
Palladium-catalyzed sequential acylation/annulation of indoles with acyl chlorides using primary amine as the directing group†
Guangbin
Jiang
 *,
Guang
Yang
,
Xinqiang
Liu
,
Shoucai
Wang
and
Fanghua
Ji
*
*,
Guang
Yang
,
Xinqiang
Liu
,
Shoucai
Wang
and
Fanghua
Ji
*
Guangxi Key Laboratory of Electrochemical and Magnetochemical Function Materials, College of Chemistry and Bioengineering, Guilin University of Technology, Guilin 541004, People's Republic of China. E-mail: jianggb@glut.edu.cn; fanghuaji@glut.edu.cn; Fax: +86-773-899-1304; Tel: +86-773-899-1304
First published on 23rd November 2020
Abstract
An attractive and convenient strategy for the direct acylation/annulation of indoles has been developed using Pd(0) as an efficient catalyst. The main feature of this protocol is the use of acyl chlorides as the acylating agents with the primary amine as the directing group. A variety of indolo[1,2-a]quinoxalines were readily obtained in reasonable efficiency and satisfactory yields with good functional group tolerance. Based on control experiments, a tentative catalytic mechanism was proposed.
1. Introduction
The reliable and convenient construction of heterocyclic compounds has been a focus of synthetic chemistry because heterocyclic molecules are very significant and widely present in functional materials, pharmaceutical agents, and numerous natural products.1–7 During the past few decades, transition-metal-catalyzed C–H bond functionalization/annulation under the assistance of the directing group has emerged as an efficient and succinct tool for the synthesis of various heterocyclic compounds.8–12 For instance, in 2017, Glorius's group reported a facile Mn-catalyzed C(sp2)–H annulation of imines.13 In the same year, a study on ruthenium-catalyzed [3+3] annulation of anilines with allyl alcohols appeared for accessing quinolone.14 Very recently, Song described the cobalt-catalyzed C(sp2)–H activation/annulations of aromatics with alkynes using an N,O-bidentate group as a directing group.15 In addition, various directing groups have been developed within recent years, such as primary amine, aminoquinoline, amino acids, carboxyl, and hydroxyl.16–21 Among them, primary amine-assisted cyclization of substrates is particularly attractive due to their regioselectivity and practicability.22,23 Despite such great achievements, investigation on the use of primary amine as a directing group to build quinoxalines is very limited.In addition, as simple and readily available building blocks, acyl chlorides have received extensive attention in recent years,24–32 and their participation as an acyl source is mainly accomplished by classic Friedel–Crafts acylation reactions.33–36 However, the classic Friedel–Crafts acylation reactions generally result in a mixture of products with poor ortho/para regioselectivity, and usually suffer from narrow substrate scope and poor functional group tolerance.37,38 The use of stoichiometric Lewis acids is also a deficiency of this type of reaction. In recent years, palladium-catalyzed directing group assisted acylations of acyl chlorides are particularly attractive due to their remarkable regioselectivity and practicability. In 2019, a Pd-catalyzed acylation of acyl chlorides was elegantly developed by Wu and co-workers (Scheme 1a).39 Inspired by this excellent work and as part of our continuing interest in straightforward transition-metal-catalyzed C–H activation,40,41 herein, we report the first palladium-catalyzed sequential acylation/annulation of indoles using acyl chlorides as the acylating agents with primary amine as the directing group (Scheme 1b).
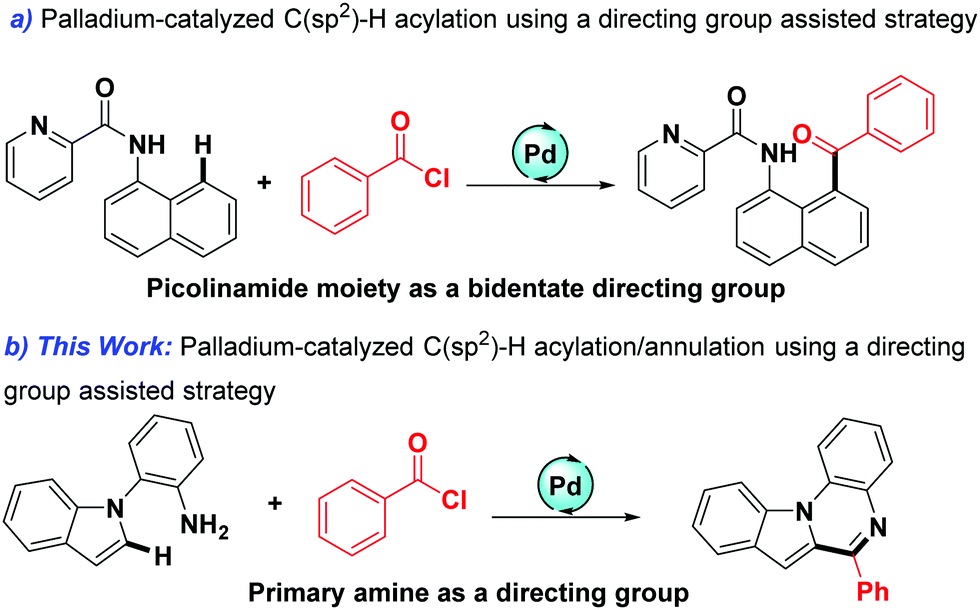 | ||
| Scheme 1 Coupling reaction of acyl chloride by using a directing group assisted strategy. | ||
2. Experimental section
2-(1H-Indol-1-yl)anilines 1 (0.1 mmol), acyl chloride 2 (0.1 mmol), Pd(dba)2 (10 mol%), AgSbF6 (25 mol%), CsOPiv (0.2 mmol) and anhydrous toluene/1,4-dioxane = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (1 mL) were sealed in a Schlenk tube under an Ar atmosphere. The mixture was then stirred at 130 °C (oil bath temperature) for 24 h. After the condensation was completed (monitored by TLC), the resulting mixture was cooled to room temperature and extracted with ethyl acetate, dried over anhydrous MgSO4, filtered and evaporated in vacuo. The desired products 3 were obtained in the corresponding yields after being purified by column chromatography on silica gel with a mixture of petroleum ether and ethyl acetate.
:1 (1 mL) were sealed in a Schlenk tube under an Ar atmosphere. The mixture was then stirred at 130 °C (oil bath temperature) for 24 h. After the condensation was completed (monitored by TLC), the resulting mixture was cooled to room temperature and extracted with ethyl acetate, dried over anhydrous MgSO4, filtered and evaporated in vacuo. The desired products 3 were obtained in the corresponding yields after being purified by column chromatography on silica gel with a mixture of petroleum ether and ethyl acetate.
3. Results and discussion
Initially, we chose 2-(1H-indol-1-yl)aniline (1a) as the model substrate and benzoyl chloride (2a) as the acylating agent to identify the optimal reaction conditions, and significant results are summarized in Table 1. During the screening of several reaction conditions, including additive, catalysts, bases, and solvents, the expected acylation/annulation proceeded smoothly to give the corresponding product 3aa in 76% isolated yield under the following reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), Pd(dba)2 (10 mol%), AgSbF6 (25 mol%) and CsOPiv (0.2 mmol) were stirred in 1 mL of solvent (toluene/1,4-dioxane = 1:1) for 24 h at 130 °C under an argon atmosphere (Table 1, entry 1). Notably, the isolated yield of the reaction was highly dependent on the palladium(II) generated in situ, and the optimal result was observed with Pd(dba)2 (Table 1, entries 2–5). Pd(dba)2 supported by other corresponding additives was inferior (Table 1, entries 6–9). Bases, such as NaOTf and K3PO4, showed a moderate reactivity, whereas NaOH was ineffective (Table 1, entries 10–12). Control experiments suggested that both Pd(dba)2 and CsOPiv were vital for this acylation/annulation reaction (Table 1, entries 13 and 14). Finally, the efficiency of this transformation could not be improved by changing the reaction solvent (Table 1, entries 15 and 16, see the ESI† for details).
|
|
||
|---|---|---|
| Entry | Variations from standard conditions | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), catalyst (10 mol%), additive (25 mol%), base (0.2 mmol) and anhydrous toluene/1,4-dioxane (1/1, v/v) (1.0 mL) were sealed in a 25 mL Schlenk tube at 130 °C for 24 h under an Ar atmosphere. b Isolated yields; n.d. = not detected. | ||
| 1 | None | 76 |
| 2 | Pd(OAc)2 instead of Pd(dba)2 | 36 |
| 3 | Pd(PPh3)4 instead of Pd(dba)2 | 48 |
| 4 | Pd(TFA)2 instead of Pd(dba)2 | 42 |
| 5 | PdCl2 instead of Pd(dba)2 | 26 |
| 6 | AgOAc instead of AgSbF6 | 47 |
| 7 | Ag2CO3 instead of AgSbF6 | 50 |
| 8 | K2S2O8 instead of AgSbF6 | 15 |
| 9 | Cu(OAc)2 instead of AgSbF6 | Trace |
| 10 | NaOTf instead of CsOPiv | 36 |
| 11 | K3PO4 instead of CsOPiv | 37 |
| 12 | NaOH instead of CsOPiv | Trace |
| 13 | Without Pd(dba)2 | n.d. |
| 14 | Without CsOPiv | Trace |
| 15 | DMF instead of toluene/1,4-dioxane | n.d. |
| 16 | CH3CN instead of toluene/1,4-dioxane | Trace |

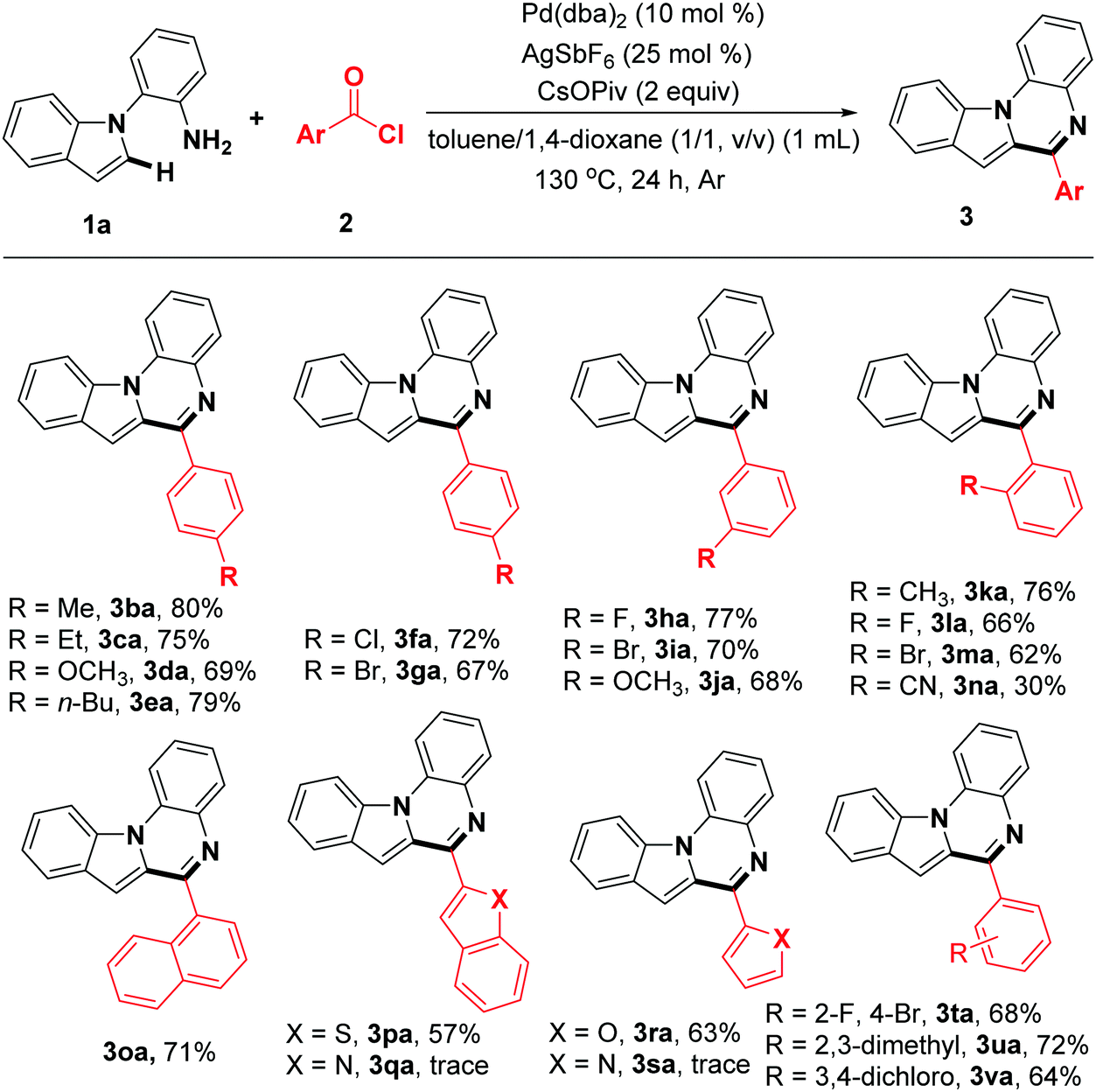
With the optimal reaction conditions in hand, the scope of the transformation with respect to the acyl chlorides was also observed (Table 2). Generally, this reaction afforded the corresponding products in modest to excellent yield and with unique regioselectivity. The acyl chlorides with para-substituted electron-donating groups gave the desired indolo[1,2-a]quinoxalines 3ba–3ea in 69–80% yields. In addition, acyl chlorides with halogen-substitution worked well and gave the expected products 3fa and 3ga in moderate yields, which could be used for further transformation via cross coupling reactions. Other functional groups, such as meta-F, meta-Br and meta-OCH3 can also be compatible with the reaction system, providing the corresponding indolo[1,2-a]quinoxalines 3ha–3ja in good yields. The reaction of 2k, 2l, 2m and 2n gave the desired products 3ka, 3la, 3ma and 3na respectively, even though the reaction site is sterically hindered. When the 1-naphthoyl chloride, benzo[b]thiophene-2-carbonyl chloride, and furan-2-carbonyl chloride were used as the substrates, the desired products 3oa, 3pa and 3ra were obtained in yields of 57–71%. Unfortunately, nitrogen-containing acyl chlorides are not compatible with the reaction system (3qa, 3sa). Finally, double-substituted acyl chlorides also gave the highly functional products 3ta–3va in reasonable yield.
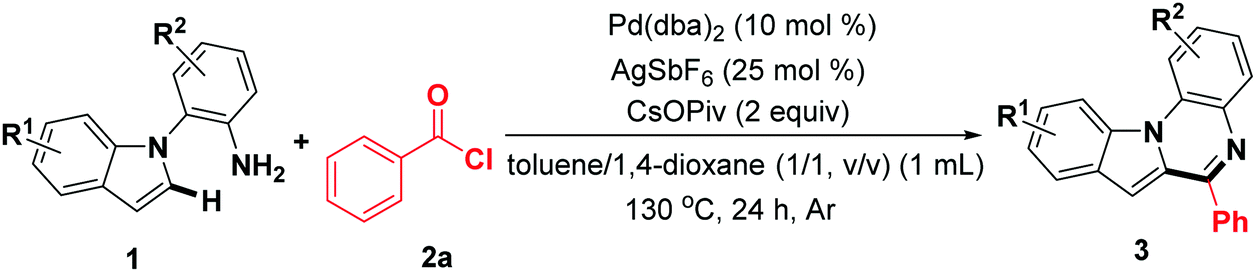
Subsequently, the scope of indoles was investigated under the optimal reaction conditions. As the data of Table 3 show, indole rings substituted with 3-Me, 4-Me, 4-OCH3, 4-benzyloxy, 5-OCH3, and 6-OCH3 groups successfully participated in this transformation to give the corresponding products in moderate to good yields (3ab–3ag). Furthermore, 2-(4,6-dimethyl-1H-indol-1-yl)aniline reacted well to afford the expected product (3ah) with excellent yield. Noticeably, the halogen atoms were compatible with this reaction system (3ai–3al). In addition, 1-(2-aminophenyl)-1H-indole-5-carbonitrile was also investigated, and to our delight, the target product 3am was synthesized, albeit in relatively lower yield (45%). Remarkably, 2-(1H-indol-1-yl)-4-methylaniline was a suitable substrate, affording the corresponding product 3an in 85% yield. The indoles 1o–1q bearing a halogen (F, Cl) at the aniline rings were compatible for the transformation, providing the expected products 3ao–3aq in 76%, 73%, and 69% yields, respectively. In addition, the dihalogen-substituted starting materials could be functionalized under standard reaction conditions. It should be noted that 1s containing a CF3 group was suitable, albeit resulting in obviously decreased reactivity (38%).
|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product | Yieldb (%) |
| a A mixture of 1 (0.1 mmol), 2a (0.2 mmol), Pd(dba)2 (10 mol%), AgSbF6 (25 mol%), CsOPiv (0.2 mmol), and anhydrous toluene/1,4-dioxane (1/1, v/v) (1.0 mL) was sealed in a 25 mL Schlenk tube at 130 °C for 24 h under an Ar atmosphere. b Yields refer to isolated yield. | ||||
| 1 | 3-CH3 | H | 3ab | 82 |
| 2 | 4-CH3 | H | 3ac | 84 |
| 3 | 4-OCH3 | H | 3ad | 72 |
| 4 | 4-Benzyloxy | H | 3ae | 76 |
| 5 | 5-OCH3 | H | 3af | 71 |
| 6 | 6-OCH3 | H | 3ag | 67 |
| 7 | 4,6-Di-CH3 | H | 3ah | 80 |
| 8 | 4-F | H | 3ai | 70 |
| 9 | 4-Cl | H | 3aj | 78 |
| 10 | 5-F | H | 3ak | 67 |
| 11 | 6-F | H | 3al | 65 |
| 12 | 5-CN | H | 3am | 45 |
| 13 | H | 4-CH3 | 3an | 85 |
| 14 | H | 4-Cl | 3ao | 76 |
| 15 | H | 5-Cl | 3ap | 73 |
| 16 | H | 4-F | 3aq | 69 |
| 17 | H | 4-Cl-6-F | 3ar | 56 |
| 18 | H | 4-CF3 | 3as | 38 |
In order to elucidate the reaction mechanism, a few preliminary control experiments were carried out (Scheme 2). First, the designed substrate 4a was conducted under the standard reaction conditions, and no corresponding product 5aa was observed (Scheme 2a), suggesting that the primary amine as a directing group should be essential in this reaction. When 2.0 equiv. of TEMPO, 1,1-diphenylethylene, and BHT were added under the optimal reaction conditions, the target product 6-phenylindolo[1,2-a]quinoxaline (3aa) was obtained in 61%, 57% and 65% yields, respectively (Scheme 2b). These results indicate that a radical process is not involved in this transformation.42–44 Furthermore, by employing deuterium-labeled compound 1a-d1 and 1a as substrates, the experiment for calculating the KIE (kinetic isotope effect) value was performed (see the ESI†),45,46 and the result (KIE = 1.1) may suggest that the C(sp2)–H bond cleavage of 2-(1H-indol-1-yl)aniline (1a) should not be involved in the rate-limiting step (Scheme 2c).
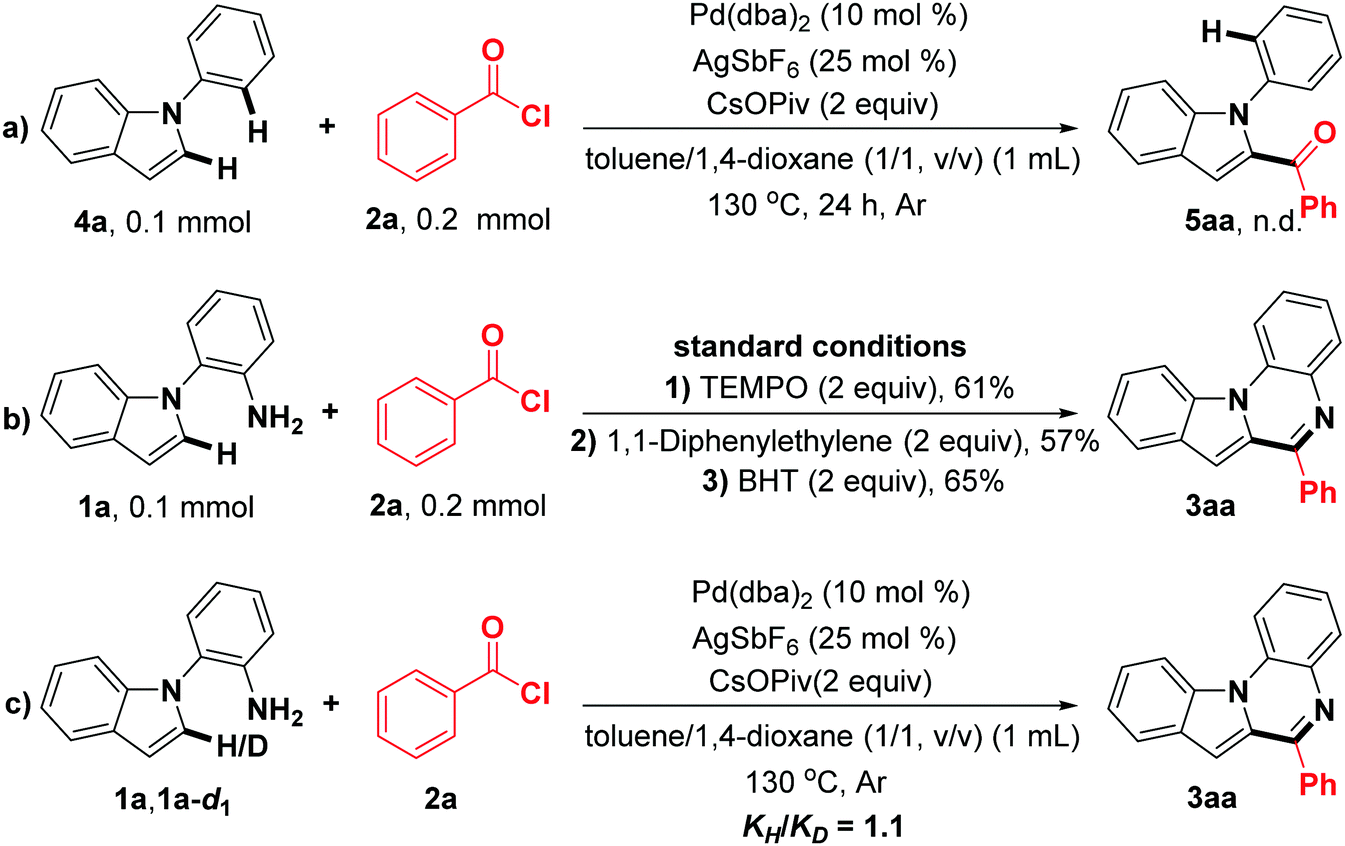 | ||
| Scheme 2 Control experiments. | ||
Based on our control experiments and previous literature reports,47–49 a possible catalytic cycle is outlined in Fig. S2 (ESI†). We propose that the acylation/annulation process may proceed via a Pd(II)/Pd(IV) catalytic cycle. First, the treatment of Pd(dba)2 with silver salts gives rise to the activated Pd(II) catalyst, which undergoes C(sp2)–H functionalization with 2-(1H-indol-1-yl)aniline (1a) to obtain a six-membered palladacyclic intermediate B.50–53 Subsequently, the oxidative addition of benzoyl chloride (2a) to intermediate B gives the probable Pd(IV) intermediate C, which undergoes reductive elimination to obtain the acylating product D along with regeneration of the activated Pd(II) species. Simultaneously, the final product 3aa was obtained by an intramolecular dehydration reaction.
4. Conclusions
In summary, we have reported a unique, directing group protocol for a one-pot synthesis of functionalized indolo[1,2-a]quinoxalines from acyl chloride and indoles, utilizing a palladium-catalyzed C(sp2)–H acylation method as the crucial step. This protocol exhibits a good functional group tolerance, high regioselectivity, and convenient operation. Dramatically, simple and readily available acyl chlorides and high step economy make this strategy particularly attractive. In addition, we believe that these transformations proceed via a classical Pd(II)/Pd(IV) catalytic cycle. Further studies on the construction of heterocyclic compounds via C–H activation as well as acylation/annulation are underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Guangxi Natural Science Foundation (2018GXNSFBA050024, 2017GXNSFBA198224 and 2018GXNSFAA281203), Guangxi Science and Technology Base and Special Fund for Talents (AD19245008 and AD19110110), PhD Scientific Research Foundation of Guilin University of Technology, and Key Laboratory of Electrochemical and Magnetochemical Function Materials.References
- M. He, N. Chen, J. Wang and S. Peng, Org. Lett., 2019, 21, 5167–5171 CrossRef CAS.
- J. Li, S. Yang, W. Wu and H. Jiang, Eur. J. Org. Chem., 2018, 1284–1306 CrossRef CAS.
- J. Zhu, W. Hu, S. Sun, J.-T. Yu and J. Cheng, Adv. Synth. Catal., 2017, 359, 3725–3728 CrossRef CAS.
- S. K. Das, S. Roy, H. Khatua and B. Chattopadhyay, J. Am. Chem. Soc., 2018, 140, 8429–8433 CrossRef CAS.
- J.-F. Tan, C. T. Bormann, F. G. Perrin, F. M. Chadwick, K. Severin and N. Cramer, J. Am. Chem. Soc., 2019, 141, 10372–10383 CrossRef CAS.
- G.-B. Jiang, W.-Y. Zhang, M. He, Y.-Y. Gu, L. Bai, Y.-J. Wang, Q.-Y. Yi and F. Du, Spectrochim. Acta., Part A, 2019, 220, 117132 CrossRef CAS.
- G.-B. Jiang, W.-Y. Zhang, M. He, Y.-Y. Gu, L. Bai, Y.-J. Wang, Q.-Y. Yi and F. Du, Polyhedron, 2019, 169, 209–218 CrossRef CAS.
- Y. Shang, K. Jonnada, S. L. Yedage, H. Tu, X. Zhang, X. Lou, S. Huang and W. Su, Chem. Commun., 2019, 55, 9547–9550 RSC.
- G. Zheng, J. Sun, Y. Xu, S. Zhai and X. Li, Angew. Chem., Int. Ed., 2019, 58, 5090–5094 CrossRef CAS.
- S. Kim, S. H. Han, N. K. Mishra, R. Chun, Y. H. Jung, H. S. Kim, J. S. Park and I. S. Kim, Org. Lett., 2018, 20, 4010–4014 CrossRef CAS.
- F. Ling, C. Ai, Y. Lv and W. Zhang, Adv. Synth. Catal., 2017, 359, 3707–3712 CrossRef CAS.
- H. Zhu, R. Zhuang, W. Zheng, L. Fu, Y. Zhao, L. Tu, Y. Chai, L. Zeng, C. Zhang and J. Zhang, Tetrahedron, 2019, 75, 3108–3112 CrossRef CAS.
- Q. Lu, S. Greβies, S. Cembellín, F. J. R. Klauck, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 12778–12782 CrossRef CAS.
- G. S. Kumar, P. Kumar and M. Kapur, Org. Lett., 2017, 19, 2494–2497 CrossRef CAS.
- X.-C. Li, G. Du, H. Zhang, J.-L. Niu and M.-P. Song, Org. Lett., 2019, 21, 2863–2866 CrossRef CAS.
- K. Jing, X.-N. Wang and G.-W. Wang, J. Org. Chem., 2019, 84, 161–172 CrossRef CAS.
- G. Jiang, S. Fang, W. Hu, J. Li, C. Zhu, W. Wu and H. Jiang, Adv. Synth. Catal., 2018, 360, 2297–2302 CrossRef CAS.
- J. Liu, J. Zou, J. Yao and G. Chen, Adv. Synth. Catal., 2018, 360, 659–663 CrossRef CAS.
- J. Liu, Z. Xue, Z. Zeng, Y.-X. Chen and G. Chen, Adv. Synth. Catal., 2016, 358, 3694–3699 CrossRef CAS.
- J. Li, S. Zhang, M. R. Lonka, J. Zhang and H. Zou, Chem. Commun., 2019, 55, 11203–11206 RSC.
- X.-S. Zhang, Y.-F. Zhang, Z.-W. Li, F.-X. Luo and Z.-J. Shi, Angew. Chem., Int. Ed., 2015, 27, 5478–5482 CrossRef.
- G. Jiang, S. Wang, J. Zhang, J. Yu, Z. Zhang and F. Ji, Adv. Synth. Catal., 2019, 358, 1798–1802 CrossRef.
- P. Villuendas and E. P. Urriolabeitia, J. Org. Chem., 2013, 78, 5254–5363 CrossRef CAS.
- W. Wang, C. Pan, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 3978–3980 RSC.
- S. Gaspa, A. Porcheddu and L. D. Luca, Adv. Synth. Catal., 2016, 358, 154–158 CrossRef CAS.
- J. Amani, E. Sodagar and G. A. Molander, Org. Lett., 2016, 18, 732–735 CrossRef CAS.
- C.-J. Lee, C.-C. Tsai, S.-H. Hong, G.-H. Chang, M.-C. Yang, L. Möhlmann and W. Lin, Angew. Chem., Int. Ed., 2015, 54, 8502–8505 CrossRef CAS.
- Y. Du, Y. Liu and J.-P. Wan, J. Org. Chem., 2018, 83, 3403–3408 CrossRef.
- S.-M. Xu, J.-Q. Chen, D. Liu, Y. Bao, Y.-M. Liang and P.-F. Xu, Org. Chem. Front., 2017, 4, 1331–1335 RSC.
- T. Kochi, A. Tazawa, K. Honda and F. Kakiuchi, Chem. Lett., 2011, 40, 1018–1020 CrossRef CAS.
- P. M. Liu and C. G. Frost, Org. Lett., 2013, 15, 5862–5865 CrossRef CAS.
- D. C. McAteer, E. Javed, L. Huo and S. Huo, Org. Lett., 2017, 19, 1606–1609 CrossRef CAS.
- R. H. Vekariya and J. Aubé, Org. Lett., 2016, 18, 3534–3537 CrossRef CAS.
- H. Y. Kim, E. Song and K. Oh, Org. Lett., 2017, 19, 312–315 CrossRef CAS.
- H. Koo, H. Y. Kim and K. Oh, Org. Chem. Front., 2019, 6, 1868–1872 RSC.
- S. K. Guchhait, M. Kashyap and H. Kamble, J. Org. Chem., 2011, 76, 4753–4758 CrossRef CAS.
- K. Jing, Z.-Y. Li and G.-W. Wang, ACS Catal., 2018, 8, 11875–11881 CrossRef CAS.
- N. A. Serratore, C. B. Anderson, G. B. Frost, T.-G. Hoang, S. J. Underwood, P. M. Gemmel, M. A. Hardy and C. J. Douglas, J. Am. Chem. Soc., 2018, 140, 10025–10033 CrossRef.
- X. Yu, F. Yang, Y. Wu and Y. Wu, Org. Lett., 2019, 21, 1726–1729 CrossRef CAS.
- S. Wang, K. Wang, X. Kong, S. Zhang, G. Jiang and F. Ji, Adv. Synth. Catal., 2019, 361, 3986–3990 CrossRef CAS.
- G. Jiang, W. Hu, J. Li, C. Zhu, W. Wu and H. Jiang, Chem. Commun., 2018, 54, 1746–1749 RSC.
- X. Wang, J. Zhang, D. Chen, B. Wang, X. Yang, Y. Ma and M. Szostak, Org. Lett., 2019, 21, 7038–7043 CrossRef CAS.
- X. Jie, Y. Shang, X. Zhang and W. Su, J. Am. Chem. Soc., 2016, 138, 5623–5633 CrossRef CAS.
- K. Natte, R. V. Jagadeesh, L. He, J. Rabeah, J. Chen, C. Taeschler, S. Ellinger, F. Zaragoza, H. Neumann, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 2782–2786 CrossRef CAS.
- C. Du, P.-X. Li, X. Zhu, J.-F. Suo, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2016, 55, 13571–13575 CrossRef CAS.
- G. Cera, T. Haven and L. Ackermann, Chem. – Eur. J., 2017, 23, 3577–3582 CrossRef CAS.
- E. Abada, P. Y. Zavalij and A. N. Vedernikov, J. Am. Chem. Soc., 2017, 139, 643–646 CrossRef CAS.
- J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70–76 RSC.
- W. Wu, S. Fang, G. Jiang and H. Jiang, Org. Chem. Front., 2019, 6, 2200–2204 RSC.
- S. Xu, R. Chen, Z. Fu, Q. Zhou, Y. Zhang and J. Wang, ACS Catal., 2017, 7, 1993–1997 CrossRef CAS.
- Y. Wu, C. Jiang, D. Wu, Q. Gu, Z.-Y. Luo and H.-B. Luo, Chem. Commun., 2016, 52, 1286–1289 RSC.
- S. S. John-Campbell, A. J. P. White and J. A. Bull, Chem. Sci., 2017, 8, 4840–4847 RSC.
- S. Choi, T. Chatterjee, W. J. Choi, Y. You and E. J. Cho, ACS Catal., 2015, 5, 4796–4802 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj04406g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |