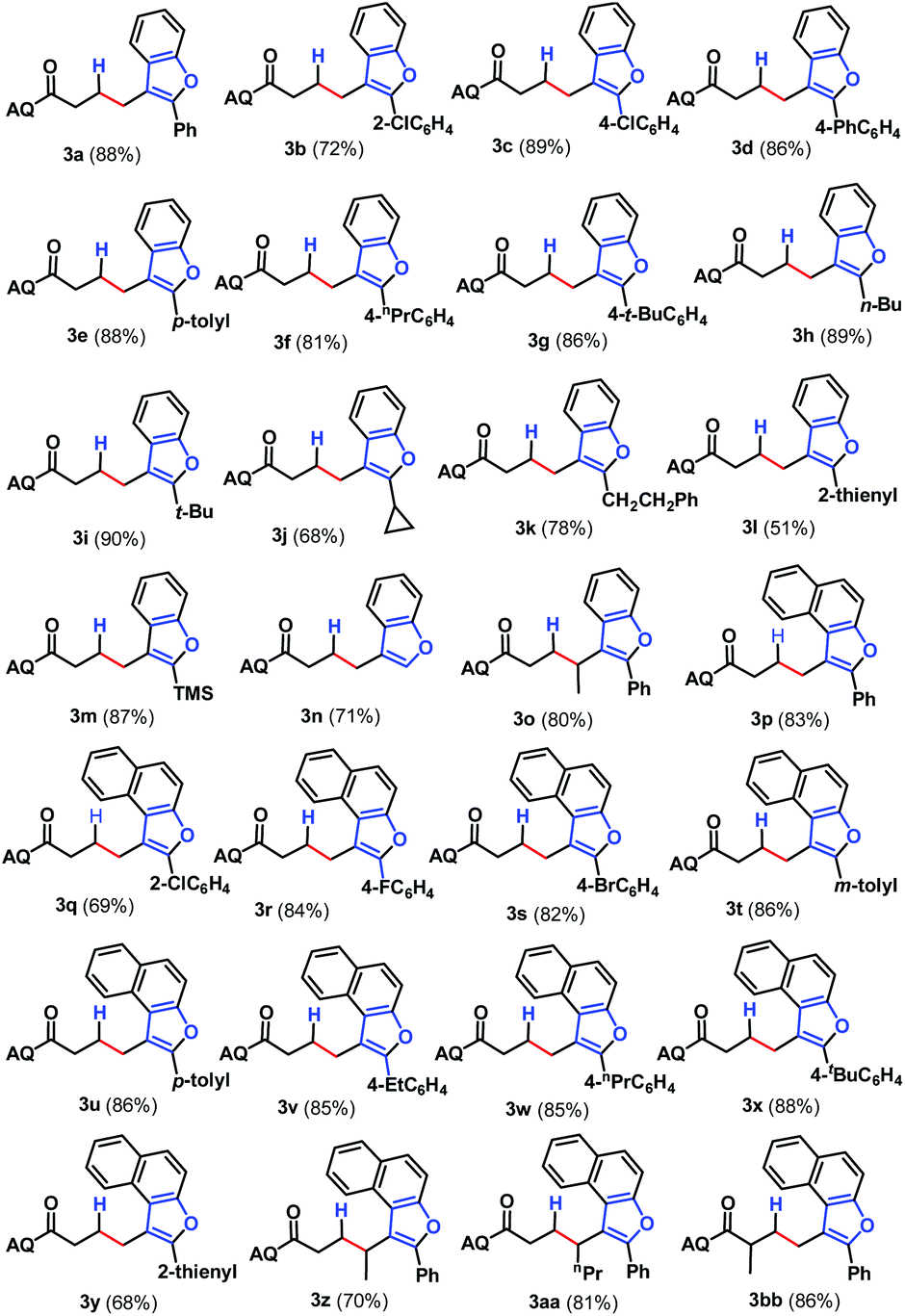
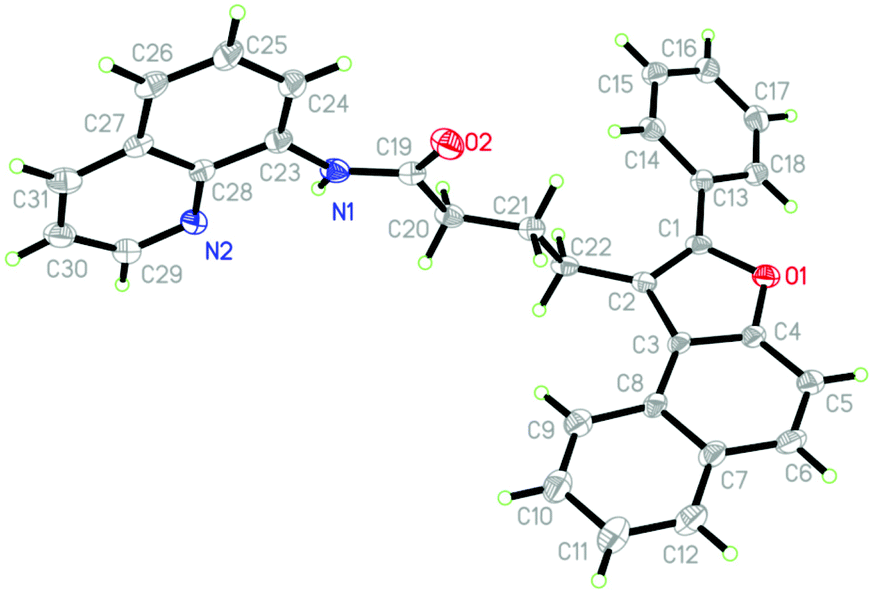
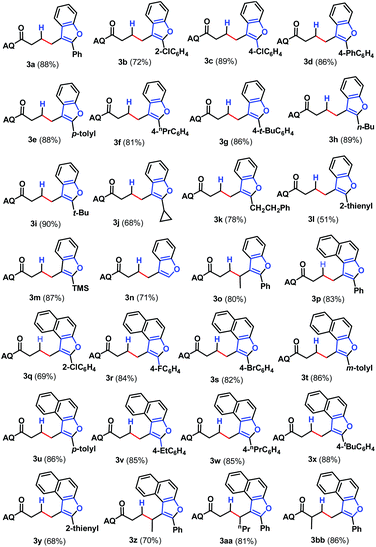
With an optimal set of catalytic conditions selected, we were then poised to examine the generality of this catalytic furan annulation cascade by exploiting various α-alkynyl arylols, and the results are summarized in Scheme 2. Firstly, o-alkynyl phenols 2 with diverse electronic natures and different substituents in the arene ring relative to the arylalkynyl moiety (R) were investigated by repeating the reaction of 1a under standard conditions. Both electron-withdrawing (e.g. chloro, 2b and 2c) and electron-donating (e.g. phenyl, 2d; methyl, 2e; propyl, 2f and t-butyl, 2g) groups were compatible in this Pd-catalysis, furnishing the corresponding products 3b–3g in 72–89% yields. Among them, sterically encumbered 2-chlorophenyl counterpart 2b was found to have no influence on the course of the reaction, as benzofuran 3b was afforded in 72% yield. Besides, the aryl fragment in the alkynyl moiety was alternated with an alkyl group such as n-butyl (2h), t-butyl (2i), cyclopropyl (2j) and 2-phenethyl (2k), and the target transformation was achieved with the optimal conditions, as demonstrated by the generation of 3h–3k in good to excellent yields (68–90%). Phenols bearing a 2-thienyl 2l, trimethylsilyl (TMS, 2m) group or H (2n) tethered by the alkynyl motif were then examined to demonstrate the generality of this transformation, allowing the catalytic furan annulation process to access the products 3l–3n in acceptable yields. Noticeably, unactivated internal alkene 1b was also workable, giving access to product 3o in 80% yield. Moreover, α-alkynylnaphthalen-2-ols with electroneutral (H 2n), electron-withdrawing (chloro 2p, fluoro 2q and bromo 2r), or electron-donating (methyl 2s–2t, ethyl 2u, n-propyl 2v and t-butyl 2w) groups linked by the arylalkynyl moiety (R3) were probed,17 and all of them proceeded smoothly to offer the target products 3p–3x in 69–88% yields. Moreover, α-alkynylnaphthalen-2-ol 2y with a 2-thienyl group was accommodated, confirming the efficiency of this Pd catalysis, as product 3y was gained with an acceptable yield. Similarly, the Pd-catalyzed reaction between internal alkenes 1b and 1c worked efficiently with 2o, affording product 3z and 3aa in 70% and 81% yields, respectively. In addition, α-methyl-substituted alkene 1d can also react with 2o, and the target product 3bb can be obtained with 88% yield. Next, o-alkynyl anilines 5 were employed as indole precursors to evaluate the feasibility of hydro-indolation. Unluckily, the desired products 4 were not detected using free or Ts-protected o-alkynyl anilines as starting materials under standard conditions (Scheme 3). In addition, the directing group removal experiment with 3x as an example was carried out (Scheme 4). The AQ group was readily removed by treatment of 3x with boron trifluoride diethyl etherate in ethanol at 100 °C, producing 6 in high yield.4c To expand the potential application of this method, an amplification reaction was conducted under the standard conditions. We were delighted to find that product 3p was isolated in 87% yield on a 2.5 mmol scale (Scheme 5). The structures of these products were fully determined by NMR spectroscopy and HRMS data (see the ESI†). The structure of 3p was identified by single-crystal X-ray diffraction (Fig. 2).
 |
| | Scheme 2 Substrate scope for the synthesis of products 3. | |
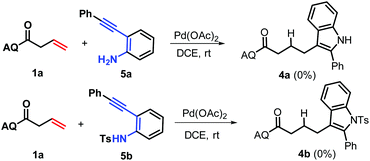 |
| | Scheme 3 Limitation of Pd-catalysis. | |
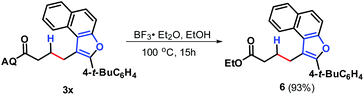 |
| | Scheme 4 Directing group removal experiment. | |
 |
| | Scheme 5 An amplification reaction of 1a with 2o. | |
 |
| | Fig. 2 X-Ray structure of product 3p. | |
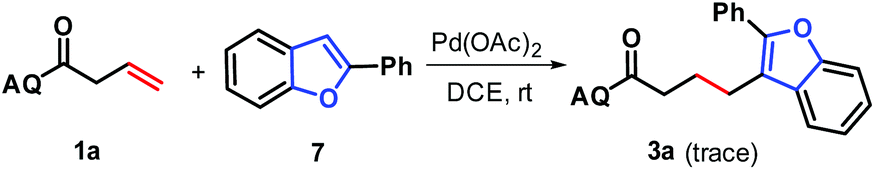
To gain a mechanistic insight into the formation of products 3, the preformed 2-phenylbenzofuran 7 was subjected to reaction with N-(quinolin-8-yl)but-3-enamide 1a under standard conditions, and only a trace amount of the desired product 3a was observed (Scheme 6). This indicates that 2-phenylbenzofuran, due to the low polarity and high bond-dissociation energy of its C–H bond at C3 position, may be not an intermediate for the generation of 3, and that the addition of in situ-generated benzofuran-Pd σ-complex to alkenes occurs prior to its protodepalladation.
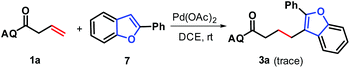 |
| | Scheme 6 Control experiments. | |
Based on the experimental results and previous reports,3 a plausible catalytic cycle is shown in Scheme 7. Initially, palladium(II) binds to the directing group, making it in the close proximity to the olefin and thus facilitating π-Lewis acid activation. In the meantime, the palladium complex A also activates triple C–C bonds of substrate 2 to form benzofuran-Pd σ-complex B, followed by nucleopalladation in an outer-sphere mechanism to give the cis-nucleopalladated intermediate C.18 The palladacycle, because of the stability and conformational rigidity imparted by the directing group, does not undergo β-hydride elimination and instead is sufficiently long-lived to be intercepted with a proton in a protodepalladation step,19 giving the hydrofuranization product and regenerating the active palladium catalyst.
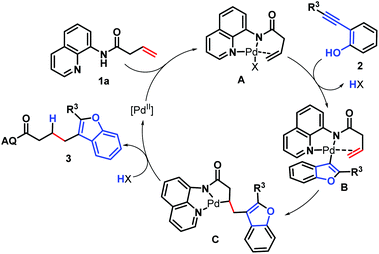 |
| | Scheme 7 Plausible reaction pathway. | |
Conclusions
In summary, we have demonstrated a new Pd-catalyzed chelation-controlled hydrofuranization of unactivated olefins with α-alkynyl arylols through a successive nucleophilic cyclization and the following addition to double C–C bonds process. The Pd-catalysis reaction provides a general and regioselective method for the synthesis of a wide range of C3-alkylated benzofurans with generally good yields. It is believed that the 8-aminoquinoline directing group dictates the regioselectivity and prevents β-hydride elimination, which is the key to form C3-alkylated benzofurans. The protocol features 100% atom utilization, good functional group tolerance, and mild conditions, and thus opens new avenues for catalytic difunctionalization of internal alkynes and unactivated olefins. Further investigation and application of this Pd-catalyzed difunctionalization of unactivated olefins is underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the NSFC (no. 21801130) and the NSF of Jiangsu Province (BK20180689).
Notes and references
-
(a) L. N. Lewis and J. F. Smith, Catalytic Carbon-Carbon Bond Formation via Ortho-metalated Complexes, J. Am. Chem. Soc., 1986, 108, 2728 CrossRef CAS;
(b) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Efficient Catalytic Addition of Aromatic Carbon-Hydrogen Bonds to Olefins, Nature, 1993, 366, 529 CrossRef CAS;
(c) X. Zeng, Recent advances in catalytic sequential reactions involving hydroelement addition to carbon–carbon multiple bonds, Chem. Rev., 2013, 113, 6864 CrossRef CAS;
(d)
Catalytic Cydroarylation of Carbon-Carbon Multiple Bonds, ed. L. Ackermann, T. B. Gunno and L. G. Habgood, Wiley-VCH, Weinheim, 2018 Search PubMed.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Transition-Metal-Catalyzed C−H Alkylation Using Alkenes, Chem. Rev., 2017, 117, 9333 CrossRef CAS.
-
(a) J. A. Jr Gurak, K.-S. Yang, Z. Liu and K. M. Engle, Directed, Regiocontrolled Hydroamination of Unactivated Alkenes via Protodepalladation, J. Am. Chem. Soc., 2016, 138, 5805 CrossRef;
(b) K.-S. Yang, J. A. Jr Gurak, Z. Liu and K. M. Engle, Catalytic, Regioselective Hydrocarbofunctionalization of Unactivated Alkenes with Diverse C-H Nucleophiles, J. Am. Chem. Soc., 2016, 138, 14705 CrossRef CAS;
(c) Z. Liu, Y.-Y. Wang, Z.-C. Wang, T. Zeng, P. Liu and K. M. Engle, Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation, J. Am. Chem. Soc., 2017, 139, 11261 CrossRef CAS;
(d) M. L. O'Duill, R. Matsuura, Y.-Y. Wang, J. L. Turnbull, J. A. Gurak, D.-W. Gao, G. Lu, P. Liu and K. M. Engle, Tridentate Directing Groups Stabilize 6-Membered Palladacycles in Catalytic Alkene Hydrofunctionalization, J. Am. Chem. Soc., 2017, 139, 15576 CrossRef;
(e) R. Matsuura, T. C. Jankins, D. E. Hill, K. S. Yang, G. M. Gallego, S. Yang, M. He, F. Wang, R. P. Marsters, I. McAlpine and K. M. Engle, Chem. Sci., 2018, 9, 8363 RSC.
-
(a) H. Wang, Z. Bai, T. Jiao, Z. Deng, H. Tong, G. He, Q. Peng and G. Chen, Palladium-catalyzed amide-directed enantioselective hydrocarbofunctionalization of unactivated alkenes using a chiral monodentate oxazoline ligand, J. Am. Chem. Soc., 2018, 140, 3542 CrossRef CAS;
(b) C. Wang, G. Xiao, T. Guo, Y. Ding, X. Wu and T.-P. Loh, Palladium-catalyzed regiocontrollable reductive Heck reaction of unactivated aliphatic alkenes, J. Am. Chem. Soc., 2018, 140, 9332 CrossRef CAS;
(c) C.-L. Tang, R. Zhang, B. Zhu, J.-K. Fu, Y. Deng, L. Tian, W. Guan and X.-H. Bi, Directed Copper-Catalyzed Intermolecular Heck-Type Reaction of Unactivated Olefins and Alkyl Halides, J. Am. Chem. Soc., 2018, 140, 16929 CrossRef CAS;
(d) Y. Li, J.-F. Gong and M.-P. Song, Palladium-catalyzed δ-selective reductive Heck reaction of alkenyl carbonyl compounds with aryl iodides and bromides, Org. Chem. Front., 2020, 7, 2216 RSC;
(e) Z.-B. Bai, Z.-Q. Bai, F.-F. Song, H. Wang, G. Chen and G. He, Palladium-Catalyzed Amide-Directed Hydrocarbofunctionalization of 3-Alkenamides with Alkynes, ACS Catal., 2020, 10, 933 CrossRef CAS.
- M. M. Heravi, V. Zadsirjan, H. Hamidi and P. H. T. Amiri, Total synthesis of natural products containing benzofuran rings, RSC Adv., 2017, 7, 24470 RSC.
-
(a) S. Shimazu, K. Takahata, H. Katsuki, H. Tsunekawa, A. Tanigawa, F. Yoneda, J. Knoll and A. Akaike, (-)-1-(Benzofuran-2-yl)-2-propylaminopentane enhances locomotor activity in rats due to its ability to induce dopamine release, Eur. J. Pharmacol., 2001, 421, 181 CrossRef CAS;
(b) B. M. Singh and E. M. V. Williams, Third class of antiarrhythmic action. Effects on atrial and ventricular intracellular potentials, and other pharmacological actions on cardiac muscle, of MJ 1999 and AH 3474 [DL-5-(2-tert-butylamino-1-hydroxyethyl)salicylamide hydrochloride], Br. J. Pharmacol., 1970, 39, 657 CrossRef CAS.
- K. Kawazu, Bioactive compounds in leaves of Viburnum odoratissimum Ker. Part 1. Isolation of vibsanines A, B, C, D, E and F from Viburnum odoratissimum, Agric. Biol. Chem., 1980, 44, 1367 CAS.
-
(a) R. Ahmed and R. Stevenson, Synthesis of eupomatenoid-7 and eupomatenoid-12, Phytochemistry, 1975, 14, 2710 CrossRef CAS;
(b) B. A. McKittrick and R. Stevenson, Natural benzofurans. Synthesis of eupomatenoids-1, -3, -4, -5, -6, -7, and -13, J. Chem. Soc., Perkin Trans. 1, 1983, 475 RSC;
(c) T. A. Engler, W. Chai and K. O. LaTessa, Lewis Acid-Controlled Regioselectivity in Reactions of Styrenyl Systems with Benzoquinone Monoimides: New Regioselective Syntheses of Substituted 2-Aryl-2,3-dihydrobenzofurans, 2-Aryl-2,3-dihydroindoles, and 2-Arylindoles, J. Org. Chem., 1996, 61, 9297 CrossRef CAS;
(d) T. A. Engler and W. Chai, Synthesis of (±)-licarin B and eupomatenoids-1 and -12: a general approach to 2-aryl-7-alkoxybenzofuranoid neolignans, Tetrahedron Lett., 1996, 37, 6969 CrossRef CAS.
-
(a) Y. Fukuyama, M. Nakahara, H. Minami and M. Kodama, Two new benzofuran-type lignans from the wood of Viburnum awabuki, Chem. Pharm. Bull., 1996, 44, 1418 CrossRef.
-
(a) X.-W. Guo, R. Yu, H.-J. Li and Z.-P. Li, Iron-Catalyzed Tandem Oxidative Coupling and Annulation: An Efficient Approach to Construct Polysubstituted Benzofurans, J. Am. Chem. Soc., 2009, 131, 17387 CrossRef CAS;
(b) U. A. Kshirsagar, R. Parnes, H. Goldshtein, R. Ofir, R. Zarivach and D. Pappo, Aerobic Iron−Based Cross−Dehydrogenative Coupling Enables Efficient Diversity−Oriented Synthesis of Coumestrol−Based Selective Estrogen Receptor Modulators, Chem. – Eur. J., 2013, 19, 13575 CrossRef CAS;
(c) E. Gaster, Y. Vainer, A. Regev, S. Narute, K. Sudheendran, A. Werbeloff, H. Shalit and D. Pappo, Significant Enhancement in the Efficiency and Selectivity of Iron−Catalyzed Oxidative Cross−Coupling of Phenols by Fluoroalcohols, Angew. Chem., Int. Ed., 2015, 54, 4198 CrossRef CAS;
(d) K.-S. Huang and E.-C. Wang, A Novel Synthesis of Substituted Naphthalenes via Claisen Rearrangement and RCM Reaction, Tetrahedron Lett., 2001, 42, 6155 CrossRef CAS;
(e) E.-C. Wang, M.-K. Hsu, Y.-L. Lin and K.-S. Huang, A New Synthesis of Substituted 2,5-Dihydrobenzo[b]oxepines, Heterocycles, 2002, 57, 1997 CrossRef CAS.
- S. B. Sapkal, K. F. Sapkal, B. B. S. Shelke and S. S. Murlidhar, An Efficient Synthesis of Benzofuran Derivatives under Conventional/Non-Conventional Method, Chin. Chem. Lett., 2010, 21, 1439 CrossRef CAS.
- A. H. Wadsworth, H. A. Mitchell, I. Fellows and D. R. Sutherland, Synthesis of a Carbon-14 Labelled Version of Benzofuran GR151004B, J. Labelled Compd. Radiopharm., 1996, 38, 863 CrossRef CAS.
- S. M. Rafiq, R. Sivasakthikumaran and A. K. Mohanakrishnan, Lewis Acid/Brönsted Acid Mediated Benz-Annulation of Thiophenes and Electron-Rich Arenes, Org. Lett., 2014, 16, 2720 CrossRef CAS.
-
(a) C.-J. Liu, W.-B. Huang, M. Wang, B. Pan and Y.-L. Gu, Expedient Synthesis of Substituted Benzoheterocycles using 2-Butoxy-2,3-dihydrofurans as [4+2] Benzannulation Reagents, Adv. Synth. Catal., 2016, 358, 2260 CrossRef CAS;
(b) C.-H. Liu, L. Zhou, D. Jiang and Y.-L. Gu, Multicomponent Reactions of Aldo-X Bifunctional Reagent α-Oxoketene Dithioacetals and Indoles or Amines: Divergent Synthesis of Dihydrocoumarins, Quinolines, Furans, and Pyrroles, Asian J. Org. Chem., 2016, 5, 367 CrossRef CAS;
(c) P. Ravichandiran, B.-B. Lai and Y.-L. Gu, Aldo-X Bifunctional Building Blocks for the Synthesis of Heterocycles, Chem. Rec., 2017, 17, 142 CrossRef CAS.
- S. Paria and O. Reiser, Visible Light Photoredox Catalyzed Cascade Cyclizations of α-Bromochalcones or α-Bromocinnamates with Heteroarenes, Adv. Synth. Catal., 2014, 356, 557 CrossRef CAS.
- J. Zhang, J.-Y. Wang, M.-H. Huang, W.-J. Hao, X.-C. Tu, S.-J. Tu and B. Jiang, Ligand-Free Pd-Catalyzed Synthesis of 3-Allylbenzofurans by Merging Decarboxylative Allylation and Nucleophilic Cyclization, J. Org. Chem., 2020, 85, 7036 CrossRef CAS.
- For recent examples of transformations of α-alkynylnaphthalen-2-ols, see:
(a) T. Xu, N. Lin, W.-J. Hao, J. Zhang, M.-F. Li, S.-J. Tu and B. Jiang, Synthesis of polycyclic indoles via an organocatalytic bicyclization of α-alkynylnaphthalen-2-ols with nitrones, Chem. Commun., 2020, 56, 11406 RSC;
(b) T. Xu, K. Chen, H.-Y. Zhu, W.-J. Hao, S.-J. Tu and B. Jiang, Yb(OTf)3-c atazlyed alkyne-carbonyl metathesis-Oxa-Michael addition relay for diastereoselective synthesis of functionalized naphtho[2,1-b]furans, Org. Lett., 2020, 22, 2414 CrossRef CAS;
(c) K. Chen, T. Xu, J. Liang, M. Zhou, J. Zhang, W.-J. Hao, J. Wang, S.-J. Tu and B. Jiang, Completely regioselective insertion of unsymmetrical alkynes into electron-deficient alkenes for the synthesis of new pentacyclic indoles, Chem. Commun., 2019, 21, 9784 Search PubMed;
(d) C.-L. Ji, W.-J. Hao, J. Zhang, F.-Z. Geng, T. Xu, S.-J. Tu and B. Jiang, Catalytic three-component synthesis of functionalized naphtho[2,1-b]oxecines via a double bond cleavage–rearrangement cascade, Org. Lett., 2019, 21, 6494 CrossRef CAS.
- For examples of similar Pd-complexes, see: R. Giri, N. Maugel, B. M. Foxman and J.-Q. Yu, Dehydrogenation of Inert Alkyl Groups via Remote C-H Activation: Converting a Propyl Group into a π-Allylic Complex, Organometallics, 2008, 27, 1667 CrossRef CAS.
- For representative C–Pd(II) protonolysis precedents, see:
(a) M. J. Gaunt and J. B. Spencer, Derailing the Wacker Oxidation: Development of a Palladium-Catalyzed Amidation Reaction, Org. Lett., 2001, 3, 25 CrossRef CAS;
(b) T. Pei and R. A. Widenhoefer, Palladium-Catalyzed Intramolecular Addition of 1,3-Diones to Unactivated Olefins, J. Am. Chem. Soc., 2001, 123, 11290 CrossRef CAS;
(c) F. E. Michael and B. M. Cochran, Room Temperature Palladium-Catalyzed Intramolecular Hydroamination of Unactivated Alkenes, J. Am. Chem. Soc., 2006, 128, 4246 CrossRef CAS;
(d) B. M. Cochran and F. E. Michael, Mechanistic Studies of a Palladium-Catalyzed Intramolecular Hydroamination of Unactivated Alkenes: Protonolysis of a Stable Palladium Alkyl Complex Is the Turnover-Limiting Step, J. Am. Chem. Soc., 2008, 130, 2786 CrossRef CAS;
(e) M. L. O'Duill and K. M. Engle, Protodepalladation as a Strategic Elementary Step in Catalysis, Synthesis, 2018, 50, 4699 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2036353 (3p). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qo01247e |
|
| This journal is © the Partner Organisations 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
De-Cai
Wang
*a and
Bo
Jiang
b,
De-Cai
Wang
*a and
Bo
Jiang