DOI:
10.1039/D0NJ04280C
(Paper)
New J. Chem., 2021,
45, 131-140
DFT study on the [4+4] domino cycloaddition of ynones with benzylidenepyrazolones to access eight-membered cyclic ethers: effects of DBU vs. Et3N†
Received
25th August 2020
, Accepted 18th November 2020
First published on 18th November 2020
Abstract
The mechanisms of [4+4] domino cycloaddition of ynones with benzylidenepyrazolones to access highly substituted eight-membered cyclic ethers are studied to explore the effects of bases (t-BuOK vs. DBU vs. Et3N) based on DFT calculations. Our theoretical calculations indicate that the bases can promote the present catalytic reaction as the H+-shuttle agents by a two-step H+-shift reaction. More importantly, the calculations reveal that the difference in the catalytic activity of bases is primarily attributed to their electron-donating ability (t-BuO− > DBU > Et3N). The catalytic activity order of bases is t-BuO− > DBU > Et3N for the present base-catalyzed domino reactions. In a word, the present calculated results can reasonably explain the experimental phenomena and provide useful guidance and advice for the synthesis of medium-sized rings via the domino reactions to select the appropriate bases.
1. Introduction
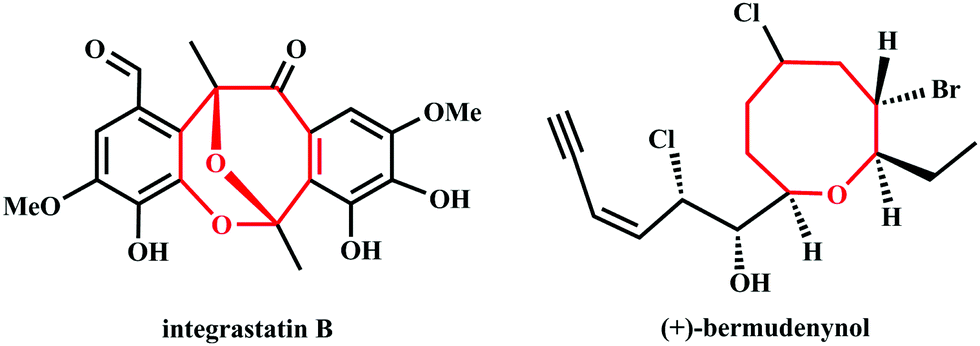
Eight-membered cyclic ethers as the key structural units are often found in natural products and biologically active molecules.1,2 For instance, integrastatin B comes from endophytic Ascochyta species3–5 and (+)-bermudenynol comes from red alga6,7(Fig. 1). According to our investigation of related literature, the ring-closing metathesis,8 ring expansion,9 intramolecular alkylation,10–13 retro-Claisen rearrangement,14–16 and other synthetic strategies17–22 have been developed to construct medium-ring structures. However, the abovementioned methods suffer from several limitations such as unsatisfactory yields, costly catalysts, poor stereoselectivity and tedious processes for purification. Therefore, the development of an efficient strategy for the construction of eight-membered cyclic ethers has received increasing attention in the fields of organic and medicinal chemistry.
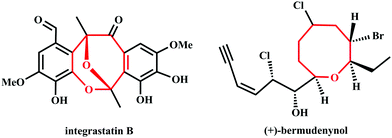 |
| | Fig. 1 The structures of integrastatin B and (+)-bermudenynol. | |
In the past few decades, bases (Brønsted base or Lewis base) have been widely employed as catalysts to effectively promote various organic catalytic reactions. For example, Guo and coauthors23 reported the DABCO (1,4-diazabicyclo[2.2.2]octane)-catalyzed [3+2] annulation reaction of sulfamate-derived cyclic imines with isocyanoacetates under mild conditions to access sulfamate-fused 2-imidazolines in moderate to excellent yields. In 2014, Wang et al.24 successfully proposed the unprecedented catalytic tandem formal [4+3] cycloaddition/decarboxylation/isomerization of methyl coumalate and imine esters with Et3N (triethylamine) as the suitable base affording a series of highly functionalized seven-membered heterocyclic azepine derivatives in good yields. DBU (1,8-diazabicyclo[5.4.0]undec-7-ene)-promoted cyclization of ortho-(3-hydroxy-1-alkynyl)benzamide was presented by Mo's group,25 providing an efficient method for the synthesis of trans-3,4-dihydroisoquinolin-1(2H)-ones and (E)-4-(1-alkenyl)isoquinolin-1(2H)-ones under mild conditions. Inspired by the above reports, DABCO, Et3N and DBU are also employed as catalysts to accelerate the synthesis of eight-membered cyclic compounds by domino reactions owing to the convenience and efficiency strategy. In 2015, an efficient method was reported by Huang and co-workers26 through DABCO-mediated [4+4] domino annulation reactions of ynones and α-cyano-α, β-unsaturated ketones for the construction of functionalized eight-membered cyclic ethers in good yields under mild conditions. It is worth mentioning that Miao et al.27 proposed that the base plays a crucial role in assisting proton migration, and that the reaction yields can be controlled by using different bases (such as DBU vs. Et3N) in the base-promoted [4+4] domino annulation reactions to access eight-membered cyclic ethers from ynones and benzylidenepyrazolones in moderate to good yields. Obviously, the usage of bases displays the positive catalytic effects in the synthesis of eight-membered rings via the domino reactions. However, the detailed mechanisms of base-mediated chemical reactions have not been illustrated clearly up to date, and the experimental technology still restricts the understanding of the reaction mechanisms. Therefore, it is necessary to do deep research studies on the roles of bases by exploring the detailed mechanisms of base-catalyzed reactions based on the theoretical calculations.
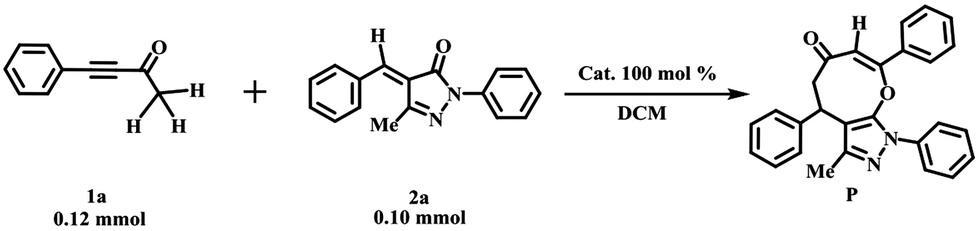
In this work, the mechanisms of [4+4] domino annulation reactions of ynones (1a) and benzylidenepyrazolones (2a) using bases as the catalysts to form eight-membered cyclic ethers27 (Table 1) are studied systematically to better explore the following issues in detail: (a) how DBU and Et3N promote the reactions. (b) Understanding the difference in the catalytic activity of bases (DBU vs. Et3N). More importantly, what causes this difference? And (c) predicting the acceleration of a strong base t-BuOK for the present domino cycloadditions. These issues have quite an important guiding significance for the exploration of the effects of bases in the synthesis of medium-sized rings via the domino annulation reactions.
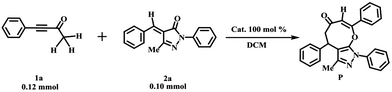
Table 1 The [4+4] domino annulation reactions of ynones (1a) and benzylidenepyrazolones (2a) with bases as the catalysts
|
|
| Entry |
Base (cat.) |
100 mol% of base |
Temp. |
Time (min) |
Yield (%) |
| 1 |
DBU |
100 |
r.t. |
60 |
74 |
| 2 |
Et3N |
100 |
r.t. |
60 |
0 |
| 3 |
t-BuOK |
100 |
r.t. |
60 |
? |
2. Computational details
All calculations reported in the present paper were carried out using the Gaussian 09 program package.28 The B3LYP method29–31 is employed to fully optimize the geometries of the species involving intermediates and transition states in DCM (dichloromethane) solvent based on density functional theory (DFT).32 The B3LYP method has been successfully applied to explore the mechanism of catalytic reactions.33,34 As a further test, CAM-B3LYP, BLYP, B3P86 and B3PW91 are used for evaluating the reliability of B3LYP calculated results to avoid the shortcomings of functions (see Table S1 in the ESI†). The double-ζ basis set (6-31G*)35–37 is employed for N, C, O and H elements. Subsequently, the basis sets of 6-31G**, 6-31+G* and 6-311G* are tested, and their calculated values are close to those of 6-31G* (see Table S2 in the ESI†). At the same theory level, the frequency calculations are adopted to verify the rationality of all stationary points (only one imaginary frequency for transition states and no imaginary frequency for equilibrium structures) and to obtain zero-point energy. The intrinsic reaction coordinate (IRC)38,39 calculations are performed to confirm the connectivity of transition states, which connects with the correct reactants and products. The key IRC analyses are described in the ESI.† The solvent-effect of dichloromethane (DCM) is simulated using the self-consistent reaction field (SCRF) method40,41 through the polarized continuum model (PCM)42–44 on the basis of the solvent-phase optimized structures. The solvation single-point energy is evaluated at the M06-2x-D3/6-311++G** levels in the solvent phase. The better treatment of dispersion interactions can be provided by D3, and M06-2x is more suited to describe non-covalent interactions and thermodynamics.45–47 Adding the gas-phase thermal correction to the Gibbs free energy and the solvation single-point energy provides the solution phase Gibbs free energy. Unless stated otherwise, the relative solution-phase Gibbs free energies (Grel, kJ mol−1) are the discussed energies in the present study.
3. Results and discussion
3.1 Roles of bases
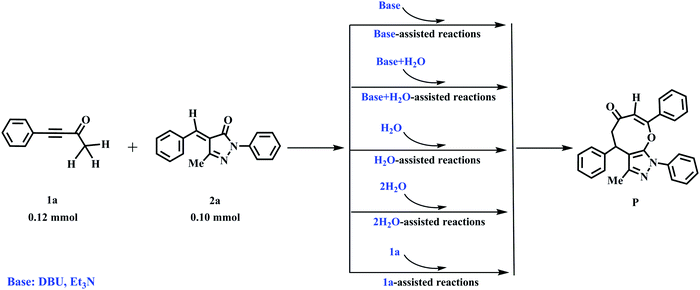
The substrates 4-phenylbut-3-yn-2-ones (1a) and 4-benzylidene-5-methyl-2-phenylpyrazolones (2a) are catalyzed by the base (DBU) to form the eight-membered cyclic ethers (P) with a yield of 74% (see entry 1 of Table 1). The reaction mechanisms of the cycloaddition of 1a with 2a are designed firstly by our group, which are shown in Scheme 1. Subsequently, the detailed discussions about the base-assisted paths are presented to study the roles of bases (DBU and Et3N) in the rate acceleration of the present reaction.
 |
| | Scheme 1 The reaction mechanisms of [4+4] domino cycloaddition of ynones (1a) with benzylidenepyrazolones (2a) to access eight-membered cyclic ethers (P) in DCM medium. | |
Mechanism of DBU-promoted reactions.
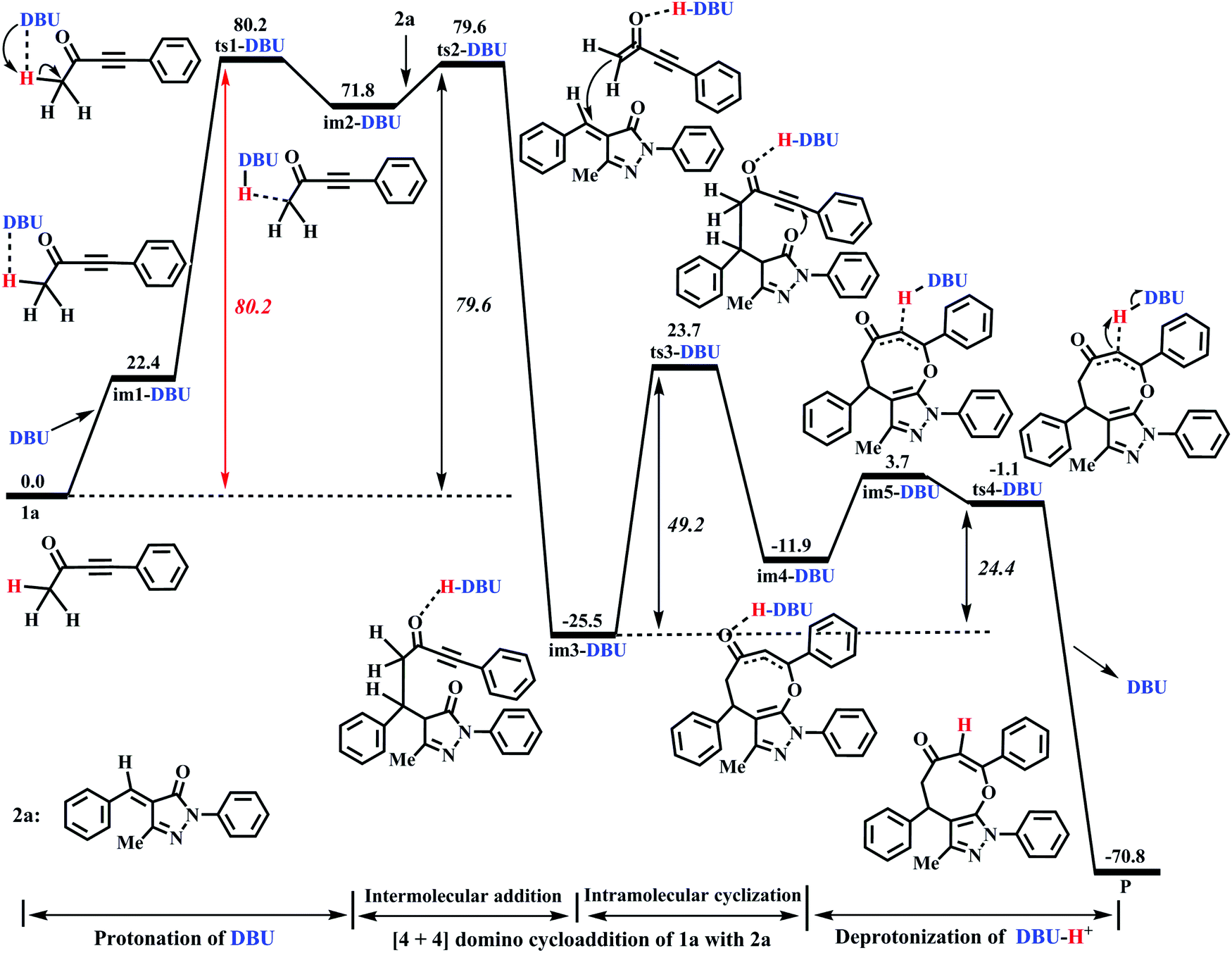
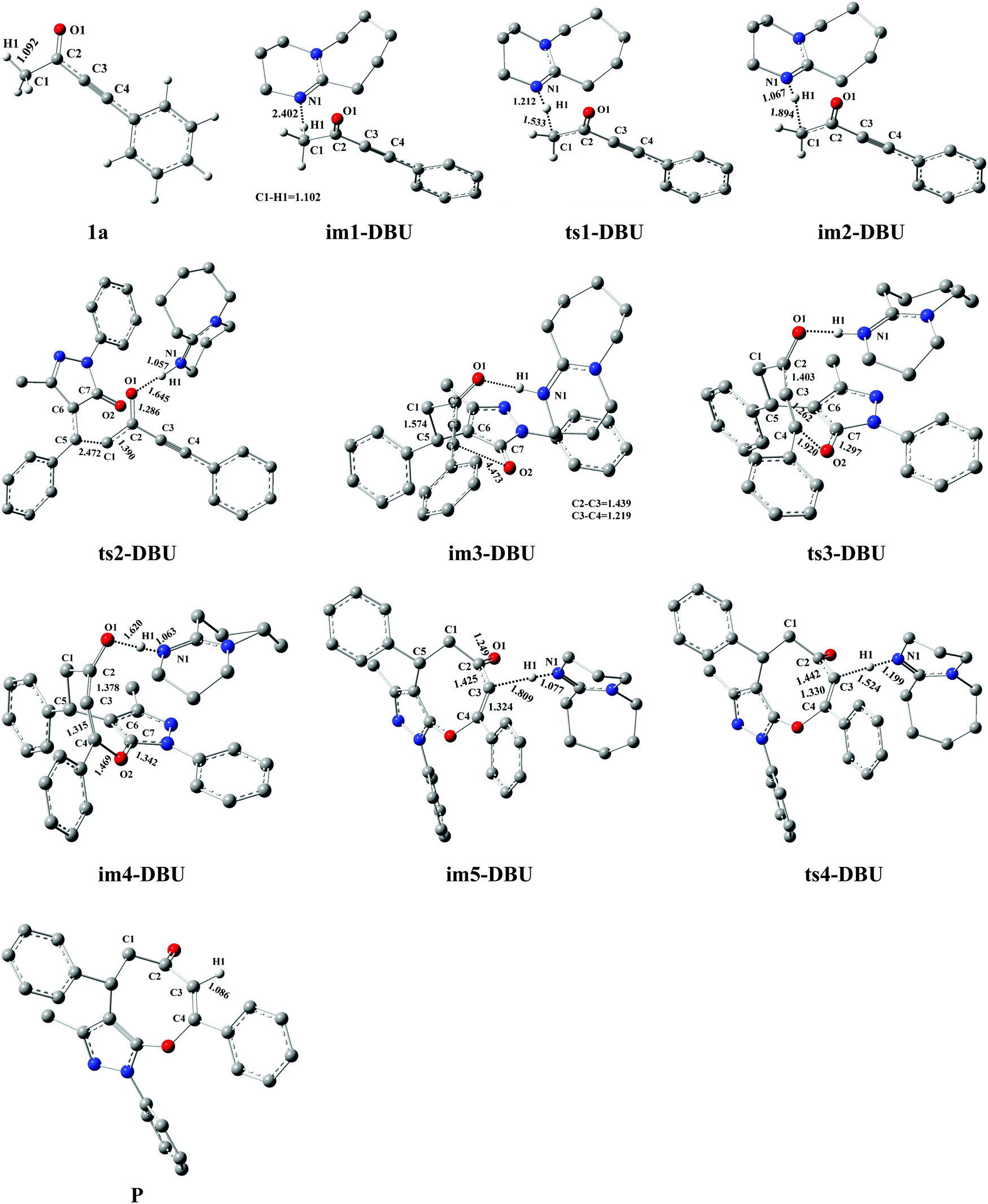
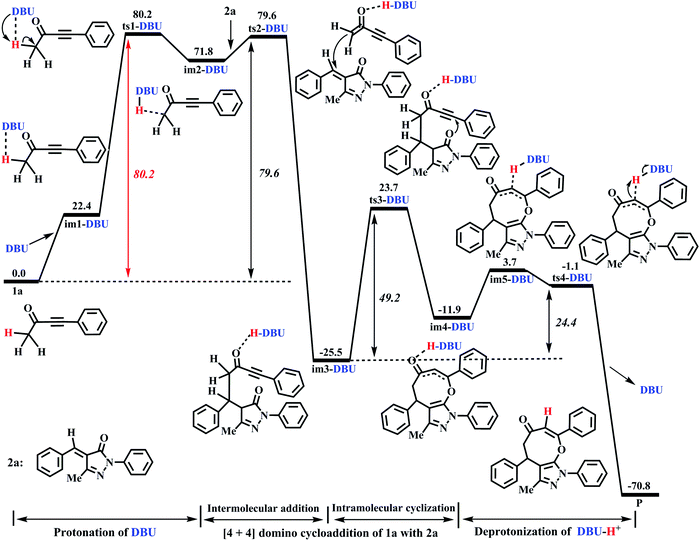
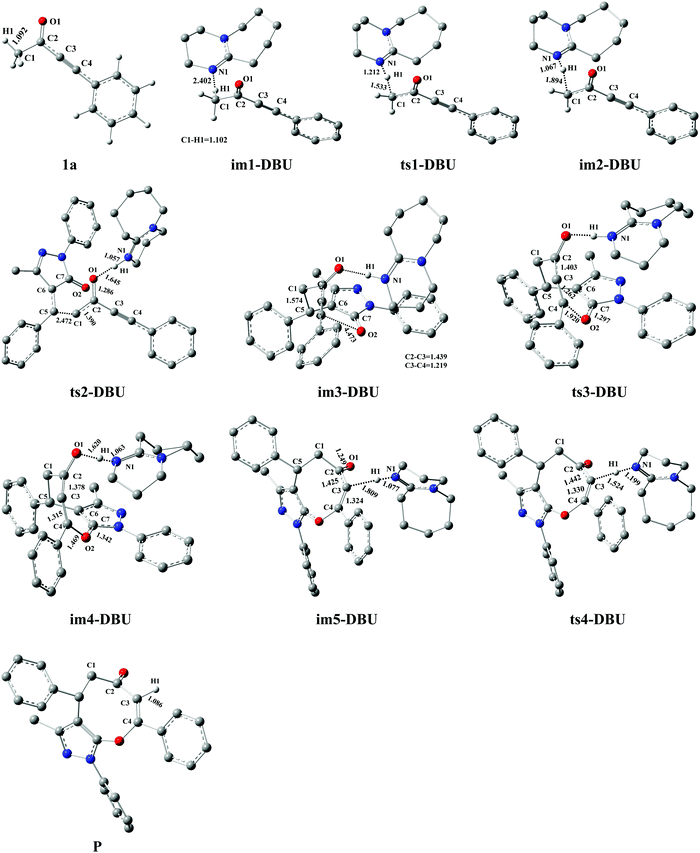
The DFT-computed energy profile of DBU-promoted [4+4] domino annulation reactions of ynones (1a) and benzylidenepyrazolones (2a) is given in Fig. 2. The optimized structures along with the reaction path are shown in Fig. 3. The H1–C1 bond length in im1-DBU is longer than that in 1a (1.102 vs. 1.092 Å, see Fig. 3). Obviously, the C1–H1 bond is activated by DBU via the hydrogen-bond of N1⋯H1–C1 (H1⋯N1 = 2.402 Å). Subsequently, the intermediate im2-DBU is formed via a new transition state ts1-DBU with the formation of a hydrogen-bond N1–H1⋯C1 (H1–N1 =1.067 Å, H1⋯C1= 1.894 Å), generating the protonated DBU (DBU-H+). In ts1-DBU, H1+ is transferred from C1 to N1 through the hydrogen-bond N1⋯H1–C1, and the C1–H1 and H1–N1 bond lengths are 1.533 and 1.212 Å, respectively. The next reaction is a [4+4] domino cycloaddition of 1a with 2a, including an intermolecular addition and an intramolecular cyclization (see Fig. 2). In the intermolecular addition, C1 as the nucleophilic site of im2-DBU attacks the C5 of 2a, providing a stable intermediate im3-DBUvia a transition state ts2-DBU, which has a C1–C5 bond length of 2.472 Å. In the intramolecular cyclization, O2 acts as the nucleophilic site that attacks C4, forming a new intermediate im4-DBUvia a transition state ts3-DBU, in which, the O2–C5 bond length is 1.920 Å. As the reaction proceeds, im4-DBU is isomerized to im5-DBU accompanied by the formation of hydrogen-bond C3⋯H1–N1 (C3⋯H1 = 1.809 Å, H1–N1 = 1.077 Å, in im5-DBU) and the fracture of hydrogen-bond O1⋯H1–N1 (O1⋯H1 = 1.620 Å, H1–N1 = 1.063 Å, in im4-DBU), and then im5-DBU goes through a transition state ts4-DBU to form a more stable product P with the shift of H1+ from N1 (in DBU–H+) to C3, regenerating the free-metal catalyst DBU. It is worth noting that the relative free energy of im5-DBU (3.7 kJ mol−1) is higher than that of ts4-DBU (−1.1 kJ mol−1), suggesting that the reaction from im5-DBU to P is a barrier-free transformation, which has been proposed in previous theoretical studies.48–50 Additionally, the IRC calculation also confirms the reliability of ts4-DBU (see Fig. S11 in the ESI†).
 |
| | Fig. 2 Energy profile of the DBU-promoted [4+4] domino cycloaddition of ynones (1a) with benzylidenepyrazolones (2a) to access eight-membered cyclic ethers (P) in DCM medium (unit: kJ mol−1). | |
 |
| | Fig. 3 Optimized structures for the DBU-promoted [4+4] domino cycloaddition of ynones (1a) with benzylidenepyrazolones (2a) to access eight-membered cyclic ethers (P) in DCM medium. The H atom that does not participate in the reactions has been omitted for clarity. Bond distance is given in Å. | |
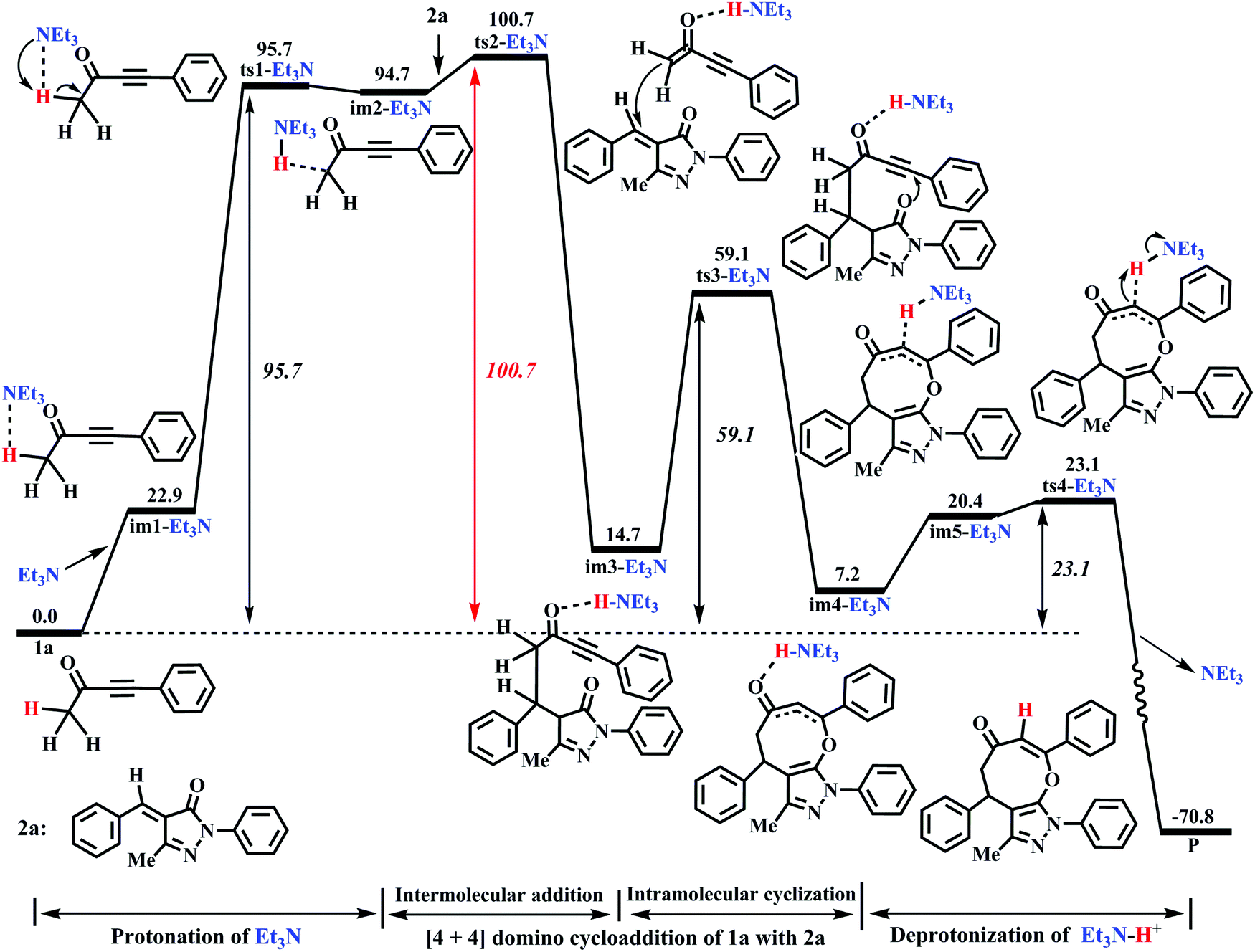
From Fig. 2, the energy profile of the DBU-catalyzed reaction involves the protonation process of DBU, the [4+4] cycloaddition process of 1a with 2a, and the deprotonation process of DBU–H+. The free energy barriers are 80.2, 79.6, 49.2, and 24.4 kJ mol−1 for the abovementioned reaction processes, respectively. The rate-determining step locates in the protonation of DBU. The rate-determining free energy barrier is 80.2 kJ mol−1, which is favorable for the construction of products (P) in DCM medium under the given experimental conditions (r.t.). More importantly, our calculations reveal that DBU could serve as a proton-shuttle to assist the shift of H1+ from C1 to C3 by the protonation of DBU and the deprotonation of DBU–H+. It is noteworthy that the [DBU + H2O]-, H2O-, and 2H2O-catalyzed reaction mechanisms are considered in this work (Fig. S1–S3 in the ESI†). The free energy barriers for ts1-[DBU + H2O], ts1-H2O and ts1-2H2O (their geometric structures are described in Fig. S4 of the ESI†) are 113.8, 194.5, and 172.1 kJ mol−1, which are 33.6, 114.3 and 91.9 kJ mol−1 higher than the rate-determining free energy barrier (80.2 kJ mol−1) of the DBU-catalyzed strategy. Furthermore, the effect of excess substrate 1a (see Table 1) as the catalyst is also considered (see Scheme S1 in the ESI†), but its catalytic activity is too weak to capture H1+ from C1 (in another 1a molecule). The scan profile shows that the energy increases sharply along with the fracture of C1–H1 bond from 1.099 to 2.599 Å (see Fig. S5 in the ESI†). The previous theoretical studies have successfully applied the scan calculation to explain the experimental results.51–53 It is clear that the [DBU + H2O]-, H2O-, 2H2O-, and 1a-catalyzed reactions are quite unfavorable for the synthesis of P when compared with the DBU-catalyzed cases.
Effects of Et3N on the reactions.
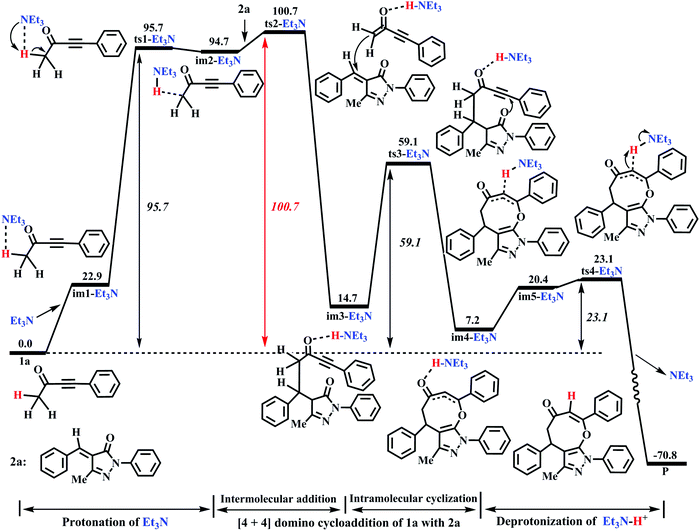
Similar to DBU, the Et3N molecule can also act as the proton-shuttle to assist H1+ transfer via the base-assisted catalysis strategy in our works. To study the roles of Et3N in detail, the energy profile of Et3N is described in Fig. 4, and the corresponding geometries are provided in Fig. 5.
 |
| | Fig. 4 Energy profile of the Et3N-promoted [4+4] domino cycloaddition of ynones (1a) with benzylidenepyrazolones (2a) to access eight-membered cyclic ethers (P) in DCM medium (unit: kJ mol−1). | |
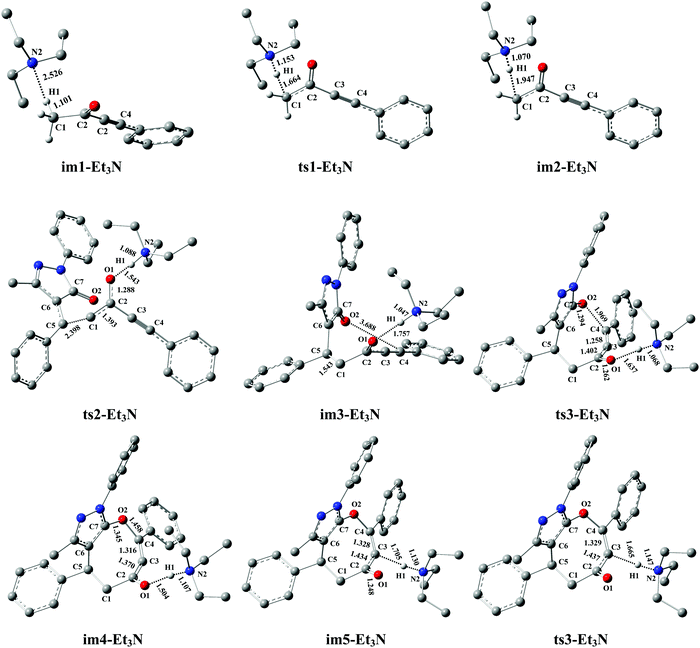 |
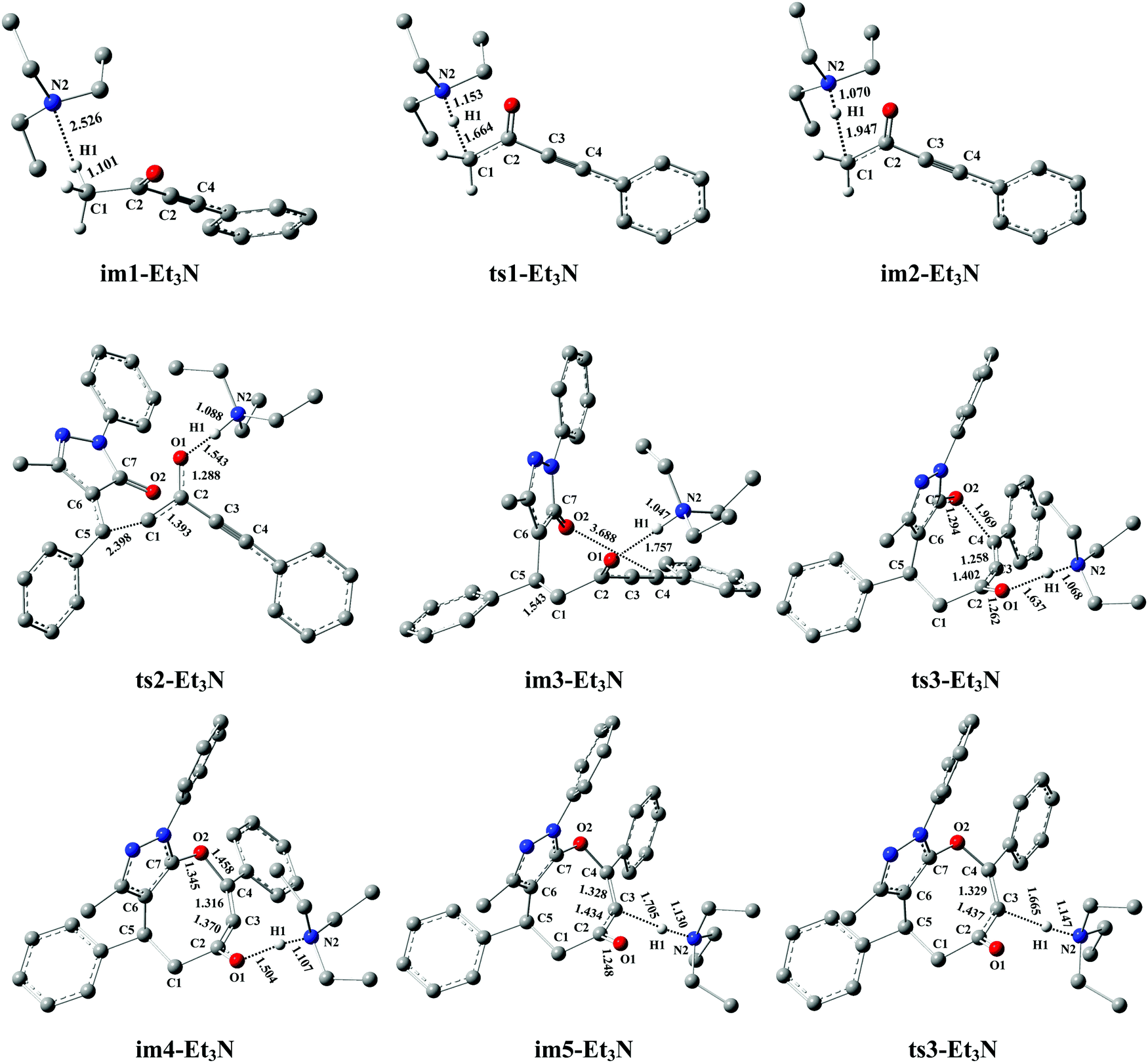
| | Fig. 5 Optimized structures for the Et3N-promoted [4+4] domino cycloaddition of ynones (1a) with benzylidenepyrazolones (2a) to access eight-membered cyclic ethers (P) in DCM medium. The H atom that does not participate in the reactions has been omitted for clarity. Bond distance is given in Å. | |
As seen from Fig. 4, the Et3N-catalyzed reaction is similar to the DBU-assisted ones (Fig. 2), including a protonation of Et3N, a [4+4] two-step domino cycloaddition of 1a with 2a, and a deprotonation of Et3N–H+. The free energy barriers of abovementioned reaction processes are 95.7, 100.7, 59.1, and 23.1 kJ mol−1, respectively (see Fig. 4). The rate-determining step changes into the process of intermolecular addition of 1a with 2a, and the rate-determining free energy barrier increases to 100.7 kJ mol−1. Clearly, 100.7 kJ mol−1 is 20.5 kJ mol−1 higher than 80.2 kJ mol−1 (the rate-determining free energy barrier) of the DBU-catalyzed reaction (see Fig. 2 and 4), which reveals that the catalytic activity of Et3N is weaker than that of DBU. Thus, the experimental results can be reasonably explained using our theoretical calculations (yield: 74% vs. 0%, see Table 1). It should be mentioned that the [Et3N + H2O]-catalyzed reaction mechanism is also considered in this paper (see Fig. S6 in the ESI†). The free energy barrier for ts1-[Et3H + H2O] is 53.8 kJ mol−1 higher than the rate-limiting energy barrier of DBU-catalyzed strategy (134.0 vs. 80.2 kJ mol−1). Clearly, the [Et3N + H2O]-catalyzed reaction is also quite unfavorable for the synthesis of cyclic ethers (P).
3.2 Understanding the catalytic differences of DBU vs. Et3N
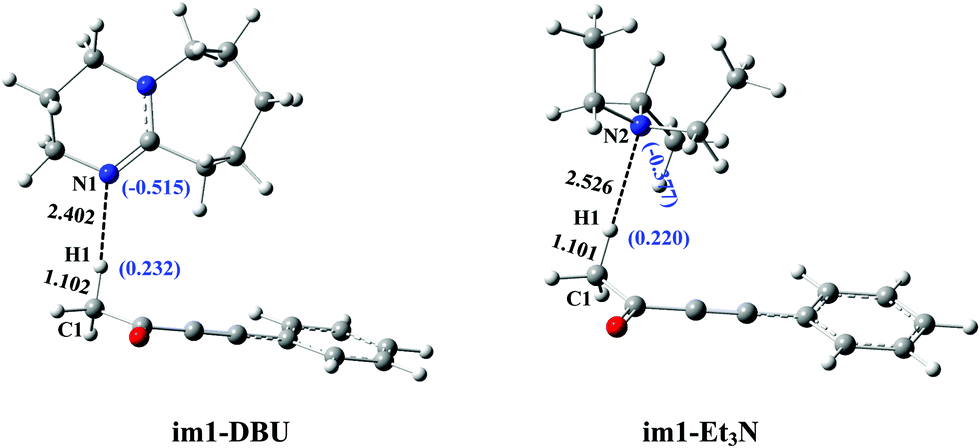
Herein, the primary origin of catalytic difference in bases (DBU vs. Et3N) for the present reactions is explored by our group. Again looking at Fig. 2 and 4, the difference in the catalytic activity of DBU and Et3N is mainly attributed to the process of protonation of DBU (and Et3N). The intermediates im1-DBU and im1-Et3N are chosen as a studied system to compare their geometric and electronic structures. From Fig. 6, the Mulliken charges of N1 (in im1-DBU) and N2 (in im1-Et3N) are −0.515 and −0.377, respectively. It is clear that N1 has a higher negative charge when compared with N2, which reveals that N1 is easier to provide the electrons than N2. The order of bases is DBU > Et3N in the electron-donating ability. In addition, the H1···N1 (in im1-DBU) and H1···N2 (in im1-Et3N) bond lengths are 2.402 and 2.526 Å, respectively, which indicate that the interaction of H1 with N1 is stronger than that of H1 with N2. Both the geometric and electronic structures of im1-DBU and im1-Et3N show that N1 as the electron-donor with the higher negative charge is more inclined to capture H1+ than N2. The similar fact can also be obtained by comparing the geometric and electronic structures of ts1-DBU and ts1-Et3N (see Fig. S8 in the ESI†). The strong electron-donating ability of DBU is beneficial to the present base-catalyzed reaction. The energy barrier of protonation of DBU is 15.5 kJ mol−1 lower than that of the protonation of Et3N (80.2 vs. 95.7 kJ mol−1). In brief, the calculated results reveal that the electron-donating ability is proved to be the primary origin that controls the catalytic activity of bases (DBU > Et3N) in the present catalytic reaction including the base-catalyzed proton-transfer process.
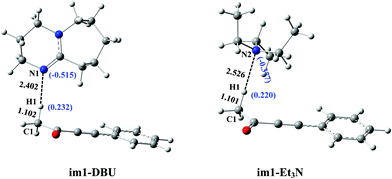 |
| | Fig. 6 The electronic and geometric structures of im1-DBU and im1-Et3N in catalytic reaction. | |
3.3 Catalytic effects of t-BuO−
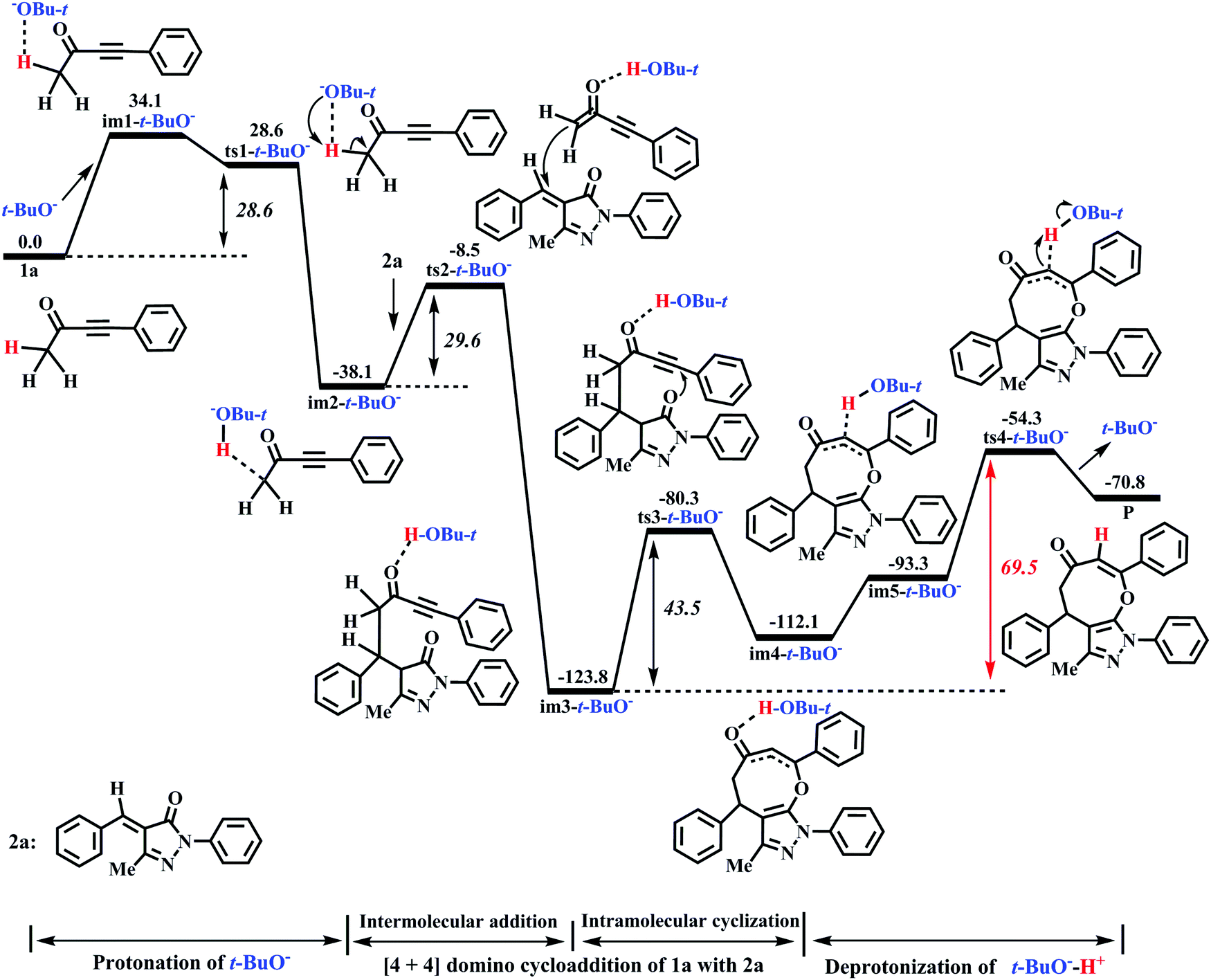
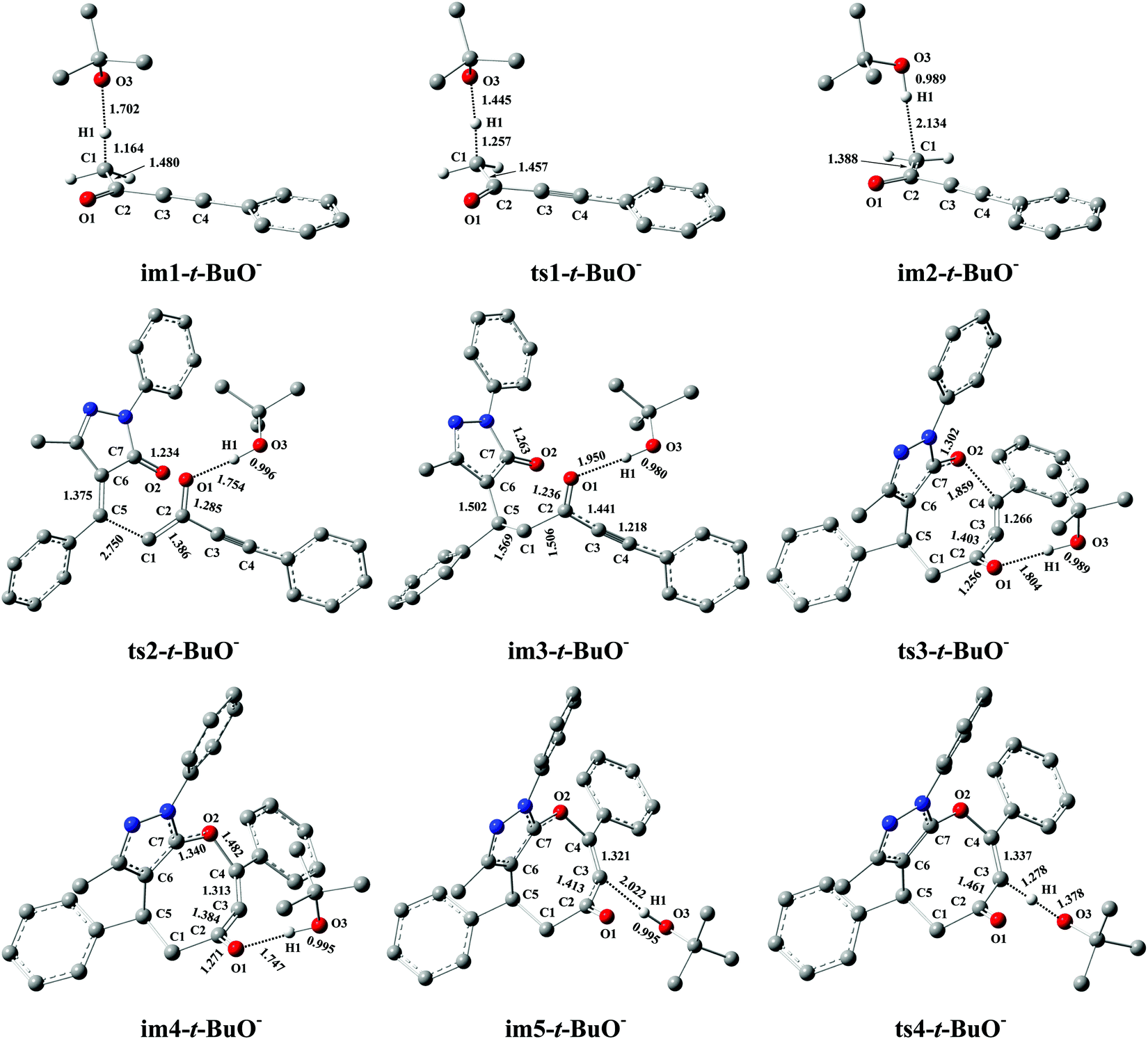
Miao and his co-workers27 stated that the eight-membered cyclic ethers (P) could be produced in moderate yield (74%) with DBU as the catalyst, which means that the yield of P can be further improved (>74%). Previous literature54–58 reported that t-BuOK (potassium tert-butylate) or t-BuONa (sodium tert-butylate) acting as the base can capture a proton from the substrate 1a to efficiently promote the catalytic reaction. Furthermore, our calculations have proved that the electron-donating ability of base is the primary origin that causes the difference in the catalytic activity of DBU and Et3N in the present base-catalyzed domino reactions, and the strong base DBU has the strong catalytic activity when compared to the weak base Et3N. Encouraged by the above research studies and our calculated results, one inference is that a good yield of P (>74%) can be obtained when t-BuOK is employed as the base because the basicity/electron-donating ability of t-BuO− is stronger than that of DBU or Et3N (see Fig. S9 and S10, ESI†). To test this speculation, the reaction mechanism of t-BuO−-assisted cycloaddition of ynones (1a) with benzylidenepyrazolones (2a) is studied in detail. The t-BuO−-assisted reaction is similar to the DBU-assisted catalytic strategy, which is also divided into a protonation of t-BuO−, a two-step intermolecular cyclization process, and a deprotonation of t-BuO−-H+, as shown in Fig. 7 (the corresponding geometric structures are described in Fig. 8). Their free energy barriers are 28.6, 29.6, 43.5, and 69.5 kJ mol−1, respectively. Clearly, the free energy barrier of the protonation process of t-BuO− is greatly reduced when t-BuOK is used as the base when compared with that of the protonation process of DBU (28.6 vs. 80.2 kJ mol−1, see Fig. 2 and 7). Importantly, the rate-limiting step of the t-BuO−-catalyzed reaction is transferred to the deprotonation process of t-BuO−-H+, and now the rate-limiting energy barrier reduces to 69.5 kJ mol−1, which is 10.7 kJ mol−1 lower than that (80.2 kJ mol−1) of the DBU-assisted strategy. Therefore, the t-BuOK as the strong base is more suitable than DBU or Et3N in helping H1+ transfer from C1 to C3. The present [4+4] cycloaddition is more likely to happen due to the assistance of t-BuO−. It is noteworthy that the relative free energy of ts1-t-BuO− is 5.5 kJ mol−1 lower than that of im1-t-BuO− (28.6 vs. 34.1 kJ mol−1). The reliability of ts1-t-BuO− is also confirmed using IRC calculations (see Fig. S12 in the ESI†). The intermediate im3-t-BuO− (−123.8 kJ mol−1) is the lowest point along with the reaction coordinate (see Fig. 7). However, the whole reaction is a dynamic equilibrium process, resulting in that im3-t-BuO− cannot exist for a long time, and the regeneration of catalyst is quite a fast process.
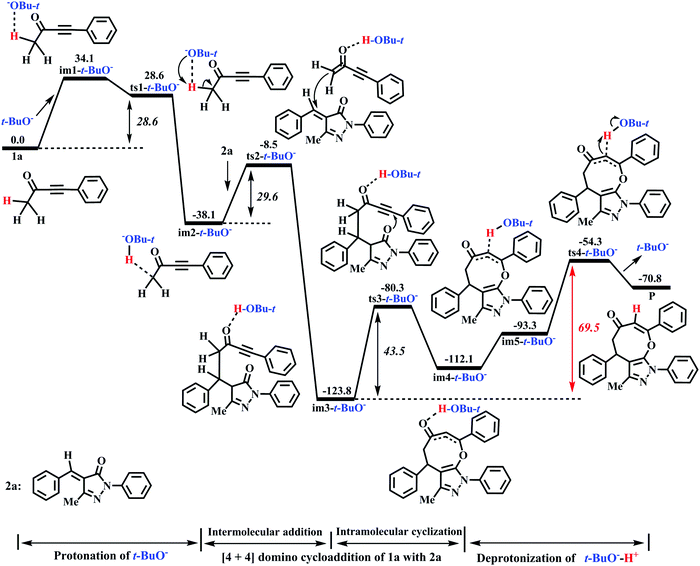 |
| | Fig. 7 Energy profile of the t-BuO−-promoted [4+4] domino cycloaddition of ynones (1a) with benzylidenepyrazolones (2a) to access eight-membered cyclic ethers (P) in DCM medium (Unit: kJ mol−1). | |
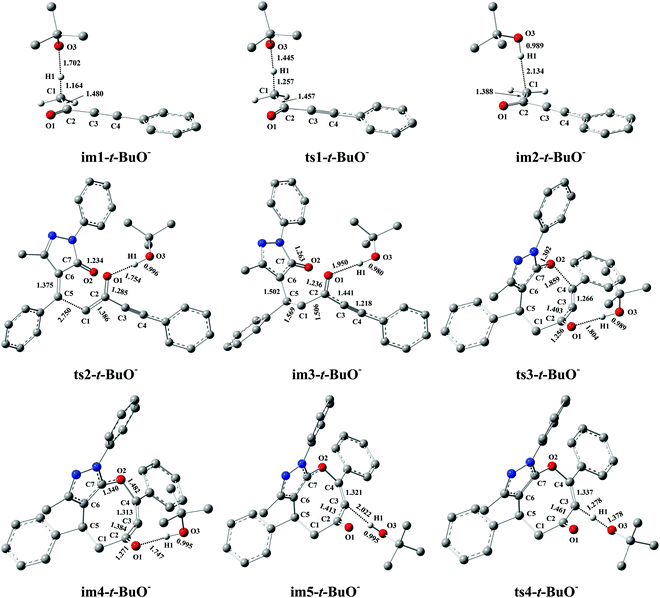 |
| | Fig. 8 Optimized structures for the t-BuO−-promoted [4+4] domino cycloaddition of ynones (1a) with benzylidenepyrazolones (2a) to access eight-membered cyclic ethers (P) in DCM medium. The H atom that does not participate in the reactions has been omitted for clarity. Bond distance is given in Å. | |
4. Conclusions
The mechanisms of base-catalyzed ynones (1a) and benzylidenepyrazolones (2a) to form the eight-membered cyclic ethers (P) are explored by means of the DFT method (PCM//M06-2x-D3/6-311++G**//B3LYP/6-31G*). The calculated results indicate that DBU and Et3N as the proton-shuttle can assist H1+ shift from C1 to C3 by the two-step proton-transfer mechanism. What's more, the calculations reveal that the difference in the catalytic activity of bases (DBU vs. Et3N) is primarily attributed to their electron-donating ability in the present base-catalyzed domino reactions. The increasing trend of electron-donating ability of bases is DBU > Et3N, which is consistent with the order of catalytic activity DBU > Et3N. When compared with Et3N, DBU exhibits a stronger electron-donating ability, resulting in that the rate-determining energy barrier of the DBU-mediated reaction is lower than that of the Et3N-catalyzed one (80.2 vs. 100.7 kJ mol−1). Therefore, the experimental phenomena (74% vs. 0%) are well explained based on our theoretical calculations. Besides, the calculations predict that t-BuO− with a stronger electron-donating ability than DBU or Et3N would further accelerate the catalytic reaction and even increase the reaction yield (>74%), because of the lower rate-determining energy barrier (69.5 kJ mol−1) of t-BuO−-assisted reaction. These interesting findings and insights are valuable in understanding the base-catalyzed synthesis of the eight-membered cyclic ethers (P) using a base as the catalyst through domino reactions, and also provide valuable guidance to design new catalysts and new catalytic systems.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21805014), the Natural Science Foundation of Chongqing of China (cstc2018jcyjAX0189), the Scientific and Technological Research Program of Chongqing Municipal Education Commission (KJQN201801418, KJQN201801424, KJ1601201), the Growth of Young Talents Program of Yangtze Normal University (2018QNRC15), and the Talent Research Start-up Project of Yangtze Normal University (2017KYQD116), and the Yangtze Normal University (2015XJXM33).
References
- S. A. Snyder, A. P. Brucks, D. S. Treitler and I. Moga, J. Am. Chem. Soc., 2012, 134, 17714–17721 CrossRef CAS.
- Z. Zhou, M. Menna, Y. Cai and Y. Guo, Chem. Rev., 2015, 115, 1543–1596 CrossRef CAS.
- P. M. Tadross, P. Bugga and B. M. Stoltz, Org. Biomol. Chem., 2011, 9, 5354–5357 RSC.
- C. V. Ramana, C. N. Reddy and R. G. Gonnade, Chem. Commun., 2008, 3151–3153 RSC.
- A. A. More and C. V. Ramana, Org. Lett., 2016, 18, 1458–1461 CrossRef CAS.
- G. Kim, T. Sohn, D. Kim and R. S. Paton, Angew. Chem., Int. Ed., 2014, 53, 272–276 CrossRef CAS.
- J. H. Cardellina, S. B. Horsley, J. Clardy, S. R. Leftow and J. Meinwald, Can. J. Chem., 1982, 60, 2675–2677 CrossRef CAS.
- M. T. Crimmins and M. T. Powell, J. Am. Chem. Soc., 2003, 125, 7592–7595 CrossRef CAS.
- S. A. Snyder, D. S. Treitler, A. P. Brucks and W. Sattler, J. Am. Chem. Soc., 2011, 133, 15898–15901 CrossRef CAS.
- H. Kim, H. Lee, J. Kim, S. Kim and D. Kim, J. Am. Chem. Soc., 2006, 128, 15851–15855 CrossRef CAS.
- H. Kim, H. Lee, D. Lee, S. Kim and D. Kim, J. Am. Chem. Soc., 2007, 129, 2269–2274 CrossRef CAS.
- B. Kim, M. Lee, M. J. Kim, H. Lee, S. Kim, D. Kim, M. Koh, S. B. Park and K. J. Shin, J. Am. Chem. Soc., 2008, 130, 16807–16811 CrossRef CAS.
- S. Baek, H. Jo, H. Kim, H. Kim, S. Kim and D. Kim, Org. Lett., 2005, 7, 75–77 CrossRef CAS.
- R. K. Boeckman, J. Zhang and M. R. Reeder, Org. Lett., 2002, 4, 3891–3894 CrossRef CAS.
- R. K. Boeckman, M. D. Shair, J. R. Vargas and L. A. Stolz, J. Org. Chem., 1993, 58, 1295–1297 CrossRef CAS.
- R. K. Boeckman and M. R. Reeder, J. Org. Chem., 1997, 62, 6456–6457 CrossRef CAS.
- A. Thakur, M. E. Facer and J. Louie, Angew. Chem., Int. Ed., 2013, 52, 12161–12165 CrossRef CAS.
- M. C. Carreno, R. D. Mazery, A. Urbano, F. Colobert and G. Solladie, Org. Lett., 2005, 7, 2039–2042 CrossRef CAS.
- N. Ortega, T. Martin and V. S. Martin, Org. Lett., 2006, 8, 871–873 CrossRef CAS.
- G. Hernandez-Torres, J. Mateo, F. Colobert, A. Urbano and M. C. Carreno, ChemistrySelect, 2016, 1, 4101–4107 CrossRef CAS.
- G. Hernandez-Torres, J. Mateo, A. Urbano and M. C. Carreno, Eur. J. Org. Chem., 2013, 6259–6262 CrossRef CAS.
- S. K. Mandal and S. C. Roy, Tetrahedron, 2007, 63, 11341–11348 CrossRef CAS.
- Z. Gao, L. Zhang, Z. Sun, H. Yu, Y. Xiao and H. Guo, Org. Biomol. Chem., 2014, 12, 5691–5697 RSC.
- K. Liu, H.-L. Teng and C.-J. Wang, Org. Lett., 2014, 16, 4508–4511 CrossRef CAS.
- W. Chen, J. Cui, Y. Zhu, X. Hu and W. Mo, J. Org. Chem., 2012, 77, 1585–1591 CrossRef CAS.
- L. Liang, E. Li, X. Dong and Y. Huang, Org. Lett., 2015, 17, 4914–4917 CrossRef CAS.
- C. Cheng, J. Y. Zhang, X. Wang and Z. W. Miao, J. Org. Chem., 2018, 83, 5450–5457 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, J. C. Burant and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
-
R. G. Parr and W. Yang, Density-functional theory of atoms and molecules, Oxford university press, 1989 Search PubMed.
- G.-Y. Ruan, Y. Zhang, Z.-H. Qi, D.-X. Ai, W. Liu and Y. Wang, Comput. Theor. Chem., 2015, 1054, 16–21 CrossRef CAS.
- J.-M. Wang, S.-B. Yu, Z.-M. Li, Q.-R. Wang and Z.-T. Li, J. Phys. Chem. A, 2015, 119, 1359–1368 CrossRef CAS.
- V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- G. Rauhut, T. Clark and T. Steinke, J. Am. Chem. Soc., 1993, 115, 9174–9181 CrossRef CAS.
- M. W. Wong, K. B. Wiberg and M. J. Frisch, J. Am. Chem. Soc., 1992, 114, 1645–1652 CrossRef CAS.
- S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS.
- T. Mineva, N. Russo and E. Sicilia, J. Comput. Chem., 1998, 19, 290–299 CrossRef CAS.
- B. Yuan, J. Chen, X. Xie, S. He, Y. Luo, X. Guo, H. Huang and R. He, New J. Chem., 2019, 43, 9265–9273 RSC.
- B. Yuan, Z. Tang, Y. Lin, G. Wang, L. Fang, X. Guo, Y. Zhao, X. Xie, J. Chen and R. He, New J. Chem., 2019, 43, 19149–19158 RSC.
- B. Yuan, X. Guo, G. Wang, H. Huang, F. Zhang, J. Xu and R. He, New J. Chem., 2019, 43, 4291–4305 RSC.
- B. Yuan, R. He, W. Shen and M. Li, Eur. J. Org. Chem., 2017, 3947–3956 CrossRef CAS.
- Y. Liu, X. Yang, L. Liu, H. Wang and S. Bi, Dalton Trans., 2015, 44, 5354–5363 RSC.
- B. Zhu, W. Guan, L.-K. Yan and Z.-M. Su, J. Am. Chem. Soc., 2016, 138, 11069–11072 CrossRef CAS.
- B. Yuan, R. He, W. Shen, W. Hu and M. Li, RSC Adv., 2016, 6, 20294–20305 RSC.
- B. Yuan, J. Chen, G. Wang, F. Zhang, L. Fang, X. Guo, H. Huang and R. He, Appl. Organomet. Chem., 2020, e5443 CAS.
- B. Yuan, R. He, W. Shen, C. Huang and M. Li, J. Org. Chem., 2015, 80, 6553–6563 CrossRef CAS.
- J. Ahmed, A. K. Swain, A. Das, R. Govindarajan, M. Bhunia and S. K. Mandal, Chem. Commun., 2019, 55, 13860–13863 RSC.
- M. Mahdavi, R. Hassanzadeh-Soureshjan, M. Saeedi, A. Ariafard, R. BabaAhmadi, P. R. Ranjbar and A. Shafiee, RSC Adv., 2015, 5, 101353–101361 RSC.
- Z. Wang, J. Zhang, J. Shia and H. Wang, RSC Adv., 2019, 9, 30139–30143 RSC.
- N. Ishida, Y. Masuda, Y. Imamura, K. Yamazaki and M. Murakami, J. Am. Chem. Soc., 2019, 141, 19611–19615 CrossRef CAS.
- T. G. Santiago, C. Urbaneja, E. Álvarez and E. Ávila, Dalton Trans., 2020, 49, 322–335 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details about the energies and Cartesian coordinates of all stationary points in the reactions. See DOI: 10.1039/d0nj04280c |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Huimin
Zhou
a,
Youqing
Yu
c,
Xiaogang
Guo
a,
Yu
Zhao
d,
Fulan
Zhang
a,
Haimei
Zhou
a,
Huisheng
Huang
*a and
Rongxing
He
*ab,
Huimin
Zhou
a,
Youqing
Yu
c,
Xiaogang
Guo
a,
Yu
Zhao
d,
Fulan
Zhang
a,
Haimei
Zhou
a,
Huisheng
Huang
*a and
Rongxing
He