
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe crystal structures of 2-(4-benzhydrylpiperazin-1-yl)-N-(4-sulfamoylphenyl)acetamide in complex with human carbonic anhydrase II and VII provide insights into selective CA inhibitor development
Katia
D’Ambrosio†
*a,
Anna
Di Fiore†
a,
Martina
Buonanno
a,
Shikha
Kumari
b,
Manisha
Tiwari
 b,
Claudiu T.
Supuran
c,
Chandra Bhushan
Mishra‡
b,
Simona Maria
Monti
*a and
Giuseppina
De Simone
a
b,
Claudiu T.
Supuran
c,
Chandra Bhushan
Mishra‡
b,
Simona Maria
Monti
*a and
Giuseppina
De Simone
a
aIstituto di Biostrutture e Bioimmagini-CNR, Via Mezzocannone 16, 80134, Napoli, Italy. E-mail: katia.dambrosio@cnr.it; marmonti@unina.it
bDr. B. R. Ambedkar Centre for Biomedical Research, University of Delhi, Delhi, India
cDipartimento NEUROFARBA, Università di Firenze, Via Ugo Schiff 6, 50019 Sesto Fiorentino, Italy
First published on 9th December 2020
Abstract
2-(4-Benzhydrylpiperazin-1-yl)-N-(4-sulfamoylphenyl)acetamide is an effective human carbonic anhydrase (hCA) inhibitor designed through the tail approach using the acetamide moiety as linker and the benzhydrylpiperazine group as tail. Here we report the crystal structures of this compound in complex both with the ubiquitous hCA II and the brain-associated hCA VII, showing that in agreement with the previously reported inhibition constants, the inhibitor is stabilized by a higher number of polar and hydrophobic interactions in the active site of hCA VII compared to hCA II. Results point out the conformational flexibility of the linker and the tail length as fundamental features to establish significant differences in the number of favorable enzyme/inhibitor interactions and consequently in the inhibition selectivity against the two hCA isoforms.
Introduction
Human carbonic anhydrases (hCAs, EC 4.2.1.1) are zinc-containing enzymes, which catalyze a very simple reaction: the reversible hydration of carbon dioxide to bicarbonate and proton ions.1 So far, 15 different hCAs have been identified which differ in their catalytic activity, subcellular localization and tissue distribution. Among the 12 catalytically active isoforms, 5 are cytosolic (hCA I–III, VII, and XIII), 4 are membrane-bound (hCA IV, IX, XII, and XIV), 2 are mitochondrial (hCA VA and VB), and 1 is secreted in milk and saliva (hCA VI).2,3 All catalytically active hCAs share a common three-dimensional structure characterized by a central β-sheet surrounded by helical connections and additional β-strands, with the active site located in a large conical cavity that spans from the protein surface to the center of the molecule. This cavity is divided into two very different environments: the first one is delimited by a cluster of hydrophobic amino acids which facilitate the binding of the CO2 substrate,4,5 while the other one consists of hydrophilic residues which assist the catalytic reaction.6–8 The catalytic zinc ion is located at the bottom of the active site cavity and is coordinated by three conserved histidine residues and one water molecule/hydroxide ion.hCA VII is one of the least understood cytosolic isoforms. This enzyme is mainly expressed in some brain tissues, such as the cortex, hippocampus and thalamus regions.9,10 It promotes the neuronal excitation9 establishing a GABAergic transmission functionally excitatory, supplying the bicarbonate gradient that results in the efflux of HCO3− ions through GABAA receptors.11 Moreover, hCA VII has been proposed to play a role in the control of neuropathic pain, thus suggesting that its inhibition may represent a novel pharmacologic mechanism for the treatment of this pathology.12 Recently, a role for hCA VII reactive cysteines towards oxidative insult has been proposed.13–15
Three different classes of CA inhibitors (CAIs) have been characterized so far: (i) molecules that bind to the catalytic zinc ion, such as primary sulfonamides and their isosteres (sulfamates and sulfamides),16,17 dithiocarbamates,18 and xanthates;19 (ii) molecules which anchor to the zinc-coordinated water molecule/hydroxide ion, as observed for phenols20,21 and polyamines;22 (iii) compounds occluding the active site entrance, as reported for coumarins23,24 and lacosamide.25 Several inhibitors belonging to these classes have been designed and tested against hCA VII,26–29 however, although showing good inhibition efficiency, they are generally poorly selective; for this reason, new molecules are continuously synthesized and tested to identify more selective hCA VII inhibitors.
A promising method to gain selective CAIs is the so-called tail approach applied to benzenesulfonamide derivatives.30,31 Such an approach consists in appending a variety of chemical frameworks (tail) to the benzenesulfonamide zinc-binding group (ZBG), by means of a suitable chemical linker, with the aim to reach the regions of the CA active site which show a major amino acid variability.32 Ureas, thioureas, secondary sulfonamides are only some examples of utilized linkers (Fig. 1A).31,33–35
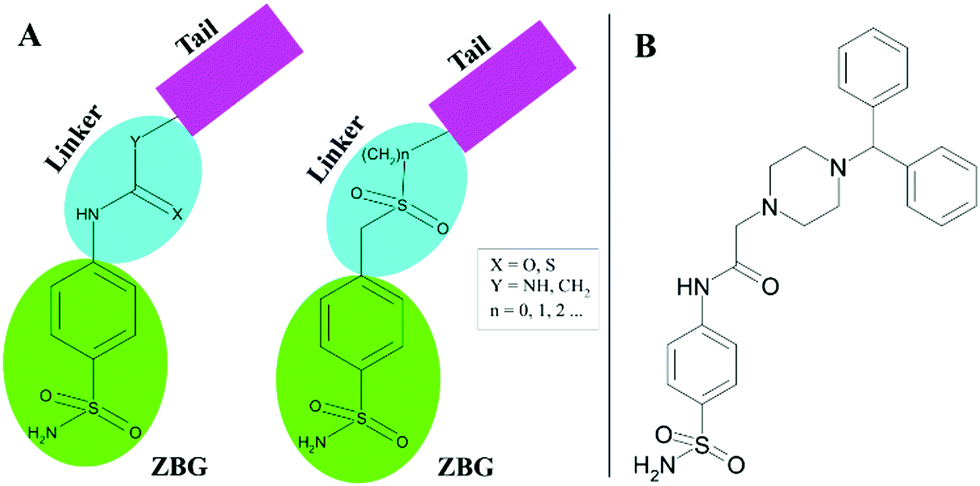 | ||
| Fig. 1 (A) Schematic representation of CAI design by the “tail approach”. (B) Chemical structure of 2-(4-benzhydrylpiperazin-1-yl)-N-(4-sulfamoylphenyl)acetamide 1. | ||
Recently, a new series of 2-(4-substituted piperazin-1-yl)-N-(4-sulfamoylphenyl)acetamide derivatives was developed using the tail approach31 where the acetamide moiety constituted the linker, whereas substituted piperazines, which are heterocyclic building blocks widely used to develop potent CAIs,36–38 represented the tail. Among these molecules, the 2-(4-benzhydrylpiperazin-1-yl)-N-(4-sulfamoylphenyl)acetamide (compound 1 in Fig. 1B) revealed to be particularly interesting, since it showed preferential inhibition of the hCA VII isoform with a KI value of 8.9 nM with respect to the ubiquitous hCA II (KI of 43.2 nM).31 To better understand the molecular basis of the observed selectivity, the crystal structures of compound 1, in complex both with the ubiquitous hCA II isozyme and the brain-associated isoform hCA VII, were determined. The obtained results open new perspectives for the structure-based drug design of novel inhibitors selective for hCA VII with respect to hCA II.
Results and discussion
Crystals of the hCA II/1 complex were prepared by co-crystallization experiments, whereas those of hCA VII/1 were obtained using the soaking technique. Data collection and refinement of both adducts were performed as reported in the Experimental section (see Table 1 for statistics).| hCA II/1 | hCA VII/1 | |
|---|---|---|
a
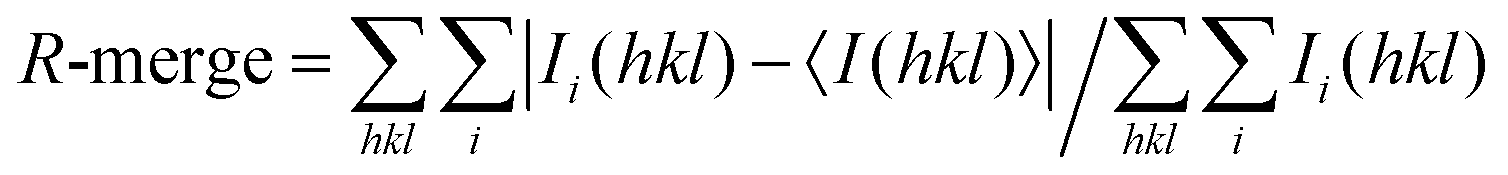 , where Ii(hkl) is the intensity of an observation and 〈I(hkl)〉 is the mean value for its unique reflection; summations are over all reflections.
b , where Ii(hkl) is the intensity of an observation and 〈I(hkl)〉 is the mean value for its unique reflection; summations are over all reflections.
b
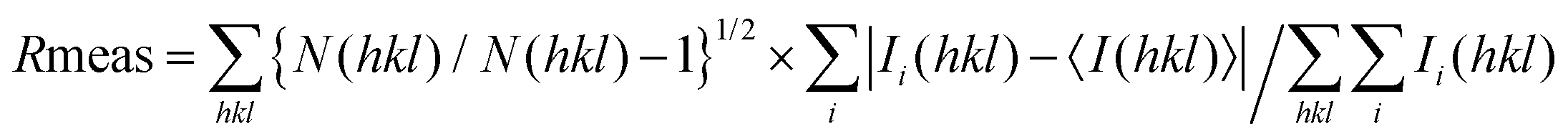 .
c .
c
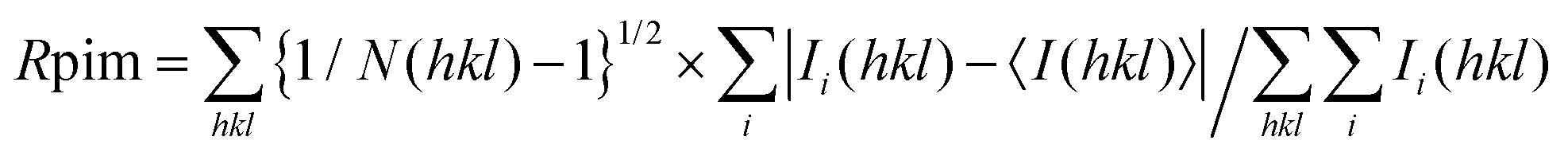 .
d CC1/2 = 〈I2〉 − 〈I〉2/〈I2〉 − 〈I〉2 + σ2ε (values refer to the highest resolution shell).
e .
d CC1/2 = 〈I2〉 − 〈I〉2/〈I2〉 − 〈I〉2 + σ2ε (values refer to the highest resolution shell).
e
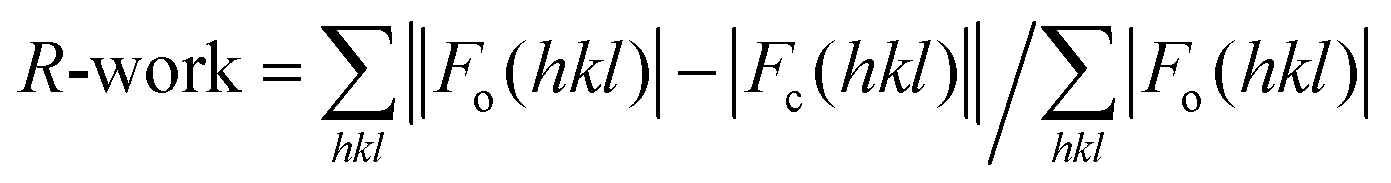 calculated for the working set of reflections. R-Free is calculated as for R-work, but from data of the test set that was not used for refinement (test set size (%) = 5.0 and 6.0 for hCA II and hCA VII adduct, respectively). Values in parentheses refer to the highest resolution shell (1.83–1.79 Å and 2.09–2.05 Å for hCA II and hCA VII adduct, respectively). calculated for the working set of reflections. R-Free is calculated as for R-work, but from data of the test set that was not used for refinement (test set size (%) = 5.0 and 6.0 for hCA II and hCA VII adduct, respectively). Values in parentheses refer to the highest resolution shell (1.83–1.79 Å and 2.09–2.05 Å for hCA II and hCA VII adduct, respectively).
|
||
| Cell parameters | ||
| Space group | P21 | P21212 |
| Cell dimensions (Å, °) | a = 42.3 | a = 66.8 |
| b = 41.3 | b = 88.6 | |
| c = 71.6 | c = 44.3 | |
| β = 104.3 | ||
| Number of independent molecules | 1 | 1 |
| Data collection statistics | ||
| Wavelength (Å) | 1.54178 | 1.54178 |
| Resolution limits (Å) | 26.6–1.79 | 35.7–2.05 |
| Total reflections | 105![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 028 028 |
61105 |
| Unique reflections | 21893 |
16529 |
| Redundancy | 4.8 | 3.7 |
| Completeness (%) | 96.9 (86.9) | 95.8 (88.0) |
| R-Mergea | 0.053 (0.206) | 0.053 (0.375) |
| Rmeasb | 0.059 (0.244) | 0.061 (0.503) |
| Rpimc | 0.024 (0.127) | 0.029 (0.330) |
| CC1/2d | 0.946 | 0.683 |
| 〈I〉/〈σ(I)〉 | 22.2 (5.8) | 17.0 (2.0) |
| Refinement statistics | ||
| Resolution limits (Å) | 26.6–1.79 | 33.4–2.05 |
| R-Worke (%) | 16.4 | 19.6 |
| R-Freee (%) | 19.9 | 24.1 |
| R.m.s.d. from ideal geometry | ||
| Bond lengths (Å) | 0.010 | 0.008 |
| Bond angles (°) | 1.6 | 1.5 |
| Number of protein atoms | 2092 | 2063 |
| Number of inhibitor atoms | 33 | 33 |
| Number of water molecules | 206 | 103 |
| Average B factor (Å2) | ||
| All atoms | 13.49 | 25.26 |
| Protein atoms | 12.84 | 25.04 |
| Inhibitor atoms | 15.88 | 28.42 |
| Waters | 19.72 | 28.71 |
| PDB accession code | 6ZR8 | 6ZR9 |
In both complexes the inspection of the initial |Fo − Fc| electron density maps clearly revealed the binding of one inhibitor molecule in the active site (Fig. 2A and B). The inhibitor binding does not alter hCA II and hCA VII three-dimensional structures as shown by the low r.m.s.d. values calculated by superposition of all the Cα atoms of the hCA II/1 and hCA VII/1 adducts with those of the native proteins (r.m.s.d. values of 0.3 Å and 0.2 Å for hCA II/1 and hCA VII/1, respectively).
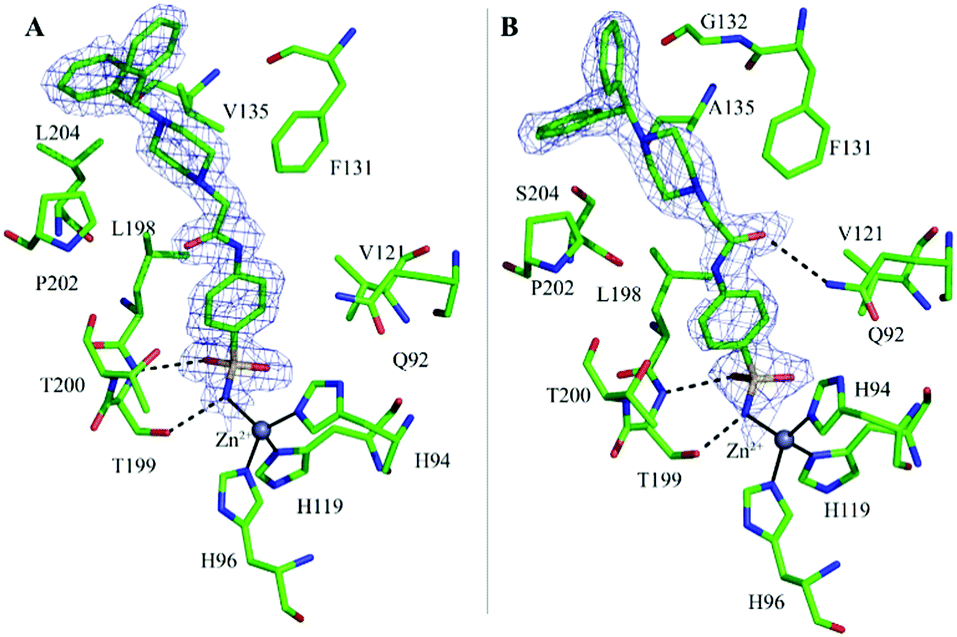 | ||
| Fig. 2 Active site region of the hCA II/1 (A) and hCA VII/1 (B) adducts showing the σA-weighted |2Fo − Fc| simulated annealing omit map (contoured at 1.0σ) relative to the inhibitor molecule. The zinc ion coordination (continuous lines), hydrogen bonds (dashed lines) and residues involved into van der Waals interactions are also depicted. | ||
A careful inspection of the two structures shows that the benzenesulfonamide moiety of the inhibitor presents a similar binding mode to both enzymes. Indeed, as also observed for other CAIs containing the same ZBG,1 the deprotonated nitrogen atom of sulfonamide moiety coordinates the catalytic zinc ion with a tetrahedral geometry, displacing the water molecule/hydroxide ion present in the native enzymes.39,40 Additional hydrogen bond interactions with Thr199 residue further stabilize the inhibitor binding. Finally, the phenyl ring establishes in both structures strong van der Waals contacts (distance <4.0 Å) with several residues (Fig. 2A and B).
In contrast, differences between the two structures are observed when the acetamide moiety is considered.
Indeed, by superimposing hCA II/1 and hCA VII/1 adducts it is possible to notice that this linker can adopt two different orientations in the enzyme active site (Fig. 3A), thanks to the different values that the dihedral angle C1–C2–N3–C4 can assume (Fig. 3B).
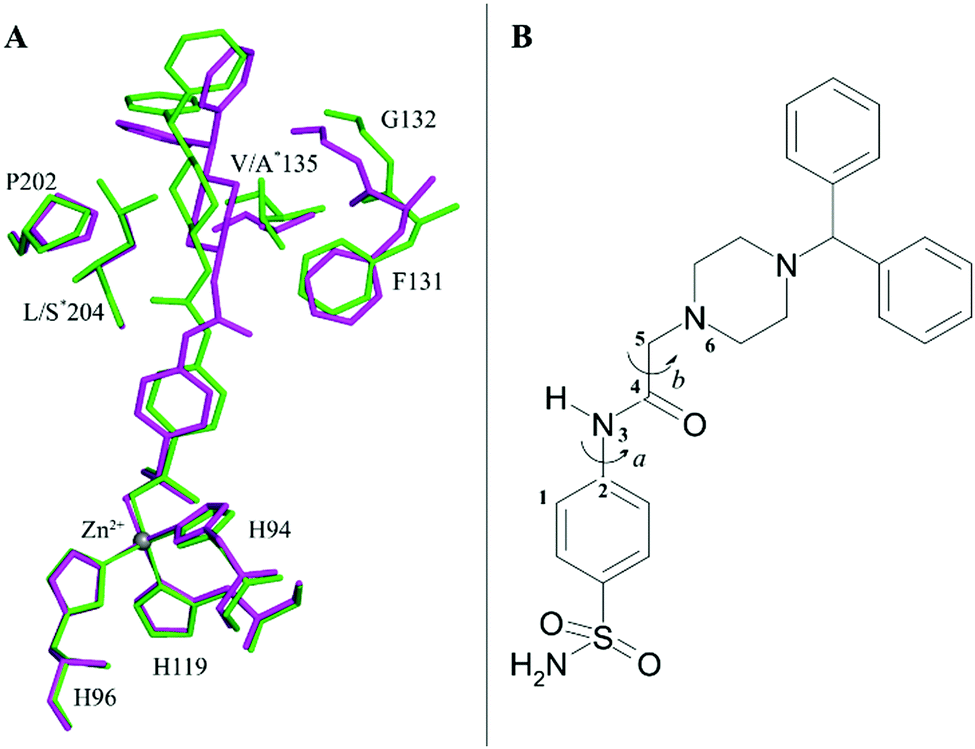 | ||
| Fig. 3 (A) Structural superposition of compound 1 when bound to hCA II (green) and hCA VII (magenta) showing residues involved into van der Waals interactions with benzhydrylpiperazine tail. The asterisk refers to residues that differ in the two isoforms. (B) Schematic representation of the two dihedral angles: a, which determines the two different orientations of the acetamide linker, and b, which is responsible for the linker plasticity. | ||
In hCA II/1 adduct this angle measures −7.6° and the carbonyl oxygen atom points towards Pro202 residue, being engaged only in water-mediated hydrogen bonds with protein residues. On the contrary, in hCA VII/1 complex, this dihedral angle measures 178.2°, with the carbonyl oxygen atom pointing towards Gln92 side chain and forming a weak hydrogen bond interaction with its NE2 atom (Fig. 2). Several differences are also observed when the benzhydrylpiperazine group is considered. Indeed, even if in both structures this moiety is oriented toward the same region of the active site, with the benzhydryl tail located on the border of the cavity, local adjustments are observed to take into account the different residues present in the two enzymes (Fig. 3A). It is well known that the rim of the CA active site cavity is the most variable region among the different isoforms.32 In particular, the small hydrophobic pocket delineated by Val135, Pro202 and Leu204 which accommodates the inhibitor tail in the hCA II/1 adduct differs for two amino acid substitutions, namely Val135 → Ala135 and Leu204 → Ser204, from hCA VII. These substitutions make the pocket wider allowing compound 1 to find a structural arrangement able to optimize protein/inhibitor interactions. As a consequence, in the hCA II/1 complex the benzhydrylpiperazine establishes 6 strong van der Waals contacts with enzyme residues, whereas in hCA VII/1 adduct this is twice the number (see Table 2) (Fig. 2).
| Residue | hCA II | hCA VII | |
|---|---|---|---|
| a Residues different between hCA II and hCA VII. | |||
| Benzenesulfonamide moiety | |||
| Gln 92 | 1 | 1 | |
| His 94 | 1 | 2 | |
| Val 121 | 1 | 1 | |
| Leu 198 | 4 | 5 | |
| Thr 199 | — | 1 | |
| Thr 200 | 2 | 3 | |
| Acetamide linker | |||
| Phe 131 | 1 | 2 | |
| Leu 198 | 1 | — | |
| Pro 202 | 1 | — | |
| Benzhydrylpiperazine tail | |||
| Phe 131 | — | 1 | |
| Gly 132 | — | 2 | |
| Val/Alaa 135 | 2 | — | |
| Pro 202 | 2 | 7 | |
| Leu/Sera 204 | 2 | 2 | |
| Total | 18 | 27 | |
These structural data are in line with the reported inhibition constants.31 Indeed, the higher affinity of 1 for hCA VII compared to hCA II can be rationalized considering that the inhibitor is stabilized by a higher number of polar and hydrophobic interactions in the active site of hCA VII compared to hCA II. Moreover, since it has been already reported that the H-bond interaction with Gln92 does not play a dominant role in the enzyme–inhibitor binding,27 the higher affinity of compound 1 towards hCA VII can be mostly attributed to van der Waals interactions, which are higher than those established with hCA II along the entire inhibitor scaffold (see Table 2).
Fig. 4 reports the structural superposition of compound 1, when bound to hCA II and hCA VII active site, with several other CAIs containing an acetamide moiety as a liker between the benzenesulfonamide ZBG and the ring tail.41–47
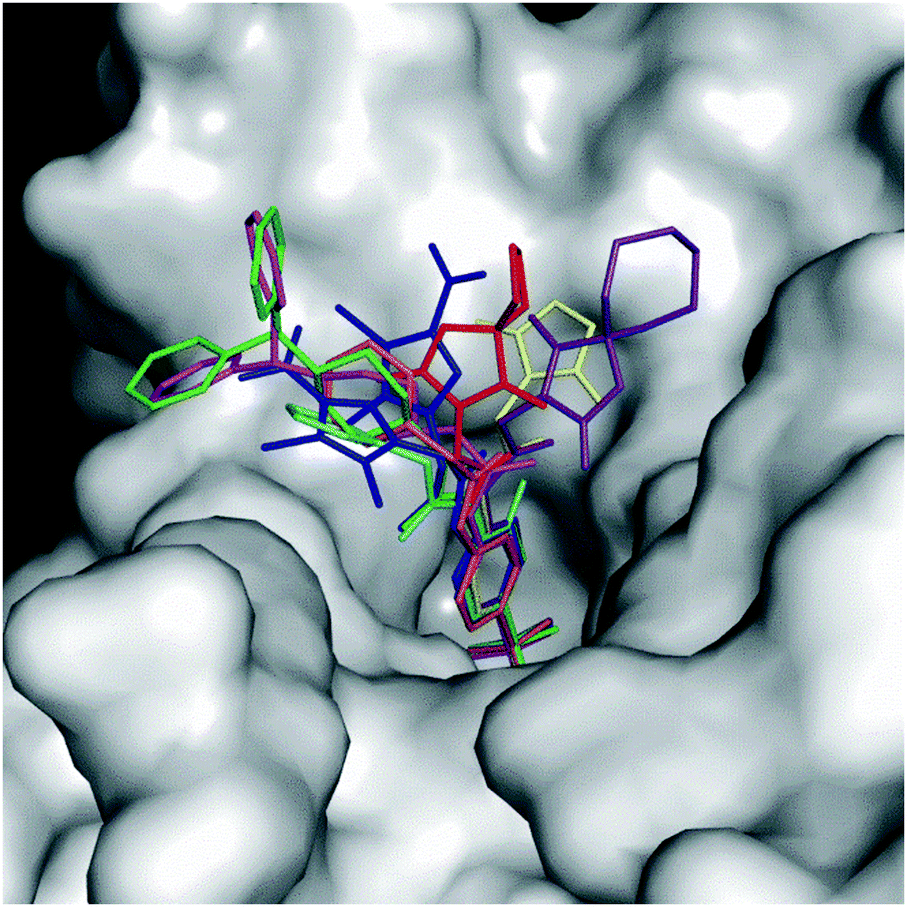 | ||
| Fig. 4 Superposition of some benzenesulfonamide derivatives containing the acetamide linker when bound to the CA active site with 1 in the conformation observed in the complex with hCA II (green) and hCA VII (magenta). 5T74 is colored in yellow, 6OE0 is colored in purple, 5NXV in blue (for this compound a double conformation is observed), 3OYS in orange, 6ODZ in red, and 3R17 in cyan. hCA II is represented as a surface-model. | ||
It is interesting to observe that, despite a substantial conservation in the benzenesulfonamide location, the orientation of both the acetamide moiety and the tail can be strongly different. In particular, the acetamide presents two main orientations rotated of about 180° like those observed for compound 1 in the active site of hCA II and hCA VII, whereas the ring tails occupy different regions of the active site thanks to the conformational plasticity of the N3–C4–C5–N6/C6 dihedral angle of the linker (in some inhibitors the N6 atom is substituted by a carbon atom indicated as C6) (see Fig. 3B and Table 3).
| PDB code | C1–C2–N3–C4 dihedral angle (°) | N3–C4–C5–N6/C6 dihedral angle (°) | Inhibitor chemical structure |
|---|---|---|---|
| a hCA II structure containing the inhibitor in conformation (a). b hCA II structure containing the inhibitor in conformation (b). | |||
| 6ZR8 | −7.6 | 154.4 |
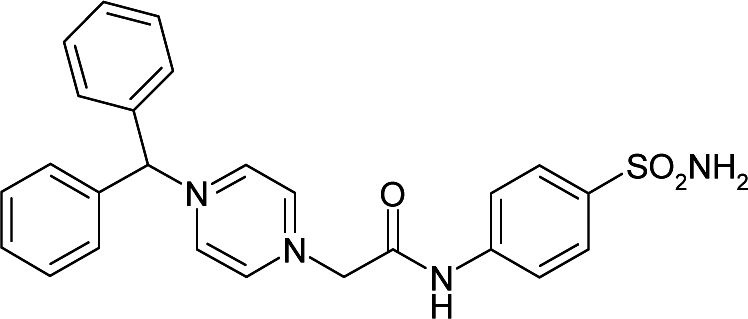
|
| 6ZR9 | 178.2 | −98.5 | |
| 3R16 | 179.1 | −91.5 |
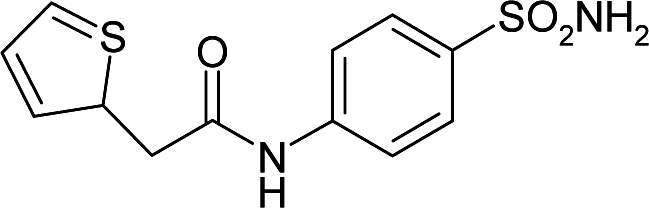
|
| 3R17 | −22.4 | 133.6 |
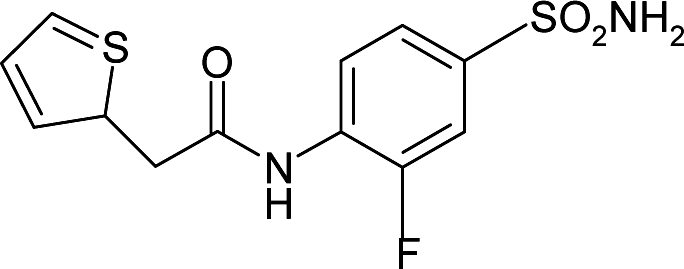
|
| 4ILX | 176.8 | −100.6 |
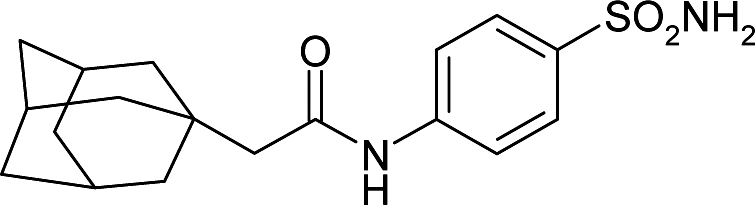
|
| 3OYS | 178.4 | −101.1 |
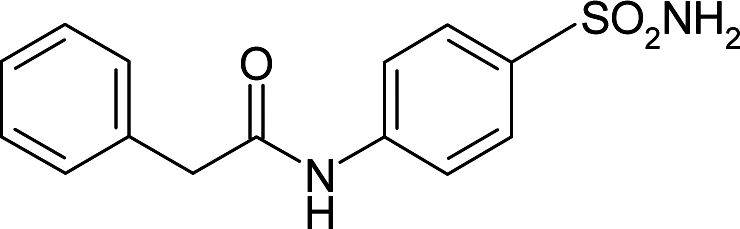
|
| 6ODZ | 165.0 | −165.4 |
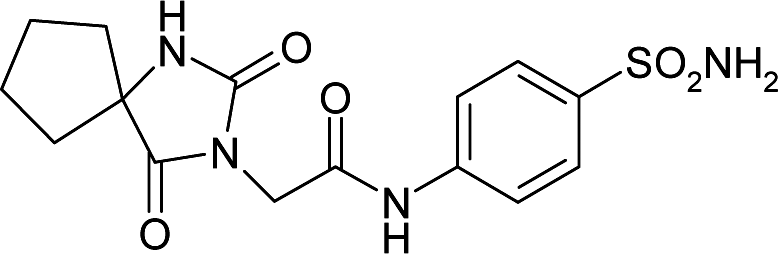
|
| 6OE0 | 150.8 | 155.3 |
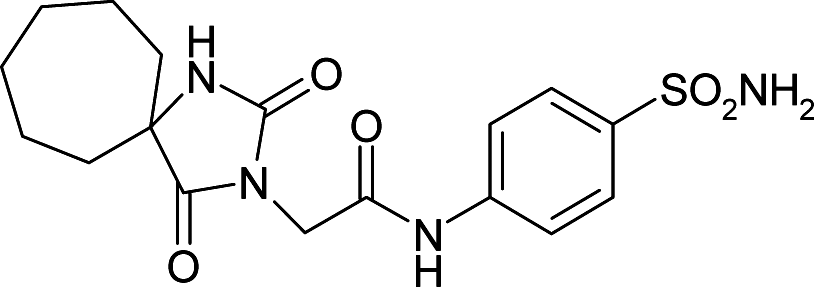
|
| 5T74 | 156.9 | −177.8 |
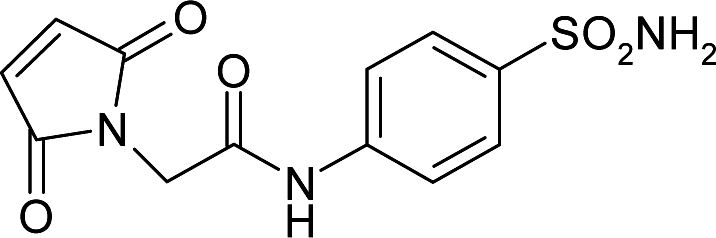
|
| 5NXV | −0.35 | −81.2 |
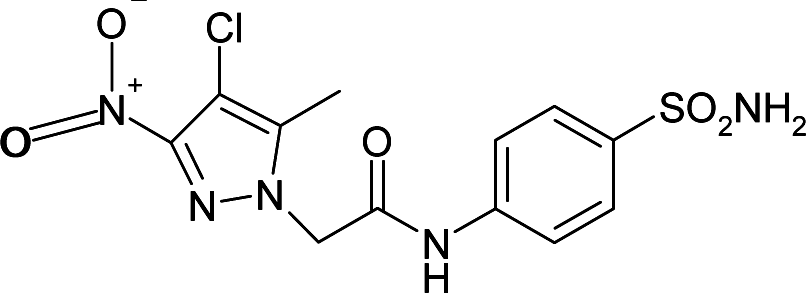
|
| 5NXV | −177.7 | 81.2 | |
| 3ML2 | 168.6 | −93.2 |
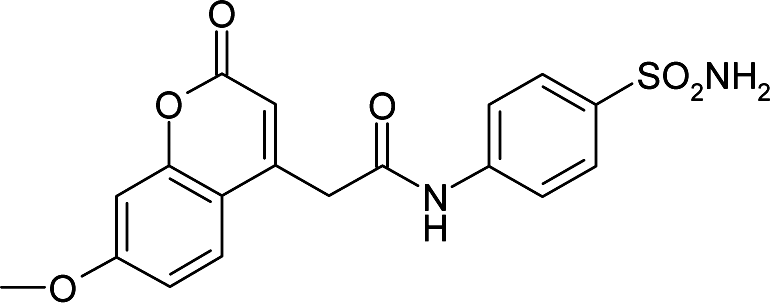
|
This dihedral angle can assume multiple values, differently from what observed for the most utilized ureido linker. Indeed, in the latter case the single C–C bond is substituted by a partial double C–N bond and the corresponding dihedral angle can only assume 0° and 180° values, thus strongly limiting the possible positions of the tail within the enzyme active site.33,48
Experimental
Protein expression and purification
Home-made hCA II and hCA VII were obtained following the experimental procedure previously reported by our group.39,49 Regarding hCA VII, a mutated form where the cysteine residues in position 183 and 217 were mutated into serines was used, since this mutant is more suitable for crystallization.39X-ray crystallography
Crystals of the hCA II/1 adduct were obtained using the co-crystallization technique. In particular, the complex was prepared by adding a 5-fold excess of the inhibitor to a 10 mg mL−1 protein solution. The mixture was equilibrated for 30 minutes at room temperature and then used for the crystallization experiments. Drops were obtained by mixing 1 μL of enzyme/inhibitor solution with 1 μL of precipitant solution containing 1.2 M sodium citrate, 0.1 M Tris–HCl, pH 8.5 and further equilibrated over a well containing 500 μL of precipitant. Crystals suitable for X-ray analysis appeared in the drops within 2–3 days and grew to a maximum dimension of 0.3 × 0.25 × 0.3 mm3 in about one week.Crystals of the hCA VII/1 adduct were obtained using the soaking technique at room temperature and the vapor diffusion hanging drop method. Equal volumes of enzyme (5 mg mL−1 in 0.02 M Tris–HCl pH 8.0 and 0.1 M NaCl) and precipitant solution (25% v/v Peg 3350, 0.2 M ammonium acetate and 0.1 M Tris pH 8.5) were mixed and equilibrated against 500 μL reservoir containing the same precipitant. Then, 0.1 μL of a solution containing 2.5 mM compound 1 dissolved in 100% DMSO was added to a 1 μL drop of hCA VII crystals, which were kept in this soaking solution overnight.
hCA II/1 and hCA VII/1 crystals were then transferred in a freshly prepared precipitant solution containing the inhibitor and glycerol as cryo-protectant at the concentration of 15% (v/v) and 25% (v/v) for hCA II and hCA VII, respectively, prior to be frozen in a gaseous nitrogen stream. Complete X-ray data were collected using a copper rotating anode generator developed by Rigaku and equipped with a Rigaku Saturn CCD detector. Diffraction data were processed and scaled using the HKL2000 program (HKL Research).50 All data collection statistics are reported in Table 1.
The initial phases of hCA II/1 and hCA VII/1 structures were calculated using the atomic coordinates of the unbound enzymes (PDB accession code 1CA2 and 6G4T for hCA II and hCA VII, respectively) with waters removed.39,40 The structures were refined using the CNS program,51 whereas model building and map inspections were performed using the program O.52 Topology files for inhibitor 1 were obtained using the PRODRG server.53 Refinement statistics for hCA II and hCA VII adducts are reported in Table 1. Coordinates and structure factors have been deposited in the Protein Data Bank (accession codes 6ZR8 and 6ZR9 for hCA II/1 and hCA VII/1, respectively).
Conclusions
Two main results can be derived from the study here reported: (i) the acetamide moiety is an ideal linker to connect a ZBG and a suitable tail in the design of CAIs. Indeed, thanks to its conformational flexibility, the tail can bind in different regions of the active site cavity establishing as many as possible interactions with enzyme residues; (ii) the molecular determinants responsible of the different inhibition constants of compound 1 against hCA VII and hCA II isoforms have been identified highlighting that, due to the tail length, inhibitor 1 can reach the border of the active site cavity which is the most variable region among the different CA isoforms. This permits significant differences in the number of favorable enzyme/inhibitor interactions and consequently, gaining selectivity in inhibiting the two hCA isoforms.In conclusion, these studies open new perspectives in the structure-based drug design of selective CA inhibitors, suggesting that an acetamide linker and very long tails are excellent tools to achieve this goal.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support for this research by MIUR-PRIN2017 (grant 201744BN5T), MIUR-PON “Ricerca e Innovazione” 2014–2020 (grant MOLIM ONCOBRAIN LAB) and Regione Campania PO FESR 2014–2020 (grant eMORFORAD). We thank Maurizio Amendola and Luca De Luca for their skillful technical assistance with X-ray measurements.References
- V. Alterio, A. Di Fiore, K. D'Ambrosio, C. T. Supuran and G. De Simone, Chem. Rev., 2012, 112, 4421–4468 CrossRef CAS.
- C. T. Supuran, A. Di Fiore and G. De Simone, Expert Opin. Emerging Drugs, 2008, 13, 383–392 CrossRef CAS.
- Carbonic Anhydrases as Biocatalysts – From Theory to Medical and Industrial Applications, ed. C. T. Supuran and G. De Simone, Elsevier B.V., Amsterdam, 2015 Search PubMed.
- J. F. Domsic, B. S. Avvaru, C. U. Kim, S. M. Gruner, M. Agbandje-McKenna, D. N. Silverman and R. McKenna, J. Biol. Chem., 2008, 283, 30766–30771 CrossRef CAS.
- K. M. J. Merz, J. Am. Chem. Soc., 1991, 113, 406–411 CrossRef CAS.
- D. N. Silverman and R. McKenna, Acc. Chem. Res., 2007, 40, 669–675 CrossRef CAS.
- A. Roy and S. Taraphder, J. Phys. Chem. B, 2007, 111, 10563–10576 CrossRef CAS.
- S. Z. Fisher, C. M. Maupin, M. Budayova-Spano, L. Govindasamy, C. Tu, M. Agbandje-McKenna, D. N. Silverman, G. A. Voth and R. McKenna, Biochemistry, 2007, 46, 2930–2937 CrossRef CAS.
- A. Thiry, J. M. Dogne, C. T. Supuran and B. Masereel, Curr. Top. Med. Chem., 2007, 7, 855–864 CrossRef CAS.
- A. Thiry, B. Masereel, J. M. Dogne, C. T. Supuran, J. Wouters and C. Michaux, ChemMedChem, 2007, 2, 1273–1280 CrossRef CAS.
- J. L. Perez Velazquez, Eur. J. Neurosci., 2003, 18, 1337–1342 CrossRef.
- M. N. Asiedu, G. L. Mejia, C. A. Hubner, K. Kaila and T. J. Price, J. Pain, 2014, 15, 395–406 CrossRef CAS.
- R. Del Giudice, D. M. Monti, E. Truppo, A. Arciello, C. T. Supuran, G. De Simone and S. M. Monti, Biol. Chem., 2013, 394, 1343–1348 CAS.
- D. M. Monti, G. De Simone, E. Langella, C. T. Supuran, A. Di Fiore and S. M. Monti, J. Enzyme Inhib. Med. Chem., 2017, 32, 5–12 CrossRef CAS.
- A. Di Fiore, D. M. Monti, A. Scaloni, G. De Simone and S. M. Monti, Oxid. Med. Cell. Longevity, 2018, 2018, 2018306 Search PubMed.
- F. Carta, A. Scozzafava and C. T. Supuran, Expert Opin. Ther. Pat., 2012, 22, 747–758 CrossRef CAS.
- A. Thiry, J. M. Dogne, C. T. Supuran and B. Masereel, Curr. Pharm. Des., 2008, 14, 661–671 CrossRef CAS.
- F. Carta, M. Aggarwal, A. Maresca, A. Scozzafava, R. McKenna and C. T. Supuran, Chem. Commun., 2012, 48, 1868–1870 RSC.
- F. Carta, A. Akdemir, A. Scozzafava, E. Masini and C. T. Supuran, J. Med. Chem., 2013, 56, 4691–4700 CrossRef CAS.
- S. K. Nair, P. A. Ludwig and D. W. Christianson, J. Am. Chem. Soc., 1994, 116, 3659–3660 CrossRef CAS.
- R. A. Davis, A. Hofmann, A. Osman, R. A. Hall, F. A. Muhlschlegel, D. Vullo, A. Innocenti, C. T. Supuran and S. A. Poulsen, J. Med. Chem., 2011, 54, 1682–1692 CrossRef CAS.
- F. Carta, C. Temperini, A. Innocenti, A. Scozzafava, K. Kaila and C. T. Supuran, J. Med. Chem., 2010, 53, 5511–5522 CrossRef CAS.
- A. Maresca, C. Temperini, H. Vu, N. B. Pham, S. A. Poulsen, A. Scozzafava, R. J. Quinn and C. T. Supuran, J. Am. Chem. Soc., 2009, 131, 3057–3062 CrossRef CAS.
- A. Maresca, C. Temperini, L. Pochet, B. Masereel, A. Scozzafava and C. T. Supuran, J. Med. Chem., 2010, 53, 335–344 CrossRef CAS.
- C. Temperini, A. Innocenti, A. Scozzafava, S. Parkkila and C. T. Supuran, J. Med. Chem., 2010, 53, 850–854 CrossRef CAS.
- S. M. Monti, C. T. Supuran, G. De Simone and A. Di Fiore, in Carbonic Anhydrases as Biocatalysts, ed. C. T. Supuran and G. De Simone, Elsevier B. V., 2015, ch. 9, pp. 151–168 Search PubMed.
- M. R. Buemi, A. Di Fiore, L. De Luca, A. Angeli, F. Mancuso, S. Ferro, S. M. Monti, M. Buonanno, E. Russo, G. De Sarro, G. De Simone, C. T. Supuran and R. Gitto, Eur. J. Med. Chem., 2019, 163, 443–452 CrossRef CAS.
- M. Bozdag, M. Ferraroni, C. Ward, F. Carta, S. Bua, A. Angeli, S. P. Langdon, I. H. Kunkler, A. S. Al-Tamimi and C. T. Supuran, Eur. J. Med. Chem., 2019, 182, 111600 CrossRef CAS.
- D. Tanini, A. Capperucci, M. Ferraroni, F. Carta, A. Angeli and C. T. Supuran, Eur. J. Med. Chem., 2020, 185, 111811 CrossRef CAS.
- A. Scozzafava, L. Menabuoni, F. Mincione, F. Briganti, G. Mincione and C. T. Supuran, J. Med. Chem., 1999, 42, 2641–2650 CrossRef CAS.
- C. B. Mishra, S. Kumari, A. Angeli, S. M. Monti, M. Buonanno, M. Tiwari and C. T. Supuran, J. Med. Chem., 2017, 60, 2456–2469 CrossRef CAS.
- V. Alterio, M. Hilvo, A. Di Fiore, C. T. Supuran, P. Pan, S. Parkkila, A. Scaloni, J. Pastorek, S. Pastorekova, C. Pedone, A. Scozzafava, S. M. Monti and G. De Simone, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16233–16238 CrossRef CAS.
- F. Pacchiano, M. Aggarwal, B. S. Avvaru, A. H. Robbins, A. Scozzafava, R. McKenna and C. T. Supuran, Chem. Commun., 2010, 46, 8371–8373 RSC.
- A. Cecchi, J. Y. Winum, A. Innocenti, D. Vullo, J. L. Montero, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem. Lett., 2004, 14, 5775–5780 CrossRef CAS.
- A. Petrou, A. Geronikaki, E. Terzi, O. O. Guler, T. Tuccinardi and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2016, 31, 1306–1311 CrossRef CAS.
- H. Turkmen, M. Durgun, S. Yilmaztekin, M. Emul, A. Innocenti, D. Vullo, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem. Lett., 2005, 15, 367–372 CrossRef CAS.
- T. Abdulkadir Coban, S. Beydemir, I. Gulcin, D. Ekinci, A. Innocenti, D. Vullo and C. T. Supuran, Bioorg. Med. Chem., 2009, 17, 5791–5795 CrossRef CAS.
- J. Slawinski, K. Szafranski, D. Vullo and C. T. Supuran, Eur. J. Med. Chem., 2013, 69, 701–710 CrossRef CAS.
- M. Buonanno, A. Di Fiore, E. Langella, K. D'Ambrosio, C. T. Supuran, S. M. Monti and G. De Simone, Int. J. Mol. Sci., 2018, 19, 1571 CrossRef.
- A. E. Eriksson, T. A. Jones and A. Liljas, Proteins, 1988, 4, 274–282 CrossRef CAS.
- J. Wagner, B. S. Avvaru, A. H. Robbins, A. Scozzafava, C. T. Supuran and R. McKenna, Bioorg. Med. Chem., 2010, 18, 4873–4878 CrossRef CAS.
- N. Hen, M. Bialer, B. Yagen, A. Maresca, M. Aggarwal, A. H. Robbins, R. McKenna, A. Scozzafava and C. T. Supuran, J. Med. Chem., 2011, 54, 3977–3981 CrossRef CAS.
- A. Angeli, L. Di Cesare Mannelli, C. Ghelardini, T. S. Peat, G. Bartolucci, M. Menicatti, F. Carta and C. T. Supuran, Eur. J. Med. Chem., 2019, 177, 188–197 CrossRef CAS.
- S. Biswas, M. Aggarwal, O. Guzel, A. Scozzafava, R. McKenna and C. T. Supuran, Bioorg. Med. Chem., 2011, 19, 3732–3738 CrossRef CAS.
- S. Biswas, F. Carta, A. Scozzafava, R. McKenna and C. T. Supuran, Bioorg. Med. Chem., 2013, 21, 2314–2318 CrossRef CAS.
- A. Pecina, J. Brynda, L. Vrzal, R. Gnanasekaran, M. Horejsi, S. M. Eyrilmez, J. Rezac, M. Lepsik, P. Rezacova, P. Hobza, P. Majer, V. Veverka and J. Fanfrlik, ChemPhysChem, 2018, 19, 873–879 CrossRef CAS.
- K. H. DuBay, K. Iwan, L. Osorio-Planes, P. L. Geissler, M. Groll, D. Trauner and J. Broichhagen, ACS Chem. Biol., 2018, 13, 793–800 CrossRef CAS.
- M. Y. Mboge, B. P. Mahon, N. Lamas, L. Socorro, F. Carta, C. T. Supuran, S. C. Frost and R. McKenna, Eur. J. Med. Chem., 2017, 132, 184–191 CrossRef CAS.
- G. De Simone, A. Angeli, M. Bozdag, C. T. Supuran, J. Y. Winum, S. M. Monti and V. Alterio, Chem. Commun., 2018, 54, 10312–10315 RSC.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- A. T. Brunger, Nat. Protoc., 2007, 2, 2728–2733 CrossRef CAS.
- T. A. Jones, J. Y. Zou, S. W. Cowan and M. Kjeldgaard, Acta Crystallogr., Sect. A: Found. Crystallogr., 1991, 47(Pt 2), 110–119 CrossRef.
- A. W. Schuttelkopf and D. M. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef.
Footnotes |
| † These authors contributed equally to the work. |
| ‡ Present address: College of Pharmacy Sookmyung Women's University, Seoul, South Korea. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |