
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilica particles with fluorescein-labelled cores for evaluating accessibility through fluorescence quenching by copper†
Samuel H.
Gallagher
,
Paul
Schlauri
,
Emanuele
Cesari
,
Julian
Durrer
and
Dominik
Brühwiler
 *
*
Institute of Chemistry and Biotechnology, Zürich University of Applied Sciences, CH-8820 Wädenswil, Switzerland. E-mail: dominik.bruehwiler@zhaw.ch
First published on 4th October 2021
Abstract
Core–shell particles with fluorescent cores were synthesised by growing silica shells on fluorescein-labelled Stöber-type particles. The porosity of the shell could be altered in a subsequent pseudomorphic transformation step, without affecting the particle size and shape. These core–shell particles constitute a platform for the evaluation of pore connectivity and core accessibility by observing the effect of a quencher on the fluorescence signal emitted by the fluorescein-labelled cores. In combination with argon sorption measurements, quenching experiments with copper provided valuable information on the porosity generated during the shell formation process. It was further observed that the introduction of well-defined mesopores by pseudomorphic transformation in the presence of a structure-directing agent reduces the core accessibility. This led to the conclusion that the analysis by conventional gas sorption methods paints an incomplete picture of the mesoporous structure, in particular with regard to pores that do not offer an unobstructed path from the external particle surface to the core.
Introduction
Core–shell particles consisting of a mesoporous silica shell and a variety of active cores have been reported. Cores include metals for catalysis,1,2 lanthanide-doped nanoparticles for up-conversion,3 Fe3O4 for cancer therapy4 and magnetic resonance imaging,5 as well as quantum dots for sensing6 and molecular imaging.7 Magnetic cores can facilitate the separation of the core–shell particles and have been used in combination with appropriately functionalised mesoporous silica shells for the removal of heavy metals,8 aromatic sulphur compounds9 and polycyclic aromatic hydrocarbons,10 for catalysis,11,12 as well as for targeted drug delivery.13,14The molecular transport within the pore systems is a key parameter in most of the applications of mesoporous materials. Recent work based on pulsed field gradient NMR (nuclear magnetic resonance) and continuous wave EPR (electron paramagnetic resonance) has provided insight into the mechanisms determining the diffusion of molecules in mesopores.15,16 The diffusion processes are highly complex, being strongly influenced by factors such as temperature, confinement, and interactions between probe, solvent, and surface. Methods that allow for an in situ monitoring of diffusion processes are desirable to optimise porous materials for their respective applications.
The accessibility of the core of a core–shell particle largely depends on the shell building procedure. Cores might be inaccessible even if the shell features sufficient pore volume to enable the uptake of a specific molecule. The pore volume, which is typically obtained from nitrogen sorption, is an insufficient measure for the accessibility. With this problem in mind, we have developed a core–shell architecture that allows for the evaluation of core accessibility in function of the shell structure. The particles consist of a fluorescein-labelled core and a silica shell. The porosity of the shell can be controlled by pseudomorphic transformation (PT), i.e., by a rearrangement of the pore structure while preserving the particle size and shape. This is achieved by carefully balancing the dissolution and reprecipitation of silica in the presence of a structure-directing agent.17,18 PT and postsynthetic functionalisation can generate a variety of pore sizes17–22 and pore surface chemistries.23,24 The concept is illustrated in Fig. 1, showing a particle with a fluorescein-labelled core and a rhodamine-labelled shell. In such a system, the accessibility of the core can be assessed with the use of compounds that are able to quench the fluorescence of the core. Herein, the synthesis and characterisation of such a core–shell architecture is reported. The resulting core–shell particles are then used to study the effect of PT on the accessibility of the core.
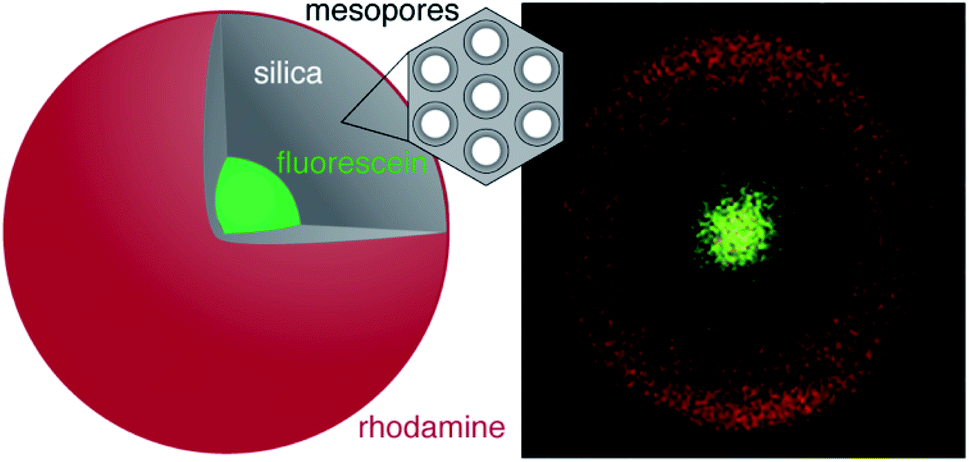 | ||
| Fig. 1 Schematic representation and confocal laser scanning microscopy (CLSM) image of a core–shell microparticle with a fluorescein-labelled core (green) and a rhodamine-labelled external surface (red). | ||
The investigation of the time-dependent quenching of the fluorescent core yields data on the pore connectivity that cannot be obtained by conventional methods such as gas sorption and electron microscopy. The probing of the pore connectivity is highly specific, due to the ability to adjust the properties of the quencher species (e.g., size or charge). Not only does this provide novel insights regarding the pore structure and connectivity, but it also advances the mechanistic understanding of postsynthetic treatments such as pseudomorphic transformation.
Results and discussion
Functionalised core particles
In order to obtain monodisperse spherical silica particles with sub-micrometre diameter, a Stöber-type synthesis was conducted,25 leading to particle growth by aggregation according to the model of Bogush and Zukoski.26 The product displayed a narrow particle size distribution centred at 0.35 μm as well as a uniform spherical morphology (Fig. 2A). Following calcination the particles featured a type II adsorption isotherm, indicative of a non-porous or macroporous material.27 The surface of the particles was functionalised with (3-aminopropyl)triethoxysilane (APTES) at approximately 50% silanol coverage based on two –OH per nm2 and a specific surface area of 12 m2 g−1.28 Following the functionalisation with APTES, fluorescein isothiocyanate (FITC) was coupled to the surface-bound amino groups.29–31 The particles displayed a pale-yellow colour after FITC labelling. These labelled particles were used as cores to build the core–shell particles. | ||
| Fig. 2 SEM images of (A) core particles, (B) core–shell particles with a single shell (C–1S), (C) core–shell particles with a double shell (C–2S) and (D) CLSM image of functionalised C–2S (fluorescein-labelled core and rhodamine-labelled external particle surface). The scale bars are 5 μm. | ||
Shell growth
Silica shell growth is suppressing the generation of stable secondary particles and particle aggregation, while the primary particle diameter is enlarged.32,33 This can be achieved by focusing on the total particle surface area per reaction volume (SA/V). It has been reported that a low SA/V results in the formation of secondary particles, while a high SA/V promotes aggregation.32,34–36 The SA/V can be regulated by the use of seed particles, the composition of the hydrolysis solution and the addition rate of tetraethoxysilane (TEOS).34,36,37In our case, the fluorescein-labelled core particles act as seed particles, allowing control of oligomer saturation, as they counteract any supersaturation by growing before nucleation starts. The size, dispersity and morphology of the core particles were found to directly influence the shell growth. To ensure maximum available surface area the core particles were dispersed and sonicated in a hydrolysis solution (containing NaCl, NH4F, aqueous ammonia and ethanol, see Experimental) prior to shell building. The hydrolysis solution facilitates the growth of the silica shells. A high pH results in quick hydrolysis (large H2O and NH4OH quantities) and condensation (small ethanol quantity).38,39 The silanol surface groups are deprotonated, leading to stabilisation of the particles. The double layer thickness, which could hinder shell building, is reduced due to the large excess of hydroxide and ammonium ions.40 Shell growth was conducted at 5 °C, because low temperature has been shown to disfavour adjacent particle crosslinking and secondary particle formation.35,41,42 However, temperatures below 5 °C should be avoided, as this results in solubility problems for TEOS. The addition of small amounts of salts (typically NaCl) has been shown to inhibit the formation of secondary particles. The concentration must be carefully adjusted, as destabilisation of the suspension occurs if the concentration is too high, leading to particle aggregation.35,43
For shell growth, TEOS and additional hydrolysis solution must be continuously added to the reaction mixture. Additional hydrolysis solution is required to maintain the SA/V. This slows down the increase in mass fraction and allows more TEOS to be dissolved.44 Maximum addition rates of TEOS for different silica seed sizes under typical conditions of silica seed growth have been put forward by Giesche.36 Particularly in the later stages of particle growth, addition of the hydrolysis solution at a constant rate can lead to a decrease of the SA/V. This dilution effect can cause the formation of secondary particles. However, the addition must be continued, because with increasing diameter, the distance between the particles decreases, thereby promoting aggregation.36 For the formation of thick shells (C–2S particles, Fig. 2C), we have therefore conducted the growth process in two steps, with a smaller and gradually decreasing addition rate of the hydrolysis solution during the second step. Fig. 2 shows the scanning electron microscopy (SEM) images of the particles at different stages of the growth process. The particles obtained after the growth of the first and second shell (C–1S and C–2S) show low polydispersity and only a minor fraction of small secondary particles.
Constant addition rates of TEOS and hydrolysis solution were used for the first shell. For C–S particles (thin shell) this was completed for 20 min (TEOS (1.5 mL) and hydrolysis solution (8.4 mL)) to obtain a particle diameter of 0.5 μm or a 75 nm thick shell (Table 1).
| Sample | Particle diameter [μm] | Shell thickness [nm] |
|---|---|---|
| Cores | 0.35 | — |
| C–S | 0.50 | 75 |
| C–1S | 0.86 | 255 |
| C–2S | 1.70 | 675 |
The constant addition of TEOS could be completed up to 9 mL (2 h), which resulted in a particle diameter of 0.86 μm or a 255 nm thick shell (Fig. 2B). An amount of 1 mL of the reaction mixture was extracted every 10 min and immediately quenched in 10 mL of ethanol. The SEM images of these samples are presented in Fig. S2 (ESI†). In the early stages, the shell growth is slow as the concentration of TEOS is low. Between 20 to 30 min the formation of unstable secondary particles (USPs) was observed. It is believed that at this stage the particles start to grow by particle aggregation and Ostwald ripening. Physisorption of the USPs on the stable primary particles was indeed observed. In addition, the temporal development of the particle size compared well with theoretical calculations of particle growth according to Chen et al. (Fig. S3†).43 However, in the early stages, the theoretical calculations were found to deviate from the experimental observations, because the theoretical model assumes uniform growth and therefore neglects the generation of secondary particles. The gradually improving agreement between the theoretical and the experimental particle size indicates a constant particle population. Additional support for the consumption of USPs for shell growth comes from IIer's conclusions regarding the physisorption of USPs.41 A concave surface, such as that formed by the aggregation of two spherical particles, has a lower solubility than a convex surface. Consequently, reprecipitation occurs at the immediate surface of the respective primary particle. It was found that the shell building could be stopped at any point to obtain different shell thicknesses, such as that for C–S, although additional agitation was required for up to 1 h to allow sufficient time for USP consumption. The products showed a narrow particle size distribution and homogeneous morphology.
The constant addition rate of TEOS and hydrolysis solution past 2 h still resulted in shell growth. However, due to the large particles present at this point of the process, the growth was found to be slow in relation to the amount of TEOS given to the system. At 2 h the theoretical and the experimental particle size are identical (Fig. S3†), in addition to TEOS starting to become immiscible. For further shell growth, a transition from a constant to a dynamic addition rate is therefore reasonable. To accelerate growth as a function of TEOS, the reaction was split at this point, with 30 mL of the reaction mixture aliquoted from the centre of the suspension. This had the result of largely eliminating the USPs. The addition rate of TEOS for the formation of the second shell, i.e., for the growth from C–1S to C–2S, remained constant, but the addition rate of the hydrolysis solution was dynamic, with the rate reducing over time, thereby allowing the SA/V to remain high as the reaction continued. This increased the probability of monomers reacting with a silica surface site rather than with each other, which was evident through the absence of USPs in the SEM images. The particles shown in Fig. 2C were obtained after an additional 4 h of reaction time. They showed regular spherical morphology and a narrow particle size distribution with an average diameter of 1.7 μm (675 nm shell thickness). However, the SEM image did show some aggregation and secondary particles (Fig. 2C).
Characterisation of the shell
The spatial distribution of the fluorescent labels was determined with the C–2S particles. Selective labelling of the external particle surface29 allowed for a convenient visualisation of the core–shell structure by CLSM (Fig. 1 and 2D). The dark region between the fluorescein-labelled core and the rhodamine-labelled external particle surface corresponds to the silica shell.To further characterise the shell, argon sorption measurements were completed. All isotherms presented in Fig. 3 are from C–S particles following 20 min of shell building. Argon was chosen as an adsorptive due to the microporosity of the shells.45 The C–S particles display a type I isotherm indicative of a microporous material.27 The almost linear increase of adsorbed volume in the high pressure region of the isotherm indicates an unordered porous network spanning the entire mesoporous range and into the macroporous range, an observation confirmed by the substantial mesopore volume (Fig. 4). The microporosity is most likely a result of particle agglomeration during shell growth.46 Most of the micropores collapse upon calcination at 550 °C, leading to a decreased micropore volume and a considerable reduction of the total pore volume (sample C–S-cal, Fig. 4, Table S1†).
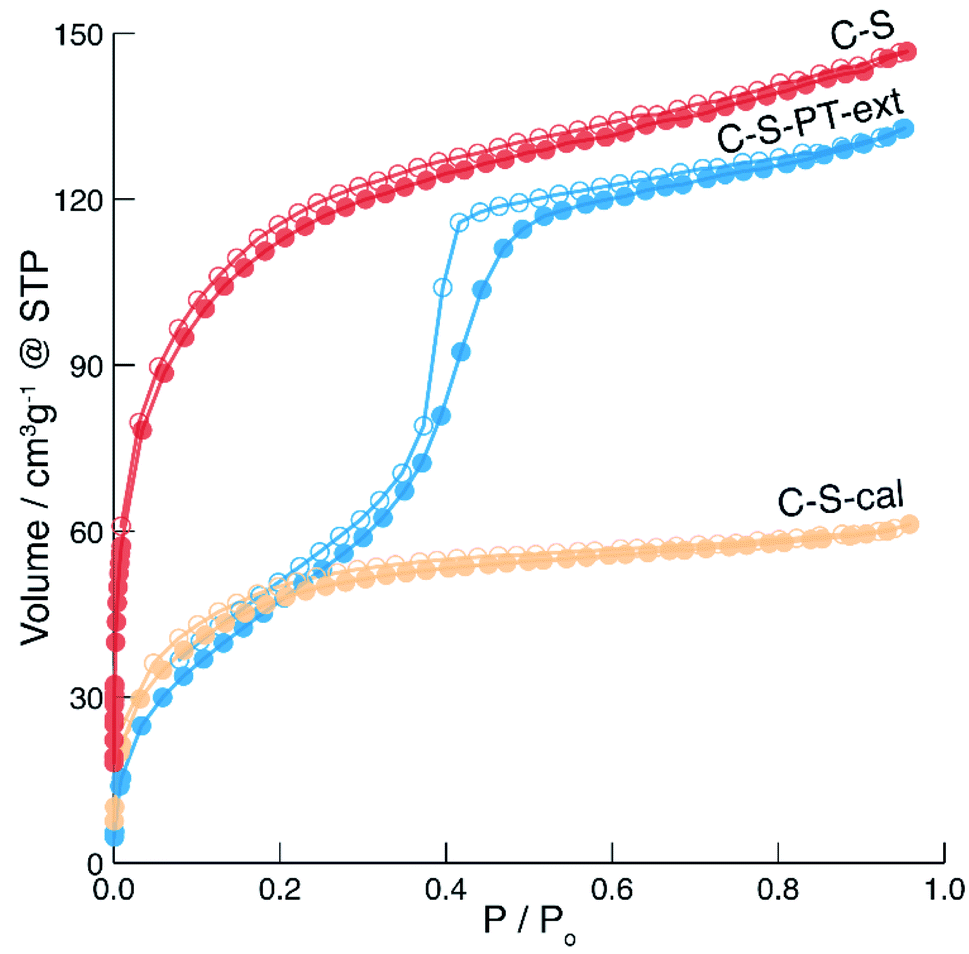 | ||
Fig. 3 Argon sorption isotherms of C–S ( ), calcined C–S (C–S-cal, ), calcined C–S (C–S-cal,  ) and pseudomorphically transformed and extracted C–S particles (C–S-PT-ext, ) and pseudomorphically transformed and extracted C–S particles (C–S-PT-ext,  ). Hollow points denote desorption. ). Hollow points denote desorption. | ||
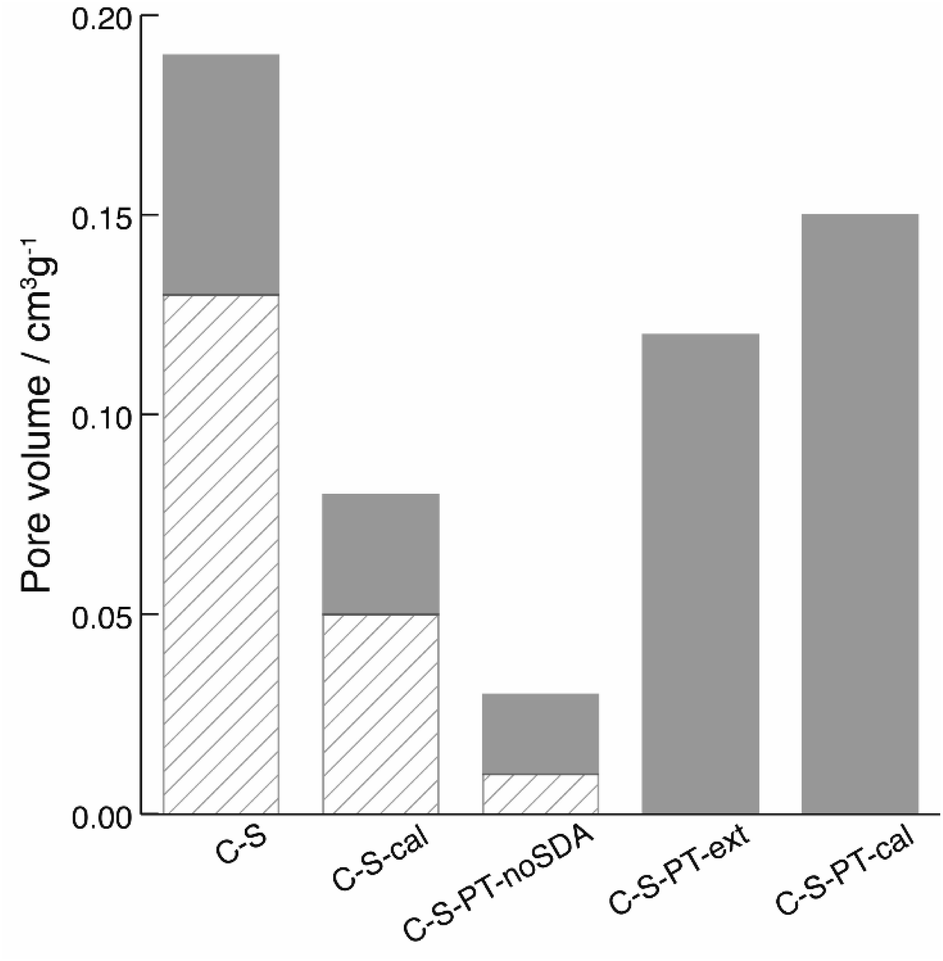 | ||
| Fig. 4 Gas sorption data taken from argon adsorption isotherms of C–S, calcined C–S (C–S-cal), C–S after PT without SDA (C–S-PT-noSDA), C–S after PT and extraction of the SDA (C–S-PT-ext), C–S after PT and calcination (C–S-PT-cal). Displayed is the volume contribution of micropores for all pores smaller than 2 nm (diagonal lines) and the volume contribution of pores with sizes between 2 and 7 nm (solid grey). | ||
Pseudomorphic transformation (PT)
In previous work, we have shown that the degree of PT in the presence of cetyltrimethylammonium bromide (CTAB) as a structure-directing agent (SDA) can be controlled by the reaction conditions and firstly affects the most accessible (i.e. outer) regions of the particle.21,22 In recent work, the effect of a partial pseudomorphic transformation on the porosity of mesoporous silica SBA-15 was further investigated, indicating a complex alteration of the initial pore structure.47 If PT does occur from the outer regions first, a transformation of the shell without degradation of the functionalised core is possible by carefully adjusting the temperature, pH and duration of the PT. In fact, the fluorescein-labelled core provides an excellent means for evaluating whether the PT has reached the particle centre, as this would lead to the removal of the covalently bound fluorescein by hydrolysis, with subsequent distribution over the entire core–shell particle. To assess the core accessibility by quenchers, the integrity of the fluorescent core is crucial. Therefore, relatively mild PT conditions were implemented, and the SEM images were observed to ensure that the particle morphology was retained (Fig. S4†).The argon sorption isotherm of the core–shell particles after PT and extraction of the SDA displays a distinct pore condensation step (Fig. 3). The isotherm is classified as type IV on account of this pore condensation step. A narrow pore size distribution centred at 4 nm was obtained (Fig. S5†), indicating that the mesoporous structure is preserved after the extraction of the SDA. Gas sorption data of different types of pseudomorphically transformed particles are presented in Fig. 4. It was found that the process of dissolution and reprecipitation during PT at high pH leads to the closing of micropores, which manifests itself by a reduction of the micropore volume. This effect can be well observed when a sample is treated under the conditions of PT but in the absence of the SDA (sample C–S-PT-noSDA, Fig. 4, Table S1†).
The SDA must be present for the generation of the 4 nm mesopores. As micropores are closed during PT, small quantities of large mesopores (>40 nm) and macropores in the C–S particles are expected to play an important role by allowing the uptake of the SDA prior to the transformation.
Accessibility studies
To determine the accessibility of the fluorescein-labelled cores with varying shell architecture, a fluorescence quencher was added. Fluorescein has been shown to be readily quenched by Cu2+ through both heavy atom and electron exchange quenching mechanisms of which both occur over short distances.48–51 To prove that no irreversible changes in the system had occurred after the quenching experiments, Ca2+ was added and the fluorescence could be fully recovered (Fig. S6†).To a cuvette, a sonicated suspension of the particles in methanol was added. Methanol was used due to its good refractive index matching52 and pore wetting.29 The amount of particles was adjusted until a set number of counts was achieved. The particles were stirred and excited slightly below their absorption maximum. All time-based measurements were recorded at λem = 519 nm, which approximately corresponds to the emission maximum of the fluorescein-labelled cores. It was found that the SDA did not hinder the diffusion of the quencher (Fig. S7†), so unless expressly stated, all results are for as-synthesised particles.
The shape of the fluorescence bands and their maxima provide information about the local environment of the fluorescein moieties53,54 as long as the number of cores is constant. The pristine core particles produced a single emission band with a maximum at approximately 520 nm (Fig. 5). The C–S particles displayed a broader band with a 260% reduction in signal intensity compared to the pristine core particles, as well as a slight blue shift, indicating an increase of the fluorescein monoanion/dianion ratio.55 The increased fluorescence intensity of the pseudomorphically transformed particles is most likely due to a high dianion/monoanion ratio caused by the basic conditions during the transformation.
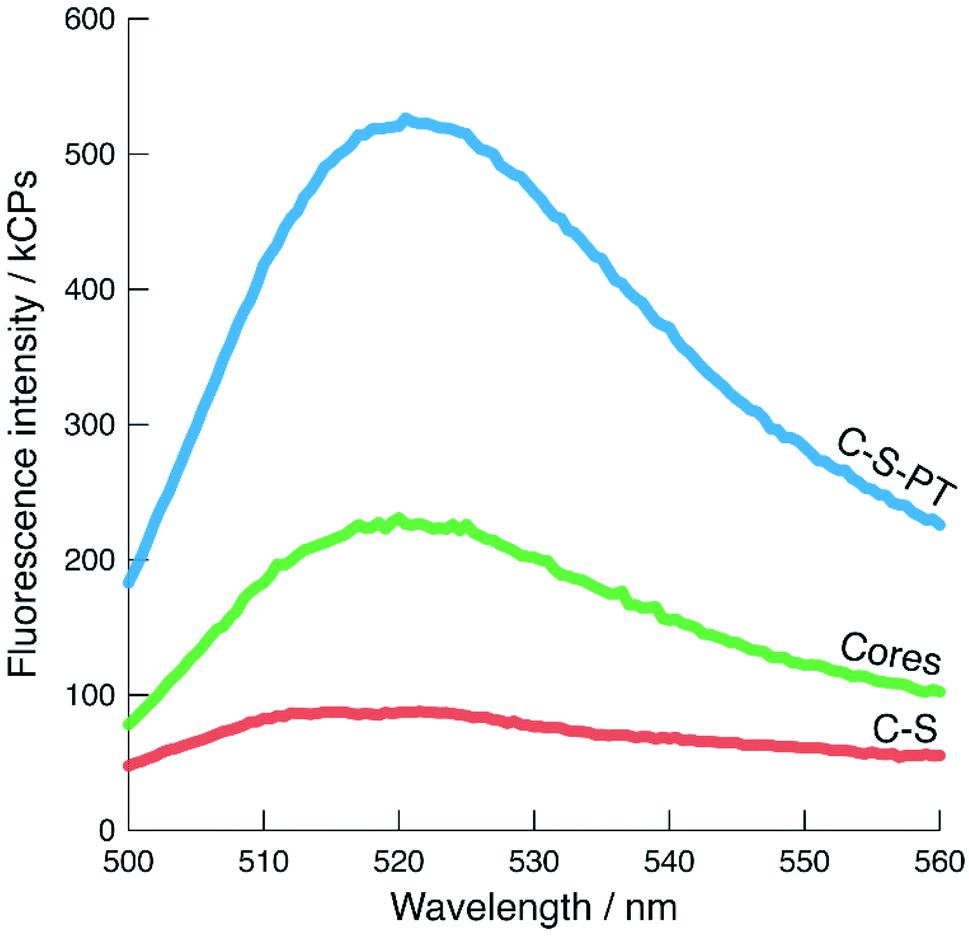 | ||
Fig. 5 Fluorescence spectra of cores (pristine,  ), C–S ( ), C–S ( ), and C–S-PT particles ( ), and C–S-PT particles ( ), excited at 485 nm. The number of fluorescein-labelled cores present in each sample is comparable. ), excited at 485 nm. The number of fluorescein-labelled cores present in each sample is comparable. | ||
The addition of CuCl2 had no significant effect on the absorbance values as monitored for λem and λex. An excess of 24 eq. of Cu2+ (relative to the amount of FITC used for the labelling of the core particles) was added as this eliminated any uncertainty regarding the number of fluorophores in the different samples. The effect of photobleaching and particle settling on the time-based measurements was evaluated by observing the fluorescence intensity of C–S particles in the absence of a quencher (Fig. 6). Over a period of 12 h, the fluorescence intensity decreased by less than 9%.
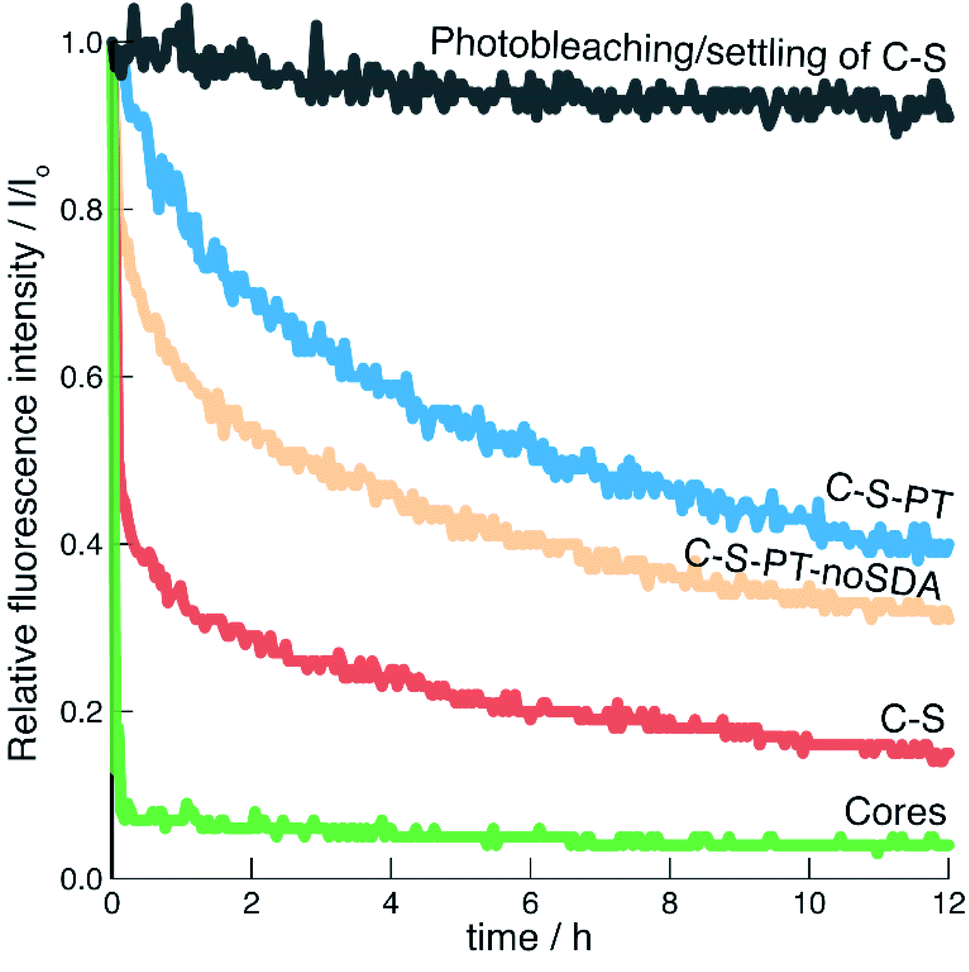 | ||
Fig. 6 Relative fluorescence intensities as a function of time. Prestened is the effect of photobleaching/settling of C–S in the absence of a quencher ( ). For the other curves, the quencher (Cu2+) was added at 0 h: cores (pristine, ). For the other curves, the quencher (Cu2+) was added at 0 h: cores (pristine,  ), C–S ( ), C–S ( ), C–S-PT ( ), C–S-PT ( ) and C–S-PT-noSDA ( ) and C–S-PT-noSDA ( ). ). | ||
The fluorescence of the pristine fluorescein-labelled core particles was efficiently quenched by Cu2+. This result is expected, as postsynthetic functionalisation of the non-porous core particles with APTES/FITC produced highly accessible fluorescein labels on the external particle surface. The C–S particles also showed rapid quenching, indicating that a porous shell was formed. This is in agreement with the results obtained from the argon sorption measurements. However, it seems that the shell formation has led to some inaccessible fluorophores. Good reproducibility was found for multiple repetitions of the same sample-quencher system (Fig. S8†).
The fluorescence of the C–S-PT particles could not be efficiently quenched, indicating that the permeability of the shell had been affected by PT. As-synthesised MCM-41 particles co-condensed with APTES/FITC were used to elucidate the possible contribution of residual SDA on the diffusion of Cu2+ through the mesopores. Co-condensed MCM-41 has thin pore walls with a thickness of approximately 0.9 nm.56 The fluorescein moieties should therefore in principle be well accessible for the quencher. We found that the fluorescence of the as-synthesised co-condensed MCM-41 sample was indeed efficiently quenched by Cu2+ even in the presence of the SDA (Fig. S7†), leading to the conclusion that neither residual SDA nor the 4 nm pores significantly limit the diffusion of the quencher.
An additional PT experiment was completed without the SDA (C–S-PT-noSDA). Quenching of the resulting particles by Cu2+ was not efficient, although slightly more efficient than for C–S-PT. As we have reported in Fig. 4, C–S-PT-noSDA shows very little porosity compared to C–S, which could be interpreted as a reason for the reduced quenching efficiency and for the decrease in shell permeability. However, the C–S-PT samples, which were transformed in the presence of the SDA, show a much larger porosity, which surprisingly does not seem to improve the accessibility of the core. The origin of this discrepancy must be sought in the connectivity of the pores. The shells of the C–S particles before PT obviously consist of a network of well-connected pores spanning the entire range from micro- to macropores. These pores are formed during the process of shell building. The highly basic conditions of PT led to the elimination of the micropores and small mesopores (in the absence of the SDA), causing a decreased shell permeability for Cu2+. PT in the presence of the SDA transformed the larger pores into small mesopores. These newly formed well-defined 4 nm mesopores further decrease the shell permeability. PT in the presence of CTAB has been found to lead to the formation of 2D-hexagonally ordered pore systems.22 Our results indicate that these are likely to contain dead-end pores and might not always effectively connect the particle core with the external particle surface or with large mesopores and macropores remaining after PT.
Experimental
Materials
Aqueous ammonia (28–30%), tetraethoxysilane (TEOS, ≥99.0%), (3-aminopropyl)triethoxysilane (APTES, 97%), fluorescein isothiocyanate (FITC, >90%), rhodamine B isothiocyanate (RBITC, mixed isomers), NaCl (≥99.99%), NH4F (≥99.99%), NaOH (≥98%), cetyltrimethylammonium bromide (CTAB, >99%), and copper(II) chloride (CuCl2, powder, 99%) were obtained from Sigma-Aldrich. Ethanol (>96%) was purchased from Reuss. 3-Aminopropyltris(methoxyethoxyethoxy)silane (APTMEES, 95%) was obtained from ABCR. Ammonium nitrate (NH4NO3, 99%) was purchased from Merck. Methanol (99.9% for analysis) was purchased from Acros. All chemicals were used as received.Synthesis of core particles
Core particles were synthesised following the procedure by Stöber.25 A solution of aqueous ammonia (14 mL, 28–30%), H2O (43 mL), and ethanol (50 mL) was magnetically stirred (300 rpm) in a 250 mL polypropylene beaker. After 10 min, stirring was increased to 500 rpm and TEOS (8 mL) was added quickly. The beaker was covered with a watch glass and the mixture was further stirred for 4 h. The resulting suspension was transferred to centrifugation tubes and centrifuged for 10 min at 4000 rpm. The particles were washed three times with 20 mL of H2O and once with 20 mL of ethanol. The product was dried in an oven at 80 °C for 2 h before being calcined at 550 °C overnight with a heating rate of 1 °C min−1.Labelling of core particles
For CLSM imaging and shell permeability studies, the core particles were labelled with FITC. Core particles (500 mg) were dispersed in a solution of ethanol (10 mL) containing APTES (2.4 μL) and FITC (1.0 mg). The suspension was briefly sonicated and then agitated for a further 30 min on a Vortex mixer set to 1400 rpm. Following centrifugation (10 min, 4000 rpm), the fluorescein-labelled core particles were washed with ethanol (20 mL) and dried overnight at 80 °C.Synthesis of core–shell particles with a thin shell (C–S)
A hydrolysis solution was made before shell synthesis, containing NaCl (170 mg), NH4F (110 mg), H2O (360 mL), aqueous ammonia (486 mL, 28–30%) and ethanol (79 mL). In a 50 mL centrifuge tube, 150 mg of labelled core particles and 10 mL of the hydrolysis solution were combined. The suspension was sonicated and occasionally agitated for 30 min and pipetted into a 250 mL polypropylene round-bottom flask equipped with an oval shape magnetic stirrer bar (ca. 3 cm in length) and a rubber septum. The flask was cooled to 5 °C and allowed to equilibrate for 30 min. TEOS (1.5 mL) and hydrolysis solution (8.4 mL) were separately added by means of two syringe pumps operating at rates of 0.075 and 0.42 mL min−1, respectively. Following completion, the suspension was transferred to a centrifuge tube and agitated for 1 h. The suspension was then centrifuged (10 min, 4000 rpm). The resulting core–shell particles (C–S) were washed three times with H2O (20 mL), once with ethanol (20 mL), and dried in an oven at 80 °C overnight.Synthesis of core–shell particles with a single shell (C–1S)
The same experimental procedure was completed as detailed for the C–S particles. This time 9 mL of TEOS and 50 mL of hydrolysis solution were added separately by means of two syringe pumps operating at rates of 0.075 and 0.42 mL min−1, respectively. The stirring speed was increased stepwise by increments of 10 rpm from 250 to 350 rpm over the two hours.Following completion, the suspension was centrifuged (10 min, 4000 rpm). The resulting core–shell particles (C–1S) were washed three times with H2O (20 mL), once with ethanol (20 mL), and dried in an oven at 80 °C overnight.
Synthesis of core–shell particles with a double shell (C–2S)
To facilitate the imaging of the core–shell structure by CLSM, an additional shell was deposited. For this purpose, 30 mL of the suspension containing the freshly prepared C–1S particles (before the centrifugation and washing step) was pipetted into a separate 250 mL polypropylene round-bottom flask, which was cooled to 5 °C and equipped with an oval shape magnetic stirrer bar (ca. 3 cm in length) and a rubber septum. Agitation was started at 250 rpm and increased stepwise by increments of 10 rpm to 350 rpm over the four hours. TEOS (18 mL) and hydrolysis solution (49 mL) were added separately by means of two syringe pumps over the course of 4 h. An addition rate of 0.075 mL min−1 was set for TEOS. The addition rate of the hydrolysis solution was gradually adjusted from 0.16 (start of the reaction) to 0.12 mL min−1 (after 4 h). Following completion, the suspension was centrifuged (10 min, 4000 rpm). The resulting core–shell particles (C–2S) were washed three times with H2O (20 mL), once with ethanol (20 mL), and dried in an oven at 80 °C overnight.Pseudomorphic transformation (PT)
Core–shell particles (C–S, 100 mg) were mixed with the structure-directing agent CTAB (65 mg, 0.18 mmol) in a PTFE-lined autoclave (10 mL). Aqueous NaOH solution (1 mL, 80 mM) was added, and the mixture was stirred for 30 min. The autoclave was closed and heated at 80 °C for 24 h. After cooling to room temperature, the product was obtained by centrifugation and washed with H2O until a neutral pH and no foaming (due to excess CTAB) was observed. After further washing with ethanol (20 mL) the product was dried in an oven at 80 °C overnight.Extraction of the structure-directing agent (SDA)
After PT, the SDA was extracted by stirring the core–shell particles in an ethanolic solution of NH4NO3 (0.05 M) at 60 °C according to a previously reported procedure.57 The extraction was repeated three times.External surface functionalisation
RBITC (4 mg, 7.5 μmol) was dissolved in ethanol (10 mL). APTMEES (31.5 μL, 75 μmol) was added to the solution and the resulting mixture was stirred under nitrogen atmosphere for 16 h. Core–shell particles (C–2S, 200 mg) were dispersed in ethanol (5 mL) and 5 mL of the solution containing RBITC and APTMEES was added to the suspension containing the core–shell particles. After stirring for 16 h, the core–shell particles were recovered by centrifugation and washed with ethanol until the washing solution was colourless. The product was dried in an oven at 80 °C overnight.Synthesis of co-condensed fluorescein-labelled MCM-41
A mixture of CTAB (1.1 g, 3 mmol), H2O (26 mL) and aqueous ammonia (12 mL, 28–30%) was heated to 35 °C and magnetically stirred until CTAB had fully dissolved. The solution was then allowed to cool to room temperature. A second solution was made containing FITC (40 mg, 0.12 mmol) in ethanol (5 mL) and APTES (16 μL, 0.07 mmol). This solution was magnetically stirred for 4 h or when approximately 3 mL of ethanol had evaporated. TEOS (5 mL, 0.94 mol) was added to this solution and the combined solution was slowly added to the original CTAB containing solution. The combined reaction mixture was stirred for 3 h at room temperature before being transferred to a PTFE-lined autoclave, where it was subsequently heated to 100 °C for 48 h. The particles were recovered by filtration, washed with 1 L of H2O and dried in an oven at 80 °C overnight.Characterisation
Confocal laser scanning microscopy (CLSM) was performed with an Olympus BX60 microscope equipped with a FluoView FV300 confocal unit and excitation at 488 nm and 543.5 nm. Scanning electron microscopy (SEM) images were acquired with a Thermo Scientific Quanta FEG 250. Fluorescence measurements were performed on a Fluorolog-3 (Horiba). Emission spectra were obtained at an excitation wavelength of λex = 485 nm and an emission wavelength range of λem = 500–560 nm with a resolution of 0.5 nm, an integration time of 0.1 s and an average of five scans. For time-based measurements λex = 485 nm and λem = 519 nm were fixed, and measurements were taken every 160 s for 12 h. Excitation and emission slits were set to 4 nm. The spectra were corrected, dark-offset, and a blank subtraction of a quartz-cuvette with methanol was completed. Stirring was applied via a magnetic stirrer. The argon sorption isotherms were measured at 87.3 K with a Quantachrome Autosorb iQ MP equipped with a CryoCooler. As-synthesised (SDA containing) samples had the SDA extracted before gas sorption measurements, according to the abovementioned procedure. The samples (C–S, C–S-PT-ext, C–S-PT-noSDA) were vacuum degassed at 80 °C for 3 h prior to the sorption measurements. Calcined samples (C–S-cal, C–S-PT-cal) were vacuum degassed at 300 °C for 3 h. The pore size distributions and average pore diameters were determined from the adsorption branch by a non-local density functional theory (NLDFT) model developed for silica exhibiting cylindrical pore geometry (Software ASiQwin v3.01, Quantachrome Instruments, Boynton Beach, FL, USA).58 The micropore volume (Vμ, pore diameters below 2.25 nm) and mesopore volume (Vmeso, 2.25–7.05 nm) were obtained from the volume histogram of the NLDFT. Vtot was taken at a relative pressure of 0.95 based on Gurvich's rule.27Conclusions
Silica shells with different architectures were synthesised on fluorescent cores. This was achieved through basic conditions and the addition of TEOS and hydrolysis solution in two steps. The first step yielded well-defined core–shell particles that were used for shell structure characterisation and accessibility studies. Further enlargement of the shell through a consecutive step provided micrometre-sized core–shell particles for convenient imaging via CLSM.The as-synthesised shells featured substantial microporosity with unordered mesopores and were permeable to Cu2+, as demonstrated by quenching experiments. It was found that the synthesis of the shell and the subsequent pseudomorphic transformation resulted in changes to the ratio of monoanionic and dianionic fluorescein at the core, thus providing information on the chemical environment at the core–shell interface.
Pseudomorphic transformation was found to affect the shell permeability. Apart from the loss of microporosity, the presence of the structure-directing agent resulted in the reshaping of large pores to small mesopores (4 nm in diameter). This transformation was found to reduce the accessibility of the core, most likely due to the formation of dead-end mesopores and pore systems not offering a direct path from the external surface to the particle core. As the process constitutes a partial pseudomorphic transformation, some well-connected larger pores remain. It is very likely that these remaining pores still provide pathways to the core of the particles.
Research on pseudomorphic transformation of silica particles in the presence of a structure-directing agent has mainly focused on the introduction of well-ordered pore systems into particles of pre-defined size and shape. While pseudomorphic transformation is undoubtedly a promising method for the synthesis of novel porous materials, effects on the connectivity of the pore system must be considered. Our results indicate that the effect of such transformations on the pore connectivity might – depending on the targeted application – not be beneficial. Applications in catalysis typically require fast diffusion, while slow diffusion is of interest for drug delivery and controlled release. Core–shell particles with fluorescent cores provide a versatile platform for the investigation of core accessibility and pore connectivity.
Author contributions
SHG and DB designed the study. SHG, PS, EC and JD conducted the experimental work. SHG and DB wrote the paper. All authors discussed and commented on the manuscript. DB acquired the funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Swiss National Science Foundation (grant no. 172805).Notes and references
- S. H. Joo, J. Y. Park, C.-K. Tsung, Y. Yamada, P. Yang and G. A. Somorjai, Nat. Mater., 2009, 8, 126–131 CrossRef CAS.
- S. Zhang, A. Han, Y. Zhai, J. Zhang, W.-C. Cheong, D. Wang and Y. Li, Chem. Commun., 2017, 53, 9490–9493 RSC.
- K. Wiercigroch-Walkosz, J. Cichos and M. Karbowiak, Colloids Surf., A, 2019, 572, 1–9 CrossRef CAS.
- K. Sun, Z. Gao, Y. Zhang, H. Wu, C. You, S. Wang, P. An, C. Sun and B. Sun, J. Mater. Chem. B, 2018, 6, 5876–5887 RSC.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS.
- R. Gui, A. Wan and H. Jin, Analyst, 2013, 138, 5956–5964 RSC.
- J. Pan, D. Wan and J. Gong, Chem. Commun., 2011, 47, 3442–3444 RSC.
- S. Hou, X. Li, H. Wang, M. Wang, Y. Zhang, Y. Chi and Z. Zhao, RSC Adv., 2017, 7, 51993–52000 RSC.
- P. Tan, J.-X. Qin, X.-Q. Liu, X.-Q. Yin and L.-B. Sun, J. Mater. Chem. A, 2014, 2, 4698–4705 RSC.
- X. Zhang, H. Niu, W. Li, Y. Shi and Y. Cai, Chem. Commun., 2011, 47, 4454–4456 RSC.
- Z. Sun, J. Yang, J. Wang, W. Li, S. Kaliaguine, X. Hou, Y. Deng and D. Zhao, J. Mater. Chem. A, 2014, 2, 6071–6074 RSC.
- Y. Deng, Y. Cai, Z. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- M. S. Moorthy, H.-B. Kim, J.-H. Bae, S.-H. Kim and C.-S. Ha, RSC Adv., 2016, 6, 29106–29115 RSC.
- W. Zhao, J. Gu, L. Zhang, H. Chen and J. Shi, J. Am. Chem. Soc., 2005, 127, 8916–8917 CrossRef CAS.
- J. Kärger and R. Valiullin, Chem. Soc. Rev., 2013, 42, 4172–4197 RSC.
- M. Wessig, M. Spitzbarth, M. Drescher, R. Winter and S. Polarz, Phys. Chem. Chem. Phys., 2015, 17, 15976–15988 RSC.
- A. Galarneau, J. Iapichella, K. Bonhomme, F. Di Renzo, P. Kooyman, O. Terasaki and F. Fajula, Adv. Funct. Mater., 2006, 16, 1657–1667 CrossRef CAS.
- S. H. Gallagher, O. Trussardi, O. Lipp and D. Brühwiler, Materials, 2020, 13, 2474 CrossRef CAS PubMed.
- T. Martin, A. Galarneau, F. Di Renzo, F. Fajula and D. Plee, Angew. Chem., Int. Ed., 2002, 41, 2590–2592 CrossRef CAS.
- M. J. Reber and D. Brühwiler, Part. Part. Syst. Charact., 2015, 32, 243–250 CrossRef CAS.
- M. J. Reber and D. Brühwiler, Dalton Trans., 2015, 44, 17960–17967 RSC.
- N. Zucchetto, M. J. Reber, L. Pestalozzi, R. Schmid, A. Neels and D. Brühwiler, Microporous Mesoporous Mater., 2018, 257, 232–240 CrossRef CAS.
- D. Brühwiler, Nanoscale, 2010, 2, 887–892 RSC.
- S. Giret, M. Wong Chi Man and C. Carcel, Chem.–Eur. J., 2015, 21, 13850–13865 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- G. H. Bogush and C. F. Zukoski, J. Colloid Interface Sci., 1991, 142, 1–18 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- L. T. Zhuravlev, Colloids Surf., A, 2000, 173, 1–38 CrossRef CAS.
- N. Gartmann and D. Brühwiler, Angew. Chem., Int. Ed., 2009, 48, 6354–6356 CrossRef CAS PubMed.
- P. Jal, S. Patel and B. K. Mishra, Talanta, 2004, 62, 1005–1028 CrossRef CAS PubMed.
- N. Gartmann, C. Schütze, H. Ritter and D. Brühwiler, J. Phys. Chem. Lett., 2010, 1, 379–382 CrossRef CAS.
- S.-L. Chen, P. Dong and G.-H. Yang, J. Colloid Interface Sci., 1997, 189, 268–272 CrossRef CAS.
- J. K. Bailey and M. L. Mecartney, Colloids Surf., 1993, 63, 151–161 CrossRef.
- K. Nozawa, H. Gailhanou, L. Raison, P. Panizza, H. Ushiki, E. Sellier, J. P. Delville and M. H. Delville, Langmuir, 2005, 21, 1516–1523 CrossRef CAS.
- B. Zhao, C. Tian, Y. Zhang, T. Tang and F. Wang, Particuology, 2011, 9, 314–317 CrossRef CAS.
- H. Giesche, J. Eur. Ceram. Soc., 1994, 14, 205–214 CrossRef CAS.
- S. M. Chang, M. Lee and W.-S. Kim, J. Colloid Interface Sci., 2005, 286, 536–542 CrossRef CAS PubMed.
- K.-S. Kim, J.-K. Kim and W.-S. Kim, Ceram. Int., 2002, 28, 187–194 CrossRef CAS.
- T. Montheil, C. Echalier, J. Martinez, G. Subra and A. Mehdi, J. Mater. Chem. B, 2018, 6, 3434–3448 RSC.
- A. Van Blaaderen, J. Van Geest and A. Vrij, J. Colloid Interface Sci., 1992, 154, 481–501 CrossRef CAS.
- R. K. Iler, The Chemistry of Silica: Solubility, Polymerization, Colloid and Surface Properties and Biochemistry, Wiley, New York, 1979 Search PubMed.
- C. G. Tan, B. D. Bowen and N. Epstein, J. Colloid Interface Sci., 1987, 118, 290–293 CrossRef CAS.
- S.-L. Chen, P. Dong, G.-H. Yang and J.-J. Yang, J. Colloid Interface Sci., 1996, 180, 237–241 CrossRef CAS.
- G. Coquerel, Chem. Soc. Rev., 2014, 43, 2286–2300 RSC.
- K. S. W. Sing, F. Rouquerol, P. Llewellyn and J. Rouquerol, in Adsorption by Powders and Porous Solids, Elsevier, 2014, pp. 303–320 Search PubMed.
- E. D. E. R. Hyde, A. Seyfaee, F. Neville and R. Moreno-Atanasio, Ind. Eng. Chem. Res., 2016, 55, 8891–8913 CrossRef CAS.
- R. Janus, M. Wądrzyk, M. Lewandowski, P. Natkański, P. Łątka and P. Kuśtrowski, J. Ind. Eng. Chem., 2020, 92, 131–144 CrossRef CAS.
- Y. Qiao and X. Zheng, Analyst, 2015, 140, 8186–8193 RSC.
- S. Seo, H. Y. Lee, M. Park, J. M. Lim, D. Kang, J. Yoon and J. H. Jung, Eur. J. Inorg. Chem., 2010, 2010, 843–847 CrossRef.
- Y. Lin, C. Wang, L. Li, H. Wang, K. Liu, K. Wang and B. Li, ACS Appl. Mater. Interfaces, 2015, 7, 27262–27270 CrossRef CAS.
- H. M. Lee, M. H. Kim, Y. I. Yoon and W. H. Park, Mar. Drugs, 2017, 15, 105 CrossRef.
- V. Cauda, A. Schlossbauer, J. Kecht, A. Zürner and T. Bein, J. Am. Chem. Soc., 2009, 131, 11361–11370 CrossRef CAS PubMed.
- M. M. Martin and L. Lindqvist, J. Lumin., 1975, 10, 381–390 CrossRef CAS.
- R. Sjöback, J. Nygren and M. Kubista, Spectrochim. Acta, Part A, 1995, 51, L7–L21 CrossRef.
- N. Klonis and W. H. Sawyer, Photochem. Photobiol., 2000, 72, 179–185 CrossRef CAS PubMed.
- C. G. Sonwane and S. K. Bhatia, J. Phys. Chem. B, 2000, 104, 9099–9110 CrossRef CAS.
- N. Lang and A. Tuel, Chem. Mater., 2004, 16, 1961–1966 CrossRef CAS.
- J. Landers, G. Y. Gor and A. V. Neimark, Colloids Surf., A, 2013, 437, 3–32 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General synthesis concept, shell growth, pseudomorphic transformation, pore volume data, reversibility of quenching with copper, accessibility of co-condensed fluorescein-labelled MCM-41, reproducibility of time-dependent fluorescence measurements. See DOI: 10.1039/d1na00599e |
| This journal is © The Royal Society of Chemistry 2021 |