
Recent progress in the construction of polymers with advanced chain structures via hybrid, switchable, and cascade chain-growth polymerizations
Guang
Chen
 a,
Lei
Xia
a,
Fei
Wang
b,
Ze
Zhang
*a and
Ye-Zi
You
*a
a,
Lei
Xia
a,
Fei
Wang
b,
Ze
Zhang
*a and
Ye-Zi
You
*a
aCAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei, Anhui 230026, People's Republic of China. E-mail: yzyou@ustc.edu.cn; zze320@mail.ustc.edu.cn
bNeurosurgical Department, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, Hefei, Anhui 230026, China
First published on 18th May 2021
Abstract
The construction of artificial polymers with a comparative degree of structural and compositional diversity/complexity similar to the biomacromolecules created by nature is a great challenge, especially for those prepared by chain-growth polymerizations. Since the development of some novel catalysts and polymerization technologies in the past decade, one-pot chain-growth polymerization strategies have been developed to polymerize structurally distinct monomers, as a result, producing block, multiblock, random, and gradient chain structures with diverse compositions. In this minireview, the recent progress on the synthesis of polymers with advanced chain structures via chain-growth polymerization has been summarized. The main part of this article is divided into three sections according to the different polymerization strategies: hybrid copolymerization, switchable polymerization, and cascade polymerization. It is anticipated that this minireview will provide a unique view for the fabrication of polymers with a high degree of structural and compositional diversity/complexity.
 Guang Chen | Guang Chen received his B.S. degree in polymer science and engineering from the University of Science and Technology of China in 2016. Currently, he is a PhD student at the University of Science and Technology of China under the supervision of Prof. Ye-Zi You. His research focuses on photopolymerization and photocatalysis. |
 Lei Xia | Lei Xia received his bachelor's degree from Hefei University of Technology, China (2015) and his Ph.D. from the University of Science and Technology of China (2020). He then joined Prof. Chunyan Hong's group at the University of Science and Technology of China as a postdoctoral fellow. His scientific interest includes the development of visible-light-induced (co)polymerization. |
 Fei Wang | Fei Wang is working as an associate chief physician at the Department of Neurosurgery, The First Affiliated Hospital of USTC. He obtained his Ph.D. degree from Capital Medical University in 2014. He became a postdoctoral researcher at the Division of Life Sciences and Medicine of USTC to study the application of nanomedicine in cancer treatment in 2019. His research interests include the development of polymeric materials and treatment of malignant glioma with functional materials. |
 Ze Zhang | Ze Zhang is currently an associate research fellow at the University of Science and Technology of China. He received his B.Sc. (2012) and Ph.D. (2017) from Anhui University and the University of Science and Technology of China, respectively. He then worked in Prof. Chun-Yan Hong's group as a postdoctoral fellow from July 2017 to January 2020. His scientific interest includes the development of novel polymerization methods. |
 Ye-Zi You | Ye-Zi You is currently a professor of polymer chemistry at the University of Science and Technology of China. He received his B.Sc. (1996) and Ph.D. (2003) from Hefei University of Technology and the University of Science and Technology of China, respectively. He then worked as a visiting scholar and postdoctoral fellow at the Tokyo Institute of Technology and Wayne State University, respectively. He joined the University of Science and Technology of China in 2008. His scientific interests include the controlled release of polymer nanomedicine and tumor treatment. |
Introduction
Synthetic polymers have much extended diversity/complexity of compositions and chain structures, benefiting from the continuous development of polymerization methods and chemical reactions in the past century. As a result, over 300 M tons of synthetic polymers are produced every year for applications ranging from commodity to specialty products with ever-increasing social benefits.1 However, considering that nature has created complex but precise biomacromolecules for millions of years,2 we believe that the progress made on synthetic polymers is still in its infancy.3,4 The construction of artificial polymers with a high degree of structural and compositional diversity/complexity via simple synthetic approaches is a persistent pursuit for polymer chemists. Recently, multicomponent polymerization, segmer assembly polymerization and other methods could contribute to design periodic chain structures with sufficient diversity/complexity and interesting properties during step-growth polymerizations,5–11 but broadening the diversity/complexity of chain structures and compositions during chain-growth polymerizations is still a great challenge.Unlike step-growth polymerizations, a chain-growth polymerization often proceeds via a specific propagation species, such as a radical, anion, or cation, using an appropriate initiator or catalyst, thus producing the desired polymer chain through repetitive reactions between the propagation species and suitable monomers. Generally, different types of monomers have different polymerization mechanisms, catalysts and propagation species.12 For example, vinyl monomers are generally polymerized by a radical or ion vinyl-addition mechanism while cyclic monomers undergo a ring-opening mechanism. Among the cyclic monomers, lactones are generally polymerized via an anionic mechanism while tetrahydrofuran can only be polymerized via a cationic mechanism. Among the vinyl monomers, ethylene can be polymerized via radical or coordination processes; however, isobutylene can only be polymerized via a cationic process. The restricted match between monomer structures and polymerization processes resulted in the fact that the complexity/diversity of chain structures and compositions produced from a single chain-growth polymerization, such as addition polymerization, ring-opening polymerization, metathesis polymerization, and coordination polymerization, was very limited.
One possible strategy to lessen this inherent restriction is the presence of two or more distinct propagation species and polymerization mechanisms in a single chain formation process, either concurrent propagation species or the conversion of a propagation species into another one during the polymerization. This alteration would enable the synthesis of novel polymers with advanced chain structures that cannot be easily accessible using a single chain-growth polymerization mechanism. However, different propagation species may be incompatible and may interfere with each other when they coexist in a system. The conversion of a propagation species into another one is also generally disfavored. Excitingly, since some novel catalysts and polymerization technologies have been explored, gradually increasing reports highlight the one-pot strategies to polymerize structurally distinct monomers in the presence of two or more distinct propagation species and polymerization mechanisms, as a result, forming block, multiblock, random, and gradient chain structures with diverse compositions.13 On the other hand, besides the above-mentioned various chain structures, introducing organic cascade reactions in chain propagating processes could produce novel polymers with complex repeat unit structures.14,15 We try to summarize these strategies into three categories: hybridization of control/living polymerizations, switchable polymerization, and cascade polymerization. Therefore, the aim of this minireview is to clearly overview the recent progress in the construction of polymers with advanced chain structures via hybrid copolymerization, switchable polymerization, and cascade polymerization strategies, especially regarding the design principles, mechanistic analyses and polymer structures. Since this contribution is not a comprehensive review, we attempted to include only the most typical scientific reports; thus, the polymerization strategies that only contribute to simple chain structures are carefully excluded. On the other hand, some seminal works on the combination of chain-growth and step-growth polymerizations are also highlighted.
Hybrid copolymerization
The development of novel catalysts and polymerization technologies has broken the barriers of various polymerization mechanisms, resulting in the fact that some distinct control/living polymerization mechanisms could be involved in a single polymerization process concurrently. During these hybridization systems of control/living polymerizations, the different propagation species can coexist and, in most cases, can interconvert into each other many times. As a result, the monomer selectivity can be constantly changed during the polymerization process to synthesize polymers with more complex chain structures.Hybrid addition/ring-opening copolymerization
Zhang et al. developed a hybridization system of anionic addition and anionic ring-opening polymerizations. Under the catalysis of the superbase t-BuP4, polymerization of methyl methacrylate and that of ε-caprolactone could proceed concurrently.16 They found that t-BuP4 could initiate anionic addition polymerization of MMA. During the polymerization of MMA, the carbon anion could alternatively attack ε-caprolactone in a nucleophilic manner. Thus, the system is interconverted to ring-opening polymerization of ε-caprolactone. The oxygen anion during ring-opening polymerization could also alternatively attack MMA. As a result, anionic addition polymerization of MMA and anionic ring-opening polymerization of ε-caprolactone could interconvert into each other catalyzed by t-BuP4 to produce an interesting copolymer with a random chain structure (Fig. 1). For a single propagating chain, the anionic addition propagation of MMA and anionic ring-opening propagation of ε-caprolactone proceeded alternatively, while for the whole polymerization system, the anionic addition polymerization and anionic ring-opening polymerization proceeded concurrently. Further studies have demonstrated that this hybrid copolymerization system is suitable for several different monomer pairs, such as lactide or cyclic carbonate ester with methyl acrylate, and CL with butyl acrylate.17,18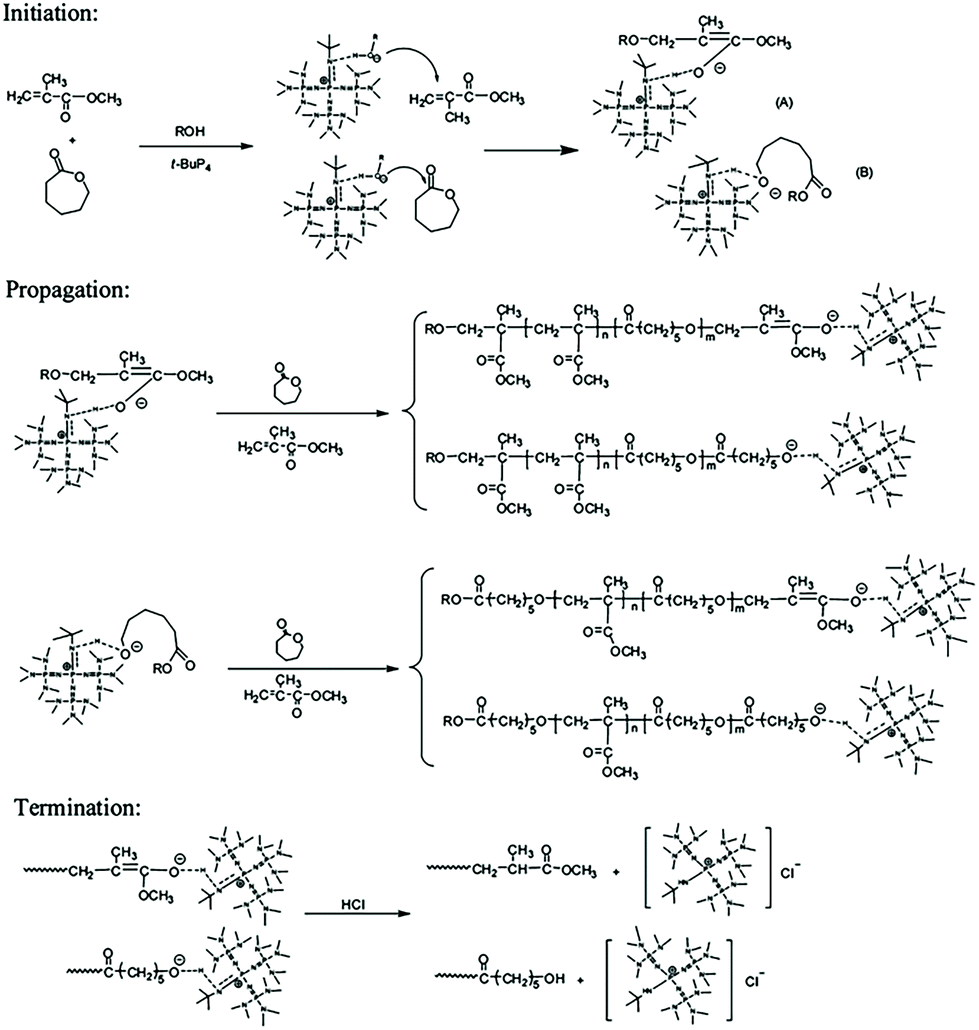 | ||
| Fig. 1 Hybridization of anionic addition polymerization of MMA and anionic ring-opening polymerization of ε-caprolactone. Reproduced from ref. 16 with permission from the American Chemical Society, copyright 2012. | ||
Besides the anionic hybrid copolymerization, the cationic hybrid systems between cationic addition polymerization of vinyl monomers and cationic ring-opening polymerization of alkylene oxide have also been developed. Aoshima et al. first reported the concurrent copolymerization of vinyl ether and alkylene oxide (Fig. 2). Generally, the cationic copolymerization of vinyl monomers and alkylene oxide is very difficult since a vinyl group cannot nucleophilically attack a stable oxonium ion and an oxonium ion is unfavorable to be transformed into a ring-opened carbocation for addition to a vinyl monomer. The key to reaching the crossover between cationic vinyl-addition polymerization of vinyl ether and cationic ring-opening polymerization of alkylene oxide is the rational design of monomer structures and initiation systems. In the copolymerization of isopropyl vinyl ether (IPVE) and isobutylene oxide (IBO), the strong Lewis acid catalyst B(C6F5)3 may form a weakly coordinating counteranion at the IBO-derived growing chain end. The IBO-derived end thus showed a moderate crossover to IPVE from a direct addition reaction between IPVE and the IBO carbocation or an addition after carbocation isomerization, resulting in favorable copolymerization to obtain multiblock-like poly(IBO-co-IPVE)s with several blocks. It is worth noting that although the copolymers obtained under the optimized reaction conditions had relatively controlled molecular weights, the molecular weight distributions were generally broad (Đ > 1.6).19 The authors further expanded the monomer and catalyst libraries for this novel cationic hybrid copolymerization system, such as styrene and cyclic acetal.20,21
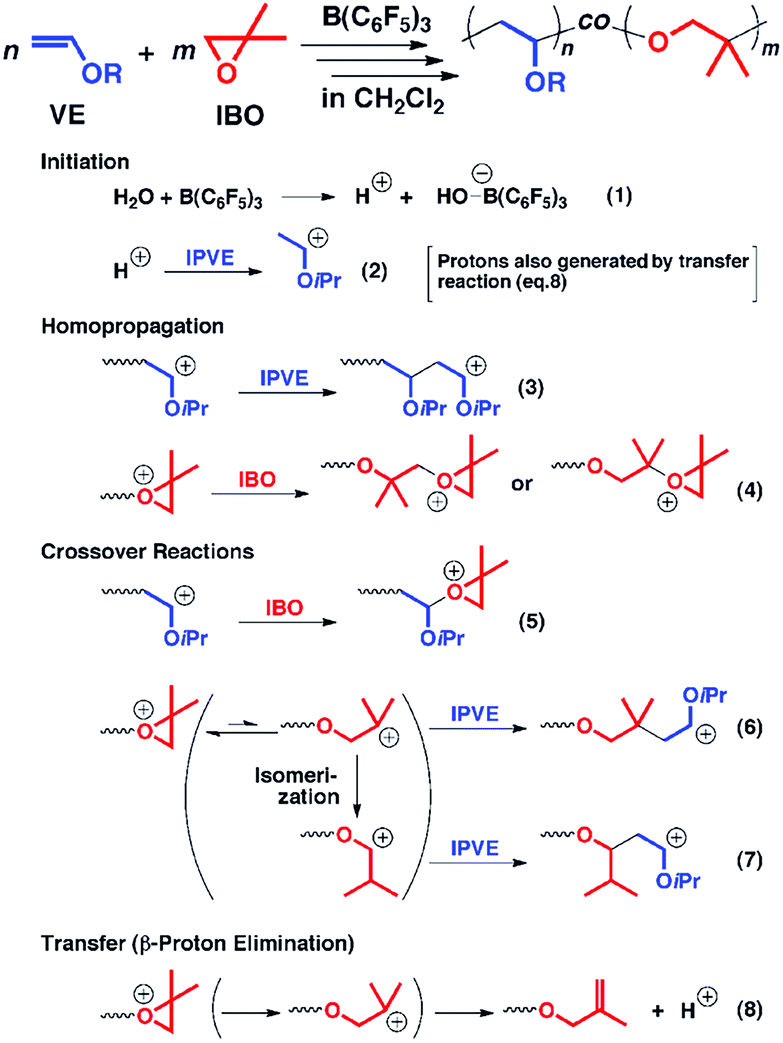 | ||
| Fig. 2 Hybridization of cationic addition polymerization of vinyl ether and cationic ring-opening polymerization of alkylene oxide. Reproduced from ref. 19 with permission from the American Chemical Society, copyright 2013. | ||
Barner-Kowollik, Boydston and coworkers introduced a visible-light-induced hybridization of radical addition polymerization of MMA and ROMP of ring-strained cyclic olefins. In this system, radical polymerization of MMA and ROMP of norbornene could proceed concurrently. During chain formation, propagation could interconvert between ROMP of norbornene and radical polymerization of MMA using 2,4,6-tris(4-methoxyphenyl) pyrylium tetrafluoroborate (MeOTPP+BF4−) as the photocatalyst22 (Fig. 3). When using ethyl-1-propenyl ether as the initiator, the polymerization commences with the initiation of norbornene and subsequent ROMP under visible light irradiation. Then, a metathesis of MMA with the active chain end and the release of an α-vinyl ether result in an intermediate that can initiate the radical propagation of MMA. Thus, the propagation interconverted from ROMP to a radical process. The released α-vinyl ether does not initiate new chains but can perform nucleophilic addition to the radical cation of the active chain end, leading to the regeneration of a vinyl ether end group allowing for further ROMP of norbornene. As a result, random copolymers of poly(norbornene-co-MMA) containing a high proportion of the norbornene repeating unit were formed.
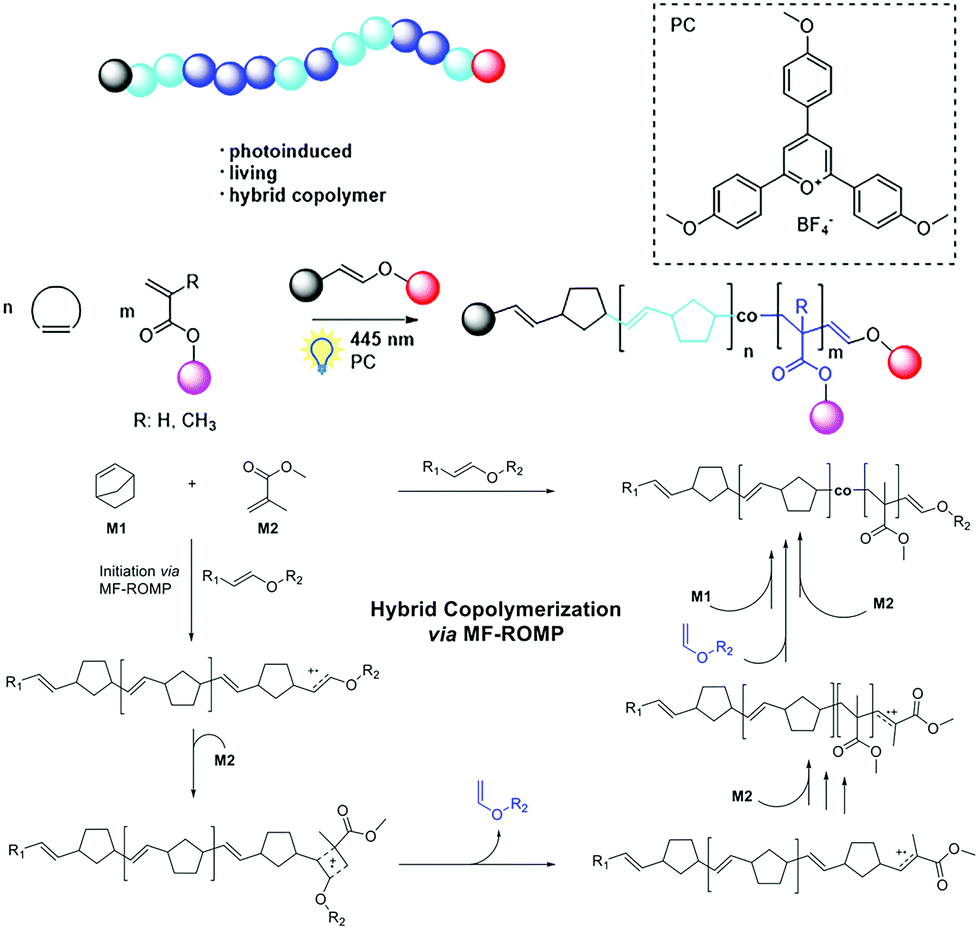 | ||
| Fig. 3 Visible-light-induced hybridization of radical addition polymerization of MMA and ROMP of ring-strained cyclic olefins. Reproduced from ref. 22 with permission from the American Chemical Society, copyright 2019. | ||
Although the above-mentioned hybrid polymerization strategies can realize the copolymerization of many distinct monomers to prepare various polymers with complex chain structures, the interconversion behavior of these hybrid systems and the molecular weight and Đ value of most of the prepared polymers are uncontrollable. You and Zhang developed the hybridization of radical addition polymerization of acrylamide monomers and anionic ring-opening polymerization of episulfides. Besides being involved in radical RAFT polymerization, it was found that trithiocarbonate could also act as the initiator for the anionic ring-opening polymerization (AROP) of thiirane monomers in the presence of a quaternary onium salt.23 Thus, new interconvertible RAFT polymerization and AROP had been achieved, in which the trithiocarbonate acts as both a chain transfer agent for radical RAFT polymerization of vinyl monomers and an initiator of AROP simultaneously and independently. The interconversion of radical polymerization and AROP can be achieved (Fig. 4A). The driving force for this interesting crossover propagation was the chain transfer process of RAFT (Fig. 4B). As a result, the copolymers with diblock, triblock, multiblock and gradient chain structures could be produced by adjusting the concentration ratio of the initiator, catalyst and monomers. This hybrid system has a moderate monomer and trithiocarbonate scope and the resulting copolymers have controlled molecular weights and narrow Đ values.24 It is worth noting that the prepared copolymers contain both vinyl segments and thioether segments, which are very difficult to obtain by any other methods. The interconversion times during this novel hybrid polymerization reached up to ∼5 and the copolymerization kinetics was easily affected by many factors.
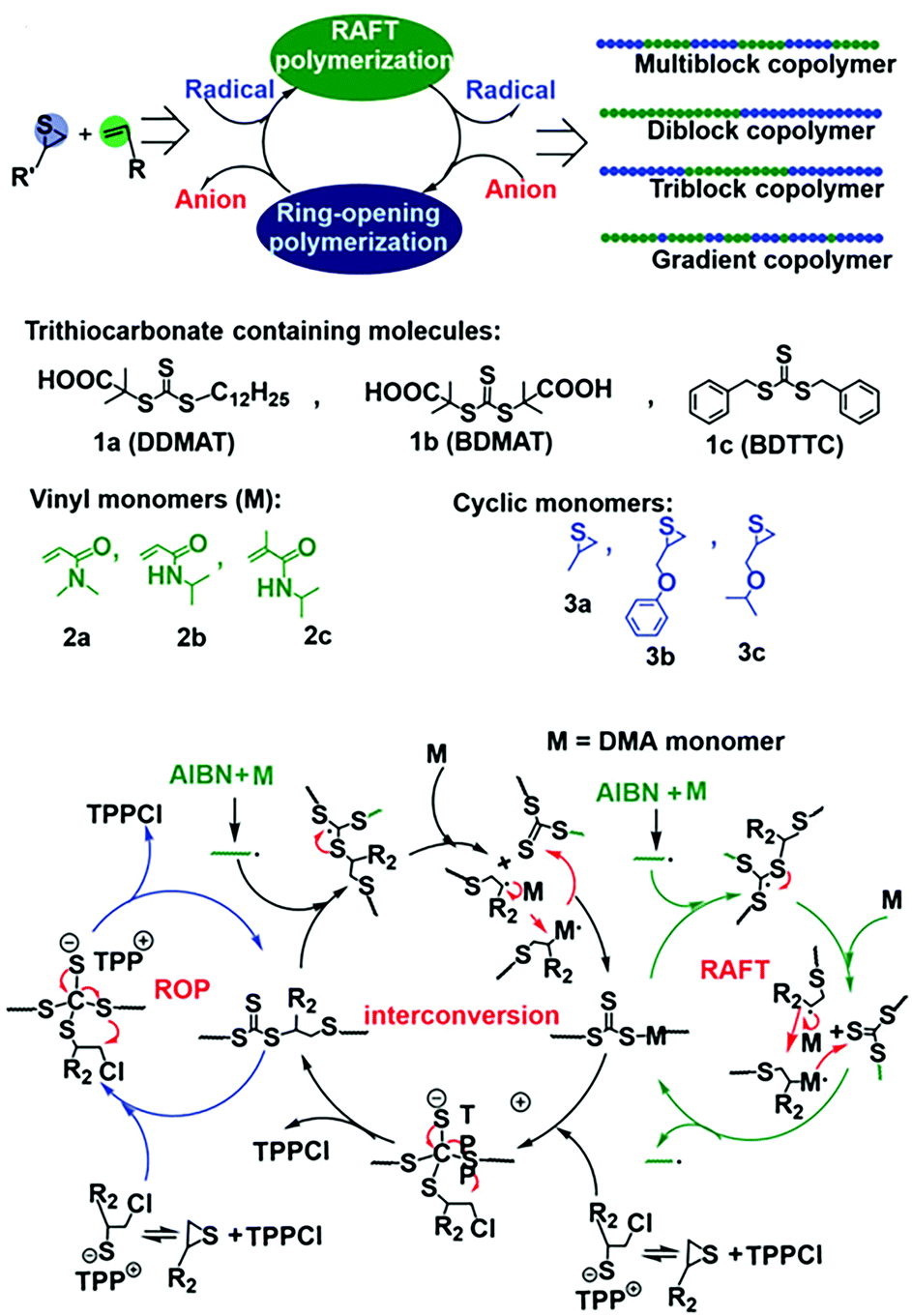 | ||
| Fig. 4 Interconvertible hybrid copolymerizations of AROP and RAFT. Reproduced from ref. 23 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Hybrid radical/anionic copolymerization
The copolymerization of acrylate and vinyl ether via the radical mechanism could only produce acrylate-dominant polymers due to the huge activity difference between these two kinds of monomers. On the other hand, the copolymerization of acrylate and vinyl ether via the cationic mechanism could only produce a homopolymer of vinyl ether. No efficient method can synthesize poly(acrylate-co-vinyl ether) with tunable compositions until Kamigaito, Satoh, et al. reported an interconvertible controlled/living radical and cationic hybrid copolymerization through reversible activation of common dormant species in 2014.12 Trithiocarbonate and dithiocarbamate can act as effective chain transfer agents for RAFT radical polymerization of various vinyl monomers. Also, the authors found that they could also be activated by Lewis acids to generate carbocation species to control the cationic polymerization of vinyl ether via a similar process to that in RAFT radical polymerization. They carefully designed the structures of trithiocarbonate or dithiocarbamate to make sure that these two mechanisms can be compatible with each other. A radical initiator and a Lewis acid were used to activate the common trithiocarbonate or dithiocarbamate into radical and cationic species. The radical propagating chain of acrylate was dormant after transferring onto one chain transfer agent and can alternatively convert to a cationic propagating species for further cationic polymerization of vinyl ether. Similarly, the cationic propagating chain of vinyl ether was dormant after transferring onto one chain transfer agent and can alternatively convert to a radical propagating species for further polymerization of acrylate. Therefore, this interconvertible radical and cationic copolymerization of acrylate and vinyl ether produced a copolymer chain that consists of radically and cationically polymerized segments (Fig. 5). In their research, the interconversion times could reach up to ∼8; as a result, multiblock poly(acrylate-co-vinyl ether)s were obtained in one shot with controlled molecular weights and narrow Đ values (<1.4).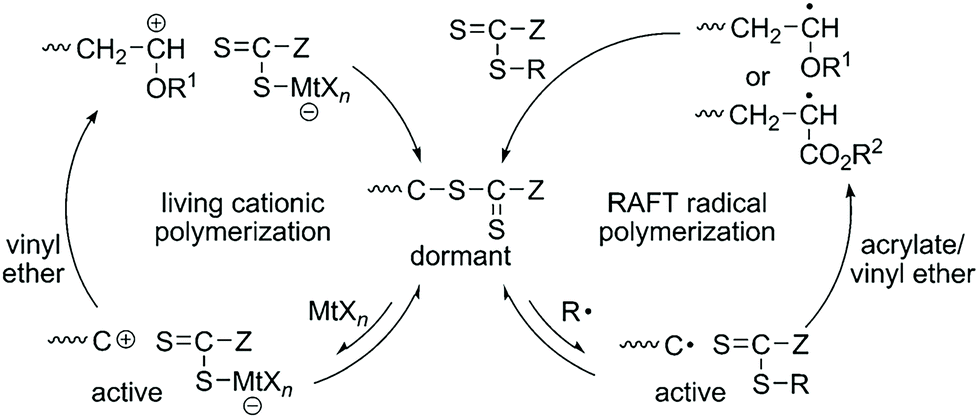 | ||
| Fig. 5 Hybridization of radical and cationic addition polymerizations through reversible activation of common dormant species. Reproduced from ref. 12 with permission from John Wiley and Sons, copyright 2014. | ||
Hybrid cationic/anionic ring-opening copolymerization
Generally, the coexistence of cationic and anionic propagation species in a single polymerization system is very difficult since they react with each other to produce dead products. In 2014, Ling et al. provided a concept of “Janus polymerization”, which refers to polymer chain propagation on both ends by cationic and anionic polymerizations simultaneously.25 In the presence of lutetium triflate/propylene oxide (Lu (OTf)3/PO) and two monomers, ε-caprolactone (CL) and tetrahydrofuran (THF), THF nucleophilically attacked the compound Lu (OTf)3/PO, generating two ends containing alkyltetrahydrofuranium and the coordinated structure of Lu (OTf)3 with alkoxide respectively. The former can initiate the cationic ROP of THF and CL, and the latter can solely initiate the anionic ROP of CL (Fig. 6). Most importantly, the cationic ROP of THF and the anionic ROP of CL have no interference up to 90% of CL conversion. This copolymerizaton kept a living manner, producing diblock copolymers of PCL-b-P(THF-co-CL) with narrow Đ values (<1.2). It is interesting that a step-growth polymerization affords a multiblock polymer of (PCL-b-P(THF-co-CL))m by an intermolecular coupling reaction of two chain ends after the complete conversion of CL. Additionally, this strategy also provided a facile one-step approach to synthesize copolymers with advanced structures like 3-miktoarm star terpolymers and branched polymers.26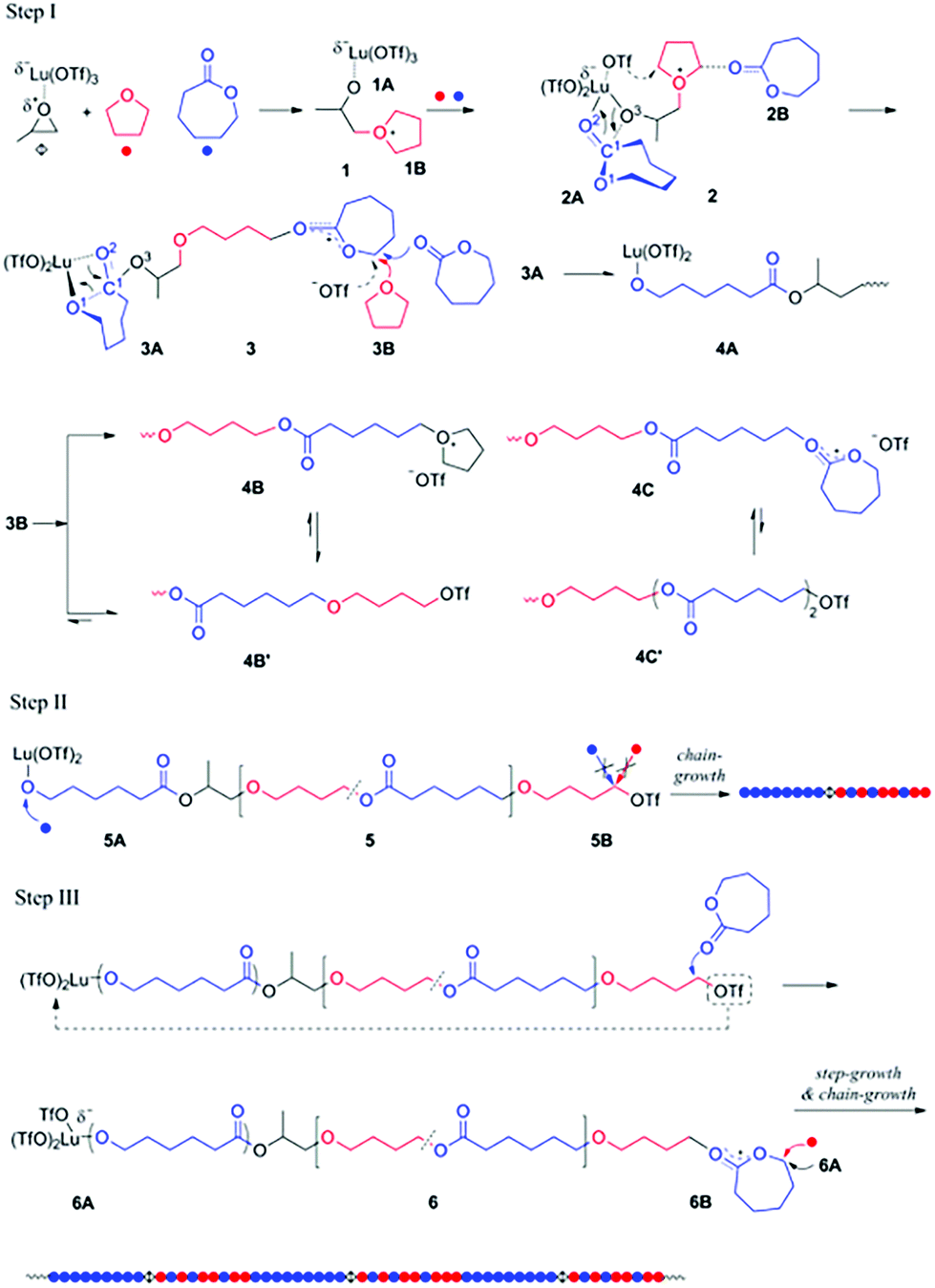 | ||
| Fig. 6 Hybridization of cationic and anionic copolymerizations catalyzed by Lu (OTf)3/PO. Reproduced from ref. 26 with permission from the American Chemical Society, copyright 2014. | ||
Hybrid chain/step-growth copolymerization
Besides the hybridization of different chain-growth polymerizations, in 2018, Jiang and Yang et al. found that a weaker superbase, t-BuP2, could rapidly catalyze the oxa-Michael addition polymerizations of diacrylates and diols via attacking the double bonds of acrylate with the alkoxide at room temperature. By combining this polymerization with t-BuP2 catalyzed anionic ring-opening polymerization of lactone initiated by alkoxide, they reported a hybrid polymerization of either ethylene glycol, neopentyl glycol diacrylate and CL or 2-hydroxyethyl acrylate and CL by concurrent oxa-Michael addition and ROP to synthesize copolymers with good hydrophilicity and degradability.27As a well-known click reaction, the thiol–ene reaction is robust in the synthesis of anti-Markovnikov adduct polymers via step-growth polymerization under radical conditions. Alternatively, Kamigaito et al. found that this thiol–ene reaction can be quantitatively and efficiently induced by organic acids, such as p-toluenesulfonic acid (PTSA) as the Markovnikov adduct mechanism (cationic process), and can be applied to synthesize novel polymers. Therefore, the thiol–ene click polymerization of dithiol and divinyl ether would produce poly(thioether) by the radical process but led to poly(thioacetal) by the cationic process.28 Thus, they developed a hybrid step-growth polymerization in which the two polymerizations could occur concurrently and construct copolymer structures consisting of both thioacetal and thioether linkages. Interestingly, some products like 16- and 18-membered cyclic thioacetals and thioethers obtained under high-dilution conditions by this system, having a similar structure to crown ether, can be used as host or ligand molecules.
Switchable polymerization
Switchable polymerization allows the consecutive formation of two or more distinct chain blocks via the controlled switch of one propagating mechanism to others triggered by external stimuli or the depletion of one of the monomers. In a hybridization system, different polymerization mechanisms and propagating species are involved concurrently; their interconversion was realized by an internal kinetic process. However, in a switchable polymerization system, the switch of the propagating mechanism and the monomer selectivity was controlled by external stimuli or the depletion of one monomer. This means that there is only one propagating mechanism involved at any moment of the polymerization system (before or after applying external stimuli), and different polymerization mechanisms are implemented sequentially rather than concurrently. As a result, the compositions, molecular weights and chain structures of polymers via switchable polymerizations could be regulated more precisely than those in hybridization systems. Switchable polymerization has been excellently summarized by Bielawski at 2016.29 In this minireview, we tried to highlight some representative studies, especially very recent reports.Switchable polymerization triggered by external stimuli
Although α-olefins and polar vinyl compounds are all alkene monomers, controlled polymerization of α-olefins primarily relies on coordination–insertion polymerization as radicals and other intermediates are not stabilized enough. In contrast, polymerization of polar monomers is difficult to achieve via a coordination–insertion method due to the poisoning of the catalysts induced by the interaction with polar monomers.30 Polar monomers are usually polymerized through radical, cationic, anionic and ring-opening metathesis polymerization. A switchable polymerization strategy has the potential to switch the mechanism between coordination–insertion polymerization and other methods to produce complex polymer chain architectures of both monomer classes. Harth, Beezer, and coworkers have reported a novel switchable polymerization strategy, in which coordination–insertion polymerization of hexene switched to radical polymerization of methacrylate in one shot under visible light irradiation31 (Fig. 7). The key to carrying out this strategy is the design and use of a cationic palladium diimine catalyst. Under darkness, the catalyst performed the insertion polymerization of hexene. Visible light irradiation resulted in metal–carbon bond homolysis of the catalyst to generate a carbon radical in situ so as to facilitate radical propagation. Thus, in a mixture of hexene and methacrylate, the polymerization was started from the coordination–insertion polymerization of hexene to the radical polymerization of methacrylate only using blue light irradiation to trigger the conversion. As a result, a large variety of diblocks were synthesized with molecular weights up to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1 and Đ < 1.6. This switchable copolymerization approach contributes to controllably prepare block copolymers of α-olefin blocks and polar olefin blocks in a one-shot process without the need for multiple steps and purification.
400 g mol−1 and Đ < 1.6. This switchable copolymerization approach contributes to controllably prepare block copolymers of α-olefin blocks and polar olefin blocks in a one-shot process without the need for multiple steps and purification.
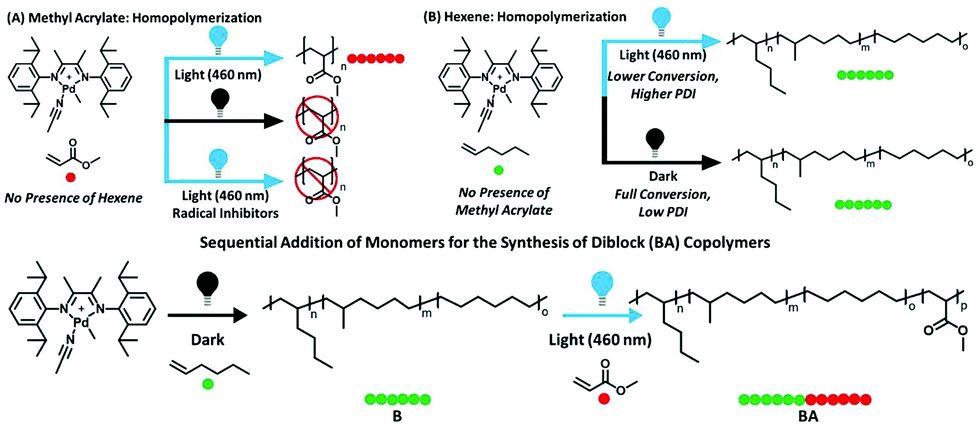 | ||
| Fig. 7 The switchable copolymerization converting from coordination–insertion polymerization of hexene to radical polymerization of methacrylate in one shot under visible light irradiation. Reproduced from ref. 31 with permission from the American Chemical Society, copyright 2018. | ||
In the above report of Kamigaito and Satoh, the interconversion behavior between the radical and cationic mechanisms during the hybridization of radical and cationic addition polymerization systems cannot be controlled artificially. Fors and coworkers found that an excited photocatalyst (2,4,6-tris(p-methoxyphenyl)pyrylium tetrafluoroborate) under green light could oxidize a special trithiocarbonate, resulting in a carbocation for cationic RAFT polymerization.32 On the other hand, using Ir(ppy)3 as a photocatalyst could regulate radical RAFT polymerization of MA under blue light with an ON/OFF switch (PET-RAFT). Therefore, the authors demonstrated the effective control of the switch behavior by simply changing the wavelength of stimulus light to selectively polymerize specific monomers by the radical or cationic mechanism so as to produce radically and cationically polymerized segments on demand (Fig. 8A).33 Thereafter, they found that this cationic RAFT polymerization of vinyl ethers could be activated by the addition of ferrocenium salts (FcX) to oxidize the trithiocarbonate and be halted by the addition of a dithiocarbamate anion to reduce FcX and chain-end carbocation. Also, this temporal control over the initiation and reversible termination of polymerization could be achieved by the addition of ferrocenium salts and dithiocarbamate anions alternately and repeatedly.34 This chemical-induced cationic polymerization is orthogonal with visible-light-induced radical polymerization, and the two mechanisms could be switched with each other by modulating the corresponding stimulus (Fig. 8B).
 | ||
| Fig. 8 The switchable controlled/living radical and cationic copolymerizations with orthogonal stimuli. Reproduced from ref. 33 and 34 with permission from the American Chemical Society, copyright 2017 and 2018. | ||
Based on the above study, Fors et al. found that ferrocene can also be electrochemically oxidized into ferrocenium for cationic RAFT polymerization of IBVE. Additionally, this electrochemically controlled cationic polymerization could also be reversibly terminated by applying a cathodic current. By pairing this electrochemical cationic polymerization of IBVE with PET-RAFT polymerization of MA in one shot, a switchable copolymerization can also be performed.35 Yan and coworkers further reported dually electrochemical conversion of cationic and radical RAFT polymerizations.36 In the presence of 2,3-dichloro-5,6-dicyanodiphenol (DDQ2−) as a catalyst precursor, applying an external oxidizing potential (+1.2 V) could oxidize DDQ2 into DDQ, which could oxidize the chain transfer agent to give a carbocation for cationic polymerization of IBVE. Additionally, the polymerization would be halted when the applied potential was removed. On the other hand, the external reducing potential could reduce nicotinamide adenine dinucleotide (NAD+) to nicotinamide adenine dinucleotide (NADH) that could promote the reduction of a chain transfer agent, triggering the generation of radicals for the RAFT polymerization of MA. Thus, they constructed an electrochemically controlled copolymerization that enabled switching between living cationic and radical RAFT polymerizations by alternative electrochemical stimuli in one shot. Varying the period or phases of voltage stimulation enables control over the polymer chain structure, including diblock, multiblock, random, and tapered copolymers.
Zhao et al. constructed a controlled/living anionic ring-opening switchable copolymerization of epoxides and lactones using a biased Lewis pair as a metal-free catalytic system that consists of a Lewis acid, triethylborane (Et3B) and a Lewis base, phosphazene base (t-BuP2).37 This system allows selective anionic polymerization of either epoxides or lactones from mixed-monomer feedstock for the preparation of multiblock copolymers with both ether segments and ester segments as well as on-demand block number and length (Fig. 9). The key to achieving the monomer selectivity and effective switch was the amount of Et3B and t-BuP2. In detail, when t-BuP2 is used in excess relative to Et3B, the ROP of lactone is only selectively turned “on” due to poor nucleophilicity of the alkoxide capped by one equivalent of Et3B and the absence of excess Et3B, as well as higher base sensitivity of lactone than that of epoxide and appropriate basicity of t-BuP2. Then, upon adding an excess amount of Et3B relative to t-BuP2, the ROP of cyclic ester is turned “off” and the ROP of epoxide is turned “on” as a result of which the alkoxide capped by two equivalents of Et3B is the actual active center, which selectively reacts with epoxides. Also, the ROP of cyclic ester would be restarted, if a catalytic amount of t-BuP2 was further added. Therefore, the switch between polymerization of lactone and that of epoxide during this system could be easily regulated by the amount of Et3B and t-BuP2. The obtained multiblock copolymers, such as (PPO-b-PVL)n, have well-controlled and relatively high molecular weights and low distributions.
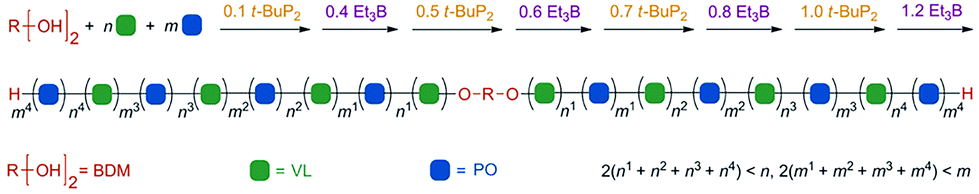 | ||
| Fig. 9 Illustration of one-pot synthesis of a multiblock copolymer from a mixture of VL and PO after several successive switches in selectivity. Reproduced from ref. 37 with permission from John Wiley and Sons, copyright 2019. | ||
You and Zhang developed the hybridization of radical addition polymerization of acrylamide monomers and anionic ring-opening polymerization of episulfides. The interconversion times during this hybrid polymerization could only reach up to ∼5 and the copolymerization kinetics was easily affected by many factors.24,38,39 To further regulate the switch behavior precisely, the authors introduced external stimuli to control this propagating process. They found that this AROP of thiiranes can proceed successfully only at high temperatures (>50 °C), while no monomer conversion was observed at lower temperatures (<20 °C). Therefore, the polymerization could be switched ON/OFF by varying the temperature. On the other hand, the PET-RAFT polymerization could be switched ON/OFF by light irradiation (Fig. 10). Therefore, a dually switchable and controlled interconvertible polymerization system can be constructed. In this system, PET-RAFT polymerization and AROP can be selectively switched ON/OFF independent of each other, and they can be interconverted artificially and promptly by changing the external stimuli so as to insert distinct monomers into the resulting copolymer chains temporally, allowing on-demand precise arrangement of chain structures in the resulting copolymers.40 By applying various well-designed programs of heating and irradiation, copolymers with common and advanced chain structures like multiblock copolymers (pentablock, heptablock, nonablock, and undecablock copolymers) and gradient copolymers with tailored compositions and chain length would be precisely produced. All the obtained complex copolymers have controlled molecular weights and narrow Đ values.
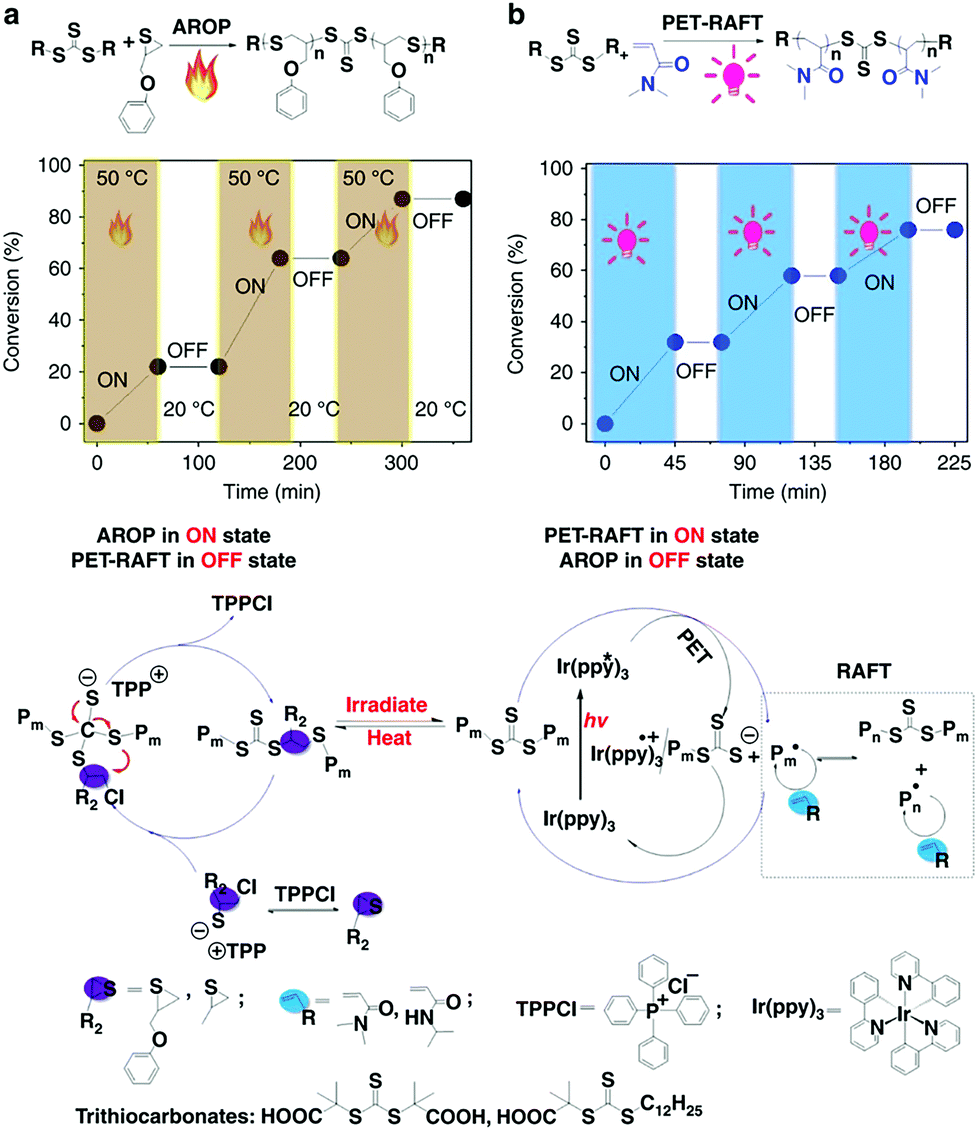 | ||
| Fig. 10 Dually switchable and controlled interconvertible copolymerizations of AROP and PET-RAFT switched ON/OFF and interconverted in response to stimuli. Reproduced from ref. 40 with permission from the Nature Publishing Group, copyright 2018. | ||
Controlled conversion between the polymerization of vinyl monomers and that of cyclic monomers has been further explored by Xie, Wang and coworkers. They reported two interesting works, in which a facile switch from radical polymerization of polar vinyl monomers to ROCOP of propylene oxide and the switch from ROCOP of propylene oxide to radical polymerization of polar vinyl monomers were successfully achieved41,42 (Fig. 11). The key to carrying out these two conversions is based on the CoIII(salen) catalyst. CoIII(salen) could serve as the Lewis acid to initiate ring-opening copolymerization of propylene oxide with CO2 upon activation with a Lewis base.43 Meanwhile, it can also control the radical polymerization of various vinyl monomers, such as acrylate, vinyl acetate and ethylene (OMRP).44,45 It seems that the conversion between ROCOP and radical polymerization could be conducted using a single CoIII(salen) compound, but the barrier is that the active species for ROCOP are difficult to generate from the (salen)CoIII–C–R chain end of radical polymerization and the active species for radical polymerization are also difficult to generate from the (salen)CoIII–O–R chain end of ROCOP. The authors found that a single oxygen atom can be inserted into (salen)CoIII–C–R to form (salen)CoIII–O–R using O2 as a trigger, thereby switching the active species for OMRP to the active species for ROCOP. Thus, the authors first used a Co compound to mediate the radical polymerization of VAc under the initiation of AIBN to obtain PVAc with the (salen)CoIII–C–R chain end. Then, after treating with O2, the formed (salen)CoIII–O–R chain end further initiated the copolymerization of propylene oxide with CO2 to produce diblock PVAc-b-PPC in one pot. The diblock copolymers possessed well-defined structures, controlled molecular weights and low dispersities (<1.2). Furthermore, the PVAc-b-PMA-b-PPC triblock copolymer could also be successfully produced. Very excitingly, the authors further expanded the Co-based switchable copolymerization from the opposite reaction direction. They found that a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group can be inserted into (salen)CoIII–O–R to form (salen)CoIII–C–(CO)–R using CO gas as a trigger, thereby switching the active species for ROCOP to the active species for OMRP. The author synthesized poly(propylene phthalate) (PPE) with the complete alternation structure of epoxides and anhydrides and then converted (salen)CoIII–O–R to form (salen)CoIII–C–(CO)–R completely by CO insertion. The (salen)CoIII–C–(CO)–R chain end further acted as a successful OMRP photoinitiator for the OMRP of methacrylate. As a result, a series of diblock PPE-b-PMA with tailored block lengths, controlled molecular weights and low dispersities (<1.2) could be easily prepared. It showed that on-demand monomer incorporation with the adjustment of CO and light could be used for the one-pot terpolymerization of epoxides, anhydrides and acrylates with a precise programmed chain structure. The importance of these results lies in the use of gas molecules as triggers to achieve switchable copolymerization between different types of monomers. Further advancement may focus on the design and production of multiblock copolymers using alternate O2 and CO treatment.
O group can be inserted into (salen)CoIII–O–R to form (salen)CoIII–C–(CO)–R using CO gas as a trigger, thereby switching the active species for ROCOP to the active species for OMRP. The author synthesized poly(propylene phthalate) (PPE) with the complete alternation structure of epoxides and anhydrides and then converted (salen)CoIII–O–R to form (salen)CoIII–C–(CO)–R completely by CO insertion. The (salen)CoIII–C–(CO)–R chain end further acted as a successful OMRP photoinitiator for the OMRP of methacrylate. As a result, a series of diblock PPE-b-PMA with tailored block lengths, controlled molecular weights and low dispersities (<1.2) could be easily prepared. It showed that on-demand monomer incorporation with the adjustment of CO and light could be used for the one-pot terpolymerization of epoxides, anhydrides and acrylates with a precise programmed chain structure. The importance of these results lies in the use of gas molecules as triggers to achieve switchable copolymerization between different types of monomers. Further advancement may focus on the design and production of multiblock copolymers using alternate O2 and CO treatment.
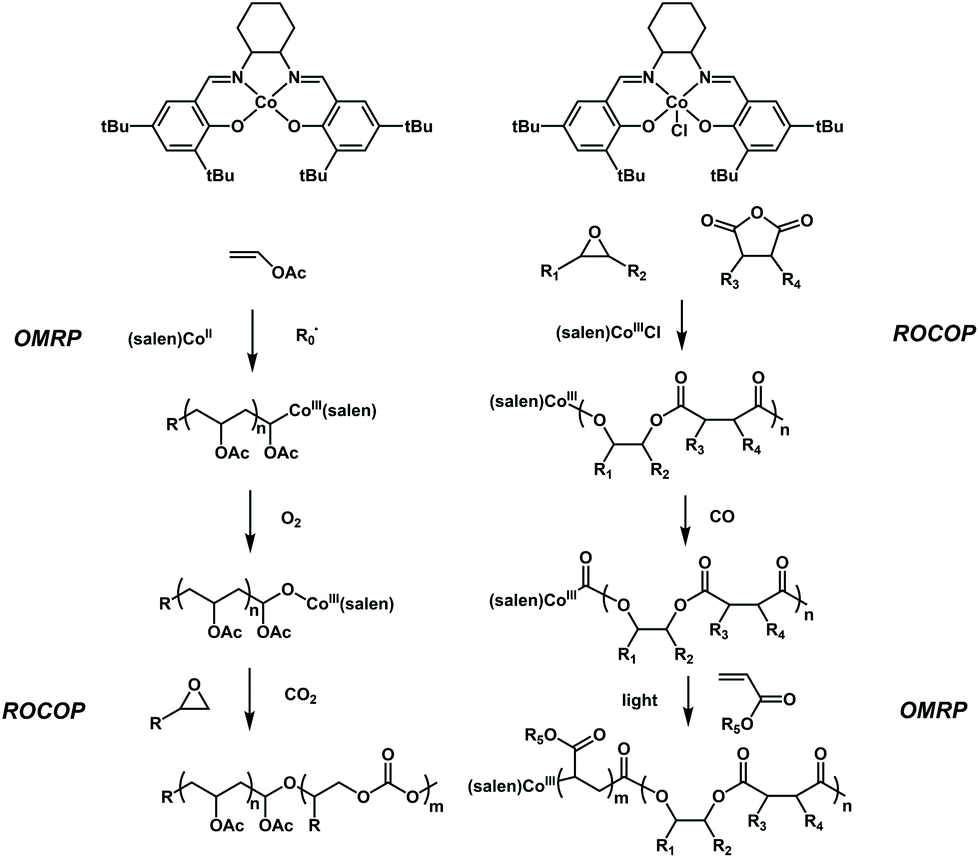 | ||
| Fig. 11 (A) O2-triggered copolymerization switched from cobalt-mediated OMRP to ROCOP for the synthesis of CO2-based diblock copolymers. (B) CO-triggered copolymerization switched from cobalt-mediated ROCOP to OMRP for the one-pot synthesis of polyester-b-polyacrylate. | ||
Switchable polymerization converting from chain-growth to step-growth polymerization could also be achieved. Zhao and coworkers designed the one-pot sequential ring-opening polymerization of ethylene oxide and step-growth polymerization of hydroxy and isocyanate by a base–acid catalyst switch, producing multiblock-like amphiphilic polyurethanes constituted by poly(ethylene oxide) and biosourced betulin for antifouling application (Fig. 12).46
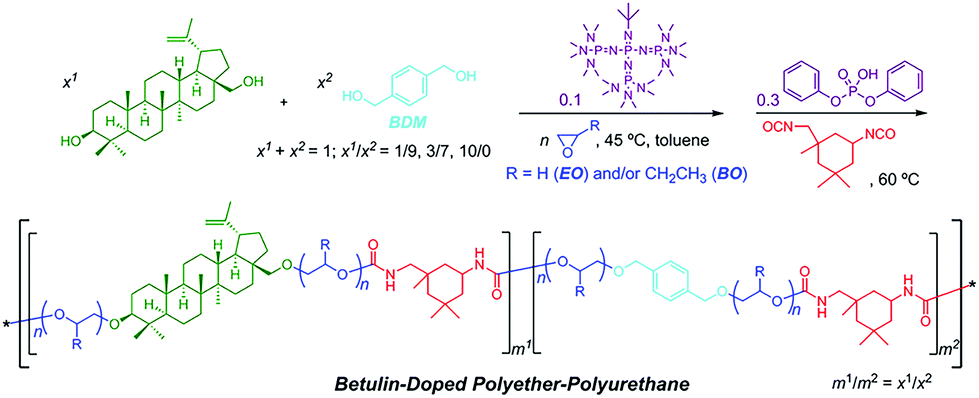 | ||
| Fig. 12 One-pot switchable ring-opening polymerization of ethylene oxide and step-growth polymerization of hydroxy and isocyanate. Reproduced from ref. 46 with permission from the American Chemical Society, copyright 2018. | ||
Self-switchable polymerization
Besides being triggered by external stimuli, switchable polymerization could also be performed by the depletion of one of the monomers. These polymerization systems are known as self-switchable polymerization in the previous literature. The ring-opening copolymerization (ROCOP) of epoxides with anhydrides produces polyesters with alternative epoxide/anhydride structures.47 The ROCOP of epoxides with CO2 could produce well-defined polycarbonates.48,49 Coates and coworkers reported the one-shot switchable copolymerization of an epoxide, anhydride and CO2 mixture, obtaining polyester-b-polycarbonate block copolymers by sequential ROCOP of epoxides/anhydrides and ROCOP of epoxides/CO2.50 The conversion from ROCOP of epoxides/anhydrides to ROCOP of epoxides/CO2 relied on the depletion of anhydride monomers. With a β-diiminate (bdi) zinc catalyst, insertion of anhydride into a zinc alkoxide intermediate is much faster than the insertion of CO2 (Fig. 13, ka ≫ kb), and insertion of the epoxide monomer is likely the rate-determining step for both polyester and polycarbonate formation. This feature promotes the polymerization system to only carry out ROCOP of epoxides/anhydrides before the anhydride is fully consumed. Once the anhydride is consumed, the polymerization mechanism will spontaneously change to ROCOP of epoxides/CO2. Williams et al. further developed the one-shot switchable copolymerization of epoxide, anhydride (CO2) and lactone using a dizinc catalyst, obtaining polyester (polycarbonate)-b-polyester block copolymers by sequential ROCOP of epoxides/anhydrides (CO2) and ROP of lactones51,52 (Fig. 14). The conversion from ROCOP of epoxides/anhydrides (CO2) to ROP of lactones also relied on the depletion of the anhydride (CO2) monomer. Insertion of anhydride (CO2) into a zinc alkoxide intermediate is much faster than the insertion of lactone, coupled with the inability of zinc carbonate or zinc ester to initiate ROP of lactone. Then, a series of metal catalysts, such as a heterodinuclear Zn(II)/Mg(II) catalyst, a [SalphenFAlCl] catalyst and a commercial Cr–salen catalyst [SalcyCrCl], were further developed to catalyze these copolymerizations.53–57 Despite the high activity and selectivity of these developed metal catalysts, the metal residual in the produced polymers are believed to have an adverse effect on polymer properties and degradation. Li and coworkers58 and Zhao and coworkers59 independently reported the organic base tBuP1 as the catalyst to mediate the switchable copolymerization, involving ROCOP of epoxides/anhydrides and ROP of lactide, to produce block copolyesters. The control ability of this organic catalyst is very close with that of metal-based catalysts, resulting in polymers with well-defined structures, designed DP, and narrow Đ (<1.2). By using multifunctional initiators, pentablock, three- and six-arm star block copolyesters have also been easily prepared. Besides the mechanism conversion from ROCOP of epoxides with anhydrides (CO2) to ROP of lactone, the switchable copolymerization between ROCOP of epoxides and anhydrides and ROP of epoxides can be achieved by both metal and organic catalysts and the switchable copolymerization between ROP of epoxide and ROP of lactide, resulting in polyester-b-polyether block copolymers.60–62 Furthermore, based on the above developments, one-shot sequential ROCOP of epoxides with anhydrides, ROP of lactone and ROP of epoxides via twice mechanism conversions could be achieved by using triethylborine and DBU pairs, resulting in polyester-b-polyester-b-polyether triblock copolymers.63 Both systems were controlled and chemoselective, and were especially free of transesterification. These methods simplify the preparation of multiblock polyester-based materials and is expected to be of value for the preparation of new types of thermoplastic elastomers and other materials.64–66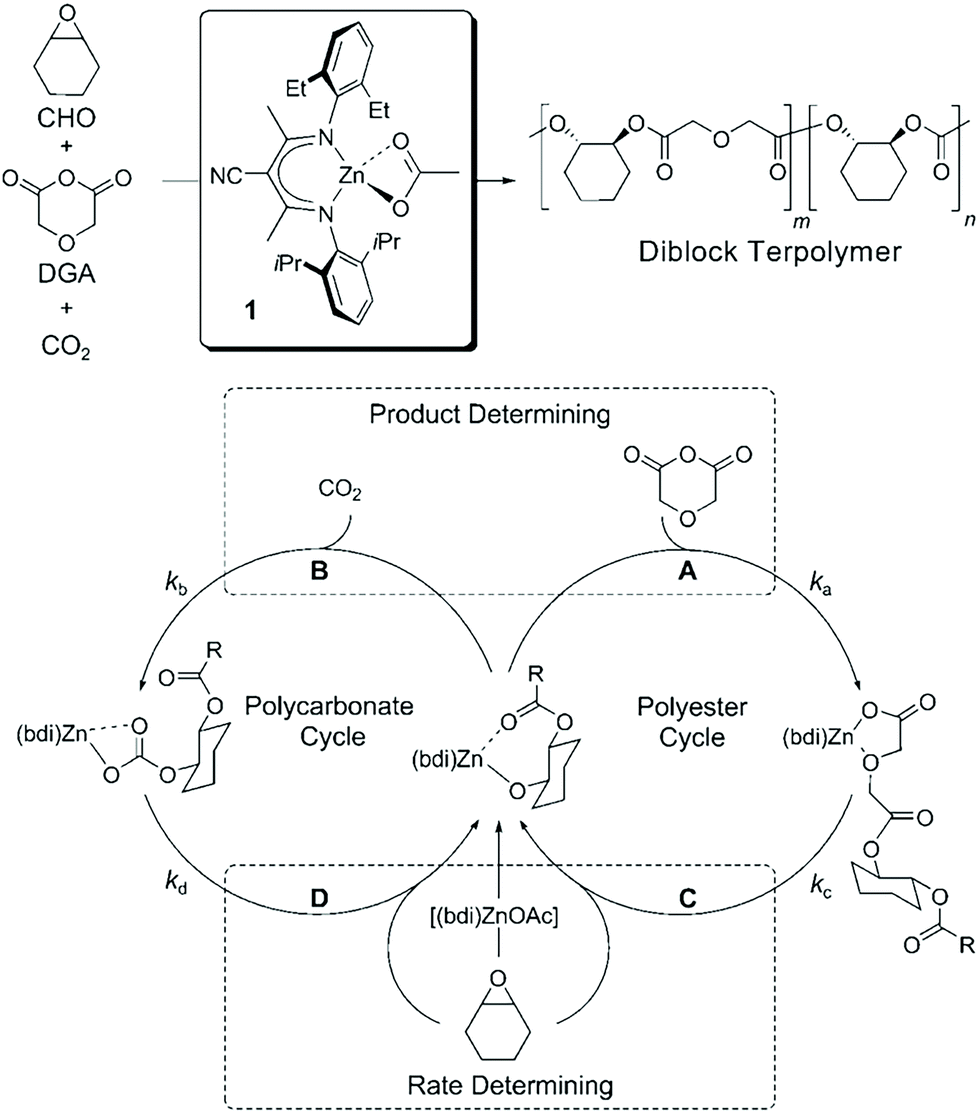 | ||
| Fig. 13 One-shot switchable copolymerization of an epoxide, anhydride and CO2 mixture, obtaining polyester-b-polycarbonate block copolymers by sequential ROCOP of epoxides/anhydrides and ROCOP of epoxides/CO2. Reproduced from ref. 50 with permission from John Wiley and Sons, copyright 2008. | ||
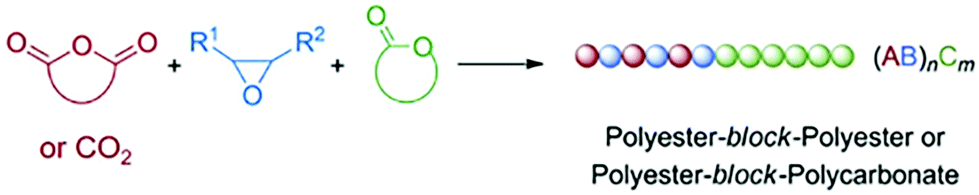 | ||
| Fig. 14 One-shot switchable copolymerization of epoxide, anhydride (CO2) and lactone using a dizinc catalyst, obtaining polyester (polycarbonate)-b-polyester block copolymers by sequential ROCOP of epoxides/anhydrides (CO2) and ROP of lactones. | ||
Cascade polymerization
Cascade polymerization is a polymerization in which the repeat unit is formed via an organic cascade reaction, as a result obtaining novel polymers with complex repeat unit structures. The chain-growth polymerization triggered by the cascade reaction is very important for the synthesis of polymers with complex repeat unit structures. The recent advances mainly focus on the radical and metathesis mechanisms.The radical polymerization of vinyl monomers produces various polymers with an all-carbon backbone, which means the absence of a functionality and which is not easy to degrade. Hawker et al. developed a radical-driven ring-opening cascade polymerization that allows the introduction of functional groups into the backbone to synthesize main chain degradable polymers.67 However, the propagating sulfur radicals generated by ring opening cannot be reversibly inactivated/controlled, resulting in low reactivity and poor control of polymerization. In 2018, Niu et al. developed a novel approach to access controlled radical-driven ring-opening cascade polymerization of low-strain macrocyclic monomers by introducing an allylic sulfone structure68 (Fig. 15A). During this radical cascade reaction, SO2 gas was extruded by β-elimination of alkyl sulfone and subsequent rapid α-fracture, thereby producing secondary alkyl radicals stabilized by adjacent carbonyl groups. Since the secondary alkyl radicals generated are similar to the chain growth radicals of acrylic monomers, the copolymerization of macrocyclic monomers with acrylate can proceed smoothly and be well controlled by performing the RAFT process. Then the authors further developed a radical-induced ring-closing/ring-opening cascade polymerization for more complex main chain structures69 (Fig. 15B). Incorporation of 1,6-diene fused allyl sulfone motifs into macrocyclic monomers could achieve cascade radical ring-closing/ring-opening processes, in which the addition of radicals to the terminal vinyl will promote five-membered cyclization of 1,6-diene and β-elimination of alkyl sulfone groups. In this process, with the opening of the macrocyclic monomer and the exhaust of SO2 gas, stable secondary carbon radicals are generated for chain growth. This ring-closing/ring-opening cascade polymerization strategy also exhibits controllable polymerization characteristics and could synthesize block polymers with low dispersities. During the polymerization process, the unsubstituted and unconjugated pendant vinyl group remains unreactive and the five-membered ring is formed in the backbone, which provides the possibility to synthesize polymers with more complex structures and degradability. All in all, the reported radical cascade reactions enable the copolymerization of vinyl monomers and designed macrocyclic monomers to degradable polymers with various functionalities in the backbone.
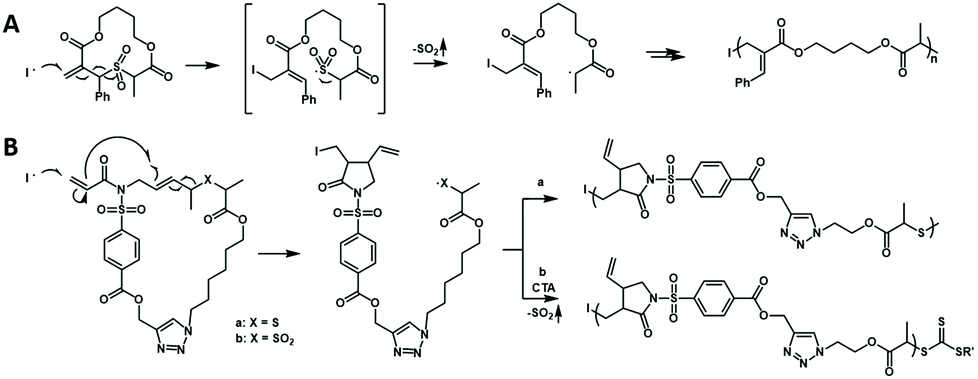 | ||
| Fig. 15 (A) Radical-driven ring-opening cascade polymerization enabled by the allylic sulfone trigger. (B) Radical-driven ring-closing/ring-opening cascade polymerization of macrocyclic monomers consisting of 1,6-diene-fused allylic sulfide and sulfone. | ||
Besides radical cascade reactions, the polymerization triggered by cascade metathesis reactions can also produce complex polymer structures, especially with unsaturated repeat units. Choi et al. developed a ring-opening/ring-closing cascade metathesis reaction by using a third-generation Grubbs catalyst to synthesize a polymer with many alkenyl groups and cycloolefins in the backbone (Fig. 16). In this cascade polymerization system, low-activity cyclohexene and terminal alkyne functional groups are integrated into one monomer to form enyne via a sulfonamide spacer. The author proposes that the Grubbs catalyst may preferentially react with terminal alkynyl groups to irreversibly form a diene. Then, the newly formed ruthenium carbene undergoes a ring-closing/ring-opening metathesis reaction with the neighboring cyclohexene. As a result, the synthesized polymer backbone has 1,3-diene units, which could be post-modified by the Diels–Alder reaction to synthesize polymers with more complex structures.70,71
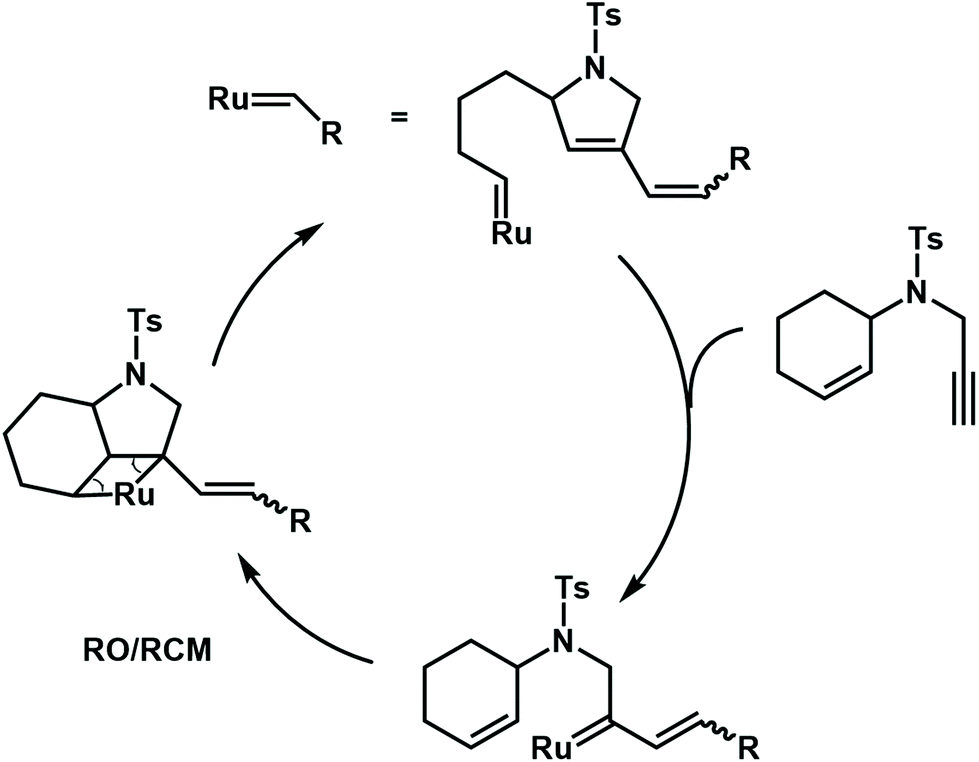 | ||
| Fig. 16 Mechanisms for cascade metathesis polymerization of monomers containing cyclohexene and a terminal alkyne. | ||
The authors further developed a new cascade polymerization containing three types of olefin metathesis transformations: ring-opening, ring-closing, and cross metathesis.72 By using the first-generation Grubbs catalyst, the monomer containing two cyclopentene moieties undergoes a ring-opening/ring-closing metathesis reaction to form a polymer with cyclopentene in the main chain. Then, the synthesized polymer was cross-metathesized with diacrylate using the second-generation Hoveyda–Grubbs catalyst to produce a completely alternating copolymer containing degradable groups (Fig. 17). Then, the authors further expanded the monomer scope of this cascade metathesis polymerization method and improved the polymerization efficiency through smart design of the monomer structure. Since cyclohexene has a lower strain energy than cyclopentene, by changing the position of the double bond in the monomer, the formation of cyclohexene after the cascade metathesis reaction could increase the polymerization efficiency. The use of these new monomers could efficiently and selectively carry out one-shot multiple olefin metathesis polymerizations to generate AB-alternating copolymers with minimum side reactions.
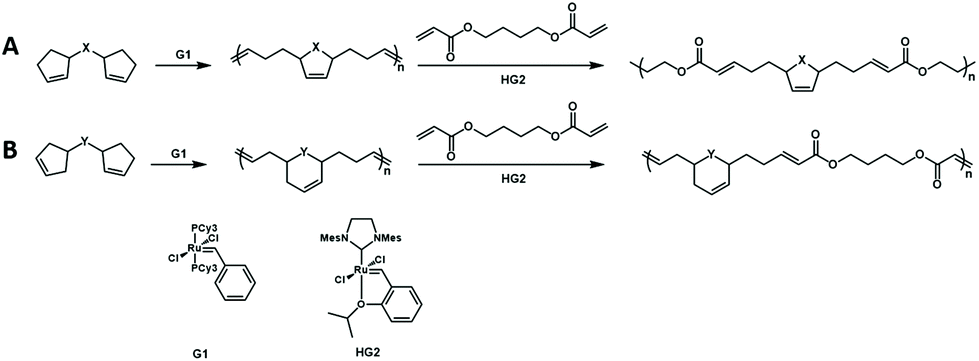 | ||
| Fig. 17 Cascade metathesis polymerization containing ring-opening, ring-closing, and cross metathesis. (A) Cyclopentene in the main chain. (B) Cyclohexene in the main chain. | ||
The cascade olefin metathesis and metallotropic 1,3-shift reactions were also used to synthesize unique polyenyne. A reasonable design of the monomer structure is very important for the successful realization of this cascade polymerization (Fig. 18). A symmetrical monomer, tetradeca-1,6,8,13-tetrayne, has an internal diacetylene structure that can successfully undergo cascade polymerization. The synthesized polymer has a special conjugated structure, including three alkenyl groups and one alkynyl group. By further tuning the monomer structure, it is possible to control the composition and sequence of alkenyl and alkynyl groups in the backbone.73,74 This strategy provides an opportunity to synthesize conjugated polymers with well-defined, complex and special electronic structures.
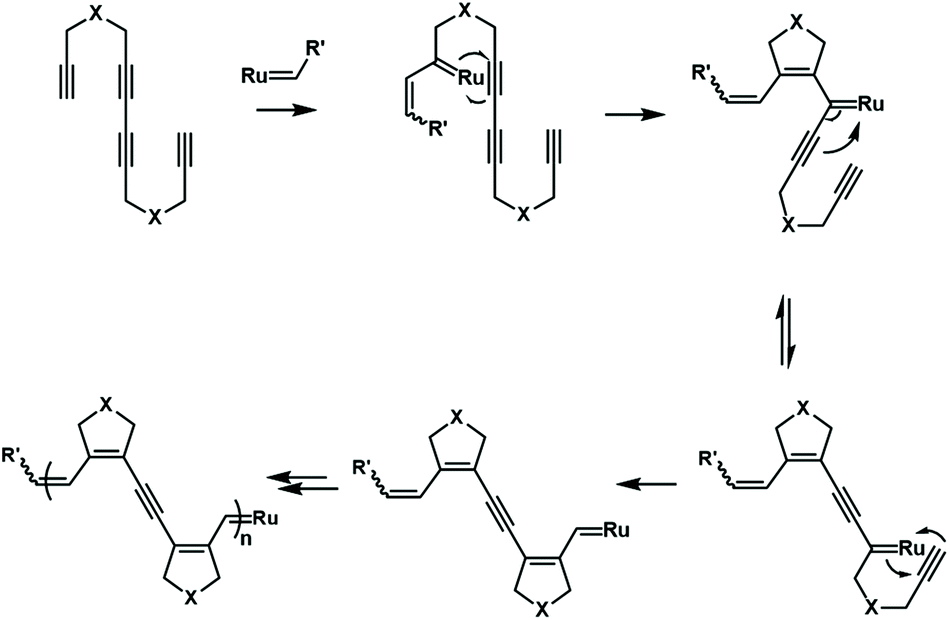 | ||
| Fig. 18 Cascade olefin metathesis/metallotropic 1,3-shift (M&M) polymerization. | ||
Besides polymerizations triggered by cascade radical reactions and metathesis reactions, those systems triggered by ionic, nucleophilic and coordination–insertion cascade reactions could also produce novel polymers with complex repeat unit architectures, which cannot be accessed by any other methods. These parts have been reviewed in several literature studies.14,15
Conclusions and outlook
In this minireview, we discussed the construction of polymers with a high degree of structural and compositional diversity/complexity via chain-growth polymerizations. Three main approaches, including hybrid copolymerization, switchable polymerization, and cascade polymerization, have been used to get rid of the inherent limitation of the single chain-growth polymerization process and polymerize structurally distinct monomers in the presence of two or more distinct propagation species and polymerization mechanisms, as a result, obtaining block, multiblock, random, and gradient chain structures with diverse compositions. Although significant advancements have been made, new and efficient methods are still needed for constructing novel polymers with sufficient diversity/complexity. Furthermore, the assembly, properties and functionalities of the produced polymers should still be explored and investigated to expand the existing applications and, more importantly, develop new and highly specialized materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial supports from the National Natural Science Foundation of China (21525420, 21801234, 22071232, 52073269) and the Fundamental Research Funds for the Central Universities (WK3450000005) are gratefully acknowledged.Notes and references
- D. J. Walsh, M. G. Hyatt, S. A. Miller and D. Guironnet, ACS Catal., 2019, 9, 11153–11188 Search PubMed.
- J. F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 6146 Search PubMed.
- J. F. Lutz, J. M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- R. B. Grubbs and R. H. Grubbs, Macromolecules, 2017, 50, 6979–6997 Search PubMed.
- J. Li, S. N. Rothstein, S. R. Little, H. M. Edenborn and T. Y. Meyer, J. Am. Chem. Soc., 2012, 134, 16352–16359 Search PubMed.
- T. Han, H. Q. Deng, Z. J. Qiu, Z. Zhao, H. K. Zhang, H. Zou, N. L. C. Leung, G. G. Shan, M. R. J. Elsegood, J. W. Y. Lam and B. Tang, J. Am. Chem. Soc., 2018, 140, 5588–5598 Search PubMed.
- T. F. Mao, G. Q. Liu, H. B. Wu, Y. Wei, Y. Z. Gou, J. Wang and L. Tao, J. Am. Chem. Soc., 2018, 140, 6865–6872 Search PubMed.
- Z. Zhang, Y. Z. You and C. Y. Hong, Macromol. Rapid Commun., 2018, 39, 1800362 Search PubMed.
- T. Tian, R. R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 6156–6163 Search PubMed.
- H. Kim, K. T. Bang, I. Choi, J. K. Lee and T. L. Choi, J. Am. Chem. Soc., 2016, 138, 8612–8622 Search PubMed.
- Z. Zhang, Y. Z. You, D. C. Wu and C. Y. Hong, Macromolecules, 2015, 48, 3414–3421 Search PubMed.
- H. Aoshima, M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2014, 53, 10932–10936 Search PubMed.
- Q. L. Song, S. Y. Hu, J. P. Zhao and G. Z. Zhang, Chin. J. Polym. Sci., 2017, 35, 581–601 Search PubMed.
- G. I. Peterson and T. L. Choi, Chem. Sci., 2020, 11, 4843–4854 Search PubMed.
- J. S. Yuan, W. Q. Wang, Z. F. Zhou and J. Niu, Macromolecules, 2020, 53, 5655–5673 Search PubMed.
- H. J. Yang, J. B. Xu, S. Pispas and G. Z. Zhang, Macromolecules, 2012, 45, 3312–3317 Search PubMed.
- H. J. Yang, J. B. Xu and G. Z. Zhang, Sci. China: Chem., 2013, 56, 1101–1104 Search PubMed.
- H. J. Yang, X. L. Qian, T. Bai, W. Y. Huang, X. Q. Xue and B. B. Jiang, Acta. Polym. Sin., 2014, 356–360, DOI:10.3724/Sp.J.1105.2014.13255.
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Am. Chem. Soc., 2013, 135, 9330–9333 Search PubMed.
- A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2014, 47, 6635–6644 Search PubMed.
- D. Hotta, A. Kanazawa and S. Aoshima, Macromolecules, 2018, 51, 7983–7992 Search PubMed.
- T. Krappitz, K. Jovic, F. Feist, H. Frisch, V. P. Rigoglioso, J. P. Blinco, A. J. Boydston and C. Barner-Kowollik, J. Am. Chem. Soc., 2019, 141, 16605–16609 Search PubMed.
- A. Nagai, N. Koike, H. Kudo and T. Nishikubo, Macromolecules, 2007, 40, 8129–8131 Search PubMed.
- Z. Zhang, L. Xia, T. Y. Zeng, D. C. Wu, W. J. Zhang, C. Y. Hong and Y. Z. You, Polym. Chem., 2019, 10, 2117–2125 Search PubMed.
- L. X. You and J. Ling, Macromolecules, 2014, 47, 2219–2225 Search PubMed.
- Y. Li, M. von der Luhe, F. H. Schacher and J. Ling, Macromolecules, 2018, 51, 4938–4944 Search PubMed.
- H. J. Yang, C. Q. Chai, Y. K. Zuo, J. F. Huang, Y. Y. Song, L. Jiang, W. Y. Huang, Q. M. Jiang, X. Q. Xue and B. B. Jiang, Chin. J. Polym. Sci., 2020, 38, 231–239 Search PubMed.
- M. Uchiyama, M. Osumi, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2020, 59, 6832–6838 Search PubMed.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969–1992 Search PubMed.
- A. Keyes, H. E. B. Alhan, E. Ordonez, U. Ha, D. B. Beezer, H. Dau, Y. S. Liu, E. Tsogtgerel, G. R. Jones and E. Harth, Angew. Chem., Int. Ed., 2019, 58, 12370–12391 Search PubMed.
- A. Keyes, H. E. B. Alhan, U. Ha, Y. S. Liu, S. K. Smith, T. S. Teets, D. B. Beezer and E. Harth, Macromolecules, 2018, 51, 7224–7232 Search PubMed.
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2016, 138, 15535–15538 Search PubMed.
- V. Kottisch, Q. Michaudel and B. P. Fors, J. Am. Chem. Soc., 2017, 139, 10665–10668 Search PubMed.
- B. M. Peterson, V. Kottisch, M. J. Supej and B. P. Fors, ACS Cent. Sci., 2018, 4, 1228–1234 Search PubMed.
- M. J. Supej, B. M. Peterson and B. P. Fors, Chem, 2020, 6, 1794–1803 Search PubMed.
- J. N. Zhu, X. Hao and Q. Yan, Sci. China: Chem., 2019, 62, 1023–1029 Search PubMed.
- S. Liu, T. W. Bai, K. Ni, Y. Chen, J. P. Zhao, J. Ling, X. D. Ye and G. Z. Zhang, Angew. Chem., Int. Ed., 2019, 58, 15478–15487 Search PubMed.
- C. H. Wang, Y. S. Fan, Z. Zhang, Q. B. Chen, T. Y. Zeng, Q. Y. Meng and Y. Z. You, Appl. Surf. Sci., 2019, 475, 639–644 Search PubMed.
- Z. Zhang, X. Nie, F. Wang, G. Chen, W. Q. Huang, L. Xia, W. J. Zhang, Z. Y. Hao, C. Y. Hong, L. H. Wang and Y. Z. You, Nat. Commun., 2020, 11, 3654 Search PubMed.
- Z. Zhang, T. Y. Zeng, L. Xia, C. Y. Hong, D. C. Wu and Y. Z. You, Nat. Commun., 2018, 9, 2577 Search PubMed.
- F. Ouhib, B. Grignard, E. Van den Broeck, A. Luxen, K. Robeyns, V. Van Speybroeck, C. Jerome and C. Detrembleur, Angew. Chem., Int. Ed., 2019, 58, 11768–11773 Search PubMed.
- Y. J. Zhao, Y. Wang, X. P. Zhou, Z. G. Xue, X. H. Wang, X. L. Xie and R. Poli, Angew. Chem., Int. Ed., 2019, 58, 14311–14318 Search PubMed.
- J. M. Longo, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 15897–15900 Search PubMed.
- C. M. Liao, C. C. Hsu, F. S. Wang, B. B. Wayland and C. H. Peng, Polym. Chem., 2013, 4, 3098–3104 Search PubMed.
- F. S. Wang, T. Y. Yang, C. C. Hsu, Y. J. Chen, M. H. Li, Y. J. Hsu, M. C. Chuang and C. H. Peng, Macromol. Chem. Phys., 2016, 217, 422–432 Search PubMed.
- Y. Chen, Q. L. Song, J. P. Zhao, X. J. Gong, H. Schlaad and G. Z. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 6593–6600 Search PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 Search PubMed.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 Search PubMed.
- S. Paul, Y. Q. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 Search PubMed.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 Search PubMed.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 Search PubMed.
- Y. Q. Zhu, C. Romain and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 12179–12182 Search PubMed.
- C. Romain, Y. Q. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 Search PubMed.
- S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 Search PubMed.
- T. Stosser and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 6337–6341 Search PubMed.
- T. Stoesser, D. Mulryan and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 16893–16897 Search PubMed.
- S. Kernbichl, M. Reiter, F. Adams, S. Vagin and B. Rieger, J. Am. Chem. Soc., 2017, 139, 6787–6790 Search PubMed.
- H. Y. Ji, B. Wang, L. Pan and Y. S. Li, Angew. Chem., Int. Ed., 2018, 57, 16888–16892 Search PubMed.
- H. Li, H. T. Luo, J. P. Zhao and G. Z. Zhang, ACS Macro Lett., 2018, 7, 1420–1425 Search PubMed.
- T. Stosser, G. S. Sulley, G. L. Gregory and C. K. Williams, Nat. Commun., 2019, 10, 2668 Search PubMed.
- H. Li, G. C. He, Y. Chen, J. P. Zhao and G. Z. Zhang, ACS Macro Lett., 2019, 8, 973–978 Search PubMed.
- A. B. Biernesser, K. R. Chiaie, J. B. Curley and J. A. Byers, Angew. Chem., Int. Ed., 2016, 55, 5251–5254 Search PubMed.
- S. S. Zhu, Y. Wang, W. Z. Ding, X. P. Zhou, Y. G. Liao and X. L. Xie, Polym. Chem., 2020, 11, 1691–1695 Search PubMed.
- G. S. Sulley, G. L. Gregory, T. T. D. Chen, L. P. Carrodeguas, G. Trott, A. Santmarti, K. Y. Lee, N. J. Terrill and C. K. Williams, J. Am. Chem. Soc., 2020, 142, 4367–4378 Search PubMed.
- S. Kernbichl, M. Reiter, J. Mock and B. Rieger, Macromolecules, 2019, 52, 8476–8483 Search PubMed.
- Y. Chen, S. Liu and J. P. Zhao, Acta. Polym. Sin., 2020, 51, 1067–1082 Search PubMed.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 Search PubMed.
- H. C. Huang, B. H. Sun, Y. Z. Huang and J. Niu, J. Am. Chem. Soc., 2018, 140, 10402–10406 Search PubMed.
- H. C. Huang, W. Q. Wang, Z. F. Zhou, B. H. Sun, M. R. An, F. Haeffner and J. Niu, J. Am. Chem. Soc., 2019, 141, 12493–12497 Search PubMed.
- H. Park and T. L. Choi, J. Am. Chem. Soc., 2012, 134, 7270–7273 Search PubMed.
- H. Park, H. K. Lee and T. L. Choi, J. Am. Chem. Soc., 2013, 135, 10769–10775 Search PubMed.
- H. K. Lee, J. Lee, J. Kockelmann, T. Herrmann, M. Sarif and T. L. Choi, J. Am. Chem. Soc., 2018, 140, 10536–10545 Search PubMed.
- C. Kang, H. Park, J. K. Lee and T. L. Choi, J. Am. Chem. Soc., 2017, 139, 11309–11312 Search PubMed.
- C. Kang, S. Kwon, J. C. Sung, J. Kim, M. H. Baik and T. L. Choi, J. Am. Chem. Soc., 2018, 140, 16320–16329 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |