
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDe-alloyed PtCu/C catalysts with enhanced electrocatalytic performance for the oxygen reduction reaction†
Zhuojie
Xiao
 a,
Hao
Wu
a,
Huichi
Zhong
a,
Ali
Abdelhafiz
*bc and
Jianhuang
Zeng
*ad
a,
Hao
Wu
a,
Huichi
Zhong
a,
Ali
Abdelhafiz
*bc and
Jianhuang
Zeng
*ad
aSchool of Chemistry and Chemical Engineering, South China University of Technology; Guangdong Key Lab for Fuel Cell Technology, Guangzhou 510641, China. E-mail: cejhzeng@scut.edu.cn
bDepartment of Nuclear Science and Engineering, Massachusetts Institute of Technology, Massachusetts 02139, USA. E-mail: ali_m@mit.edu
cDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Massachusetts 02139, USA
dZhongke of Hydrogen Power Co. Ltd, Nansha District, Guangzhou 511458, China
First published on 12th July 2021
Abstract
In electrochemical reactions, interactions between reaction intermediates and catalytic surfaces control the catalytic activity, and thereby require to be optimized. Electrochemical de-alloying of mixed-metal nanoparticles is a promising strategy to modify catalysts’ surface chemistry and/or induce lattice strain to alter their electronic structure. Perfect design of the electrochemical de-alloying strategy to modify the catalyst's d-band center position can yield significant improvement on the catalytic performance of the oxygen reduction reaction (ORR). Herein, carbon supported PtCu catalysts are prepared by a simple polyol method followed by an electrochemical de-alloying treatment to form PtCu/C catalysts with a Pt-enriched porous shell with improved catalytic activity. Although the pristine PtCu/C catalyst exhibits a mass activity of 0.64 A mg−1Pt, the dissolution of Cu atoms from the catalyst surface after electrochemical de-alloying cycling leads to a significant enhancement in mass activity (1.19 A mg−1Pt), which is 400% better than that of state-of-the-art commercial Pt/C (0.24 A mg−1Pt). Furthermore, the de-alloyed PtCu/C-10 catalyst with a Pt-enriched shell delivers prolonged stability (loss of only 28.6% after 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles), which is much better than that of Pt/C with a loss of 45.8%. By virtue of scanning transmission electron microscopy and elemental mapping experiments, the morphology and composition evolution of the catalysts could clearly be elucidated. This work helps in drawing a roadmap to design highly active and stable catalyst platforms for the ORR and relevant proton exchange membrane fuel cell applications.
000 cycles), which is much better than that of Pt/C with a loss of 45.8%. By virtue of scanning transmission electron microscopy and elemental mapping experiments, the morphology and composition evolution of the catalysts could clearly be elucidated. This work helps in drawing a roadmap to design highly active and stable catalyst platforms for the ORR and relevant proton exchange membrane fuel cell applications.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) with high power density, low operating temperature and zero carbon dioxide emissions have widely been considered as one of the most efficient clean energy generation systems compared to conventional internal combustion engines.1 Currently, the commercialization of this technology is mainly limited by the prohibitive cost of platinum (Pt), which is used as the electrocatalyst in the anodes and cathodes.2 Reactions at both the anode and cathode, especially the oxygen reduction reaction (ORR) at the cathode, have sluggish kinetics, which has triggered extensive research to improve ORR activity and lower the cost of electrocatalysts in the past decade.3 One desirable protocol to reduce the amount of Pt required and address the performance challenge is to alloy Pt with transition metals (e.g., Ni, Co, Fe, Cu, and so forth).4–13 Early studies came up with various key factors that determine the ORR activity of Pt-based electrocatalysts, including, but not limited to, the geometric structure,14 the electronic structure15,16 and the adsorption energy of oxygenated species on the surface Pt atoms.17 Experimental studies18,19 in combination with theoretical approaches15,20 reported that the ORR activity of alloy nanoparticles was given by the strength of the oxygen–metal bond interaction, which depends on the position of the Pt d-band center relative to the Fermi level.21Interestingly, the shift of the electronic band structure of alloy nanoparticles and weakening of the chemisorption energy of oxygenated species can also be achieved by using electrochemical de-alloying for forming a Pt-rich shell to exhibit compressive strain, due to the dissolution of non-noble metal atoms from the alloy surface layer. For instance, Strasser et al. reported that the de-alloyed PtCu nanoparticles demonstrated higher ORR activity originating from the controllable compressive strain formed in Pt-enriched surface layers via potential cycling.17 Stimulated by this finding, numerous studies have been dedicated to developing de-alloyed Pt-based nanoparticles, especially PtCu nanoparticles,22–25 in which Cu is removed from the alloy surface layers, forming a Pt-rich shell and a PtCu core structure with enhanced ORR activity compared with pure Pt nanoparticles. Generally, the electronic structure of a Pt-rich surface can be perturbed by a PtCu alloy core with differing composition or structure, which can further affect the adsorption energy of oxygenated species.26 Although, immense efforts have been previously devoted to improving the ORR performance of de-alloyed PtCu nanoparticles, the structural and componential observation, especially the change of lattice strain in the process of electrochemical de-alloying and accelerated durability test (i.e. ADT), is not perfectly clear to the best of our knowledge. Thus, the correlation among the lattice strain, composition and enhanced catalytic activity of the de-alloyed PtCu catalyst was investigated after different potential cycling in perchloric acid electrolyte.
Herein, we report the synthesis of de-alloyed PtCu/C catalysts with high electrocatalytic performance and low Pt-loading, which can be prepared via a simple polyol method followed by an electrochemical de-alloying treatment for improving the ORR activity. The de-alloyed PtCu/C-10 catalyst demonstrated enhanced ORR performance (1.19 A mg−1Pt) with 28.6% loss after 30000 cycles at 30 °C, surpassing the DOE 2020 target in both Pt mass activity and durability, compared with the Pt/C-10 catalyst (0.24 A mg−1Pt) with 45.8% loss. For PtCu/C catalysts, the compositional and morphological evolution was determined after potential cycling. A Pt-rich shell is formed and covers the PtCu alloy core. Moreover, it was found that the de-alloying treatment results in structural and lattice parameter changes which can be observed in the shift of the diffraction peak in the X-ray powder diffraction patterns. This study provides visual and valuable insight into the rational design of de-alloyed Pt-based electrocatalysts with a modified electronic structure.
2. Experimental section
2.1. Chemicals and materials
Copper sulfate pentahydrate (CuSO4 5H2O) and potassium hydroxide (KOH) were purchased from Damao Chemical Reagent Factory (Tianjin, China). Chloroplatinic acid hexahydrate (H2PtCl6 6H2O) was purchased from Jinke Chemical Reagent Co., Ltd (Shenyang, China). Trisodium citrate dihydrate (C6H5Na3O7 2H2O), ethylene glycol (EG), perchloric acid (HClO4) and isopropanol were all supplied by Guangzhou Chemical Reagent Factory (Guangzhou, China). Nafion solution (5.0 wt%, D520) was provided by DuPont. The abovementioned reagents were of analysis grade (A.R.) and used without further purification. Commercial Pt/C (Hispec-3000, 20 wt% loading) was purchased from Johnson Matthey. Deionized water (18.2 MΩ cm) was used for preparing the solutions.2.2. Synthesis of PtCu/C catalysts
Carbon-supported PtCu/C catalysts were prepared by using a polyol method, in which EG served as the reducing agent and solvent. Initially, 360.0 mg of C6H5Na3O7 2H2O was dispersed in 150 mL of EG solution in a 250 mL flask container with three necks. After ultrasonic agitation for 30 min, 4.00 mL of 0.038 M H2PtCl6/EG solution and 50.0 mg of CuSO4 5H2O were added into the above dispersion and stirred for 2 h to obtain a pale green solution. Sequentially, 120.0 mg of Vulcan XC-72R carbon was dispersed in the above solution under stirring condition for 30 min, followed by another 30 min of ultrasonic dispersion. The pH of the final mixture was adjusted to be 10 with 1.0 M KOH/EG solution. The final mixture was heated up to 120 °C and maintained for 3 h under stirring conditions, before it was cooled to room temperature. After that, the sample was filtered, washed with deionized water and dried in a vacuum oven at 50 °C for 12 h. The synthesis yielded PtCu/C catalysts. The as-prepared PtCu/C catalysts were further electrochemical de-alloyed between 0.05 to 1.10 V (vs. RHE) in a N2-purged 0.1 M HClO4 solution at a scan rate of 50 mV s−1. Based on different voltammetric cycles, the de-alloyed catalysts are hereafter referred to as PtCu/C-x (x represented as the voltammetric cycles). For comparison, the Pt/C catalysts were treated under the same electrochemical conditions.2.3. Physical characterization
X-ray diffraction (XRD) patterns were collected on a TD-3500 power diffractometer (Tongda, China) with Cu Kα radiation (λ = 1.5406 Å) in the scan range from 20° to 90°, operated at 40 kV and 30 mA. Transmission electron microscopy (TEM) images were obtained with JEM-2100F operated at 200 kV. Scanning transmission electron microscopy (STEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) were carried out on a TALOS F200 field emission transmission electron microscope (FEI) operating at 200 kV. Amorphous carbon-coated nickel grids were used during all the EDS experiments. The composition of the catalysts was determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Agilent 730, Japan). X-ray photoelectron spectroscopy (XPS) was performed on a LAB250 ESCA system (Thermo Fisher, America) with Al Kα radiation (1486.6 eV). The binding energy (BE) was calibrated by referencing the major C 1s peak at 284.8 eV.2.4. Electrochemistry test of catalysts
All electrochemical characteristics were performed in a conventional three-electrode cell system at 30 °C on a potentiostat/galvanostat (Autolab PGSTNT302N). A glassy carbon rotating disk electrode (RDE, 5 mm in diameter, 0.196 cm2, Pine Research Instrumentation, AFE5T050GC), reversible hydrogen electrode (RHE, Hong Kong Phychemi Co. Ltd) and platinum gauze were used as the working electrode (WE), reference electrode (RE) and counter electrode (CE), respectively. The catalyst ink was prepared by mixing 5.0 mg of the catalyst and 5.00 mL of a mixture which consisted of 1.00 mL of H2O, 4.00 mL of isopropanol and 10 μL of Nafion solution. 8 μL of homogeneous ink was dropped onto a RDE using a micropipette and dried to form the catalyst layer at room temperature. The commercial Pt/C catalyst served as a benchmark to compare the catalytic activity of PtCu/C catalysts. Cyclic voltammetry (CV) curves were recorded in a N2-purged 0.1 M HClO4 solution from 0.05 to 1.10 V (vs. RHE) at a scan rate of 50 mV s−1. Accelerated durability tests (ADTs) were performed by applying 5000, 15000 and 30000 potential cycles between 0.60 to 1.00 V (vs. RHE) at a sweep rate of 100 mV s−1 in an O2-purged 0.1 M HClO4 solution. CO-stripping cyclic voltammograms were recorded in a CO-saturated 0.1 M HClO4 electrolyte at a scan rate of 50 mV s−1. The catalytic ORR performance was evaluated using linear sweep voltammetry (LSV) from 0.15 to 1.05 V (vs. RHE) in an O2-saturated 0.1 M HClO4 solution with a rotation rate of 1600 rpm at a sweep rate of 10 mV s−1.
Single cell tests were performed on a fuel-cell test system (Scribner Associates Inc., Model 850e). The catalyst ink (2 mg mL−1) was prepared by blending the catalyst, Nafion solution, H2O and isopropanol at an ionomer/carbone weight ratio of 0.5/1 and an isopropanol/H2O volume ratio of 1/4, and then sprayed on a Nafion 211 membrane.27 The active area was 25 cm2. The anode catalyst was 20 wt% Pt/C-Pristine and the cathode catalyst was Pt/C-10 or PtCu/C-10. The Pt loading of the anode and the cathode was fixed at 0.10 mgPt cm−2 and 0.20 mgPt cm−2, respectively. The single cell electrochemical characterization was analyzed at a cell temperature of 80 °C, H2/Air (Stoichiometric of 1.2/2.5) with 100% relative humidity (RH) and 150 kPa of back pressure. The accelerated durability tests (ADTsc) of single cells were conducted in the potential range of 0.6–1.1 V with the sweep rate of 100 mV s−1 for 10000 cycles at a condition of cathode feed of fully humidified N2.
3. Results and discussion
Carbon supported PtCu nanoparticles were successfully synthesized via a simple polyol method. Fig. 1a shows the XRD spectra of PtCu nanoparticles synthesized under different reaction conditions in comparison to monometallic Pt nanoparticles. The broad and weak diffraction peaks at ca. 25° were ascribed to the (002) plane of the carbon support.28 XRD spectra of PtCu nanoparticles showed no Cu–Cu signal. The absence of any Cu peaks implied the complete diffusion of Cu atoms into the Pt lattice, forming PtCu alloy.29 In addition, the characteristic Pt peak at 40° was shifted to higher angles for all PtCu samples, compared to pure Pt nanoparticles. This further confirmed the formation of PtCu alloy, where Pt lattice contraction was observed when Cu with shorter atomic radii was present within the platinum lattice. Furthermore, XRD spectra suggested that PtCu nanoparticles maintained a face-centered cubic (fcc) crystal orientation. The electrochemical de-alloying effect was confirmed by a gradual shift of the Pt peak to lower angles, with Cu dissolution out of the PtCu lattice, which in return relieved the lattice compression on PtCu nanoparticles.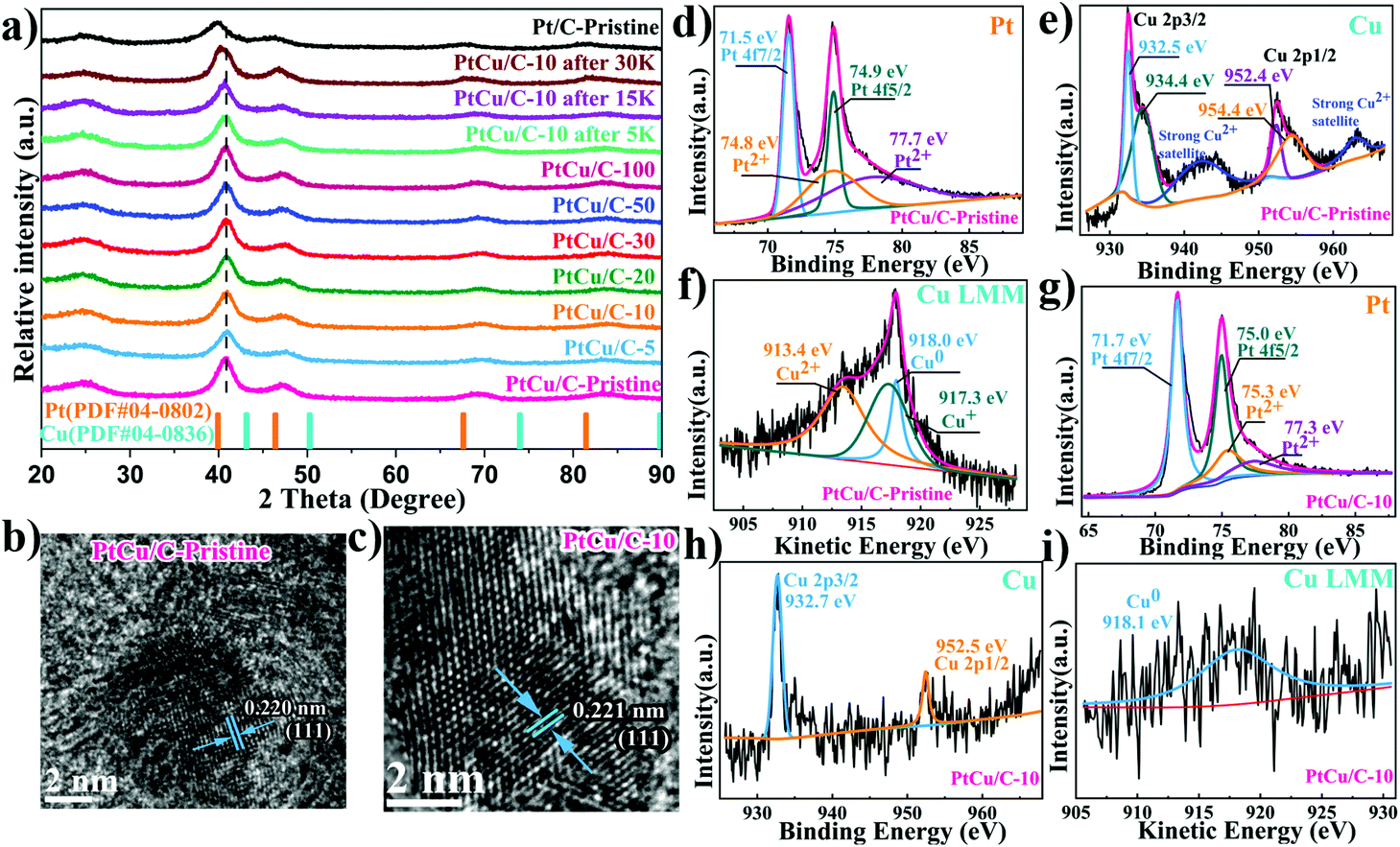 | ||
| Fig. 1 X-ray diffraction patterns of all catalysts (a). Below are peak positions of standard pure metallic Pt and Cu. HR-STEM images of PtCu/C-Pristine (b) and PtCu/C-10 (c). XPS spectra of Pt 4f, Cu 2p and Cu LM2 for PtCu/C-Pristine (d–f) and D-PtCu/C (g–i). | ||
TEM images (Fig. S1†) revealed that the PtCu/C nanoparticles were well dispersed on the carbon supports, similar to the commercial Pt/C catalyst. EDX elemental mapping showed a high entropic alloying of PtCu nanoparticles, where both Pt and Cu dispersion were even across the whole nanoparticle. This further indicated that Cu atoms were infused within the Pt lattice to form a uniform PtCu alloy, which agrees with XRD results as discussed earlier. Fig. 1b and c show atomic resolution STEM images of PtCu/C and Pt/C nanoparticles, where the lattice parameter exhibits lattice fringes of 0.220 and 0.221 nm, where the lattice parameter of PtCu is further compressed by 0.45% compared to the monometallic Pt/C catalyst.30,31
The chemical states of the surface component were determined by XPS to observe changes in the d-band center position, as an indication on strain induced on Pt atoms.32–34Fig. 1d shows that the Pt 4f7/2 peak shifts towards a higher binding energy (71.5 eV) compared with pure Pt (71.3 eV),35,36 revealing the downshift in the center of the d-band for PtCu/C-Pristine catalysts. As shown in Fig. 1e, the obvious satellite peak located at the higher binding energy of 942.1 eV corresponding to that of Cu 2p3/2 peaks, suggesting the presence of Cu(II) species.37 In addition, the Cu 2p peaks can be fitted by two peaks with a close binding energy, which are assigned to Cu(I) or Cu(0).38 Therefore, the Auger Cu LMM spectra were used to confirm the presence of Cu(0), Cu(I) or Cu(II) in the pristine PtCu/C-Pristine catalyst.39Fig. 1f shows the Auger spectrum of Cu LMM, where the peak deconvolution resulted in three sub-peaks at 913.4, 917.3 and 918.0 eV, corresponding to Cu(II), Cu(I) and Cu(0) states. Auger analysis indicated the co-existence of metallic and oxide Cu states. Cu oxide is believed to form naturally due to oxidation under ambient conditions, when Cu atoms at the surface were exposed to air. Thus, the thickness of the native Cu oxide layer was too thin to be detected by XRD.40 Based on different electrochemical dealloying conditions (i.e., no# cycles), we found that PtCu/C-10 showed the highest electrocatalytic activity, as displayed in Fig. S2 and Table S1.† Thus, the characterization data shown in the manuscript hereon will be dedicated to PtCu/C-10, as a representative sample for dealloyed catalysts. Fig. 1g shows Pt 4f duplets, where Pt 4f7/2 peaks shift towards a higher binding energy (71.7 eV) compared to that of the pristine PtCu/C-Pristine catalyst, implying an improvement in ORR performance due to the downshift of the Pt d-band center.41 Meanwhile, Cu 2p peak intensities became significantly weak, owing to the dissolution of Cu surface atoms under electrochemical de-alloying conditions. It is worth noting that the absence of satellite peaks and the Auger peak of Cu(II) and Cu(I) species indicates the formation of a Pt-rich shell, implying the dissolution of Cu at the surface under electrochemical de-alloying conditions. Electrochemical dealloying treatment did not affect the particles’ size or morphology, as can be seen in Fig. S1.†
In addition, Fig. 2 displays the HAADF-STEM image and the corresponding elemental mapping of the PtCu/C-10 catalyst. Imaging clearly revealed the formation of core–shell nanostructures with a Pt-rich shell and a PtCu alloy core. Compared with PtCu/C-Pristine, the composition of PtCu/C-10 was further determined by EDS spectra. As displayed in Fig. S3,† the results of the molar ratio in both Pt and Cu indicate that the Pt composition concentered at the surface of metallic nanoparticles, suggesting the presence of a Pt-rich shell.
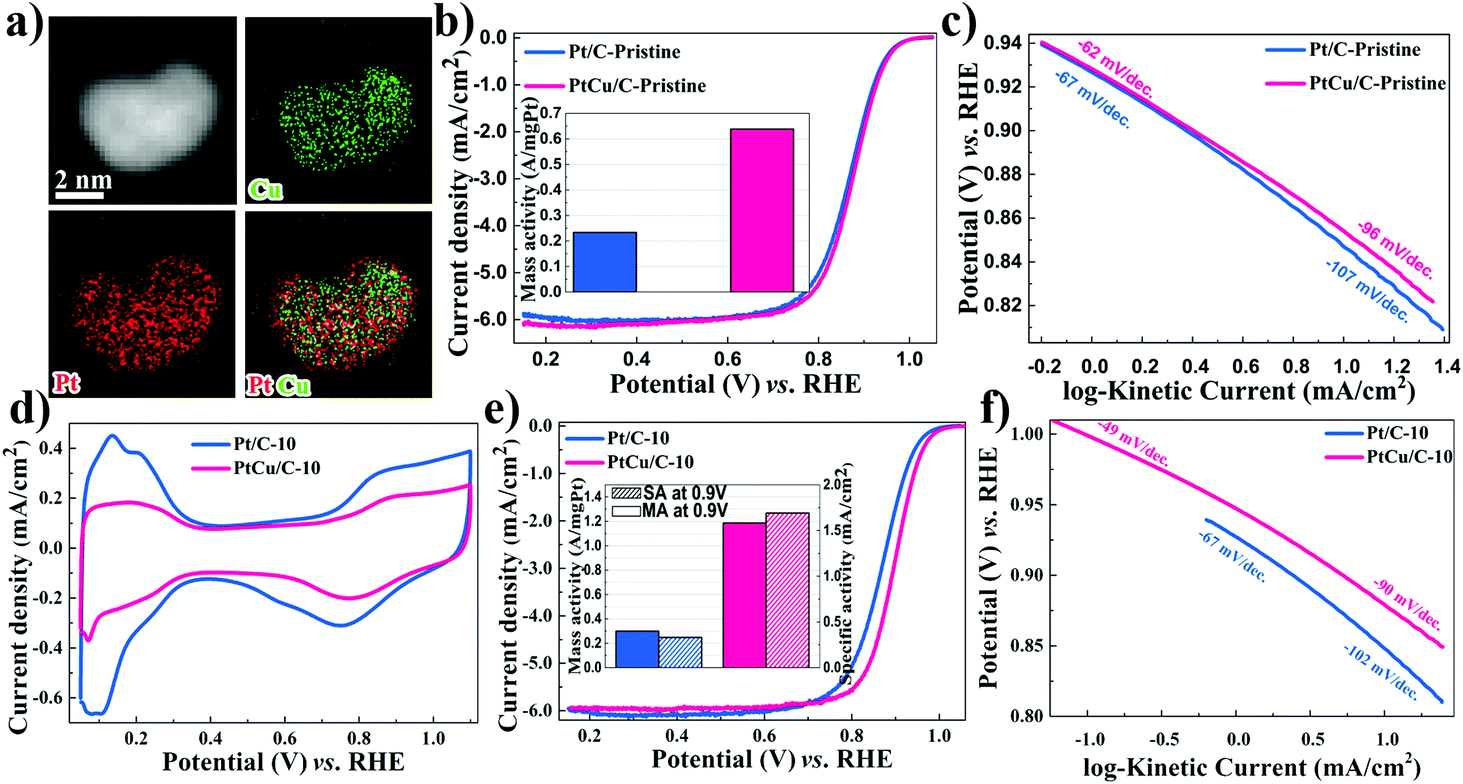 | ||
| Fig. 2 HAADF-STEM overview images and EDS-HAADF-STEM element mapping images of PtCu/C-10 (a). ORR polarization curves in O2-saturated 0.1 M HClO4 at a rotation rate of 1600 rpm and a scan rate of 10 mV s−1 for Pt/C-Pristine and PtCu/C-Pristine (b). Inset: Pt mass activities at 0.90 V (vs. RHE). Tafel plots for these pristine catalysts (c). Cyclic voltammograms in deoxygenated 0.1 M HClO4 for Pt/C-10 and PtCu/C-10 in tenth cycles at a scan rate of 50 mV s−1 (d). ORR polarization curves and Tafel plots for Pt/C-10 and PtCu/C-10 (e–f). | ||
The catalytic performance of PtCu/C-Pristine for the ORR was investigated in an O2-saturated 0.1 M HClO4 solution.42 As can be seen in Fig. 2b, the MA of Pt/C-Pristine is 0.23 A mg−1Pt which is three times lower than the MA of PtCu/C-Pristine (0.64 A mg−1Pt). Tafel slopes represented in Fig. 2c are −62 and −96 mV per decade in low and high overpotential regions, respectively, which implies that the charge transfer and removal of oxygenated intermediates are the rate-limiting steps.43,44 As shown in Fig. S2a,† under different de-alloying conditions, the CV scans of PtCu/C-5, recorded in a N2-purged 0.1 M HClO4 solution, displayed a weaker hydrogen under potential deposition wave (Hupd), compared with that of PtCu/C-10. This indicated the formation of a Pt-rich shell in PtCu/C-10 due to the dissolution of Cu atoms on the surface. The electrochemical surface area (ECSA) of all catalysts, calculated by integrating the area of Hupd peaks, is listed in Table 1. Furthermore, the calculated ratio of ECSACO:ECSAHupd (Fig. S3†), a descriptor of surface structure in the case of the Pt alloy, is 1.47 on the de-alloyed PtCu/C-10, implying the nature of the Pt-rich shell structure.45 As is evident from Fig. 2e, the ORR activity of PtCu/C-10 manifests higher than that of Pt/C-10 (1.19 versus 0.24 A mg−1Pt). However, the MA of Pt/C before and after de-alloying, almost, remained intact. The de-alloyed PtCu/C-10 catalyst delivers a higher SA of 1.69 mA cm−2, with a 6.5-fold enhancement relative to the Pt/C-10 catalyst (0.26 mA cm−2). Furthermore, the ORR polarization curves of PtCu/C-10 demonstrated the half-wave potential (E1/2) at 30 mV more positive compared to Pt/C-10. In Fig. 2f, the higher kinetic current on the de-alloyed PtCu/C-10 catalyst indicated that the PtCu/C-10 catalyst maintains a higher ORR performance across a wide range of potentials.46 Such an enhanced electrocatalytic activity is attributed to the modified electronic structure of the Pt-rich shell and the optimal lattice compression. Both effects would affect the bonding strength between Pt and oxygenated species.47,48 Compared with PtCu/C-Pristine and PtCu/C-5, a large number of active sites were formed on the surface of PtCu/C-10 (higher ECSA) after cycling. However, gradual cycling resulted in the further loss of Cu, so that the lattice parameter of PtCu was similar to that of Pt/C, indicating the degradation of catalytic performance. To further understand the effects of morphological changes on ORR activity, the loss ratio of Cu of PtCu alloy nanoparticles was investigated after different voltage cycles. ICP-OES experiments were conducted to determine the atomic ratio of Cu, as shown in Table S2.† At the 5th voltage cycle, the Cu fraction in PtCu/C dropped significantly from 58.2% to 37.2% due to the dissolution of outer layer's Cu atoms, in agreement with the XRD and XPS analyses. After further electrochemical de-alloying and stability tests, a slight decay in Cu atomic% was observed, indicating that interior Cu atoms can be greatly preserved intact from dissolution.
| Samples | ECSA (m2 g−1) | ORR at 0.9 V | ||
|---|---|---|---|---|
| H upd | MA (A mg−1Pt) | MA loss (%) | SA (mA cm−2) | |
| Pt/C-10 | 90.3 | 0.24 | N/A | 0.27 |
| Pt/C-10 after 5 K | 77.0 | 0.23 | 4.2 | 0.30 |
| Pt/C-10 after 15 K | 69.9 | 0.19 | 20.8 | 0.27 |
| Pt/C-10 after 30 K | 49.2 | 0.13 | 45.8 | 0.26 |
| PtCu/C-10 | 70.5 | 1.19 | N/A | 1.69 |
| PtCu/C-10 after 5 K | 52.0 | 1.13 | 5.0 | 2.17 |
| PtCu/C-10 after 15 K | 41.5 | 1.05 | 11.8 | 2.53 |
| PtCu/C-10 after 30 K | 39.9 | 0.85 | 28.6 | 2.13 |
Apart from excellent ORR activities, the stability of catalysts is a crucial factor affecting PEMFCs’ lifetime. Consequently, the accelerated durability tests were performed to evaluate the stability of the dealloyed PtCu/C-10 and Pt/C-10 catalysts. In Fig. 3a, CV scans obtained using PtCu/C-10 showed a significant loss of ECSA after 30000 cycles (from 70.5 to 39.9 m2 g−1Pt), likely due to the aggregation of PtCu nanoparticles.49 Nevertheless, no obvious change was observed in LSV polarization curves (a slight negative shift of E1/2 from 0.90 to 0.89 V), as displayed in Fig. 3b. It should be noted that after 30000 cycles, a slight change in the amount of copper was observed for PtCu/C-10 (from 35.8% to 26.9%), suggesting that a Pt-rich shell can perhaps protect the interior Cu atoms from leaching.50 After 30000 cycles, the MA of the Pt/C-10 catalyst showed a loss of 45.8%, which is much worse than that of 28.6% for the PtCu/C-10 catalyst (listed in Table 1).
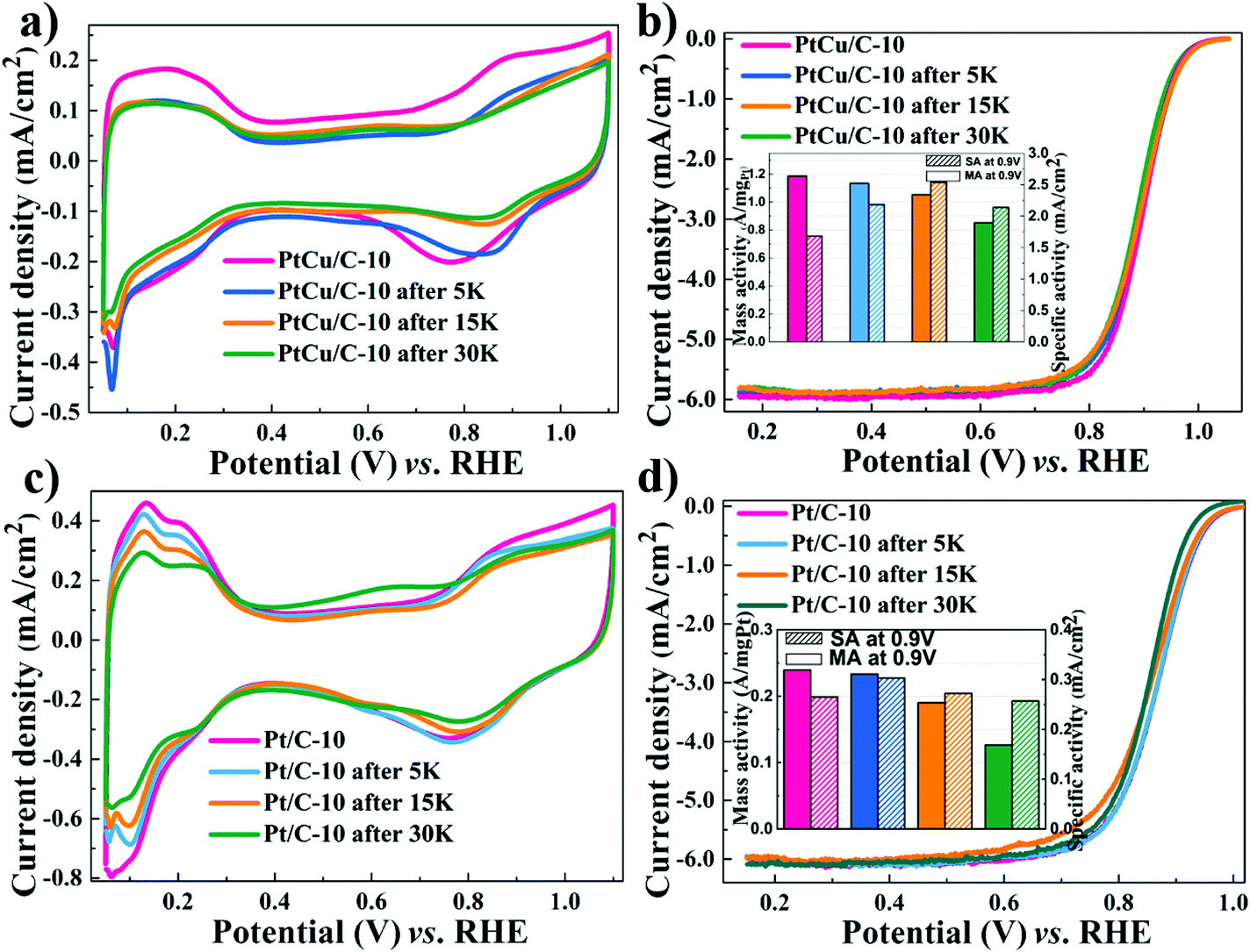 | ||
| Fig. 3 Cyclic voltammograms and ORR polarization curves for PtCu/C-10 (a–b) and Pt/C-10 (c–d) during ADT. Insert: Pt mass activities and specific activities at 0.90 V (vs. RHE). | ||
TEM analysis was conducted to observe the morphological and compositional changes to obtain more insight into the reasons for the electrocatalytic activity loss on the nanoscale.51 It can be observed from Fig. 4 that the morphology of all catalysts after ADT showed obvious aggregation, which agrees with ECSA analyses discussed earlier. Most remarkably, TEM images of Pt/C after ADT showed a dramatic increase in nanoparticles size compared to the PtCu/C-10 catalyst. Furthermore, the change in the composition of PtCu/C-10 catalysts after ADT is revealed in Fig. 5, and the atomic ratios of Pt and Cu were also detected and are displayed in Fig. S4.† From the elemental mapping analysis and EDS spectra, it can be seen that there is an obvious change in the atomic ratio of Cu. In Table 2, the atomic concentrations of Pt in the surface composition increased after ADT, while the atomic concentration of Cu decreased. The observation in composition suggests that the Pt-rich shell gradually thickened during ADT. Fig. 6a shows a schematic image of the formation of a thicker Pt-rich shell. Therefore, it can be supposed that the decrease in electrocatalytic performance of PtCu/C-10 catalysts mainly resulted from the aggregation and relieved lattice compression due to the thicker Pt-rich shell.
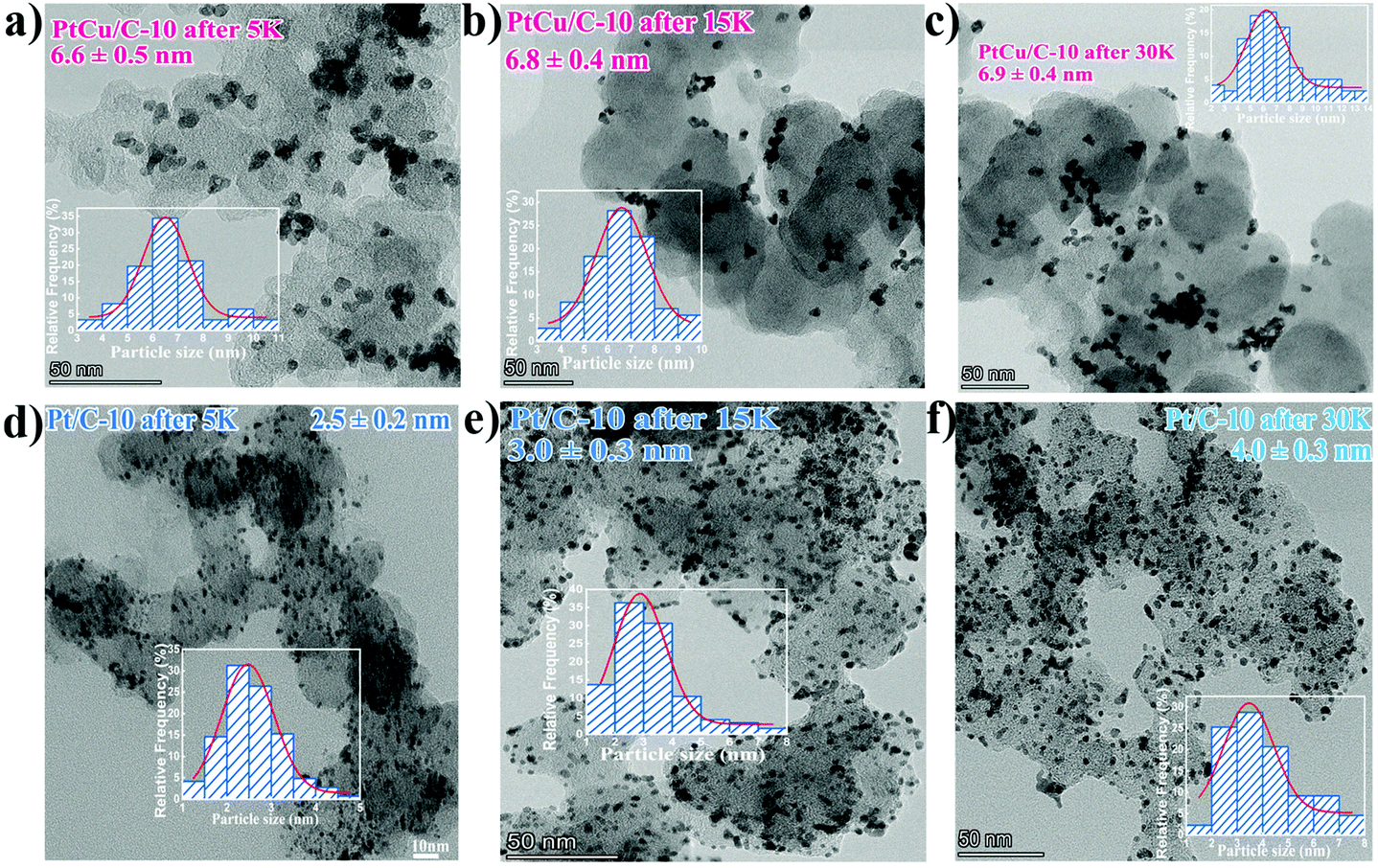 | ||
| Fig. 4 TEM images of PtCu/C-10 after 5 K (a), 15 K (b) and 30 K (c), as well as Pt/C-10 after 5 K (d), 15 K (e) and 30 K (f), Insert: the corresponding particle size distribution histogram. | ||
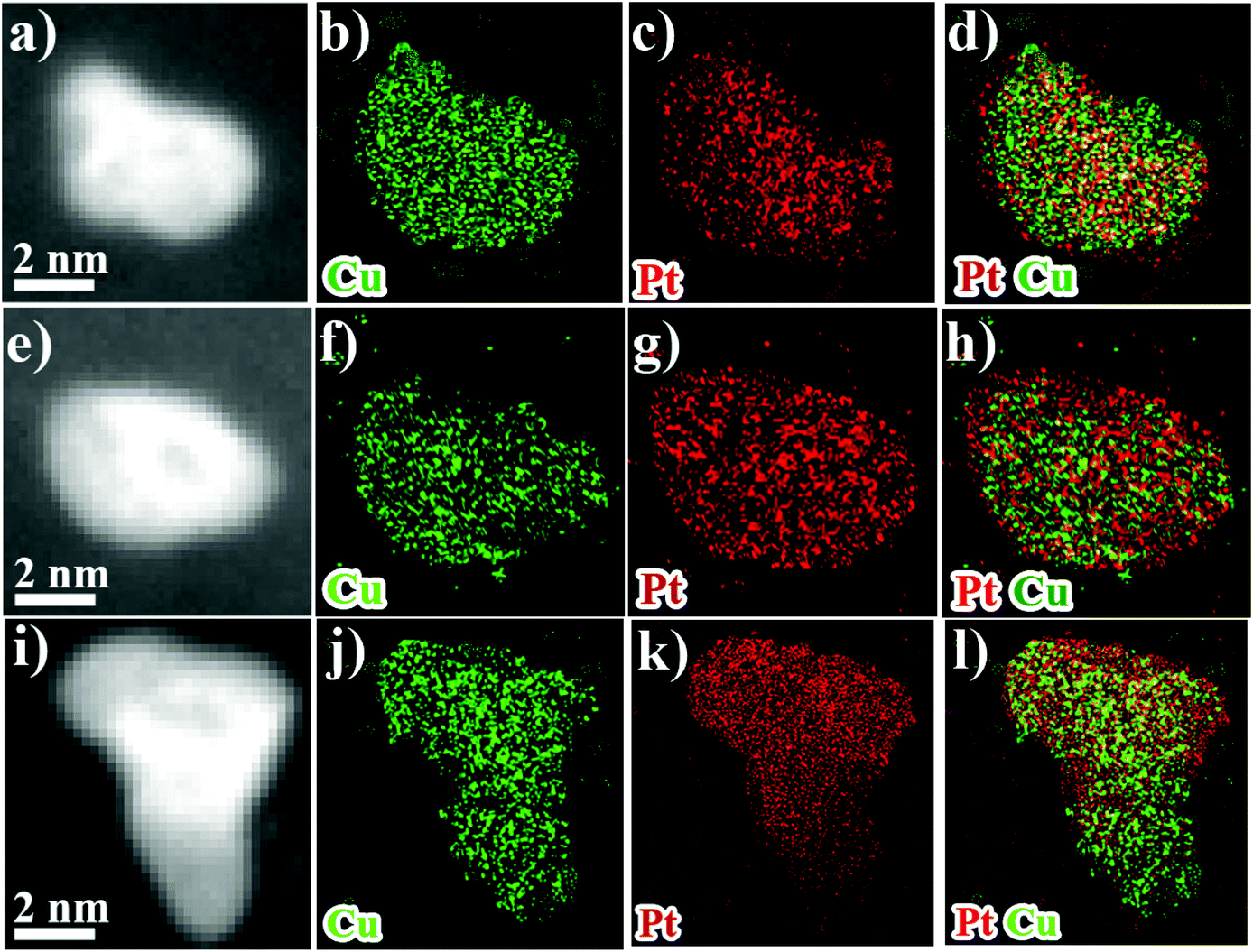 | ||
| Fig. 5 HAADF-STEM overview images and EDS-HAADF-STEM element mapping images of PtCu/C after 5 K (a–d), 15 K (e–h) and 30 K (i–l). | ||
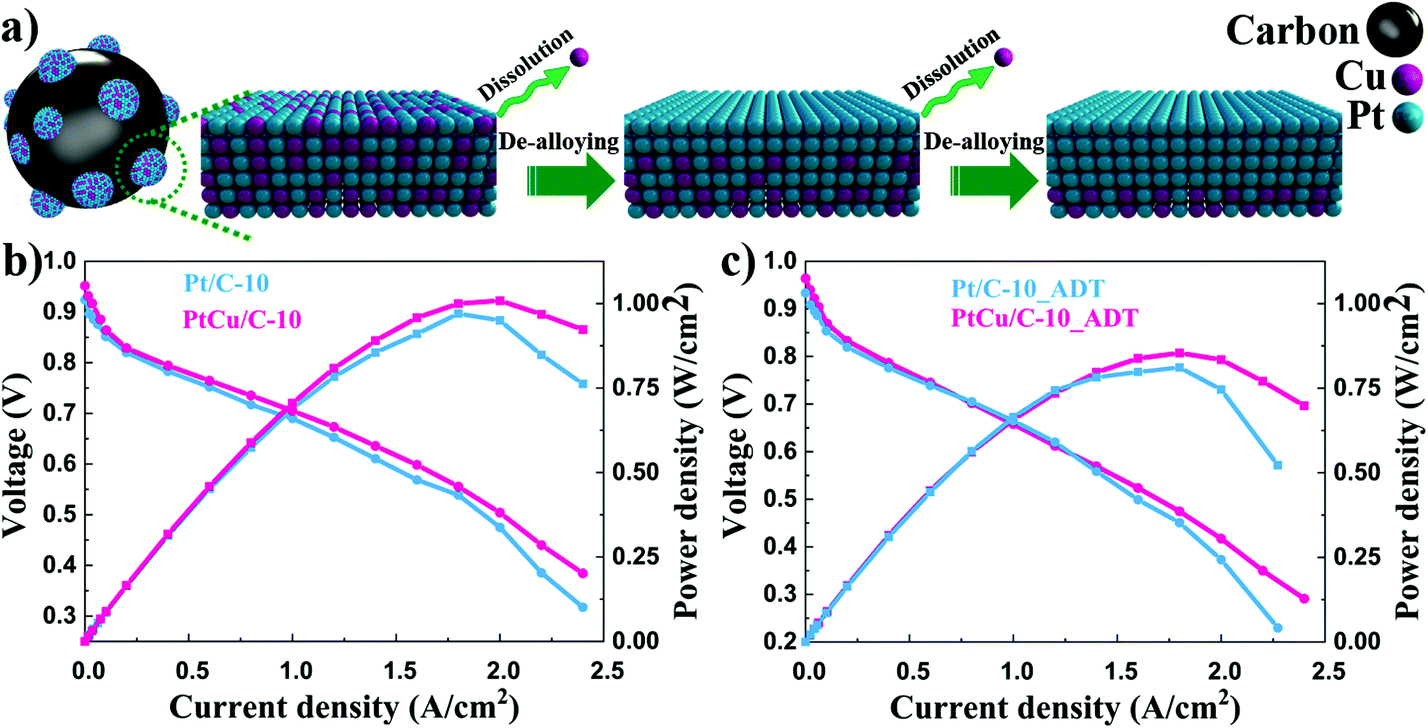 | ||
| Fig. 6 (a) Schematic image illustrating the formation of the Pt-rich shell of carbon-supported PtCu alloy catalysts. Black, blue and red spheres represent carbon, Pt and Cu, respectively. Polarization curves and power density curves of PtCu/C-10 and Pt/C-10 before (b) and after (c) ADTsc. | ||
| Samples | Atomic concentration (at%) | |
|---|---|---|
| Pt | Cu | |
| PtCu/C-Pristine | 17.2 | 82.8 |
| PtCu/C-10 | 58.5 | 41.5 |
| PtCu/C-10 after 5 K | 60.3 | 39.7 |
| PtCu/C-10 after 15 K | 67.5 | 32.5 |
| PtCu/C-10 after 30 K | 67.6 | 32.4 |
Single cell performances of PtCu/C-10 and Pt/C-10 catalysts before and after durability tests are shown in Fig. 6. In Fig. 6b, the PtCu/C-10 catalyst provided higher catalytic performance than Pt/C-10. This suggests that the existence of Cu in the cathode catalyst layer may enhance the proton conductivity of the electrode, thus improving the charge transfer performance of the cell.52 The PtCu/C-10 catalyst exhibited a maximum power density of 1.00 W cm−2, whereas the Pt/C-10 showed 0.95 W cm−2. At 0.8 V, the current densities were 0.35 and 0.30 A cm−2 for PtCu/C-10 and Pt/C-10 catalysts before ADTsc, respectively. Consistent with the observation from half-cell tests, the PtCu/C-10 catalyst delivered higher current density compared to Pt/C-10. After ADTsc, the current density and maximum power density were 0.33 A cm−2 and 0.85 W cm−2 for PtCu/C-10, and 0.29 A cm−2 and 0.83 W cm−2 for Pt/C-10. The loss of the maximum power density for PtCu/C-10 is 15.0%, which is much worse than that of 12.6% for the Pt/C-10 catalyst. Furthermore, a clear drop in the polarization curve of the PtCu/C-10 catalyst after ADTsc was observed in the low current density region. This suggests that the dissolved Cu ion may reduce the proton conductivity due to contaminating the membrane,53 thus indicating the necessity of the de-alloying process. The performance of Pt/C-10 exhibited a sharp decline in the high current density region, presumably due to the aggregation of Pt nanoparticles.54 Therefore, de-alloyed PtCu/C catalysts provide an ideal pretreatment method to improve the activity and durability of Pt based alloy catalysts for PEM fuel cell applications.
4. Conclusion
In summary, the de-alloyed PtCu/C catalysts have been well prepared by a simple polyol synthesis protocol and subsequently electrochemically activated in an acid electrolyte. The lattice contraction of PtCu nanoparticles was observed using HRSTEM. XRD demonstrated that the lattice compression on PtCu nanoparticles was relieved with Cu dissolution away from the PtCu lattice. XPS experiments showed that the electronic structure of the PtCu/C-10 catalyst has been altered by the electrochemical de-alloying treatment. The dissolution of Cu atoms from the Pt lattice relieved the lattice compressive strain, and altered the electronic structure in a manner which lowered the Pt d-band center. This enhanced the electrocatalytic activity by ∼400% for the ORR. The morphological and compositional analysis indicated that the activity loss of PtCu/C catalysts was mainly derived from the aggregation of nanoparticles and relieved lattice contraction after prolonged AST. Compared with PtCu/C and commercial Pt/C catalysts, the de-alloyed PtCu/C-10 catalyst with the Pt-rich shell shows the best electrocatalytic activity (1.19 A mg−1Pt) and stability (28.6% of activity loss after 30000 cycles) towards the oxygen reduction reaction. ORR activity test results suggested that dealloyed PtCu catalysts with a Pt-rich shell structure may be considered as a great replacement catalyst for application in fuel cells due to the optimized surface catalytic properties.
Conflicts of interest
The authors declare that there is no conflict of interest in this paper.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2018YFB0105500).References
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279–289 CrossRef.
- F. T. Wagner, B. Lakshmanan and M. F. Mathias, J. Phys. Chem. Lett., 2010, 1, 2204–2219 CrossRef CAS.
- R. Chattot, T. Asset, P. Bordet, J. Drnec, L. Dubau and F. Maillard, ACS Catal., 2017, 7, 398–408 CrossRef CAS.
- J. Li, S. Sharma, X. Liu, Y.-T. Pan, J. S. Spendelow, M. Chi, Y. Jia, P. Zhang, D. A. Cullen and Z. Xi, Joule, 2019, 3, 124–135 CrossRef CAS.
- Q. Wang, S. Chen, F. Shi, K. Chen, Y. Nie, Y. Wang, R. Wu, J. Li, Y. Zhang, W. Ding, Y. Li, L. Li and Z. Wei, Adv. Mater., 2016, 28, 10673–10678 CrossRef CAS PubMed.
- I. Dutta, M. K. Carpenter, M. P. Balogh, J. M. Ziegelbauer, T. E. Moylan, M. H. Atwan and N. P. Irish, J. Phys. Chem. C, 2010, 114, 16309–16320 CrossRef CAS.
- C. Dai, Y. Yang, Z. Zhao, A. Fisher, Z. Liu and D. Cheng, Nanoscale, 2017, 9, 8945–8951 RSC.
- J. Song, Z. Xiao, Y. Jiang, A. A. Abdelhafiz, I. Chang and J. Zeng, Int. J. Hydrogen Energy, 2019, 44(23), 11655–11663 CrossRef CAS.
- X. Ao, W. Zhang, B. Zhao, Y. Ding, G. Nam, L. Soule, A. Abdelhafiz, C. Wang and M. Liu, Energy Environ. Sci., 2020, 13, 3032–3040 RSC.
- M. Wu, X. Wu, L. Zhang, A. Abdelhafiz, I. Chang, C. Qu, Y. Jiang, J. Zeng and F. Alamgir, Electrochim. Acta, 2019, 306, 167–174 CrossRef CAS.
- K. Eid, H. Wang, V. Malgras, S. M. Alshehri, T. Ahamad, Y. Yamauchi and L. Wang, J. Electroanal. Chem., 2016, 779, 250–255 CrossRef CAS.
- K. Eid, H. Wang, V. Malgras, Z. A. Alothman, Y. Yamauchi and L. Wang, Chem. – Asian J., 2016, 11, 1388–1393 CrossRef CAS PubMed.
- S. Koh and P. Strasser, J. Am. Chem. Soc., 2007, 129, 12624–12625 CrossRef CAS PubMed.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed.
- J. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., Int. Ed., 2005, 44, 2132–2135 CrossRef CAS PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS PubMed.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed.
- F. Kong, Z. Ren, M. Norouzi Banis, L. Du, X. Zhou, G. Chen, L. Zhang, J. Li, S. Wang, M. Li, K. Doyle-Davis, Y. Ma, R. Li, A. Young, L. Yang, M. Markiewicz, Y. Tong, G. Yin, C. Du, J. Luo and X. Sun, ACS Catal., 2020, 4205–4214 CrossRef CAS.
- D. Wang, Y. Yu, J. Zhu, S. Liu, D. A. Muller and H. D. Abruna, Nano Lett., 2015, 15, 1343–1348 CrossRef CAS PubMed.
- J. A. Wittkopf, J. Zheng and Y. Yan, ACS Catal., 2014, 4, 3145–3151 CrossRef CAS.
- D. Wang, Y. Yu, H. L. Xin, R. Hovden, P. Ercius, J. A. Mundy, H. Chen, J. H. Richard, D. A. Muller, F. J. Disalvo and H. D. Abruña, Nano Lett., 2012, 12, 5230–5238 CrossRef CAS PubMed.
- Z. Xiao, Y. Jiang, H. Wu, H. Zhong, H. Song, A. Abdelhafiz and J. Zeng, J. Alloys Compd., 2021, 877, 160221 CrossRef CAS.
- I. Dutta, M. K. Carpenter, M. P. Balogh, J. M. Ziegelbauer, T. E. Moylan, M. H. Atwan and N. P. Irish, J. Phys. Chem. C, 2010, 114, 16309–16320 CrossRef CAS.
- A. Abdelhafiz, B. Zhao, Z. Xiao, J. Zeng, X. Deng, L. Lang, Y. Ding, H. Song and M. Liu, ACS Appl. Mater. Interfaces, 2020, 12(44), 49510–49518 CrossRef CAS PubMed.
- M. Gong, Z. Deng, D. Xiao, L. Han, T. Zhao, Y. Lu, T. Shen, X. Liu, R. Lin and T. Huang, ACS Catal., 2019, 9, 4488–4494 CrossRef CAS.
- Z. Zhang, Z. Luo, B. Chen, C. Wei, J. Zhao, J. Chen, X. Zhang, Z. Lai, Z. Fan, C. Tan, M. Zhao, Q. Lu, B. Li, Y. Zong, C. Yan, G. Wang, Z. J. Xu and H. Zhang, Adv. Mater., 2016, 28, 8712–8717 CrossRef CAS.
- L. Cao, G. Zhang, W. Lu, X. Qin, Z. Shao and B. Yi, RSC Adv., 2016, 6, 39993–40001 RSC.
- J. X. Wang, C. Ma, Y. Choi, D. Su, Y. Zhu, P. Liu, R. Si, M. B. Vukmirovic, Y. Zhang and R. R. Adzic, J. Am. Chem. Soc., 2011, 133, 13551–13557 CrossRef CAS PubMed.
- A. Vitale, H. Murad, A. Abdelhafiz, P. Buntin and F. M. Alamgir, ACS Appl. Mater. Interfaces, 2019, 11(1), 1026–1032 CrossRef CAS PubMed.
- A. Abdelhafiz, A. Vitale, C. Joiner, E. Vogel and F. M. Alamgir, ACS Appl. Mater. Interfaces, 2015, 7(11), 6180–6188 CrossRef CAS PubMed.
- J. il Choi, A. Abdelhafiz, P. Buntin, A. Vitale, A. W. Robertson, J. Warner, S. S. Jang and F. M. Alamgir, Adv. Funct. Mater., 2019, 29(46), 1902274 CrossRef.
- X. Wang, L. Zhang, H. Gong, Y. Zhu, H. Zhao and Y. Fu, Electrochim. Acta, 2016, 212, 277–285 CrossRef CAS.
- A. Abdelhafiz, A. Vitale, P. Buntin, B. Deglee, C. Joiner, A. Robertson, E. M. Vogel, J. Warner and F. M. Alamgir, Energy Environ. Sci., 2018, 11, 1610–1616 RSC.
- F. Severino, J. L. Brito, J. Laine, J. L. G. Fierro and A. L. Agudo, J. Catal., 1998, 177, 82–95 CrossRef CAS.
- T. Ghodselahi, M. A. Vesaghi, A. Shafiekhani, A. Baghizadeh and M. Lameii, Appl. Surf. Sci., 2008, 255, 2730–2734 CrossRef CAS.
- E. J. Coleman and A. C. Co, J. Catal., 2014, 316, 191–200 CrossRef CAS.
- I. Mintsouli, J. Georgieva, S. Armyanov, E. Valova, G. Avdeev, A. Hubin, O. Steenhaut, J. Dille, D. Tsiplakides, S. Balomenou and S. Sotiropoulos, Appl. Catal., B, 2013, 136–137, 160–167 CrossRef CAS.
- M. Wakisaka, S. Mitsui, Y. Hirose, K. Kawashima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2006, 110, 23489–23496 CrossRef CAS PubMed.
- Z. Liu, A. A. Abdelhafiz, Y. Jiang, C. Qu, I. Chang, J. Zeng, S. Liao and F. M. Alamgir, Mater. Chem. Phys., 2019, 225, 371–378 CrossRef CAS.
- A. Holewinski and S. Linic, J. Electrochem. Soc., 2012, 159, H864–H870 CrossRef CAS.
- M. Li, Y. Lei, N. Sheng and T. Ohtsuka, J. Power Sources, 2015, 294, 420–429 CrossRef CAS.
- C. Cui, M. Ahmadi, F. Behafarid, L. Gan, M. Neumann, M. Heggen, B. R. Cuenya and P. Strasser, Faraday Discuss., 2013, 162, 91–112 RSC.
- D. C. Martínez-Casillas, H. A. Calderon, V. Collins-Martínez and O. Solorza-Feria, Int. J. Hydrogen Energy, 2013, 38, 12674–12680 CrossRef.
- Y. Kang, J. Snyder, M. Chi, D. Li, K. L. More, N. M. Markovic and V. R. Stamenkovic, Nano Lett., 2014, 14, 6361–6367 CrossRef CAS.
- W. Xiao, M. A. Liutheviciene Cordeiro, M. Gong, L. Han, J. Wang, C. Bian, J. Zhu, H. L. Xin and D. Wang, J. Mater. Chem. A, 2017, 5, 9867–9872 RSC.
- Y. Yu, H. L. Xin, R. Hovden, D. Wang, E. D. Rus, J. A. Mundy, D. A. Muller and H. D. Abruña, Nano Lett., 2012, 12, 4417–4423 CrossRef CAS PubMed.
- A. Sarkar and A. Manthiram, J. Phys. Chem. C, 2010, 114, 4725–4732 CrossRef CAS.
- Y. Zhang, S. Chen, Y. Wang, W. Ding, R. Wu, L. Li, X. Qi and Z. Wei, J. Power Sources, 2015, 273, 62–69 CrossRef CAS.
- R. Lin, L. Che, D. Shen and X. Cai, Electrochim. Acta, 2020, 330, 135251 CrossRef CAS.
- D. A. J. Rand and R. Woods, J. Electroanal. Chem. Interfacial Electrochem., 1974, 55, 375–381 CrossRef CAS.
- N. Ramaswamy, W. Gu, J. M. Ziegelbauer and S. Kumaraguru, J. Electrochem. Soc., 2020, 167, 64515 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr02820k |
| This journal is © The Royal Society of Chemistry 2021 |