
Bimetallic RuPd nanoparticles in ionic liquids: selective catalysts for the hydrogenation of aromatic compounds†
Gabriel
Abarca
 ab,
Wellington D. G.
Gonçalves
a,
Brunno L.
Albuquerque
a,
Jairton
Dupont
*a,
Martin H. G.
Prechtl
*cd and
Jackson D.
Scholten
*a
ab,
Wellington D. G.
Gonçalves
a,
Brunno L.
Albuquerque
a,
Jairton
Dupont
*a,
Martin H. G.
Prechtl
*cd and
Jackson D.
Scholten
*a
aInstituto de Química, UFRGS, Av. Bento Gonçalves, 9500, Agronomia, CEP 91501-970, Porto Alegre, RS, Brazil. E-mail: jairton.dupont@ufrgs.br; jackson.scholten@ufrgs.br; Tel: +55 51 3308 9633
bUniversidad Bernardo O’Higgins, Escuela de Obstetricia y Puericultura, Centro Integrativo de Biología y Química Aplicada (CIBQA), Santiago 8370993, Chile
cUniversität zu Köln, Department of Chemistry, Greinstr. 6, D-50939 Köln, Germany. E-mail: martin.prechtl@uni-koeln.de
dInstituto Superior Técnico, Universidade de Lisboa, Av. Roviso Pais 1, 1049-001 Lisboa, Portugal
First published on 8th December 2020
Abstract
Bimetallic RuPd nanoparticles (NPs) immobilized in ionic liquids (ILs) were shown to be a highly active medium for the selective hydrogenation of benzene and phenol under mild conditions (4 bar H2, 60 °C) in a biphasic system (n-heptane/IL). The equimolar combination of Ru and Pd into a bimetallic particle generated a synergistic catalyst that allowed the selective production of cyclohexane (>99% selectivity, 94% conversion) and cyclohexanol (99% selectivity, >98% conversion) from the reduction of benzene and phenol, respectively. Moreover, the catalytic results revealed that the activity and selectivity are dependent on the Ru![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd ratio into the bimetallic NPs.
:Pd ratio into the bimetallic NPs.
The hydrogenation of aromatic compounds is one of the most investigated and attractive catalytic processes.1–3 Due to the considerable stability provided by the aromaticity, it is an up-hill reaction and needs high energy to occur. It is well-known that the hydrogenation of aromatic substrates is a typical reaction promoted by heterogeneous catalysts, taking into account the aspects required, such as energy and the presence of multi-sites for the effective coordination of the molecule.2 Indeed, homogeneous complexes are usually employed as sources for heterogeneous species in this transformation. However, rare examples of their use as active catalysts can be found in the literature.4 In particular, the hydrogenation of benzene and phenol is attractive for investigation since the products are of industrial interest for the production of fine chemicals. In fact, cyclohexane (a total hydrogenated product from benzene) is a crucial starting material for the synthesis of adipic acid and ε-caprolactam, which are used in the manufacture of nylon. On the other hand, the selective reduction of phenol to cyclohexanol affords a suitable building block precursor for the preparation of useful chemicals. Also, the hydrodeoxygenation (HDO) reaction of biomass-derived compounds is a sustainable way to produce valuable compounds.5 Several nanostructured materials in different media have been used to carry out these reactions. In particular, Ru,5–7 Rh,5,8 Ir,8 Pt,5,9 and Pd5 metal NPs in ILs were tested as the catalytic phase used to hydrogenate aromatic compounds, where the catalytic activities are dependent on the reaction conditions employed in each case. However, the understanding of a rational design of active heterogeneous catalysts to achieve high conversions with tunable selectivities remains a challenge. In this context, the development of bimetallic nanomaterials has attracted attention due to the possibility of combining unique catalytic properties, which are generally superior to those observed for monometallic analogues.10,11 Encouraged by our previous work on the use of Ru@Pt core–shell NPs in ILs for the selective generation of 1,3-cyclohexadiene from benzene hydrogenation,12 herein, we demonstrate that the synthesis of bimetallic RuPd NPs in ILs produces highly active and selective catalysts for the hydrogenation of benzene and phenol under biphasic conditions. Surprisingly, the equimolar addition of Pd (less active metal for arene hydrogenation)13–15 to Ru produces a superior catalyst, reaching almost complete conversion (94%) and preferred cyclohexane selectivity (>99%) for benzene hydrogenation at 60 °C. In the case of phenol, conversion >98% and selectivity of 99% in cyclohexanol were attained under the same conditions. The preparation of RuPd bimetallic NPs in ILs was carried out by a similar method applied for monometallic nanoparticles using two metal precursors, [Ru(Me-allyl)2(COD)] (COD = 1,5-cyclooctadiene) and [Pd2(dba)3] (dba = dibenzylideneacetone) (detailed synthetic procedure is available in the ESI†). The bimetallic NPs were characterized by transmission electron microscopy (TEM), X-ray powder diffraction (XRD), and X-ray photoelectron spectroscopy (XPS) analyses.
Fig. 1 shows the TEM micrographs and the particle size distribution histogram of the colloidal RuPd NPs immediately after preparation. TEM observations showed well-dispersed RuPd particles with a spherical shape and average diameter of 2.2 ± 1.1 nm (RuPd 1:1), 2.2 ± 0.5 nm (RuPd 1:9) and 3.6 ± 1.8 nm (RuPd 9:1), although few counts of larger NPs were found in the RuPd samples as similarly observed in the literature.16–18 More than 300 randomly chosen nanoparticles were measured to ensure statistical representation of the nanoparticle sizes (Fig. 1).19,20 The lattice spacing measured by the FFT of a single particle showed different values for the different ratios of RuPd nanoparticles. The RuPd 1:1 lattice spacing of 0.22 nm can be attributed to the Pd spacing in (111) planes.21 The 0.21 nm fringe corresponds to the Ru (101) lattice planes in agreement with previous analysis.22 The RuPd 1:9, which is rich in Pd, showed two lattice fringes with the same spacing, 0.18 nm, which corresponds to the (200) Pd plane.23 In addition, for the Ru rich particle, RuPd 9:1, the lattice spacing value is closer to that observed for Ru. These HRTEM results strongly suggest the coexistence of Ru and Pd to form a single bimetallic particle with an alloy-like structure. Since the d-spacing were measured by the FFT of a single particle, a small deviation can occur by the pixel positions.
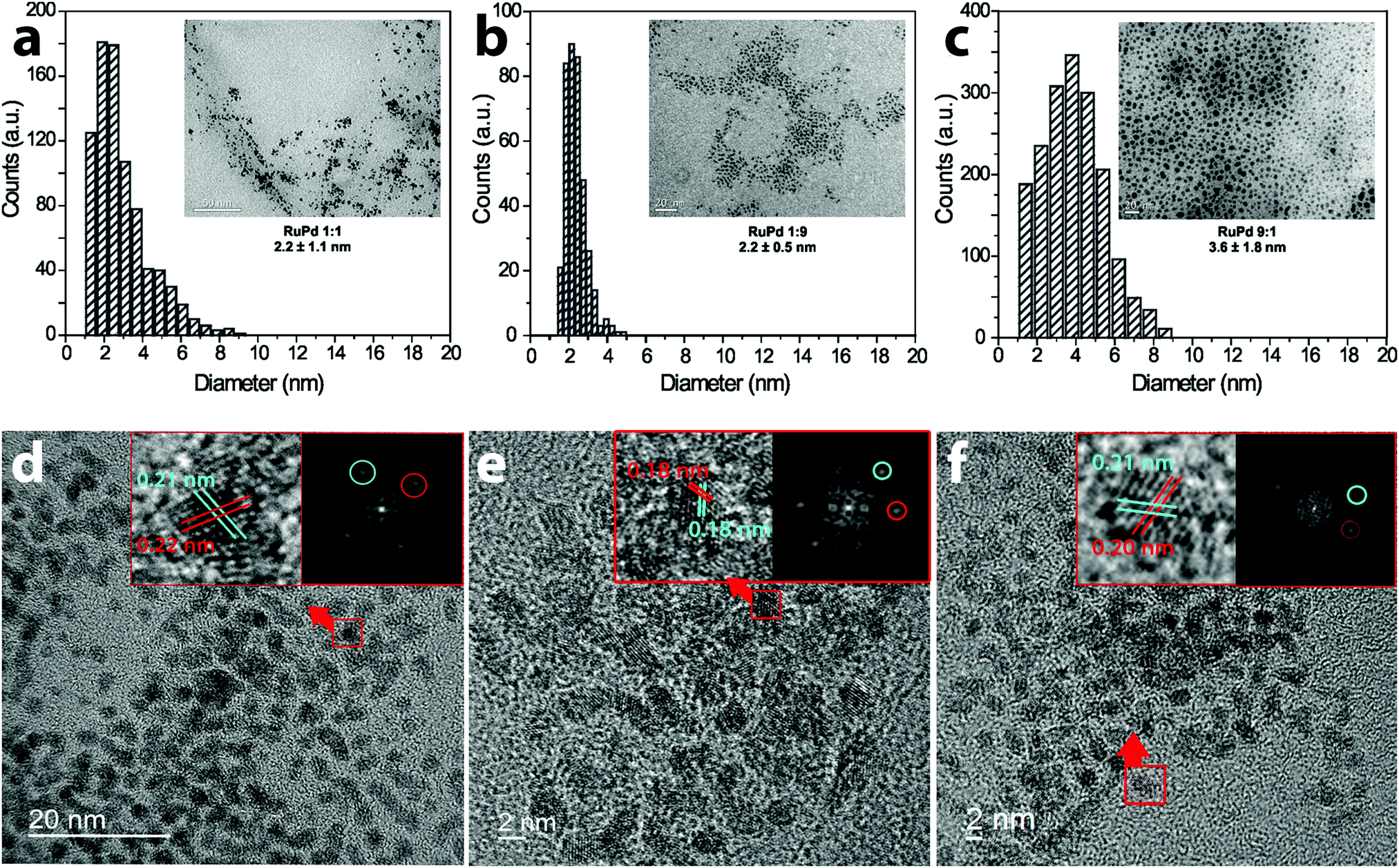 | ||
| Fig. 1 TEM (a)–(c) of the RuPd NPs and their corresponding histogram of size. HRTEM (d)–(f) of the RuPd NPs and the FFT of a single NP showing the lattice spacing (inset). | ||
Fig. 2 shows the comparison between Ru and Pd XRD patterns and experimental XRD patterns of RuPd 1:1 NPs. The three characteristic reflections at 40°, 46°, and 67° correspond to (111), (200), and (220) of the palladium fcc crystalline structure (Pd pattern ICSD 76148, space group fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). For Ru, it is possible to determine the main reflections at 38°, 42°, and 44°, which corresponded to (100), (002), (101), respectively, confirming the hcp structure (Ru pattern ICSD 40354, space group P63/mmc). Since the elements structurally belong to different space groups, despite the similarity of ionic radii (Goldschmidt's criterion and Vegard's law), there are no conditions to form bulk phase solid solutions. However, at the nanoscale both phases can coexist in a single nanoparticle.24,25 Thus, we may conclude that the formation of RuPd 1:1 nanostructure belongs to an alloy-like system containing the Ru hcp and Pd fcc phases.26,27
m). For Ru, it is possible to determine the main reflections at 38°, 42°, and 44°, which corresponded to (100), (002), (101), respectively, confirming the hcp structure (Ru pattern ICSD 40354, space group P63/mmc). Since the elements structurally belong to different space groups, despite the similarity of ionic radii (Goldschmidt's criterion and Vegard's law), there are no conditions to form bulk phase solid solutions. However, at the nanoscale both phases can coexist in a single nanoparticle.24,25 Thus, we may conclude that the formation of RuPd 1:1 nanostructure belongs to an alloy-like system containing the Ru hcp and Pd fcc phases.26,27
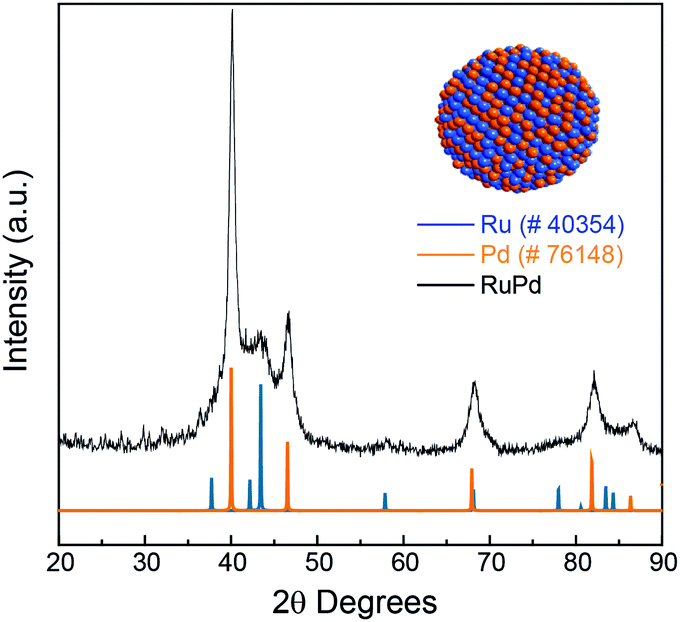 | ||
| Fig. 2 XRD measurement of the RuPd 1:1 NPs compared with the Ru (ICSD 40354) and Pd (ICSD 76148) patterns. | ||
XPS was performed to determine the surface chemical composition of the bimetallic RuPd NPs with two incident photon energies (1840 eV and 3000 eV) before the catalytic reaction (Fig. 3 and Table S1 in ESI†).12,28 At 1840 eV, the Pd 3d region showed three peaks for 3d5/2 components at binding energies (BE) of 334.71 eV (Pd0), 338.46 eV (PdO), and 340.82 eV (Pd–X), which displayed a Pd0:Pdδ+ ratio of 23:77. Comparing this result with previously obtained data for Pd NPs prepared under similar conditions (74% Pd0),29 we concluded that the Pd surface is more electrophilic in the bimetallic RuPd NPs than in Pd NPs. The Pd–X (X = N, F, O2) component at lower binding energies was ascribed to the interaction of the Pd surface atoms with the IL atoms (N and F), which produce an IL layer that provides partial protection of Pd atoms against oxidation (Pd–X, X = O2).28,30,31 The presence of the oxygen atom is probably due to the oxidation of the metal surface during experimental manipulation. The Ru 3d and C 1s regions were also analyzed at 1840 eV.32–34 These peaks suffered a strong overlap; nevertheless, the relative quantification could be determined by peak fitting.35 The Ru 3d region showed two peaks for the 3d5/2 components at BE of 279.77 eV and 282.02 eV, corresponding to Ru0 (36%) and Ru4+ (64%), respectively. Therefore, the XPS analysis at 1840 eV indicated an oxidized surface of RuPd NPs. In comparison with a previous work,6 XPS of Ru NPs synthesized in IL afforded 53% Ru0 and 47% Ruδ+, which confirms the high electron-deficient surface of the present bimetallic RuPd NPs. To understand the ratio between Ru and Pd in different depths in the nanoparticle, we performed a study comparing the XPS intensities from Pd 3d and Ru 3d at beam energies of 1840 eV and 3000 eV (see Table S1, ESI†). After normalizing the corresponding cross-section and inelastic mean free path by each incident flux,36,37 we observed a small change in the Pd:Ru ratio from 0.6 (1840 eV) to 0.8 (3000 eV). In other words, in greater depth (higher energy) there is almost the same Pd and Ru distribution than in the surface region. That is, there is clear evidence that does not have a region rich in Ru or Pd in these RuPd bimetallic nanoparticles. This result provides strong evidence for the existence of an alloy-like structure in the bimetallic RuPd NPs,12,38,39 which is in agreement with the HRTEM analysis. In addition, at higher energy (3000 eV) the presence of Ru0 and Pd0 is predominant, suggesting that the inner atoms are in the zero oxidation state. Moreover, when comparing the Ru 3p and the Pd 3d regions, the Ru:Pd atomic ratio in the bimetallic NPs was estimated as 1.2:1 (Ru:Pd), akin to those already observed for bimetallic NPs in ILs.12,31 This result was confirmed by ICP-OES measurement where a ratio of 1.1:1.0 (Ru:Pd) was obtained. H2-TPR analysis was performed to investigate the reducible behaviors of the bimetallic RuPd NPs, Ru NPs, and Pd NPs samples. TPR analysis spectra of Ru, Pd, and RuPd samples exhibited a single peak at 137 °C, 109 °C, and 116 °C, respectively (Fig. S1 in ESI†). Typically, bimetallic samples provide intermediate reductive characteristics compared to their monometallic samples. This information suggests the formation of a RuPd bimetallic interaction and also confirms the possibility of the proximity of Ru and Pd atoms, which provides a synergistic effect in the reduction temperature.40,41
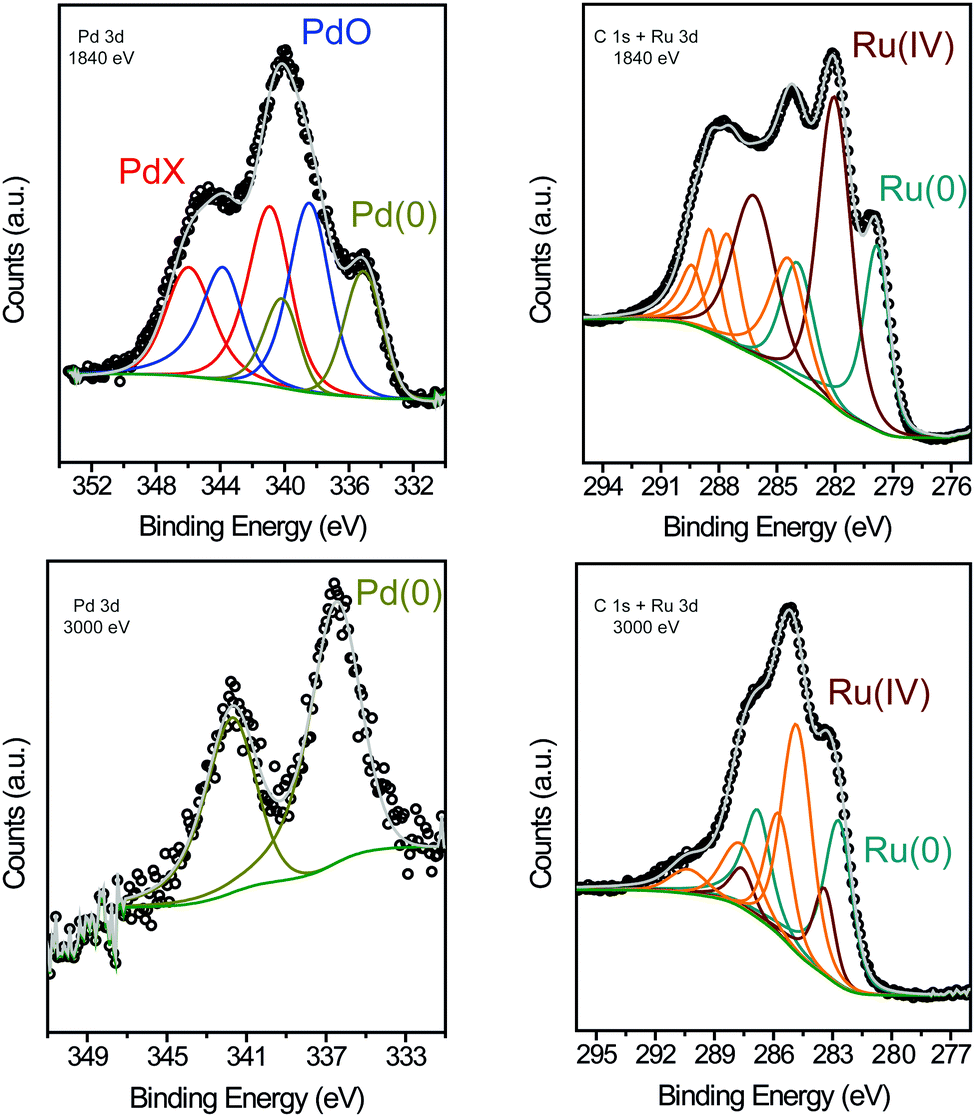 | ||
| Fig. 3 XPS of the RuPd 1:1 NPs showing the Pd 3d and C 1s + Ru 3d regions at 1840 eV (a), (b) and 3000 eV (c), (d). | ||
The catalytic activity of these bimetallic NPs was evaluated in the hydrogenation of benzene and phenol under biphasic conditions (Schemes 1 and 2). Similar to our previous work,12 we decided to use a co-solvent in order to remove the products from the IL phase, which would reduce their contact with the active metal sites tuning their selectivities based on different solubilities in n-heptane/IL mixture. Moreover, our intention with the addition of Pd was to modulate the catalyst activity enough to observe some selectivity in the intermediate compounds. As observed earlier, the activity of RuPd NPs was dependent upon the particle composition for the hydrogenation of benzene. To our surprise, an equimolar ratio of Ru:Pd generates particles with high activity (94% conversion) and selectivity (>99%) for the complete hydrogenation of benzene to cyclohexane (entry 1, Table 1). However, by adding a small amount of Pd (Ru:Pd 9:1), the conversion decreased to 68% under the same reaction conditions (entry 2, Table 1). This result indicates that the presence of 10 mol% Pd decreases the catalyst activity, and the bimetallic material is acting preferably as a pure Ru NP, akin to previous observations for Ru NPs in ILs6 (entry 5, Table 1). Increasing the Pd content (Ru:Pd 1:9) demonstrates a drastic drop in the conversion (entry 3, Table 1), which indicates preferable Pd catalytic behavior, corroborating with the expected reduced activity of Pd NPs in the hydrogenation of arenes. Although the intention of using a co-solvent was to tune the selectivity of the products, there is a small amount formation of the intermediates 2 and 3 for the benzene hydrogenation using RuPd 1:1 NPs (entry 1, Table 1). These intermediates were not detected even at very low conversion in the reaction catalyzed by RuPd 1:9 NPs (entry 3, Table 1), where the main product was cyclohexane (4). In other words, even using a higher amount of Pd (a relative less active metal for arene hydrogenation),13–15 there is no formation of intermediates (2 and 3) or since they are formed, a rapid conversion to the total hydrogenated compound occurs. Therefore, the synergistic effect in the bimetallic RuPd 1:1 NPs (entry 1, Table 1) afforded a superior catalyst for the hydrogenation of benzene when compared to the monometallic Ru and Pd particles. Indeed, the activity and selectivity may also be related to the electrophilic surface of the bimetallic NPs, evidenced by XPS. These electron-deficient sites are a benefit for catalysis because they allow effective coordination of the electron-rich benzene molecule at the metal surface, facilitating the hydrogenation reaction.24
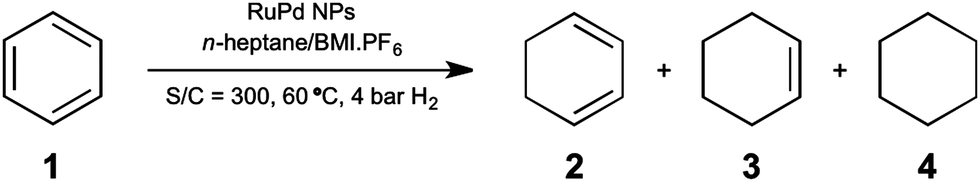 | ||
| Scheme 1 Hydrogenation of benzene catalyzed by the RuPd NPs under n-heptane/IL biphasic conditions. | ||
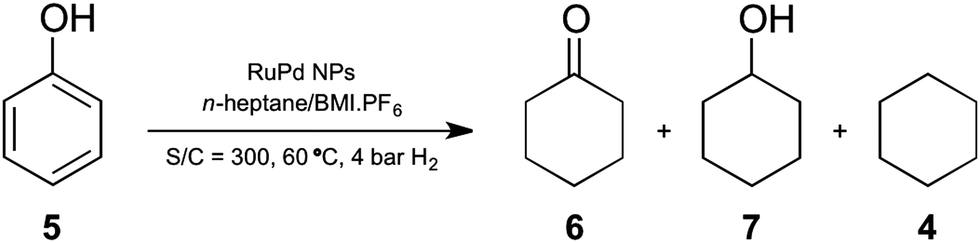 | ||
| Scheme 2 Hydrogenation of phenol catalyzed by the RuPd NPs under n-heptane/IL biphasic conditions. | ||
| Entry | Ru:Pd (molar ratio) |
Substrate | Ionic liquid | Additive | T (°C) | Products | Time (h) | |
|---|---|---|---|---|---|---|---|---|
| Conversion (%) | Selectivity (%) | |||||||
| Reaction conditions: benzene (5.24 g, 67.2 mmol), phenol (6.32 g, 67.2 mmol), substrate/catalyst = 300 (molar ratio), ionic liquid (1 mL), PH2 (4 bar). For reactions in the presence of additives: n-heptane (1 mL), H2O (200 μL), H2O/HF (100 μL H2O/100 μL HF). Conversion and selectivity determined by GC and GC-MS analysis. | ||||||||
| 1 | 1:1 |
Benzene | BMI·PF6 | n-Heptane | 60 | 94 | 4 (>99) | 18 |
| 2 | 9:1 |
Benzene | BMI·PF6 | n-Heptane | 60 | 68 | 4 (100) | 18 |
| 3 | 1:9 |
Benzene | BMI·PF6 | n-Heptane | 60 | 2 | 4 (100) | 18 |
| 4 | Ru6 | Benzene | BMI·PF6 | — | 75 | 10 | 3 (15) | 2 |
| 5 | Ru6 | Benzene | BMI·PF6 | — | 75 | 73 | 4 (100) | 18.5 |
| 6 | 1:1 |
Phenol | BMI·PF6 | n-Heptane | 60 | >98 | 7 (99) | 18 |
| 7 | 1:1 |
Phenol | BMI·PF6 | — | 60 | 12 | 6 (23), 7 (77) | 2 |
| 8 | 1:1 |
Phenol | BMI·PF6 | — | 60 | >93 | 7 (99) | 18 |
| 9 | 1:1 |
Phenol | BMI·PF6 | — | 130 | 49 | 4 (100) | 18 |
| 10 | 1:1 |
Phenol | BMI·PF6 | n-Heptane | 25 | 45 | 6 (15), 7 (85) | 18 |
| 11 | 1:1 |
Phenol | BMI·PF6 | H2O | 60 | 20 | 6 (34), 7 (66) | 18 |
| 12 | 1:1 |
Phenol | BMI·PF6 | H2O/HF | 60 | 7 | 6 (32), 7 (68) | 18 |
| 13 | Ru | Phenol | BMI·PF6 | — | 60 | 21 | 6 (2), 7 (98) | 2 |
| 14 | Ru | Phenol | BMI·PF6 | — | 60 | 86 | 7 (99) | 18 |
| 15 | Ru | Phenol | BMI·PF6 | H2O | 60 | 76 | 6 (13), 7 (74), 4 (11) | 18 |
| 16 | Ru | Phenol | BMI·PF6 | H2O/HF | 60 | 71 | 6 (45), 7 (13), 4 (42) | 18 |
| 17 | Pd | Phenol | BMI·PF6 | — | 60 | >5 | 6 (78), 7 (22) | 18 |
| 18 | Pd | Phenol | BMI·PF6 | H2O | 60 | >11 | 6 (62), 7 (38) | 18 |
| 19 | Pd | Phenol | BMI·PF6 | H2O/HF | 60 | >12 | 6 (65), 7 (45) | 18 |
| 20 | 1:1 |
Phenol | BMI·NTf2 | — | 60 | 85 | 7 (99) | 18 |
| 21 | 1:1 |
Phenol | BMI·NTf2/BMI·PF6 | — | 60 | 82 | 7 (99) | 18 |
| 22 | 1:1 |
Phenol | BMI·NTf2/BMI·PF6 | H2O | 60 | 55 | 6 (9), 7 (91) | 18 |
| 23 | 1:1 |
Phenol | BMI·NTf2/BMI·PF6 | H2O/HF | 60 | 40 | 6 (28), 7 (72) | 18 |
| 24 | Ru5 | Phenol | BMI·BF4 | Acidic IL | 130 | 77 | 7 (3), 4 (97) | 4 |
| 25 | Pd5 | Phenol | BMI·BF4 | Acidic IL | 130 | 15 | 6 (5.6), 7 (14), 4 (80) | 4 |
| 26 | Pd/C43 | Phenol | — | AlCl3 | 30 | >99 | 6 (>99) | 12 |
To investigate the scope of the reactions, the RuPd NPs were examined for the selective hydrogenation of phenol. Indeed, the bimetallic RuPd NPs (1:1) promoted the hydrogenation of phenol to exclusively form cyclohexanol (entry 6, Table 1) in a biphasic system (n-heptane/BMI·PF6). It is assumed that in the first step, the cyclohexanone is formed, and then the ketone is rapidly hydrogenated to cyclohexanol. In this case, the presence of n-heptane has a slight positive effect on the reaction (entry 8, Table 1). Notably, the dehydration reaction was not observed; therefore, one possible interaction between phenolic groups and the HF formed from the decomposition of the IL anion (PF6−) is not sufficient to promote the HDO reaction.42 However, increasing the temperature induces the dehydration process (entry 9, Table 1), but this was not detected when water and/or HF were added (entries 11 and 12, Table 1). The presence of water inhibited the conversion and increased the selectivity of the ketone product. Lower temperature causes a decrease in catalytic activity, but even at 25 °C conversion of 45% and selectivity of 85% in cyclohexanol and 15% in cyclohexanone were achieved (entry 10, Table 1), which is a remarkable performance. For the monometallic NPs, the reaction employing Ru also showed selectivity to cyclohexanol (entry 14, Table 1), although the catalytic performance was inferior when compared to the bimetallic RuPd NPs. In contrast to the bimetallic catalyst, the addition of water and/or HF to the Ru NPs produced up to 42% of the HDO product (entries 15 and 16, Table 1). As expected, the Pd NPs were not able to promote the hydrogenation of phenol efficiently (entries 17–19, Table 1), but the preferable selectivity for the cyclohexanone product is in agreement with previous reports using Pd catalysts.43–47
A previous study on the hydrogenation of phenol showed that the classically supported Pd catalysts (Pd/C, Pd/Al2O3, Pd/NaY) in the presence of Lewis acids could selectively produce cyclohexanone at low temperatures, inhibiting further hydrogenation of the ketone.43 An interesting catalytic system based on supported monometallic Ru NPs (Ru/HZSM-5) for the direct transformation of phenol into cyclohexane in an aqueous-phase process was described.5 Ru and Pd NPs combined with Brønsted acidic ILs can be active systems for the reduction of phenol into cyclohexane (entries 24 and 25, Table 1). Recently, the HDO reaction of phenol over supported Pd catalysts showed that benzene is the primary product formed for the catalysts Pd/TiO2 and Pd/ZrO2, while cyclohexanone was generated using Pd/SiO2, Pd/Al2O3, Pd/CeO2, and Pd/CeZrO2.44 Recent work described a high conversion and selectivity for cyclohexanone in the hydrogenation of phenol by an alkali-metal-Pd/TiO2 catalyst in aqueous medium at 80 °C.48 In this case, the presence of the alkali-metal plays a fundamental role in the high catalyst activity. The present bimetallic RuPd NPs can be regarded as potential highly active catalysts to perform the hydrogenation of phenol to cyclohexanol under mild conditions. By employing this bimetallic catalyst, it was possible to selectively achieve the alcohol, which is a different behavior when compared to supported Pd catalysts. The observed properties of the Pd NPs relative to the other metal NPs in the formation of cyclohexanone may be ascribed to the favorable desorption of cyclohexanone on the Pd surface, inhibiting further hydrogenation to cyclohexanol. However, in the case of RuPd NPs, they behave as a tandem catalytic system where the affinity of the ketone and the metal surface favors the reaction step of cyclohexanol formation, inhibiting the dehydration process.
Generally, the efficient catalytic activity of the bimetallic NPs may be related to the electronic synergism between both metals that modify the electron density of one metal against the other,41 making bimetallic catalysts more active than monometallic ones. In the case of RuPd NPs, the electron-deficient surface also contribute to the catalytic activity, since the coordination of unsaturated substrates is favored at the electrophilic surface.49,50 A proposed mechanism for the hydrogenation of benzene is based on the high affinity between the electron-rich benzene molecule and the electrophilic surface of RuPd NPs. First, it is assumed that benzene is strongly adsorbed on the metal surface, which favors the insertion of the surface hydrides formed from the oxidative addition of H2. The high electrophilicity of RuPd NPs difficult the desorption of the generated intermediates (2 and 3) and facilitate the formation of the total hydrogenated product (4). This would explain the preferred selectivity in cyclohexane. In the case of phenol hydrogenation, the first step is the conversion of phenol into cyclohexenol, which is fast transformed in cyclohexanone (6). At this stage, the high Lewis acidic character of the metal surface might stabilize the cyclohexanone, but not sufficient to avoid its reduction to cyclohexanol (7). We suggest that in both cases (benzene and phenol hydrogenation) the IL has an important role for the electronic and steric stabilization of the nanoparticles as well as a suitable catalytic phase for hydrogenation reactions. On the other hand, mainly for benzene hydrogenation, our expectation with the use of n-heptane was to gain selectivity in the intermediate compounds, as observed in a previous work using Ru@Pt NPs,12 but this effect was not detected in the present study. Maybe, one reason is that the high electrophilic surface of the RuPd NPs produced a very active catalyst, avoiding a considerable formation of the intermediates.
Conclusions
In summary, we have demonstrated that bimetallic RuPd NPs (1:1) 2 nm in size stabilized by BMI·PF6 IL have been proved to be highly active catalysts for the hydrogenation of both benzene and phenol at 60 °C under biphasic conditions. Our preliminary results showed conversions up to 94% and selectivity >99% in cyclohexane for benzene hydrogenation, while a conversion >98% with a selectivity of 99% in cyclohexanol was observed during phenol reduction. The equimolar ratio of Ru and Pd affords the best bimetallic catalyst under the related conditions. Notably, the addition of a less active arene hydrogenation metal (Pd) to the Ru provides a synergistic effect in producing a highly competent catalyst. These results indicate the significant electronic change in the bimetallic materials, which can produce unique catalytic properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank CNPq (449758/2014-1), FAPERGS (16/2551-0000373-4), Fondecyt Iniciación (No. 11170879), DFG (Heisenberg-Program), Ministerium für Innovation, Wissenschaft und Forschung (NRW-returnee award), Alexander-von-Humboldt Foundation, DAAD, COST Actions CARISMA, and CHAOS for the financial support. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001. Authors are also grateful to LNLS (Campinas-Brazil, proposal SXS-20160887) for the SXS beamline facilities and LNNano (Campinas-Brazil, proposal TEM-C1-25367) for HRTEM analysis.Notes and references
- J. Struijk and J. J. F. Scholten, Appl. Catal., A, 1992, 82, 277–287 CrossRef CAS.
- P. J. Dyson, Dalton Trans., 2003, 2964–2974 RSC.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS.
- W. X. Gu, M. M. Stalzer, C. P. Nicholas, A. Bhattacharyya, A. Motta, J. R. Gallagher, G. H. Zhang, J. T. Miller, T. Kobayashi, M. Pruski, M. Delferro and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 6770–6780 CrossRef CAS.
- N. Yan, Y. Yuan, R. Dykeman, Y. Kou and P. J. Dyson, Angew. Chem., Int. Ed., 2010, 49, 5549–5553 CrossRef CAS.
- E. T. Silveira, A. P. Umpierre, L. M. Rossi, G. Machado, J. Morais, G. V. Soares, I. L. R. Baumvol, S. R. Teixeira, P. F. P. Fichtner and J. Dupont, Chem. – Eur. J., 2004, 10, 3734–3740 CrossRef CAS.
- M. H. G. Prechtl, M. Scariot, J. D. Scholten, G. Machado, S. R. Teixeira and J. Dupont, Inorg. Chem., 2008, 47, 8995–9001 CrossRef CAS.
- G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner, S. R. Teixeira and J. Dupont, Chem. – Eur. J., 2003, 9, 3263–3269 CrossRef CAS.
- C. W. Scheeren, G. Machado, J. Dupont, P. F. P. Fichtner and S. R. Texeira, Inorg. Chem., 2003, 42, 4738–4742 CrossRef CAS.
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. N. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS.
- A. K. Singh and Q. Xu, ChemCatChem, 2013, 5, 652–676 CrossRef CAS.
- A. Weilhard, G. Abarca, J. Viscardi, M. H. G. Prechtl, J. D. Scholten, F. Bernardi, D. L. Baptista and J. Dupont, ChemCatChem, 2017, 9, 204–211 CrossRef CAS.
- R. R. Deshmukh, J. W. Lee, U. S. Shin, J. Y. Lee and C. E. Song, Angew. Chem., Int. Ed., 2008, 47, 8615–8617 CrossRef CAS.
- T. Maegawa, A. Akashi, K. Yaguchi, Y. Iwasaki, M. Shigetsura, Y. Monguchi and H. Sajiki, Chem. – Eur. J., 2009, 15, 6953–6963 CrossRef CAS.
- J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184–200 CrossRef CAS.
- J. Zhang, K. Gao, S. Wang, W. Li and Y. Han, RSC Adv., 2017, 7, 6447–6456 RSC.
- A. V. Ruban, H. L. Skriver and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15990–16000 CrossRef.
- X. Ma, R. Lin, R. Y. Ofoli, Z. Mei and J. E. Jackson, Mater. Chem. Phys., 2016, 173, 1–6 CrossRef CAS.
- S. Chung, D. N. Leonard, V. Altoe, S. Aloni, J. J. De Yoreo and S. Franzen, Part. Part. Syst. Charact., 2013, 30, 280–286 CrossRef CAS.
- G. Chacón, C. Pradel, N. Saffon-Merceron, D. Madec and M. Gomez, J. Chem., 2016, 4, 37–50 Search PubMed.
- M. Shao, T. Yu, J. H. Odell, M. Jin and Y. Xia, Chem. Commun., 2011, 47, 6566–6568 RSC.
- W. Chen, D. Ghosh, J. Sun, M. C. Tong, F. Deng and S. Chen, Electrochim. Acta, 2007, 53, 1150–1156 CrossRef CAS.
- C. Sun, Z. Cao, J. Wang, L. Lin and X. Xie, New J. Chem., 2019, 43, 2567–2574 RSC.
- K. Kusada, H. Kobayashi, R. Ikeda, Y. Kubota, M. Takata, S. Toh, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 1864–1871 CrossRef CAS.
- T. Sato, S. Takagi, S. Deledda, B. C. Hauback and S.-i. Orimo, Sci. Rep., 2016, 6, 23592 CrossRef CAS.
- M. Liu, W. Yu, H. Liu and J. Zheng, J. Colloid Interface Sci., 1999, 214, 231–237 CrossRef CAS.
- D. Wu, K. Kusada and H. Kitagawa, Sci. Technol. Adv. Mater., 2016, 17, 583–596 CrossRef CAS.
- L. Luza, C. P. Rambor, A. Gual, F. Bernardi, J. B. Domingos, T. Grehl, P. Brüner and J. Dupont, ACS Catal., 2016, 6, 6478–6486 CrossRef CAS.
- A. P. Umpierre, G. Machado, G. H. Fecher, J. Morais and J. Dupont, Adv. Synth. Catal., 2005, 347, 1404–1412 CrossRef CAS.
- L. Luza, C. P. Rambor, A. Gual, J. A. Fernandes, D. Eberhardt and J. Dupont, ACS Catal., 2017, 7, 2791–2799 CrossRef CAS.
- M. I. Qadir, A. Weilhard, J. A. Fernandes, I. de Pedro, B. J. C. Vieira, J. C. Waerenborgh and J. Dupont, ACS Catal., 2018, 8, 1621–1627 CrossRef CAS.
- I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797–2808 RSC.
- K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H. P. Steinruck and P. Licence, Chem. Rev., 2010, 110, 5158–5190 CrossRef CAS.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- K. Baranowska, J. Okal and W. Tylus, Appl. Catal., A, 2016, 511, 117–130 CrossRef CAS.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1991, 17, 911–926 CrossRef CAS.
- J. A. Rodriguez, R. A. Campbell and D. W. Goodman, J. Phys. Chem., 1991, 95, 5716–5719 CrossRef CAS.
- D. Wu, M. Cao and R. Cao, Chem. Commun., 2014, 50, 12970–12972 RSC.
- A. B. Yousaf, M. Imran, P. Kasak, F. S. Zavahir, S. J. Zaidi and C. Fernandez, Catal. Sci. Technol., 2017, 7, 3283–3290 RSC.
- J. Chen, X. Liu and F. Zhang, Chem. Eng. J., 2015, 259, 43–52 CrossRef CAS.
- S. A. Katsyuba, M. V. Vener, E. E. Zvereva, Z. Fei, R. Scopelliti, G. Laurenczy, N. Yan, E. Paunescu and P. J. Dyson, J. Phys. Chem. B, 2013, 117, 9094–9105 CrossRef CAS.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250–1252 CrossRef CAS.
- P. M. de Souza, R. C. Rabelo-Neto, L. E. P. Borges, G. Jacobs, B. H. Davis, D. E. Resasco and F. B. Noronha, ACS Catal., 2017, 7, 2058–2073 CrossRef CAS.
- Z. L. Li, J. H. Liu, C. G. Xia and F. W. Li, ACS Catal., 2013, 3, 2440–2448 CrossRef CAS.
- X. Xu, H. R. Li and Y. Wang, ChemCatChem, 2014, 6, 3328–3332 CrossRef CAS.
- R. F. Nie, H. Z. Jiang, X. H. Lu, D. Zhou and Q. H. Xia, Catal. Sci. Technol., 2016, 6, 1913–1920 RSC.
- H. Zhou, B. B. Han, T. Z. Liu, X. Zhong, G. L. Zhuang and J. G. Wang, Green Chem., 2017, 19, 3585–3594 RSC.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- J. A. Rodriguez and D. W. Goodman, Science, 1992, 257, 897–903 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures. See DOI: 10.1039/d0nj02674c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |