
Fabrication of porous ultrathin carbon nitride nanosheet catalysts with enhanced photocatalytic activity for N- and O-heterocyclic compound synthesis†
Yancong
Li
,
Jiliang
Ma
 *,
Zhendong
Liu
,
Dongnv
Jin
,
Gaojie
Jiao
,
Yanzhu
Guo
,
Qiang
Wang
,
Jinghui
Zhou
and
Runcang
Sun
*
*,
Zhendong
Liu
,
Dongnv
Jin
,
Gaojie
Jiao
,
Yanzhu
Guo
,
Qiang
Wang
,
Jinghui
Zhou
and
Runcang
Sun
*
Center for Lignocellulosic Chemistry and Biomaterials, College of Light Industry and Chemical Engineering, Dalian Polytechnic University, Dalian, 116034, China. E-mail: jlma@dlpu.edu.cn; rcsun3@dlpu.edu.cn
First published on 19th November 2020
Abstract
A simple and efficient photocatalytic method for the synthesis of dihydropyrimidinones (DHPMs) and their derivatives via porous ultrathin carbon nitride nanosheets (p-CNNs) without solvents was demonstrated. The yields of 3,4-dihydropyrimidin-2(1H)-ones/thiones and their derivatives were up to 97.0%. Furthermore, the yield of 5-ethoxycarbonyl-6-methyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one in the 10th cycle retained 98.9% of its 1st value. Considering the environmental and economic benefits, this work exhibits various merits including excellent yields, environmental friendliness, inexpensiveness, and avoiding the use of solvents and metal-based photocatalysts. In addition, the excellent performance of the p-CNNs and environmentally benign reaction system have also been checked by the photocatalytic synthesis of 12-phenyl-9,9-dimethyl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one and 5-phenyl-1(4-methoxyphenyl)-3[(4-methoxy-phenyl)-aminol]-1H-pyrrol-2(5H)-one. This work paves a new way for carrying out a three-component reaction using metal-free photocatalysts under mild reaction conditions.
Introduction
Green organic synthesis has become a matter of great concern in recent years. To achieve this goal, various strategies have been developed involving three criteria: solvent, catalyst, and synthesis method.1–3 Photocatalysis as a green synthesis method has attracted much attention due to its high efficiency, easy operation, mild reaction conditions, wide application, low cost, low energy consumption and so on.4–6 The key to proper design of a photocatalytic reaction is to first design and prepare an appropriate photocatalyst. The traditional photocatalysts, such as metal oxides (ZnO, TiO2)7,8 and metal sulfides (CdS, ZnS),9,10 exhibited high photocatalytic performance. However, some of them suffered from the narrow absorption range of visible light or light corrosion, which limited their application. Recently, a series of novel photocatalysts have been reported, such as, BiOX (X = Cl, Br, I), metal–organic frameworks (MOFs), covalent organic frameworks (COFs) or functional carbon nitride.11–16 These functionalized photocatalysts have made great achievements in the fields of solar water splitting,14 CO2 reduction,17–19 N2 fixation,20 biorefinery21 and organic synthesis.22 High cost and complex preparation processes of other photocatalysts as compared with functional carbon nitride makes these photocatalysts inconsistent with green chemistry development requirements. Thus, taking environmental and economic benefits into consideration, the development of various metal-free, environmentally friendly and inexpensive functional carbon nitride photocatalysts for use in organic synthesis is of great significance.Dihydropyrimidinones (DHPMs) and their derivatives are a series of heterocyclic compounds, which are widely distributed in nature. Since the DHPMs were discovered, they have attracted much attention due to their wide range of biological activities, such as antiviral, antimitotic, anticarcinogenic and antihypertensive effects.23–25 Furthermore, some functionalized DHPMs are the constituents of neuropeptide Y antagonists, calcium channel modulators and α-1a antagonists.26–28 Especially Batzelladine A and B play an important role in the HIV gp-120-CD4 treatment.29 Therefore, developing a mild and efficient strategy is significant for the synthesis of DHPMs.
The first straightforward method for the synthesis of DMPHs via condensation of aldehydes, β-dicarbonyl compounds and urea or thiourea reported by P. Biginelli (the reaction was then named as Biginelli reaction)30 inspired a new wave of research on the synthesis and application of DHPMs. And after that, various strategies for carrying out Biginelli reaction were developed.31–36 However, most of them suffered from longer reaction time, harsher reaction conditions, high cost, and tedious work-up or purification. Currently, several new protocols with natural catalysts (e.g. citric acid,37 baker's yeast,38 phytic acid,39D-xylonic acid,2etc.) for carrying out Biginelli reaction have been demonstrated, which gave good yields. The only problem is that the recovery and reuse of these catalysts are relatively complicated. In this regard, efficient heterogeneous catalysts have aroused great interest among researchers. Taking into account the advantages of photocatalysis, developing a metal-free and efficient heterogeneous photocatalyst for carrying out Biginelli reaction has vital significance.
In this study, p-CNN photocatalyst was prepared by direct calcination of copper oxide nanobelts (CuO NBs) and melamine, which could effectively synthesize 3,4-dihydropyrimidin-2(1H)-ones/thiones and their derivatives photocatalytically without solvents under mild conditions (Scheme 1). The performance of p-CNNs in the photothermal mediated Biginelli reaction was assessed at varying irradiation time (30–120 min), temperature (30–100 °C), photocatalyst dosage (0.56–5.60 wt%) and feed ratios. Furthermore, the stability and recycling of p-CNNs were also investigated. In addition, the excellent performance of p-CNNs and the environmentally benign reaction system were also checked by the photocatalytic synthesis of 12-phenyl-9,9-dimethyl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one and 5-phenyl-1(4-methoxyphenyl)-3[(4-methoxy-phenyl)-aminol]-1H-pyrrol-2(5H)-one (Schemes 2 and 3).
 | ||
| Scheme 1 p-CNNs photocatalytic synthesis of DHPMs under solvent-free conditions. | ||
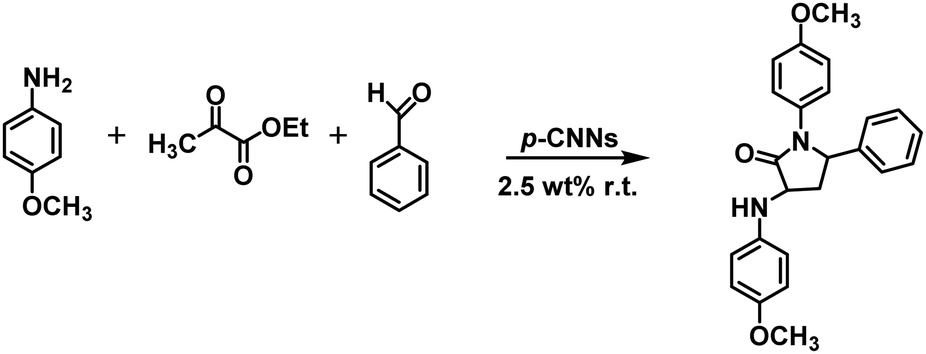 | ||
| Scheme 2 Photocatalytic p-CNNs for the synthesis of 5-phenyl-1(4-methoxyphenyl)-3[(4-methoxyphenyl)-amino]-1H-pyrrol-2(5H)-one under solvent-free conditions. | ||
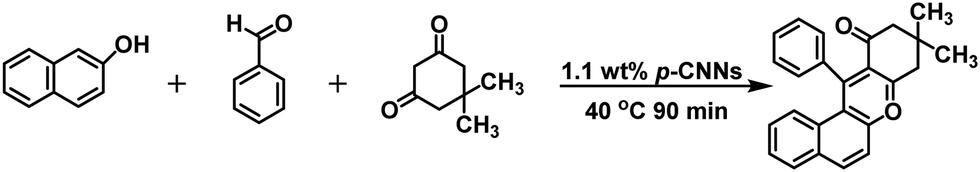 | ||
| Scheme 3 p-CNNs photocatalytic for the synthesis of 12-phenyl-9,9-dimethyl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one under solvent-free conditions. | ||
Experimental section
Materials
Melamine, sodium hydroxide, cetyltrimethylammonium bromide and copperdiacetatehydrate were provided by Macklin Industrial Corporation (Shanghai, China). Aldehydes and 1,3-dicarbonyl compounds were purchased from Aladdin Industrial Corporation. Urea, thiourea, ethanol and other reagents were obtained from Dalian Chemical Reagent Factory, China. All the chemicals were analytical grade and used directly without further purification.Preparation of photocatalyst
In a typical procedure, a certain amount of sodium hydroxide (NaOH) was dissolved in a certain volume of deionized water under ice water bath. Subsequently, cetyltrimethylammonium bromide (CTAB) was added into the above solution with a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.5 (CTAB:NaOH). The system was then heated from room temperature to 60 °C under stirring to give a NaOH–CTAB solution. In addition, a certain amount of copperdiacetatehydrate was dissolved in deionized water, and the solution was then added into the NaOH–CTAB solution drop by drop. The obtained mixed system was heated at 60 °C for 60 min. After the reaction was complete, the reaction system was filtered while hot and washed three times with deionized water and ethanol, respectively. The black solid products were dried at 50 °C for 12 h. Finally, the CuO NB was calcined at 350 °C for 120 min under N2 atmosphere before use.
:5.5 (CTAB:NaOH). The system was then heated from room temperature to 60 °C under stirring to give a NaOH–CTAB solution. In addition, a certain amount of copperdiacetatehydrate was dissolved in deionized water, and the solution was then added into the NaOH–CTAB solution drop by drop. The obtained mixed system was heated at 60 °C for 60 min. After the reaction was complete, the reaction system was filtered while hot and washed three times with deionized water and ethanol, respectively. The black solid products were dried at 50 °C for 12 h. Finally, the CuO NB was calcined at 350 °C for 120 min under N2 atmosphere before use.
The method for the preparation of multilayer carbon nitride (ml-CN) was based on the reported literature,40 with a slight difference. First, 2.0 g of melamine and 3 mL of deionized water were taken in a crucible; the mixed system was then stirred for 30 min. Subsequently, the mixture was calcined at 250 °C for 60 min under N2 atmosphere. After the calcination process was complete, the system was heated to 560 °C with a rate of 5 °C min−1 and then kept for 180 min. The obtained ml-CN powder was calcined at 560 °C for another 115 min before use.
In a typical procedure: first, 2.0 g of melamine, 10 mg of CuO NB and 3 mL of deionized water were taken in a crucible; the mixed system was then stirred for 30 min. Subsequently, the mixture was calcined at 250 °C for 60 min under N2 atmosphere. After the calcination process was complete, the system was heated to 560 °C with a rate of 5 °C min−1 and then kept for 180 min. The obtained powder was fully ground and then calcined at 560 °C for 115 min under N2 atmosphere to give p-CNNs.
Characterization
The investigation of morphology by scanning electron microscopy (SEM), transmission electron microscopy (TEM) and atomic force microscopy (AFM) was conducted on Hitachis-4800 SEM, Nanoscope III AFM and JEM-2100 CXII TEM, respectively. X-ray photoelectron spectroscopy (XPS) was studied using a Kratos Axis Ultra DLD spectrometer equipped with a monochromatic A1KR X-ray source. X-ray diffraction (XRD) patterns of the ml-CN and p-CNNs were explored by a Bruker D8 Focus diffractometer. Fourier transform infrared (FT-IR) spectra of the catalysts were investigated by a Bruker Tensor 27 spectrophotometer in the range of 400–4000 cm−1. N2 adsorption–desorption isotherm measurements were conducted using a physisorption analyzer, and the Brunauer–Emmett–Teller (BET) method was used to calculate the specific surface area, as well as the desorption branch of the isotherm in line with the Barrett–Joyner–Halenda method was utilized to evaluate the average pore diameter and pore size distribution of p-CNNs. The ultraviolet-visible diffuse reflectance spectra (UV-vis DRS) of ml-CN and p-CNNs were recorded on a Cary 5000 spectrophotometer fitted with an integrating sphere attachment from 200 to 800 nm. Inductively coupled plasma atomic emission spectroscopy (ICP-MS) was performed using an Agilent 7800 equipment. 1H NMR spectral measurements were performed at 400 MHz using TMS as the internal standard, and 13C NMR spectral measurements were carried out at 100 MHz with complete proton decoupling.General procedure for the synthesis of dihydropyridine-2(1H)-ones/thiones by using p-CNN photocatalyst
In a typical procedure, a mixture of aldehydes, 1,3-dicarbonyl compounds, urea or thiourea and p-CNNs was added into a reactor. The mixture was stirred at dark condition for 30 min. Subsequently, the reaction system was conducted at different temperatures for different irradiation time. Once the reaction was complete, ice water was added. The obtained crude product was then washed with ice water several times, filtered and dried at 100 °C for 10 h. Finally, the pure products were obtained by recrystallization of the crude products in ethanol.In the cycling experiment, the p-CNNs was recovered during the recrystallization process. The obtained p-CNNs was washed with hot ethanol several times and then dried at 100 °C for 10 h before reuse.
General procedure for the synthesis of 12-phenyl-9,9-dimethyl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one using p-CNN photocatalyst
In a typical procedure, a mixture of benzaldehyde (1.0 mmol), 2-naphthol (1.0 mmol), trimethadione (1.2 mmol) and p-CNNs (1.1 wt%) was added into a reactor. The mixture was stirred at dark condition for 30 min. Subsequently, the reaction system was performed at 40 °C for 90 min under visible-light irradiation. Once the reaction was complete, the system was cooled to room temperature and ethyl acetate was added. The reaction system was then filtered and washed with ethyl acetate several times, and the obtained filtrate was collected. The pure product was afforded by evaporation of the collected filtrate, followed by column chromatography on silica gel using ethyl acetate/petroleum ether as the eluent.General procedure for the synthesis of 5-phenyl-1(4-methoxyphenyl)-3[(4-methoxyphenyl)-amino]-1H-pyrrol-2(5H)-one by using p-CNN photocatalyst
In a typical procedure, a mixture of benzaldehyde (1.0 mmol), p-methoxyaniline (2.0 mmol), ethyl pyruvate (1.5 mmol) and p-CNNs (2.5 wt%) was added into a reactor. The mixture was stirred at dark condition for 30 min. Subsequently, the reaction system was performed at room temperature for 0.5 h under visible-light irradiation. Once the reaction was complete, 5 mL of ethanol was added and the obtained reaction system was stirred for 3–4 min. Subsequently, the reaction system was filtered and washed with ethanol and ether several times, respectively. The pure product was finally obtained by drying at 80 °C for 10 h.Results and discussion
The characterization of samples
The morphology of ml-CN and p-CNNs was investigated by scanning electron microscopy (SEM), transmission electron microscopy (TEM) and atomic force microscopy (AFM). The ml-CN prepared by the direct calcination of the melamine slurry without the addition of CuO NB displayed obvious multi-layer structure (Fig. 1A), while the p-CNNs prepared by the simple calcination of CuO NB (Fig. S1, ESI†) and melamine exhibited typical two-dimensional structure (Fig. 1B). This finding shows that CuO NB can assist the p-CNNs to form nanosheet structure during the calcination process. The lamellar structure and amorphous structure of the p-CNNs was also observed by TEM results, as well as the amounts of holes existing in the nanosheet (Fig. 1C and D). To further investigate the porous structure of p-CNNs, AFM measurement was performed, and the obvious pores were found (Fig. 1E). Meanwhile, the p-CNN was hundreds of nanometers in width and ∼1.5 nm in thickness (Fig. 1F). All the results indicated that the p-CNNs prepared in this work owns porous and flaky structure, which is beneficial for the adsorption and mass transfer of the reactants. | ||
| Fig. 1 SEM images of ml-CN (A) and p-CNNs (B); TEM images of p-CNNs (C and D); AFM images of p-CNNs (E) and its thickness distribution curves (F). | ||
To investigate the chemical composition and status of the elements in p-CNNs, XPS measurement was carried out. As shown in Fig. 2A, three peaks were observed in the C 1s spectra, which were located at 288.3 eV, 286.5 eV and 284.8 eV, corresponding to N = C–N, C–N and C–C, respectively.41 The narrow scan N 1s spectra of p-CNNs are shown in Fig. 2B, and the peaks were observed at 401.4 eV, 400.3 eV and 398.8 eV, attributed to N–C, N–(C)3 and C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, respectively.42 Meanwhile, no Cu related species were detected in the XPS spectra. Furthermore, the results of ICP-MS show that there is no Cu loading in the p-CNNs. Fig. 2C displays the XRD patterns of ml-CN and p-CNNs, which is similar to that of pure graphitic carbon nitride (JCPDS Card No. 87-1526). The only difference is that the intensity of the (100) and (002) planes of p-CNNs was lower than that of ml-CN, leading to the reduction of interlayer distance of the graphitic like planar p-CNNs. As shown in Fig. 2D, the FT-IR spectra of ml-CN and p-CNNs were similar with those reported;43 besides the change in signal peak intensity, no obvious difference in the FT-IR spectra of ml-CN and p-CNNs was observed. The N2 sorption isotherms of p-CNNs exhibited type IV isotherm patterns, suggesting that lots of mesopores exist in p-CNNs, and the surface area was 85.2 m2 g−1 (Fig. 2E). To investigate the visible-light absorption regions of ml-CN and p-CNNs, UV-vis DRS was performed. As shown in Fig. 2F, the spectrum of the sample showed blue-shift with the decrease of thickness, indicating that the excitations by photocarriers with energy is lower as compared with the intrinsic band-band transition. Furthermore, their bandgaps reduced with the decrease in sample thickness, resulting in an enhanced photocatalytic activity.
C, respectively.42 Meanwhile, no Cu related species were detected in the XPS spectra. Furthermore, the results of ICP-MS show that there is no Cu loading in the p-CNNs. Fig. 2C displays the XRD patterns of ml-CN and p-CNNs, which is similar to that of pure graphitic carbon nitride (JCPDS Card No. 87-1526). The only difference is that the intensity of the (100) and (002) planes of p-CNNs was lower than that of ml-CN, leading to the reduction of interlayer distance of the graphitic like planar p-CNNs. As shown in Fig. 2D, the FT-IR spectra of ml-CN and p-CNNs were similar with those reported;43 besides the change in signal peak intensity, no obvious difference in the FT-IR spectra of ml-CN and p-CNNs was observed. The N2 sorption isotherms of p-CNNs exhibited type IV isotherm patterns, suggesting that lots of mesopores exist in p-CNNs, and the surface area was 85.2 m2 g−1 (Fig. 2E). To investigate the visible-light absorption regions of ml-CN and p-CNNs, UV-vis DRS was performed. As shown in Fig. 2F, the spectrum of the sample showed blue-shift with the decrease of thickness, indicating that the excitations by photocarriers with energy is lower as compared with the intrinsic band-band transition. Furthermore, their bandgaps reduced with the decrease in sample thickness, resulting in an enhanced photocatalytic activity.
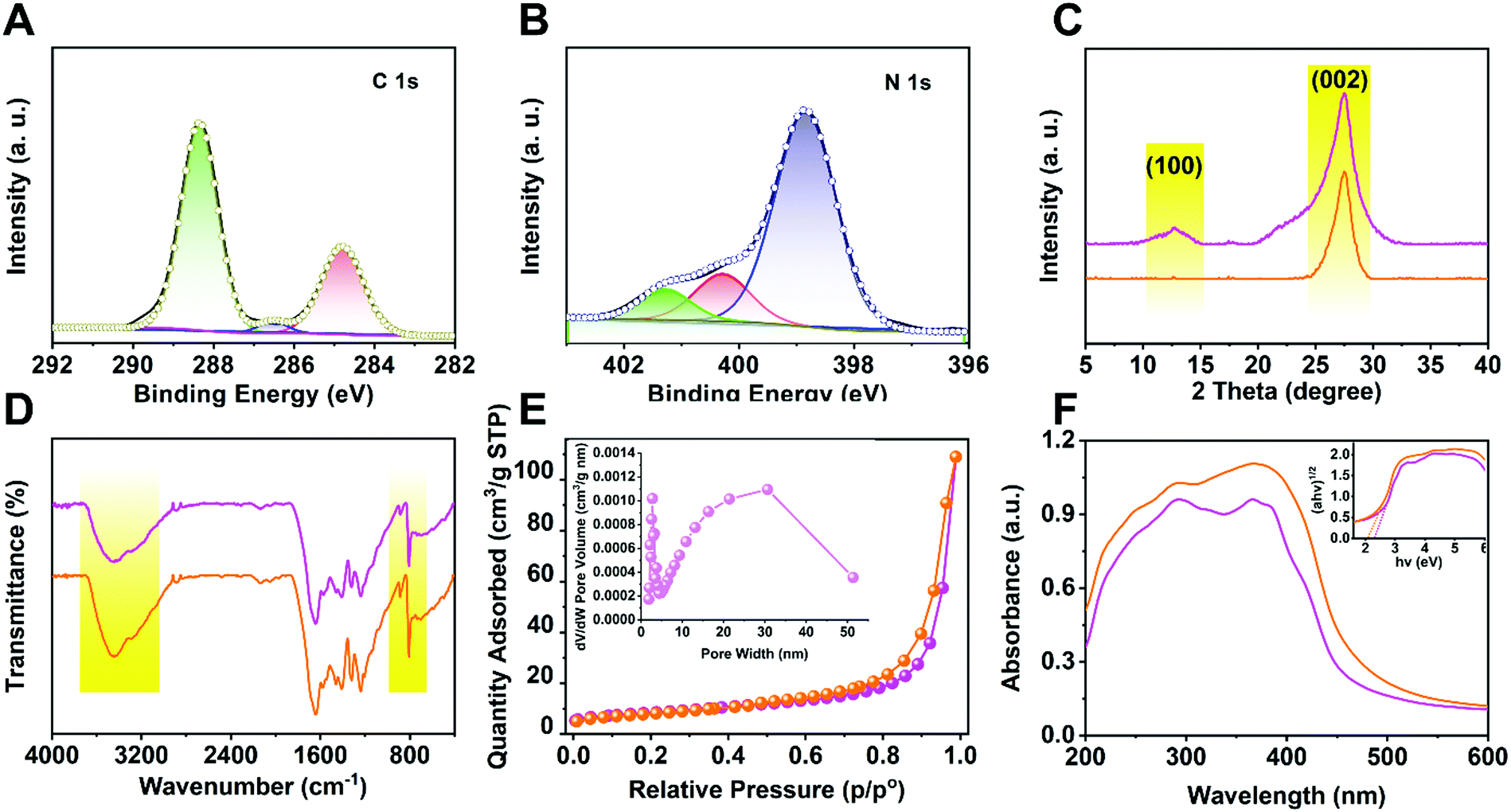 | ||
| Fig. 2 XPS spectra of p-CNNs, (A): C 1s, (B): N 1s; (C): XRD patterns of ml-CN (magenta) and p-CNNs (orange); (D): FT-IR spectra of ml-CN (magenta) and p-CNNs (orange); (E): N2 adsorption isotherm, inset in E shows the BJH pore size distribution; (F): UV-vis DRS of ml-CN and p-CNNs, inset in F shows the plot of transformed Kubelka–Munk function versus photo energy for ml-CN and p-CNNs. | ||
The photocatalytic activity of samples
Initially, the influence of visible light or dark condition on Biginelli reaction catalyzed by p-CNNs was investigated. The product of the Biginelli condensation reaction of benzaldehyde (5 mmol) with ethyl acetoacetate (5 mmol) and urea (5 mmol) was named as 3a. When the reaction catalyzed by p-CNNs was carried out at 40 °C or 90 °C for 60 min in the dark conditions, only trace amounts of 3a was obtained. Once visible light was applied in the above system at 40 °C, 19.0% yield of 3a was observed, suggesting that visible light is beneficial for the synthesis of 3a. Subsequently, the effects of reaction time and reaction temperature on the synthesis of 3a photocatalyzed by p-CNNs were also investigated, and the results are given in Table 1. Interestingly, the yield of 3a increased at first and then decreased (Table 1, entries 1–4). The maximum yield of 3a was achieved in 90 min, and thus, this period of time was used as the optimum reaction time for further investigations. As we know, temperature plays an important role in organic synthesis. In this study, the influence of different reaction temperatures on the synthesis of 3a was also explored. As the reaction temperature was increased from 30 to 90 °C, the yield of 3a increased from 17.9 to 84.5% (Table 1, entries 3 and 5–8). However, the yield of 3a appeared to decrease slightly when the reaction temperature was increased from 90 to 100 °C (Table 1, entries 8 and 9). This finding indicates that proper temperature is instrumental in the synthesis of 3a in the p-CNN catalytic system irradiated by visible light.|
|
|||||
|---|---|---|---|---|---|
| Entry | p-CNNs dosage (wt%) | Time (min) | Temperature (°C) | Ratiob | Yieldc (%) |
| a 3a is 5-ethoxycarbonyl-6-methyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one. b The order of the reactants ratio is benzaldehyde to ethyl acetoacetate to urea (5 mmol benzaldehyde, 1 equiv.). c Isolated yield. | |||||
| 1 | 1.12 | 30 | 40 | 1:1:1 |
14.8 |
| 2 | 1.12 | 60 | 40 | 1:1:1 |
19.0 |
| 3 | 1.12 | 90 | 40 | 1:1:1 |
26.6 |
| 4 | 1.12 | 120 | 40 | 1:1:1 |
25.2 |
| 5 | 1.12 | 90 | 30 | 1:1:1 |
17.9 |
| 6 | 1.12 | 90 | 50 | 1:1:1 |
48.1 |
| 7 | 1.12 | 90 | 70 | 1:1:1 |
72.7 |
| 8 | 1.12 | 90 | 90 | 1:1:1 |
84.5 |
| 9 | 1.12 | 90 | 100 | 1:1:1 |
82.0 |
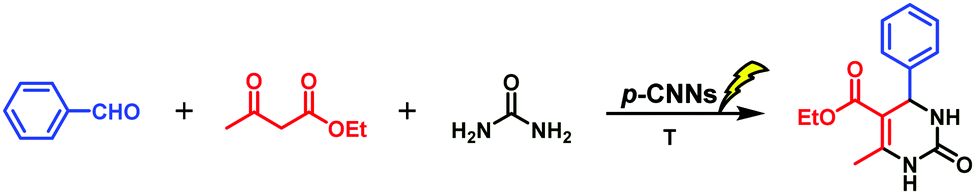
To investigate the effects of the p-CNN photocatalyst dosage and the stoichiometry of the reactants for the synthesis of 3a, a series of experiments was performed and the results are presented in Table 2. Firstly, the yield of 3a increased along with the increase in the p-CNNs dosage from 0.56 to 1.68 wt% (Table 2, entries 1–3). However, no obvious increase in 3a yield was observed when the photocatalyst dosage was 2.80, 3.92, and 5.60 wt%, respectively (Table 2, entries 4–6). This is probably because the adsorption of benzaldehyde, ethyl acetoacetate and urea on the surface of p-CNNs formed unstable species, leading to a lower activation energy of the entire reaction. Table 2 also shows the influence of the stoichiometry of the reactants on the synthesis of 3a. Apparently, as the urea dosage increased, the yield of 3a increased (Table 2, entries 3 and 8, 7 and 9). Conversely, the yield of 3a decreased when the content of ethyl acetoacetate was increased (Table 2, entries 7, 8, 9 and 10). According to the aforementioned analysis, two points can be reflected. One is that the optimum reaction condition was observed at 90 °C for 90 min with the reaction ratio of 1:1.2:1.5 (benzaldehyde:ethyl acetoacetate:urea) under the irradiation of visible light with the p-CNNs photocatalyst (1.68 wt%). The other is that the Biginelli reaction for the efficient synthesis of 3a was achieved with the assistance of photo-thermo catalysis.
|
|
|||||
|---|---|---|---|---|---|
| Entry | p-CNNs dosage (wt%) | Time (min) | Temperature (°C) | Ratio | Yieldb (%) |
| a 3a is 5-ethoxycarbonyl-6-methyl-4-phenyl-3,4-dihydropyrimidin-2(1H)-one. b Isolated yield. | |||||
| 1 | 0.56 | 90 | 90 | 1:1:1 |
75.3 |
| 2 | 1.12 | 90 | 90 | 1:1:1 |
84.5 |
| 3 | 1.68 | 90 | 90 | 1:1:1 |
88.4 |
| 4 | 2.80 | 90 | 90 | 1:1:1 |
87.8 |
| 5 | 3.92 | 90 | 90 | 1:1:1 |
87.3 |
| 6 | 5.60 | 90 | 90 | 1:1:1 |
80.1 |
| 7 | 1.68 | 90 | 90 | 1:1.2:1.2 |
84.7 |
| 8 | 1.68 | 90 | 90 | 1:1:1.2 |
94.2 |
| 9 | 1.68 | 90 | 90 | 1:1.2:1.5 |
95.4 |
| 10 | 1.68 | 90 | 90 | 1:1.5:1.5 |
68.1 |
To further understand the merits of the synthesis of 3a photocatalyzed by p-CNNs, the system was compared with that of the earlier reported ones. As shown in Table 3, p-CNNs exhibited excellent activity in a shorter reaction time as compared to D-xylonic acid, PPH3, B(C6F5)3 and SBNBSA.2,46–48 The SFP catalyst also gave a good yield of 3a, but suffered from the high reaction temperature.45 SBNPSA and ZrOCl2/mont K10 can work at relatively lower temperature and shorter time.48,49 However, additional solvents or metal catalysts were employed. The ZSPS catalyst exhibited excellent activity at shorter reaction time, but the catalyst preparation process is complex and expensive when compared with p-CNNs.45 Obviously, with respect to the efficiency or environmental economic benefit, the p-CNN system for the synthesis of 3a is superior to most reported systems. To investigate the relationship between the structure of p-CNNs and its performance, the photocatalytic activities of ml-CN and p-CNNs on the synthesis of 3a were investigated, and the photocatalytic performance of ml-CN was lower than that of p-CNNs. According to the characterization of SEM and AFM, the possible active sites of p-CNNs in the synthesis of 3a was the porous structure of the p-CNNs.
| Entry | Catalyst | Time (min) | Temperature (°C) | Solvent | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a ZSPS represents zirconium(IV)–salophen perfluorooctanesulfonate. b SFP represents sulfated polyborate. c SBNPSA represents silica-bonded N-propyl sulfamic acid. | ||||||
| 1 | p-CNNs (1.68 wt%) | 90 | 90 | — | 95.4 | This work |
| 2 | D-Xylonic acid (6.5 mol%) | 300 | 100 | D-Xylonic acid | 87 | 2 |
| 3 | ZSPSa (5 mol%) | 30 | 90 | — | 96 | 41 |
| 4 | SFPb (5 wt%) | 20 | 100 | — | 94 | 42 |
| 5 | PPH3 (2.78 mol%) | 720 | 100 | — | 70 | 43 |
| 6 | B(C6F5)3 (2.78 mol%) | 150 | Reflux | EtOH | 95 | 44 |
| 7 | SBNPSAc (1 mol%) | 240 | 80 | EtOH | 95 | 45 |
| 8 | ZrOCl2/mont K10 (40 wt%) | 20 | 80 | — | 78 | 46 |
To investigate the substrate universality, Biginelli reaction of various aldehydes with ethyl acetoacetate or methyl acetoacetate and urea or thiourea was performed. From the perspective of green chemistry, the stoichiometric ratio of the reactants was changed from 1:1.2:1.5 to 1:1:1.2, and the other optimum conditions were not changed. Firstly, the influences of aromatic aldehydes with different electro-donating groups and electro-withdrawing groups on the Biginelli reaction were studied. As shown in Fig. 3, excellent yields were achieved (3b–3h), and no obvious difference in the yields of different products was observed, indicating that the electron-donating groups and electron-withdrawing groups in the aromatic aldehyde (p-) are all beneficial for the Biginelli reaction. Subsequently, the effect of spatial hindrance on the Biginelli reaction was explored. The yield of the corresponding product was 90.3 (3c), 87.1 (3i) and 80.3% (3j) when p-methoxybenzaldehyde, m-methoxybenzaldehyde and o-methoxybenzaldehyde were employed, respectively. This finding suggests that larger the steric hindrance of the aromatic aldehyde, the more unfavorable the Biginelli reaction. As compared with aromatic aldehydes, fatty aldehydes used in the Biginelli reaction with ethyl acetoacetate and urea gave good yields, (3k, 3l) but lower than that of the aromatic aldehydes. Furthermore, the effect of methyl acetoacetate on the Biginelli reaction was investigated. No obvious difference in the yields of corresponding products was observed when compared with ethyl acetoacetate (3f–3m, 3h–3n, 3d–3o, 3b–3p). In addition, the influence of thiourea on the reaction was also investigated, and 89.7% yield of 3q was achieved. Other investigations (3r and S-3a–3e) were similar with the above ones, and all the results indicated that the photo-thermo catalysis system using p-CNNs is appropriate for the Biginelli reaction.
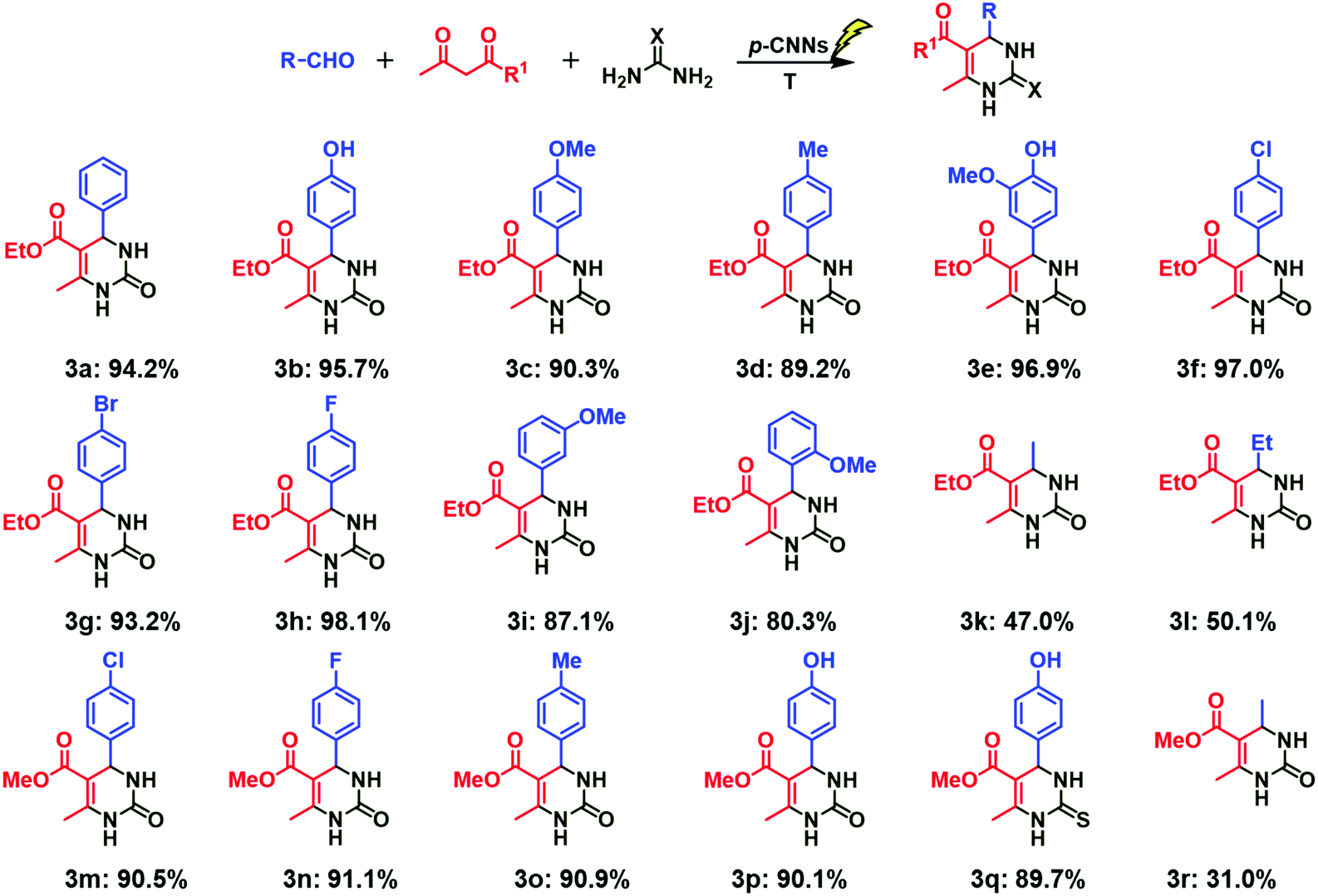 | ||
| Fig. 3 Photo-thermo catalysis for the synthesis of dihydropyrimidin-2(H)-ones and thiones photocatalyzed by p-CNNs. | ||
With regard to the excellent performance and mild reaction conditions of the reaction system, it is worth exploring for the synthesis of N- and O-heterocyclic compounds. Pyrroles and their analogs are a series of N-heterocyclic compounds, which play an important role in the synthesis of pharmacologically significant molecules and natural products.50 Therefore, the synthesis of pyrroles is of great significance. In addition, benzoxanthene derivatives are a class of O-heterocyclic compounds. They have favorable biological and pharmaceutical properties of analgesic, antiviral and antibacterial.51,52 Thus, the synthesis of benzoxanthene derivatives has vital significance. For 5-phenyl-1(4-methoxyphenyl)-3[(4-methoxyphenyl)-amino]-1H-pyrrol-2(5H)-one synthesis, the reaction was performed at room temperature by mixing p-CNNs, benzaldehyde, 4-methoxyaniline and ethyl pyruvate in the presence of visible-light irradiation, and the yield of the product was up to 93%. However, for 12-phenyl-9,9-dimethyl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one synthesis, the reaction was carried out at 50 °C by mixing p-CNNs, benzaldehyde, 2-hydroxynaphthalene and 5,5-dimethyl-1,3-cyclohexanedione in the presence of visible-light irradiation, and the yield of the product was up to 97%.
Due to the excellent activity of p-CNNs, it is worth exploring its cycle stability. In this work, the cycling experiment was performed using benzaldehyde, ethyl acetoacetate and urea (1:1:1.2) at 90 °C for 90 min in the presence of p-CNNs (1.68 wt%). In the cycling experiment, the p-CNNs was recovered during the recrystallization process. The obtained p-CNNs was washed with hot ethanol several times and then dried at 100 °C for 10 h before reuse. As shown in Fig. 4, no obvious decrease in the yield of 3a was observed, indicating that the p-CNNs has excellent stability.
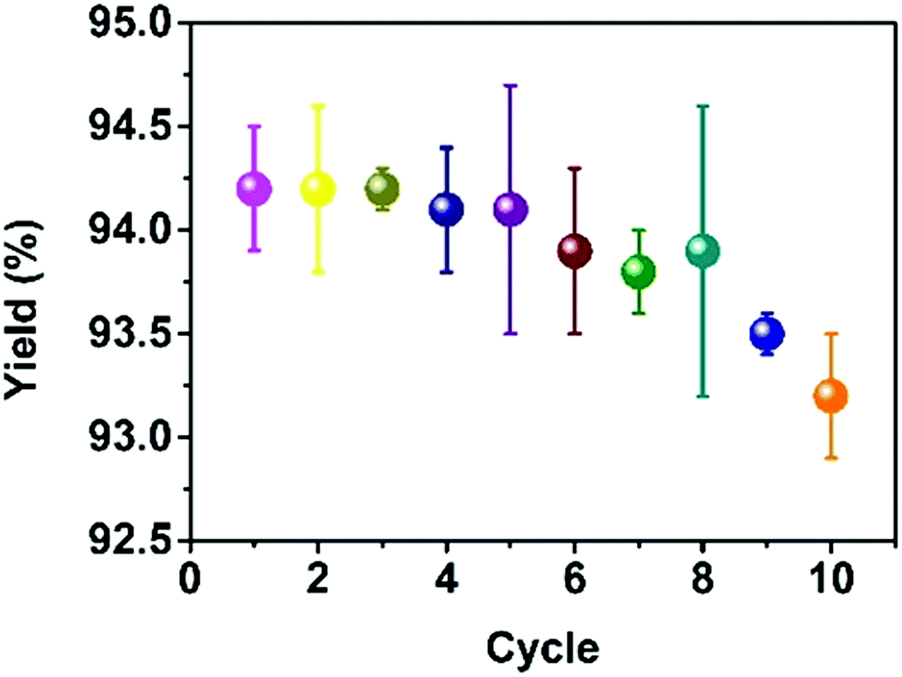 | ||
| Fig. 4 Recycling of p-CNNs photocatalyst. | ||
Conclusions
In summary, this work provides a novel and green photocatalytic system for the synthesis of N- and O-heterocyclic compounds. The p-CNN photocatalyst showed excellent activity for the “one-pot” three-component Biginelli reaction to give DHPMs and their derivatives. The ease of use, environmental friendliness as well as air, water, and substrate tolerances make it an excellent system for carrying out Biginelli reaction. Meanwhile, 5-phenyl-1(4-methoxyphenyl)-3[(4-methoxyphenyl)-amino]-1H-pyrrol-2(5H)-one and 12-phenyl-9,9-dimethyl-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one were all obtained via this reaction system with 93% and 97% yields, respectively, under mild reaction conditions. This work paves a novel way for the organic synthesis of heterocyclic compounds, especially the N- and O-heterocyclic compounds.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We wish to thank for the Foundation of NSFC-CONICFT Joint Project (No. 51961125207), Liaoning Province “Xingliao Talent Plan” Outstanding Talent Project (XLYC1901004), Innovation Support Program for High-level Talents of Dalian (Top and Leading Talents) (201913), National Natural Science Foundation of China (22008018), Natural Science Foundation of Liaoning Province (2020-MS-272), China Postdoctoral Science Foundation (2020M670716), Start-up Fund for Doctoral Research of Dalian Polytechnic University (2020-07) and the Foundation (No. KF201914) of State Key Laboratory of Biobased Material and Green Papermaking, Qilu University of Technology, Shandong Academy of Sciences.Notes and references
- C. Y. Guan, S. S. Chen, T. H. Lee, C. P. Yu and D. C. W. Tsang, J. Cleaner Prod., 2020, 260, 121097 CrossRef CAS.
- J. L. Ma, L. X. Zhong, X. W. Peng and R. C. Sun, Green Chem., 2016, 18, 1738–1750 RSC.
- R. H. Mei, U. Dhawa, R. C. Samanta, W. B. Ma, D. J. Wencel and L. Ackermann, ChemSusChem, 2020, 13(13), 3306–3356 CrossRef CAS.
- L. Chen, D. Y. Zhu, J. T. Li, X. X. Wang, J. F. Zhu, P. S. Francis and Y. H. Zheng, Appl. Catal., B, 2020, 273, 119050 CrossRef CAS.
- B. Xu, G. L. Troian, R. Dykstra, R. T. Martin, O. Gutierrez and U. K. Tambar, J. Am. Chem. Soc., 2020, 142(13), 6206–6215 CrossRef CAS.
- C. H. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- S. I. G. Costa, V. D. Cauneto, F. L. D. Fiorentin, P. B. Almeida, R. C. Oliveira, E. Longo, A. N. Modenes and V. Slusarski-Santana, Chem. Eng. J., 2020, 392, 123737 CrossRef CAS.
- M. Motola, M. Baudys, R. Zazpe, M. Krbal, J. Michalicka, J. Rodriguez-Pereira, D. Pavlinak, J. Prikryl, L. Hromadko, H. Sopha, J. Krysa and J. M. Macak, Nanoscale, 2019, 11, 23126–23131 RSC.
- P. Zhou, Q. H. Zhang, Z. K. Xu, Q. Y. Shang, L. Wang, Y. G. Chao, Y. J. Li, H. Chen, F. Lv, Q. Zhang, L. Gu and S. J. Guo, Adv. Mater., 2020, 32, 1904249 CrossRef CAS.
- A. Serra, R. Artal, A. J. Garcia, B. Sepulveda, E. Gomez, J. Nogues and L. Philippe, Adv. Sci., 2019, 7(3), 1902447 CrossRef.
- E. Bardos, A. K. Kiraly, Z. Pap, L. Baia, S. Garg and K. Hernadi, Appl. Surf. Sci., 2019, 479, 745–756 CrossRef CAS.
- M. L. Luo, Q. Yang, W. B. Yang, J. H. Wang, F. F. He, K. W. Liu, H. M. Cao and H. J. Yan, Small, 2020, 16(20), 2001100 CrossRef CAS.
- L. Qin, R. Ru, J. W. Mao, Q. Meng, Z. Fan, X. Li and G. L. Zhang, Appl. Catal., B, 2020, 269, 118754 CrossRef CAS.
- L. W. Zhang, R. Long, Y. M. Zhang, D. L. Duan, Y. J. Xiong, Y. J. Zhang and Y. P. Bi, Angew. Chem., Int. Ed., 2020, 59(15), 6224–6229 CrossRef CAS.
- X. W. Zhu, J. M. Yang, X. J. She, Y. H. Song, J. C. Qian, Y. Wang, H. Xu, H. M. Li and Q. Y. Yan, J. Mater. Chem. A, 2019, 7(10), 5209–5213 RSC.
- C. K. Yao, A. L. Yuan, Z. S. Wang, H. Lei, L. Zhang, L. M. Guo and X. P. Dong, J. Mater. Chem. A, 2019, 7(21), 13071–13079 RSC.
- F. He, B. C. Zhu, B. Cheng, J. G. Yu, W. K. Ho and W. Macyk, Appl. Catal., B, 2020, 272, 119006 CrossRef CAS.
- X. W. Zhu, S. Q. Huang, Q. Yu, Y. B. She, J. M. Yang, G. L. Zhou, Q. D. Li, X. J. She, J. J. Deng, H. M. Li and H. Xu, Appl. Catal., B, 2020, 269, 118760 CrossRef CAS.
- Z. S. Wang, H. J. Yu, L. Zhang, L. M. Guo and X. P. J. Dong, J. Taiwan Inst. Chem. Eng., 2020, 107, 119–128 CrossRef CAS.
- X. L. Xue, R. P. Chen, C. Z. Yan, Y. Hu, W. J. Zhang, S. Y. Yang, L. B. Ma, G. Y. Zhu and Z. Jin, Nanoscale, 2019, 11, 10439–10445 RSC.
- X. J. Wu, X. T. Fan, S. J. Xie, J. C. Lin, J. Cheng, Q. H. Zhang, L. Y. Chen and Y. Wang, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- S. Cailotto, M. Negrato, S. Daniele, R. Luque, M. Selva, E. Amadio and A. Perosa, Green Chem., 2020, 22, 1145–1149 RSC.
- C. O. Kappe, Mol., 1998, 3(1), 1–9 CAS.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043–1052 CrossRef CAS.
- M. Kidwai, S. Saxena, M. K. R. Khan and S. S. Thukral, Eur. J. Med. Chem., 2005, 40(8), 816–819 CrossRef CAS.
- K. S. Atwal, G. C. Rovnyak, S. D. Kimball, D. M. Floyd, S. Moreland, B. N. Swanson, J. Z. Gougoutas, J. Schwartz, K. M. Smillie and M. F. Malley, J. Med. Chem., 1990, 33(9), 2629–2635 CrossRef CAS.
- K. S. Atwal, B. N. Swanson, S. E. Unger, D. M. Floyd, S. Moreland, A. Hedberg and B. C. O’Reilly, J. Med. Chem., 1991, 34, 806–811 CrossRef CAS.
- B. B. Snider and Z. Shi, J. Org. Chem., 1993, 58, 3828–3839 CrossRef CAS.
- A. D. Patil, N. V. Kumar, W. C. Kokke, M. F. Bean, A. J. Freyer, C. Debrosse, S. Mai, A. Truneh, D. J. Faulkner, B. Carte, A. L. Breen, R. P. Hertzberg, R. K. Johnson, J. W. Westley and B. C. M. Potts, J. Org. Chem., 1995, 60(5), 1182–1188 CrossRef CAS.
- P. Biginelli, Ital, 1893, 23, 360–416 Search PubMed.
- K. S. Atwal, G. C. Rovnyak, B. C. O’Reilly and J. Schwartz, J. Org. Chem., 1989, 54, 5898–5907 CrossRef CAS.
- B. Jauk, F. Belaj and C. O. Kappe, J. Chem. Soc., Perkin Trans. 1, 1999, 307–314 RSC.
- N. A. Liberto, S. d. P. Silva, A. de Fatima and S. A. Fernandes, Tetrahedron, 2013, 69, 8245–8249 CrossRef CAS.
- W. Su, J. J. Li, Z. G. Zheng and Y. C. Shen, Tetrahedron Lett., 2005, 46, 6037–6040 CrossRef CAS.
- J. Safari and Z. Zarnegar, New J. Chem., 2014, 38, 358–365 RSC.
- M. K. Raj, H. S. P. Rao, S. GManjunatha, R. Sridharan, S. Nambiar, J. Keshwan, J. Rappai, S. Bhagat, B. S. Shwetha, D. Hegde and U. Santhosh, Tetrahedron Lett., 2011, 52(28), 3605–3609 CrossRef.
- A. de Vasconcelos, P. S. Oliveira, M. Ritter, R. A. Freitag, R. L. Romano, F. H. Quina, L. Pizzuti, C. M. P. Pereira, F. M. Stefanello and A. G. Barschak, J. Biochem. Mol. Toxicol., 2012, 26(4), 155–161 CrossRef CAS.
- C. Jiang and Q. D. You, Chin. Chem. Lett., 2007, 18(6), 647–650 CrossRef CAS.
- Q. Zhang, X. Wang, Z. Li, W. Wu, J. Liu, H. Wu, S. Cui and K. Guo, RSC Adv., 2014, 4, 19710–19715 RSC.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8(1), 76–80 CrossRef CAS.
- J. W. Fu, B. C. Zhu, C. J. Jiang, B. Cheng, W. You and J. G. Yu, Small, 2017, 13(15), 1603938 CrossRef.
- Q. D. Qin, X. Gao, X. Wu and Y. H. Liu, Mater. Lett., 2019, 256, 126623 CrossRef CAS.
- Q. Liu, T. X. Chen, Y. R. Guo, Z. G. Zhang and X. M. Fang, Appl. Catal., B, 2017, 205, 173–181 CrossRef CAS.
- N. B. Li, Y. J. Wang, F. J. Liu, X. Zhao, X. H. Xu, Q. An and K. M. Yun, Appl. Organomet. Chem., 2020, 34(3), e5454 CAS.
- C. K. Khatri, D. S. Rekunge and G. U. Chaturbhuj, New J. Chem., 2016, 40(12), 10412–10417 RSC.
- H. Slimi, Y. Moussaoui and R. ben Salem, Arabian J. Chem., 2016, 9, 510–514 CrossRef.
- S. K. Prajapti, K. K. Gupta and B. N. Babu, J. Chem. Sci., 2015, 127(6), 1047–1052 CrossRef.
- S. R. Jetti, A. Bhatewara, T. Kadre and S. Jain, Chin. Chem. Lett., 2014, 25(3), 469–473 CrossRef CAS.
- B. Vijayakumar and G. R. Rao, J. Porous Mater., 2012, 19(4), 491–497 CrossRef CAS.
- N. R. Candeias, P. M. P. Gois and C. A. M. Afonso, J. Org. Chem., 2006, 71(15), 5489–5497 CrossRef CAS.
- H. N. Hafez and M. I. Hegab, Bioorg. Med. Chem. Lett., 2008, 18(16), 4538–4543 CrossRef CAS.
- H. Wang, L. Lu, S. Y. Zhu, Y. H. Li and W. M. Cai, Curr. Microbiol., 2006, 52(1), 1–5 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj05101b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |