
Investigation on [OH−]-responsive systems for construction of one-dimensional hydroxyapatite via a solvothermal method†
Yin-chuan
Wang
 ab,
Zi-chen
Wang
c,
Gui-yong
Xiao
*ab,
Wei-li
Xu
ab,
Kai
Wang
ab,
Yan
Jiao
d,
Mei-li
Qi
ab and
Yu-peng
Lu
*ab
ab,
Zi-chen
Wang
c,
Gui-yong
Xiao
*ab,
Wei-li
Xu
ab,
Kai
Wang
ab,
Yan
Jiao
d,
Mei-li
Qi
ab and
Yu-peng
Lu
*ab
aKey Laboratory for Liquid–Solid Structural Evolution and Processing of Materials, Ministry of Education, Shandong University, Ji’nan 250061, China. E-mail: biosdu@sdu.edu.cn; xiaoguiyong@sdu.edu.cn
bSchool of Materials Science and Engineering, Shandong University, Ji’nan, 250061, China
cShandong Liming Polytechnic Vocational College, Ji’nan 250061, China
dFood and Drug Department, Shandong Institute of Commerce and Technology, Ji’nan 250061, China
First published on 24th November 2020
Abstract
An oleic acid-assisted solvothermal method has been utilized to prepare hydroxyapatite (HA) nanofibers with a high flexibility and aspect ratio. Understanding the role of oleic acid in the synthesis of HA may provide effective routes for the design of advanced biomaterials. The concentration of OH− ions ([OH−]) has a great impact on the reaction system, which in turn affects the nucleation and crystal growth of HA. In this work, the effect of oleic acid on the morphology and structure of the as-prepared products under different concentrations of NaOH was investigated by XRD, FTIR and FE-SEM. The formation process of HA was also explored to better explain the role of oleic acid based on structured characteristics and morphology evolution. These results showed that oleic acid molecules absorbed on the surface of HA would effectively induce a one-dimensional crystal growth, even if the crystal growth habit had been changed under different [OH−]. For [OH−] < 1.50 M, [OH−]-responsive oleic acid and its corresponding behavior as a pH buffer can regulate pH at a relatively constant value. Besides, the flexible ultralong HA nanofibers can be formed at [OH−] = 1.25–1.50 M, while the crystallite size and preferred growth along the c-axis were both improved with an increase in [OH−] added.
1. Introduction
Hydroxyapatite (HA, Ca10(PO4)6(OH)2), the major inorganic constituents of vertebrate bone and tooth,1 has been considered as an outstanding candidate for applications in drug/gene delivery,2 bone repair, and tissue engineering3 due to its excellent bioactivity, biocompatibility, and osteoconductivity.4 However, the low mechanical strength of typical HA bioceramics has severely restricted their applications in load-bearing sites. To attain the strength for clinical use, extensive efforts have been made to synthesize HA with superior mechanical properties.5 As we all know, HA crystals are rod-shaped with c-axis orientation in living bone, while they are plate-shaped with a-axis orientation in the dental enamel surfaces.6,7 Highly c-axis oriented HA crystals have higher biomechanical properties than other oriented HA.6,8 Besides, one-dimensional (1D) nanomaterials have attracted great interest due to their special physical properties.9–12 Thus, 1D HA nanofibers have been considered as an effective strategy to improve the above problem and have shown great application potential in various biomedical fields.13Synthesis methods of HA nanofibers have been widely reported in recent decades, including homogeneous precipitation,7,14 hydrothermal,15,16 hydrothermal homogeneous precipitation17,18 and so on19,20 (for more detailed information see Table S1, ESI†). However, as can be seen from Table S1, the lengths of HA nanofibers reported in literature reports are usually less than 30 μm. Thus, synthesized HA nanofibers with high flexibility and aspect ratio are still a huge challenge. Based on previous reports,21–23 crystal growth intimately depends on the properties of the starting materials, amongst which surfactants are remarkable materials to manufacture HA with well-controlled structure, size, morphology and array. Besides, the initial pH (ipH) of the reaction solution has an important influence on the preferred growth orientation of HA crystal. Over-low ipH values may cause the formation of impurities.24 Over-high ipH values favor HA growth along the a-axis to form lath-like instead of fibrous crystals.23 Thus, by choosing suitable surfactants and controlling the ipH, flexible HA nanofibers with a regular and controlled morphology may be obtained. Oleic acid, a fatty acid that occurs naturally in various animal and vegetable fats, has been proved to synthesize 1D nanostructured HA with controlled size and morphology based on previous reports.21,25,26 As a surfactant, oleic acid can adjust the interaction with HA surfaces to favor an oriented growth along the c-axis by complexing with Ca2+ on the crystal surfaces.27 Moreover, the presence of a carboxyl group in the oleic acid molecule leads to its [OH−]-responsive property, which has a direct influence on the pH of the reaction system.28 Unfortunately, to the best of our knowledge, no systematic study of the effect of [OH−]-responsive oleic acid on the pH of the reaction environment and its influence on the HA crystal growth under different pH environments has been reported.
Herein, we report a solvothermal synthesis of 1D HA materials using oleic acid as the surfactant under different ipH. Sodium hydroxide (NaOH) with different concentrations is used to control the ipH of the reaction solution. The effect of oleic acid on the phases, structures and morphologies of the products and the pH of the reaction system is analyzed. The aim of this study is to elucidate the effect of oleic acid on the crystal growth of HA in more detail to identify, if possible, optimum synthesis conditions.
2. Materials and methods
2.1 Materials
Anhydrous calcium chloride (CaCl2, AR), sodium hydroxide (NaOH, AR), sodium dihydrogen phosphate dihydrate (NaH2PO4·2H2O, AR) and ethanol (AR) were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China). Oleic acid was purchased from Shanghai Aladdin Bio-Chem Technology Co. Ltd. (China). All chemical reagents were used as received and without any further purification.2.2 Preparation
The products of HA were synthesized by a solvothermal method as previously reported.19,25 First, oleic acid (12 g), ethanol (11 g) and deionized water (10 g) were mixed to form mixture A in a closed system under room temperature. Then, 20 ml of NaOH aqueous solution with different concentrations from 1.0 to 2.0 M was prepared and added dropwise into the above mixture A to form the reaction solution B. In this process, sodium oleate was formed through the reaction between NaOH and oleic acid. And the molar ratio of oleic acid to NaOH has been listed in Table 1. Subsequently, 20 ml of CaCl2 aqueous solution (0.2 M) and 20 ml of NaH2PO4·2H2O aqueous solution (0.19 M) were prepared and added separately into the above reaction solution B to form the resulting mixture C. All the above processes were carried out under vigorous magnetic stirring. Finally, the resulting mixture C was moved into a 100 ml Teflon-lined stainless steel autoclave and heated at 180 °C for 23 h. After the autoclave cooled to room temperature, the white product was washed with ethanol and deionized water three times and collected by centrifugation at 2000 rpm, 649× for further use.| Sample ID | The concentration of NaOH (M) | Oleic acid/NaOH molar ratio | ipH | Final pH | Crystallinity (%) | Lattice parameter | |
|---|---|---|---|---|---|---|---|
| a (Å) | c (Å) | ||||||
| a Note: the data were tested in the water phase of the reaction solution before heating, because the solution was in the separated state of the oil phase and the water phase. | |||||||
| NF1 | 1.000 | 2.12 | 6.032a | 7.611 | 56.28 | 9.3849 | 6.9083 |
| NF2 | 1.125 | 1.89 | 6.691 | 7.617 | 53.39 | 9.3881 | 6.8489 |
| NF3 | 1.250 | 1.70 | 6.728 | 7.590 | 90.95 | 9.4062 | 6.9521 |
| NF4 | 1.375 | 1.54 | 6.822 | 7.600 | 89.05 | 9.3113 | 7.0999 |
| NF5 | 1.500 | 1.42 | 7.456 | 7.781 | 92.18 | 9.3479 | 7.2613 |
| NF6 | 1.625 | 1.31 | 7.697 | 7.956 | 95.19 | 9.3723 | 7.1898 |
| NF7 | 1.750 | 1.21 | 7.911 | 8.283 | 86.82 | 9.3516 | 7.1051 |
| NF8 | 1.875 | 1.13 | 8.026 | 8.269 | 91.11 | 9.4527 | 6.7982 |
| NF9 | 2.000 | 1.06 | 8.092 | 8.289 | 87.98 | 9.5032 | 6.8310 |
2.3 Characterization
The X-ray powder diffraction (XRD) patterns of the prepared products were characterized using an X-ray diffractometer (DMAX-2500PC, Rigaku, Tokyo, Japan) with a Cu Kα radiation source (λ = 1.5418 Å). The chemical composition of the products was collected using Fourier transform infrared spectroscopy (FTIR) (Nexus 670, Thermo Nicolet, Waltham, USA) with a resolution of 4 cm−1. Morphology was observed using a field-emission scanning electron microscope (FESEM) (JSM-7800F, JEOL, Tokyo, Japan) equipped with an energy dispersive spectrometer (EDS) (X-max 80, Oxford Instruments, Abingdon, UK). The pH values were measured by a pH meter (PHSJ-4F, INESA, Shanghai, China) at room temperature, first for the resulting mixture C (ipH) before heating; then after solvothermal treatment, that of the mixture containing the white product was taken as representing the final pH.The crystallite sizes (Dhkl) of the as-prepared HA products were calculated by using the ‘Debye–Scherrer equation’.29–31 The degree of crystallinity (XC) was also calculated using:32
| XC = 1 − (V112/300/I300) |
3. Results
3.1 Crystal phase analysis of the HA products
Fig. 1 shows the XRD patterns of all the samples by comparison with the strand diffraction data of HA (JCPDS No. 09-0432). All the peaks can be indexed to HA, indicating that oleic acid has no detrimental effect on the phase purity of the products under the different concentrations of NaOH. The peak intensity ratio I(300)/I(211) first increased then decreased, and the ratio I(300)/I(211) dropped rapidly when the concentration of NaOH ([OH−]) was 1.875 M, indicating that the preferential growth direction of the crystals changed with increased [OH−]. Besides, the strongest peak intensity of sample NF9 (Fig. S1, ESI†) was for the (211) lattice plane, rather than (300) as was similar to other samples, suggesting that the high [OH−] was not conducive to the growth of HA along the c axis. | ||
| Fig. 1 XRD patterns of HA synthesized with different concentrations of NaOH. (a) 1.125 M; (b) 1.375 M; (c) 1.625 M; (d) 1.875 M. | ||
In order to better investigate the effect of oleic acid on the crystal growth of HA under different [OH−], the ‘Debye–Scherrer equation’ was used to calculate the crystallite size (Dhkl) by measuring the full width at half maximum (FWHM), which is shown in Fig. 2. The crystallite sizes and their ratio both showed a parabola trend with increased [OH−]. Sample NF6 with [OH−] = 1.625 M had the largest D value, but its values of D300/D210 and D300/D211 was smaller than that of sample NF4, indicating that its preferred growth orientation shifted from the c-axis to the a-axis.
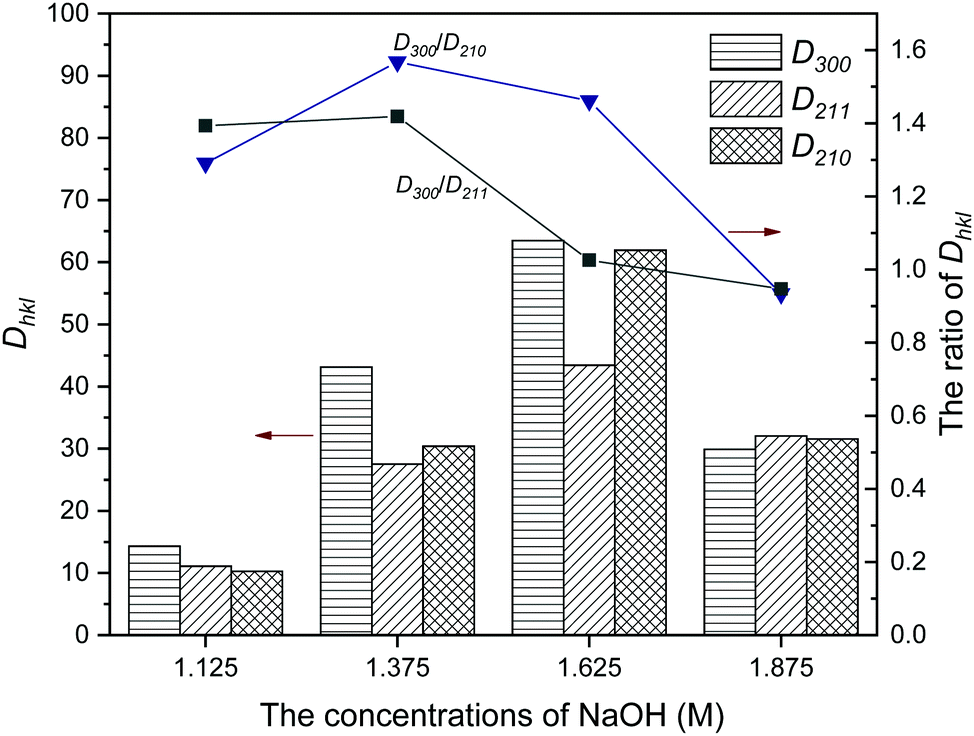 | ||
| Fig. 2 The crystallite sizes and ratios of Dhkl of HA synthesized with different concentrations of NaOH. (Dhkl: the crystallite size in the direction perpendicular to the lattice planes.) | ||
The characteristics of the products are summarized in Table 1. For [OH−] < 1.25 M, the samples NF1 and NF2 synthesized using oleic acid as the surfactant had relatively low crystallinity. The crystallinity of the other samples presented a slightly irregular change with increasing [OH−], which kept at an approximate 90%. The reason for the sudden change in crystallinity between samples NF2 and NF3 needs to be further explored combined with other characterization results. The lattice parameters varied regularly: a first decreased and then increased, and the changing trend of c was opposite to a. It is worth pointing out that the lattice parameters of samples NF4 to NF7 showed obvious preferential growth along the c-axis compared with that of stoichiometric HA (a = 9.418 Å, c = 6.884 Å).23,33
3.2 FTIR analysis of the HA products
FTIR analysis was performed to determine the constitution of the products. In the FTIR spectra (Fig. 3), all samples synthesized under different [OH−] had similar patterns. The absorption bands observed at 3572 and 635 cm−1 were ascribed to the vibrational mode of the OH− groups. The broad bands detected at 3425 cm−1 belonged to the O–H vibration of H2O, which absorbed on the surface of the samples. The absorption bands appearing in the regions of 1093, 1027, 962, 604, and 561 cm−1 are the characteristic bands of the PO43− group.34 Briefly, the above characteristic bands were as follows: 1093 and 1027 cm−1 as ν3 antisymmetric P–O stretching, 962 cm−1 as ν1 symmetric P–O stretching, and finally 604 and 561 cm−1 as ν4 O–P–O bending vibrations. In addition, the bands that appeared at about 2922, 2854 and 1460 cm−1 were attributed to the stretching and bending vibrations of –CH2–, and the characteristic bands of –COO− were detected at 1560 cm−1, suggesting that the oleate group may be absorbed on the surface of the samples.35 It was noted that the oleate group nearly disappeared for sample NF8, showing that the presence of it in the as-prepared products obviously decreased with higher [OH−].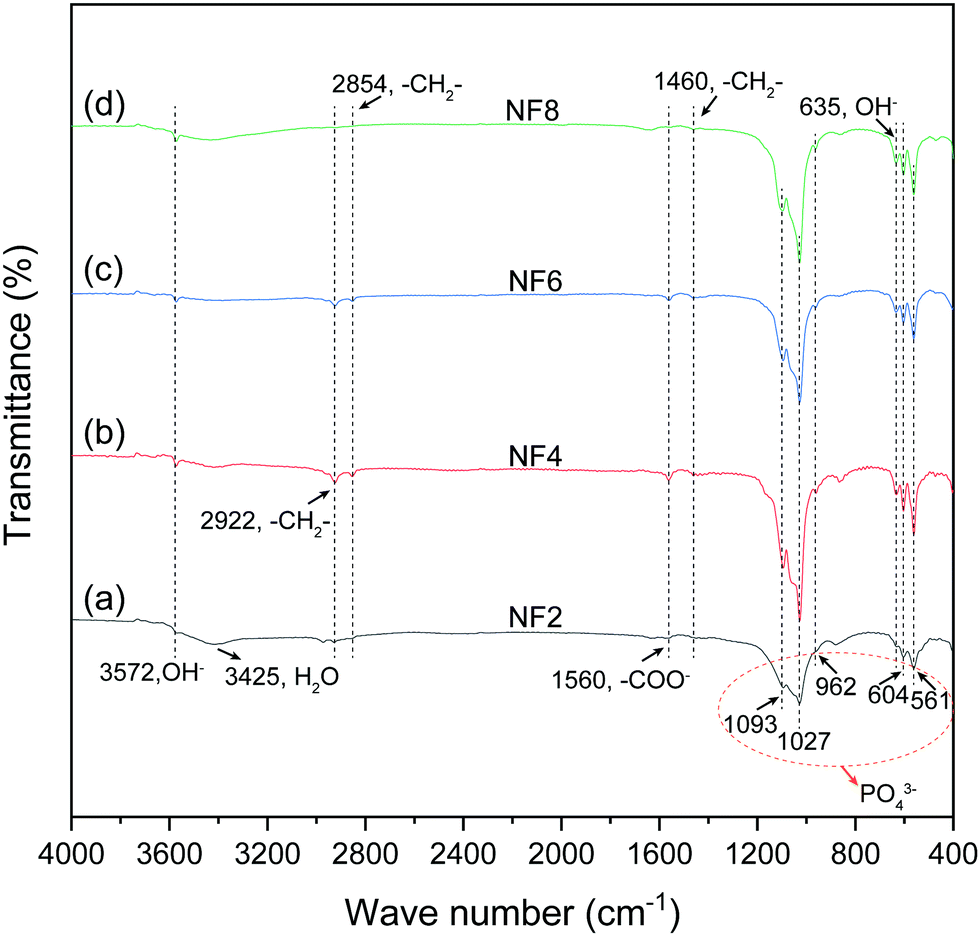 | ||
| Fig. 3 FTIR spectra of HA synthesized with different concentrations of NaOH. (a) 1.125 M; (b) 1.375 M; (c) 1.625 M; (d) 1.875 M. | ||
3.3 Morphologies of the HA products
Fig. 4 shows the FESEM photographs and EDS spectra of the obtained products synthesized with different [OH−]. As shown in Fig. 4 and Fig. S2 (ESI†), the morphologies of the HA products synthesized at different [OH−] exhibit a gradual change. The morphology evolves from nanofibers with white fungus-like clusters to flexible ultralong nanofibers, then to nanofibers with a few nanorods, and finally to fusiform products composed of nanorods. The sample NF2 obtained with low [OH−] (Fig. 4a and b) consists of nanofibers and white fungus-like substances. The corresponding EDS result showed that the Ca/P ratio of the white fungus-like substance was much lower than that of nanofibers or stoichiometric HA (Fig. 4c). When [OH−] was increased to 1.375 M, the white fungus-like substance disappeared, and nanofibers with ultralong length (>500 μm) exhibited higher aspect ratios and a more highly flexible 1D nanostructure without being broken than the HA fiber as summarized in Table S1, ESI† (Fig. 4d and e). Moreover, the paper prepared by these nanofibers also exhibits excellent flexibility, which can be bent without breaking (Fig. S3, ESI†). The magnified FESEM micrograph in Fig. 4e indicated that an ultralong nanofiber was assembled by one or more finer nanofibers. Extending [OH−] to 1.625 M, HA micro/nano-rods with uneven sizes were observed (Fig. 4g and h). Combined with its XRD result and analysis, HA micro/nano-rods have a larger grain size, but have a smaller preferred growth orientation along the c-axis than nanofibers. As [OH−] continued to increase (Fig. 4j and k), flexible HA nanofibers were completely transformed into fusiform assemblies of nanorods.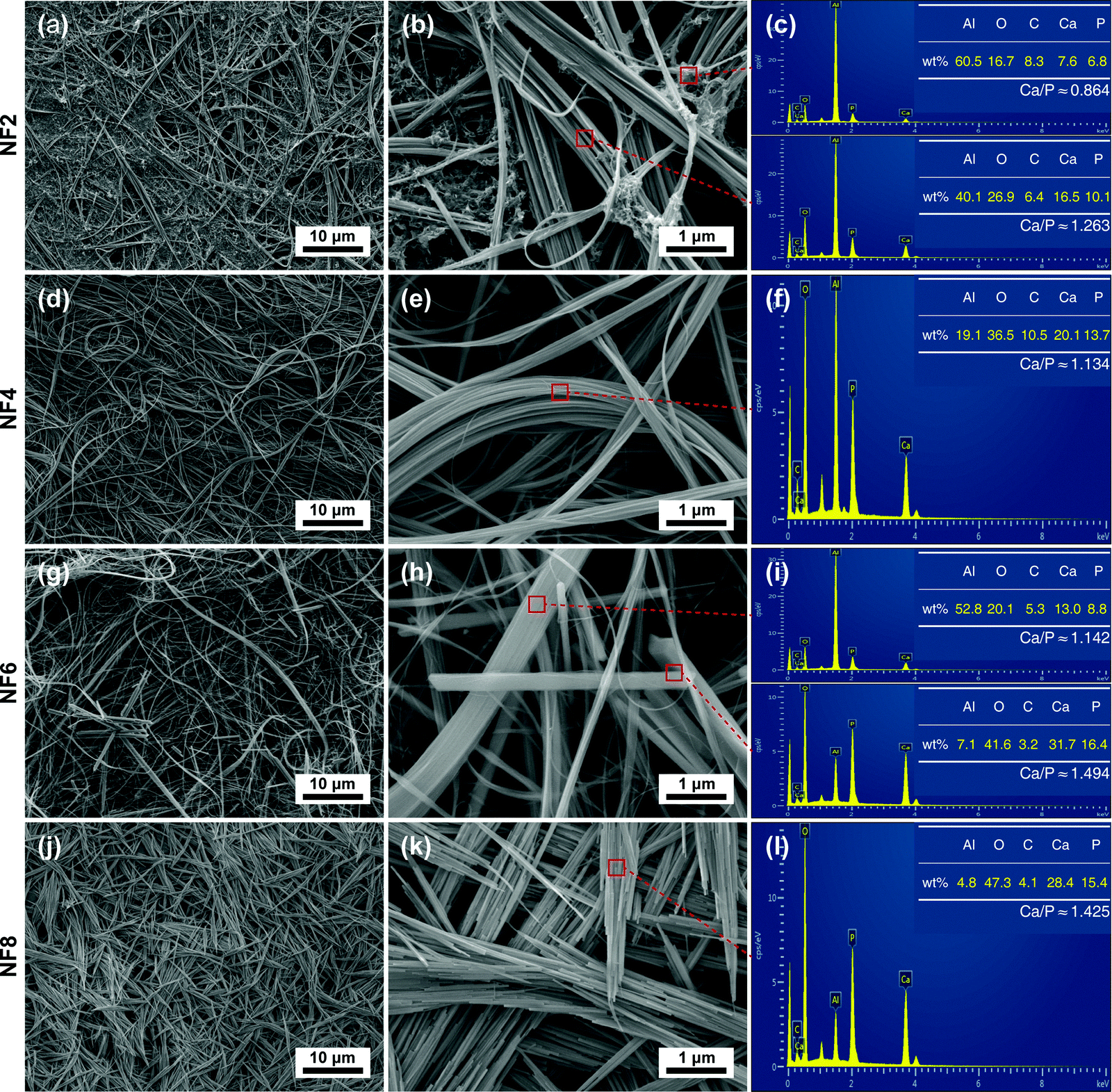 | ||
| Fig. 4 FESEM photographs and EDS spectra of HA synthesized with different concentrations of NaOH. (a)–(c) 1.125 M; (d)–(f) 1.375 M; (g)–(i) 1.625 M; (j)–(l) 1.875 M. | ||
The corresponding EDS analyses of HA nanofibers and micro/nano-rods showed that similar morphologies of HA in different samples had similar calculated Ca/P ratios, and the calculated Ca/P ratios of the nanofibers were lower than that of micro/nano-rods. Furthermore, it was noted that the calculated Ca/P ratios of HA nanofibers and micro/nano-rods were lower than the theoretical Ca/P ratio (1.67) of HA, which may be attributed to a deficiency in the crystal lattice of calcium. In addition, the lattice defect may be one of the reasons for the high flexibility of HA nanofibers.
4. Discussion
ipH, an indicator of supersaturation, can indirectly reflect the rate of formation of crystal nuclei, while it can affect the crystal growth of HA.23 Besides, fewer nuclei formed at low supersaturation of the solution, which favored the formation of larger crystals.23 As can be seen from Table S1 (ESI†), HA nanofibers were easier to synthesize in the ipH range of 6–10. Thus, low and suitable ipH or low concentration of OH− ions was beneficial for the preparation of HA nanofibers. In addition, the concentration of Ca2+ and PO43− ions also affected the degree of HA supersaturation (S) according to the following equation:36where α is the concentration of ions and Ks is the solubility of HA, which is a constant at a specific temperature. The concentration of Ca2+ ions in the oleic acid-assisted solvothermal system depended markedly on the Ca2+ release rate of calcium oleate, as the high pH tended to accelerate the hydrolysis of calcium oleate due to the nucleation and crystal growth of HA. Considering these, ipH should be strictly controlled, and the effect of [OH−]-responsive oleic acid on the pH of the reaction system and crystal growth of HA need to be systematically investigated. To better explain the effect of oleic acid on the crystal growth of HA, NaOH was used to regulate the ipH of the reaction solution and the formation process of the as-prepared products based on the structural characteristics and morphology evolution is shown in Fig. 5.
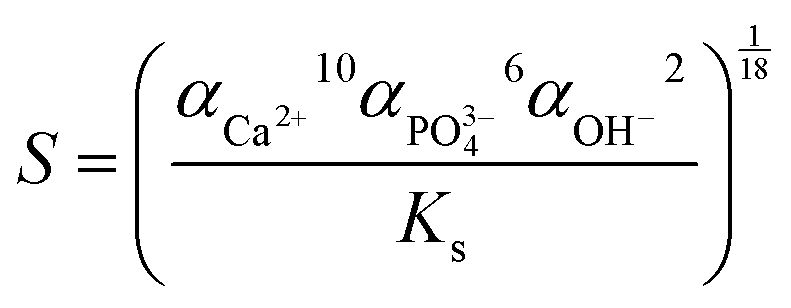
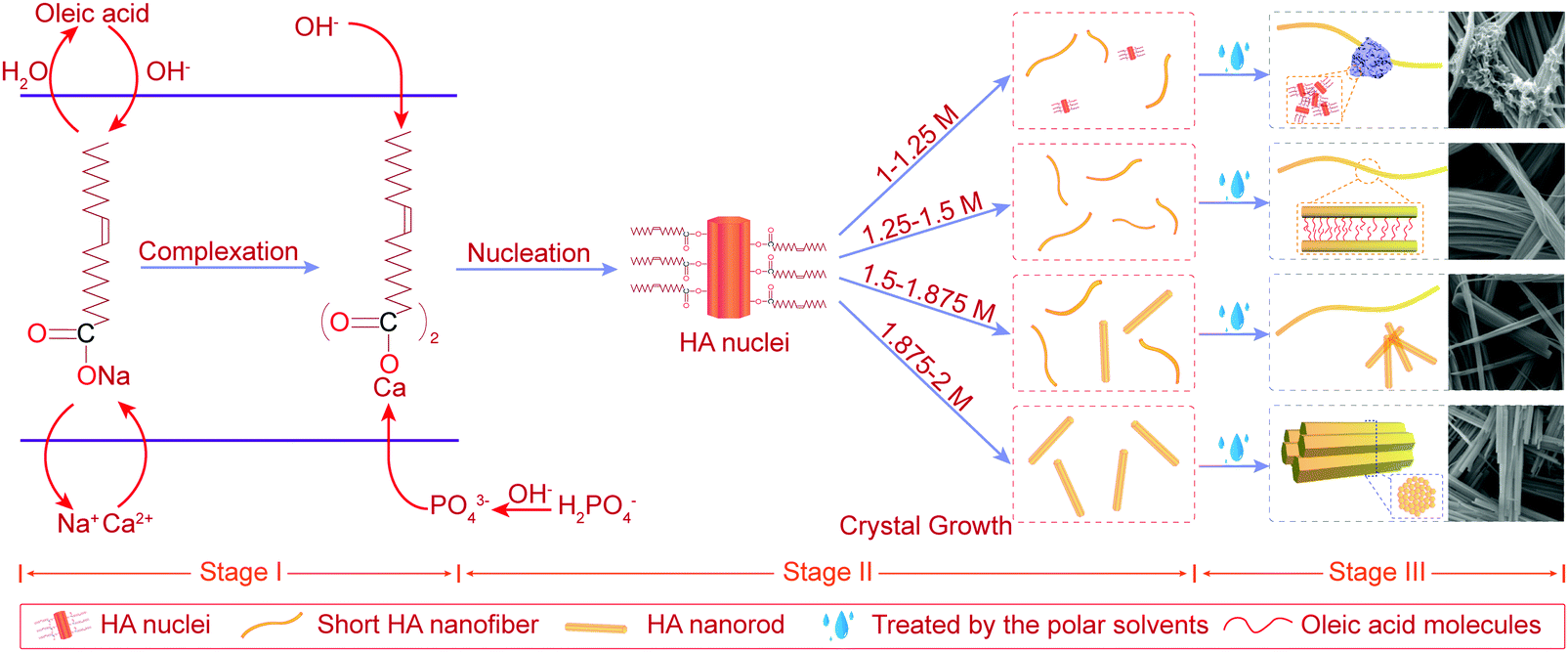 | ||
| Fig. 5 Schematic illustration of the formation process of the as-prepared products synthesized with different concentrations of NaOH from 1 M to 2 M. | ||
4.1 Stage I – the beginning of the synthetic process
As shown in the stage I of Fig. 5, sodium oleate was formed by the chemical reaction between oleic acid and OH− ions and its concentration increased with an increase in [OH−]. Then, calcium oleate was prepared with the addition of CaCl2, acting as a precursor and a calcium source.4.2 Stage II – nucleation and crystal growth
As shown in stage II of nucleation and crystal growth of Fig. 5, with the process of a solvothermal reaction, the hydrolysis of oleates began and continuously provided OH− ions to meet the nucleation and crystal growth of HA, while calcium oleate also slowly released Ca2+ ions. Both these routes may assist in maintaining the supersaturation of the reaction solution at a constant value, and favor the formation of HA with uniform morphology. Subsequently, the nucleation of HA formed by the charge attraction among Ca2+, PO43− and OH− ions under the conditions of high temperature and high pressure. During the process of nucleation and crystal growth, oleic acid molecules bound to the surface of newly generated HA, and induced the preferred growth direction of HA crystals along the c-axis. Besides, it was noted that the reaction environment with higher ipH had a higher degree of supersaturation due to a higher concentration of sodium oleate. Therefore, a higher ipH drove the system to become quickly supersaturated to form more nuclei during the solvothermal process.When the reaction solution was no longer in a supersaturated state, HA nuclei grew into 1D nanostructured crystals with different morphologies under different [OH−]. For [OH−] = 1.0–1.25 M, some HA nuclei grew into short flexible nanofibers under the induction of oleic acid molecules bound to their surface, as others may stop growing due to the lower ipH that did not provide sufficient OH− ions to meet the growth of HA. Combining the results of the corresponding FTIR and EDS, the white fungus-like clusters may be a mixture of HA nuclei, oleate, and other ions (Fig. 3a and 4c). And its existence may be responsible for the low crystallinity of samples N1 and N2 (Table 1, Fig. 1a and Fig. S1a, ESI†). For [OH−] = 1.25–1.50 M, the as-prepared products consisted of short HA nanofibers with flexibility. And short HA nanofibers with a disorderly distribution were deposited on the bottom of the reaction solution in the solvothermal process. Besides, from the changing trend of the final pH in Table 1, it can be inferred that oleic acid and oleate act as pH buffers to maintain a stable pH for [OH−] < 1.50 M, which may assist in the growth of highly flexible HA nanofibers. For [OH−] = 1.50–1.875 M, HA micro/nano-rods began to appear and their numbers increased with an increase in [OH−], indicating that 1.50 M was a critical concentration affecting the preferred growth orientation of HA along the a-axis or c-axis. Although the changing trends of the lattice parameter showed that the preferential growth direction of HA shifted from the c-axis to the a-axis (Table 1), the morphologies of the products still maintained a 1D nanostructure, suggesting that oleic acid had a remarkable effect on inducing the HA crystal growth into 1D morphologies. For [OH−] = 1.875–2.0 M, HA nanofibers were completely transformed into nanorods. Besides, the crystallite size and diameter of sample NF6 were larger than that of sample NF8 (Fig. 2 and 4h, k). Undoubtedly, high ipH or high supersaturation would tend to drive more HA nuclei, and the crystal growth of HA would be limited, leading to short and thin nanorods.
4.3 Stage III – the end of the synthetic process
In the solvothermal process, oleate ions can absorb on the surface of HA through the interplay between the calcium ions of HA and carboxyl groups, and the alkyl chains exposed to the reaction solution. When the as-prepared products transferred from the reaction solution to the polar solvents (water and ethanol), they would self-assemble into the ordered structures due to the incompatibility of the alkyl chains of oleate ions with the polar solvents (ethanol and water).37 Thus, as shown in stage III of Fig. 5, under the interaction between oleate ions absorbed on their surface and the polar solvents, the as-prepared products synthesized at different [OH−] exhibited a similar self-assembly process. For [OH−] = 1.0–1.25 M, HA nuclei and other ions formed the white fungus-like clusters. For [OH−] = 1.25–1.875 M, a large number of short HA nanofibers self-assembled into long nanofibers with a higher aspect ratio. For [OH−] = 1.875–2.0 M, short and thin HA nanorods self-assembled into fusiform products.5. Conclusions
The morphology and structural characteristics of the as-prepared HA exhibited a gradual change using oleic acid as the surfactant under different [OH−], but the constitution was unaffected. When [OH−] < 1.50 M, oleic acid molecules bound to the surface of HA effectively induced the preferential growth along the c-axis. Meanwhile, [OH−]-responsive oleic acid and its corresponding oleate in the solution acted as pH buffers to maintain the stability of the supersaturated system. And the crystalline size and the c-axial preferential growth direction were both improved with the increase of [OH−]. When [OH−] > 1.50 M, oleic acid absorbed on the surface of HA can still remarkably induce the crystal growth into 1D nanostructured morphologies. However, with the increase in [OH−], the preferred growth of HA began to shift from the c-axis to the a-axis, and the crystalline size of HA also decreased. In this work, HA nanofibers with high flexibility and high aspect ratio can be obtained at [OH−] 1.25–1.50 M or ipH 6.72–6.82. These results could offer some views to synthesize flexible HA nanofibers using oleic acid as a surfactant.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Shandong Provincial Natural Science Foundation (ZR2017MEM014), and Project of Shandong Province Higher Educational Science and Technology Program (J17KA004).References
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Chem. Rev., 2008, 108, 4754–4783 CrossRef CAS.
- V. Uskokovic and D. P. Uskokovic, J. Biomed. Mater. Res., Part B, 2011, 96, 152–191 CrossRef.
- H. Zhou and J. Lee, Acta Biomater., 2011, 7, 2769–2781 CrossRef CAS.
- S. V. Dorozhkin, Acta Biomater., 2010, 6, 715–734 CrossRef CAS.
- M. Sadat-Shojai, M. T. Khorasani, E. Dinpanah-Khoshdargi and A. Jamshidi, Acta Biomater., 2013, 9, 7591–7621 CrossRef CAS.
- W. Chen, B. Tian, Y. Lei, Q. F. Ke, Z. A. Zhu and Y. P. Guo, Mater. Sci. Eng., C, 2016, 67, 395–408 CrossRef CAS.
- Z. Zhuang, T. J. Fujimi, M. Nakamura, T. Konishi, H. Yoshimura and M. Aizawa, Acta Biomater., 2013, 9, 6732–6740 CrossRef CAS.
- H. Kim, R. P. Camata, S. Chowdhury and Y. K. Vohra, Acta Biomater., 2010, 6, 3234–3241 CrossRef CAS.
- C. M. Lieber, Solid State Commun., 1998, 107, 607–616 CrossRef CAS.
- Y. Xia, P. Yang, Y. Sun, Y. Wu, B. Mayers, B. Gates, Y. Yin, F. Kim and H. Yan, Adv. Mater., 2003, 15, 353–389 CrossRef CAS.
- M.-L. Qi, K. He, Z.-N. Huang, R. Shahbazian-Yassar, G.-Y. Xiao, Y.-P. Lu and T. Shokuhfar, JOM, 2017, 69, 1354–1360 CrossRef CAS.
- M.-l. Qi, Z. Huang, W. Yao, F. Long, M. Cheng, B. Song, D. Banner, R. Shahbazian-Yassar, Y.-p. Lu and T. Shokuhfar, CrystEngComm, 2018, 20, 1031–1036 RSC.
- B.-Q. Lu and Y.-J. Zhu, Can. J. Chem., 2017, 95, 1091–1102 CrossRef CAS.
- K. Kandori, N. Horigami, A. Yasukawa and T. Ishikawa, J. Am. Ceram. Soc., 1997, 80, 1157–1164 CrossRef CAS.
- D. O. Costa, S. J. Dixon and A. S. Rizkalla, ACS Appl. Mater. Interfaces, 2012, 4, 1490–1499 CrossRef CAS.
- M.-l. Qi, G.-y. Xiao, T. Shokuhfar and Y.-p. Lu, Surf. Innovations, 2017, 5, 75–81 CrossRef.
- M. Aizawa, H. Ueno, K. Itatani and I. Okada, J. Eur. Ceram. Soc., 2006, 26, 501–507 CrossRef CAS.
- H. Zhang and B. W. Darvell, Acta Biomater., 2010, 6, 3216–3222 CrossRef CAS.
- Y.-c. Wang, W.-l. Xu, Y.-p. Lu, W.-h. Xu, H. Yin and G.-y. Xiao, Mater. Sci. Eng., C, 2020, 108, 110408 CrossRef CAS.
- F. Mohandes, M. Salavati-Niasari, Z. Fereshteh and M. Fathi, Ceram. Int., 2014, 40, 12227–12233 CrossRef CAS.
- C. Qi, S. Musetti, L. H. Fu, Y. J. Zhu and L. Huang, Chem. Soc. Rev., 2019, 48, 2698–2737 RSC.
- N. K. Nga, L. T. Giang, T. Q. Huy, P. H. Viet and C. Migliaresi, Colloids Surf., B, 2014, 116, 666–673 CrossRef CAS.
- H. Zhang and B. W. Darvell, Acta Biomater., 2011, 7, 2960–2968 CrossRef CAS.
- M. Sadat-Shojai, M.-T. Khorasani and A. Jamshidi, J. Cryst. Growth, 2012, 361, 73–84 CrossRef CAS.
- B. Q. Lu, Y. J. Zhu and F. Chen, Chemistry, 2014, 20, 1242–1246 CrossRef CAS.
- Z. C. Xiong, Y. J. Zhu, F. F. Chen, T. W. Sun and Y. Q. Shen, Chemistry, 2016, 22, 11224–11231 CrossRef CAS.
- X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Adv. Mater., 2006, 18, 2031–2034 CrossRef CAS.
- W. Tao, J. Wang, W. J. Parak, O. C. Farokhzad and J. Shi, ACS Nano, 2019, 13, 4876–4882 CrossRef CAS.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS.
- M. Ghiyasiyan-Arani, M. Salavati-Niasari and S. Naseh, Ultrason. Sonochem., 2017, 39, 494–503 CrossRef CAS.
- B. Ma, S. Zhang, F. Liu, J. Duan, S. Wang, J. Han, Y. Sang, X. Yu, D. Li, W. Tang, S. Ge and H. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 33717–33727 CrossRef CAS.
- E. Landi, A. Tampieri, G. Celotti and S. Sprio, J. Eur. Ceram. Soc., 2000, 20, 2377–2387 CrossRef CAS.
- M. Sivakumar, T. S. S. Kumar, K. L. Shantha and K. P. Rao, Biomaterials, 1996, 17, 1709–1714 CrossRef CAS.
- A. Diez-Escudero, M. Espanol, S. Beats and M. P. Ginebra, Acta Biomater., 2017, 60, 81–92 CrossRef CAS.
- Z. Xu and G. Hu, RSC Adv., 2012, 2, 11404–11409 RSC.
- T. Ruikang and H. N. George, Pure Appl. Chem., 2002, 74, 1851–1857 Search PubMed.
- F. Chen and Y. J. Zhu, ACS Nano, 2016, 10, 11483–11495 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj04476h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |