
Synthesis, crystal molecular structure, and magnetic characteristics of coordination polymers formed by Co(II) diketonates with pentaheterocyclic triphenodioxazines†
Sergey
Aldoshin
 *a,
Eugeny
Ivakhnenko
b,
Gennadii
Shilov
a,
Valerii
Tkachev
a,
Andrei
Utenyshev
a,
Andreii
Palii
a,
Pavel
Dorovatovskii
c,
Anastasiia
Kovalenko
b,
Roman
Morgunov
a,
Anatoly
Metelitsa
b and
Vladimir
Minkin
b
*a,
Eugeny
Ivakhnenko
b,
Gennadii
Shilov
a,
Valerii
Tkachev
a,
Andrei
Utenyshev
a,
Andreii
Palii
a,
Pavel
Dorovatovskii
c,
Anastasiia
Kovalenko
b,
Roman
Morgunov
a,
Anatoly
Metelitsa
b and
Vladimir
Minkin
b
aInstitute of Problems of Chemical Physics, Russian Academy of Sciences, 1 Acad. Semenov Av., 142432 Chernogolovka, Russia. E-mail: sma@icp.ac.ru
bInstitute of Physical and Organic Chemistry, Southern Federal University, 194/2 Stachki St., 344090 Rostov on Don, Russia. E-mail: ivah@ipoc.rsu.ru
cNational Research Center “Kurchatov Institute”, 1, Academician Kurchatov square, Moscow, 123182, Russia
First published on 21st November 2020
Abstract
Stable crystalline complexes of Co(II) acetylacetonate [Co(II)(acac)2], trifluoacetylacetonate [Co(II)(tfac)2] and hexafluoroacetylacetonate [Co(II)(hfac)2] with triphenodioxazines (TPDOs) were synthesized and their structures studied using X-ray crystallography. In the crystal, complexes [Co(II)(tfac)2]TPDO and [Co(II)(hfac)2]TPDO form infinite ⋯N⋯Co⋯N⋯ chains featuring 1D coordination polymeric structures, whereas in the [Co(II)(acac)2]TPDO complex, the Co(acac)2 units fill only half of the possible crystallographic positions. The electron accepting trifluoro substituents in the diketonate moieties significantly enhance the thermal stability of the complexes with TPDO. Of all the complexes, only [Co(II)(hfac)2]TPDO does not dissociate into the components in solution. In all studied complexes, the Co(II) atom is in a high-spin state and has distorted octahedral surroundings. Distortion of the octahedral polyhedrons appears as axial stretching of the octahedrons along the Co–N bonds; it is due to the specific features of the crystalline structure of the metal polymeric chain in the compounds.
1. Introduction
Coordination polymers (CPs) composed of organic bridging ligands coordinatively bonded to metal ions1–3 and commonly referred to as two-dimensional metal organic frameworks (2D MOFs) have currently gained significant attention as a prospective class of solid state materials with unique magnetic,4 thermoelectric,5 optoelectronic,6 and mechanical7 properties. The relatively flexible coordination bonds formed by metal linkers with organic ligands provide for diverse modes of self-assembly of CP macromolecules and manifold types of their crystal structures. The crystal structure and dimensionality of CPs are strongly determined by the capability of the ligand to form multiple coordination bonds. In the extensive search for multiatom bridges ensuring formation of CPs with immense potential for a range of applications, significant attention is currently drawn to electrically neutral N-donor terminal bidentate linkers offering a platform for very stable 1D and 2D networks,3,4,8 where the dimensionality is determined by the number of directions in space the array extends.9 Generally, these platforms include derivatives of dipyridines, phenanthroline, pyrazine and various 5-membered ring systems such as imidazole, pyrazole, triazole or tetrazole.8,10 It has been shown that a good number of frameworks of CPs constructed on the basis of these bidentate ligands can efficiently transmit magnetic interactions between paramagnetic metal centers, Mn(II), Fe(II), and Co(II).4In the present work, we report on the synthesis of and investigation into the structure and magnetic properties of a new group of 1D cobalt CPs featuring previously unexplored N-heterocyclic bridging ligands – sterically crowded triphenodioxazines TPDOs I,11 in which the bidentate p-phenylene diamine fragment is embedded into the rigid π-conjugated pentacyclic fragment. The ability of triphenodioxazines I to function as an efficient linker in the formation of 1D CPs has been demonstrated by the readily occurring reaction of 2,4-di-(tert-butyl)benzo[5,6][1,4]oxazine[2,3-b]phenoxazines I with Co(II) acetylacetonate [Co(II)(acac)2], trifluoacetylacetonate [Co(II)(tfac)2] and hexafluoroacetylacetonate [Co(II)(hfac)2]. A series of stable complexes formed by Co(II) diketonates and salicylaldiminates with N-heterocyclic compounds has been previously prepared and structurally characterized (Scheme 1).12,13
 | ||
| Scheme 1 Synthesis of CPs II by the reaction of triphenodioxazines I with Co(II) diketonates. | ||
2. Results and discussion
2.1. Spectral studies
The relative stability of complexes IIa–c with respect to dissociation critically depends on the structure of the diketonate ligand. The electronic absorption spectra of IIa and IIb with acetylacetonate and trifluoroacetylacetonate ligands are almost identical to those of the related TPDO I,11 whereas the spectrum of IIc containing the hexafluoroacetylacetonate ligand is sharply different (Fig. 1) and remains practically unchanged upon heating of the solution. This observation is, obviously, indicative of dissociation of complexes IIa and IIb into the components. | ||
| Fig. 1 Electronic absorption spectra of compounds IIa–c (toluene, C = 1 × 10−5 M, T = 293 K). | ||
2.2. Crystalline structure
Complexes IIb and IIc are isostructural (Table 1), though in the TPDO ligand of complex IIc one of the H atoms in the aromatic ring is replaced by the Cl atom. The Co atom has distorted pseudo-octahedral coordination. In the equatorial plane, the Co atom is coordinated by two oxygen atoms O(3) and O(4) of the hfac ligand (IIc) and the tfac ligand (IIb), while in the apical position it is coordinated by the N(1) and N(1A) atoms of two TPDO ligands located above and beneath the equatorial plane at a 80.3 degree angle (Fig. 2). The N(2) and N(2A) atoms of the TPDO ligands coordinate with other Co atoms to form a polymeric chain ⋯Co⋯N(TPDO)N⋯Co⋯ (Fig. 2). Accordingly, the chemical structure of IIc and IIb can be represented as 1D CP: ⋯[2(hfac)Co(TPDO)]⋯ and ⋯[2(tfac)Co(TPDO)]⋯ (Fig. 3).| Parameters | Compound I | Compound IIa | Compound IIb | Compound IIc |
|---|---|---|---|---|
| a For compound IIa, the composition corresponding to the unit cell is indicated (Z = 1). | ||||
| Chemical formula | C26H25ClN2O2 | C114H114Cl4CoN8O12 | C36H34CoF6N2O6 | C36H27ClCoF12N2O6 |
| Crystal color | Colourless | Clear red | Clear red | Purple |
| Molecular weight, g mol−1 | 432.93 | 1988.01 | 763.58 | 905.98 |
| Temperature, K | 100 | 100 | 100 | 100 |
| CCDC | 2036311 | 2036044 | 2036045 | 2036046 |
| Crystal symmetry | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group | P21/c | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| Unit cell dimensions | a = 14.332(3) Å | a = 20.251(4) Å | a = 9.416(2) Å | a = 9.511(2) Å |
| b = 10.0370(2) Å | b = 6.596(1) Å | b = 13.569(3) Å | b = 13.682(3) Å | |
| c = 15.245(3) Å | c = 18.852(4) Å | c = 14.140(3) Å | c = 14.743(3) Å | |
| β = 95.01(3)° | β = 91.14(3)° | α = 94.41(3)° | α = 94.97(3)° | |
| β = 97.43(3)° | β = 95.59(3)° | |||
| γ = 105.87(3)° | γ = 104.55(3)° | |||
| Volume (V), Å3 | 2184.6(6) | 2517.7(9) | 1711.2(6) | 1836.0(6) |
| Z | 4 | 1a | 2 | 2 |
| Density calc. (ρ), g cm−3 | 1.316 | 1.313 | 1.482 | 1.639 |
| μ(MoKα), mm−1 | 0.795 | 0.462 | 0.785 | 0.869 |
| Crystal size, mm | 0.18 × 0.15 × 0.13 | 0.10 × 0.10 × 0.05 | 0.15 × 0.05 × 0.02 | 0.15 × 0.12 × 0.05 |
| Theta range for data collection | ≤31.06 | ≤30.98 | ≤31.01 | ≤30.78 |
| Measured reflections | 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 209 209 |
32055 |
20422 |
30614 |
| Independent reflections >2Θ(I) | 4963/2937 | 5738/4387 | 7564/5491 | 8321/6465 |
| Index ranges | −18 < h < 18 | −26 < h < 26 | −12 < h < 12 | −12 < h < 12 |
| −13 < k < 12 | −8 < k < 8 | −17 < k < 15 | −17 < k < 17 | |
| −19 < l < 19 | −24 < l < 24 | −18 < l < 18 | −19 < l < 19 | |
| Number of parameters refined | 287 | 337 | 580 | 557 |
| Goodness-of-fit | 1.006 | 1.032 | 1.031 | 1.037 |
| Final R indices | R 1 = 0.065 | R 1 = 0.059 | R 1 = 0.055 | R 1 = 0.045 |
 | ||
| Fig. 2 Structure of the coordination polyhedron in complex IIc. | ||
 | ||
| Fig. 3 1D CP formation in the crystalline structure of IIb (hydrogen atoms are not shown). | ||
In IIc one of the CF3 groups of the hfac ligand is disordered over two positions in a 1:1 ratio, while in IIb CH3 and CF3 of the tfac ligand are mutually disordered in a 1:1 ratio. The X-ray density of IIc and IIb is 1.639 and 1.482 g cm−3, respectively.
The crystalline structures of IIb and IIc are formed of two independent coordination units Co(1) and Co(2) located at the inversion centers, and therefore the Co–N and Co–O bond lengths and valence angles at the Co(1) and Co(2) atoms are equivalent in pairs. In the IIc structure, the average values of the Co–O bonds at the Co(1) and Co(2) centers are equal to 2.042 Å and 2.019 Å, and those of the Co–N bonds are equal to 2.357 Å and 2.447 Å. In the IIb structure, the average values of the Co–O bonds for Co(1) and Co(2) are equal to 2.028 Å and 2.026 Å, and the Co–N bonds are equal to 2.387 Å and 2.363 Å (Table S2, ESI†). The average values for Co–O and Co–N are 2.027 and 2.375 Å. Thus, the Co–O bond lengths in IIb and IIc are identical within the margin of error, while the Co–N bond in IIc is longer than in IIb. All bond lengths have a margin of error of 0.002 Å. As far as the cobalt atoms are located at the inversion centers, the valence angles between the symmetric atoms in the Co polyhedrons are equal to 180°; other angles lie in the range of 85–94°.
The structure of complex IIa is essentially different. As follows from X-ray analysis, the complex composition is 0.25[Co(acac)2](TPDO). In the crystalline structure of the complex, coordination units Co(acac)2 occupy only 50% of their crystallographic positions. The Co atoms cannot occupy the adjacent positions because in this case the acac ligands would overlap, which is physically impossible and forbidden by the rules of molecular packing in the crystal (Fig. 4). The distance between the Co⋯Co positions is 6.596 Å.
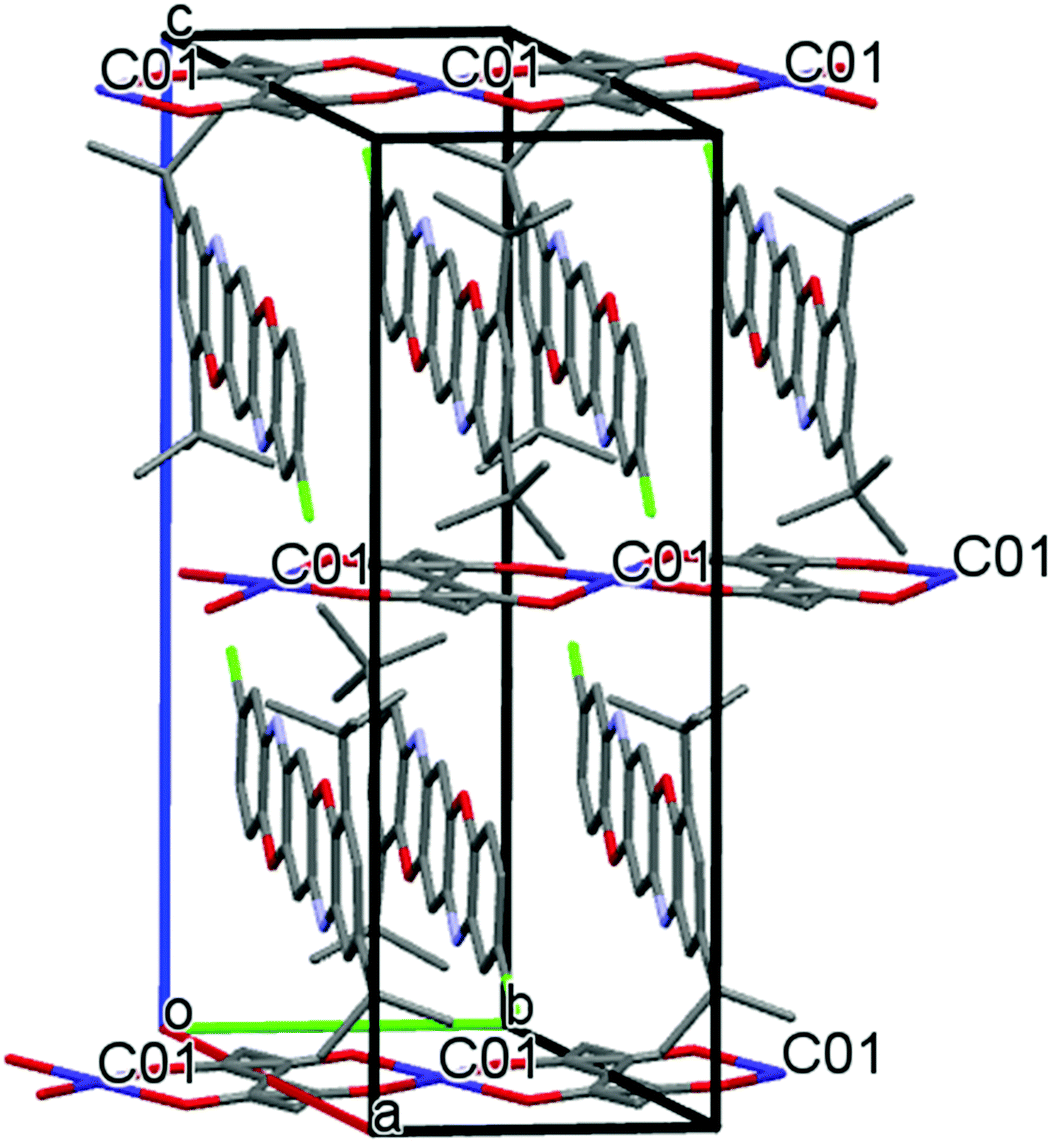 | ||
| Fig. 4 Crystalline structure of IIa. The hydrogen atoms are not shown. | ||
Fig. 4 shows the crystalline structure of compound IIa. TPDO molecules are parallel to the ac plane, and they are stacked in the “head to tail” mode along the b axis of the unit cell. There are many shortened intermolecular contacts between the atoms of TPDO molecules (see Table S1, ESI†). The unit cell contains two TPDO stacks, with the Co(acac)2 coordination units between them, which form chains along the b axis.
The Co atom has increased mean-square shifts along the unit cell c axis as compared to the other atoms. The ellipsoid of Co thermal vibrations is stretched towards the Co–N bond (Fig. 5).
 | ||
| Fig. 5 Fragment of the crystalline structure of IIa. Atoms are shown as ellipsoids, hydrogen atoms are not shown. | ||
The amplitude of the cobalt atom displacement is 0.30 Å, and that of the ligand oxygen atoms is 0.17 Å. Based on the difference in the displacement values, the assumption can be made that the Co atom shifts from the ligand ac plane towards the N atom of the TPDO ligand. This displacement is due to Co thermal vibrations, or to static disordering, or to both. In any case, it results in distortions of the coordination polyhedron. In the case of static disordering, at least three options of the complex can be available in the crystal. For the first option, the cobalt atom is located in a mean square position and its planar square coordination is complemented to an octahedral one by the ligand's nitrogen atoms N(1) and N(2), which are located at a distance of 2.67 Å. For the second and third options, the Co atom shifts up or down from the ligand plane. In these cases, identical distorted octahedrons form, and the distance from the Co atom to one nitrogen atom decreases, and that to the other increases.
Due to the 50% occupancy of the Co(acac)2 coordination units, the neighboring mutually exclusive positions of the Co atoms are linked by translation along the b axis; hence, this translational symmetry for the complexes is forbidden. However, if a unit cell with a doubled b period is considered, coordination units can be placed in the crystalline structure in a consistent mode without violation of the translational symmetry. One of the options is shown in Fig. 6a. The period is doubled; the unit cell contains eight TPDO molecules and two Co(acac)2 coordination units. By multiplying the content of this unit cell by translations, a crystalline structure with a consistent arrangement of complexes can be obtained. Fig. 6b shows the second option of the consistent arrangement of the complexes in the crystalline structure.
 | ||
| Fig. 6 (a and b) Projection of the crystalline structure on the bc plane. The b parameter of the unit cell is doubled. (a) The first option of the structure; and (b) the second option of the structure. | ||
Considering the experimental reflection intensities with a doubled b period, an inconsiderable quantity of very weak reflections with odd k indexes is present. This rather corroborates the assumption of doubling the b parameter of the unit cell. However, the crystalline structure in this unit cell cannot be refined because of a very small number of additional reflections and their weakness. If the distribution of the electron density is plotted for one of these suggested structures, there will be peaks of the electron density corresponding to the crystalline structure involving both options of the complex arrangement.
The question is how these options exist in the crystal. For example, they can be twins. In this case, squares of structural amplitudes F2 are added, i.e.,
| F2 = F12 + F22. | (1) |
Both options yield identical powder theoretical spectra (see Fig. 7a, spectrum I). There are reflections with odd k indexes in this spectrum.
 | ||
| Fig. 7 (a) Theoretical powder patterns for two crystal types. I – for the twin crystal. II – two packing options in the same coherent block. (b) Experimental spectrum (I) and the theoretical one for the initial structure (II). | ||
For the second option, both components are present in the same coherent block; the structural amplitudes are added
| F = F1 + F2, | (2) |
Accordingly, with a high degree of probability, the crystals under study consist of blocks containing both structural types; simultaneously, the unit cell should have the b period twice as high as that determined from the experiment. Only in such a way can the cobalt atoms be distributed consistently in the structure, with the translational symmetry being kept. To transform from one structure type to another in the layers where the cobalt atoms are located, at least one additional position of the complex in the chains along the b axis should be empty. Then, in the other part of the crystallite block, Co complexes can be allocated in the position corresponding to the second structure type. We can assume that there are a few additional empty positions, and then they can be occupied by smaller complexes, for example, Co(OH)2. The Co atoms can be arranged in chains in adjacent positions to form a stratum. Let's consider a mathematical model describing such a crystal type. For our specific case the structural amplitude will look like
| F(H) = F1(H) + 1/2F2(H) + 1/2F2′(H), | (3) |
As follows from the studies, the phenomena we observe are due to the specific features of the crystalline structure arrangement. TPDO molecules form a large number of mutual intermolecular contacts, thus forming stacks along the b axis. This part of the structure is well described in a small unit cell. The Co(acac)2 position is determined by the arrangement geometry of TPDO molecules, whose nitrogen atoms complement the Co atom coordination to give an octahedral one by interacting with it. Exactly due to these interactions, a flat-square Co(acac)2 is kept in the structure because no other interactions in the chain along the b axis are available.
In complex IIa, the Co–N distances (2.674(3) Å) appear to be much longer than in complexes IIc (2.402 Å) and IIb (2.375 Å), and the Co–O bonds shorter (1.994(3) and 1.997(3) Å) than in IIc (2.030 Å) and IIb (2.027 Å). The Co–N distance appears to be the longest among the available Co(II) complexes, which is the evidence of the planar square coordination of Co, which is complemented to a square-bipyramidal one (4 + 2) by weak interactions with the nitrogen atoms of TPDO ligands. In the crystalline structure of IIa, no infinite chains of ⋯N⋯Co⋯N⋯ type form, and the complex structure is Co(acac)2(TPDO)4. The N(2) atom of the TPDO ligand, which does not coordinate with the Co atom, has nearest contacts N(2)⋯O(2′) and N(2)⋯O(2′′) = 3.83 and 3.87 Å with the adjacent molecules. Positions not occupied by the Co(acac)2 units stay vacant. The intermolecular distances N⋯N between the neighboring TPDO molecules are: N(2)⋯N(2′) = 5.34 and 5.408 Å; and N(2)⋯N(1′) = 4.57 and 4.68 Å. The X-ray density of the IIa crystals is 1.32 g cm−3, which is much lower than that of the IIc and IIb crystals (1.639 and 1.482 g cm−3).
To find out whether the crystalline structure of the TPDO ligand is responsible for the structure of complex IIa, an X-ray study of TPDO crystals was performed. However, neither the parameters of the crystalline unit cell (Table 1) nor the mode of molecular packing in the crystals of the TPDO ligand (Fig. 8) correspond to the crystalline structure of IIa.
 | ||
| Fig. 8 Chemical structure (a) and crystal structure (b) of 2,4-di-(tert-butyl)-9-chloro-benzo[5,6][1,4]-oxazine[2,3-b]phenoxazine. | ||
The obtained data suggest that formation of diacetylacetonate Co(II) complexes with TPDO ligands is strongly affected by the CF3 groups of acac-ligands, which strengthen the electron accepting properties and are responsible for the formation of Co–N bonds of different strength, and for various distortions of the coordination polyhedron of the Co atom. Distortion of the coordination polyhedron of the high-spin Co(II) can lead to a difference in the magnetic properties of the complexes under study.
2.3. Magnetic studies
At room temperature χMT for complex IIc is around 3.5 cm3 K mol−1 (Fig. 9), which is considerably higher than the spin only value of IIc (1.875 cm3 K mol−1), due to a significant orbital contribution to the magnetic moment accompanied by the presence of a non-zero temperature independent paramagnetic contribution (TIP). Upon cooling, χMT almost linearly decreases in the temperature range from 300 to 100 K due to the TIP; then the decrease becomes more pronounced, and χMT reaches a value of ≈1.535 cm3 K mol−1 at 3 K. Apparently, the low-temperature decrease of χMT can be due to single-ion magnetic anisotropy of the Co(II) ion. The low-temperature (T = 2 K) magnetization as a function of magnetic field almost saturates at 5 T, reaching a value of around 2NAμB (Fig. 10), which is significantly lower than the value of 3NAμB corresponding to the pure spin S = 3/2 ground state with g = 2.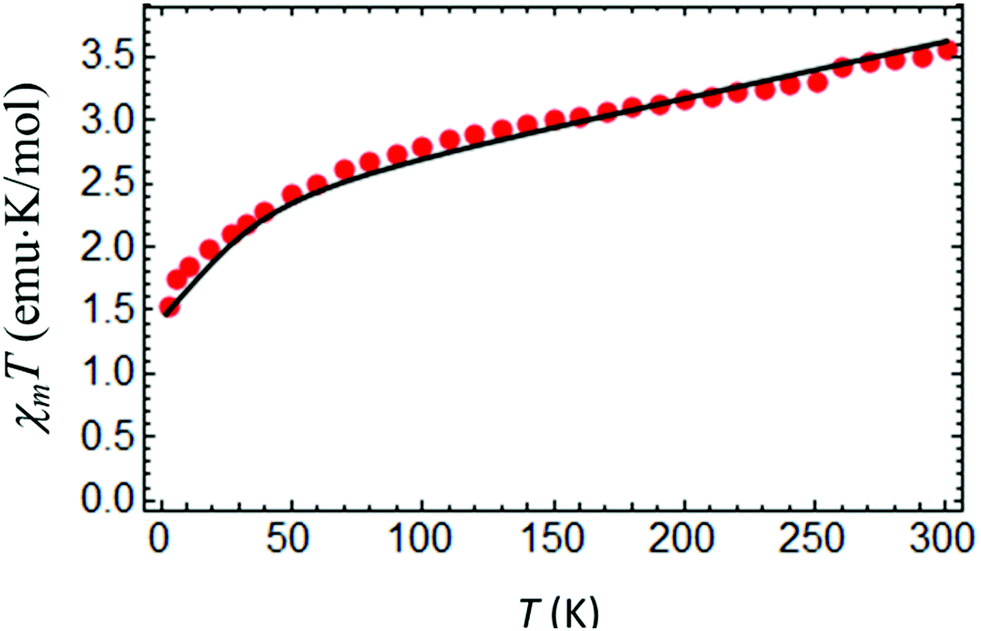 | ||
| Fig. 9 Temperature dependence of χMT for IIc measured at BDC = 0.1 T. Theoretical curve (solid line) calculated with gZ = 2.06, gX,Y = 2.32, D = 35.09 cm−1, E = 4.89 cm−1 and χtip = 4.7 × 10−3 emu K mol−1. | ||
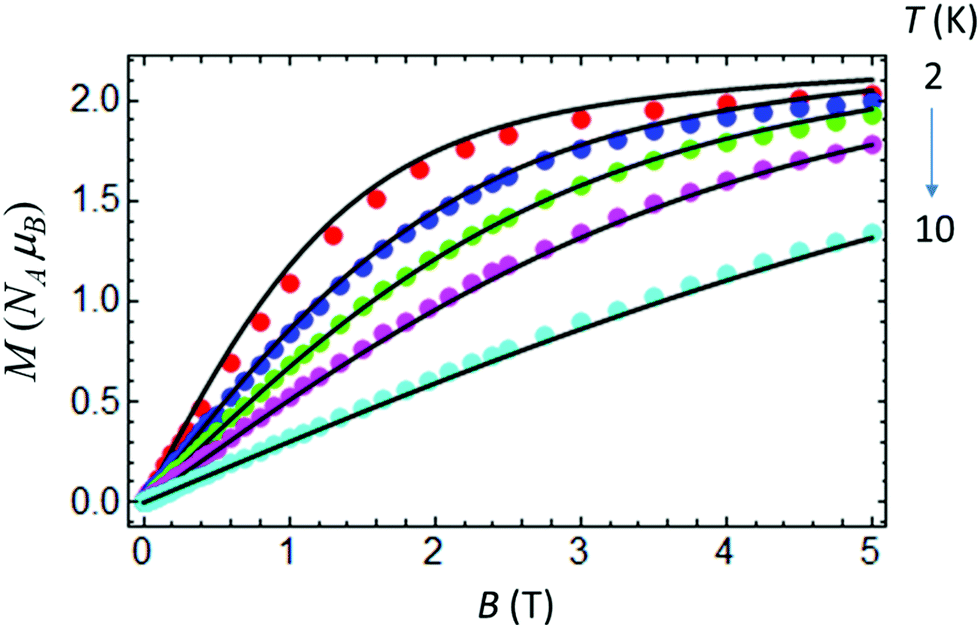 | ||
| Fig. 10 Magnetization vs. field for IIc measured at T = 2, 3, 4, 5.5 and 10 K. Theoretical curves (solid lines) calculated with gZ = 2.06, gX,Y = 2.32, D = 35.09 cm−1 and E = 4.89 cm−1. | ||
In order to choose an adequate theoretical model for the description of these DC magnetic properties, one should take into account that the octahedral surroundings of the Co(II) ion are stretched along one of the tetragonal axes (see detailed discussion in the structural part of the article). For such distortion, one can expect that the ground tetragonal crystal-field term of the Co(II) ion is a singlet orbital 4A2g, and spin–orbit splitting of this term into two Kramers doublets can be described by the following ZFS spin-Hamiltonian:
| Ĥ = D[ŜZ2 − 1/3S(S + 1)] + E(ŜX2 − ŜY2) + μB(BXgXŜX + BYgYŜY + BZgZŜZ) | (4) |
The set of best-fit parameters is found to be: D = 35.09 cm−1, E = 4.89 cm−1, gZ = 2.06, and gX,Y = 2.32. It can be seen that the system exhibits strong easy-plane type magnetic anisotropy with a weak rhombic component. Also, in order to reproduce the slope of the χMT vs. T curve, one has to introduce the TIP susceptibility χtip = 4.7 × 10−3 emu K mol−1. The found parameters allow both the observed temperature dependence of χMT (Fig. 9) and the field dependences of magnetization at different temperatures to be satisfactorily described (Fig. 10).
Fig. 11 shows the temperature dependences of χMT for the two other complexes, whose field dependences of magnetization do not obey the formalism described above. In complex IIb (violet), the magnetization has non-typical low values, which are not consistent with reasonable parameters, while, for complex IIa (green), the field dependence of the magnetization demonstrates hysteresis. Taking into account specific features of the complex IIa structure, we can assume that part of the Co vacancies are occupied by a ferromagnetic admixture, for example, cobalt hydroxide Co(OH)2. The presence of such an admixture is responsible for the observed magnetization hysteresis. Investigation of the surface of the IIa crystals by X-ray photoelectron spectroscopy (XPS) corroborates the presence of Co with OH hydroxyl groups and a Co concentration excess in the near-surface layer. The Co:Cl ratio in the near-surface layer is 1:2 instead of 1:4, as follows from the crystalline structure of IIa (2, ESI†).
 | ||
| Fig. 11 Temperature dependence of χMT for IIb (violet circles) and IIa (green circles) measured at BDC = 0.1 T. Theoretical curves (solid lines) are calculated with gZ = 1.92, gX,Y = 2.47, D = 59.34 cm−1, and E = 5.36 cm−1 for complex IIb and with gZ = 2.0, gX,Y = 2.42, D = 28.31 cm−1, E = 3.65 cm−1, and χtip = 2.1 × 10−3 emu K mol−1 for complex IIa. | ||
The initial temperature dependence of the effective magnetic moment μeff = 2.828(χMT)1/2 contained a small peak in the temperature dependence, which confirms the presence of the Co based nanoparticles in the synthesized powder (3, Fig. 3S1, ESI†). This peak in the χMT dependence was extracted by modeling of the ZFC curve by the standard formula applicable to ferromagnetic nanoparticles with parameters consistent with the peak position and amplitude. The resulting χMT(T) dependence is presented in Fig. 11. The solid line demonstrates a good approximation with reasonable parameters gZ = 2.0, gX,Y = 2.42, D = 28.31 cm−1, E = 3.65 cm−1, and χtip = 2.1 × 10−3 emu K mol−1 for complex IIa.
3. Experimental
3.1. Synthesis
The initial 2,4-di-(tert-butyl)-benzo[5,6][1,4]oxazine[2,3-b]phenoxazines were prepared according to the previously described procedure11 and characterized by 1H and 13C NMR spectra and mass spectroscopy. All other reagents and solvents were purchased from commercial sources (Aldrich) and used without additional purification. IR spectra were recorded on a Varian Excalibur 3100 FT-IR instrument using the attenuated total internal reflection technique (ZnSe crystal). UV-vis spectra were registered on a Varian “Cary 100” spectrophotometer. Complexes IIa–c were synthesized by refluxing heptane or o-xylene solutions of the heteropentacene ligands I and Co(II) diketonates.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cl) in heptane (10 mL), and the common solution was stirred under reflux for 70 min. The solution changes its color from the initial red to green. The deposited crystals were filtered off and washed away with water from the unreacted acetylacetonate. The precipitate was dried and recrystallized from toluene to yield 190 mg (83%) of brown crystals of IIa. M.p.: 248–250 °C. IR spectrum (cm−1): 3399.9, 2952.0, 2907.46, 2868.6, 1581.8, 1561.6, 1518.7, 1440.1, 1391.4, 1376.9, 1361.5, 1266.1, 1240.5, 1176.4, 1018.2, 930.0, 866.4, 800.3, 767.5, 581.4.
H) in heptane (10 mL), and the common solution was stirred under reflux for 30 min. The deposited reddish crystals (200 mg, yield 79%) were purified in the same way as those of IIa. M.p.: 272–274 °C. IR spectrum (cm−1): 3108.2, 2955.9, 2869.6, 1553.9, 1515.8, 1463.2, 1389.5, 1359.6, 1295.0, 1223.6, 1182.2, 1135.9, 857.7, 774.3, 756.4, 743.0.
Cl) in xylene (10 mL), and the common solution was stirred under reflux for 40 min. In the course of the reaction, the solution color changes from red to violet. The deposited light brown crystals (195 mg, yield 61%) were purified in the same way as those of IIa and b. M.p. > 275 °C. IR spectrum (cm−1): 3483.3, 3368.1, 3275.5, 1637.8, 1563.5, 1536.5, 1477.7, 1252.1, 1200.0, 1139.3, 1093.0, 804.7, 673.5, 588.7.
Cl) in heptane (10 mL), and the common solution was stirred under reflux for 70 min. The solution changes its color from the initial red to green. The deposited crystals were filtered off and washed away with water from the unreacted acetylacetonate. The precipitate was dried and recrystallized from toluene to yield 190 mg (83%) of brown crystals of IIa. M.p.: 248–250 °C. IR spectrum (cm−1): 3399.9, 2952.0, 2907.46, 2868.6, 1581.8, 1561.6, 1518.7, 1440.1, 1391.4, 1376.9, 1361.5, 1266.1, 1240.5, 1176.4, 1018.2, 930.0, 866.4, 800.3, 767.5, 581.4.
H) in heptane (10 mL), and the common solution was stirred under reflux for 30 min. The deposited reddish crystals (200 mg, yield 79%) were purified in the same way as those of IIa. M.p.: 272–274 °C. IR spectrum (cm−1): 3108.2, 2955.9, 2869.6, 1553.9, 1515.8, 1463.2, 1389.5, 1359.6, 1295.0, 1223.6, 1182.2, 1135.9, 857.7, 774.3, 756.4, 743.0.
Cl) in xylene (10 mL), and the common solution was stirred under reflux for 40 min. In the course of the reaction, the solution color changes from red to violet. The deposited light brown crystals (195 mg, yield 61%) were purified in the same way as those of IIa and b. M.p. > 275 °C. IR spectrum (cm−1): 3483.3, 3368.1, 3275.5, 1637.8, 1563.5, 1536.5, 1477.7, 1252.1, 1200.0, 1139.3, 1093.0, 804.7, 673.5, 588.7.
3.2. X-ray crystallography
Single crystals of complexes IIa–c were grown from heptane or xylene solutions. The compounds can be stored in air for a long time without any signs of decomposition.The structures of the three Co(II) diacetylacetonate complexes with pentacyclic ligand triphenodioxazine (TPDO) (hexafluoroacetylacetonate complex “hfac” (IIc), trifluoroacetylacetonate complex “tfac” (IIb), and acetylacetonate complex “acac” (IIa)) and of the TPDO ligand (I) have been studied on single crystals on the ‘Belok’ beamline diffractometer of the National Research Center “Kurchatov Institute” (Moscow, Russia) using a Rayonix SX165 detector at λ = 0.79272 Å (for IIb and IIa), 0.78790 Å (for IIc), and 0.79475 Å (for I); synchrotron radiation at 100 K was used. The data were indexed and integrated in the CCP4 program using the iMOSFLM utility,14 and then scaled with absorption correction by means of the Scala program.15 The crystallographic data and main refinement parameters are shown in Table 1. The structures were solved by the direct method.16 Positions and temperature parameters of non-hydrogen atoms were refined in the anisotropic approximation by the full-matrix least squares method.16 Positions of all hydrogen atoms were calculated geometrically, and then were refined by setting restrictions according to the “riding” model. All calculations were performed using the SHELXTL program complex.16 Main bond lengths and valence angles for complexes I and IIa–c are presented in the ESI,† 1 (Tables S1–S4, respectively). The X-ray crystal structure data have been deposited with the Cambridge Crystallographic Data Center, with reference codes CCDC 2036311, 2036044, 2036045, and 2036046.†
3.3. Magnetic measurements
A powder sample of ∼10 mg was tightly packed in a diamagnetic capsule, its contribution being extracted. Samples were mounted in an MPMS XL (Quantum Design) SQUID magnetometer operating in the highly sensitive RSO (reciprocating sample option) mode. The dependences of magnetic moment M on temperature (T = 2–300 K) were recorded in a DC magnetic field H = 1000 Oe. The field dependences of the magnetic moment (H = 0–50 kOe) were recorded at a few temperatures in the range of 1.8–5 K. The experimental values of the magnetic moment obtained for each point of the M(T) dependence were used to calculate the values of the effective magnetic moment μeff using the following formula: μeff = (3χMT/NAμB)1/2 ≈ (8MT/νH)1/2μb, where χMT = M/νH is the molar static magnetic susceptibility, k is the Boltzmann constant, T is temperature, NA is the Avogadro constant, and μB is the Bohr magneton.4. Conclusions
In the search for stable and magnetically active coordination polymers formed by transition metal ions and multicenter organic ligands, new bidentate pentaheterocyclic bridging ligands – sterically crowded 2,4-di-(tert-butyl)-benzo[5,6][1,4]oxazine[2,3-b]phenoxazine I (TPDO) and its 9-chloro derivative – were studied. When reacting with Co(II) acetylacetonate, trifluoroacetylacetonate, and hexafluoroacetylacetonate, the TPDOs form stable crystalline complexes [Co(II)(tfac)2]TPDO, and [Co(II)(hfac)2]TPDO. In the crystal, complexes [Co(II)(tfac)2]TPDO and [Co(II)(hfac)2]TPDO form infinite ⋯N⋯Co⋯N⋯ chains featuring 1D coordination polymeric structures, whereas in the [Co(II)(acac)2]TPDO complex, the Co(acac)2 units fill only half of the possible crystallographic positions. The electron accepting trifluoro substituents in the diketonate moieties significantly enhance the thermal stability of the complexes with TPDO. Of all the complexes, only [Co(II)(hfac)2]TPDO does not dissociate into the components in solution.In all studied complexes IIa–c, the Co(II) atom is in a high-spin state and has distorted octahedral surroundings. Distortion of the octahedral polyhedrons appears as axial stretching of the octahedrons along the Co–N bonds; it is due to the specific features of the crystalline structure of the metal polymeric chain in IIb, IIc, and complex IIa. The maximal distortion of the coordination polyhedron is observed for IIa, and this is due to the weak accepting properties of acac-ligands as compared to tfac- and hfac-ligands. The Co atom in the crystalline structure of IIa has abnormally high thermal vibrations along the Co–N bonds, which could be due to dynamic and static disorder of the Co atom. Its DC magnetic properties could not be described satisfactorily.
The studied IIa–c complexes show a typical temperature–frequency dependence of the magnetic susceptibility in an external magnetic field, which points to slow magnetic relaxation of magnetization (AC properties); this will be the subject of further thorough research.
Author contributions
Conceptualization, S. M. A.; methodology, E. P. I. and G. V. S.; synthesis, E. P. I.; validation, A. A. K. and A. V. P.; investigation, G. V. S., V. V. T., A. N. U., R. B. M., A. V. P., and P. V. D.; resources, S. M. A. and V. I. M.; data curation, S. M. A.; writing—original draft preparation, V. I. M., and S. M. A.; writing—review and editing, S. M. A.; supervision, S. M. A.; project administration, S. M. A. and A. V. M.; funding acquisition, S. M. A. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by the Ministry of Science and Higher Education of the Russian Federation (Grant No. 075-15-2020-779). The authors acknowledge Oksana V. Koplak (Multi-User Analytical Center of the Institute of Problems of Chemical Physics, RAS) for XPS investigations.References
- O. L. Ortiz and L. D. Ramırez, Coordination Polymers and Metal Organic Frameworks: Properties, Types, and Applications, Nova Science Publishers, 2012 Search PubMed.
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O’Keeffe, M. P. Suh and J. Reedijk, IUPAC Terminology of metal-organic frameworks and coordination polymers, Pure Appl. Chem., 2013, 85, 1715–1724, DOI:10.1351/PAC-REC-12-11-20.
- M. Tran, K. Kline, Y. Qin, Y. Shen, M. D. Green and S. Tongay, 2D coordination polymers: design guidelines and materials perspective, Appl. Phys. Rev., 2019, 6, 041311, DOI:10.1063/1.5110895.
- A. E. Thorarinsdottir and T. D. Harris, Metal–Organic Framework Magnets, Chem. Rev., 2020, 120, 8716–8789 CrossRef CAS PubMed.
- Y. Lu and D. J. Young, Coordination Polymers for n-Type Thermoelectric Applications, Dalton Trans., 2020, 49, 7644–7657, 10.1039/D0DT00872A.
- W. Xinga, P. Yea, J. Lub, X. Wua, Y. Chena, T. Zhua, A. Penga and H. Huanga, Tellurophene-based metal–organic framework nanosheets for high-performance organic solar cells, J. Power Sources, 2018, 401, 13–18 CrossRef.
- G. I. Dzhardimalieva and I. E. Uflyand, Design and synthesis of coordination polymers with chelated units and their application in nanomaterials science, RSC Adv., 2017, 7, 42242–42288, 10.1039/C7RA05302A.
- A. V. Desai, S. Sharma, S. Let and S. K. Ghosh, N-Donor Linker Based Metal-Organic Frameworks (MOFs): Advancement and Prospects as Functional Materials, Coord. Chem. Rev., 2019, 395, 146–192 CrossRef CAS.
- X. Chen, B. Ye and M. Tong, Metal–organic molecular architectures with 2,2-bipyridyl-like and carboxylate ligands, Coord. Chem. Rev., 2005, 249, 545–565 CrossRef.
- A. Y. Robin and K. M. Fromm, Coordination polymer networks with O- and N-donors. What they are and how they are made, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS.
- E. P. Ivakhnenko, G. V. Romanenko, N. A. Makarova, A. A. Kovalenko, P. A. Knyazev, I. A. Rostovtseva, A. G. Starikov and V. I. Minkin, A new approach to the synthesis of the sterically crowded photostable and fluorescent phenodioxazines, Dyes Pigm., 2020, 176, 108174 CrossRef CAS.
- M. Y. Antipin, E. P. Ivakhnenko, Y. V. Koshchienko, P. A. Knyazev, M. S. Korobov, A. V. Chernyshev, K. A. Lyssenko, A. G. Starikov and V. I. Minkin, Adducts of cobalt(II) bis(salicylaldiminates) and redox-active Phenoxazin-1-one: Synthesis, structure, and magnetic properties, Russ. Chem. Bull., 2013, 62, 1744–1751 CrossRef CAS.
- V. I. Minkin and A. A. Starikova, Molecular design of the valence tautomeric mixed-ligand adducts of CoII diketonates with redox-active ligands, Mendeleev Commun., 2015, 25, 83–92 CrossRef CAS.
- T. G. G. Battye, L. Kontogiannis, O. Johnson, H. R. Powell and A. G. W. Leslie, IMosflm: a new graphical interface for diffraction-image processing with MOSFLM, Acta Crystallogr., 2011, D67, 271–281 CrossRef PubMed.
- P. R. Evans, Scaling and assessment of data quality, Acta Crystallogr., 2006, D62, 72–82 CrossRef CAS PubMed.
- G. M. Sheldrick, (8/06/2000). SHELXTL v. 6.14. Structure Determination Software Suite, Bruker AXS: Madison, Wisconsin, USA, 2000 CrossRef PubMed; G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary materials 1, 2, and 3 are available. 1. X-ray data: Table S1: Bond lengths [Å] and angles [deg] for IIc, Table S2: Bond lengths [Å] and angles [deg] for IIb; Table S3: Bond lengths [Å] and angles [deg] for IIa; Table S4: Bond lengths [Å] and angles [deg] for I. 2. XPS investigations. 3. Magnetic data. CCDC 2036311, 2036044–2036046. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0nj05279e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |