
Transition-metal ion-mediated morphological transformation of pyridine-based peptide nanostructures†
Narendra
Singh
 a,
Ramesh
Singh
b,
Swati
Sharma
a,
Khushboo
Kesharwani
b,
Khashti Ballabh
Joshi
*b and
Sandeep
Verma
*a
a,
Ramesh
Singh
b,
Swati
Sharma
a,
Khushboo
Kesharwani
b,
Khashti Ballabh
Joshi
*b and
Sandeep
Verma
*a
aDepartment of chemistry, Indian Institute of Technology, Kanpur-208016, India. E-mail: sverma@iitk.ac.in
bDepartment of Chemistry, School of Chemical Science and Technology, Dr HarisinghGour Central University, Sagar, MP 470003, India. E-mail: kbjoshi77@gmail.com
First published on 10th November 2020
Abstract
Inspired by natural metallopeptides, we have rationally designed two pyridine-conjugated short peptides. These two peptide conjugates formed a pair of constitutional isomers that helped us describe their structure–function relationship. Both the isomers consisted of an equal number of aromatic amino acid residues, but shuffling was observed in the position of two key amino acids, viz; tyrosine and phenylalanine, which brought a remarkable change in their self-assembling behavior. The presence of specific functional groups and chemical diversity in both conjugates made them very active towards metal coordination. Both the constitutional isomers adopted different pathways of self-assembly, which could be further controlled or transformed by the use of transition metal ions. Interestingly, it was observed that the metal ions could precisely control the morphology of these metallopeptide nanostructures and make them more stable. Therefore, such artificial metallopeptides possess remarkable advantages over the natural counterparts primarily due to their tailor-made chemical structures.
Smartly designed short-peptide conjugates with appropriate functionality help us understand the structures and functions of several biologically relevant metal-binding proteins, which may unlock ways to develop promising materials, devices, and technologies of potential biotechnological interest.1–7 For biological functioning, approximately one-third of biomolecules (proteins and enzymes) are activated by appropriate co-factors i.e. metal ions5,6 because of their distinctive periodic properties, including the presence of various coordinating sites, electron-accepting ability, specific ligand affinity, positive charge and different electronic structures.6–9 Short peptides equipped with a wide range of functional groups or metal chelating sites exhibit variable interactions with metal ions.1,2,8–12 Such metal-peptide composite biomaterials can mimic the functioning of natural metallopeptides, which possess various metal-binding sites, and therefore, are highly attractive systems for foreseeable future investigations.1,5,8,13,14
Rationally designed short peptide conjugates along with small covalent linkers increase the ability to mimic the hierarchical organization of natural proteins.8,15–17 The conjugation of a flexible or rigid scaffold to a short peptide is one of the useful methods that provide thermodynamic and kinetic control during the self-assembly process.16–18 By applying natural principles, several advances have already been made in the construction of nanostructures, including the self-assembly of nanofibrils, nanotubes, nanorods, nanorings, nanosheets, nanovesicles and helical ribbons, to name a few.20–26
The formation of a specific secondary or three-dimensional structure primarily depends on the design of the basic skeleton of the molecular building blocks that self-assemble into well-defined and stable structures through covalent/non-covalent bonds.20,25,27
The literature and our previous research work on synthesizing meaningful nanostructures using peptides for various applications28–32 demonstrate that the rational insertion of C2 symmetric flexible linkers, such as 1,2-diaminoethane,33 1,4-diaminobutane34 and 6-aminocaproic acid,35 has been extensively utilized to increase the ordering and transformation of non-fibrillating short peptides into fibrillating peptides.16,33–35 Further, the conjugation of a C3 symmetric scaffold, namely tris(2-aminoethyl)amine (TREN), with the dipeptide Trp-Trp, biotinylated ditryptophan peptide and others36–42 has been exploited to form fascinating and highly precise nanocages or rings,41 nanotorus37 and gold nanobangle36 structures. Other than C2 symmetric flexible linkers, 2,6-pyridine dicarboxylic acid and picolinic acid are known for their structural rigidity and therefore, can control the symmetry of the conjugated peptide fragments.16,17 Such pre-defined rigidity in peptide fragments symmetrically appended on both sides can be used for numerous bio(nano)technological applications.17 Therefore, in this milieu, this aromatic scaffold was conjugated to peptide conjugates of aromatic amino acids. In the first report, a ditryptophan peptide was linked and used to rescue clinically relevant bacterial cells from mercury toxicity.43 In another report, the di-phenylalanine peptide was used to control coinage metal-mediated self-assembly modification and could be used as a novel metallopeptide material to exploit interfacial metal ion interactions.19
It can be predicted that a rigid linker minimizes the conformational entropy changes, which results in the formation of highly organized supramolecular structures. It is well-known that 2,6-pyridyl di-substituted type rigid linkers, such as 2,6-pyridine-dicarboxamide, have frequently been utilized to produce a helical conformation or a curvature in supramolecular structures.17,44–46
Therefore, inspired by the reported literature and based on our past research, we have rationally synthesized two isomeric compounds 1 and 2 (Scheme 1). The tripeptide conjugates Tyr-Phe-Phe(N → C) and Phe-Phe-Tyr(N → C) were synthesized by solution-phase peptide synthesis47,48 (ESI) followed by conjugation to 2,6-pyridine dicarboxylic acid, forming conjugates 1 and 2, respectively (Scheme 1). The resulting compounds were purified by HPLC, followed by NMR and mass spectral characterization (ESI). The insertion of Tyr in the previously designed pyridine-diphenylalanine (Phe–Phe)19 conjugate increased the aromatic π–π interactions, H-bonding through the sidechain and the ability to interact with specific metal ions due to the presence of a phenolic group.
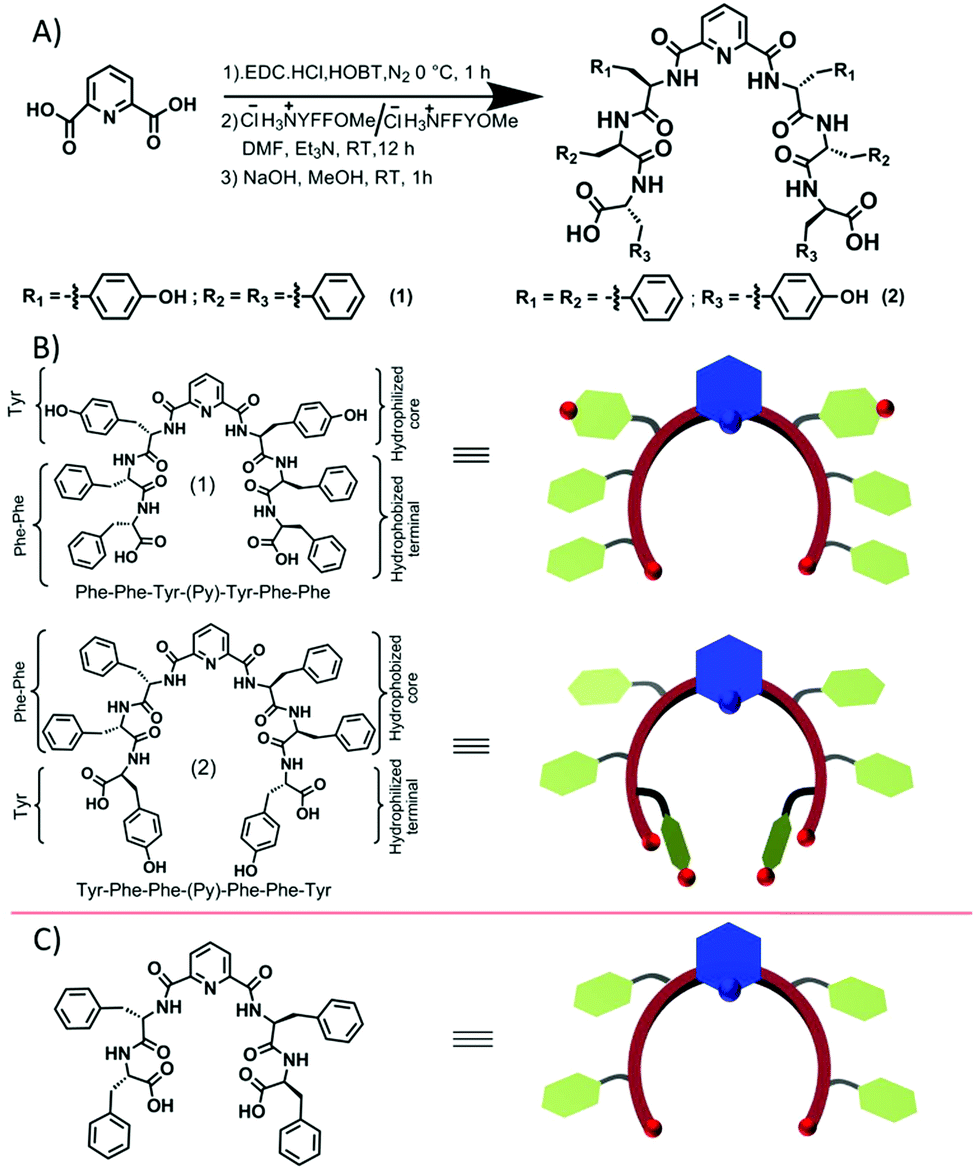 | ||
| Scheme 1 (A) The representative synthesis scheme of the designed isomeric short peptide conjugates 1 and 2; (B) structures of conjugates 1 and 2 and the corresponding predicted curvatures, which are supported by (C) the reported molecule and its corresponding cartoon.19 | ||
These isomeric compounds 1 and 2 differ in the peptide sequence only by the position of the tyrosine residue. Compound 1 has the tyrosine residue close to the pyridyl-scaffold, whereas compound 2 has the tyrosine residue far from the scaffold i.e. at the terminal position (Scheme 1B bottom). Such a molecular level change probably leads to a substantial conformational difference between these two peptides and hence their self-assembly.
We performed the structural analysis of both compounds by using spectroscopy techniques. 1H-NMR spectra were recorded for both the compounds (Fig. 1D). We observed that the aromatic and amide proton regions were the most sensitive; therefore, any degree of structural changes between 1 and 2 can easily be distinguished by the difference in the chemical shift of these protons. Interestingly, it was observed that the signal for the ortho protons of the Tyr side chain in peptide 1 was shifted downfield compared to that of peptide 2 (Fig. 1D, blue box). The nature and splitting patterns of the Tyr and Phe aromatic protons from 6.85 to 7.25 ppm (marked by blue arrows) were found different for peptides 1 and 2, revealing that peptide 2 existed in a more symmetric conformation compared with 1. Further, the signal corresponding to the pyridyl proton (C4–H) at 8.05 ppm was broader for 1 compared with that of 2 (Fig. 1D, black circle),49,50 depicting the presence of an overcrowded environment around the pyridine ring because the bulky Tyr group is directly attached with the pyridine ring in 1. The signal for other pyridyl protons (C3–H and C5–H) also showed a slight difference in chemical shift value (Fig. 1D, black box).49,50 The conformational differences between the aromatic moieties in 1 and 2 were also confirmed by the Fourier transform Infrared (FT-IR) spectral analysis. A clear difference in the out-of-plane (Ar C–H) vibration band was observed (Fig. 1C) in the fingerprint region (1400 cm−1 to 600 cm−1). The FT-IR spectra B for the free and H-bonded N–H vibrations in the amide regions A and also showed clear differences in the nature of H-bonding interactions (Fig. 1A). On the other hand, the amide regions I and II, which disclose information about the secondary structure contribution and differences in the amide backbone, showed very similar band positions at 1721, 1650 and 1521 cm−1 for 1 and 1720, 1647, 1519 cm−1 for 2 due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carboxylic acid bond and amide I and II, respectively, which support a similar kind of participation from both molecules. Further deconvolution of the IR spectra in the amide band I region revealed the quantitative analysis of the secondary structural contribution in both the peptides (Fig. 2). It was found that peptide 1 showed 100% contribution from the α-helical conformation (Fig. 2B), while in peptide 2, it was distributed between two structures, i.e. α-helix-64.45% and β-sheet-34.55% (Fig. 2D).26,27
O carboxylic acid bond and amide I and II, respectively, which support a similar kind of participation from both molecules. Further deconvolution of the IR spectra in the amide band I region revealed the quantitative analysis of the secondary structural contribution in both the peptides (Fig. 2). It was found that peptide 1 showed 100% contribution from the α-helical conformation (Fig. 2B), while in peptide 2, it was distributed between two structures, i.e. α-helix-64.45% and β-sheet-34.55% (Fig. 2D).26,27
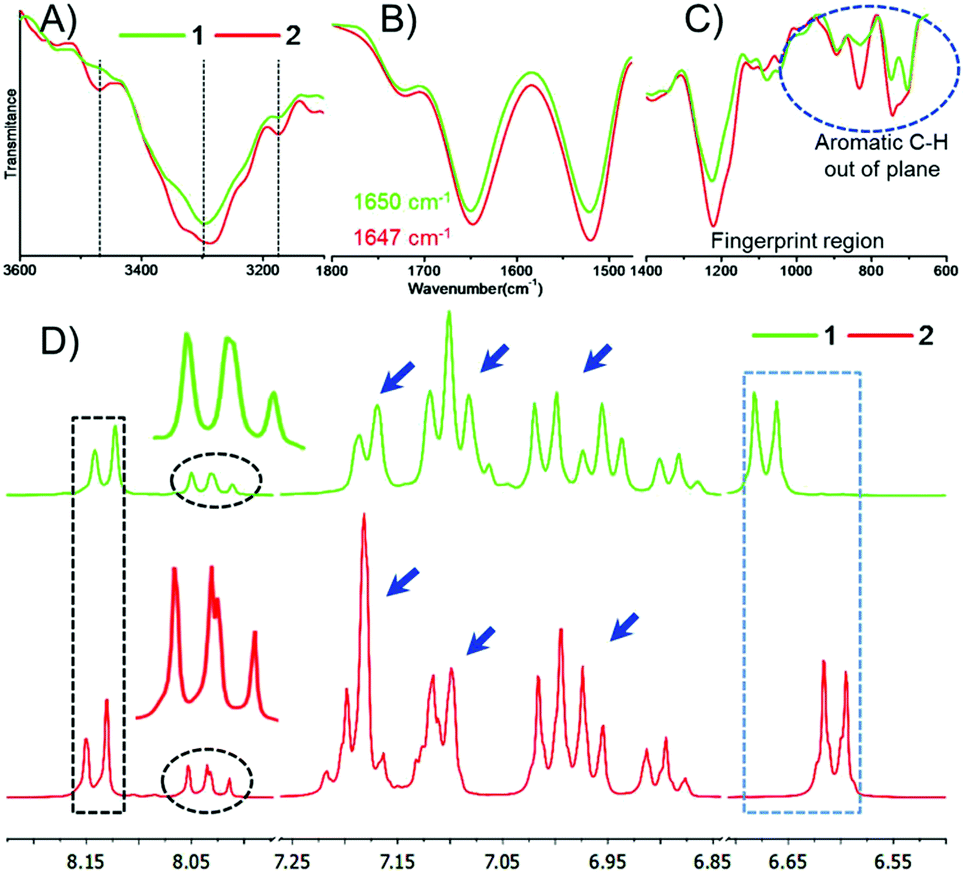 | ||
| Fig. 1 Top: FT-IR spectra of compounds 1 and 2. (A) Amide A and B regions ranging from 3600–3000 cm−1, representing N–H bond stretching; (B) amide I and II regions ranging from 1800 to 1500 cm−1; (C) fingerprint region ranging from 1400 to 600 cm−1. Bottom: (D) The 1H-NMR spectra of compounds 1 and 2 in CD3OD. | ||
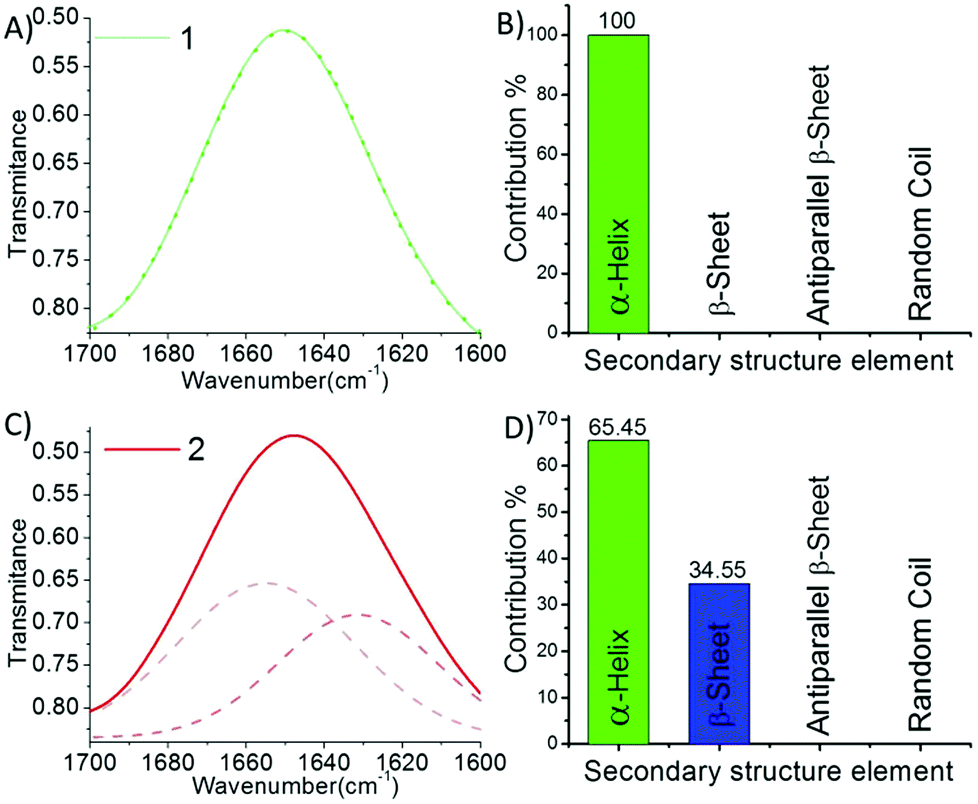 | ||
| Fig. 2 (A) The FT-IR spectrum of 1 (solid line) and its deconvolution (dashed line), and (B) the corresponding secondary structure contributions to self-assembly. (C) The FT-IR spectrum of 2 (solid line) and its deconvolution (dashed line), and (D) the corresponding secondary structure contributions to self-assembly. Deconvolution was done by fitting multiple Gaussian peaks in the amide I region ranging from 1600 to 1700 cm−1. | ||
Fresh solutions of 1 and 2 (200 μM) in ethanol revealed the formation of spherical self-assembled structures when deposited over a freshly cleaved mica surface (Fig. 3). Noticeably, microscopic (AFM and OM) observations revealed that these spherical structures of 1 and 2 were different from each other in shape, size and uniformity (Fig. 3). The SEM observations also corresponded well with the AFM and OM observations.
 | ||
| Fig. 3 (A) AFM image of the self-assembled structures of 1 in ethanol, (B) the corresponding 3D image, and (C) the fluorescence optical image of the fluorescein-stained sample of 1. (D) AFM image of the self-assembled structures of 2, (E) the corresponding 3D image, and (F) the fluorescence optical image of the fluorescein-stained sample. These images confirm that 1 and 2 follow the different self-assembly processes. | ||
The particle size distribution (PSD) histogram and subsequent Gaussian fitting revealed that the self-assembled structures of compound 1 were irregular and possessed diameters ranging from 50–350 nm (Fig. 4A, right) with an average value of 181.8 ± 17.5 nm (Fig. 4A, right). However, the diameter of the spherical structures of conjugate-2 ranged from 300–400 nm with an average value of 345.8 ± 2.0 nm (Fig. 3B, right). The transmission electron microscopy (Fig. 5) and liquid cell AFM imaging (ESI) observations also corresponded well with other microscopy results and supported the formation of spherical nanostructures with varied diameters. Therefore, it can be envisaged that the obtained self-assembled nanostructures are consistent in both liquid and air medium, showing the negligible effect of the used hydrophilic and hydrophobic surfaces, such as mica (AFM), carbon-coated copper grid (TEM), copper surface (SEM) and glass surface (OM).
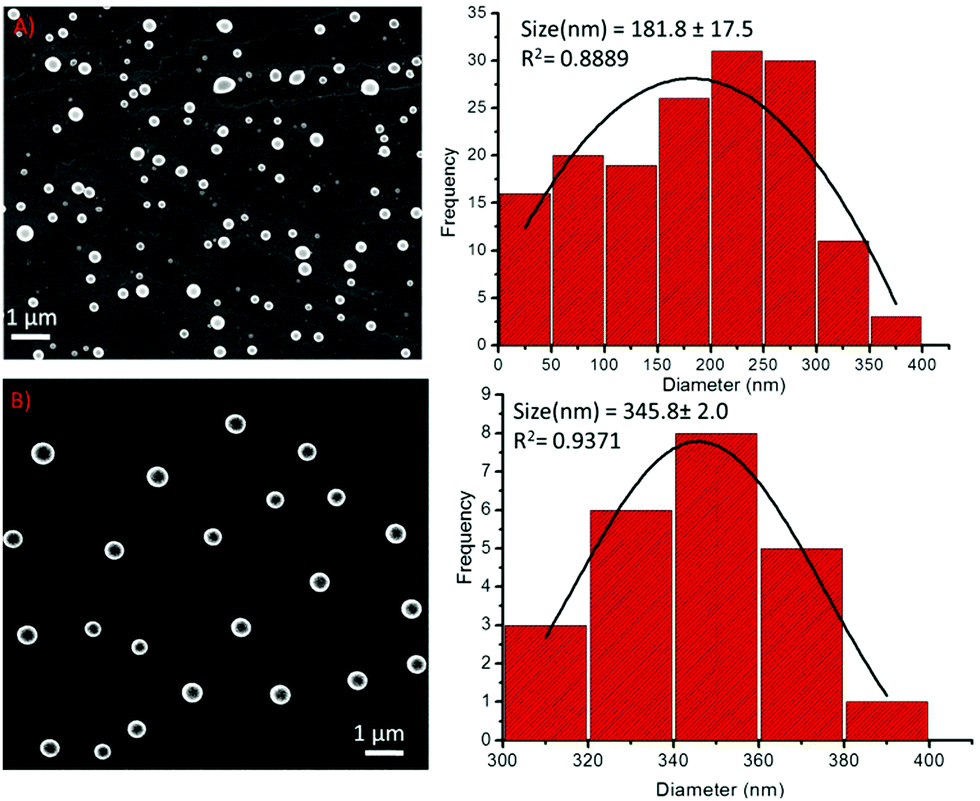 | ||
| Fig. 4 (A) Left: SEM images of the self-assembly of compound 1; Right: The corresponding particle size distribution histogram of compound 1, (B) Left: SEM images of the self-assembly of compound 2; Right: The corresponding particle size distribution histogram of compound 2. | ||
 | ||
| Fig. 5 TEM images of the self-assembled structures of (A and B) peptide 1 and (C and D) peptide 2. | ||
Both tyrosine and phenylalanine are well-known to produce ordered structures in solutions.51,52 Based on the literature,17,19 we also predicted that both 1 and 2 may engender a ‘crescent-like’ rigid conformation with more metal ion co-ordination sites for the interrogation of the peptide self-assembly process.19 An appropriate position of tyrosine due to its π–π and π–OH group interactions also provides a guiding effect27,53 on the assembling structures in addition to the crescent-like structure of the 2,6-pyridinedicarboxylic template,17,19,43,45 thereby fostering a unique self-assembly pathway. Further, the intrinsic fluorescence of the tyrosine residue provides an additional advantage in monitoring the self-assembly process in the presence of external stimuli.27,30 The presence of phenylalanine along with tyrosine residues may lead to transient collocation in solution due to the π–π interactions and hydrogen bonds with adjacent residues and the amide backbone. Such interactions would reduce the lateral chain fluctuations and increase the incidences of metal ion coordination.
Fluorescence titration measurements with different transition metal ions were carried out (Fig. S9–S11, ESI†). Both 1 and 2 had the emission wavelength (λem) centred at 336 nm. The phenolic–OH group has a very good binding affinity for Fe3+ ions.27,30,54,55 Therefore, a gradual decrease was observed in the fluorescence intensity at 336 nm for both 1 and 2 on increasing the concentration of the Fe3+ ions (Fig. 6A and B and ESI†). In the case of the addition of Ni2+, Cu2+ and Zn2+ ions to 1, quenching was observed to a lesser extent (Fig. 6A and ESI†), while Co2+ slightly increased the fluorescence intensity of 1 and 2. The Stern–Volmer plots for these quenching spectra revealed that the order of complexation between 1 and the metal ions was Fe3+ ≫ Cu2+ > Ni2+ > Zn2+ > Co2+ (Fig. 6C). In the case of compound-2, the addition of Fe3+ and Cu2+ followed similar trends of quenching, whereas the addition of Ni2+ showed fluorescence quenching to a lesser extent (Fig. 6B and ESI†). Interestingly, opposite trends were observed when 2 was titrated with Co2+ and Zn2+ ions, as these enhanced the fluorescence intensity of 2 (Fig. 6B and ESI†). The corresponding Stern–Volmer plots revealed that the order of complexation between 2 and the metal ions was Fe3+ ≥ Cu2+ > >Ni2+ > Zn2+ > Co2+ (Fig. 6D). The effective quenching of the fluorescence intensity of both the peptides by the Fe(III) and Cu(II) ions can be explained by charge transfer.30,53 Very less or negligible quenching of both the peptides in the presence of Ni(II) and Zn(II) ions is perhaps due to the condensed aggregation of the chromophores.27,56 Metal-ion enhanced fluorescence (MEF) emission30,57 was also observed in the presence of Co(II) for both 1 and 2, whereas changes in the fluorescence of peptide 2 in the presence of Zn(II) displayed an electric field enhancement effect.30,57
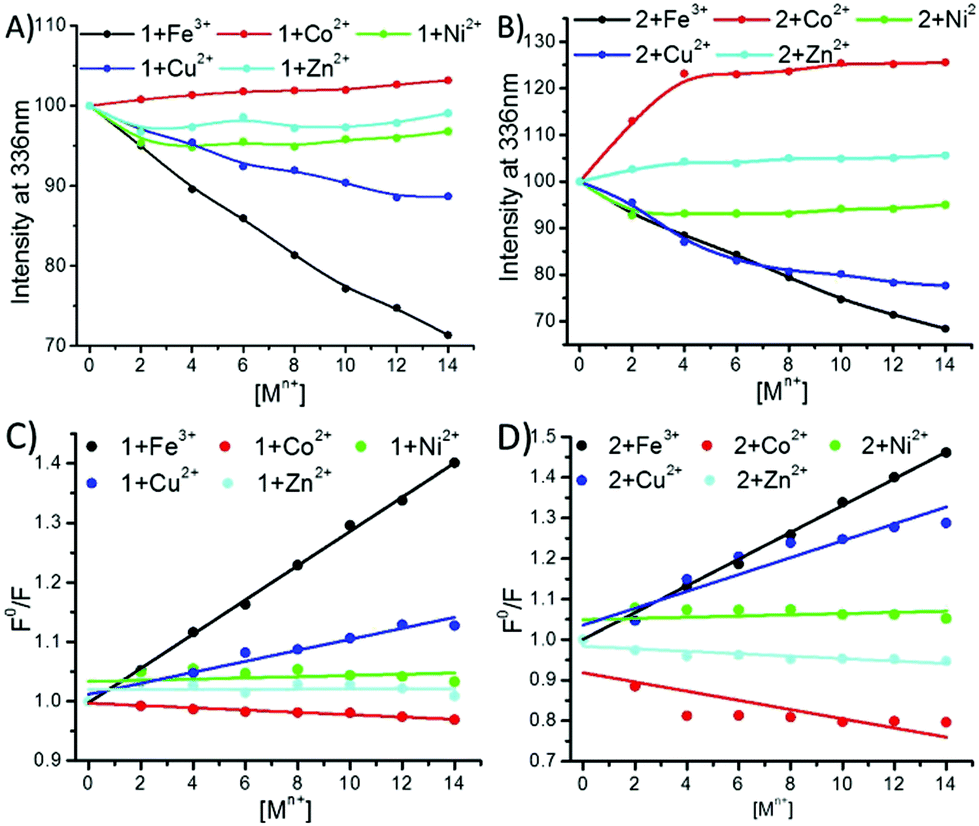 | ||
Fig. 6 Fluorescence spectra of compounds 1 and 2 with the gradual addition of metal ions. (A) Plot of the fluorescence intensity of compound 1 at 336 nm versus the concentration of added Fe3+ (●), Co2+ ( ), Ni2+ ( ), Ni2+ ( ), Cu2+ ( ), Cu2+ ( ) and Zn2+ ( ) and Zn2+ ( ); (B) plot of the fluorescence intensity of compound 2 at 336 nm versus the concentration of added Fe3+ (●), Co2+ ( ); (B) plot of the fluorescence intensity of compound 2 at 336 nm versus the concentration of added Fe3+ (●), Co2+ ( ), Ni2+ ( ), Ni2+ ( ), Cu2+ ( ), Cu2+ ( ) and Zn2+ ( ) and Zn2+ ( ). (C) Stern–Volmer plots of compound 1 with Fe3+, Co2+, Ni2+, Cu2+ and Zn2+; (D) Stern–Volmer plots of compound 2 with Fe3+, Co2+, Ni2+, Cu2+ and Zn2+. ). (C) Stern–Volmer plots of compound 1 with Fe3+, Co2+, Ni2+, Cu2+ and Zn2+; (D) Stern–Volmer plots of compound 2 with Fe3+, Co2+, Ni2+, Cu2+ and Zn2+. | ||
UV-Vis analysis was also done to support the obtained fluorescence results. A continuous increase in the absorbance intensity of 1 and 2 at 275 nm (λmax Tyr sidechain) was observed with the gradual addition of Fe(III) and Cu(II) solutions, which revealed that almost all the added metal ions participated in complexation (Fig. 7 and ESI†).
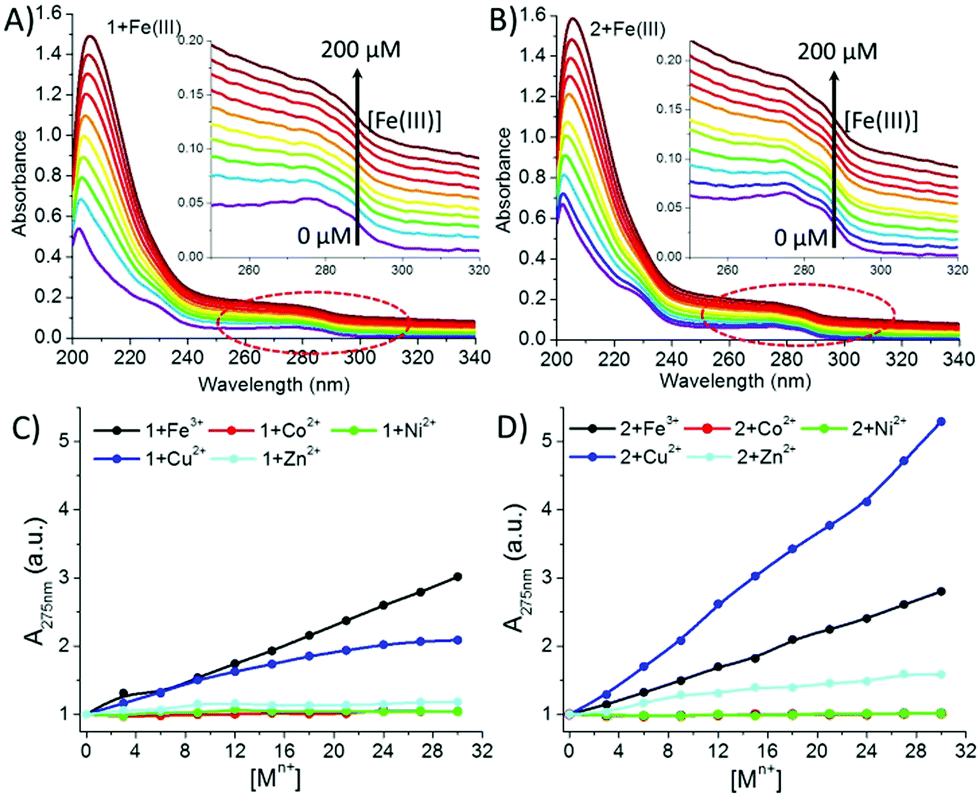 | ||
| Fig. 7 UV-Vis titration spectra of (A) 1 (40 μM) and (B) 2 (40 μM) with Fe(III) as a representative; (C and D) increase in absorbance intensity of the aromatic Tyr sidechain at 275 nm with the gradual addition of different metal ions: Fe(III), Co(II), Ni(II), Cu(II), and Zn(II) to 1 and 2, respectively. | ||
Interestingly, the isomers showed different complexation behaviour with Fe(III) and Cu(II). Peptide 1 presented the order Fe(III) > Cu(II), while for peptide 2, it was reversed, i.e. Cu(II) > Fe(III) (Fig. 7C and D), which is probably due to the close location of the tyrosine sidechain to the terminal –COO group, indicating that a certain amount of Cu(II) was bound to the terminal –COO group. On the other hand, in the case of Co(II), Ni(II) and Zn (II), a negligible increase in the intensity at 275 nm was observed for both the peptides. This early-stage saturation of absorbance indicated that a small fraction of the Co(II), Ni(II) and Zn (II) ions interacted with the Tyr residues. However, a gradual increase in the absorbance intensity at 203 nm due to the n → π* transition of the amidic and carboxylic –CO– groups indicated that all the metal ions were also coordinating with the amidic or carboxylic group. Further, from our previous study19 and the fluorescence and UV data analysis, we could predict that the Co(II), Ni(II) and Zn(II) ions were weakly interacting with Tyr; however, they were preferably binding with the terminal carboxylic and amide groups.
Transition-metal ions are well-known to play an important role in the structural transformations of chelating biomolecules.10–12,30,36 Therefore, after examining the nature of the fluorescence of 1 and 2 in the presence of these transition-metal ions and the UV-Vis titration observations, we recorded the 1H-NMR spectra of peptides 1 and 2 in the presence of Fe(III) ions-(as a representative) in order to know the positions of the peptide that predominantly coordinate with the metal ions (Fig. S19, ESI†). The 1H-NMR spectra of 1 and 2 in the presence of iron depicted that the nature of the aromatic-H signals was highly affected, along with a slight downfield shift. This perhaps is due to the aromatic side chain of the aromatic amino acids, including pyridine, that mainly participate in metal-ion coordination. In addition to this, interestingly, the signal of the ortho protons of the Tyr side chain in peptide 2 converted into a broad triplet-like signal, while in the case of peptide 1, it remained a doublet but with some broadening. This difference in the NMR signals between the peptides indicates the role of the aromatic sidechain in the metal-ion interactions, with the tyrosine aromatic sidechain of peptide 2 showing significant binding with the metal ions due to its feasible position.
Then, the effect of these metal ions on the self-assembly process of the sequence-shuffled isomeric peptides was examined by scanning electron microscopy (SEM). Iron (Fe3+) has been previously reported to combine easily with the phenolic-OH group of Tyr residues,27,30,54,55 and hence, we first investigated the effect this metal ion had on the self-assembly of 1. The addition of Fe3+ ions to 1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 composition) instantaneously increased the aggregation of 1 and minimized the diameter of the spheres to the range of 40–80 nm (Fig. 8A). The corresponding PSD analysis demonstrated that the diameter of these spherical structures significantly decreased, and the average diameter of the compound 1-Fe3+ hybrid structures was calculated to be 68.3 ± 0.9 nm (Fig. 8B). In the presence of Co2+ and Ni2+ ions, interesting results were obtained. SEM observations revealed that the shape and size of the self-assembled spherical peptide-metal hybrids had a high degree of uniformity compared with those made of the peptide alone (Fig. 8C and E). The particle size distribution histogram analysis demonstrated that the size of these spherical structures had increased several folds but with high precision and uniformity. The calculated average diameters of the 1-Co2+ and 1-Ni2+ hybrid structures were found to be 580.5 ± 5.0 nm (Fig. 8D) and 466 ± 3.4 nm (Fig. 8F), respectively.
:1 composition) instantaneously increased the aggregation of 1 and minimized the diameter of the spheres to the range of 40–80 nm (Fig. 8A). The corresponding PSD analysis demonstrated that the diameter of these spherical structures significantly decreased, and the average diameter of the compound 1-Fe3+ hybrid structures was calculated to be 68.3 ± 0.9 nm (Fig. 8B). In the presence of Co2+ and Ni2+ ions, interesting results were obtained. SEM observations revealed that the shape and size of the self-assembled spherical peptide-metal hybrids had a high degree of uniformity compared with those made of the peptide alone (Fig. 8C and E). The particle size distribution histogram analysis demonstrated that the size of these spherical structures had increased several folds but with high precision and uniformity. The calculated average diameters of the 1-Co2+ and 1-Ni2+ hybrid structures were found to be 580.5 ± 5.0 nm (Fig. 8D) and 466 ± 3.4 nm (Fig. 8F), respectively.
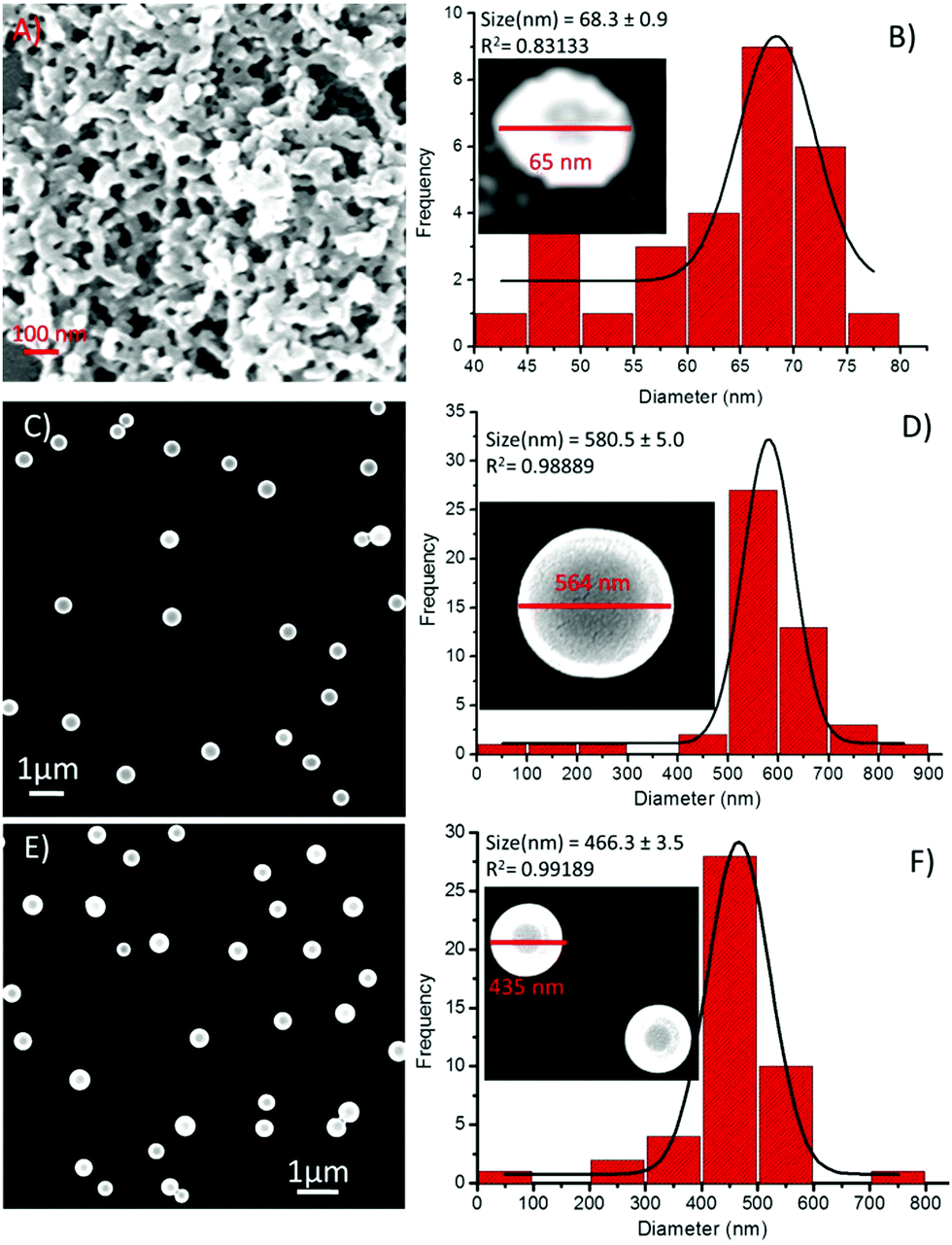 | ||
| Fig. 8 SEM images of compound 1 in the presence of metal ions and the corresponding particle size distribution histograms, respectively, (A and B) in the presence of Fe3+, (C and D) in the presence of Co2+, and (E and F) in the presence of Ni2+. | ||
On the other hand, in the presence of Cu2+ ions, a coalescence of soft structures was observed, and they subsequently self-arranged into an elongated chain-like assembly (Fig. 9A). This type of coalescence is perhaps due to coordination between the peptide and the metal ions,19 followed by one-dimensional growth (elongation into chains), the nucleation of which is mediated via the liquid–liquid separation pathway.21 The PSD analysis of the 1-Cu2+ hybrid structures revealed that the average diameter of these spherical morphologies was 536.5 ± 12.1 nm (Fig. 9B). Similarly, the presence of Zn2+ ions resulted in the dimerization of the spheres (Fig. 9C), presenting an average diameter of 481.8 ± 8.7 nm (Fig. 9D). Likewise, the effect of metal ions on the self-assembly of compound 2 was also examined by using electron microscopy. We noticed remarkable changes in the self-assembly patterns of these hybrids. The addition of a Fe3+ solution into the solution of 2 in a 1:1 ratio composition led to minimized diameters of the spheres by several folds (Fig. 10A). The particle size distribution histogram revealed that the average diameter of the 2-Fe3+ hybrid structures was 74.5 ± 5.0 nm (Fig. 10B). Unlike other metal ions, Co2+ completely transformed the spherical morphology into a circular multilayer (or stacked) sheet-like morphology (Fig. 10C and D). Further, the addition of Ni2+ ions also decreased the size of the spheres (Fig. 10E), and the average diameter of the 2-Ni2+ hybrid structures was calculated to be 53.3 ± 6.0 nm (Fig. 10F).
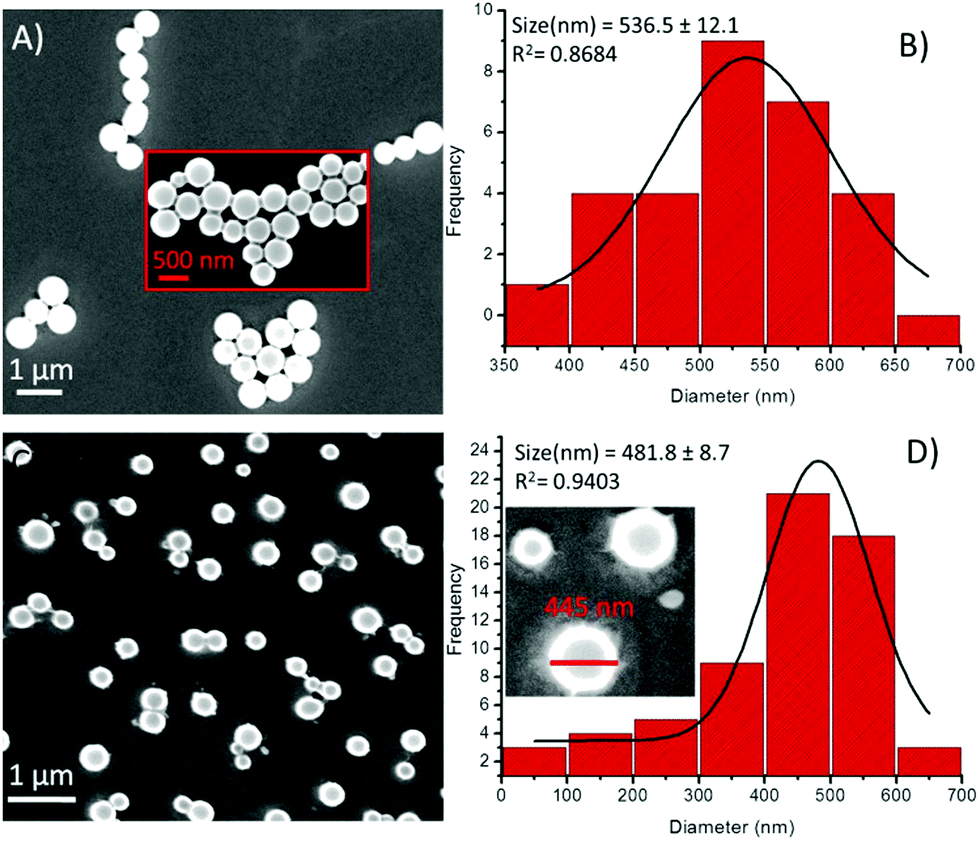 | ||
| Fig. 9 SEM images of compound 1 in the presence of metal ions and the corresponding particle size distribution histogram, respectively, (A and B) in the presence of Cu2+ and (C and D) in the presence of Zn2+ ions. | ||
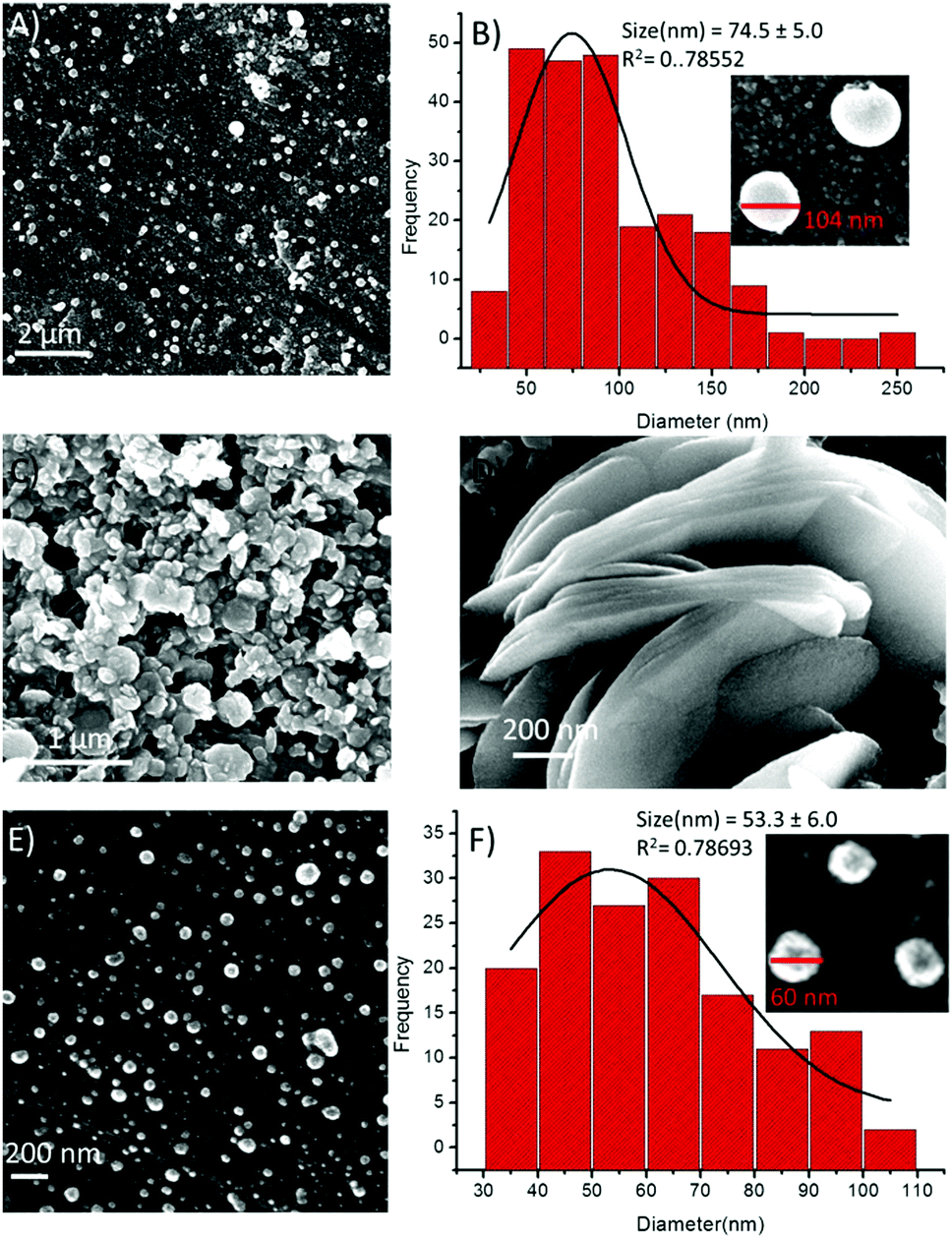 | ||
| Fig. 10 SEM images of compound 2 in the presence of metal ions and the corresponding particle size distribution, respectively (A and B) in the presence of Fe3+, (C and D) in the presence of Co2+, and (E and F) in the presence of Ni2+. | ||
A slight or almost no change in the shape and size of spheres of 2 was seen in the presence of Cu2+ ions (Fig. 11A). The particle size distribution histogram revealed that the average diameter of the hybrid structures of 2-Cu2+ was 406.7 ± 6.3 nm (Fig. 11B). On the other hand, in the presence of Zn2+ ions, the spherical morphology was transformed into an aggregated sheet-like morphology (Fig. 11C and D).
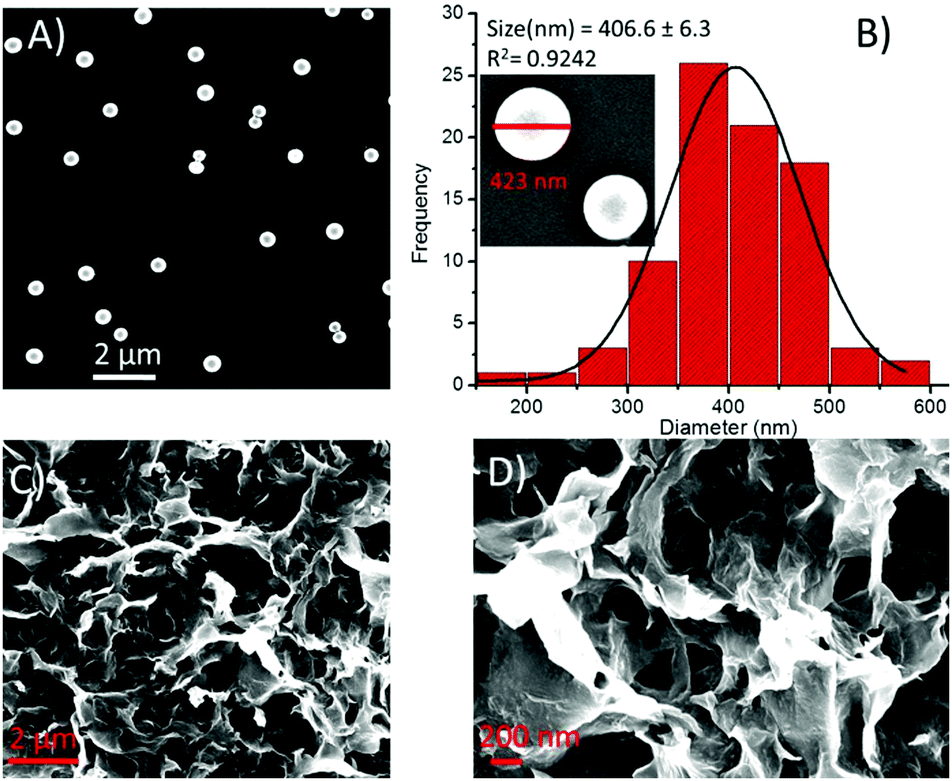 | ||
| Fig. 11 SEM images of compound 2 in the presence of metal ions and the corresponding particle size distribution, respectively, (A and B) in the presence of Cu2+ and (C and D) In the presence of Zn2+ ions. | ||
To understand the underlying mechanism of the metal-ion-induced structural transformation, we recorded the FT-IR spectra of both the peptides with and without metal ions. The Fourier transform infrared spectroscopy (FT-IR) analysis validated the structural transformation in the presence of Co2+/Zn2+ ions. The FT-IR spectra showed a strong band in the amide I region due to the amide CO stretching in all the tested samples of 1 and 2 (Fig. S16, ESI†). Deconvolution calculation was performed by fitting multiple peaks in the spectra ranging from 1600 to 1700 cm−1. The deconvoluted spectra in the amide I region revealed that peptide 1 showed a very strong band centred at 1650 cm−1 (Fig. S16, panel ‘A’ green dotted line, ESI†) that could not be further disintegrated, which represented that the main contribution was from α-helix-type secondary structures. On the other hand, deconvolution of the amide I region of the peptide 2 spectrum showed disintegration into (Fig. S16, panel ‘B’ red dotted line, ESI†) β-sheet-(1631 cm−1) and α-helix (1654 cm−1)-type secondary structures. The quantitative analysis of the FT-IR spectra of peptides 1 and 2 revealed that the obtained nano-assemblies of 1 had 100% α-helix contribution; however, for 2, 34.55% and 65.45% contribution from β-sheets and α-helices, respectively, was observed (Fig. S16 bar graph, ESI†). Interestingly, the deconvolution analysis of the peptide 1 spectrum in the presence of Co2+/Zn2+ ions showed negligible effect (Fig. S16 panel A, ESI†) of these metal ions, and hence, the morphology of the nanoassemblies was not affected much (Fig. 8C, D and 9C, D). On the other hand, the deconvolution analysis of the peptide 2 spectrum in the presence of Co2+/Zn2+ ions showed an entirely different nature of peaks with a complete transition of the α-helix/β-sheet-type secondary structures into antiparallel β-sheet/random coil-like secondary structures, which probably is the reason for the Co2+/Zn2+-induced structural transformation that results in a sheet-like morphology (Fig. 11C, D and 10C, D).
Thus, based on these results, the following possibilities could be envisaged for metallopeptides 1 and 2. Microscopic and spectroscopic evidence reveals that the crescent-shape of the monomer engendered by the pyridyl linker helps in the self-organization of 1 and 2.19,43 They also reveal that the presence of tyrosine residue at its appropriate position perhaps is the main guiding and controlling factor for achieving self-assembly with different metal ions. The different and unique electronic features of these transition-metal ions cannot be neglected. Since both the molecules are rich in aromatic sidechains with properly positioned functionalities, aromatic pi-interactions become additional predominant guiding factors. The presence of amides and free carboxylic groups and the use of hydrophilic surfaces i.e. mica with lower contact angles play a secondary role to further stabilize these structures. Further, the unique photo-physical property of the tyrosine residue provides an additional advantage in monitoring the self-assembly process in the presence of external stimuli. Therefore, based on the above experimental observations, the self-assembly processes of 1 and 2 with and without metal ions were predicted to a certain extent, as proposed in the model (Fig. 12). Both the molecules may adopt a crescent-like shape, which subsequently self-assembles into a vesicular construct. The position of tyrosine in 1 destabilizes the proper aromatic interaction engendered between/by the pyridyl ring and FF residues; therefore, irregular shapes and sizes are initially obtained for the vesicular assemblies. However, the addition of appropriate transition metal ions help stabilize the electronic contribution of the phenolic –OH of tyrosine; therefore, the aromatic interaction between the pyridyl and FF residues is in proper agreement, leading to a uniform spherical morphology and also a substantial increase in the vesicle diameter. Since Fe3+ preferably and strongly coordinates with Tyr–OH,27,30 a very high degree of coalescence along with a decrease in vesicle size was observed. However, in the presence of Cu2+ ions, beautifully arranged coalesced vesicles were visible.
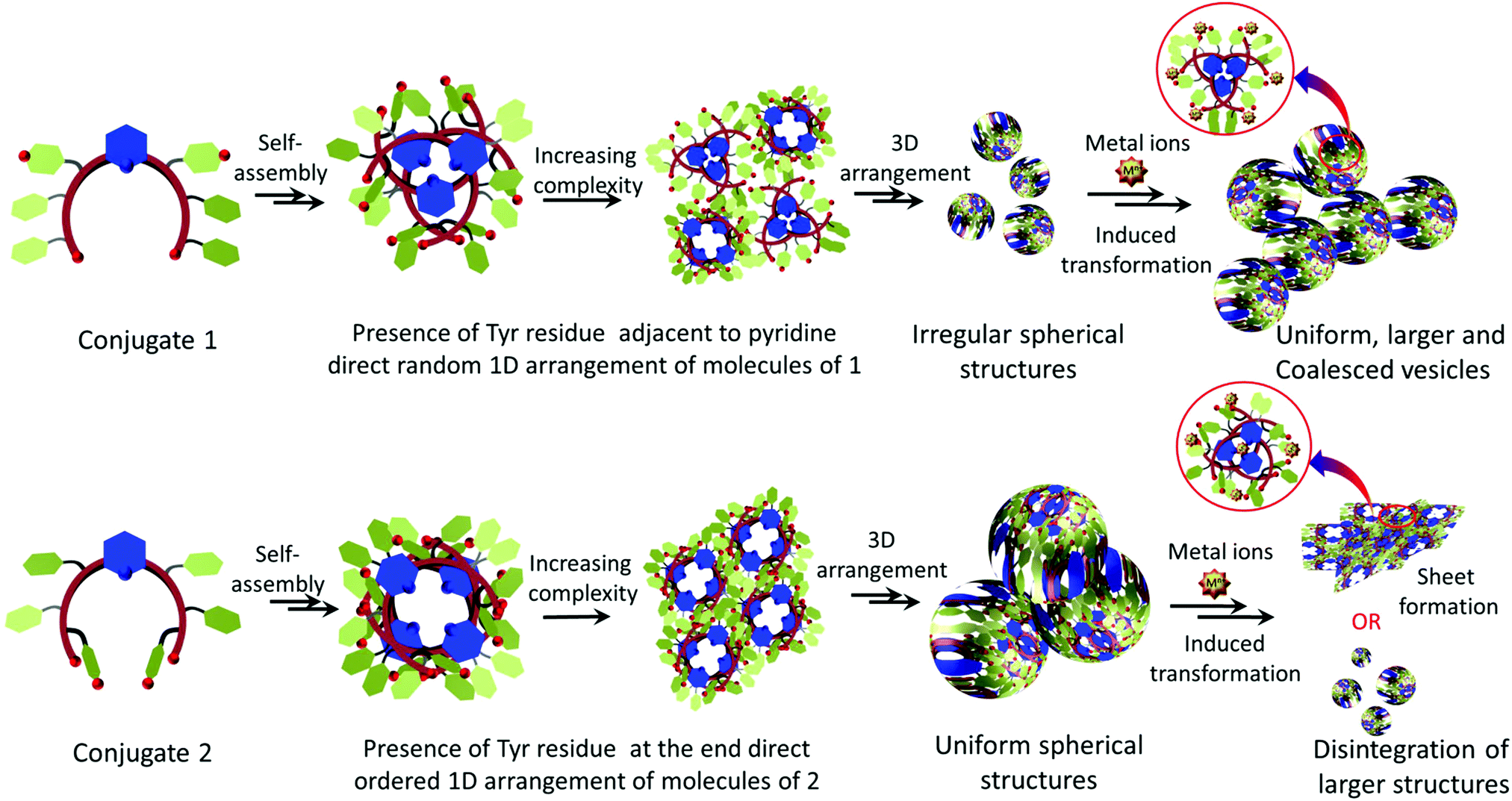 | ||
| Fig. 12 Proposed model of the metal-peptide interactions and the different self-assembly processes followed by conjugates 1 and 2 alone and in the presence of transition-metal ions. | ||
On the other hand, in conjugate 2, the position of the tyrosine residue is fixed at the terminal position of the crescent. Therefore, perhaps the presence of tyrosine at this position is more appropriate and stabilizes the aromatic pi-interactions. The phenolic –OH group may participate in intermolecular or intramolecular hydrogen bond formation with the carboxyl/phenolic –OH groups of its counterpart residues, thereby leading to the formation of structures with high precision. However, in the presence of metal ions, the end tyrosine residue preferably coordinates with the metal ions and hence tries to transform the morphology too. In the presence of Fe3+ and Ni2+, the larger vesicles disintegrate into smaller ones; however, in the presence of Cu2+, vesicles of almost the same size were obtained with some degree of coalescence. However, stacked and aggregated sheet-like morphologies were obtained in the presence of Co2+ and Zn2+, respectively (Fig. S16, ESI†).
These self-assembled nanostructures of the metallopeptides are governed by the functional properties of the peptides and metal-peptide interactions. Therefore, the coordination of specific metal ions further provides novel metallopeptides of choice, which can be considered as bioactive metal ion reservoirs for potential biotechnological applications. For example, the order of the binding abilities may be utilized for the capture, displacement and delivery of clinically relevant metal ions. This study on metallopeptides may be useful for enhancing the ability of drug delivery systems through the drug displacement strategy.7,58 Because of their metal-binding strength, these peptide structures can also be utilized to disrupt the cell walls of metal ion-containing pathogens, which would result in subsequent cell lysis.59 Such metallopeptide-based nanostructures can also be utilized for bio-mimicking numerous metalloenzymes involved in catalysis.60,61
Conclusions
Two pyridyl-based isomeric peptide conjugates 1 and 2 were designed. Based on the literature, it was believed that both the conjugates will have crescent-like molecular structures. The self-assembly of these crescent-type structures and their interactions with bioactive transition-metal ions were studied. It was demonstrated that changing the position of the desired functionally rich amino acid(s) is a very useful technique for manipulating the self-assembly of the peptide conjugates. Both the crescent-type constitutional isomers followed different self-assembly pathways and possessed substantial structural differences, as confirmed by 1H-NMR and FT-IR spectroscopy. The transition-metal ions were used to control and transform the three-dimensional hierarchical self-assembled structures. The first isomer, in which the position of the Tyr residue was near the pyridyl group, self-assembled into random aggregates. The use of transition-metal ions led to the minimization of the randomness of these structures and produced coalesced structures with iron and copper, while large structures with a high degree of regularity were obtained with cobalt, nickel and zinc. The second isomer, wherein, the Tyr residues were at the terminal of the crescent structure produced more refined and regular nanostructures compared with 1 due to the aromatic homogeneity/symmetry in the structure. However, this homogeneity was perturbed in the presence of metal ions, and the vesicular morphology transformed into stacked and aggregated sheet-like morphologies in the presence of cobalt and zinc, respectively. In the presence of nickel and iron, the large vesicular morphology disintegrated, and with copper, a smaller degree of coalescence was observed. Thus, it can be summarized that the shuffling of one or more key amino acids in rationally designed peptide conjugates can be of great significance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NS, RS and SS thank to CSIR-UGC, UGC and MHRD for pre-doctoral research fellowship. KK thanks to DHSGSU for doctoral fellowship. NS and RS contributed equally to this work. SV would like to thank the J. C. Bose Fellowship (SERB, India) for financial support. This work is partially supported by DST Nano Mission and CSIR. We acknowledge Sophisticated Instrument Centre (SIC) Dr Harisingh Gour Central University Sagar, M.P., India for AFM. Our acknowledgement is incomplete without thanking Jai Kumar, IIT-Kanpur, for TEM.Notes and references
- R. Zou, Q. Wang, J. Wu, J. Wu, C. Schmuck and H. Tian, Chem. Soc. Rev., 2015, 44, 5200–5219 RSC.
- S. L. Kuan, F. R. G. Bergamini and T. Weil, Chem. Soc. Rev., 2018, 47, 9069–9105 RSC.
- E. Degtyar, M. J. Harrington, Y. Politi and P. Fratzl, Angew. Chem., Int. Ed., 2014, 53, 12026–12044 CrossRef CAS.
- H. Sun, Q. Luo, C. Hou and J. Liu, Nano Today, 2017, 14, 16–41 CrossRef CAS.
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef CAS.
- K. J. Waldron and N. J. Robinson, Nat. Rev. Microbiol., 2009, 7, 25–35 CrossRef CAS.
- P. Das, I. Pan, E. Cohen and M. Reches, J. Mater. Chem. B, 2018, 6, 8228–8237 RSC.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS.
- K. L. Haas and K. J. Franz, Chem. Rev., 2009, 109, 4921–4960 CrossRef CAS.
- R. Singh, V. Suryavashi, V. Vinayak and K. B. Joshi, ChemistrySelect, 2019, 4, 5810–5816 CrossRef CAS.
- A. S. Knight, J. Larsson, J. M. Ren, R. Bou Zerdan, S. Seguin, R. Vrahas, J. Liu, G. Ren and C. J. Hawker, J. Am. Chem. Soc., 2018, 140, 1409–1414 CrossRef CAS.
- R. Singh, S. Gupta, V. Kumar and K. B. Joshi, ChemNanoMat, 2017, 3, 620–624 CrossRef CAS.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- R. Wakabayashi, A. Suehiro, M. Goto and N. Kamiya, Chem. Commun., 2019, 55, 640–643 RSC.
- K. B. Joshi and S. Verma, Angew. Chem., Int. Ed., 2008, 47, 2860–2863 CrossRef CAS.
- S. Mondal, A. K. Barman and S. Verma, Chimia, 2012, 66, 930–935 CrossRef CAS.
- D. W. Zhang, X. Zhao, J. L. Hou and Z. T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS.
- J. Wang, K. Liu, R. Xing and X. Yan, Chem. Soc. Rev., 2016, 45, 5589–5604 RSC.
- G. Kaur, L. A. Abramovich, E. Gazit and S. Verma, RSC Adv., 2014, 4, 64457–64465 RSC.
- C. Yuan, W. Ji, R. Xing, J. Li, E. Gazit and X. Yan, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS.
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 CrossRef CAS.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263 RSC.
- A. Dasgupta and D. Das, Langmuir, 2019, 35, 10704–10724 CrossRef CAS.
- X. Yan, P. Zhu and J. Li, Chem. Soc. Rev., 2010, 39, 1877 RSC.
- H. Cui, A. G. Cheetham, E. T. Pashuck and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 12461–12468 CrossRef CAS.
- R. Singh, N. K. Mishra, N. Singh, P. Rawal, P. Gupta and K. B. Joshi, New J. Chem., 2020, 44, 9255–9263 RSC.
- R. Singh, N. K. Mishra, P. Gupta and K. B. Joshi, Chem. – Asian J., 2020, 15, 531–539 CrossRef CAS.
- S. Sharma, C. Kulkarni, M. M. Kulkarni, R. Ali, K. Porwal, N. Chattopadhyay, D. Tewari and S. Verma, Chem. Commun., 2020, 56, 3043–3046 RSC.
- G. Kaur, S. Kumari, P. Saha, R. Ali, S. Patil, S. Ganesh and S. Verma, ACS Omega, 2019, 4, 4376–4383 CrossRef CAS.
- R. Singh, N. Kumar Mishra, V. Kumar, V. Vinayak and K. Ballabh Joshi, ChemBioChem, 2018, 19, 1630–1637 CrossRef CAS.
- N. K. Mishra, V. Kumar and K. B. Joshi, Nanoscale, 2015, 7, 20238–20248 RSC.
- N. K. Mishra, K. B. Joshi and S. Verma, J. Colloid Interface Sci., 2015, 455, 145–153 CrossRef CAS.
- K. K. Prasad, C. S. Purohit, A. Jain, R. Sankararamakrishnan and S. Verma, Chem. Commun., 2005, 2564–2566 RSC.
- C. Madhavaiah and S. Verma, Chem. Commun., 2004, 638–639 RSC.
- S. Ghosh and S. Verma, J. Phys. Chem. B, 2007, 111, 3750–3757 CrossRef CAS.
- S. Gupta, R. Singh, V. Kumar, P. Shukla and K. B. Joshi, Chem. – Asian J., 2018, 13, 3285–3295 CrossRef CAS.
- V. Kumar, R. Singh and K. B. Joshi, New J. Chem., 2018, 42, 3452–3458 RSC.
- A. K. Barman and S. Verma, Chem. Commun., 2010, 46, 6992–6994 RSC.
- S. Mondal, S. Ghosh and S. Verma, Tetrahedron Lett., 2010, 51, 856–859 CrossRef CAS.
- S. Ghosh and S. Verma, Tetrahedron, 2008, 64, 6202–6208 CrossRef CAS.
- S. Ghosh and S. Verma, Chem. – Eur. J., 2008, 14, 1415–1419 CrossRef CAS.
- S. Ghosh, M. Reches, E. Gazit and S. Verma, Angew. Chem., Int. Ed., 2007, 46, 2002–2004 CrossRef CAS.
- S. Mondal, S. Swaroop, R. Gurunath and S. Verma, Tetrahedron Lett., 2010, 51, 6111–6115 CrossRef CAS.
- T. Moriuchi, M. Nishiyama, K. Yoshida, T. Ishikawa and T. Hirao, Org. Lett., 2001, 3, 1459–1461 CrossRef CAS.
- T. Kawamoto, O. Prakash, A. S. Borovik and R. Ostrander, Inorg. Chem., 1995, 34, 4294–4295 CrossRef CAS.
- S. J. Geib, C. Vicent, E. Fan and A. D. Hamilton, Angew. Chem., Int. Ed. Engl., 1993, 32, 119–121 CrossRef.
- N. Singh, R. Singh, M. Shukla, G. Kaul, S. Chopra, K. B. Joshi and S. Verma, ACS Infect. Dis., 2020, 6, 2441–2450 CrossRef CAS.
- N. Singh, R. Singh, K. B. Joshi and S. Verma, ChemPlusChem, 2020, 85, 2001–2009 CrossRef CAS.
- A. D. Gift, S. M. Stewart and P. Kwete Bokashanga, J. Chem. Educ., 2012, 89, 1458–1460 CrossRef CAS.
- M. H. Ghalia, M. A. El-Hamid, M. A. Zweil, A. E.-G. E. Amr and S. A. Moafi, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 806–818 Search PubMed.
- C. Ménard-Moyon, V. Venkatesh, K. V. Krishna, F. Bonachera, S. Verma and A. Bianco, Chem. – Eur. J., 2015, 21, 11681–11686 CrossRef.
- D. Tomar, S. Chaudhary and K. C. Jena, RSC Adv., 2019, 9, 12596–12605 RSC.
- J. Lee, M. Ju, O. H. Cho, Y. Kim and K. T. Nam, Adv. Sci., 2019, 6, 1801255 CrossRef.
- S. A. Cantalupo, H. E. Ferreira, E. Bataineh, A. J. King, M. V. Petersen, T. Wojtasiewicz, A. G. DiPasquale, A. L. Rheingold and L. H. Doerrer, Inorg. Chem., 2011, 50, 6584–6596 CrossRef CAS.
- S. A. Koch and M. Millar, J. Am. Chem. Soc., 1982, 104, 5255–5257 CrossRef CAS.
- C. Duy and J. Fitter, Biophys. J., 2006, 90, 3704–3711 CrossRef CAS.
- W. Deng, F. Xie, H. T. M. C. M. Baltar and E. M. Goldys, Phys. Chem. Chem. Phys., 2013, 15, 15695 RSC.
- S. Tabassum, W. M. Al-Asbahy, M. Afzal, F. Arjmand and V. Bagchi, Dalton Trans., 2012, 41, 4955 RSC.
- J. L. Alexander, Z. Thompson and J. A. Cowan, ACS Chem. Biol., 2018, 13, 844–853 CrossRef CAS.
- A. Mantion, L. Massüger, P. Rabu, C. Palivan, L. B. McCusker and A. Taubert, J. Am. Chem. Soc., 2008, 130, 2517–2526 CrossRef CAS.
- G. Licini and P. Scrimin, Angew. Chem., Int. Ed., 2003, 42, 4572–4575 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj04260a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |