
The remarkable selectivity of the 2-arylquinoline-based acyl hydrazones toward copper salts: exploration of their catalytic applications in the copper catalysed N-arylation of indole derivatives and C1-alkynylation of tetrahydroisoquinolines via the A3 reaction†
Carlos A.
Echeverry-Gonzalez
 ,
Marlyn Catalina
Ortiz Villamizar
and
Vladimir V.
Kouznetsov
*
,
Marlyn Catalina
Ortiz Villamizar
and
Vladimir V.
Kouznetsov
*
Laboratorio de Química Orgánica y Biomolecular, CMN, Universidad Industrial de Santander, Parque Tecnológico Guatiguará, Km 2 vía refugio, Piedecuesta, A.A. 681011, Colombia. E-mail: kouznet@uis.edu.co
First published on 16th November 2020
Abstract
Ligands promoting copper-catalysed coupling reactions have received increasing attention because of their ability to enhance the catalytic activity of copper, making these reactions applicable in different fields such as drugs, pharmaceutically interesting molecules, and organic materials. Herein, we reported the synthesis and characterization of different selective 2-arylquinoline-based acyl hydrazones toward copper(I) salts. We explored the scope of the catalytic system based on copper/acyl hydrazones in the catalysis of C–N bond formation between N-heterocycles, specifically indoles, pyrroles and carbazoles, and aryl iodides as well as redox-A3 coupling of tetrahydroisoquinolines, terminal alkynes, and aldehydes.
Introduction
Acyl hydrazones (ACs) are widely tunable photo-switches that have received increasing attention owing mainly to their high stabilities, facile preparation,1 biological activity,2 and excellent absorption and fluorescence properties.3 These compounds containing a carbon–nitrogen double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) possess attractive properties, such as the configurational dynamics resulting from the photochemical and thermal E–Z isomerization of CN, and therefore, they could be applied as photoactive molecular motors.4
N) possess attractive properties, such as the configurational dynamics resulting from the photochemical and thermal E–Z isomerization of CN, and therefore, they could be applied as photoactive molecular motors.4
Likewise, considering their ability for metal coordination, ACs have also been applied as chelating ligands of transition metal ions in environmental sensors,5 medicinal chemistry,6–8 and catalysis.9 ACs have shown selectivity to a broad range of transition metals including Ni,9 Co,6 Ru,7 Pb,5 and Cu,8 and their selectivity to one specific metal can be tuned by structural changes.5 This ability for metal coordination is very important for improving the catalytic properties for some metals, including copper (Cu), which is one of the most studied and versatile catalytic systems. According to several investigations, the ligands play a key role in the mechanism of the Cu-catalysed reactions, leading to the formation of complexed species of Cu and an increase of reaction yields. The catalytic enhancement could be attributed to the electronic nature of the ligands as well as the increased solubility of the Cu catalyst enhancing the oxidized species of Cu in solution.10 Advances, regarding the increase of the reaction yields with lower amounts of wasted copper and more environmentally friendly reaction conditions, have been achieved through the use of chelating ligands in Cu-catalysed coupling reactions, as can be seen for the Cu-catalysed C–N,11 C–C,12 and C–O13 bond-forming reactions (Ullmann-type reactions), and the A3 coupling reactions (Fig. 1).14,15
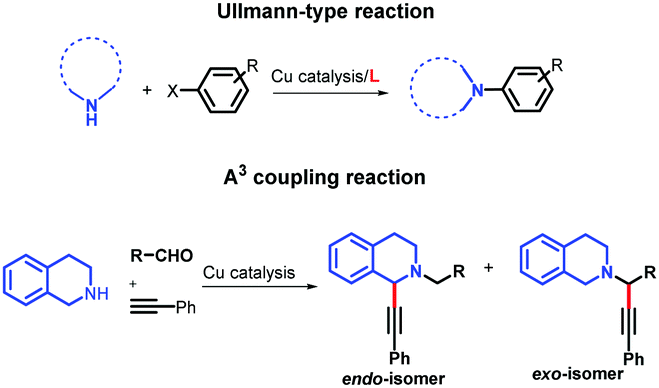 | ||
| Fig. 1 Examples of Cu-catalysed coupling reactions. | ||
For the Ullmann-type reaction, different chelating ligands have been reported including pyrrolidine-2-phosphonic acid derivatives,16,17L-proline,18N,N′-dimethyl-ethylenediamine,19 1,10-phenanthroline,20,21 carbo-hydrazides and hydrazones,12,22,23 and 8-hydroxyquinoline.24 In recent years, it has been found that ligands with more electron-rich substituents and an increased steric hindrance (e.g., naphthalene–oxalamide derivatives) give better yields in the coupling reactions of aryl halides with different nitrogen nucleophiles using greener conditions.25,26 In the case of A3 coupling reactions, to the best of our knowledge, the use of ligands for improving the catalytic activity of Cu ensuring mild reaction conditions has been more underexplored. Considering that the yields, regioselectivity (endo- or exo-isomer) and stereoselectivity (in C1) depend on both the nature of the Cu source and ligands used, different groups have reported good results employing Cu(I) or Cu(II) 2-ethylhexanoate as catalysts,27 or triphenylphosphine15,28 and N-PINAP as ligands.29 In this work, we report the use of ACs based on 2-arylquinolines with high selectivity specifically toward Cu cations as ligands, which will be demonstrated by their photophysical characterization. For the first time, these ligands will be used in a catalytic system to promote both Ullmann-type and A3 coupling reactions (Fig. 2).
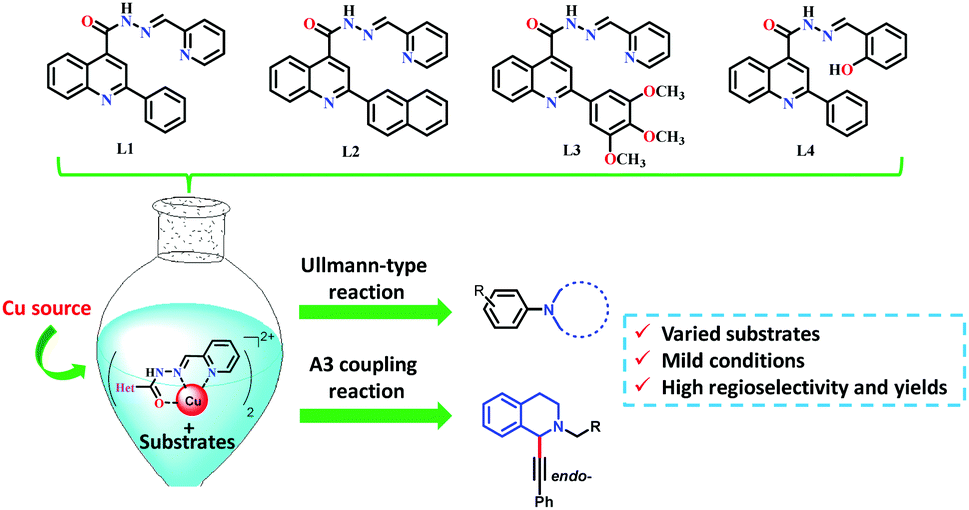 | ||
| Fig. 2 Chemical structure of the ligands L(1–4) and their use in Cu-catalysed reactions. | ||
The ligands shown in Fig. 2 were synthesised using the quinoline ring, introducing varied substituents into their structure. Chelating ligands were obtained following the relevant points: (i) the use of green, mild and efficient methodology employing only class 3 solvents without further purification; (ii) the use of moieties showing some demonstrated pharmacological activity and synthetic versatility in order to obtain environmentally friendly and varied ligands; and (iii) the use of ligands with electron-donor and bulky substituents to figure out whether they could affect the catalytic activity of Cu positively.
Results and discussion
Synthesis and photophysical characterization of the chelating ligands
As mentioned before, ACs show configurational dynamics (E–Z isomerization), and it has been demonstrated that ACs with increased steric interaction result in the formation of significant yields of the Z form. Thus, in order to obtain only the E form of L(1–4) (Fig. 2), different synthetic methodologies, including conventional heating and grinding using condensation of 2-arylquinoline-4-carbohydrazides and 2-pyridinecarboxaldehyde were used (see Scheme S1, ESI†). However, in all cases, ligands L(1–3) with the same E/Z ratio (3.3/1) were obtained. This ratio is attributed to the hydrogen bond formation between pyridine (Py) and the first nitrogen of the acyl hydrazone group (Fig. S1, ESI†). To determine whether Py ring has an effect on the catalyst, we prepared ligand L4 using the 2-hydroxyphenyl fragment rather than the Py ring, resulting in only the E form as previously reported.6Detection of Cu using chelating ligands is a relevant aim considering its key role in many biological process,30 resulting in neurodegenerative diseases when its cellular homeostasis is altered.31,32 For achieving the improvement of the catalytic ability of the Cu is commonly necessary to coordinate its oxidised species with chelating ligands resulting in a larger amount of those in solution. Considering that, we proposed to figure out whether the obtained ligands selectively recognize Cu cations by means of their photophysical characterization (Fig. 3).
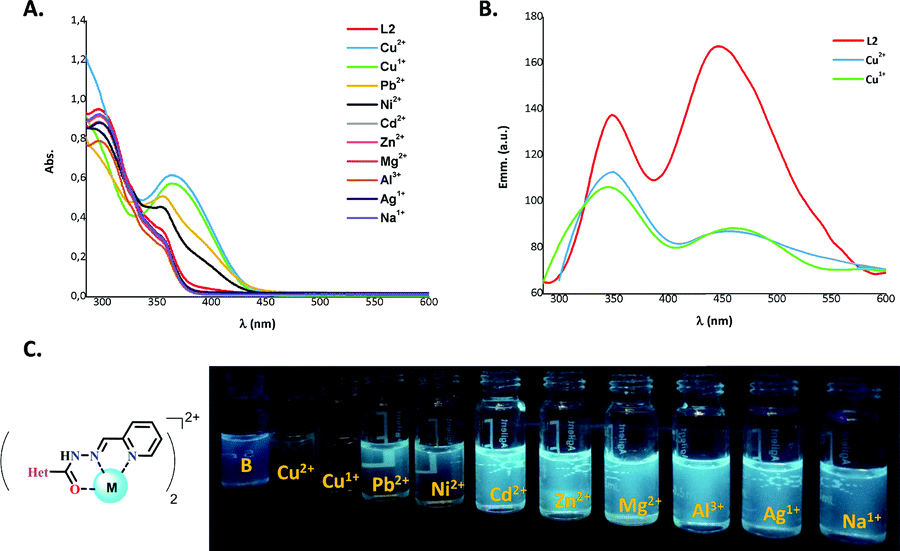 | ||
| Fig. 3 Photophysical characterization: (A) UV-vis spectra of L2 (3.0 × 10−5 M) in the presence of metallic salts (3.0 × 10−4 M); (B) fluorescence emission spectra of L2 in the presence of Cu salts, λexc = 270 nm; and (C) photograph of L2 with each metallic cation under a long wavelength (λ = 365 nm). | ||
The UV-vis and fluorescence emission spectra of L2 were recorded in dimethyl sulfoxide (DMSO) at a concentration of 3.0 × 10−5 M, and each corresponding salt was at a concentration of 3.0 × 10−4 M. A stock solution of L2 (1.4 × 10−3 M) and the corresponding salts of Cu2+, Cu1+, Pb2+, Ni2+, Cd2+, Zn2+, Mg2+, Al3+, Ag1+, and Na1+ was also prepared in DMSO. The UV-vis spectrum in solution exhibits one strong absorption band around 297 nm that corresponds to the π → π* electronic transition, with a shoulder at 355 nm corresponding to a weak intramolecular charge transfer (ICT) band between the quinoline core and the Py moiety. After addition of Cu2+ or Cu1+ to the ligand solution, the strong interaction between L2 and the Cu cations enhanced the π delocalization with an increase of the ICT, resulting in a bathochromic shift to 364 nm of the corresponding ICT band (Fig. 3A). It has also been proved that ACs can weakly coordinate the cations Pb2+ and Ni2+ which resulted in a slight increase of the absorption of the ICT band for L2. For the case of the emission behaviour, L2 showed two maxima peaks around 349 and 447 nm that were similar to those of naphthalene derivatives.33 The addition of Cu2+ or Cu1+ ions led to a substantial diminution of the fluorescence intensity up to 2-fold due to the increasing of the ICT when solutions were excited at 270 nm (Fig. 3B and C), thus demonstrating the effectiveness of L2 in coordinating Cu ions. Considering this selectivity toward Cu1+, and its widespread use in the catalysis of Ullmann-type and A3 coupling reactions, we envisioned that L(1–4) could be good ligands for improving the catalytic ability of Cu in these reactions.
N-Arylation of indole derivatives via the Ullmann-type reaction
The synthesis of indole derivatives is important because their structural motifs are a common occurrence in numerous drugs,34 pharmaceutically interesting molecules,35 natural products,36,37 and organic materials.38 We first conducted the Cu-catalysed Ullmann reaction using milder conditions than some previous reported studies,39 by coupling indole (1) and 4-iodoanisole (2) using 10 mol% of CuI at 110 °C in DMSO for 24 h, affording 3 with a yield of 33% (Table 1, entry 1).|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst | L | Solvent | Base | Yieldb (%) |
| a Reaction conditions: 4-iodoanisole (1.0 mmol), indole (1.4 mmol), Cu catalyst (0.1 mmol), ligand (0.1 mmol), base (1.4 mmol) in DMSO (2 mL mmol−1). b The uncertainty of the yield was calculated as the standard deviation of two reaction and purification using SiO2. c 1,4-Dioxane was used as the solvent. NR: no reaction. | |||||
| 1 | CuI (10 mol%) | — | DMSO | K2CO3 | 33 ± 0.7 |
| 2 | CuI (10 mol%) | — | DMSO | t-BuOK | 25 ± 0.5 |
| 3 | X-Phos-Pd-G2 | — | DMSO | t-BuOK | NRc |
| 4 | CuI (10 mol%) | L1 | DMSO | K2CO3 | 65 ± 4.9 |
| 5 | CuI (10 mol%) | L2 | DMSO | K2CO3 | 86 ± 1.4 |
| 6 | CuI (10 mol%) | L3 | DMSO | K2CO3 | 74 ± 2.8 |
| 7 | CuI (10 mol%) | L4 | DMSO | K2CO3 | 38 ± 2.7 |
| 8 | — | L2 | DMSO | K2CO3 | NR |
| 9 | CuI (10 mol%) | L2 | Toluene | K2CO3 | NR |
| 10 | CuI (5 mol%) | L2 | DMSO | K2CO3 | 44 ± 1.5 |
| 11 | CuI (15 mol%) | L2 | DMSO | K2CO3 | 99 ± 0.7 |
| 12 | CuBr (15 mol%) | L2 | DMSO | K2CO3 | 90 ± 2.1 |
| 13 | CuI (10 mol%) | L2 | DMSO | Cs2CO3 | 79 ± 4.2 |
This reaction proceeded well in the presence of K2CO3 as a base, giving a lower yield when t-BuOK was used (entry 2). In the case of palladium-catalysed cross-coupling reactions, we found that N-arylation of indole was incompatible with the Buchwald amination methodology when XPhos-Pd-G2 was used as the catalyst (entry 3).40 The catalytic system based on CuI/L(1–3) showed high yields (entries 4–6), and amongst them, the system based on L2 displayed the highest yield of 86% (entry 5). As mentioned above, factors as the increased solubility of the Cu catalyst and the electronic nature of the ligand affected the yields positively. The better yield given by L2 in comparison with L1 showed that increased steric hindrance was necessary to get more effective ligands as previously reported.25 In the case of L4, its catalytic ability was limited showing a yield of 38% (entry 7) that was almost similar to the yield without ligand (entry 1). That demonstrates the importance of the Py ring for enhancing the catalytic ability of Cu. Likewise, to the best of our knowledge, owing to the configurational dynamics showed by ACs, the E/Z ratio may not affect the catalytic ability of the system CuI/L.
It is worth noting that Cu catalyst plays a key role in the catalysis of this reaction, and thus, when only ligand L2 is used as catalytic system the reaction does not proceed (entry 8). Considering that depending on the reaction the best results can be obtained with polar or non-polar solvents, and the non-polar solvent commonly used in the Cu-catalysed Ullmann reaction corresponds to toluene,41 we evaluated it using our catalytic system (L2/CuI). However, as can be seen in entry 9, the reaction does not proceed with toluene using our standardised conditions. We varied the catalyst loading from 5 to 15 mol% (entries 10 and 11), finding the best yield (almost quantitative) was achieved when CuI was used at 15 mol%. In addition, it is worth noting that the variation of the Cu source mildly affected the reaction yield, affording 3 in 90% yield when CuBr was used (entry 12). The reaction yield was practically unaffected with the replacement of the base K2CO3 by Cs2CO3 (entry 13).
We explored the scope of the catalytic system based on CuI/L2 in the N-arylation of unsaturated N-heterocycles (Table 2). Under our optimised conditions, the reaction of indole and aryl iodide as 4-iodotoluene and 6-iodo-2-phenylquinoline led to the desired products 4 and 5 with 93% and 99% yields, respectively. Unfortunately, the catalytic system was not able to access the less reactive aryl bromide (see 5, Table 2).
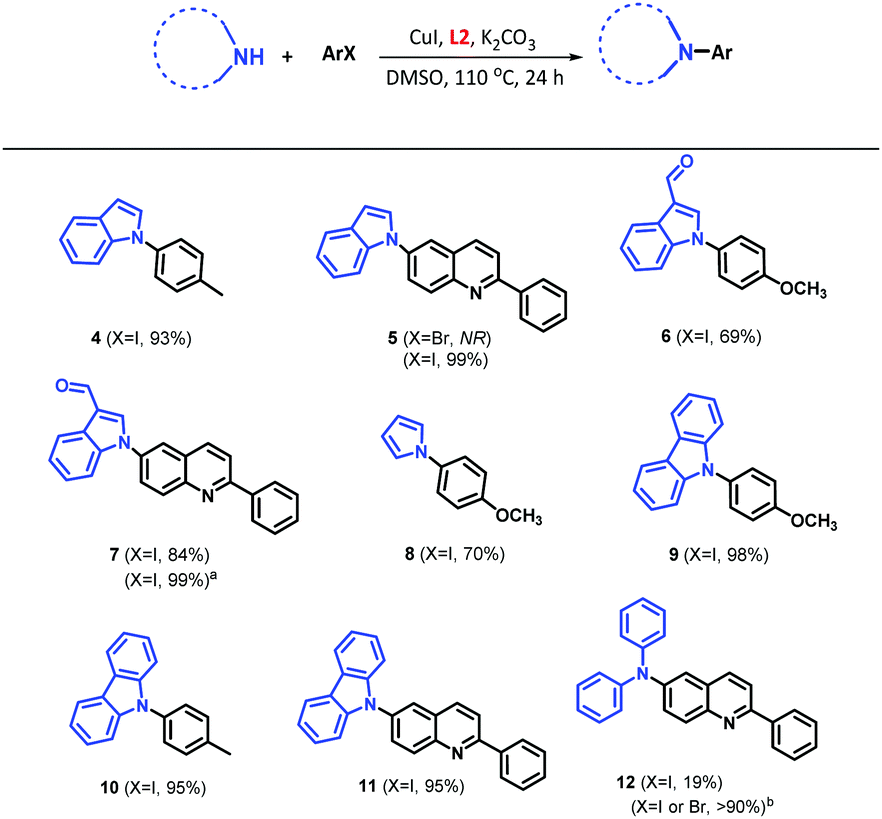
The use of nitrogen nucleophiles with electron-withdrawing substituents affected the reaction efficiency negatively, as can be seen for 6 and 7. However, an increase of the reaction time to 48 h led to an enhanced yield of 99% for 7. Additionally, the system CuI/L2 allowed for an efficient coupling of pyrrole (product 8) with a yield of 70%, which could be attributed to the formation of polypyrrole via a side polymerization reaction. In the case of the carbazole substrate, we obtained the compounds 9, 10 and 11 with higher yields (almost quantitative) than the reported methodologies using CuI/1,10-phenantroline (20 mol%),42 and palladium catalysis.43 On the other hand, the reaction did not proceed well under optimised conditions with aromatic amines (i.e., diphenylamine), affording 12 with a 19% yield; however, 12 was obtained in high yield using the Buchwald amination methodology using XPhos-Pd-G2 catalyst (1 mol%) with both aryl iodides and bromides, as we have previously we reported.44 It is worth mentioning that we had problem for the coupling reaction using azole derivatives such as imidazole and pyrazole using our catalytic system. Purification of the reaction crude and NMR analysis showed a possible interaction between nitrogen nucleophile with the ligands (data not shown).
Ligands L(1–3) have shown great versatility and efficiency with different catalytic activities depending on the nucleophilic nitrogen nature. According to Table 2, the reactivity order would be carbazole > indole > pyrrole > diphenylamine, which could be attributed to a higher acidity of carbazole and the subsequent greater ease of ionization (pKa = 19.9 in DMSO), in comparison with indole (pKa = 20.9), pyrrole (pKa = 23.0), and diphenylamine (pKa = 24.9).45
Redox-A3 reaction of tetrahydroisoquinolines, terminal alkynes, and aldehydes
Multi-component condensation, commonly known as the A3 coupling reaction, is an efficient synthetic route for the synthesis of varied products including 1,2-disubstituted tetrahydroisoquinolines (THIQs).46 Two different regioisomers of THIQs (endo- and exo-), depending on iminium ion isomerization, may be obtained by A3 coupling of THIQ, terminal alkynes, and aldehydes (Fig. 1).28 It has been demonstrated that the regioselectivity of this reaction can be well-tuned by the effect of the copper catalysts, obtaining a larger ratio of the endo/exo isomers when CuI47 or the catalytic systems based on CuBr/PPh328 are used. Considering the large class of natural and unnatural compounds based on THIQ with a stereogenic centre at the C1 position (endo-isomer),48–50 we have optimised a synthetic methodology for the synthesis of only endo-isomer in higher yields using our developed ligands shown in Fig. 2.We first conducted the A3 reaction by coupling THIQ 13, 2-bromobenzaldehyde (14), and phenylacetylene (15) using 10 mol% of CuI at 50 °C in toluene for 24 h, affording 16a with a yield of 58% (Table 3, entry 1). According to NMR, the use of CuI gave only the endo-isomer 16a but with other side products. Purification of the reaction crude by SiO2 gave an unknown impurity in 20% yield corresponding to (phenylethynyl)copper, 16a in 58% yield, and retained side products in the column head (Fig. S4, ESI†). Surprisingly, when the catalytic system based on CuI/L1 (10 mol%) was used, the desired compound 16a was obtained almost with a quantitative yield (entry 2). In contrast to the Ullmann-type reaction, ligands with steric hindrance (L2) and electron-rich quinolines (L3) showed lower yields with 16a as the only isomer (entries 3 and 4). Interestingly, when the Py group in L1 was replaced by the 2-hydroxyphenyl group in L4, the reaction yield decreased (entry 5), showing once again the importance of the Py ring in the coordination of the Cu salt. Likewise, when CuI loading was reduced to 5 mol%, 16a was obtained in 30% yield without the presence of 16b (entry 6).
|
|
|||
|---|---|---|---|
| Entrya | Catalyst | L (10 mol%) | Yield 16ab (%) |
| a Reaction conditions: tetrahydroisoquinoline 13 (1.4 mmol), 2-bromobenzaldehyde 14 (1.4 mmol), phenylacetylene 15 (1.0 mmol), Cu catalyst (0.1 mmol), ligand (0.1 mmol), in toluene (0.2 M). b Yields are those of the purified compounds using SiO2. | |||
| 1 | CuI (10 mol%) | — | 58 |
| 2 | CuI (10 mol%) | L1 | 99 |
| 3 | CuI (10 mol%) | L2 | 62 |
| 4 | CuI (10 mol%) | L3 | 72 |
| 5 | CuI (10 mol%) | L4 | 83 |
| 6 | CuI (5 mol%) | L1 | 30 |
| 7 | CuBr (10 mol%) | L1 | 94 |
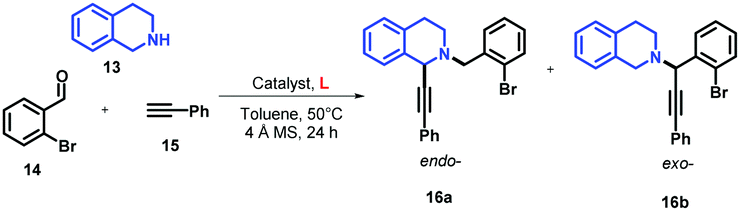
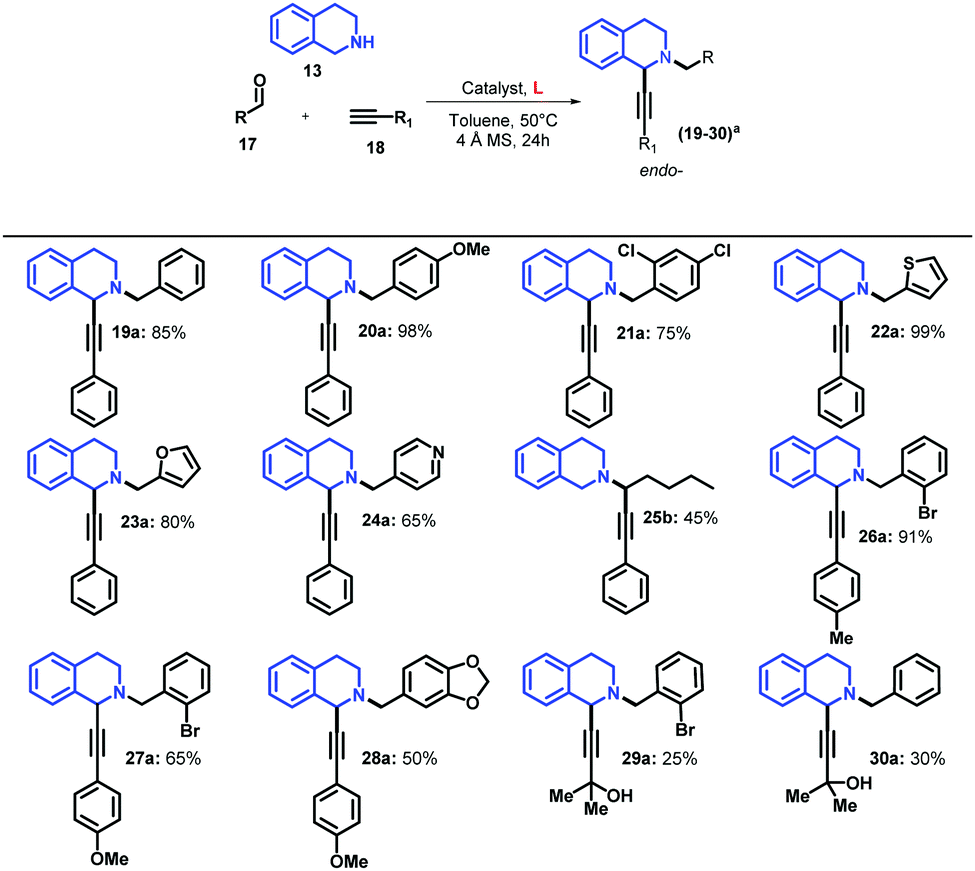
As mentioned before, Ma and co-workers reported that the use of the catalytic system based on CuBr/PPh3 at 80 °C in toluene improves considerably the yield of the endo-isomer.28 We demonstrated that the use of AC-based ligands and CuBr leads to the formation of exclusively the endo-isomer, affording 16a in 94% yield using a catalytic loading of 10 mol% with milder reaction conditions (entry 7). Considering that the catalytic system based on CuI/L1 showed the highest yields, we explored its catalytic scope using different aldehydes and terminal alkynes (Table 4).
| a Yields are those of the purified compounds using SiO2. All assays gave only the endo-isomers, except for 25a. |
|---|
|
When benzaldehyde was used, 19a was obtained with a 85% yield, which was similar to the yield previously reported by Yu for this compound,47 but we isolated only the endo-isomer. When aldehydes 17 with either electron-donor or electron withdrawing substituents and phenylacetylene were employed, higher yields were obtained in the coupling reaction in comparison with benzaldehyde (16a and 20a, Tables 3 and 4, respectively). However, the replacement of 2-bromobenzaldehyde in 16a by 2,4-chlorobenzaldehyde in 21a affected the reaction yield negatively, which could be attributed to the reduction of steric hindrance by replacing bromine by chlorine. This steric effect on the A3 reaction had already been studied by Seidel and co-workers, demonstrating that electronically similar aldehydes with smaller steric hindrance result in lower yields with a reduction of the endo-/exo-isomer ratio.27
The replacement of the aromatic aldehyde by a heterocycle aldehyde showed a high dependence of the A3 coupling on the electronic nature of these substrates, resulting in high yields in the case of electron-donor heterocycles (22a and 23a), and moderate yield for electron-withdrawing such as as 4-pyridine (24a). Interestingly, the replacement of the aromatic aldehyde by an aliphatic aldehyde led to the formation of exo-isomer in 45% yield (25b), which is in contrast to previous reports, in which the endo-isomer was predominant,47 demonstrating that the regioselectivity of this reaction can be well-tuned not only by the effect of the Cu catalysts, but also by the nature of the employed aldehyde. Likewise, the replacement of phenylacetylene by 4-ethynyltoluene affected slightly the reaction yield affording 26a in 91% yield; however, the use of alkynes with stronger electron-donor substituents led to a dramatic decrease of the reaction yield affording 27a and 28a in 65% and 50% yields, respectively.
According to the recent literature, the use of aliphatic terminal alkynes can affect the reaction yield negatively.28,47 We evaluated this effect using 2-methyl-3-butyn-2-ol as the terminal alkyne 18 (Table 4), affording the desired products 29a and 30a with yields of 25% and 30%, respectively, thus demonstrating that the catalytic systems based on CuI/ACs are very susceptible to the nature of all the substrates employed in the coupling reaction.
In order to further demonstrate the relationship of the catalytic enhancement for the A3 coupling reaction in the presence of the quinoline ring in L1, we performed the synthesis of 16a using CuI and (E)-N′-((pyridin-2-yl)methylene)benzohydrazide (L5) as the ligand. L5 was synthesised in 65% yield as previously reported,3,51 giving only its E-isomer. Surprisingly, the compound 16a was obtained with a moderate yield (66%, Scheme 1), indicating that the enhancement of the reaction yield depends on the electronic nature of the chelating ligands.
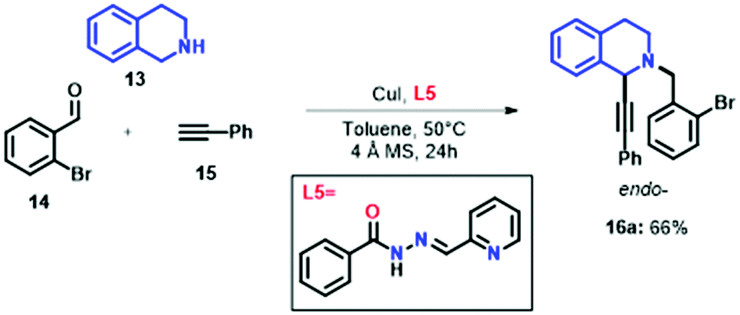 | ||
| Scheme 1 Synthesis of 16a using (E)-N'-((pyridin-2-yl)methylene)benzohydrazide. | ||
It is important to note that although the use of the Py group in the ligands improves the catalytic ability of the Cu in the A3 coupling reaction, the relevance of the 2-phenylquinoline for the catalysis become clearly more evident. In addition, previous studies have also revealed that steric hindrance affects the yield and selectivity of the A3 coupling reaction.27 Likewise, the use of alkynyl substrates with electron-donor groups dramatically reduces the reaction yield, which also occurs using our standardised conditions (see Table 4). According to our results, we can state that effectively the use of bulky ligands based on ACs leads to only endo-isomer because of the steric hindrance, learning that when non-sterically hindered aldehyde was used only the exo-isomer was obtained (see 25b in Table 4). These findings correlate with the results shown in Table 3 in different ways, namely (i) The reduction of reaction yield when ligand L2 was used is not driven by electronic factors. It has been demonstrated that the presence of naphthyl groups does not facilitate the formation of oxidised species in certain molecules in comparison with the phenyl group;52 therefore, naphthyl would not affect electronic density of Cu. This low yield could be attributed to a limitation regarding the steric hindrance of the A3 coupling reaction, and therefore, L2 with a bulky group in the quinoline ring had no effect on the reaction yield; (ii) ligand L3 with electron-donating ability showed lower yield because of electronic factors increasing the electronic density of Cu. It was also observed for alkynyl groups with electron-donor groups. In addition, this effect was similar for L4 in which an electron-deficient group such as Py was replaced by an electron-donor group as 2-hydroxyphenyl; and (iii) the replacement of the electron-deficient quinoline in L1 by the phenyl group in L5 would obviously affect the reaction yields negatively.
Conclusions
We have reported the use of stable ligands of 2-arylquinoline-based acyl hydrazones, which were very easily synthesised from cheap starting materials with high yields. These ligands were highly selective toward Cu in different oxidation states, as was demonstrated by the photophysical characterization, and they also showed a secondary selectivity toward Pb2+ and Ni2+. In the case of L2, the presence of the naphthyl group allowed us to evaluate this selectivity using fluorescence emission, finding that the addition of Cu2+ or Cu1+ to the ligand solution led to a substantial diminution of its fluorescence intensity up to 2-fold due to the increase of the ICT. Considering this selectivity, these synthesised ligands were employed to develop efficient catalytic systems based on CuI/L2 and CuI/L1 for affording a broad library of N-arylated heterocycles and THIQ derivatives, respectively. New nitrogen-containing heterocycles of both series are suitable precursors needed in organic chemistry, materials science and medicinal chemistry. For the Ullmann-type coupling reaction, our strategy was limited to the activation of aryl iodides; thereby, a main future goal must be to discover novel catalytic systems that can efficiently access less reactive aryl bromides and chlorides. Likewise, for the case of the A3 coupling reaction, further studies are important to understand the role of ligands in the catalytic process, considering that the best catalytic system based on CuI/L1 was very susceptible to the nature of all the used substrates.Experimental section
General
All reagents and solvents were used as purchased. Flash chromatography was performed using silica gel (Merck, Kieselgel 60, 230–240 mesh or Scharlau 60, 230–240 mesh). Analytical thin layer chromatography (TLC) was performed using aluminum coated Merck Kieselgel 60 F254 plates. NMR spectra were recorded on a Bruker Avance 400 (1H: 400 MHz; 13C: 100 MHz) spectrometer at 298 K using partially deuterated solvents as internal standards. Coupling constants (J) are denoted in Hz and chemical shifts (δ) in ppm. Multiplicities are denoted as follows: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad. UV-Vis spectra were recorded on a Genesys 10 s spectrophotometer using dimethyl sulfoxide (DMSO) as the solvent. Emission spectra were recorded on a Photon Technology International spectrophotometer using DMSO as the solvent. MALDI experiments were carried out on a Bruker ultrafleXtreme MALDI TOF-TOF instrument (Bruker Daltonics, Billerica, MA) equipped with a 1 kHz Smart Beam Nd:YAG laser (355 nm), 6 ns pulse and spot size of 100 μm – according to the manufacturer's specifications using the FlexAnalysis software. High-resolution ESI-mass spectra (HRMS) were measured on a Bruker ESI-micro Q-TOF III (Bruker Daltonics) apparatus.Synthesis of acyl hydrazone-based ligands
The general procedure is shown in Scheme S1 (ESI†). Ligands L1, L4 and L5 were synthesised as described previously, L1 is even commercially available.3,6 Briefly, the corresponding 2-arylquinoline-4-carbohydrazine (0.2 mmol) and 2-pyridincarbaldehyde (0.4 mmol) were heated in ethanol at 78 °C and five drops of trifluoroacetic acid (TFA) were added. The mixture was stirred for 10 min. Then, the solution was cooled, and the precipitate was filtered and used without further purification.Ligand L2
Beige solid (88%). M.p. 220–223 °C. 1H NMR (400 MHz, CDCl3) δ: 12.52 (s, 1H), 9.00 (s, 1H), 8.67–8.56 (m, 16.8, 6.5 Hz, 3H), 8.45 (d, J = 9.7 Hz, 1H), 8.26–8.21 (m, 2H), 8.12 (dd, J = 12.3, 8.1 Hz, 3H), 8.04–7.87 (m, 3H), 7.72 (t, J = 7.6 Hz, 1H), 7.67–7.58 (m, 2H), 7.48 (t, J = 6.4 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ: 163.6, 156.1, 153.4, 150.1, 149.6, 148.5, 137.5, 135.8, 134.2, 133.6, 131.0, 130.2, 129.3, 129.0, 128.1, 128.1, 127.2, 125.6, 125.2, 125.0, 123.9, 120.7, 117.9 ppm. Calculated for C26H18N4O m/z 402.148; found: [M − H] m/z 401.1482.Ligand L3
Beige solid (88%). Yellow solid (78%). M.p. 228–230 °C. 1H NMR (400 MHz, CDCl3) δ: 12.47 (s, 1H), 8.65 (d, J = 4.0 Hz, 1H), 8.50 (s, 1H), 8.40 (s, 1H), 8.20–8.16 (m, 2H), 8.08 (d, J = 7.9 Hz, 1H), 7.97–7.93 (m, 1H), 7.89–7.85 (m, 1H), 7.71–7.66 (m, 3H), 7.47 (dd, J = 7.5, 5.0 Hz, 1H), 3.96 (s, 6H), 3.77 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ: 163.6, 156.0, 153.8, 153.4, 150.1, 149.6, 148.2, 141.7, 140.0, 137.5, 134.0, 130.8, 130.1, 127.8, 125.4, 125.2, 123.7, 120.6, 117.8, 105.5, 105.3, 60.7, 56.7 ppm. Calculated for C25H22N4O4m/z 442.1641; found: [M − H] m/z 441.1605.N-Arylation of indole derivatives via the Ullmann-type reaction
Characterization of the reported compounds 3,394,398,189,42 and 1043 can be consulted in the ESI.† The synthetic procedure and characterization summary of unknown products are outlined below.Briefly, the aryl iodide (0.5 mmol), indole (0.7 mmol), K2CO3 (0.7 mmol), CuI (0.075 mmol), and ligand L2 (0.075 mmol) were dissolved in 2 mL of DMSO and heated at 110 °C for 24 h. The cooled solution was partitioned between H2O and EtOAc. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude was purified by SiO2 using dichloromethane/petroleum ether (2/1).
Compound 5
Beige solid (99%). M.p. 145–147 °C. 1H NMR (400 MHz, CDCl3) δ: 8.30 (d, J = 8.0 Hz, 1H), 8.23 (d, J = 8.7 Hz, 1H), 8.20–8.17 (m, 2H), 7.95–7.89 (m, 3H), 7.73 (d, J = 7.7 Hz, 1H), 7.67 (dd, J = 8.2, 1.0 Hz, 1H), 7.57–7.53 (m, 2H), 7.50–7.46 (m, 1H), 7.45 (d, J = 4.0 Hz, 1H), 7.29–7.25 (m, 1H), 7.23–7.19 (m, 1H), 6.75 (dd, J = 3.2, 0.9 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ: 157.4, 146.7, 139.4, 137.6, 136.5, 136.0, 131.5, 129.5, 129.0, 128.0, 127.7, 127.6, 126.8, 122.7, 121.3, 121.0, 120.7, 119.9, 110.5, 104.3 ppm. Calculated for C23H16N2m/z 320.1313; found: m/z 320.1308.Compound 6
Beige solid (69%). M.p. 105–107 °C 1H NMR (400 MHz, CDCl3) δ: 10.09 (s, 1H), 7.40 (d, J = 8.0 Hz, 1H), 7.85 (s, 1H), 7.46–7.38 (m, 3H), 7.37–7.30 (m, 2H), 7.07 (d, J = 8.0 Hz, 2H), 3.90 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ: 184.9, 159.5, 138.5, 138.0, 131.0, 126.4, 125.3, 124.5, 123.3, 122.2, 119.3, 115.1, 111.0, 55.7 ppm. Calculated for C16H13NO2m/z 251.0946; found: m/z 251.0944.Compound 7
Beige solid (99%). M.p. 141–143 °C. 1H NMR (400 MHz, CDCl3) δ: 10.15 (s, 1H), 8.44–8.40 (m, 1H), 8.36 (d, J = 8.9 Hz, 1H), 8.28 (d, J = 8.6 Hz, 1H), 8.21 (d, J = 7.0 Hz, 2H), 8.02 (s, 1H), 8.00 (d, J = 8.6 Hz, 1H), 7.96 (d, J = 2.4 Hz, 1H), 7.88 (dd, J = 8.9, 2.4 Hz, 1H), 7.58–7.55 (m, 3H), 7.53–7.59 (m, 1H), 7.42–7.36 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ: 185.0, 158.4, 147.4, 139.1, 138.1, 137.6, 136.7, 135.8, 132.0, 129.9, 129.0, 127.6, 127.5, 126.5, 125.7, 124.9, 123.7, 122.5, 122.4, 120.3, 120.1, 111.0 ppm. Calculated for C24H16N2O m/z 348.1263; found: m/z 348.1259.Compound 11
Beige solid (95%). M.p. 168–171 °C. 1H NMR (400 MHz, CDCl3) δ: 8.38 (d, J = 8.9 Hz, 1H), 8.27–8.14 (m, 5H), 7.99 (d, J = 2.3 Hz, 1H), 7.94 (d, J = 8.6 Hz, 1H), 7.91 (dd, J = 8.9, 2.3 Hz, 1H), 7.60–7.52 (m, 2H), 7.52–7.44 (m, 3H), 7.44–7.40 (m, 2H), 7.33–7.29 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ: 157.9, 147.2, 140.9, 139.4, 136.6, 135.5, 131.7, 129.6, 129.0, 127.8, 127.6, 126.2, 124.7, 123.6, 120.5, 120.3, 119.8, 109.8 ppm. Calculated for C27H18N2m/z 370.1470; found: m/z 370.1465.Compound 12
It was synthesised in 19% yield under our standardised conditions. However, it was obtained in high yield using the general procedure for the Buchwald reaction. Briefly, 6-iodo-2-phenylquinoline (0.5 mmol), diphenylamine (0.7 mmol), t-BuOK (0.7 mmol), and X-Phos-Pd-G2 (0.005 mmol) were dissolved in 1 mL of 1,4-dioxane and heated at 110 °C for 24 h. The cooled solution was partitioned between H2O and DCM. The aqueous layer was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude was purified by SiO2 using dichloromethane/petroleum ether (2/1). Yellow solid (96%). 1H NMR (400 MHz, CDCl3) δ: 8.15–8.10 (m, 2H), 8.02 (d, J = 9.1 Hz, 1H), 7.94 (dd, J = 8.7, 0.8 Hz, 1H), 7.78 (d, J = 8.6 Hz, 1H), 7.55–7.49 (m, 3H), 7.47–7.41 (m, 1H), 7.36–7.28 (m, 5H), 7.20–7.15 (m, 4H), 7.12–7.07 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ: 155.6, 147.5, 145.9, 135.5, 130.5, 129.5, 129.0, 128.8, 128.2, 127.4, 127.3, 124.8, 123.5, 119.3, 118.1 ppm. Calculated for C27H20N2m/z 372.1626; found: m/z 372.1622.Redox-A3 reaction of tetrahydroisoquinolines, terminal alkynes, and aldehydes
The synthetic procedure and characterization summary of unknown products are outlined below. The full characterization of known products can be consulted in our recently publication.53Compound 25b
A crimper vial equipped with a magnetic stir bar was charged with 1,2,3,4-tetrahydroisoquinoline 13 (1.4 mmol), pentanal (1.4 mmol), phenylacetylene (1 mmol), CuI (10 mol%), L1 (10 mol%) and 4 Å molecular sieves. The vial was sealed and was purged three times with argon and degassed toluene (0.2 M) was added. Then, the reaction mixture was heated for over 24 h at 100 °C. After cooling to room temperature, the crude mixture was loaded directly onto Celite; then, purified by column chromatography (silica gel) using hexane/ethyl acetate mixtures as the eluent. Yellow oil (45%). 1H NMR (400 MHz, CDCl3) δ: 7.48–7.44 (m, 2H), 7.33–7.30 (m, 3H), 7.16 (d, J = 3.8 Hz, 3H), 7.10 (dd, J = 6.3, 2.2 Hz, 1H), 3.98 (d, J = 14.7 Hz, 1H), 3.86–3.75 (m, 2H), 3.10–3.04 (m, 1H), 3.02–2.95 (m, 1H), 2.87–2.81 (m, 1H), 1.92–1.85 (m, 2H), 1.67–1.41 (m, 5H), 0.99 (t, J = 7.2 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ: 135.3, 134.5, 131.8(2), 128.7, 128.3(2), 128.0, 126.8, 126.0, 125.6, 123.4, 87.4, 86.1, 58.0, 52.1, 47.5, 33.4, 29.7, 29.1, 22.6, 14.2 ppm. ESI calcd for C22H25N (M + H)+: 304.2060, found: 304.2091.Conflicts of interest
There are not conflicts to declare.Acknowledgements
This work was supported by the Colombian Institute for Science and Research (COLCIENCIAS) under the project no. RC-007-2017, Cod. 110274558597. CAEG acknowledges the postdoctoral program VIE-UIS-Conv. 2018 and 2019. MCOV are thankful for the scholarship given by the doctoral program Colciencias-Conv. 617.References
- J. M. dos Santos Filho and S. M. Pinheiro, Green Chem., 2017, 19, 2212–2224 RSC.
- M. C. Mandewale, U. C. Patil, S. V. Shedge, U. R. Dappadwad and R. S. Yamgar, Beni-Suef Univ. J. Basic Appl. Sci., 2017, 6, 354–361 CrossRef.
- D. J. van Dijken, P. Kovařiček, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS.
- M. N. Chaur, D. Collado and J.-M. Lehn, Chem. – Eur. J., 2011, 17, 248–258 CrossRef CAS.
- Y.-T. Wang, S. Hu, Y. Zhang, H. Gong, R. Sun, W. Mao, D.-H. Wang and Y. Chen, J. Photochem. Photobiol., A, 2018, 355, 101–108 CrossRef CAS.
- Z. Xu, X. Zhang, W. Zhang, Y. Gao and Z. Zeng, Inorg. Chem. Commun., 2011, 14, 1569–1573 CrossRef CAS.
- P. Anitha, N. Chitrapriya, Y. J. Jang and P. Viswanathamurthi, J. Photochem. Photobiol., B, 2013, 129, 17–26 CrossRef CAS.
- Y. Li, Z.-Y. Yang and M.-F. Wang, J. Fluoresc., 2010, 20, 891–905 CrossRef CAS.
- P. Zhou, Y.-G. Zhao, Y. Bai, K.-L. Pang and C. He, Inorg. Chim. Acta, 2007, 360, 3965–3970 CrossRef CAS.
- H. Weingarten, J. Org. Chem., 1964, 29, 3624–3626 CrossRef CAS.
- P. H. Patil, J. L. Nallasivam and R. A. Fernandes, Asian J. Org. Chem., 2015, 4, 552–559 CrossRef CAS.
- H.-J. Cristau, P. P. Cellier, J.-F. Spindler and M. Taillefer, Chem. – Eur. J., 2004, 10, 5607–5622 CrossRef CAS.
- D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450–1460 CrossRef CAS.
- H. Xu, J. Wang, P. Wang, X. Niu, Y. Luo, L. Zhu and X. Yao, RSC Adv., 2018, 8, 32942–32947 RSC.
- N. Sudhapriya, A. Manikandan, M. R. Kumar and P. T. Perumal, Bioorg. Med. Chem. Lett., 2019, 29, 1308–1312 CrossRef CAS.
- H. Rao, H. Fu, Y. Jiang and Y. Zhao, J. Org. Chem., 2005, 70, 8107–8109 CrossRef CAS.
- H. Rao, Y. Jin, H. Fu, Y. Jiang and Y. Zhao, Chem. – Eur. J., 2006, 12, 3636–3646 CrossRef CAS.
- D. Ma and Q. Cai, Synlett, 2004, 128–130 CrossRef.
- J. C. Antilla, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 11684–11688 CrossRef CAS.
- R. A. Altman and S. L. Buchwald, Org. Lett., 2006, 8, 2779–2782 CrossRef CAS.
- Y. Zhang, R. P. Hsung, M. R. Tracey, K. C. M. Kurtz and E. L. Vera, Org. Lett., 2004, 6, 1151–1154 CrossRef CAS.
- J. Xie, X. Zhu, M. Huang, F. Meng, W. Chen and Y. Wan, Eur. J. Org. Chem., 2010, 3219–3223 CrossRef CAS.
- L. Li, L. Zhu, D. Chen, X. Hu and R. Wang, Eur. J. Org. Chem., 2011, 2692–2696 CrossRef CAS.
- H. Wang, Y. Li, F. Sun, Y. Feng, K. Jin and X. Wang, J. Org. Chem., 2008, 73, 8639–8642 CrossRef CAS.
- W. Zhou, M. Fan, J. Yin, Y. Jiang and D. Ma, J. Am. Chem. Soc., 2015, 137, 11942–11945 CrossRef CAS.
- J. Gao, S. Bhunia, K. Wang, L. Gan, S. Xia and D. Ma, Org. Lett., 2017, 19, 2809–2812 CrossRef CAS.
- D. Das, A. X. Sun and D. Seidel, Angew. Chem., 2013, 125, 3853–3857 CrossRef.
- W. Lin, T. Cao, W. Fan, Y. Han, J. Kuang, H. Luo, B. Miao, X. Tang, Q. Yu and W. Yuan, et al. , Angew. Chem., Int. Ed., 2014, 53, 277–281 CrossRef CAS.
- T. F. Knöpfel, P. Aschwanden, T. Ichikawa, T. Watanabe and E. M. Carreira, Angew. Chem., Int. Ed., 2004, 43, 5971–5973 CrossRef.
- A. Tigreros and J. Portilla, RSC Adv., 2020, 10, 19693–19712 RSC.
- L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10–11 CrossRef CAS.
- M. Saleem and K. H. Lee, RSC Adv., 2015, 5, 72150–72287 RSC.
- H. Maeda, T. Maeda and K. Mizuno, Molecules, 2012, 17, 5108–5125 CrossRef CAS.
- S. D. Banister, M. Moir, J. Stuart, R. C. Kevin, K. E. Wood, M. Longworth, S. M. Wilkinson, C. Beinat, A. S. Buchanan and M. Glass, et al. , ACS Chem. Neurosci., 2015, 6, 1546–1559 CrossRef CAS.
- H. Liu and A. Dömling, J. Org. Chem., 2009, 74, 6895–6898 CrossRef CAS.
- L. D. Luca, Curr. Med. Chem., 2006, 13, 1–23 Search PubMed.
- W. Gul and M. T. Hamann, Life Sci., 2005, 78, 442–453 CrossRef CAS.
- Y. Wang, Z. Wan, C. Jia and X. Yao, Synth. Met., 2016, 211, 40–48 CrossRef CAS.
- Y. Zhang, Z.-Y. Hu, X.-C. Li and X.-X. Guo, Synthesis, 2019, 1803–1808 CAS.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 15914–15917 CrossRef CAS.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- F. Chen, Y. Liu, J. Pan, A. Zhu, J. Bao, X. Yue, Z. Li, S. Wang and X. Ban, Opt. Mater., 2020, 101, 109781 CrossRef CAS.
- X. Liu, H. Sheng, Y. Zhou and Q. Song, Chem. Commun., 2020, 56, 1665–1668 RSC.
- C. A. Echeverry-Gonzalez, C. E. Puerto-Galvis, C. H. Borca, M. A. Mosquera, A. F. Luis-Robles and V. V. Kouznetsov, Org. Chem. Front., 2019, 6, 3374–3382 RSC.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- A. B. Dyatkin and R. A. Rivero, Tetrahedron Lett., 1998, 39, 3647–3650 CrossRef CAS.
- Q.-H. Zheng, W. Meng, G.-J. Jiang and Z.-X. Yu, Org. Lett., 2013, 15, 5928–5931 CrossRef CAS.
- Q. Zhang, G. Tu, Y. Zhao and T. Cheng, Tetrahedron, 2002, 58, 6795–6798 CrossRef CAS.
- K. Ye, Y. Ke, N. Keshava, J. Shanks, J. A. Kapp, R. R. Tekmal, J. Petros and H. C. Joshi, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1601–1606 CrossRef CAS.
- L. Rout, B. B. Parida, J.-C. Florent, L. Johannes, S. K. Choudhury, G. Phaomei, J. Scanlon and E. Bertounesque, Chem. – Eur. J., 2016, 22, 14812–14815 CrossRef CAS.
- N. R. Babu, S. Subashchandrabose, M. S. A. Padusha, H. Saleem, V. Manivannan and Y. Erdougdu, J. Mol. Struct., 2014, 1072, 84–93 CrossRef.
- A. M. Fraind, G. Sini, C. Risko, L. R. Ryzhkov, J.-L. Brédas and J. D. Tovar, J. Phys. Chem. B, 2013, 117, 6304–6317 CrossRef CAS.
- M. C. O. Villamizar, C. E. P. Galvis and V. V. Kouznetsov, Synthesis, 2020 DOI:10.1055/s-0040-1707370.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj04516k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |