
Fabrication of sea urchin-like hierarchical porous SAPO-11 molecular sieves toward hydrogenation of lipid to jet fuel†
Lingmei
Yang
 abc,
Shiyou
Xing
d,
Wen
Luo
abc,
Gai xiu
Yang
*abc,
Zhongming
Wang
abc and
Pengmei
Lv
*abc
abc,
Shiyou
Xing
d,
Wen
Luo
abc,
Gai xiu
Yang
*abc,
Zhongming
Wang
abc and
Pengmei
Lv
*abc
aGuangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, China. E-mail: yanggx@ms.giec.ac.cn; lvpm@ms.giec.ac.cn
bCAS Key Laboratory of Renewable Energy, Guangzhou 510640, China
cGuangdong Provincial Key Laboratory of New and Renewable Energy Research and Development, Guangzhou 510640, China
dInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
First published on 18th November 2020
Abstract
Hydrogention of lipid to jet fuel especially iso-alkanes is considered as a promising technology to improve the cold-flow properties of bio-jet fuel. In this study, we successfully synthesized a hydroisomerization catalyst that is based on a SAPO-11 molecular sieve (SAPO-11-A-6) with a sea urchin-like and hierarchical porous structure. SAPO-11-A-6 was fabricated with an Al![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cetyltrimethylammonium bromide (CTAB) ratio of 0.006 after pre-crystallization using an in situ seed-induced steam-assisted method (SISAC). The SAPO-11-A-6 molecular sieves exhibited abundant mesopores, a larger external surface area, and more acidic sites than SAPO-11-S molecular sieves without CTAB. After loading nickel nanoparticles by using a citric-acid-assisted impregnation method, the hydrogenation of oleic acid results indicated that among the series of Ni/SAPO-11 catalysts, a maximum of 78.2% isomer selectivity was achieved by the Ni/SAPO-11-A-6 catalyst under reaction conditions of 360 °C, 4 h, and 4.0 MPa H2. The crystal formation route of the sea urchin-like hierarchical porous SAPO-11 was proposed: after pre-crystallization, the cationic surfactant CTAB entered the layered semi-crystalline phase, contributing to the formation of the novel sea urchin-like mesoporous SAPO-11 structure.
:cetyltrimethylammonium bromide (CTAB) ratio of 0.006 after pre-crystallization using an in situ seed-induced steam-assisted method (SISAC). The SAPO-11-A-6 molecular sieves exhibited abundant mesopores, a larger external surface area, and more acidic sites than SAPO-11-S molecular sieves without CTAB. After loading nickel nanoparticles by using a citric-acid-assisted impregnation method, the hydrogenation of oleic acid results indicated that among the series of Ni/SAPO-11 catalysts, a maximum of 78.2% isomer selectivity was achieved by the Ni/SAPO-11-A-6 catalyst under reaction conditions of 360 °C, 4 h, and 4.0 MPa H2. The crystal formation route of the sea urchin-like hierarchical porous SAPO-11 was proposed: after pre-crystallization, the cationic surfactant CTAB entered the layered semi-crystalline phase, contributing to the formation of the novel sea urchin-like mesoporous SAPO-11 structure.
Introduction
Recently, the depletion of fossil fuels and environmental pollution have urged the use of sustainable renewable energy sources. Biofuels such as bioethanol, biodiesel, and bio-jet fuels have attracted extensive attention and are considered as the most promising replacements for fossil fuels.1–3 Bio-jet fuels including normal alkanes, iso-alkanes, and aromatics are ideal to meet the requirements of jet fuel and can be synthesized by the catalytic conversion of vegetable oil or waste oil.4,5 To meet the lower freezing point requirements of jet fuel, iso-alkanes (>70%) are added as the main components in bio-jet fuels. Therefore, one-step hydroisomerization of lipids to iso-alkanes is an important research direction in this context.In recent years, the catalytic hydroisomerization of lipids has used bifunctional catalysts, i.e., the combination of a metal center and acid sites. Usually, the precious metals Pt and Pd6–8 and transition metals Ni,9 Co,10 and Cu11,12 are used as metal centers for hydro/dehydrogenation and molecular sieves such as H-beta,13–15 ZSM-22,16 and SAPO-1117–19 are used as the acid supports when preparing bifunctional catalysts for the hydroisomerization reaction. The SAPO-11 molecular sieve as the support, with a one-dimensional ten-membered ring (0.39 nm × 0.63 nm) structure and moderate acidity,20 has shown good catalytic performance in hydroisomerization reactions.21
However, with the same metal loadings, considering mass transport, the active sites and the acidic support were less accessible to bulkier reactants such as triglycerides and fatty acids, which are the main component of vegetable oil and waste oil. This is due to the molecular sieves’ microporous structure and small external area. Thus, the pores and specific surface area of SAPO-11 molecular sieves need to be enlarged. Common strategies for molecular sieve reaming include post-treatment methods.22 However, post-treatment methods do not produce ordered mesoporous materials. Thus, ordered mesoporous molecular sieves are prepared by adding a secondary template such as cetyltrimethylammonium bromide (CTAB),23,24 carbon,25 organosilane,26 or glucose27 to a hard template. Blasco et al.28 revealed that SAPO-11 with higher external surface areas could be obtained using hexadecylamine as a surfactant. Varma et al.17 reported a hierarchically porous SAPO-11 using octadecyl dimethyl(3-trimethoxysilylpropyl) ammonium chloride as a structure-directing agent. Kim et al.25 fabricated a mesoporous SAPO-11 using carbon for secondary templating. Han et al.29 synthesized an ultrafine SAPO-11 using CTAB as a secondary template by the steam-assisted method.
According to a previous report,28,29 the secondary template was added to the starting gel used in the molecular sieve synthesis process. It remains to be clarified if the morphology and crystallization process of the molecular sieves would be affected if the secondary template was added at a different stage of the process. In this work, novel sea urchin-like hierarchical porous SAPO-11 molecular sieves were fabricated by adding the cationic surfactant agent CTAB after forming the pre-crystallized gel using the in situ seed-induced steam-assisted method. The optimal amount of CTAB and its effect on the physicochemical properties of the sea urchin-like hierarchical porous SAPO-11 were investigated. The formation process and crystallization mechanism of the sea urchin-like hierarchical porous SAPO-11 molecular sieves, and their catalytic hydroisomerization performance toward oleic acid were further investigated.
Experimental section
Materials
Tetraethyl orthosilicate (TEOS), aluminum isopropoxide (AIP), di-n-propylamine (DPA), cetyltrimethylammonium bromide (CTAB), nickel nitrate hexahydrate, citric acid monohydrate, absolute ethanol, cyclohexane and oleic acid were obtained from Aladdin Chemical Reagent. Phosphoric acid (85% H3PO4) was obtained from the Sinopharm Chemical Reagent Company, P. R. China. H2 gas was obtained from Guangzhou Yuejia Gas Co., Ltd.Synthesis of hierarchical SAPO-11 molecular sieve and catalyst
SAPO-11-S was synthesized according to a method reported by Chen et al.,30 wherein they used the SISAC method. The typical mole composition of the synthesis gel was 1.0 Al2O3:1.0 P2O5:0.3 TEOS:2.0 DPA:50 H2O. The initial gel was first pre-crystallized in an autoclave at 160 °C for 24 h. Then, appropriate amounts of CTAB were added according to the mole ratio of Al:xCTAB, where x was 0.003, 0.006, 0.009, 0.015, or 0.03. After stirring and evaporating the water in a 60 °C water bath, 50 mL of absolute ethanol was added. Then, the mixture was stirred at 60 °C to evaporate the ethanol, and a dry gel was obtained by drying at 60 °C for 48 h. The obtained dry gel was placed in a 20 mL crucible and 20 mL of deionized water was added. The crucible was then placed in a 100 mL crystallization vessel for crystallization at 200 °C for 24 h. The resulting product was filtered, washed, and dried at 110 °C and calcined at 600 °C: the final products were defined as SAPO-11-A-X, where X was 3, 6, 9, 15, or 30. Meanwhile, the dry gel obtained without adding CTAB was named DG-16 and the dry gel with an Al:CTAB = 0.006 content was named DG-160-C. For comparison, SAPO-11-C was synthesized by the hydrothermal synthesis method.
The SAPO-11 samples were loaded with 7 wt% nickel via a citric-acid-assisted impregnation method as reported by us previously.31 2.00 g of Ni (NO3)2·6H2O and 1.98 g of citric acid (Ni/C6H8O7 molar ratio of 1:1.5) were dissolved in 100 mL of absolute ethanol. Then, 5.32 g of different SAPO-11 samples was added to the mixture. The mixture was stirred in an oil bath at 60 °C for 4 h and then stirred at 80 °C for 12 h to evaporate ethanol. The catalyst was calcined in air at 550 °C for 5 h. Finally, the highly dispersed nano-7Ni/SAPO-11 catalyst was obtained after reduction under a 10% H2/N2 atmosphere at 550 °C for 2 h.
Catalyst characterization
X-ray diffraction (XRD) was performed using a PANalytical X’Pert Pro MPD diffractometer with CuKα radiation (λ = 0.15406 nm) at 40 kV and 40 mA from 5° to 80° with a scanning step of 0.02.The specific surface area and pore structures of the samples were characterized by nitrogen physisorption at 77 K using a Micromeritics ASAP Model 2020 (vacuum degassed at 300 °C for 10 h). The specific surface area was calculated based on the Brunauer–Emmett–Teller (BET) equation over a pressure range P/P0 of 0.05–0.3. The t-plot method was applied to obtain the microporous surface area and microporous volume. The total pore volume was calculated by measuring the nitrogen adsorbed at a relative pressure of 0.99.
The samples were analyzed by scanning electron microscopy (SEM) using an S-4800 scanning electron microscope (Hitachi, Japan) at an accelerating voltage of 2.0 kV. Prior to the SEM analysis, the samples were placed under an E1010 ion sputtering instrument to improve their conductivity. Transmission electron microscopy (TEM) and energy dispersive X-ray (EDX) experiments were performed using a H-7560 electron microscope (Hitachi, Japan) with an accelerating voltage of 100 kV.
Raman spectra were obtained on a LabRAM HR800 Reman system with CCD detectors. The line at 532 nm from a He–Cd laser was used as an excitation source with an output of 5.6 mW.
The acid types of the catalysts were detected using a pyridine-infrared spectrometer (model: Nicolet 6700). The sample was first evacuated to 10−2 Pa at 200 °C and then exposed to pyridine for 15 min at room temperature, followed by degassing at 200 °C and 350 °C, respectively.
The hydrogen temperature programmed reduction (H2-TPR) test was performed using a Micromeritics AutoChem 2950 HP instrument using a thermal conductivity detector.
29Si, 27Al, and 31P nuclear magnetic resonance (MAS NMR) experiments were performed using a Bruker Ascend-500 solid-state nuclear magnetic resonance spectrometer (Germany) using a 4 mm three-resonance nuclear magnetic probe at a sample rotation speed of 15 kHz.
Catalyst evaluation
Catalyst evaluation was performed in a high-pressure autoclave with a volume of 150 mL, as described in our previous work.31 In a typical reaction, 15 g of oleic acid and 0.7 g of catalyst were added into the reactor. Before the reaction, the reactor was purged with H2 at ambient temperature three times and the pressure was regulated to 4 MPa. The hydrogenation reaction was conducted for 4 h at different temperatures (300, 320, 340, 360, and 380 °C), with a mechanical agitation speed of 200 rpm. After the reaction, the reactor was cooled to ambient temperature using tap water. Then, the gaseous products (i.e., methane (CH4), carbon monoxide (CO), CO2, and C2–C6 hydrocarbons) were quantified using a 7890 A (Agilent) gas chromatograph (GC) equipped with a GS-GASPRO capillary column (Agilent; 60 m × 0.32 m), a thermal conductivity detector (TCD), and a flame ionization detector (FID). The liquid products were filtered using a 0.22 μm organic membrane and diluted with cyclohexane (a volume ratio of 1:100). The liquid products were tested using a GC-mass spectrometer (GC/MS, Agilent 7890A-5975C) equipped with a HP-5 MS column (5% phenyl methyl silox, Agilent, 30 m × 0.25 mm × 0.25 μm). All the experiments were repeated twice and the average value of the two values was recorded.
The oleic acid conversion (xA) and product selectivity (si) were calculated using eqn (1) and (2):
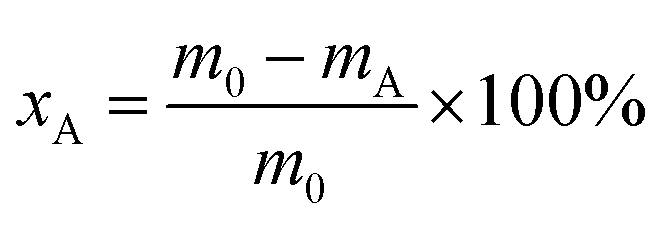 | (1) |
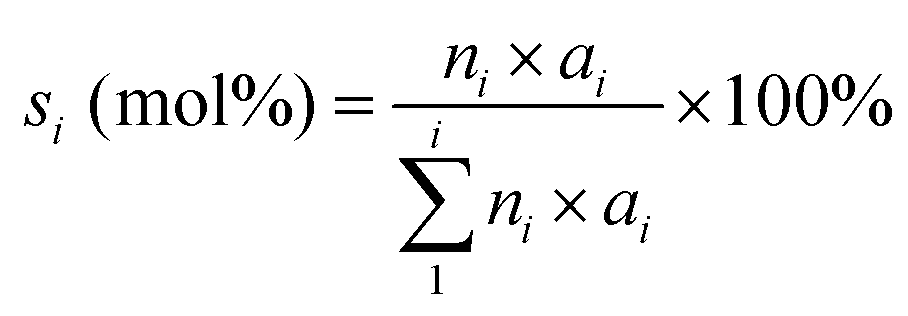 | (2) |
Results and discussion
Physicochemical characterization of sea urchin-like hierarchical porous SAPO-11
The XRD patterns of the different SAPO-11 species are shown in Fig. 1. All the species exhibit peaks at 8.1°, 9.4°, 13.1°, 15.6°, 20.3°, 21.2°, and 22.1° to 23.2°, corresponding to the classical SAPO-11 phase of the AEL-type,28 indicating that SAPO-11 crystals were successfully synthesized. Compared to that of the hydrothermally synthesized SAPO-11-C, the diffraction peaks of SAPO-11-S prepared by the SISAC method were lower in intensity and the line widths were broader. After adding an amount of CTAB (Al:CTAB = 0.006) to the pre-crystallite gel, the diffraction peaks broadened further, indicating that the crystallite size of SAPO-11-A-6 decreased. The relative crystallinity (RC) of different SAPO-11 samples is shown in Table 1. Compared with that of SAPO-11-C, the RC value of SAPO-11-A-6 was 84.4%. This was due to the surfactant CTAB with a tetrahedral structure, which played an important role as a crystal nucleus, while the cation of the tetrahedral structure entered the molecular sieve layer structure, resulting in recrystallization and reducing the crystallite size.
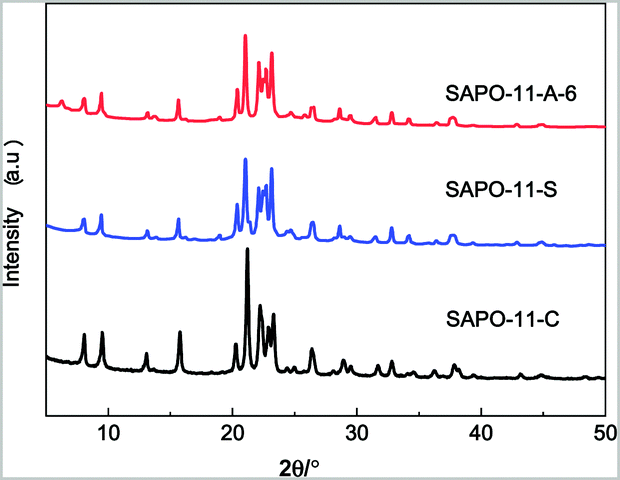 | ||
| Fig. 1 XRD patterns of different SAPO-11 species. | ||
| Catalysts | S BET (m2 g−1) | S mic (m2 g−1) | S ext (m2 g−1) | V meso (cm3 g−1) | V mic (cm3 g−1) | V toal (cm3 g−1) | R (%) |
|---|---|---|---|---|---|---|---|
| a Surface area calculated by the BET method. b Micropore volume and surface area calculated by the t-plot method. c Mesopore volume calculated by the BJH method. | |||||||
| SAPO-11-C | 193.1 | 150.8 | 42.3 | 0.136 | 0.060 | 0.196 | 100 |
| SAPO-11-S | 217.9 | 107.5 | 110.4 | 0.153 | 0.046 | 0.232 | 90.5 |
| SAPO-11-A-3 | 226.1 | 163.6 | 62.5 | 0.102 | 0.066 | 0.181 | 83.4 |
| SAPO-11-A-6 | 283.9 | 133.2 | 150.7 | 0.201 | 0.057 | 0.260 | 84.4 |
| SAPO-11-A-9 | 241.1 | 131.3 | 109.8 | 0.126 | 0.053 | 0.185 | 80.1 |
| SAPO-11-A-15 | 202.9 | 141.1 | 61.8 | 0.101 | 0.057 | 0.167 | 70.1 |
| SAPO-11-A-30 | 197.8 | 141.1 | 56.7 | 0.096 | 0.062 | 0.165 | 53.2 |
The relativity crystallinity of SAPO-11 species with different CTAB contents is shown in S1 (ESI†). For different CTAB contents, the line of relative crystallinity was denoted as I and II: they indicate the influence on the crystallization process when adding the surfactant CTAB. The ratio of Al:xCTAB should be maintained between 0.003 and 0.009 (within the critical micelle) so that the added surfactant CTAB can form crystal defects, which influence the relative crystallinity. As the amount of CTAB increases and exceeds the critical micelle concentration, in region II, the surfactant CTAB would ionize and form a tetrahedral cationic structure in the crystallization process, which can combine tetrahedral phosphate anions, and the relative crystallinity varies greatly, which is the same as the results reported by Seo et al.32
Table 1 gives the textural properties of the SAPO-11 species. As can be seen in Table 1, compared with SAPO-11-C, the specific surface area of SAPO-11-S increased from 193.1 to 217.9 m2 g−1; in particular, the external surface area increased from 42.3 to 110.4 m2 g−1, and the pore volume also increased significantly from 0.196 to 0.232 cm3 g−1, indicating that SAPO-11 prepared by the SISAC method has an increased external surface area and pore volume. When the Al:CTAB ratio was 0.006 after the pre-crystalline process, the largest external surface area and mesopore volume were obtained as 150.7 and 0.201 cm3 g−1, respectively. As is known, the addition of tetrahedral CTAB induces the formation of hydrogen bonds with tetrahedral P–OH and PO functional groups. The CTAB was removed from the molecular sieve framework after calcining, thus expanding the mesoporous structure. Meanwhile, the CTAB also is a foaming agent, which can increase the external surface area. If the amount of added CTAB is too much, the SAPO-11 grains will aggregate, blocking the molecular sieve pores and reducing the specific surface area.
Fig. 2 shows the N2 adsorption and desorption curves of different SAPO-11 samples. The hysteresis loop and relative pressure of SAPO-11-C prepared using the hydrothermal method were not completely vertical, indicating that, according to the IUPAC classification, they were a typical H1 type. The N2 adsorption–desorption curve initially showed a slight increase and then bent at a relative pressure of P/P0 < 0.01. This demonstrates a micropore volume adsorption and desorption, indicating that the prepared SAPO-11-C had an ordered microporous structure. The hysteresis loop of the SAPO-11-S sample prepared using the SISAC method presents typical H1 adsorption and desorption and a type IV annular hysteresis loop. After adding the surfactant agent CTAB, the adsorption and desorption curves of these samples varied greatly, and the inflection points were slightly different. The inflection point was ∼0.4 when the Al:CTAB ratio was 0.003. It moved to ∼0.5 when the Al:CTAB ratio reached 0.006 and then went back to a point close to 0.4 when the Al:CTAB ratio increased to 0.015 and 0.03. This shift in the inflection point indicates changes in the pore size similar to the case of the mesopores observed in Table 1. This indicates that adding too much CTAB causes the collapse or blockage of the void channel, so only an appropriate amount of CTAB should be added. According to a report by Seo et al.,32 CTAB is an intermediate templating agent that can easily aggregate and form micelles with a specific morphology, which increases the spatial structure between the molecular sieves and results in the formation of mesopores and macropores after calcination.
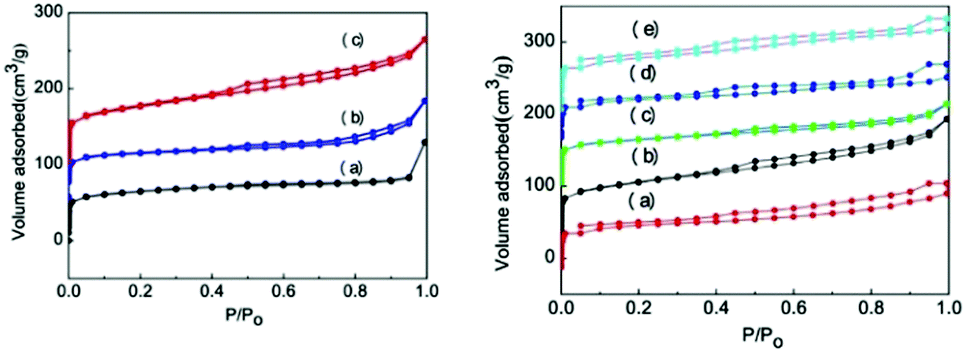 | ||
| Fig. 2 N2 isotherms of the SAPO-11 samples (left): (a) SAPO-11-C, (b) SAPO-11-S, and (c) SAPO-11-A-6, with various pore-broadening agent CTAB contents (right): (a) 0.003, (b) 0.006, (c) 0.009, (d) 0.015, and (e) 0.03. | ||
The SEM images and TEM images of different SAPO-11 species are shown in Fig. 3. The SAPO-11-C molecular sieves had a roughly spherical shape (Fig. 3a). Tiny grains were observed outside the sphere, which may be scattered crystal nuclei or micelles that form during the crystallization of the SAPO-11 molecular sieves. Nanometer rectangular crystals were observed using transmission electron microscopy (Fig. 3e), which indicated that SAPO-11 synthesized using the hydrothermal method was present as spheres formed by the accumulation of small rectangular particles, as previously reported.33 The SAPO-11-S molecular sieves also had spherical structures, with a grain size of 2–3 μm (Fig. 3b), but their surfaces were relatively smooth. No tiny grains were scattered outside the crystal, indicating that the SAPO-11-S molecular sieves had no irregular amorphous phase. This result can be attributed to the rapid nucleation of the quasi-solid dry gel systems obtained using the SISAC method. After adding the CTAB, the SAPO-11-A-6 molecular sieves showed very rough surfaces and more needle-like branches (Fig. 3c, d, g and h). The SEM images clearly showed that the SAPO-11-A-6 molecular sieves had globular structures with diameters of about 2 μm, with many thorn-like structures on the outside of the particles, so that they looked like sea urchins. A rod-shaped edge 20–30 nm long can also be observed in the enlarged image in Fig. 3h, indicating that the sea urchin-like SAPO-11-A-6 molecular sieve was stacked with nano-rod-like structures. The addition of CTAB results in a smaller crystal phase, the special sea urchin-like morphology, pore channels between the crystal pores, and an increased outer surface area. The EDX mapping (Fig. 3i) spectrum analysis of the SAPO-11-A-6 molecular sieves shows that silicon, aluminum, phosphorus, and oxygen were evenly distributed on the surface of the molecular sieves.
 | ||
| Fig. 3 SEM and TEM images of SAPO-11-C (a, e), SAPO-11-S 11 (b, f), SAPO-11-A-6 (c, d, g, h), and EDX elemental mapping of T SAPO-A-6 (i). | ||
The Brønsted (B) and Lewis (L) acid types and amounts of acid sites in different SAPO-11 samples were characterized using pyridine infrared spectroscopy, and the results are shown in Fig. 4a and b and Table S1 (ESI†). The peaks at ca. 1450, 1540, and 1490 cm−1 correspond to the L acid center, B acid center, and the coefficient peak of B and L acid sites, respectively.34 The SAPO-11-S molecular sieves contained mainly B acid sites with few L acid sites desorbed at 200 °C. Compared with SAPO-11-S, the total number of acid sites of SAPO-11-A-6 increased; especially, the concentrations of medium and strong acid sites and B acid sites reached 171 and 140 μmol g−1 desorbed at 360 °C. Thus, the SAPO-11-A-6 molecular sieve showed better catalytic hydroisomerization performance because of the strong acidic sites and mesoporous structure.
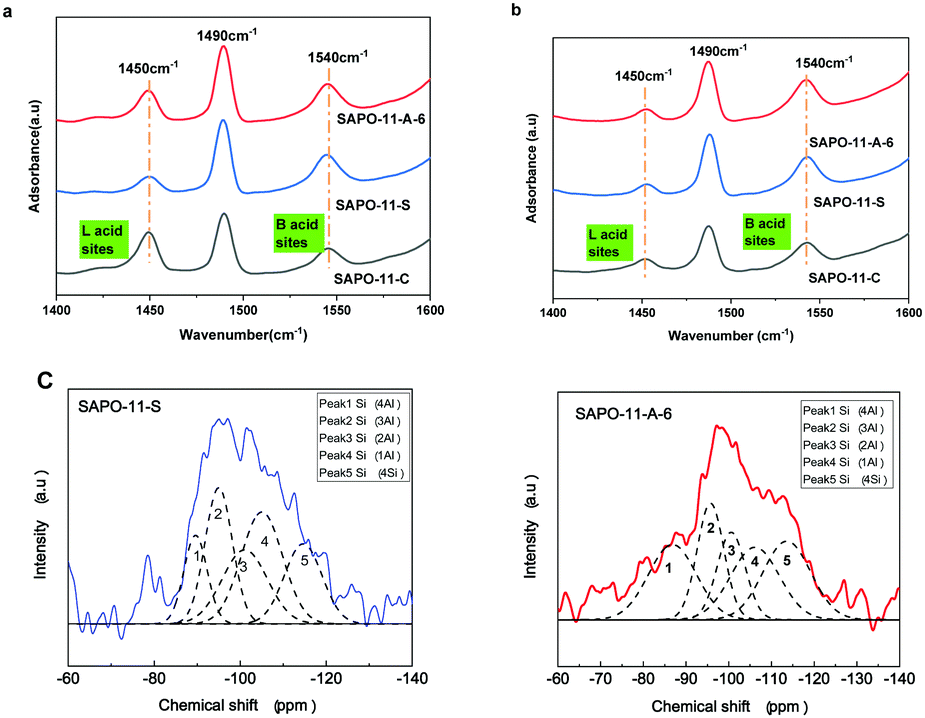 | ||
| Fig. 4 The pyridine IR absorption spectrum measured at 200 °C (a) and 360 °C (b) and 29Si MAS NMR spectra of different SAPO-11 species (c). | ||
29Si MAS NMR spectra and their deconvolution of the different SAPO-11 species are shown in Fig. 4c and Table S2 (ESI†). According to Fig. 4c, the main peak was at about −98 ppm, indicating that the Si atoms mainly form Si(2Al) sites in the synthesized SAPO-11 by the SISAC method. Table S2 (ESI†) shows the deconvolution results of different Si species. Although SAPO-11-S had the same initial Si content as SAPO-11-A-6, SAPO-11-A-6 has a larger peak area and a higher concentration of Si(nAl) (0 < n < 4) sites than SAPO-11-S. Consequently, there are more medium-strong B acid sites than in SAPO-11-S, which is consistent with the pyridine-IR results. Huang et al.35 reported that the role of surfactants in synthetic gels enhances Si dispersion, increasing the number of medium-strong B acid sites in the Si(nAl) (0 < n < 4) environment.
Effects of adding CTAB on the crystallization of SAPO-11 molecular sieves
The powder XRD patterns, Raman spectra, and 27Al and 31P solid-state NMR spectra were used to characterize the dry gel to determine the effect of adding the surfactant CTAB on the structure of the powder formation. The specific results are discussed below.Fig. 5A shows the XRD patterns of DG-160 and DG-C-160. For the unwashed sample, a very high peak at 2θ = 6.2° was found for DG-160, which is a long-chain-ordered layered intermediate, forming a (100) layered semicrystalline structure.36 The peaks at 2θ = 8.1°, 20.4°, and 21–23° correspond to the (110), (310), and (240) crystal planes of SAPO-11 (◆ represents the crystal plane that appeared). Thus, the initial SAPO-11 molecular sieve AEL structure had already formed, indicating that the dry gel forms an embryo for the SAPO-11 after pre-crystallization at 160 °C for 24 h. The small peak at 2θ = 13.0° is the transitional intermediate structure of the SAPO-11 molecular sieve.37 According to Bao's report,23 due to electrostatic attraction, the cationic surfactants can be easily adsorbed on the negatively charged SAPO-11 surface. With increasing crystallization time, the negative charges of the precursor are bridged and neutralized by CTAB, then the CTAB in solution will further bridge on the precursor surface to form bilayer micelles, which can increase the electrostatic repulsion in the solution, resulting in increased space between the silicoaluminophosphate layers. Compared with DG-160, the peak at 2θ = 6.2° was weakened and no peak was observed at 2θ = 13.0°, indicating that the CTAB was introduced into the silicoaluminophosphate layered structure.
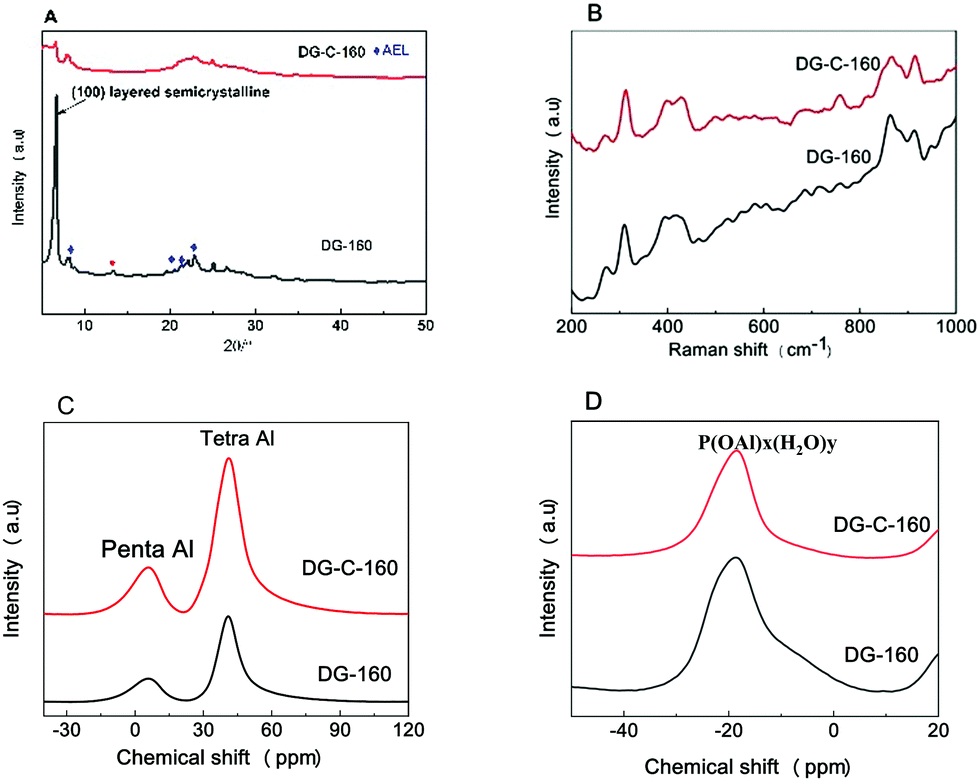 | ||
| Fig. 5 XRD patterns (A), Raman spectra (B), and 27 Al (C) and 31 P (D) MAS NMR spectra of the synthesized DG-160 and DG-C-160. | ||
The effect of adding CTAB on the structure of the dry gel was further analysed by Raman spectroscopy (with excitation at 532 nm), and the results are shown in Fig. 5B. The peak at 262 cm−1 in DG-160 is due to the ten-membered ring structure of SAPO-11. The peak in the Raman spectrum of the molecular sieves at 300–600 cm−1 can be assigned to the movement of oxygen atoms in the plane perpendicular to the T–O–T bond (where T refers to silicon or aluminum).38 The peaks in the Raman spectrum at 397 and 427 cm−1 can be attributed to the four-membered and six-membered ring structures in the molecular sieve framework. The DG-C-160 dry gel has the same peaks at 262, 397, and 427 cm−1, indicating that the addition of CTAB did not change the structure of the SAPO-11 (ten-membered ring, four-membered ring, and six-membered ring).
The 27Al solid-state nuclear magnetic resonance (NMR) spectrum of the dry gel is shown in Fig. 5C. The peaks at 41 and 8 ppm are produced by the AlO4 structural unit (4-coordinate aluminum) and the AlO5 structural unit (5-coordinate aluminum), respectively.39 In the dry DG-160 powder prepared by pre-crystallization, most of the aluminum ions are 4-coordinate, indicating that the aluminum was successfully incorporated into the molecular sieve skeleton. Compared with DG-160, the peak area of DG-C-160 at 41 ppm increased significantly, indicating that adding the surfactant CTAB favoured the formation of the 4-coordinate aluminum framework. In the 31P NMR spectrum (Fig. 5D), there is a large broad peak at −19 ppm for the two SAPO-11 samples, which is due to the P(OAl)x(H2O)y coordination structure of the aluminum–phosphorus layered intermediate.40 For the dry DG-C-160 sample, the 31P NMR peak at −19 ppm was weaker than that of DG-160, suggesting a loss of layered intermediate structure, which is consistent with the XRD results.
Furthermore, the crystallization process and the formation mechanism of the hierarchical porous SAPO-11 molecular sieves after the dry gel formed were investigated by using XRD and SEM.
The XRD patterns of the unwashed SAPO-11 molecular sieve samples after different crystallization times at 200 °C (Fig. 6A and B) illustrate the crystallization process of the two samples, SAPO-11-S and SAPO-11-A-6. As can be seen in Fig. 6A, the SAPO-11-S dry gel has a strong diffraction peak at 2θ = 6.2°, which corresponds to a layered semi-crystalline phase.36,37 The diffraction peaks for the dry gel at 2θ = 8.1°, 21.2°, and 22.1–23.2° can be attributed to the formation of the AEL structure,28 suggesting that the prototype structure of the SAPO-11 molecular sieves formed after pre-crystallization. After longer crystallization times, the intensity of the diffraction peak at 2θ = 6.2° for SAPO-11-S first increased and then decreased. The diffraction peaks of the AEL structure were obvious after 3 h of crystallization. After 6 h of crystallization, the diffraction peak at 2θ = 6.2° disappeared, implying that the layered semi-crystalline phase structure had completely transformed into an AEL structure. As can be seen in Fig. 6B, for the dry gel of SAPO-11-A-6, the peak for the (100) layered semi-crystalline phase became weaker and broader because the cationic surfactant of CTAB was easily adsorbed on the surface of the negatively charged SAPO-11 precursors due to the electrostatic repulsion.23 After 2 h of crystallization, diffraction peaks appeared at 2θ = 6.2°, 8.1°, 9.4°, 13.1°, 15.6°, 20.3°, 21.2°, and 22.1–23.2°, indicating that the SAPO-11-A-6 molecular sieves have an AEL structure and a layered semi-crystalline phase structure. The diffraction peak of the AEL structure was observed, and the layered semi-crystalline phase structure peak at 2θ = 6.2° broadened in 3 h, indicating that the layered semi-crystalline intermediate gradually transformed into the AEL structure. However, a completely formed AEL structure was observed after 6 h. The addition of CTAB after pre-crystallization can hinder the formation of the layered semi-crystalline phase structure, but it does not affect the formation of the final AEL structure, which suggests that CTAB participates in the molecular sieve crystallization process, affecting the silicon, aluminum, and phosphorus arrangement.
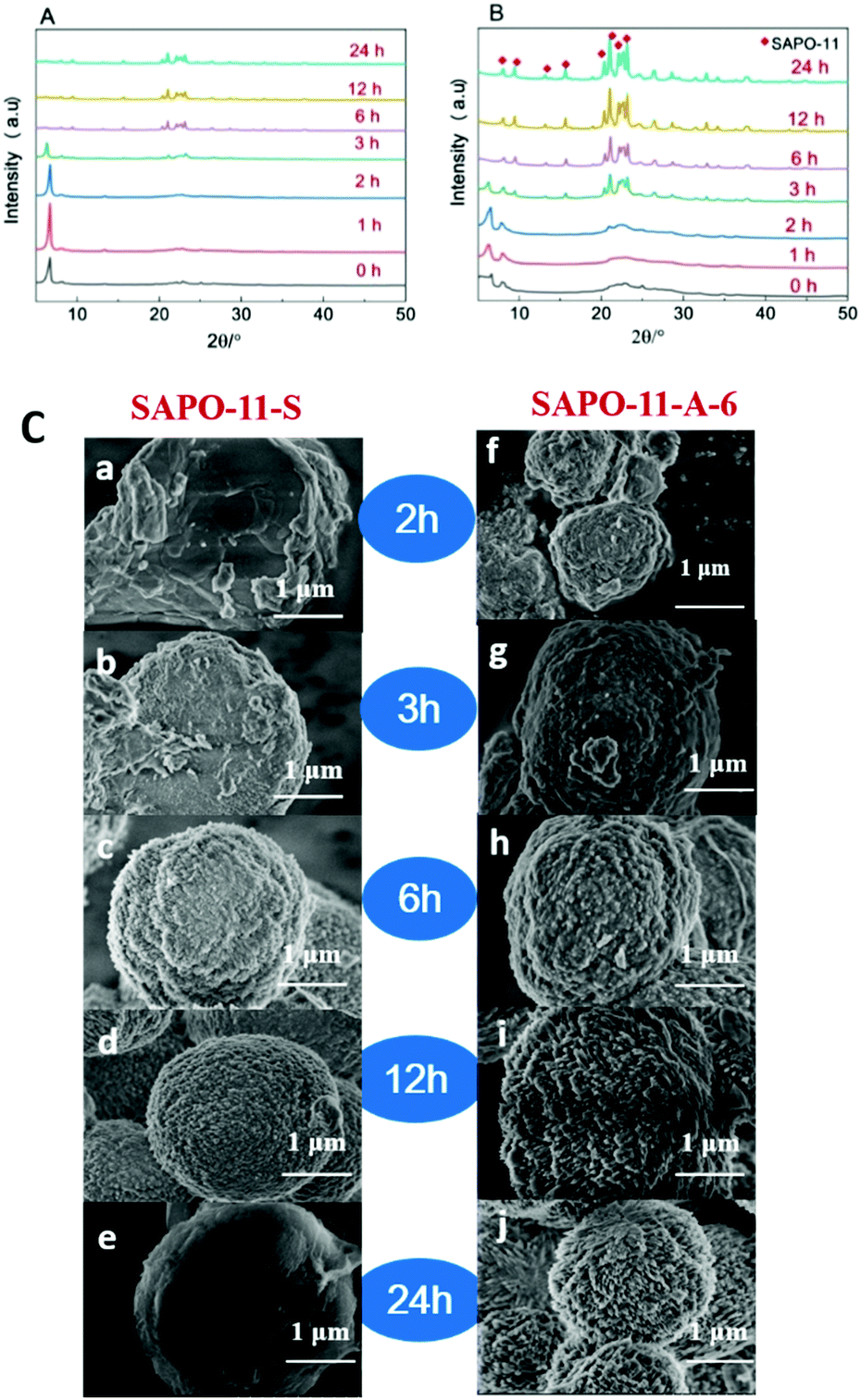 | ||
| Fig. 6 XRD patterns of SAPO-11-S (A) and SAPO-11-A-6 (B) and SEM images (C) at different crystallization times at 200 °C after pre-crystallization. | ||
The SEM images of SAPO-11-S and SAPO-11-A-6 at different crystallization times at 200 °C after pre-crystallization are shown in Fig. 6C. For the SAPO-11-S molecular sieve, a semi-elliptical structure was observed at a crystallization time of 2 h, for which the XRD pattern showed that the sample has a semi-crystalline phase and an AEL phase. After 3 h, the semi-elliptical structure had smoother edges. After 6 h of crystallization, the molecular sieves displayed a regular circular structure with a relatively rough surface. When the crystallization time was furthermore prolonged to 12 or 24 h, the molecular sieve surface became smoother. After adding an amount of CTAB (Al:CTAB = 0.006), the morphological changes in the molecular sieves at different crystallization times at 200 °C were examined by SEM. After 2 h of crystallization, a semi-elliptical shape was observed, indicating that the molecular sieves were undergoing crystallization and had not completely crystallized. Compared with that of SAPO-11-S, the surface molecular sieve structure was more apparent. SAPO-11-A-6 had elliptic structures with irregular fine crystal grains on the surface at 3 h, for which the XRD results showed that the molecular sieves had mainly AEL structures. When the crystallization time reached 6, 12, or 24 h, the surface roughness of the SAPO-11-A-6 molecular sieves increased significantly. After crystallization for 24 h, a sea urchin-like SAPO-11 molecular sieve was finally obtained.
According to the experimental results and non-classical molecular sieve orientation growth mechanism, a proposed sea urchin-like hierarchical porous SAPO-11 crystal formation route is illustrated in Scheme 1: at the first stage, silicate, aluminate, and phosphate in the gel pre-crystallized at 160 °C for 24 h to form negatively charged SAPO-11 nanocrystal particles. Then, the required amounts of cationic surfactant CTAB were added, which adsorbed on the surface of the negatively charged SAPO-11 nanocrystal particles and entered the layered semi-crystalline phase; secondary nucleation occurred between the CTAB and polysilicate, aluminate, and phosphate. The dry powder was steam-treated at 200 °C for 24 h, and the cationic surfactant CTAB bridged onto the precursor surface and recrystallization occurred to form the sea urchin-like hierarchical porous SAPO-11 molecular sieve.
 | ||
| Scheme 1 Schematic illustration of the proposed SAPO-11 crystal growth route. | ||
Catalytic performance in hydrogenation of oleic acid and structural characteristic of the catalysts
Oleic acid hydrogenation was used as a typical model reaction to study the catalytic properties of three different SAPO-11 molecular sieves loaded with 7 wt% nickel (Ni/SAPO-11-C, Ni/SAPO-11-S, and Ni/SAPO-11-A-6). According to previous studies,41 the main transformation for oleic acid is hydrodeoxygenation and hydroisomerization reactions. Heptadecane and octadecane are formed by hydrodeoxygenation (hydrodehydration, hydrodecarboxylation, and hydecarbonylation) of oleic acid. The HDO pathway for oleic acid transformation is as follows: oleic acid is first hydrogenated into stearic acid on metal centers (Ni NPs), followed by three-step hydrodehydration to form octadecane with octadecaldehyde and octadecanol as the first and second intermediates. Heptadecane (C17H36) can be formed via the hydrodecarboxylation and hydecarbonylation of stearic acid on metal or metal-acid sites. There are mainly two pathways to produce iso-alkanes: the first pathway is the formation of the alkene by the hydrodecarboxylation of oleic acid and then the formation of the iso-alkane at the metal-acid sites. Another pathway is the further hydroisomerization reaction of the heptadecane and octadecane via metal-acid sites to form the iso-alkane.31Oleic acid hydrogenation over the three different Ni/SAPO-11 catalysts was evaluated using the results shown in Fig. 7 and Table 2. As can be seen from Fig. 7 and Table 2, the hydroconversion of oleic acid mainly led to CO, CO2, and CH4 as gaseous products, while the main liquid products consisted of n-C17, n-C18, iso-C17, and iso-C18. In addition, the C2–C16 hydrocarbons were observed. Notably, the conversion of oleic acid reached 99% in the hydrogenation reaction over all Ni/SAPO-11 catalysts at a reaction temperature of 360 °C, reaction time of 4.0 h, and H2 pressure of 4 MPa. As shown in Fig. 7a, as the temperature increased, the isomer selectivity was firstly improved and then decreased. On the basis of the classical protonated cyclopropane (PCP) isomerization and β cracking mechanism: firstly, the produced alkane is dehydrogenated into alkene on metal sites. Then, the formed alkene transfers to the acid site on which it is protonated to give a carbenium ion intermediate. This is the same for the isomerization and cracking reactions. After isomerization of carbon chains, the carbenium ion shifts to the metal center to be hydrosaturated into iso-alkanes. The isomerization and cracking reactions are cleavage reactions, and they could form the same carbenium ion intermediate. The hydroisomerization of n-heptane is a slightly exothermic reaction, and cracking is an endothermic reaction; therefore, the isomer selectivity decreased at high temperature.
 | ||
| Fig. 7 The isomer selectivity, conversion (a) and liquid product compositions (b) of oleic acid hydroisomerization over different Ni/SAPO-11 catalysts; (c) the reusability of the Ni/SAPO-11-A-6 catalyst in the hydrogenation of oleic acid (reaction conditions: oleic acid (15 g), catalyst (0.7 g), T = 360 °C, t = 4.0 h, P = 4 MPa H2). | ||
| Catalysts | Ni/SAPO-11-C | Ni/SAPO-11-S | Ni/SAPO-11-A-6 |
|---|---|---|---|
| DB multibranched iso-alkanes. | |||
| The yield of gaseous products (wt%) | |||
| CH4 | 1.42 | 2.29 | 1.08 |
| C2H6–C6H12 | 0.26 | 0.32 | 0.25 |
| CO | 1.95 | 0.28 | 1.47 |
| CO2 | 0.02 | 0.02 | 0.03 |
| The composition of the liquid product (%) | |||
| Conversion (%) | 99.7 | 99.8 | 99.9 |
| Cracking products (C7–16) | 9.6 | 7.8 | 2.2 |
| Di-branched C17 | 31.6 | 33.8 | 37.1 |
| Di-branched C18 | 28.9 | 31.7 | 35.4 |
| DB-C17,C18 | 0 | 0 | 5.7 |
| Iso alkanes selectivity | 60.5 | 65.5 | 78.2 |
According to previous investigations, the hydroisomerization reaction relies, to a great extent, on the metal center and the acidic support of the catalyst. Firstly, we characterized the dispersion of nickel nanoparticles (Fig. 8a); the Ni nanoparticles are highly dispersed on the three different SAPO-11 samples, with average size distributions of 10.2, 8.1, and 7.8 nm, respectively, which are larger than the SAPO-11 microporous channel (0.39 nm × 0.63 nm), which showed that the Ni nanoclusters are mainly on the external surface of the molecular sieves as the size of the main isomer alkanes is larger than the pore diameter of SAPO-11 (0.63 nm). Therefore, according to the “pore mouth and key-lock” concept, the isomer alkanes are formed on the “pore mouth” of the SAPO-11. As a result, the SAPO-11 molecular sieves with many medium-strong acidic sites and a mesopore structure should be beneficial for hydroisomerization reaction. As can be seen from Table 2, surprisingly, Ni/SAPO-11-A-6 exhibits higher isomer selectivity (78.2%) and lower cracking selectivity (2.2%) than the Ni/SAPO-11-S catalyst, which is due to the fact that SAPO-11-A-6 has a larger external surface area, mesopore structure, and more strong Brønsted acid sites than SAPO-11-A. Meanwhile, it can be seen that isomer production was significantly different; especially, the multibranched iso-C17 and iso-C18 were obtained over the Ni/SAPO-11-A-6 catalyst according to GC-MS analysis.
 | ||
| Fig. 8 TEM images, particle size (a) and H2-TPR spectrum (b) of different Ni/SAPO-11 catalysts. | ||
Fig. 7c further shows the reusability of the Ni/SAPO-11-A-6 catalyst in the hydrogenation of oleic acid. It can be concluded that the Ni/SAPO-11-A-6 catalyst could maintain 99% conversion of oleic acid after six reusability tests, while the isomer selectivity reached 45.6%.
Furthermore, we made a comparison to the state-of-the-art catalysts in the hydrogenation of lipid to jet fuel, as listed in Table 3. From Table 3, it is noted that the Ni/SAPO-11 catalysts in the present work have an excellent isomer selectivity of 78.2%, which is similar to that of Pt/ZSM-22 (84.3%), and is better than that of the Pt, Ni, and Co loaded on other supports of SAPO-11, HMCM-49, etc. for catalytic lipid hydrogenation.
| Entry | Catalyst | Feed | Reaction conditions | Conv. (%) | Maxium of isomer yields (%) | Ref. |
|---|---|---|---|---|---|---|
| 1 | Pt–Sn/SAPO-11 | Castor oil | T = 380 °C, 4 MPa, LHSV = 1.2−1 | 90.6 | 14.36 | 42 |
| 2 | Pt/SAPO-11 | Oleic acid | T = 325 °C, P = 20 bar, t = 2 h | 97.5 | 6.8 | 43 |
| 3 | SDBS-Pt/SAPO-11 | Jatropha oil | T = 400 °C, 5 MPa, LHSV = 11.2 h−1, H2/feed = 1000 (v/v) | 99.5 | 25.4 | 44 |
| 4 | Pt/SAPO-11 | Soybean oil | T = 357 °C, P = 4 MPa, LHSV = 1 h−1 | 100 | 63 | |
| 5 | Pt/ZSM-22 | Soybean oil | T = 357 °C, P = 4 MPa, LHSV = 1 h−1 | 100 | 84.3 | 45 |
| 6 | Pt/Al2O3/SAPO-11 | Vegetable oils | T = 375 °C, 3 MPa, LHSV = 1 h−1 | 100 | 63.8 | 46 |
| 7 | Ni/SAPO-11G | Stearic acid | T = 290 °C, P = 4 MPa, t = 3 h | 98 | 15 | 47 |
| 8 | Ni-Mo/SAPO-11 | Castor oil | T = 350 °C, P = 3 MPa, t = 3 h, LHSV = 1.0 h−1 | 97.2 | 74.2 | 48 |
| 9 | Mo/ZSM-22 | Palmitic acid | T = 260 °C, P = 4 MPa, t = 4 h 0.5 g palmitic acid | 100 | 57.6 | 16 |
| 10 | Co/HMCM-49 | Palmitic acid | T = 260 °C, 4 MPa, 4 h | 100 | 34.3 | 49 |
| 11 | Pt/SAPO-34 | Oleic acid | T = 325 °C, 20 bar, 2 h | 98 | 50 | |
| 12 | Ni/SAPO-11 | Palm oil | T = 280 °C, 4 MPa, 6 h, LHSV = 2.0 h−1 | 100 | 56.4 | 51 |
| 13 | Ni/SAPO-11 | Oleic acid | T = 360 °C, 4 MPa, 4 h | 99 | 78.2 | In present |
The H2-TPR curves of different Ni/SAPO-11 catalysts are presented in Fig. 8b. As displayed in Fig. 8b, for the Ni/SAPO-11-C catalyst, it showed two hydrogen consumption regions: I and II, indicating that there were two different nickel species present on the surface of the catalysts. Region I started at 310 °C and ended at 500 °C and region II was observed at a higher temperature. For the Ni/SAPO-11 catalysts, the lower temperature reduction peak was assigned to the reduction of free NiO particles; and the higher one was assigned to the interacting Ni2+ species covalently combined with the support by bridge oxygen atoms.52,53 For the three Ni/SAPO-11 catalysts, it is obvious that the temperatures of the reduction peaks were Ni/SAPO-11-C > Ni/SAPO-11-S > Ni/SAPO-11-A-6, indicating that the small NiO nanoparticles supported on sea urchin-like hierarchical porous SAPO-11-A-6 are more easily reduced than on the other Ni/SAPO-11 catalysts.
Conclusions
In summary, a sea urchin-like hierarchical porous SAPO-11 was successfully synthesized using the SISAC method, by adding the CTAB after in situ seed formation. The optimal SAPO-11 molecular sieves were obtained at an Al:CTAB ratio of 0.006. The sea urchin-like SAPO-11 had a large external surface area, a mesoporous structure, and more acidic sites. The effects of CTAB on the SAPO-11 molecular sieve showed that the CTAB weakened the layered semi-crystalline phase. Importantly, after loading 7 wt% nickel, the Ni/SAPO-11-A-6 catalyst exhibited good catalytic performance in the hydroisomerization of oleic acid; especially, multibranched iso-C17 and iso-C18 were obtained, which is due to the presence of small Ni nanoparticles and a larger external surface area, mesoporous structure, and strong Brønsted acid sites of the sea urchin-like hierarchical porous SAPO-11 support.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support received from the National Natural Science Foundation of China (No. 51806225), National Key Research and Development Plan (No. 2019YFB1504005), Guangdong special support program (2017TX04Z109), and Natural Science Foundation of Guangdong Province (No. 2018A030310078) is much appreciated.Notes and references
- Y. Nakagawa, M. Tamura and K. Tomishige, Fuel Process. Technol., 2019, 193, 404–422 CrossRef CAS.
- N. Bálsamo, S. Mendieta, A. Heredia and M. Crivello, Mol. Catal., 2020, 481, 110290 CrossRef.
- E. G. Fawaz, D. A. Salam, L. Pinard and T. J. Daou, Catal. Sci. Technol., 2019, 9, 5456–5471 RSC.
- F. P. de Sousa, C. C. Cardoso and V. M. Pasa, Fuel Process. Technol., 2016, 143, 35–42 CrossRef CAS.
- I. Nikolopoulos, G. Kogkos, E. Kordouli, K. Bourikas, C. Kordulis and A. Lycourghiotis, Mol. Catal., 2020, 482, 110697 CrossRef CAS.
- J. Fu, X. Lu and P. E. Savage, ChemSusChem, 2011, 4, 481–486 CrossRef CAS.
- J. Fu, X. Lu and P. E. Savage, Energy Environ. Sci., 2010, 3, 311–317 RSC.
- J. Han, H. Sun, Y. Ding, H. Lou and X. Zheng, Green Chem., 2010, 12, 463–467 RSC.
- A. Kostyniuk, D. Key and M. Mdleleni, J. Energy Inst., 2020, 93, 552–564 CrossRef CAS.
- T. M. Sankaranarayanan, A. Berenguer, C. Ochoa-Hernández, I. Moreno, P. Jana, J. M. Coronado, D. P. Serrano and P. Pizarro, Catal. Today, 2015, 243, 163–172 CrossRef CAS.
- Z. Wang, D. Brouri, S. Casale, L. Delannoy and C. Louis, J. Catal., 2016, 340, 95–106 CrossRef CAS.
- S. Janampelli, G. Sethia and S. Darbha, Catal. Sci. Technol., 2020, 10, 268–277 RSC.
- B. Ma and C. Zhao, Green Chem., 2015, 17, 1692–1701 RSC.
- B. Peng, Y. Yao, C. Zhao and J. A. Lercher, Angew. Chem., Int. Ed., 2012, 51, 2072–2075 CrossRef CAS.
- P. S. F. Mendes, C. Chizallet, J. Pérez-Pellitero, P. Raybaud, J. M. Silva, M. F. Ribeiro, A. Daudin and C. Bouchy, Catal. Sci. Technol., 2019, 9, 5368–5382 RSC.
- Y. Shi, Y. Cao, Y. Duan, H. Chen, Y. Chen, M. Yang and Y. Wu, Green Chem., 2016, 18, 4633–4648 RSC.
- D. Verma, B. S. Rana, R. Kumar, M. G. Sibi and A. K. Sinha, Appl. Catal., A, 2015, 490, 108–116 CrossRef CAS.
- G. Lv, C. Wang, K. Chi, H. Liu, P. Wang, H. Ma, W. Qu and Z. Tian, Catal. Today, 2018, 316, 43–50 CrossRef CAS.
- X. Song, X. Bai, W. Wu, O. V. Kikhtyanin, A. Zhao, L. Xiao, X. Su, J. Zhang and X. Wei, Mol. Catal., 2017, 433, 84–90 CrossRef CAS.
- P. Mériaudeau, V. A. Tuan, V. T. Nghiem, S. Y. Lai, L. N. Hung and C. Naccache, J. Catal., 1997, 169, 55–66 CrossRef.
- L. Yang, W. Wang, X. Song, X. Bai, Z. Feng, T. Liu and W. Wu, Fuel Process. Technol., 2019, 190, 13–20 CrossRef CAS.
- X. Wang, Z. Liu, F. Guo, Y. Wang, X. Wei, P. Li, Y. Xue, Y. Wang, S. Guo and Y. Yu, RSC Adv., 2018, 8, 243–250 RSC.
- L. Guo, X. Bao, Y. Fan, G. Shi, H. Liu and D. Bai, J. Catal., 2012, 294, 161–170 CrossRef CAS.
- L. Guo, Y. Fan, X. Bao, G. Shi and H. Liu, J. Catal., 2013, 301, 162–173 CrossRef CAS.
- M. Y. Kim, K. Lee and M. Choi, J. Catal., 2014, 319, 232–238 CrossRef CAS.
- M. Choi, R. Srivastava and R. Ryoo, Chem. Commun., 2006, 4380–4382, 10.1039/B612265E.
- Z. Liu, L. Liu, H. Song, C. Wang, W. Xing, S. Komarneni and Z. Yan, Mater. Lett., 2015, 154, 116–119 CrossRef CAS.
- T. Blasco, A. Chica, A. Corma, W. J. Murphy, J. Agúndez-Rodríguez and J. Pérez-Pariente, J. Catal., 2006, 242, 153–161 CrossRef CAS.
- L. Han, Y. Liu, F. Subhan, X. Liu and Z. Yan, Microporous Mesoporous Mater., 2014, 194, 90–96 CrossRef CAS.
- Z. Chen, W. Song, S. Zhu, W. Lai, X. Yi and W. Fang, RSC Adv., 2017, 7, 4656–4666 RSC.
- L. Yang, S. Xing, H. Sun, C. Miao, M. Li, P. Lv, Z. Wang and Z. Yuan, Fuel Process. Technol., 2019, 187, 52–62 CrossRef CAS.
- Y. Seo, S. Lee, C. Jo and R. Ryoo, J. Am. Chem. Soc., 2013, 135, 8806–8809 CrossRef CAS.
- Y. Liu, W. Qu, W. Chang, S. Pan, Z. Tian, X. Meng, M. Rigutto, A. V. D. Made, L. Zhao, X. Zheng and F.-S. Xiao, J. Colloid Interface Sci., 2014, 418, 193–199 CrossRef CAS.
- A. Sakthivel, S. E. Dapurkar, N. M. Gupta, S. K. Kulshreshtha and P. Selvam, Microporous Mesoporous Mater., 2003, 65, 177–187 CrossRef CAS.
- X. Huang, L. Wang, L. Kong and Q. Li, Appl. Catal., A, 2003, 253, 461–467 CrossRef CAS.
- B. Chen and Y. Huang, J. Phys. Chem. C, 2007, 111, 15236–15243 CrossRef CAS.
- B. Chen and Y. Huang, Microporous Mesoporous Mater., 2011, 143, 14–21 CrossRef CAS.
- P. K. Dutta, D. C. Shieh and M. Puri, Zeolites, 1988, 8, 306–309 CrossRef CAS.
- Y. Zhang, Z. Deng, K. Zhu and X. Zhou, CrystEngComm, 2015, 17, 3214–3218 RSC.
- L. Zhang, J. Bates, D. Chen, H.-Y. Nie and Y. Huang, J. Phys. Chem. C, 2011, 115, 22309–22319 CrossRef CAS.
- S. Xing, P. Lv, H. Yuan, L. Yang, Z. Wang, Z. Yuan and Y. Chen, Green Chem., 2017, 19, 4157–4168 RSC.
- Y. Chen, X. Li, S. Liu, W. Zhang, Q. Wang and W. Zi, Ind. Crops Prod., 2020, 146, 112182 CrossRef CAS.
- M. Ahmadi, E. E. Macias, J. B. Jasinski, P. Ratnasamy and M. A. Carreon, J. Mol. Catal. A: Chem., 2014, 386, 14–19 CrossRef CAS.
- X. Li, Y. Chen, Y. Hao, X. Zhang, J. Du and A. Zhang, Renewable Energy, 2019, 139, 551–559 CrossRef CAS.
- C. Wang, Z. Tian, L. Wang, R. Xu, Q. Liu, W. Qu, H. Ma and B. Wang, ChemSusChem, 2012, 5, 1974–1983 CrossRef CAS.
- M. Rabaev, M. V. Landau, R. Vidruk-Nehemya, V. Koukouliev, R. Zarchin and M. Herskowitz, Fuel, 2015, 161, 287–294 CrossRef CAS.
- Y. Liu, D. Zheng, H. Yu, X. Liu, S. Yu, X. Wang, L. Li, J. Pang, X. Liu and Z. Yan, Microporous Mesoporous Mater., 2020, 303, 110280 CrossRef CAS.
- G. Xing, S. Liu, Q. Guan and W. Li, Catal. Today, 2019, 330, 109–116 CrossRef CAS.
- Y. Shi, R. Li, Q. Shen, M. Yang and Y. Wu, Chem. Commun., 2019, 55, 12096–12099 RSC.
- M. Ahmadi, A. Nambo, J. B. Jasinski, P. Ratnasamy and M. A. Carreon, Catal. Sci. Technol., 2015, 5, 380–388 RSC.
- Q. Liu, H. Zuo, T. Wang, L. Ma and Q. Zhang, Appl. Catal., A, 2013, 468, 68–74 CrossRef CAS.
- J. M. Rynkowski, T. Paryjczak and M. Lenik, Appl. Catal., A, 1993, 106, 73–82 CrossRef CAS.
- C. W. Hu, J. Yao, H. Q. Yang, Y. Chen and A. M. Tian, J. Catal., 1997, 166, 1–7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj03848b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2021 |