
Oxidative cleavage of cycloalkenes using hydrogen peroxide and a tungsten-based catalyst: towards a complete mechanistic investigation†
Tony
Cousin
 ab,
Gregory
Chatel
a,
Bruno
Andrioletti
*b and
Micheline
Draye
*a
ab,
Gregory
Chatel
a,
Bruno
Andrioletti
*b and
Micheline
Draye
*a
aLCME, Univ. Savoie Mont Blanc, 73000 Chambéry, France. E-mail: micheline.draye@univ-smb.fr
bUniv. Lyon, Université Claude Bernard Lyon 1, INSA-Lyon, CPE-Lyon, ICBMS UMR CNRS 5246, Campus Lyon-Tech la Doua, Bât. Lederer, 43 Boulevard du 11 Novembre 1918, 69622 Villeurbanne, France. E-mail: bruno.andrioletti@univ-lyon1.fr
First published on 17th November 2020
Abstract
The identification of the intermediates and by-products produced during the oxidative cleavage of cycloalkenes in the presence of H2O2 and a tungsten-based catalyst for the production of dicarboxylic acids has been carried out under various experimental conditions. On the basis of this mechanistic investigation and previous studies from the literature, a complete reaction scheme for the formation of the reaction products and by-products is proposed. In this hypothetical mechanism, the production of a hydroperoxyalcohol intermediate accounts for the two pathways proposed by Noyori and Venturello for the formation of the targeted dicarboxylic acid. In addition, Baeyer–Villiger oxidation of the mono-aldehyde intermediate allows explaining the formation of short chain diacids observed as by-products during the reaction. Hence, the proposed mechanism constitutes a real tool for scientists looking for a better understanding and those heading to set up environmentally friendly conditions for the oxidative cleavage of cycloalkenes.
Introduction
The oxidative cleavage of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds has witnessed a growing interest for several decades as a key transformation to access value-added mono- and dicarboxylic acids from olefins.1–5 More specifically, dicarboxylic acids are major intermediates for the manufacture of important polyesters and polyamide materials but also for the production of daily products found in a wide range of applications such as lubricants, fibers, cosmetics, pesticides, pharmaceuticals and plasticizers.6,7 In the chemical industry, this class of compounds is mainly obtained by the oxidative cleavage of unsaturated carboxylic acids and cycloalkenes.6,7 One of the oldest oxidative cleavage processes was patented by DuPont in the 1950s for the production of adipic acid. Since, this dicarboxylic acid is still manufactured via the aerobic oxidation of cyclohexane to cyclohexanol and cyclohexanone followed by the oxidative cleavage of either one or a mixture of both (ketone + alcohol = KA Oil) with nitric acid.8 This industrial process leads to the formation of adipic acid in 95% yield with an annual production of 4 million tons.7,9 Adipic acid production is mainly dedicated to the synthesis of nylon 6,6 but also to plasticizers, lubricants and polyurethane resins.7,10
C bonds has witnessed a growing interest for several decades as a key transformation to access value-added mono- and dicarboxylic acids from olefins.1–5 More specifically, dicarboxylic acids are major intermediates for the manufacture of important polyesters and polyamide materials but also for the production of daily products found in a wide range of applications such as lubricants, fibers, cosmetics, pesticides, pharmaceuticals and plasticizers.6,7 In the chemical industry, this class of compounds is mainly obtained by the oxidative cleavage of unsaturated carboxylic acids and cycloalkenes.6,7 One of the oldest oxidative cleavage processes was patented by DuPont in the 1950s for the production of adipic acid. Since, this dicarboxylic acid is still manufactured via the aerobic oxidation of cyclohexane to cyclohexanol and cyclohexanone followed by the oxidative cleavage of either one or a mixture of both (ketone + alcohol = KA Oil) with nitric acid.8 This industrial process leads to the formation of adipic acid in 95% yield with an annual production of 4 million tons.7,9 Adipic acid production is mainly dedicated to the synthesis of nylon 6,6 but also to plasticizers, lubricants and polyurethane resins.7,10
Despite the major importance of adipic acid for key applications, the DuPont process results in the production of large volumetric amounts of nitrous oxide. Actually, up to 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tons of N2O arise from the oxidative cleavage of KA oil by nitric acid, representing 8% of the total anthropogenic production of this greenhouse gas.11,12 Nitrous oxide is particularly harmful to the environment as it causes ozone depletion, smog, and acid rains and its global warming potential is 300 times higher than that of CO2.13–16 Even if efforts are made to reduce the amount of released N2O through recycling, thermal and catalytic decomposition10,14,16,17, the energy consumption for adipic acid production remains high because this multi-step reaction is performed at temperatures reaching up to 160 °C.7,8,13
000 metric tons of N2O arise from the oxidative cleavage of KA oil by nitric acid, representing 8% of the total anthropogenic production of this greenhouse gas.11,12 Nitrous oxide is particularly harmful to the environment as it causes ozone depletion, smog, and acid rains and its global warming potential is 300 times higher than that of CO2.13–16 Even if efforts are made to reduce the amount of released N2O through recycling, thermal and catalytic decomposition10,14,16,17, the energy consumption for adipic acid production remains high because this multi-step reaction is performed at temperatures reaching up to 160 °C.7,8,13
These observations have prompted researchers to develop more environmentally friendly routes for the production of dicarboxylic acids through cycloolefin oxidative cleavage. In this context, hydrogen peroxide has been considered to replace commonly used oxidants. Indeed, H2O2 presents many advantages for the development of greener oxidation reactions: it is a widely available oxidant with 47% of active oxygen content, non-toxic, only releases water as a theoretical by-product and does not require temperatures of above 100 °C to perform oxidations.18,19 The H2O2-mediated oxidative cleavage of cycloalkenes has been widely documented in the literature. The latest trends in the development of sustainable catalytic systems for this reaction have been recently reviewed.5 In particular, the use of hydrogen peroxide in combination with tungsten-based catalysts has been proven promising in reducing the environmental impact of cycloalkene oxidative cleavage. Indeed, compared to other transition metals used for this reaction, tungsten-based catalysts are low cost,20,21 non-toxic22,23 and react with H2O2 to produce peroxotungstate compounds highly active toward olefins.24,25
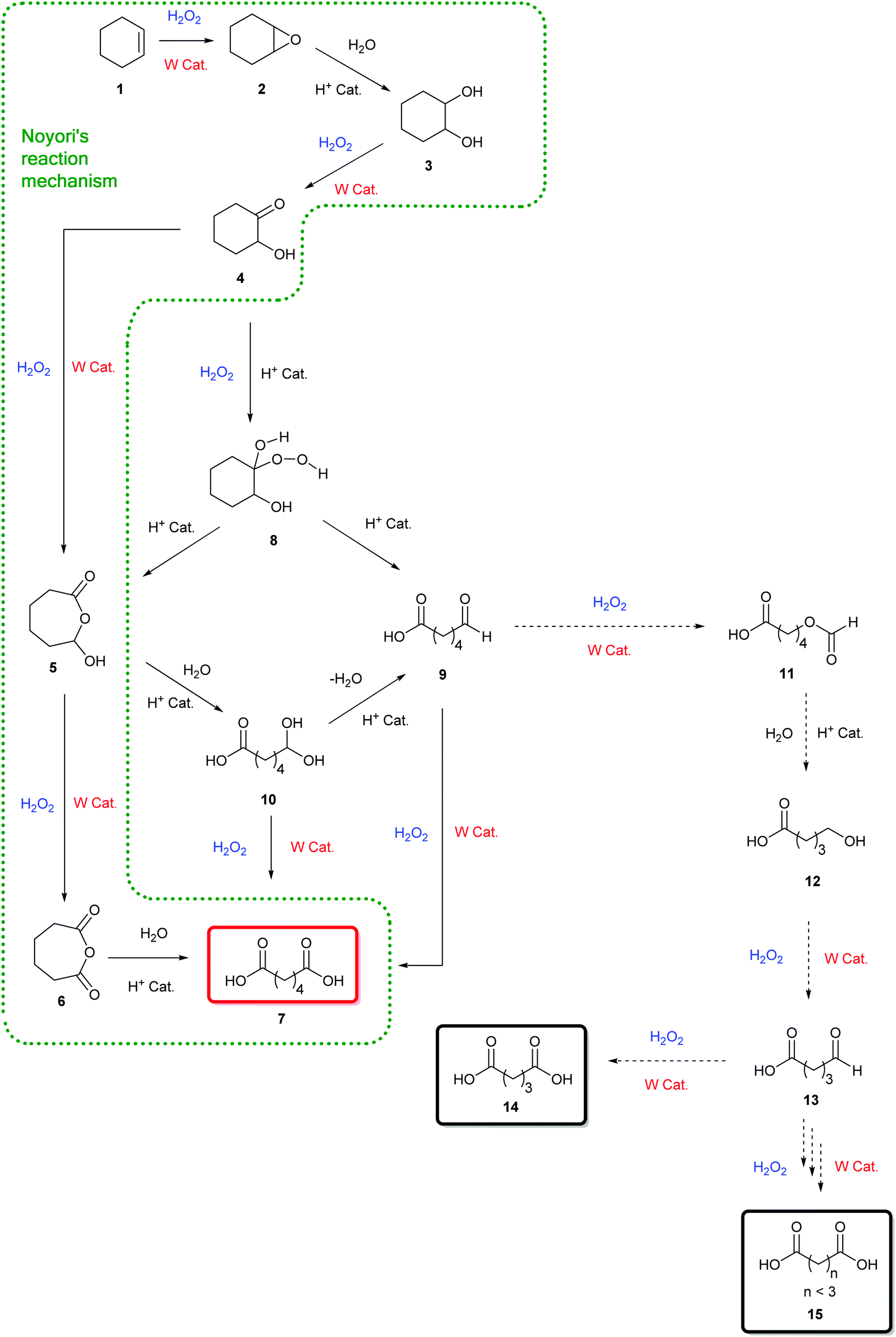
Among the numerous studies dealing with H2O2-mediated cycloalkene oxidative cleavage with tungsten-based catalysts, a large majority of publications exposes the reaction mechanism suggested by Noyori et al.11,26 for rationalizing the formation of dicarboxylic acids from cycloalkenes (Scheme 1).
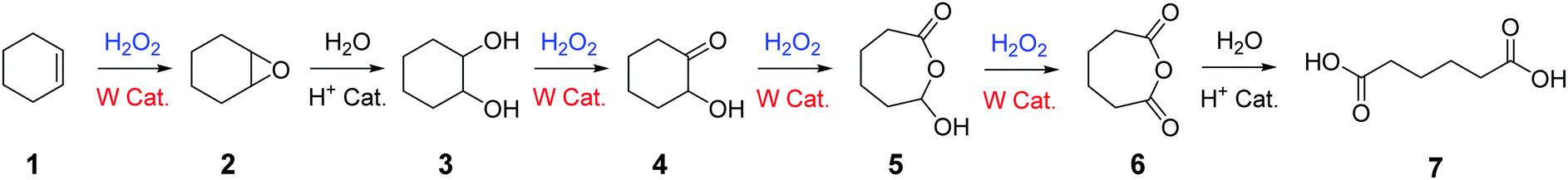 | ||
| Scheme 1 Oxidative cleavage of cycloalkenes with H2O2 and a tungsten-based catalyst according to Noyori.11,26 | ||
This mechanism involves six reaction steps including an alkene epoxidation, an epoxide hydrolysis, a first alcohol oxidation, a Baeyer–Villiger oxidation, a second alcohol oxidation and a cyclic anhydride hydrolysis (Scheme 1). Even if intermediates 2 to 4 have been unambiguously identified and described in the literature, the production of intermediates 5 and 6 through Baeyer–Villiger enlargement have not been highlighted yet. Their presence in the oxidation mechanism has been so far justified through indirect studies and computational modelling.27–29
No complete mechanism has been established to date to explain the formation of the targeted dicarboxylic acid from the hydroxy-ketone intermediate 4. Moreover, among the studies on the oxidative cleavage of cycloolefins, several by-products are often observed along with the expected dicarboxylic acid. Indeed, 1 to 5% of additional dicarboxylic acids, shortened by 1 to 3 carbon atoms compared to the targeted one, are observed at the end of the reaction.11,30,31 Despite their occurrence in studies, no possible mechanism for their formation has been suggested so far.
The accurate comprehension of the reaction pathways for product and by-product formation is of prime interest to determine the conditions favouring the production of the targeted dicarboxylic acid and minimizing the formation of these by-products.
In this study, we aim at confirming and completing the mechanisms proposed in the literature for the oxidative cleavage of cycloalkenes with H2O2 and a tungsten-based catalyst through the structural determination of intermediates and by-products of the reaction. Studies of the reaction at different stages and under different experimental conditions allow highlighting the structure and amount of by-products formed during the reaction.
Experimental
All chemicals were used without further purification and purchased from Acros, Aldrich, Alfa Aesar, Honeywell and Fluka.Gas chromatography was performed on a HP 6890 Series gas chromatograph from Hewlett Packard using a flame-ionization detector and equipped with an Optima-5MS Accent capillary column (dimethylpolysiloxane, 30 m × 0.25 mm × 0.25 μm) from Macherey-Nagel. All analyses were undertaken with helium as the carrier gas (24 mL min−1) in split mode (split ratio: 1:40) and an injector and a detector both heated at 290 °C.
Mass spectra were recorded on an Agilent 5973 N mass selective detector coupled to an Agilent 6890 GC and equipped with an Optima 5 capillary column (dimethylpolysiloxane 30 m × 0.32 mm × 0.25 μm) from Macherey-Nagel.
Cyclohexene oxidative cleavage
The reactions were performed following the procedure described in the reference study.11,26 Hydrogen peroxide (35 wt%, 2.6–525 g, 4.4 eq.) was added to a mixture of a tungsten-based catalyst (1 mol%) and a phase transfer catalyst (PTC: Aliquat 336® or Oct3MeN+ HSO4−, 1 mol%) in a round bottom flask. The solution was vigorously stirred using a magnetic stirring bar at room temperature for 10 min to solubilize as much of the catalyst as possible. After addition of cyclohexene (0.5–100 g, 1 eq.), the round bottom flask was equipped with a condenser and successively heated to 75 °C for 30 min, 80 °C for 30 min, 85 °C for 30 min, and 90 °C for 6 h 30 min while stirring at 1000 rpm. After 8 h, the reaction medium was cooled to room temperature before being placed at 5 °C overnight. Then the resulting white precipitate was separated from the aqueous solution by filtration, washed 3 times with cold deionized water and dried under reduced pressure. Sodium sulphite was added portion-wise to the aqueous filtrate until complete decomposition of the remaining hydrogen peroxide. After acidification with sulfuric acid to pH = 1, water was then evaporated under reduced pressure and 20 mL acetone was added to the solid residue. After separation of the organic solution from inorganic salts by filtration, acetone was eliminated under reduced pressure. Solid residues from the aqueous filtrate and precipitated adipic acid were analyzed by gas chromatography for quantification of all reaction products using the internal standard method (dodecane was used as the internal standard) after derivatization with N-O-bis(trimethylsilyl)trifluoroacetamide for 2 h at 50 °C. The reaction by-products were identified by GC/MS (characterization is available in the ESI†).Cycloalkene epoxidation
The cyclopentene, cyclohexene and cyclooctene epoxidations were performed as follows. Into a 250 mL round bottom flask, an aqueous solution of hydrogen peroxide (280 mmol, 26.90 g, 1.5 eq., 35 wt%) was added to a mixture of H2WO4 (1.4 mmol, 0.346 g, 0.75 mol%) and Aliquat 336® (1.4 mmol, 0.56 g, 0.75 mol%). The aqueous solution was stirred at room temperature using a stirring bar for 10 min before adding the cycloalkene (185 mmol). The round bottom flask was equipped with a condenser before stirring the biphasic mixture at 80 °C without any incubation period at 1000 rpm for 30 min.At the end of the reaction, the aqueous phase was separated from the organic layer, thoroughly extracted by CH2Cl2 or diethyl ether (3 × 30 mL) in order to recover remaining cycloalkene and newly formed products, and concentrated under reduced pressure. Isolation and analysis of the non-extracted epoxidation by-products contained in the aqueous phase were undertaken according to the same procedure as previously described. The reaction by-products were identified by GC/MS (the characterization is available in the ESI†).
Results and discussion
Within the scope of our study, we decided to limit our investigations to cyclohexene, cyclopentene and cyclooctene for the epoxidation and to cyclohexene for the oxidative cleavage. Indeed, the oxidative cleavage of cycloolefins is well-described in the literature on these small-sized cycloalkenes and affords the dicarboxylic acids with over 75% yields.5 Besides, the reaction performed on larger rings such as cyclooctene and cyclododecene results in low yields, generally below 30% because of transannular reactions occurring on the epoxide derivatives of these medium-sized cycloalkenes.5,32 Among the studies on the oxidative cleavage of cyclic alkenes with H2O2 and a tungsten-based catalyst, those from Noyori and co-workers are still a reference in terms of green chemistry. Indeed, the reaction conditions involve 4.4 equivalents of 30% H2O2, 1 mol% of Na2WO4 and Oct3MeN+HSO4− as a phase transfer catalyst (PTC). These conditions allow performing the oxidative cleavage of 100 g of cyclohexene from 75 to 90 °C in 8 h and without any organic solvent. Adipic acid is isolated in 93% yield.11,26During our studies, the oxidative cleavage of cyclohexene was investigated under the conditions described by Noyori and co-workers. The production of adipic acid as well as the by-products was followed by gas chromatography (GC). At first, the reaction was studied under the conditions used for the oxidative cleavage of 100 g of cyclohexene, corresponding to 600 mL of the reaction volume,11,26 and on 8 and 3 mL scales in order to decrease the amounts of reactants used per run. The obtained results are gathered in the Table 1.
| Entrya | Reaction volume (mL) | Mass of cyclo-hexene (g) | Conv.b (%) | Yieldc (%) | Main by-productsd (%) |
|---|---|---|---|---|---|
| a Experimental conditions: cyclohexene, H2O2 (35%, 4.4 eq.), Na2WO4 (1 mol%), Oct3MeN+HSO4− (1 mol%), 75–90 °C, 1000 rpm, 8 h. b Based on the amount of converted cyclohexene and calculated by GC with the internal standard method. c Based on the initial amount of cyclohexene and calculated by GC using the internal standard method. d By-products detected by GC/MS. e Results from the reference publication.11,26 | |||||
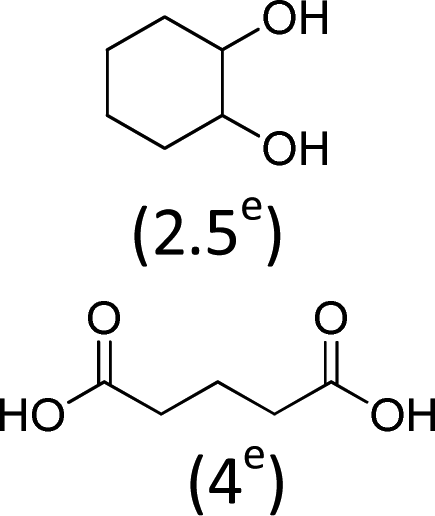
| 1 | 600 | 100 | 100e | 93e |
|
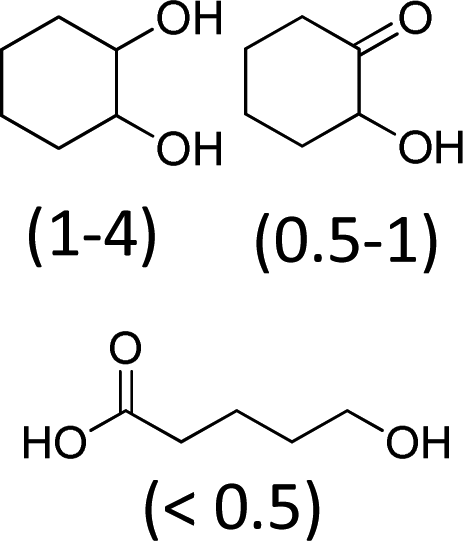
| 2 | 600 | 100 | 100 | 83 |
|
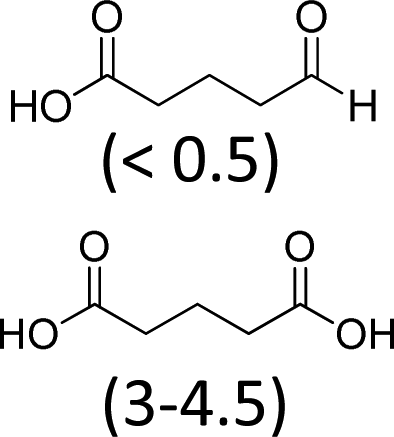
| 3 | 8 | 1.3 | 100 | 47 |
|
| 4 | 3 | 0.5 | 89 | 39 |
|
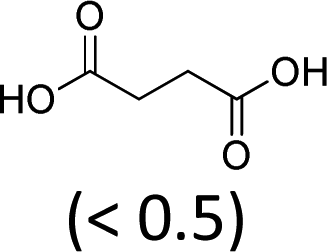
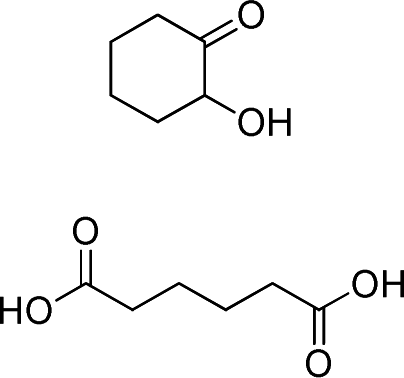
At 600 mL reaction scale, the oxidative cleavage of cyclohexene leads to 83% adipic acid yield (Table 1, entry 2) instead of the 93% published in the literature (Table 1, entry 1). These results can be ascribed to the formation of 1,2-cyclohexanediol, 2-cyclohexanone and glutaric acid observed in 0.5 to 4.5% yields. Moreover, the reaction performed under Noyori's conditions revealed the formation of by-products that were not described in the original work. Thus, small amounts (<0.5%) of 5-hydroxypentanoic, 5-oxopentanoic and succinic acid were detected during our study. When the reaction volume was decreased from 600 mL to 8 mL and 3 mL, the yield of adipic acid decreased from 83% to 47% and 39% respectively (Table 1, entries 2–4). However, the same by-products were produced in similar yields compared to 600 mL reaction volume, leading to an unsatisfactory mass balance. Hence, the oxidative cleavage yields and selectivities lower than 50% observed with reaction volumes below 10 mL can be explained by a cyclohexene loss by volatilization during the 8 h of reaction. Indeed, despite the experimental care, some of the solution has been volatilized. This would explain the choice of 100 g of starting cycloalkene chosen in the reference study,11,26 that ensured a minimum impact of volatilization of the reactant on the yield of the reaction. A reaction volume of 8 mL was chosen for the rest of the study, guaranteeing a reasonable yield and the use of a minimum amount of reactants (Table 1, entry 3).
Then the oxidative cleavage of cyclohexene was studied using different associations of tungsten-based catalysts and PTC to compare their performance with the Na2WO4/Oct3MeN+HSO4− system developed by Noyori, while following the formation of by-products. In addition, H2WO4/Aliquat 336® was chosen as catalytic system as it affords yields of above 80% for the oxidative cleavage of cyclohexene and cyclopentene in the literature.30,33 Different combinations of the W-based catalyst and PTC were studied in order to reach pH < 3 in the reaction medium. Indeed, a pH below 3 is required to ensure the formation of the active catalytic species26,34–36 and to prevent H2O2 from catalytic and thermal decomposition.18,19,37 Thus, the combination Na2WO4/Aliquat 336® was not selected as it affords a pH of 6 in the reaction medium. The results obtained for the different catalytic compositions are summarized in the Table 2.
| Entrya | Cat. W | PTC | pHb | Conv.c (%) | Yieldd (%) | Main by-productse (%) |
|---|---|---|---|---|---|---|
| a Experimental conditions: cyclohexene (16.5 mmol), H2O2 (35%, 4.4 eq.), Cat. W (1 mol%), PTC (1 mol%), 75–90 °C, 1000 rpm, 8 h. b pH of the aqueous phase during the reaction. c Based on the amount of converted cyclohexene and calculated by GC using internal standard method. d Based on the initial amount of cyclohexene and calculated by GC using internal standard method. e By-products detected by GC/MS. f 1,2-Cyclohexanediol yield. | ||||||
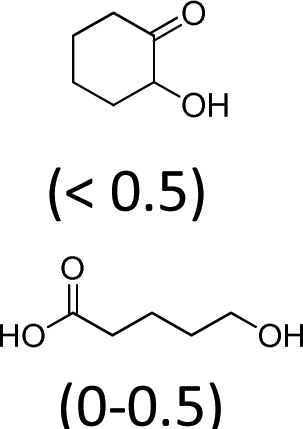
| 1 | Na2WO4· 2H2O | Oct3MeN+ HSO4− | 2 | 100 | 47 (4)f |
|
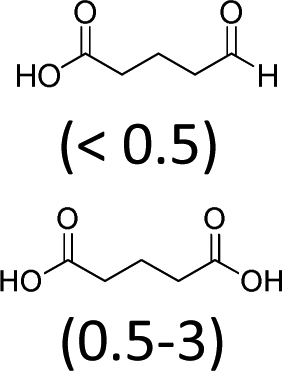
| 2 | H2WO4 | Aliquat 336® | 3 | 100 | 59 (15)f |
|
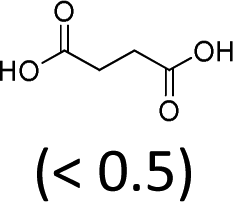
| 3 | H2WO4 | Oct3MeN+ HSO4− | <1 | 100 | 45 (5)f |
|
Among the catalytic systems that have been studied, the combination of tungstic acid with Aliquat 336® led to the best results with an adipic acid yield of 59% (Table 2, entry 2).
Under these conditions, the major intermediate is the 1,2-cyclohexandiol with a yield of 15%. The GC/MS analyses of the different runs revealed the formation of by-products of an identical structure and in a similar amount to that previously obtained (Tables 1 and 2). In order to assess the complete mechanism, the initial alkene epoxidation using H2WO4 and Aliquat 336® has been investigated.
In a very recent study, we showed that this system combined with high frequency ultrasound particularly improved the epoxidation of cis-cyclooctene and revealed important mechanistic insights into the studied reaction.38 Hence, under 800 kHz ultrasonic irradiation, in the presence of 0.75 mol% of H2WO4 and Aliquat 336® and 1.5 eq. of 35% H2O2 related to the substrate, 96% yield of epoxide was obtained with 98% selectivity within only 30 min. In addition, the non-radical nature of the cis-cyclooctene epoxidation mechanism has been demonstrated.38 In the present study, under magnetic stirring, the chosen oxidizing system leads to the formation of epoxycyclooctane in 89% yield and 91% selectivity (Table 3, entry 1). The same conditions have been then used for the epoxidation of cyclohexene and cyclopentene (Table 3, entries 2 and 3) and the structural identification of the products obtained from the oxidation of these three cycloalkenes have been performed (Table 3).
| Entrya | Olefin | Temp. (°C) | Conv.b (%) | Yieldc (%) | Main-by-productsd |
|---|---|---|---|---|---|
| a Experimental conditions: cycloalkene (185 mmol), H2O2 (35%, 1.5 eq.), H2WO4 (0.75 mol%), Aliquat 336® (0.75 mol%), 25–80 °C, 1000 rpm, 30 min. b Based on the amount of converted cycloalkene and calculated by GC using the internal standard method. c Based on the initial amount of cycloalkene and calculated by GC using the internal standard method. d Determined by GC/MS analysis, yields < 2%. e 1,2-Cycloalkanediol yield. f Temperature limited by cycloalkene boiling point. | |||||
| 1 |
|
25–85 | 99 | 89 |
|
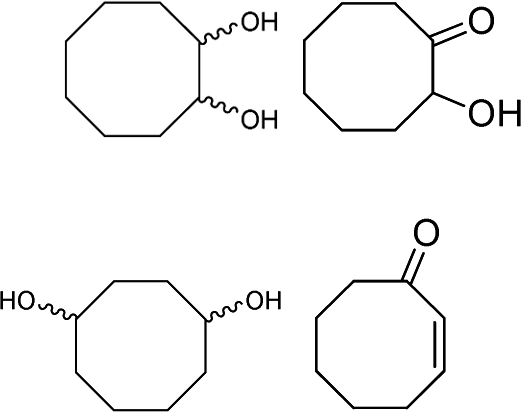
| 2 |
|
25–70f | 75 | 0 (39)e |
|
| 3 |
|
25–50f | 48 | 3 (32)e |
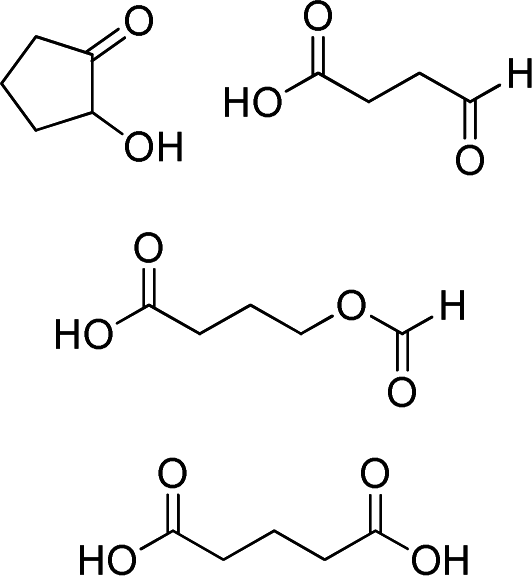
|
Unfortunately, in spite of the excellent epoxidation results observed with cis-cyclooctene, the oxidizing system was not efficient for the selective conversion of cyclohexene and cyclopentene to their corresponding epoxides in 30 min. Indeed, while cyclooctene is almost fully transformed to epoxycyclooctane in 89% yield (Table 3, entry 1), conversion of smaller cycloalkenes did not exceed 75% and their epoxides were observed in 0 and 3% yields (Table 3, entries 2 and 3). These results are explained by the higher reactivity of the double bond of cyclooctene toward electrophilic oxidants compared to that of other cycloalkenes and the strong resistance of its epoxide toward hydrolysis under acidic conditions.11,32,39 Indeed, this particular reactivity of medium-sized cycloalkenes such as cyclooctene has been attributed to transannular reactions occurring during the solvolysis of their oxides, and which is related to the spatial proximity of the opposite sides of their medium-sized ring.32,40 The few by-products observed for cyclooctene epoxidation are its oxidative cleavage intermediates 1,2-diol and α-hydroxyketone, a 1,4-diol formed from the transannular reaction, and a product of allylic oxidation, 2-cyclooctenone (Table 3, entry 1). With cyclohexene and cyclopentene, the reaction conditions mainly led to the production of their corresponding 1,2-diols with 39 and 32% yield respectively (Table 3, entries 2 and 3). Numerous by-products arising from the oxidation of these cycloolefins were also formed with yields below 2%. Among them, oxidative cleavage products and known intermediates, previously observed in this work, such as α-hydroxyketone and dicarboxylic acid have also been detected (Table 3, entries 2 and 3).
The oxidation of cyclopentene also revealed the formation of additional by-products such as 4-oxobutanoic and 4-formyloxobutanoic acids (Table 3, entry 3). In order to understand the formation of the by-products observed during the different steps of the oxidative cleavage of cycloalkenes, their structure was determined and compared to the results described in the literature.11,26,30,41
The present study on the oxidative cleavage and epoxidation of cyclohexene and cyclopentene allows highlighting the formation of 2 categories of compounds: oxidative cleavage intermediates and by-products.
On the one hand, expected reaction intermediates such as epoxide 2, 1,2-diol 3, and hydroxy-ketone 4 have been identified (Tables 1–3). However, despite the different reaction conditions and analyses carried out within the frame of our studies, hydroxy-lactone 5 and cyclic anhydride 6 proposed by Noyori have never been detected. (Scheme 1).11,26
Pioneering studies reported by Venturello et al.42 led us to consider other intermediates in this mechanism (Scheme 1). In particular, the production of a hydroperoxyalcohol intermediate 8 arising from the nucleophilic addition of H2O2 on the hydroxy-ketone 4 has been proposed, following the identification of the mono-aldehyde 9 (Scheme 2).42 The production of a hydroperoxyalcohol from the addition of H2O2 on a ketone has already been justified in earlier examples.43,44 Studies of the literature on aldehyde oxidation45,46 also support the hypothesis of the formation of the carboxylic acid 7 through the oxidation of the mono-aldehyde intermediate 9. Interestingly, both mechanisms suggested by Noyori and Venturello are connected via the hydroperoxyalcohol 8 intermediate suggested by Venturello. Indeed, compound 8, also known as the Criegee intermediate,44,47 is able to rearrange to form both the mono-aldehyde 9 and the hydroxylactone 5via a Baeyer–Villiger mechanism (Schemes 2 and 3).29,44 Mechanisms proposed by Noyori and Venturello have been combined to complete the reaction scheme suggested by Noyori (Scheme 1).
 | ||
| Scheme 2 Mechanism of the oxidative cleavage of cyclohexene and the production of dicarboxylic acids of shorter carbon chain in the presence of H2O2 and a tungsten-based catalyst. | ||
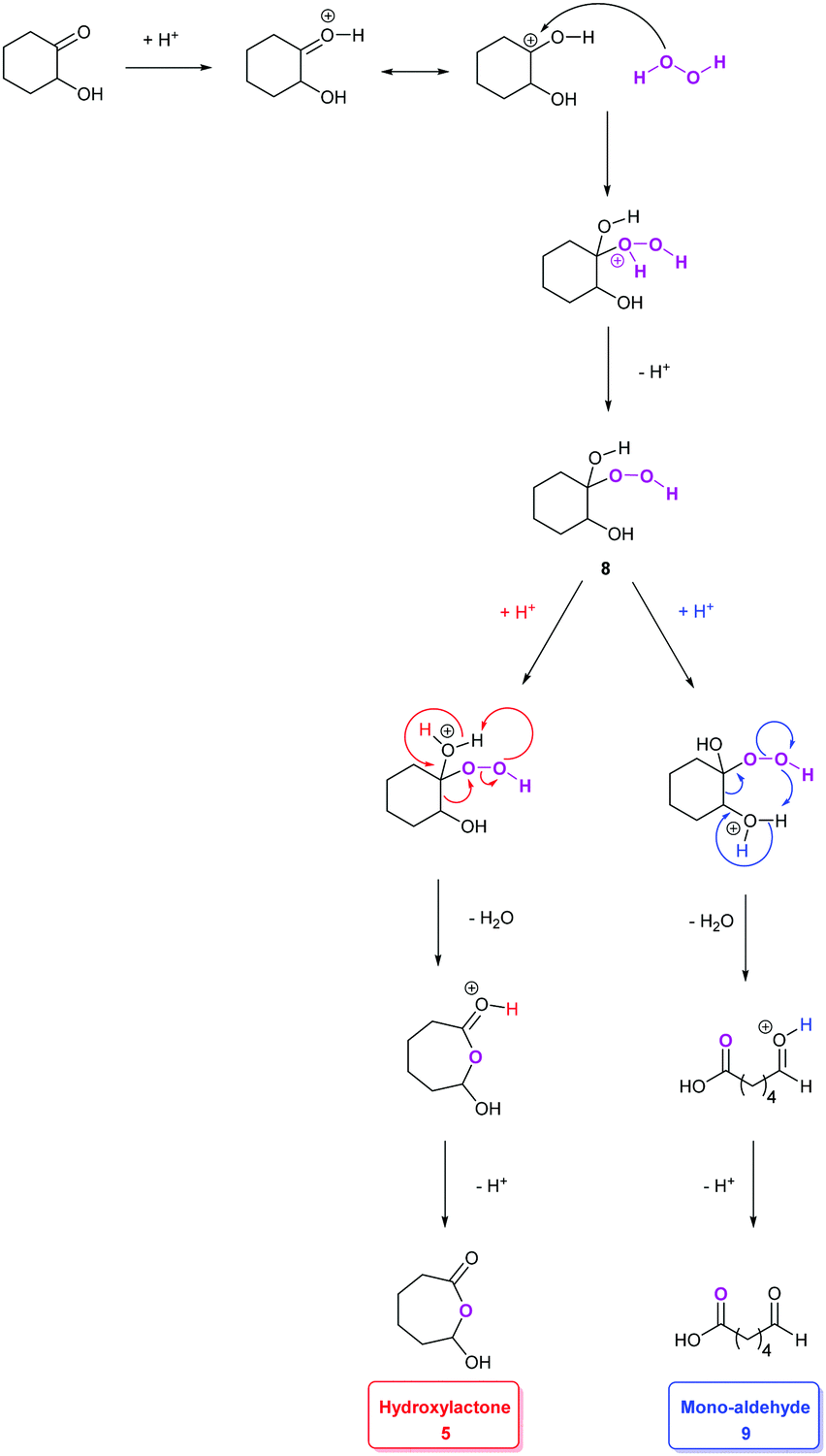 | ||
| Scheme 3 Hypothetical mechanism of the acid-catalyzed rearrangement of hydroperoxyalcohol intermediate 8 into hydroxy-lactone 5 or mono-aldehyde 9 (adapted from ref. 29, 31 and 45). | ||
In their studies on the oxidative cleavage of 1,2-cyclohexanediol derivatives, Fujitani and co-workers41 proposed the production of a gem-dihydroxy acid (10 in Scheme 2) to explain the presence of the Baeyer–Villiger product 5 suggested by Noyori, and the formation of the hydroperoxyalcohol 8 and mono-aldehyde 9 suggested by Venturello.42 Nevertheless, none of these three intermediates were detected in our studies. It is worth noting that Noyori's hydroxy-lactone 5 is the result of Venturello's mono-aldehyde 9 cyclization.
Thus, regarding the numerous interconnections existing between the above-mentioned intermediates, both pathways described in Scheme 2 should be considered to rationalize the formation of dicarboxylic acids from the cycloolefin oxidative cleavage.
On the other hand, the by-products formed from side-reactions on oxidative cleavage intermediates have also been identified in this work during the epoxidation and oxidative cleavage of cycloalkenes (Tables 1–3). More specifically, dicarboxylic acids with 1 to 3 less carbon atoms compared to the targeted product have been mainly identified during epoxidation and oxidative cleavage studies. This category of by-product is often observed in the literature but no explanation has been suggested to date to justify their formation. One reaction pathway that could lead to these shorter diacids could involve a decarboxylation of the desired dicarboxylic acid.4 Nevertheless, thermal decarboxylation of dicarboxylic acids mainly occurs at temperatures above 290 °C6 and usually leads to the formation of the corresponding alkanes. Besides, it was shown in the literature that α-oxoacids can afford diacids shortened by one carbon by decarboxylation in the presence of H2O2.48
The production of shorter dicarboxylic acids could be explained by a H2O2-mediated Baeyer–Villiger oxidation of the mono-aldehyde intermediate 9. Indeed, while the reaction between an aldehyde and hydrogen peroxide generally leads to the formation of the corresponding carboxylic acid, the nucleophilic addition of H2O2 to the aldehyde gives rise to a high-energy hydroperoxyalcohol intermediate that rearranges into formate ester (Scheme 4).18,43,44,47 The formation of the carboxylic acid and the formate ester from the rearrangement of the Criegee intermediate can take place via the nucleophilic addition of either H2O2 or peroxotungstate complexes to the mono-aldehyde.45 Once formed, the formate ester (Scheme 2, 11) is next hydrolysed to the alcohol 12 (Scheme 2) that is directly oxidized to the corresponding mono-aldehyde and dicarboxylic acid (Scheme 2, 13 and 14). The consecutive Baeyer–Villiger oxidations, ester hydrolyses and alcohol oxidations from the oxo-carboxylic acid intermediate 13 (Scheme 2) also lead to dicarboxylic acids with shorter chains (Scheme 2, 15). In this study on cycloalkenes oxidation, the formate ester 11 has been detected (Table 3, entry 3) as well as the hydroxy-acid 12 and oxo-acid 13 (Tables 1–3). In the literature, the formation of intermediates such as 12, 13 and 15 has been detected during alcohols and aldehydes oxidation using H2O2 and phosphotungstic acid.45 In addition to the expected carboxylic acids obtained by oxidation of linear and aromatic aldehydes, the presence of formate esters and their alcohol derivatives has been detected, thus supporting the hypothesis of the implementation of Baeyer–Villiger oxidation.45 In addition, Venturello and co-workers also described the presence of this side reaction during the oxidative cleavage of 1-methylcyclohexene. Indeed, 5-acetoxypentanoic acid was shown to be formed as a by-product along with the targeted 6-oxoheptanoic acid.30
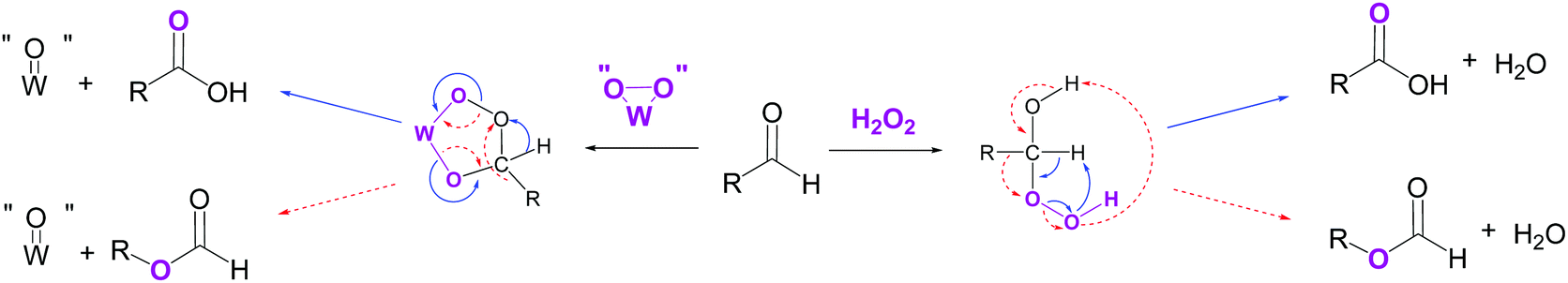 | ||
| Scheme 4 Competitive oxidation pathways occurring during aldehyde oxidation in the presence of H2O2 and a tungsten-based catalyst. In blue: aldehyde oxidation into carboxylic acid. In red dotted lines: Baeyer–Villiger oxidation (adapted from ref. 18 and 45). | ||
Thus, the mechanism disclosed in the Scheme 2 for the production of shorter chain diacids confirms the formation of the mono-aldehyde intermediate 9 as a key intermediate in the oxidative cleavage mechanism of cycloalkenes. Finally, this mechanism also explains the formation of shorter chain diacids observed during linear alkene oxidative cleavage, for which the formation of aldehyde intermediates is well-established in the literature.3,4,49
Conclusions
To summarize, this work proposes a complete mechanism of formation of the products and by-products obtained during the oxidative cleavage of cycloalkenes with H2O2 and H2WO4. The reaction has been studied for cyclohexene and cyclopentene under various conditions. The structures and proportions of the identified by-products were studied from epoxidation/dihydroxylation and oxidative cleavage stages. The present study shows that, along with targeted compounds, dicarboxylic acids shortened by 1 to 3 carbon atoms compared to the expected ones are mainly formed. The production of these by-products is explained by a Baeyer–Villiger oxidation on a mono-aldehyde intermediate that leads to the formation of a formate ester, which is hydrolysed and then oxidized to shorter chain diacids. This hypothesis is supported by the identification of formate esters and hydroxyacids but also by results on H2O2-mediated alcohol and aldehyde oxidation published in the literature, illustrating the formation of a mono-aldehyde intermediate in the cycloalkene oxidative cleavage pathway. The mechanism proposed in this study constitutes a real support for researchers aiming at developing efficient and environmentally friendly methodologies for the oxidative cleavage of olefins with H2O2 and tungsten-based catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Région Auvergne Rhône-Alpes for the financial support through an ARC Environnement doctoral research grant (ARC 2016).Notes and references
- M. Rüsch, G. Klaas, P. Bavaj and S. Warwel, Fett Wiss. Technol., 1995, 97, 359–367 Search PubMed.
- P. Spannring, P. C. A. Bruijnincx, B. M. Weckhuysen and R. J. M. Klein Gebbink, Catal. Sci. Technol., 2014, 4, 2182 RSC.
- A. Enferadi Kerenkan, F. Béland and T.-O. Do, Catal. Sci. Technol., 2016, 6, 971–987 RSC.
- A. Soutelo-Maria, J.-L. Dubois, J.-L. Couturier and G. Cravotto, Catalysts, 2018, 8, 464 CrossRef.
- T. Cousin, G. Chatel, N. Kardos, B. Andrioletti and M. Draye, Catal. Sci. Technol., 2019, 9, 5256–5278 RSC.
- B. Cornils and P. Lappe, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
- M. T. Musser, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
- C. H. Hamblet and M. Amborse, US Pat., US2557282A, 1951 Search PubMed.
- J. H. Teles, I. Hermans, G. Franz and R. A. Sheldon, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, pp. 1–103 Search PubMed.
- J. C. J. Bart and S. Cavallaro, Ind. Eng. Chem. Res., 2015, 54, 1–46 CrossRef CAS.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1647 CrossRef CAS.
- C. Bolm, O. Beckmann and O. A. G. Dabard, Angew. Chem., Int. Ed., 1999, 38, 907–909 CrossRef CAS.
- R. E. Dickinson and R. J. Cicerone, Nature, 1986, 319, 109–115 CrossRef CAS.
- R. A. Reimer, C. S. Slaten, M. Seapan, M. W. Lower and P. E. Tomlinson, Environ. Prog., 1994, 13, 134–137 CrossRef CAS.
- R. A. Reimer, C. S. Slaten, M. Seapan, T. A. Koch and V. G. Triner, in Non-CO2 Greenhouse Gases: Scientific Understanding, Control and Implementation, ed. J. van Ham, A. P. M. Baede, L. A. Meyer and R. Ybema, Springer Netherlands, Dordrecht, 2000, pp. 347–358 Search PubMed.
- F. Cavani and S. Alini, in Sustainable Industrial Chemistry, ed. F. Cavani, G. Centi, S. Perathoner and F. Trifir, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009, pp. 367–425 Search PubMed.
- A. Shimizu, K. Tanaka and M. Fujimori, Chemosphere: Global Change Sci., 2000, 2, 425–434 CrossRef CAS.
- C. W. Jones, Applications of hydrogen peroxide and derivatives, Royal Society of Chemistry, Cambridge, 1999 Search PubMed.
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374–3381 CrossRef CAS.
- C. P. Horwitz, in Innovations in Green Chemistry and Green Engineering, ed. P. T. Anastas and J. B. Zimmerman, Springer New York, NY, 2013, pp. 247–295 Search PubMed.
- E. Lassner, W.-D. Schubert, E. Lüderitz and H. U. Wolf, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
- National Research Council (U.S.)., Handbook of toxicology., Saunders, Philadelphia, 1956 Search PubMed.
- P. E. Leffler and G. Kazantzis, Handbook on the Toxicology of Metals, Elsevier, 2015, pp. 1297–1306 Search PubMed.
- W. P. Griffith, Transition Met. Chem., 1991, 16, 548–552 CrossRef CAS.
- L. Salles, C. Aubry, R. Thouvenot, F. Robert, C. Doremieux-Morin, G. Chottard, H. Ledon, Y. Jeannin and J. M. Bregeault, Inorg. Chem., 1994, 33, 871–878 CrossRef CAS.
- R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC.
- L. Knof, Justus Liebigs Ann. Chem., 1962, 656, 183–189 CrossRef CAS.
- G. M. Rubottom, J. M. Gruber, R. K. Boeckman, M. Ramaiah and J. B. Medwid, Tetrahedron Lett., 1978, 19, 4603–4606 CrossRef.
- P. Jin, L. Zhu, D. Wei, M. Tang and X. Wang, Comput. Theor. Chem., 2011, 966, 207–212 CrossRef CAS.
- E. Antonelli, R. D’Aloisio, M. Gambaro, T. Fiorani and C. Venturello, J. Org. Chem., 1998, 63, 7190–7206 CrossRef CAS.
- K. Fujitani, T. Mizutani, T. Oida and T. Kawase, J. Oleo Sci., 2009, 58, 37–42 CrossRef CAS.
- A. C. Cope, M. M. Martin and M. A. McKervey, Q. Rev., Chem. Soc., 1966, 20, 119 RSC.
- P. U. Maheswari, P. De Hoog, P. G. Enamorado, R. Hage and J. Reedijk, WO2009109857, 2009.
- N. J. Campbell, A. C. Dengel, C. J. Edwards and W. P. Griffith, J. Chem. Soc., Dalton Trans., 1989, 1203 RSC.
- A. J. Bailey, W. P. Griffith and B. C. Parkin, J. Chem. Soc., Dalton Trans., 1995, 1833 RSC.
- A. C. Dengel, W. P. Griffith and B. C. Parkin, J. Chem. Soc., Dalton Trans., 1993, 2683–2688 RSC.
- G. Strukul, Catalytic Oxidations with Hydrogen Peroxide as Oxidant, Springer, Netherlands, 1992 Search PubMed.
- T. Cousin, G. Chatel, N. Kardos, B. Andrioletti and M. Draye, Ultrason. Sonochem., 2019, 53, 120–125 CrossRef CAS.
- S. A. Hauser, M. Cokoja and F. E. Kühn, Catal. Sci. Technol., 2013, 3, 552–561 RSC.
- A. C. Cope, A. Fournier and H. E. Simmons, J. Am. Chem. Soc., 1957, 79, 3905–3909 CrossRef CAS.
- K. Fujitani, T. Mizutani, T. Oida and T. Kawase, J. Oleo Sci., 2009, 58, 323–328 CrossRef CAS.
- C. Venturello and M. Ricci, J. Org. Chem., 1986, 51, 1599–1602 CrossRef CAS.
- A. Rieche, Angew. Chem., 1958, 70, 251–266 CrossRef CAS.
- C. H. Hassall, Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1957, pp. 73–106 Search PubMed.
- C. Venturello and M. Gambaro, J. Org. Chem., 1991, 56, 5924–5931 CrossRef CAS.
- K. Sato, M. Aoki, J. Takagi and R. Noyori, J. Am. Chem. Soc., 1997, 119, 12386–12387 CrossRef CAS.
- G. R. Krow, Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1993, pp. 251–798 Search PubMed.
- A. Lopalco, G. Dalwadi, S. Niu, R. L. Schowen, J. Douglas and V. J. Stella, J. Pharm. Sci., 2016, 105, 705–713 CrossRef CAS.
- A. Godard, P. de Caro, E. Vedrenne, Z. Mouloungui and S. Thiebaud-Roux, OCL, 2016, 23, D510 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj03592k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |