
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A supramolecular cavitand for selective chromatographic separation of peptides using LC-MS/MS: a combined in silico and experimental approach†
Subbareddy
Mekapothula
,
A. D. Dinga
Wonanke
,
Matthew A.
Addicoat
 ,
John D.
Wallis
,
David J.
Boocock
and
Gareth W. V.
Cave
*
,
John D.
Wallis
,
David J.
Boocock
and
Gareth W. V.
Cave
*
School of Science and Technology, Nottingham Trent University, Clifton Campus, Nottingham, NG11 8NS, UK. E-mail: gareth.cave@ntu.ac.uk
First published on 25th November 2020
Abstract
The effective separation of a mixed peptide sample has been achieved via a computational strategy towards the bespoke design of a new silica bound supramolecular host capable of selectively interacting with different peptides via their morphology and amino acid functionalities. The experimental and in silico chromatographic behaviour of a sample of mixed peptides on a new co-pillar[4+1]arene bound silica HPLC stationary phase column was compared with RP-C18 and unfunctionalized silica (normal phase) column via liquid chromatography coupled with mass spectrometry (LC-MS/MS) and demonstrates superior separation selectivity and sensitivity for the mixed peptide solution being investigated.
1. Introduction
Proteomics is the global analysis of proteins in biological systems.1 This typically utilizes mass spectrometry (MS) to probe protein spatial distribution, response to environmental stimuli, as well as the differences between their morphology in diseased and non-diseased states. To better resolve the proteome, following on from either a top-down or bottom-up proteolytic digestion, both conventional gel and liquid chromatography-based separations are typically utilised prior to MS characterization.1Routinely, high-resolution liquid chromatography (LC) separation, coupled in-line with mass spectrometry, is the central component of a bottom-up proteomics platform due to its sensitivity and productivity.2 Over the past decade, there have been significant developments in LC separations, improved mass spectrometer acquisition speeds and mass accuracy.3,4 These combined developments have contributed to the exponential progress of proteomics over the last decade.5 However, even with such progress the most advanced proteomics workflows still cannot map an entire proteome from a single analysis. The rate limiting factor in effective proteomics is now the chromatographic separation, which still relies on generic alkyl functionalized stationary phases e.g. C18.6 Advances in modern chromatographic techniques aim towards increasing peak capacity via the utilisation of ultra-pressure liquid chromatography (UPLC) packing materials,7 such as core–shell silica,8 and instrumentational development; including multi-dimensional chromatographic techniques.9
Despite the significant progress in modern chromatographic techniques, there is still an increasing need for universal chromatographic stationary phases with the potential to separate selectively a broad range of analytes. In an attempt at designing new chromatographic stationary phases, we have incorporated supramolecular macrocycles onto the stationary phase that provide enhanced separation via non-covalent host–guest molecular recognition.10,11
In a recent study, we reported a new high yield microwave assisted synthesis of co-pillar[4+1]arene incorporating two bromo-octyl substituents.11 The co-pillar[4+1]arene selector was covalently bound to flash column grade silica, to provide a chromatographic stationary phase which demonstrated selective separation of xylene isomers and their impurities from a commercial xylene mixture using flash column chromatography. Moreover, the selective separation of these isomers was observed to match the computationally calculated binding energies.11 To demonstrate the versatility of this new class of stationary phases, we have further extended our work to explore their potential within the field of proteomics. Herein, we report a molecular binding model study that compares the host–guest interactions of a library of peptides with the dimethoxy-pillar[5]arene macrocycle. The results of these in silico studies were realised experimentally by the synthesis of a co-pillar[4+1]arene HPLC grade stationary phase, and subsequent LC-MS/MS separation and characterisation of a standard calibration mix of peptides. These assessments serve as proof of principal for further exploration of silica-bound co-pillar[4+1]arene stationary phase in the separation of biomolecules.
2. Computational methodology
The probable performance of a macrocyclic dimethoxy-pillar[5]arene as a potential chromatographic stationary phase was assessed by computing host–guest interactions for a series of peptides selected from a standard Sciex PepCal mixture (Sciex UK Limited). The sequence for the five peptides used in this study were IGNEQGVSR, SAEGLDASASLR, AVGANPEQLTR, VGNEIQYVALR and VFTPLEVDVAK.To determine the likely binding motifs and energies of the five peptides a semi-stochastic approach was employed as follows.12 Due to the size and flexibility of the peptides, geometry optimisations were possible, but frequency calculations were considered infeasible.13 To counter this, an ensemble of low energy (<50 kcal mol−1) conformers of each peptide was generated and each end of each peptide conformer was inserted semi-stochastically in the pillar[5]arene cavity, generating a minimum of 50 peptide⊂dimethoxy-pillar[5]arene complexes for each peptide. Geometries of each complex were optimised in the gas phase following which, the 20 lowest energy complexes for each conformer were selected and reoptimized in implicit solvent (acetonitrile). The peptide⊂dimethoxy-pillar[5]arene binding energies (BE) were then computed for these 20 energy minima with the formula given in eqn (1),
| BE = Ec − (Ep + Em) | (1) |
3. Experimental section
To substantiate the computational studies, the co-pillar[4+1]arene was synthesized according to our previously reported microwave assisted protocol.11 Subsequently, co-pillar[4+1]arene was attached to HPLC grade chromatographic silica particles (5 μm particle size, YMC Europe) with a mass loading of the co-pillar[4+1]arene at 19% w/w for the HPLC stationary phase; as determined by thermogravimetric analysis, ESI.† The morphological characterization of the modified silica was assessed via Scanning Electron Microscopy (SEM), ESI.† The silica functionalized co-pillar[4+1]arene stationary phase material was packed in a capillary column (1/32′′ fitting, 150 × 0.3 mm) by YMC-Europe. To condition the new co-pillar[4+1]arene stationary phase and to assess the stability of the covalent attachment to the surface of silica, the column was eluted with over 100 column volumes (water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile 1:1, 0.1% formic acid), after which time the macrocycle was shown to remain fully attached and with no change in resolution or efficiency. This is in line with traditional C18 and can be stored wet with 0.1% formic acid in water:acetonitrile (30:70).
:acetonitrile 1:1, 0.1% formic acid), after which time the macrocycle was shown to remain fully attached and with no change in resolution or efficiency. This is in line with traditional C18 and can be stored wet with 0.1% formic acid in water:acetonitrile (30:70).
Chromatographic conditions were as follows: Solvent A – water + 0.1% formic acid; Solvent B – acetonitrile + 0.1% TFA at a flow rate of 7.5 μL min−1, runtime 13 min. Gradient elution: Solvent B – 3% ramped to 8% in 5.5 minutes; to 80% in 7 min and isocratic to 11 min and re-equilibrated at 13 min.
4. Results and discussion
4.1 Computational results
The morphologies of the geometry optimised dimethoxy-pillar[5]arene cavity and the five peptides are illustrated in Fig. 1 and the binding energies for the interaction of these peptides with the macrocyclic cavities are presented in Fig. 2. The strengths of the host–guest interactions are dependent on the intramolecular interactions occurring within the peptides, which lead to their respective morphologies. It should be noted that the focus here is neither to provide answers to the thermodynamic question corresponding to the balance of the various intramolecular forces that dictate the structures of the peptides, nor to the kinetic question of the various folding routes in each peptide, which will be answered in a subsequent paper. However, it has been shown from previous studies that folding in peptides is determined by the amino acid sequence and is governed by intramolecular hydrogen bonding interactions as well as other long-range van der Waals interactions.17,18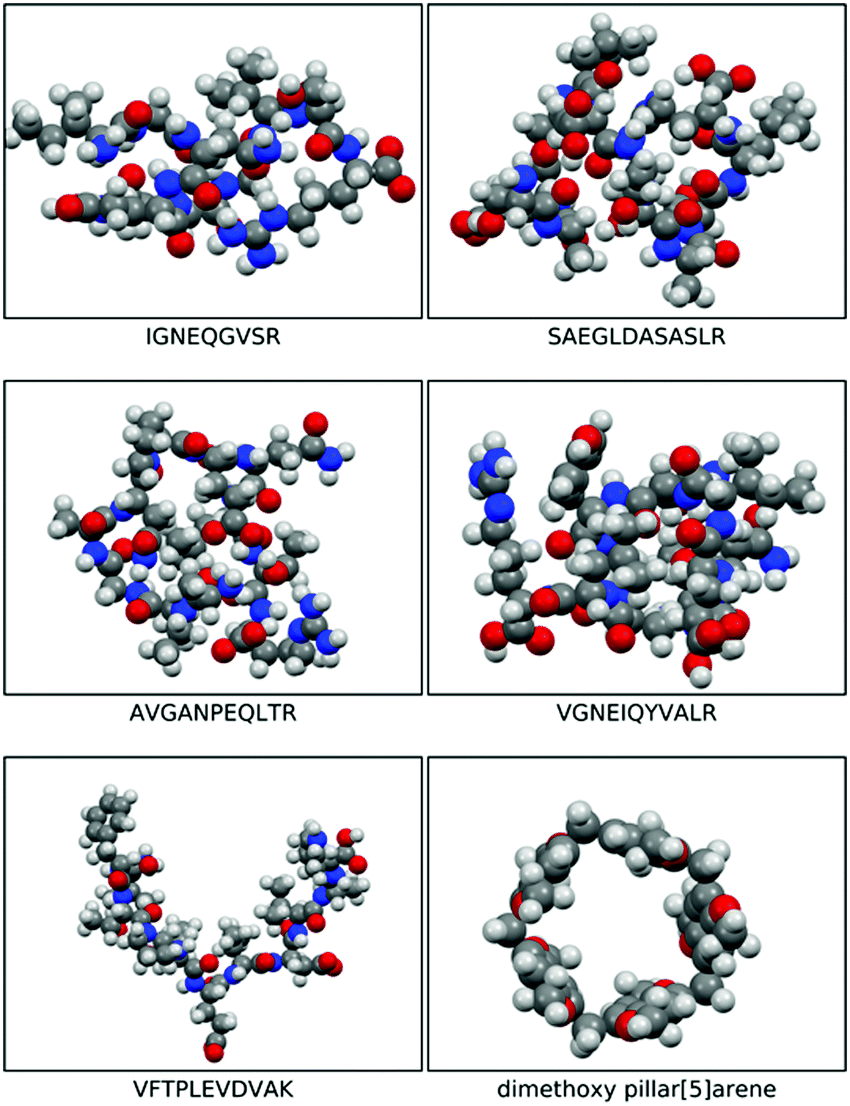 | ||
| Fig. 1 Space filled optimised structures of IGNEQGVSR, SAEGLDASASLR, AVGANPEQLTR, VGNEIQYVALR, VFTPLEVDVAK, and dimethoxy pillar[5]arene. | ||
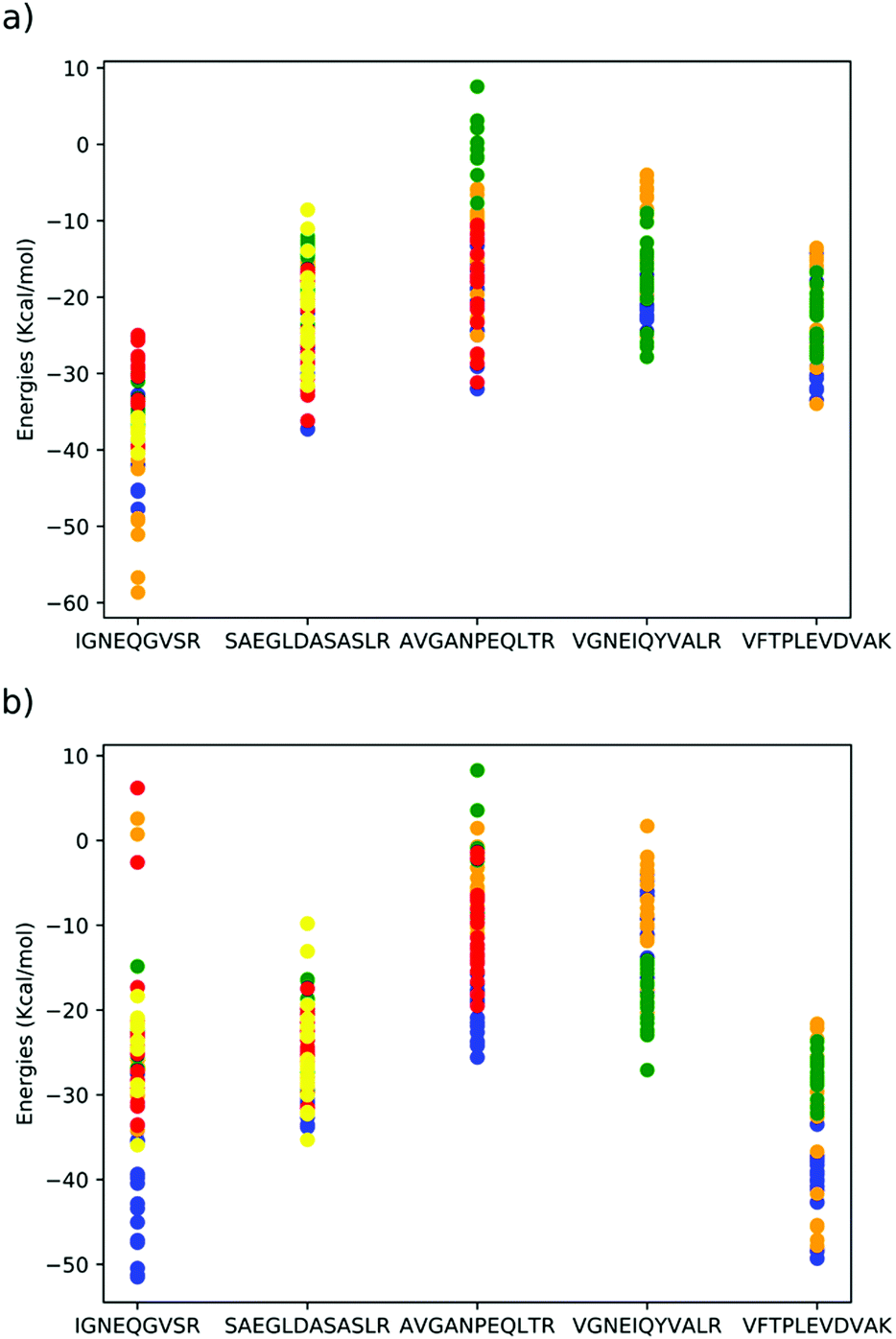 | ||
| Fig. 2 DFTB mio-1-1 binding energies of peptides⊂dimethoxypillar[5]arene in (a) gas phase, (b) implicit acetonitrile solvent. Each colour indicates a different peptide conformer, structures of each conformer are included in the ESI.† | ||
The three conformers of peptide VFTPLEVDVAK, all interact strongly with the cavity, with either the glutamic acid or valine units occupying the cavity (Fig. 3). The conformers of peptides SAEGLDASASLR and AVGANPEQLTR and VGNEIQYVALR were all predicted to have comparatively weaker interactions with the cavity. In peptide SAEGLDASASLR, the binding energies span approximately 25 kcal mol−1, the strongest interactions with the cavity are via either the leucine adjacent to the terminal arginine or via the terminal serine groups. In the case of conformers binding using the terminal serine, strong interaction is also observed with the lip of the cavity, without the peptide penetrating the cavity. In peptide AVGANPEQLTR, the strongest binding interactions were observed with either the central proline or the threonine residues. Interaction via the alanine was found to be less stable. Peptide VGNEIQYVALR, largely interacts with the cavity via its terminal valine and the adjacent glycine residues, but also strong interactions were found with the glutamic acid. In peptide IGNEQGVSR, strong binding energies were found when the glutamic acid penetrates the cavity and permits the remainder of the peptide to interact with the outside of the cavity. However, the majority of conformers of this peptide interact far more weakly with the cavity.
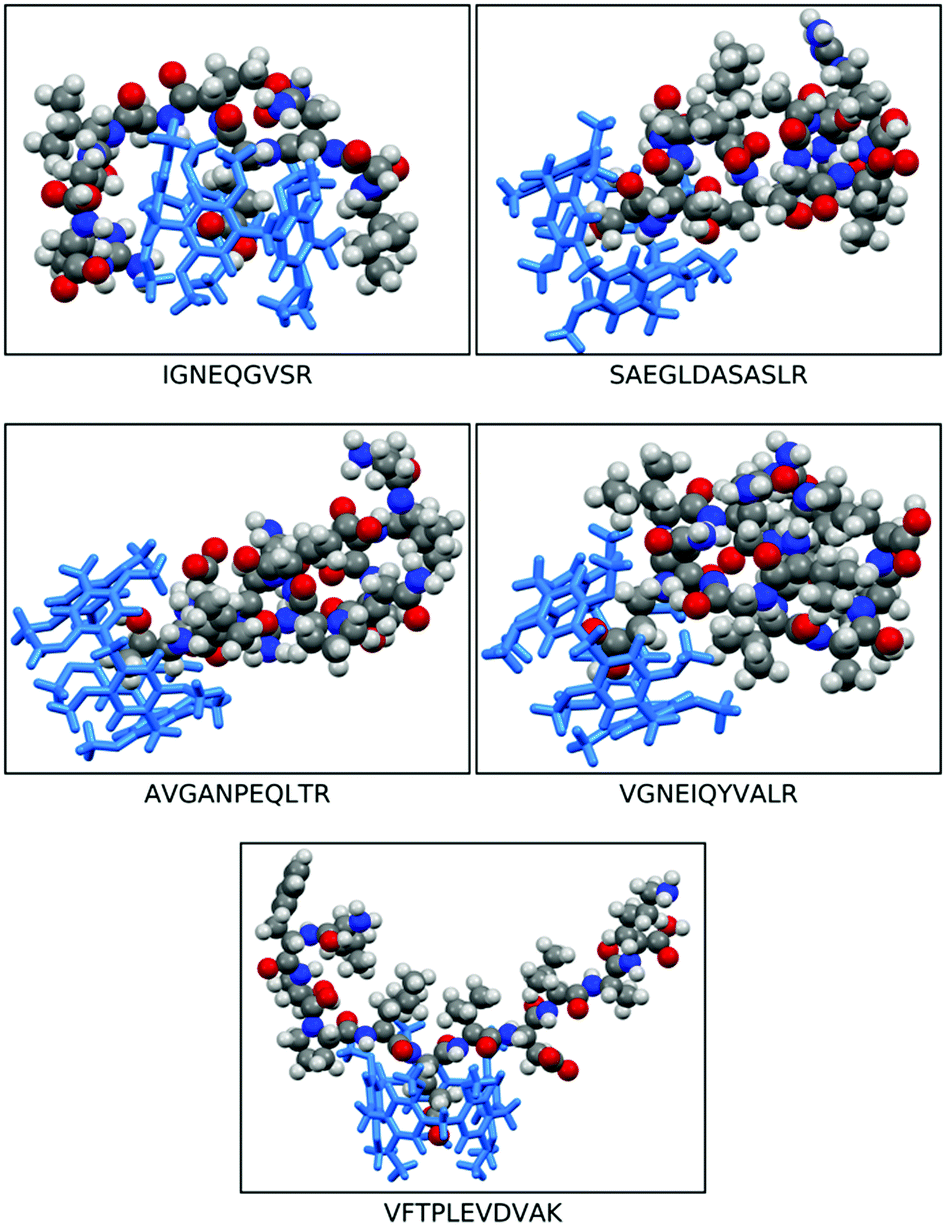 | ||
| Fig. 3 Illustration of the most stable peptide⊂dimethoxy-pillar[5]arene (DMP[5]A) interactions of: IGNEQGVSR, SAEGLDASASLR, AVGANPEQLTR, VGNEIQYVALR, and VFTPLEVDVAK, embedded in pillar[4+1]arene. The space filled molecular models represent the peptides meanwhile the sticks structures (blue) represent the DMP[5]A cavitand. | ||
4.2 Chromatographic results
The structural integrity of the modified silica with co-pillar[4+1]arene was assessed via Scanning Electron Microscopy (SEM) and demonstrated to be comparable to the starting spherical silica particles as shown in the Fig. 4. The newly functionalized silica-bound co-pillar[4+1]arene HPLC column packing materials were observed to have smooth surfaces without any structural flaws and preserved well defined cavities; necessary for the free flow of mobile phase and analytes.19 Consequently, we observed a notably low back pressure during the conditioning of the column and chromatographic evaluation process. | ||
| Fig. 4 SEM images showing silica 5 μm particles at (a) 10 and (b) 1 μm magnification and silica functionalized with co-pillar[4+1]arene stationary phase particles at (c) 10 and (d) 1 μm magnification. | ||
Following the conditioning of the new HPLC column with mobile phase (water: acetonitrile 1:1, 0.1% formic acid), the retention behaviour of the silica functionalised co-pillar[4+1]arene stationary phase was screened using reverse phase mode conditions, utilising a mobile phase gradient to evaluate the chromatographic behaviour of the five peptide mixture. Identical capillary columns (1/32′′ fitting, 150 × 0.3 mm) containing C18 (YMC Triart RP-C18, 5 μm particle size silica column) and normal phase silica (5 μm particle size silica, packed by YMC – Europe) were used as comparisons to the co-pillar[4+1]arene modified stationary phase. Normal phase silica was used to assess whether the co-pillar[4+1]arene functionalised silica was degrading or leaching from the column.
The peptide mixture (2 μL, 20 fmol μL−1) was loaded onto each of the three columns. The peptides were eluted on the columns using gradient mobile phase conditions (water:acetonitrile, 3–80% over 13 min, as shown in Fig. 5) and detected via mass spectrometry (Sciex Triple-TOFTM 5600). All five peptides eluted between 9.28 and 10.31 min on the commercial reverse phase C18 column. However, only the IGNEQGVSR peptide was resolved, while the SAEGLDASASLR, AVGANPEQLTR, VGNEIQYVALR and VFTPLEVDVAK peptides eluted together and could not be resolved. On the commercially packed normal phase column, the VFTPLEVDVAK peptide was fully resolved but the IGNEQGVSR, AVGANPEQLTR, SAEGLDASASLR and VGNEIQYVALR peptides eluted together (2.6–4.1 min). However, when the same peptide standard was eluted on the new supramolecular column all the peptides were individually resolved (IGNEQGVSR 4.0–4.4 min, SAEGLDASASLR 4.8–5.4 min, AVGANPEQLTR 5.4–5.8 min, VGNEIQYVALR 5.7–6.3 min and VFTPLEVDVAK 7.7–8.2 min).
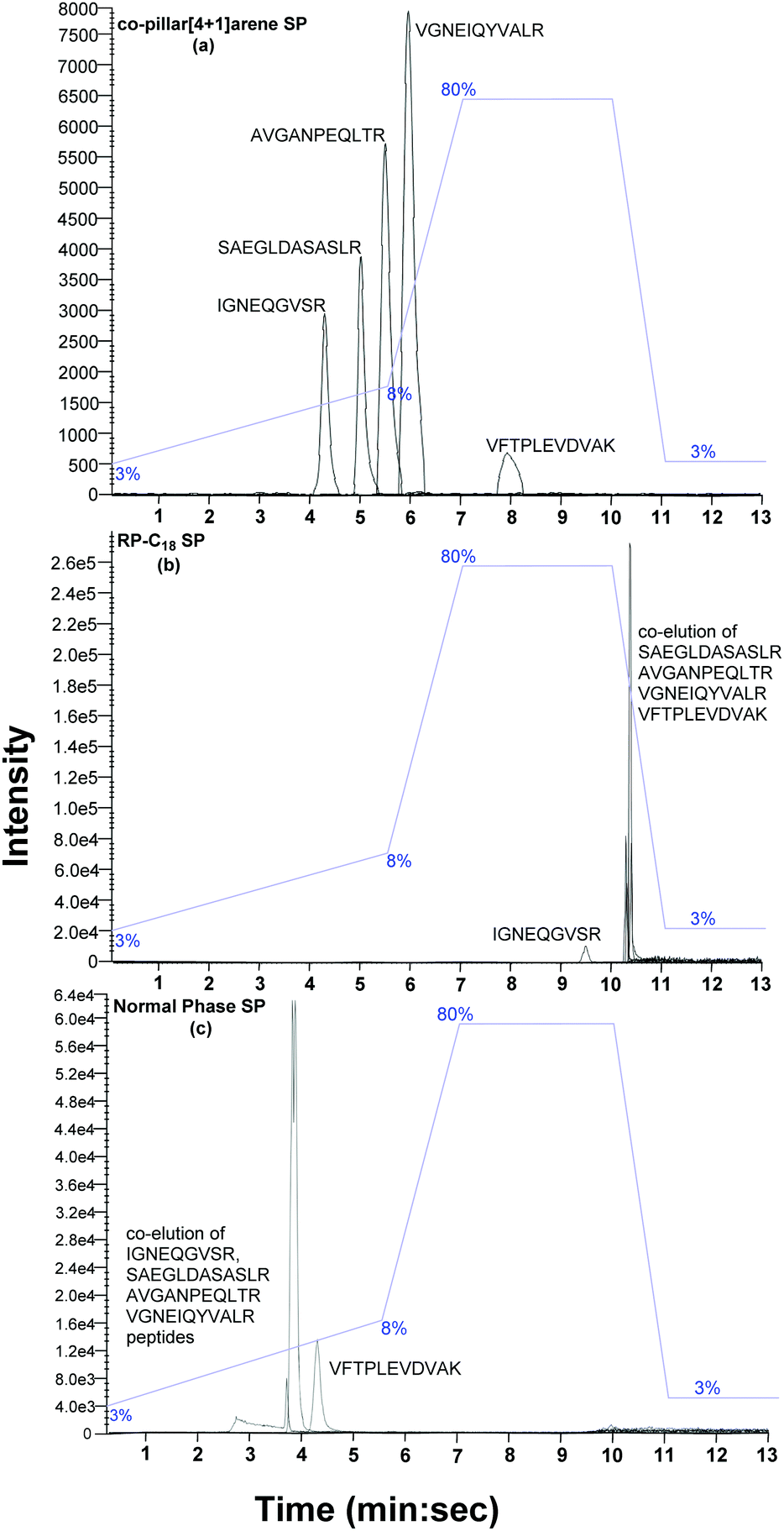 | ||
| Fig. 5 LC-MS/MS chromatographic separation of mixed peptide standard (IGNEQGVSR, SAEGLDASASLR, AVGANPEQLTR, VGNEIQYVALR and VFTPLEVDVAK) on (a) silica bound co-pillar[4+1]arene; (b) reverse phase-C18 silica; and (c) normal phase silica stationary phases. Flow rate: 7.5 μL min−1, column dimensions: 12 nm S–5 μm 150 × 0.3 mm with 1/32′′ fitting, column temperature: 30 °C, mobile phase: solvent A – water + 0.1% formic acid and solvent B – acetonitrile + 0.1% formic acid. The column dead volume is 1.92 min. | ||
Under ideal conditions, a chromatographic peak will be symmetrical and follow a Gaussian curve distribution. However, experimental peaks are more asymmetric in shape, often exhibiting some degree of tailing. Peak tailing can therefore be characterised by the tailing factor (TF).20 A value of TF = 1.00 corresponds to perfectly symmetrical peaks. An increase in peak tailing, therefore results in poorer separation. Major peak tailing can be significantly detrimental for both separation and quantification. The data described above demonstrates that the new silica bound co-pillar[4+1]arene column meets the experimental requirement for routine separation for all peaks (TF < 2). Although a resolution, Rs > 2 is desirable in chromatographic separation, this is not practical for larger peak areas. Peptides IGNEQGVSR and VFTPLEVDVAK were significantly resolved, with a resolution of 6.09, on the supramolecular column. By comparison, the C18 reverse phase silica column was only capable of a resolution of 5.66 (Fig. 5) under the same conditions. The peak resolutions of SAEGLDASASLR-AVGANPEQLTR, SAEGLDASASLR-VGNEIQYVAL and AVGANPEQLTR-VGNEIQYVALR peptides on the supramolecular column were 1.20, 2.24 and 1.26 respectively, compared to 0.29, 0.88 and 1.32 from the C18 column. Therefore, the silica bound co-pillar[4+1]arene column shows better resolution, compared to C18 column, across the standard peptide mix. In theory there is no correlation between % loading and separation resolution between 5% to 18% w/w, however, this will be subject of a further studies. The analogous flash column chromatographic stationary phase was previously observed to be effective in pure organic solvents (ethyl acetate and methanol), while resolving xylene isomers and their impurities via flash column chromatography.12
4.3 Synthetic relevance
The elution order of the peptides fits broadly with the computational binding energy predictions. For example, all conformers calculated of VFTPLEVDVAK, bind strongly to the cavity with binding energies between −20 and −50 kcal mol−1. This prediction of consistently strong interaction between the peptide and cavity was supported experimentally as this peptide was observed to possess the highest retention time of 7.93 minutes. In the strongest binding modes (blue and orange dots in Fig. 2), the glutamic acid residue penetrates the cavity fully and the neighbouring leucine and valine residues form weak hydrogen bonds with the lip of the cavity, whereas in the weaker binding modes (green dots in Fig. 2), it is the smaller valine residue penetrating the cavity, which is responsible for the weaker interaction. Details of the most important long-range intermolecular interactions, broken down by interaction type21 of all the studied peptides are presented via zenodo in the ESI.†The retention times of AVGANPEQLTR and VGNEIQYVALR also correlate well with the range of binding energies for these peptides. Peptide SAEGLDASASLR, is predicted to bind more strongly to the cavity (−10 to −35 kcal mol−1), the conformer structures insert either the terminal serine (yellow dots in Fig. 2) or the arginine-adjacent leucine residue (orange, blue and red dots in Fig. 2). However, several optimizations (green dots in Fig. 2) resulted in the peptide not penetrating the cavity at all, suggesting that while strong binding is possible between this peptide and cavity, it is less likely to be observed, which aligns with the relatively low retention time of this peptide. The peptide with the lowest retention time, IGNEQGVSR has one binding mode in common with VFTPLEVDVAK, where the glutamic acid residue is inserted into the cavity (blue dots in Fig. 2) and several hydrogen bonds are formed between the remainder of the peptide and the lip of the cavity, however, all other calculated conformers of this peptide interact weakly with the cavity using either the terminal arginine (green dots) or isoleucine residues (red, orange and yellow dots in Fig. 2). Given the specificity of the conformer required to insert the glutamic acid residue and the number of peptide–cavity interactions required to achieve the strong binding energy means this mode of interaction is unlikely to contribute to the experimentally observed weak retention.
In order to successfully conduct binding studies in the solution phase, both the upper and lower rims of the pillar[5]arene are commonly modified to enable solubilization of both the host and guest moieties.22 Unfortunately, all attempts to find a suitable solvent system that solubilized both the individual amino acids and the 1,4-dimethoxypillar[5]arene macrocycle were unsuccessful. However, general amino acid binding selectivity studies, with a modified pillar[5]arene host, will be subject to further studies.
5. Conclusions
In conclusion, computational supramolecular host–guest binding energies have been successfully used, for the first time, to design and predict a new class of chromatographic stationary phases towards the efficient and effective separation of peptides known to be of high significance in proteomic studies. This work will facilitate sample preparation prior to mass spectrometric analysis and lead to bespoke chromatographic systems that can be designed in silico to optimise the separation of physiological salts and peptides directly from trypsin digested proteins.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We would like to acknowledge YMC Europe for packing the silica bound co-pillar[4+1]arene stationary phase HPLC column. M. A. A. acknowledges HPC resources on THOMAS via membership of the UK's HEC Materials Chemistry Consortium, funded by EPSRC (EP/P020194). The authors wish to acknowledge the support of Dr Nitin Seetohul.Notes and references
- (a) K. Hixson, D. Lopez-Ferrer, E. Robinson and L. Paša-Tolić, Encycl. Spectrosc. Spectrom., 2017, 766–773 CrossRef CAS; (b) N. L. Anderson and N. G. Anderson, Electrophoresis, 1998, 19, 1853–1861 CrossRef CAS.
- Z. Fang, Y. Z. Baghdady, K. A. Schug and S. M. Chowdhury, J. Proteome Res., 2019, 18, 1916–1925 CrossRef CAS.
- (a) Y. Yang, H. Chen, M. A. Beckner, P. Xiang, J. J. Lu, C. Cao and S. Liu, Anal. Chem., 2018, 90, 10676–10680 CrossRef CAS; (b) P. Yu, S. Petzoldt, M. Wilhelm, D. P. Zolg, R. Zheng, X. Sun, X. Liu, G. Schneider, A. Huhmer and B. Kuster, Anal. Chem., 2017, 89, 8884–8891 CrossRef CAS; (c) Y. Wang and S. V. Olesik, Anal. Chem., 2018, 91, 935–942 CrossRef.
- (a) Q. Hu, R. J. Noll, H. Li, A. Makarov, M. Hardman and R. Graham Cooks, J. Mass Spectrom., 2005, 40, 430–443 CrossRef CAS; (b) G. L. Andrews, B. L. Simons, J. B. Young, A. M. Hawkridge and D. C. Muddiman, Anal. Chem., 2011, 83, 5442–5446 CrossRef CAS; (c) N. M. Riley, C. Mullen, C. R. Weisbrod, S. Sharma, M. W. Senko, V. Zabrouskov, M. S. Westphall, J. E. Syka and J. J. Coon, J. Am. Soc. Mass Spectrom., 2016, 27, 520–531 CrossRef CAS.
- M. Kim and A. Pandey, Proteomics, 2012, 12, 530–542 CrossRef CAS.
- K. M. Grinias, J. M. Godinho, E. G. Franklin, J. T. Stobaugh and J. W. Jorgenson, J. Chromatogr. A, 2016, 1469, 60–67 CrossRef CAS.
- (a) R. M. Wimalasinghe, Z. S. Breitbach, J. T. Lee and D. W. Armstrong, Anal. Bioanal. Chem., 2017, 409, 2437–2447 CrossRef CAS; (b) P. Tsai, T. Sung, C. Chong, S. Huang and S. Chen, Anal. Methods, 2018, 10, 4756–4764 RSC; (c) A. F. Gargano, L. S. Roca, R. T. Fellers, M. Bocxe, E. Domínguez-Vega and G. W. Somsen, Anal. Chem., 2018, 90, 6601–6609 CrossRef CAS.
- H. S. Berg, K. E. Seterdal, T. Smetop, R. Rozenvalds, O. K. Brandtzaeg, T. Vehus, E. Lundanes and S. R. Wilson, J. Chromatogr. A, 2017, 1498, 111–119 CrossRef CAS.
- A. L. Capriotti, C. Cavaliere, A. Cavazzini, F. Gasparrini, G. Pierri, S. Piovesana and A. Laganà, J. Chromatogr. A, 2017, 1498, 176–182 CrossRef CAS.
- (a) M. Śliwka-Kaszyńska and M. Ślebioda, J. Sep. Sci., 2014, 37, 543–550 CrossRef; (b) K. Hu, J. Liu, C. Tang, C. Wang, A. Yu, F. Wen, W. Zhao, B. Ye, Y. Wu and S. Zhang, J. Sep. Sci., 2012, 35, 239–247 CrossRef CAS; (c) W. Nowik, M. B. De Bellaistre, A. Tchapla and S. Héron, J. Chromatogr. A, 2011, 1218, 3636–3647 CrossRef CAS.
- S. Mekapothula, M. A. Addicoat, D. J. Boocock, P. Cragg, J. D. Wallis and G. W. V. Cave, Chem. Commun., 2020, 56, 1792–1794 RSC.
- M. A. Addicoat and G. F. Metha, J. Comput. Chem., 2009, 30, 57–64 CrossRef CAS.
- J. Kleinjung and F. Fraternali, Curr. Opin. Struct. Biol., 2014, 25, 126–134 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- C. Schönbeck, H. Li, B.-H. Han and B. W. Laursen, J. Phys. Chem. B, 2015, 119, 6711–6720 CrossRef.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 58, 7260 CrossRef.
- S. S. Sung, Protein Sci., 2015, 24, 1383–1388 CrossRef CAS.
- K. A. Dill, S. B. Ozkan, M. S. Shell and T. Weikl, Annu. Rev. Biophys., 2008, 37, 289–316 CrossRef CAS.
- (a) J. DeStefano, T. Langlois and J. Kirkland, J. Chromatogr. Sci., 2008, 46, 254–260 CrossRef CAS; (b) T. Hara, H. Kobayashi, T. Ikegami, K. Nakanishi and N. Tanaka, Anal. Chem., 2006, 78, 7632–7642 CrossRef CAS; (c) J. Kirkland, Anal. Chem., 1992, 64, 1239–1245 CrossRef CAS; (d) R. E. Majors, Anal. Chem., 1972, 44, 1722–1726 CrossRef CAS.
- (a) About Resolution, Part 1: SHIMADZU (Shimadzu Corporation), https://www.shimadzu.com/an/hplc/support/lib/lctalk/resol-1.html, accessed May 2020; (b) How Is Peak Asymmetry Caluclated in Analyst® Software Quantitate Mode? https://sciex.com/support/knowledge-base-articles/how-peak-asymmetry-value-caluclated-in-analyst-quant-mode, accessed May 2020; (c) “Peak Tailing in HPLC”, https://www.crawfordscientific.com/technical/chromatography-blog/hplc-chromatography-tips/hplc-troubleshooting/peak-tailing-in-hplc, accessed may 2020.
- (a) N. Gospodinova and E. Tomsik, Prog. Polym. Sci., 2015, 42, 33–47 CrossRef; (b) J. S. Mugride, G. Szigethy, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 16256–16264 CrossRef.
- (a) T. Ogoshi, M. Hashizume, T. Yamagishi and Y. Nakamoto, Chem. Commun., 2010, 46, 3708–3710 RSC; (b) N. L. Strutt, R. S. Forgan, J. M. Spruell, Y. Y. Botros and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 5668–5671 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The interatomic distances for all the 20 complexes from each peptide conformer can be downloaded from http://doi.org/10.5281/zenodo.3995081. See DOI: 10.1039/d0nj03555f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |