
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnisotropic growth of ZnO nanoparticles driven by the structure of amine surfactants: the role of surface dynamics in nanocrystal growth†
Yinping
Wang
ab,
Yannick
Coppel
 a,
Christine
Lepetit
a,
Jean-Daniel
Marty
b,
Christophe
Mingotaud
*b and
Myrtil L.
Kahn
*a
a,
Christine
Lepetit
a,
Jean-Daniel
Marty
b,
Christophe
Mingotaud
*b and
Myrtil L.
Kahn
*a
aLaboratoire de Chimie de Coordination, CNRS, UPR-8241, 205 route de Narbonne, 31077 Toulouse Cedex 04, France. E-mail: myrtil.kahn@lcc-toulouse.fr
bLaboratoire des IMRCP, CNRS UMR 5623, University of Toulouse, 118 route de Narbonne, 31062 Toulouse Cedex 9, France. E-mail: cmingo@chimie.ups-tlse.fr
First published on 30th August 2021
Abstract
Herein, we elucidate the key role of amine surfactants in the controlled anisotropic growth of ZnO nanoparticles that is achieved under mild conditions by organometallic hydrolysis. The structuring influence of alkyl substituents on the nitrogen atom of amines is jointly analyzed theoretically by DFT modeling, and experimentally by multinuclear NMR (1H, 13C and 17O) spectroscopy. We demonstrate that in initial steps leading to the growth of colloidal ZnO particles, the nature of molecular species that are involved in the solution strongly depends on the structure of the amine surfactant. By using tertiary, secondary or primary amines, no or weak adducts between the amine and zinc, or stable adducts, or adduct oligomers were identified, respectively. Afterwards, following the course of the reaction, the dynamic behavior of the amines on the grown ZnO nanocrystal surfaces is also strongly correlated with their structure. We identified that in the presence of tertiary, secondary or primary amines, no significant [Zn⋯N] adsorption, or surface adsorption with notable surface mobility, or a very strong adsorption is achieved, respectively. The last case, primary amines, significantly involves the structuring of a hydrogen bonding network. Therefore, such surface dynamic behavior has a predominant role in driving the nanocrystal growth, and orienting the ZnO material final morphology. By forming hydrogen bonds at the nanoparticle surface during the growth process, primary amines specifically lead to the formation of nanorods. Conversely, isotropic nanoparticles and aggregates are obtained when secondary and tertiary amines are used, respectively. These findings shed light on the role of weak surface interactions, herein H-bonding, that rule the growth of nano-objects and are as such crucial to identify, study, and control for achieving progress in nanoscience.
Introduction
Metal oxides and ZnO in particular are essential materials in applications with high societal impact such as solar cells,1,2 opto-electronics,3–5 catalysts6,7 for CO2 hydrogenation to MeOH8 or water splitting,9 tumor chemotherapy,10 H2 gas sensing,11 treatment of nuclear waste effluents,12 or improved wear protection.13 Many research studies are directed towards the elaboration of such materials in the form of nanoparticles which can then bring additional properties (i.e. different from those of the bulk) such as transparency, higher reactivity, and increased ease of processing.2,6,8,14In addition to nanometric sizing, controlling the morphology of nano-objects is essential since their properties can be drastically affected by their shape, core and surface structure.15–19 The use of surfactants is common for synthesis performed in solution.20–24 Even though their role throughout the synthesis is roughly known, detailed pathways and precise mechanisms controlling the morphology of nanoparticles are not always well understood and fully explained.25 This topical issue thus deserves special attention to improve current synthetic procedures.
During the synthesis of nano-objects in solution, surfactants are generally assumed to play different roles, and can primarily act as a ligand for the molecular species present in solution.26–28 The metal precursor may thus be modified after its introduction into the reaction media before the nucleation process. The surfactants are also seen as a blocking agent (or a surface stabilizing agent) during the nanocrystal (NC) growth process.29–38 In this latter case, interactions existing between the surfactant's functional group and the surface of the growing object are often invoked to explain the obtained final size, shape, and core/surface structure of the NC. Since most surfactants can not only act as ligands for molecular species present in the reaction medium, but can also coordinate to the surfaces of growing NCs, understanding their role in real systems becomes complicated. Clearly, for a better control of nano-object growth and its structure, all the interactions with the surfactants should be considered throughout the NC formation process.37 Still, discerning at each stage of the NC formation process what is important for their morphology and structure and what is not, remains elusive, especially with regard to the dynamic aspects.39
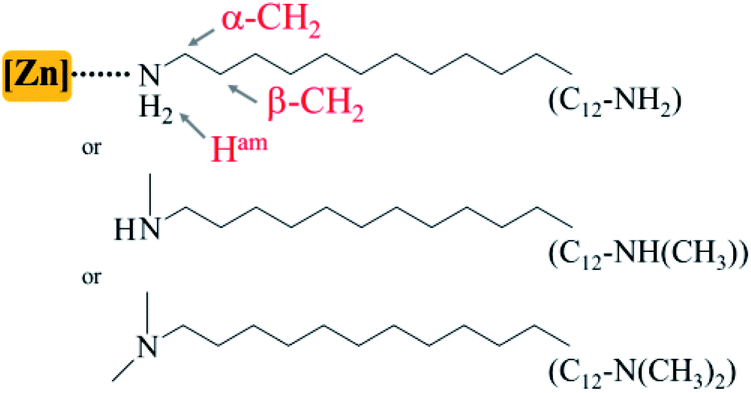
In this context, the use of amines with long fatty chains as surfactants, such as the archetypal oleylamine,40 is very popular to achieve good control of the morphology of NCs. The high boiling point of oleylamine, above 360 °C, allowed using it in thermolytic reduction processes whether in a “heat up” approach,41–49 or through the so-called “hot-injection”48–51 technique. Hexadecylamine, dodecylamine and octylamine are also extensively used under milder conditions for the formation of metallic, II–VI or III–V semiconductor NCs.33,35,52–57 Primary amines appear to be effective in controlling the morphology of NCs but the reasons for this remain unclear. Classically, the influence of the length of the amine aliphatic chain on the NC morphology is considered, but the role of the general structure of the amine and especially the number of substituents around the nitrogen atom – i.e. primary vs. secondary or tertiary amine – was not investigated in detail.19 Herein, based on the controlled synthesis of ZnO NCs, we achieved the first comparative study establishing the different roles played by primary, secondary and tertiary amine surfactants. These amines of identical aliphatic chain-lengths were mixed with a zinc precursor to get ZnO NCs of very different morphologies: either 2D defined nanorods, or isotropic spherical nanoparticles, or aggregates of ill-defined shape. In the course of nanoparticle formation, we analyzed the role of the amines at each step of the synthesis both experimentally by multinuclear NMR (1H, 13C, 17O)58–64 and theoretically by DFT modeling, including pertinent surface dynamics. We evidenced the key role of weak interactions as a lever for the control of the NC final morphology, while in the case of ZnO NCs it is the decisive role of the H-bonding network during the hydrolytic process in the presence of primary amine surfactants that results in two dimensional-defined nanorod formation.
Results and discussion
We prepared ZnO NCs following the method reported by our group.65 Compared to other recently reported synthesis of anisotropic ZnO nanoparticles,66–72 this synthesis consists of the controlled hydrolysis of the dicyclohexyl zinc compound, [ZnCy2], in the presence of surfactants. Classically, the precursor and the surfactants are mixed in a vial and two equivalents of water are added in a homemade reactor allowing water vapor to diffuse into the reaction medium (see the Experimental section for details). Until now, parameters such as temperature,73 the mixing time before hydrolysis,74 the aliphatic chain length, and the nature and concentration of reagents57,74–78 were identified as parameters allowing the control of the morphology of the NCs, and especially their aspect ratio. In all these studies, the nature of the surfactant remained the same and corresponded to primary amines. The effect of the amine structure had not yet been studied. Fig. 1 shows the TEM images and the associated 2D size plots65 of the ZnO NCs obtained following this procedure when dodecylamine (referred to as C12-NH2 hereafter), N-methyldodecylamine (referred to as C12-NH(CH3) hereafter), or N,N-dimethyldodecylamine, (referred to as C12-N(CH3)2 hereafter) is used as the surfactant. Surprisingly, the morphology specifically depends on the number of substituents on the amine nitrogen.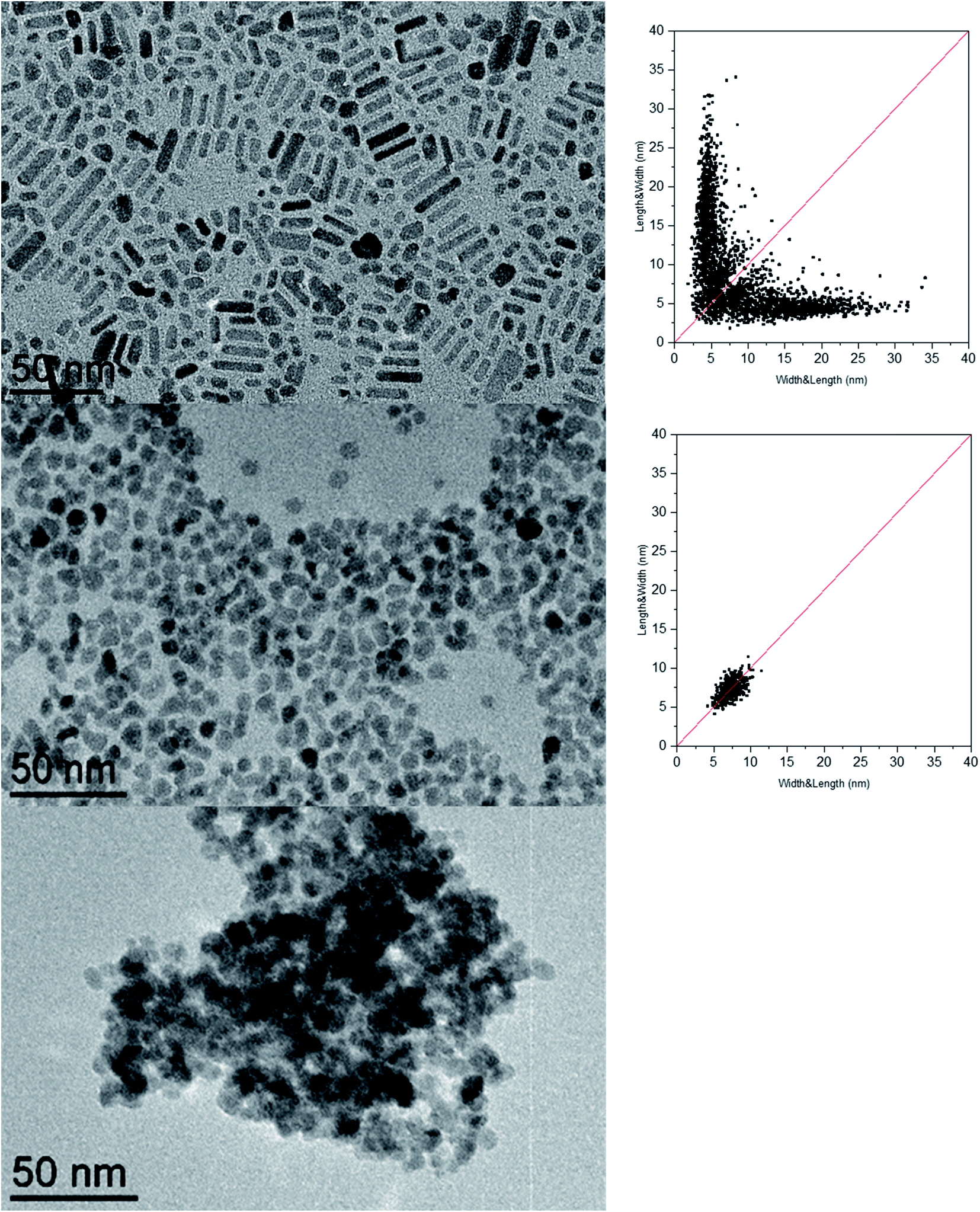 | ||
| Fig. 1 TEM images and associated 2D size plots65 of the ZnO NCs obtained when dodecylamine (C12-NH2) (top), N-methyldodecylamine (C12-NH(CH3)) (middle), or N,N-dimethyldodecylamine, (C12-N(CH3)2) (bottom) is used as a stabilizing agent. | ||
Nanorods with a diameter of 5.0 ± 0.7 nm and a length of 16 ± 7 nm are predominantly obtained when a primary amine is used, while isotropic NCs of 7.3 ± 1.1 nm and aggregates are obtained for secondary and tertiary amines, respectively (see the ESI for details and Table S1†). The ZnO nanoparticles obtained following this synthesis approach adopt a hexagonal structure as evidenced by PXRD (see the ESI for details and Fig. S1†).
To rationalize these results, we analyzed the role of amines at all stages of NC synthesis. In the following section, the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct between the zinc precursor and the amine is first described. Then, the composition of the reaction medium as a function of time is determined and the formation of amido complexes is shown for primary and secondary amines. Finally, the interaction of the different amines with the surface of the NCs is systematically reported.
:1 adduct between the zinc precursor and the amine is first described. Then, the composition of the reaction medium as a function of time is determined and the formation of amido complexes is shown for primary and secondary amines. Finally, the interaction of the different amines with the surface of the NCs is systematically reported.
Adduct formation
Regardless of the alkylamine, mixing of the surfactant and the zinc precursor leads to the formation of a 1:1 adduct as evidenced notably through the shifts of the 1H NMR signals for the aliphatic chain (Fig. S2–S4† and Table 1). This is supported by the change in multiplicity of the proton signal corresponding to the protons at the α-position relative to nitrogen that rises from coupling of the protons to both the β-methylene group of the alkyl chain, and eventually to the amino group protons Ham. The 1H NMR spectra of the free alkylamine show a triplet signal which changes for a triplet of triplet and a doublet of triplet for C12-NH2 and C12-NH(CH3), respectively. This coupling is visible because of the coordination of the surfactant to the zinc atom that slows down the exchange phenomena of the labile amino hydrogen. Changes of the self-diffusion coefficients D measured by PGSE-NMR spectroscopy also confirm the formation of this adduct (Fig. S5–S7† and Table 1). Under the same experimental conditions, the self-diffusion coefficients of the surfactant and the zinc precursor are significantly higher than those measured for the mixtures. Such observations indicate a fast exchange (on the NMR timescale) between free and coordinated amine molecules and substantiate the reversible coordination of the amine to the metal center of the zinc complex. The solutions are therefore composed of the 1:1 adduct in rapid equilibrium with the free surfactants and [ZnCy2] complex. The association constants (Ka) for the different amines| [ZnCy2] + amine ⇆ [ZnCy2(amine)]Ka | (1) |
|
|
|||
|---|---|---|---|
| Sample | α-CH2 chemical shift (ppm) | α-CH2 multiplicity, JHH (Hz) | Self-diffusion coefficient, D1h, (×1010 m2 s−1) |
| [ZnCy2] | — | — | 10.7 ± 0.4 |
| C12-NH2 | 2.55 | Triplet | 12.0 ± 0.3 |
| J αβ = 6.6 | |||
| C12-NH(CH3) | 2.45 | Triplet | 13.0 ± 0.3 |
| J αβ = 7.1 | |||
| C12-N(CH3)2 | 2.18 | Triplet | 13.0 ± 0.3 |
| J αβ = 7.3 | |||
| [ZnCy2] in mixtures | — | — | 10.0 ± 0.2 |
| C12-NH2 in mixture | 2.39 | Triplet of triplet | 8.0 ± 0.2 |
| J αβ = 6.6, | |||
| J αam = 6.8 | |||
| C12-NH(CH3) in mixture | 2.35 | Doublet of triplet | 8.0 ± 0.2 |
| J β = 6.6, | |||
| J αam = 6.8 | |||
| C12-N(CH3)2 in mixture | 2.18 | Triplet | 13.0 ± 0.2 |
| J αβ = 7.3 | |||
Note that equivalent values of the self-diffusion coefficients are measured regardless of the nature of the amine (i.e. 12.0 × 1010 m2 s−1) as well for the self-diffusion coefficients of the adducts formed with the primary and secondary amines (i.e. 8.0 × 1010 m2 s−1). The self-diffusion coefficient for the tertiary amine is equivalent to that of the free amine (i.e. 13.0 × 1010 m2 s−1) which is in agreement with the measured association constant, one order of magnitude lower than for the primary and secondary amines. The self-diffusion coefficient is defined as the weighted sum of the contributions of the free amine in solution and that involved in the adduct; the identical values measured for the primary and secondary amines confirm that the reaction medium before hydrolysis is of the same composition with species diffusing in a similar manner regardless of the amine. There is therefore no diffusion effect of the species at the origin of the observed difference in morphology.
The experimental results were corroborated by DFT simulations. The structure and stability of the various [ZnCy2(amine)] complexes were investigated at the DFT level, using a model hexyl chain instead of the C12 alkyl chain, in order to reduce the computational cost. Geometries of the corresponding [ZnCy2(C6H13NHxMe2−x)] complexes (x = 0–2), calculated at the PBE-D3/DGDZVP level in vacuum are shown in Fig. 2.
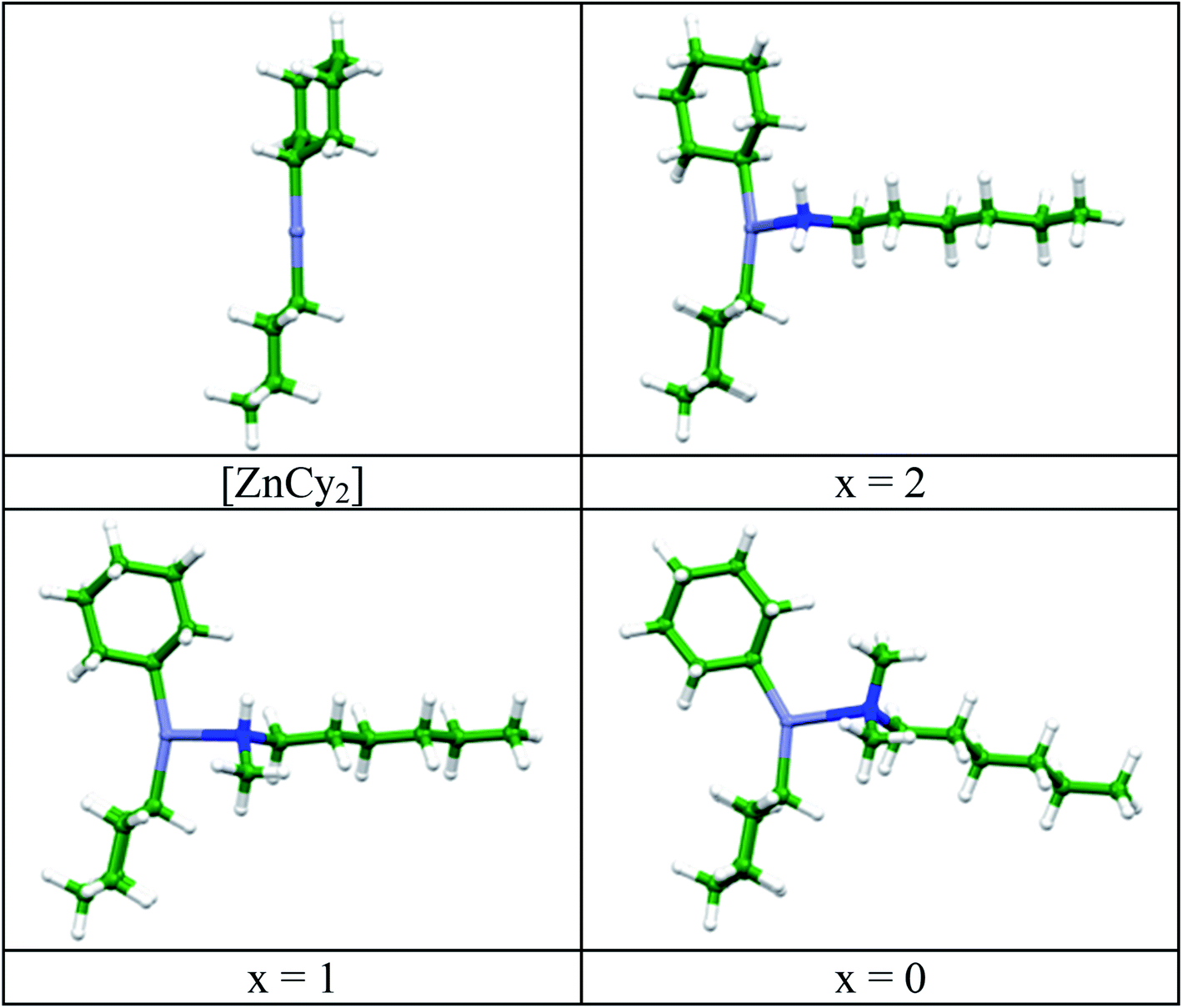 | ||
| Fig. 2 Geometries of [ZnCy2] and [ZnCy2(C6H13NHxMe2−x)] complexes (x = 0–2) calculated at the PBE-D3/DGDZVP level. | ||
In order to get as close as possible to the experimental synthesis conditions, calculations have also been made in a polarizable continuum medium (PCM). The zinc coordination sphere remains almost unchanged when taking into account an octylamine solvent (Table S2†). Zn–N and both Zn–C bonds are lengthened in the [ZnCy2(C6H13NHxMe2−x)] complex compared to the complex with the hexylamine. The C–Zn–C angle initially at 180° in [ZnCy2] decreases upon rotation of the cyclohexyl ring to accommodate the steric hindrance provided by the methyl substituent(s) of the amine (Fig. 2).
The nature and strengths of the Zn–C and Zn–N bonds can be described from both QTAIM and ELF topological analyses (see the detailed description given in the ESI†). Relevant QTAIM and ELF descriptors are given in Tables S3 and S4,† respectively. For the three Zn–amine complexes, the QTAIM description of the Zn–N bonds is in agreement with dative bonding with a weak covalence degree (V/G ≈ 1, DI ≈ 0.3). The bond strength, as indicated by the Espinosa's interaction energy Eint,80–82 is of the same order of magnitude regardless of the amine. Eint is equal to 14.7, 13.8, and 12.0 kcal mol−1 for primary, secondary, and tertiary amines, respectively. Similarly, the QTAIM description of the Zn–C bond is very close for all three complexes and corresponds to a polar covalent bond (V/G close to 2, DI ≈ 0.8). Eint is also comparable for [ZnCy2(C6H13NH2)] (42.0 and 41.5 kcal mol−1) and [ZnCy2(C6H13NHMe)] (42.1 and 41.4 kcal mol−1) and decreases very slightly for the tertiary amine [ZnCy2(C6H13NMe2)] complex (40.9 and 40.8 kcal mol−1) possibly because of the steric hindrance. The ELF analysis is in agreement with the above QTAIM description. The ELF descriptors of Zn–N (respectively Zn–C) bonds are almost the same regardless of the amine type. The oxidation state of the zinc atom is +II. The weak atomic contribution of Zn to the V(N) ELF basin (lower than 3%) and the weak covariance (|cov. (V(N), C(Zn))| ≤ 0.08) are in favor of a dative Zn–N bond of weak covalence degree, while the large atomic contribution of Zn to V (Zn, C) and the large negative covariance (cov. (V(Zn, C), C(Zn)) ≈−0.36) are in favor of a polar covalent Zn–C bond comparable to the one of the [ZnCy2] complex (Fig. S8†). The Zn–C bonds of the [ZnCy2] fragment are only slightly affected by the amine coordination and may be described by the similar most representative mesomeric forms of about equal weight of the [ZnCy2] complex (Fig. S9†).83
The Gibbs energies (ΔG) of the formation of [ZnCy2(C6H13NHxMe2−x)] complexes (x = 0–2) and the corresponding association constants (K) were then calculated at 298 K at the PBE-D3/DGDZVP level both in vacuum and in octylamine solvent medium following eqn (2). The results are reported in Table 2.
| [ZnCy2] + C6H13NHxMe2−x ⇆ [ZnCy2(C6H13NHxMe2−x)] | (2) |
| ΔG = G([ZnCy2(C6H13NHxMe2−x)]) − G([ZnCy2]) − G(C6H13NHxMe2−x) |
| ΔG = −RTlogK |
| K = [ZnCy2(C6H13NHxMe2−x)]/([ZnCy2][C6H13NHxMe2−x]) |
| [ZnCy2(C6H13NHxMe2−x)] | x = 2 | x = 1 | x = 0 |
|---|---|---|---|
| ΔG | −1.36 | −0.95 | +1.52 |
| ΔG* | +1.58 | +1.21 | +4.06 |
| K | 9.92 | 4.97 | 0.08 |
| K* | 0.07 | 0.13 | 0.001 |
Regardless of the calculation conditions (i.e. in a vacuum or in octylamine solvent medium), ΔG (resp. ΔG*) values are little negative or positive but close to zero, except for the dimethylhexyl amine for which a significant positive value of ca. +4 kcal mol−1 is calculated in octylamine solvent. This suggests that coordination of the tertiary amine to the zinc atom is not favorable despite a recognized higher Lewis basicity.84 The strong steric hindrance induced by the two methyl groups thus disfavors the coordination; the electronic effects are – according to the ELF and QTAIM topological analyses of the Zn–N bond described above – identical regardless of the structure of the amine. Interestingly, the association constants of the primary and secondary amines to [ZnCy2] are close to each other, whatever the reaction medium is, while for the tertiary amine, the association constant is much lower, in agreement with the experimental results.
These results confirmed our hypothesis that tertiary amines have low propensity to form adducts, while primary and secondary amines do not. For these latter amines, we have shown here, both experimentally and theoretically, that the association constants were comparable. Therefore, the difference found in the NC morphology when using primary and secondary amine surfactants cannot originate from an initial difference in coordination to zinc. The reactivity of the zinc–amine complexes was thus investigated from Fukui indices condensed on QTAIM and ELF basins.85 For all three Zn–amine complexes and [ZnCy2], the largest fELF− (resp. fQTAIM−) values are only found for Zn–C bonds (resp. C atomic basins) and are very similar (Tables S5 and S6†). The Zn–C bonds (resp. C atoms bound to Zn) appear therefore to be the most sensitive to electrophilic attack such as the one occurring in acid–base reactions with water or amine. The largest fELF+ values are also found for Zn–C bonds but fQTAIM+ values suggest that Zn will be the most sensitive to nucleophilic attack (Tables S5 and S6†), for example during the coordination of the amine to [ZnCy2]. Before studying the role of the interaction of amines on the surface of the NCs, another reaction must therefore be considered, namely the acid–base reaction between the precursor and the surfactant that will give rise to amido complexes for primary or secondary amines only (eqn (3), x = 0 or 1).
| [ZnCy2] + C6H13NH1+xMe1−x → [ZnCy(C6H13NHxMe1−x)] + CyH | (3) |
Acid–base reaction
The acid–base reaction was investigated by computational DFT studies (PBE-D3/DGDZVP level of calculation). Based on the reaction Gibbs energies calculated at 298 K, this acid–base reaction is only slightly more favorable for the secondary amine than for the primary amine (−11.3 versus −9.7 kcal mol−1, respectively). This suggests that the acid–base reaction is not expected to play a key role in the change of the NC morphology between primary and secondary amines. These results are consistent with the experimental pKa values that are close for primary and secondary amines (10.62 and 10.64, respectively); this parameter thus does not appear relevant to explain the difference in morphology. However, our modeling does not take into account the possible formation of oligomers through this acid–base reaction. Indeed, in a previous study, we showed that for primary amines, this reaction leads to the formation of mono and bis-amido complexes that oligomerized over time and are responsible for the formation of processable gels through entanglement of the oligomers after several hours.74 In the context of the present study, this oligomerization could be prevented by steric hindrance in the case of the secondary amine. Multinuclear NMR measurements, describe below, were therefore performed to confirm such hypothesis.Consistent with the DFT computational approach, the 1H NMR spectra as well as the D values for C12-N(CH3)2 and [ZnCy2] alone (13.0 ± 0.3 and 10.7 ± 0.4, respectively) or in the mixture (13.0 ± 0.3, 10.0 ± 0.2, Table 1) evidence that the [ZnCy2]/C12-N(CH3)2 mixture consists mostly of free surfactants and the [ZnCy2] complex. The 1H NMR spectrum recorded after two weeks shows no change (Fig. S10c†). In contrast, for C12-NH(CH3), the 1H NMR spectrum of the mixture exhibits features similar (i.e. broadening of the signals especially for α-CH2 and NCH3 resonances, an additional peak at 2.9 ppm with a self-diffusion coefficient D of 5.7 ± 0.4 × 10−10 m2 s−1, Fig. S11†) to the ones observed over time for C12-NH2 (D of 6.6 and 4.3 × 10−10 m2 s−1)86 that are characteristic of mono and bis-amido molecular complexes (Fig. S10a and b†). Importantly, by integration of 1H liquid NMR signals, an amount of roughly 10% of amido species could be estimated. The reaction mixture is thus primarily composed of the free [ZnCy2] complex and the C12-NH(CH3) surfactant. The sharp signal at 1.44 ppm corresponding to evolving cyclohexane molecules from cyclohexyl protonation is observed in agreement with the acid–base reaction taking place between the amine and the zinc precursor. Note that this peak is much more important in the case of C12-NH2 than for C12-NH(CH3) (Fig. S10a and b,† respectively) which substantiates that the acid–base reaction is more effective for primary amines and suggests that oligomerization occurs only in this case.
This result was confirmed by a 13C MAS NMR study evidencing that no formation of Zn amido oligomers occurs when [ZnCy2] and 2 equiv. C12-NH(CH3) are mixed (Fig. S12–S14†). After 55 h of mixing, the only change observed in the 13C INEPT experiment is a small increase of the cyclohexane signal at 26.2 ppm. After 8 days, in addition to a notable increase of the cyclohexane signal, new 13C INEPT signals appear notably in the 57–59 and 38–44 ppm ranges. These signals are characteristic of the formation of different molecular species made of monomeric and dimeric Zn amido complexes. However, no 13C CPMAS signal is observable stating that such molecular species remain small and exhibit high mobility, which assert that they are not oligomers. After 28 days, these signals just slightly increase and only very weak 13C CPMAS signals could be detected.
Thus, while both primary and secondary amines lead to Zn amido complexes, a notable difference is observed between primary and secondary amines: secondary amines are too hindered and only primary amines form oligomers. However, our NMR study showed that such oligomerization takes place on a timescale of several hours74 and on the other hand, the preparation of ZnO nanorods requires hydrolysis to be conducted for three days to achieve a maximum aspect ratio. In order to know whether the oligomers observed with the primary amines are responsible for the formation of the nanorods, it is therefore important to know which of the two oligomerization or hydrolysis has the higher rate.
[ZnCy2] hydrolysis vs. the acid–base reaction
Controlled hydrolysis of [ZnCy2] in the presence of C12-NH2 was performed using 17O-enriched water (40%). Samples were taken at different times (between 10 minutes and 6.5 hours) and 1H, DOSY, 13C, and 17O NMR experiments were conducted. First, 1H spectra evidenced a decrease of the [ZnCy2] resonances until their complete disappearance after a reaction time of about 2 h (Fig. S15†). The [ZnCy2] starting precursor is completely consumed after this time. Concomitantly, the 1H resonances of C12-NH2 changed, particularly the ones of NH and α-CH2 groups, which shows that the C12-NH2 electronic environment changes over time. Importantly, broad and weak signals were detected around 3 ppm, which were superimposed on the ones previously assigned to mono- and dimeric zinc amido complexes and zinc amido oligomers (Fig. S16†) exhibit diffusion coefficient values between 5.7 and 4.0 × 10−10 m2 s−1 (Fig. S17†), which is consistent with D values measured for mono- and dimeric zinc amido complexes but definitely not with oligomeric species74 (1.3 × 10−10 m2 s−1).13C INEPT and CP MAS experiments confirmed the formation of the Zn–amido complexes only. Until 2 h, characteristic 13C signals of amido α-CH2 and β-CH2 groups are indeed observed at 48.1 and 37.5 ppm only in INEPT spectra (Fig. S18†) but not in the CP MAS ones (Fig. S19†). After 2 h, these INEPT signals disappear and no signal associated with oligomers shows up in the CP experiment. Finally, after 7 h, both CP and INEPT 13C MAS spectra show signals typical of C12-NH2 stabilizing ZnO NCs.87
Fig. 3 shows 17O NMR spectra recorded over time. After 10 min, weak and broad signals are observed in the 10–40 ppm range. Their intensity and resolution increase significantly for 30 min of hydrolysis time and signals at 14.6 and 30.0 ppm are clearly observed. These values can tentatively be assigned to hydroxyl groups and water molecules within zinc molecular complexes as they definitely do not correspond to ZnO for which classical 17O signals are observed between −18 and −30 ppm.88 They cannot be attributed to Zn–O–Cy species as well, since no correlating 1H resonance can be detected above 3.5 ppm (Fig. S16†).
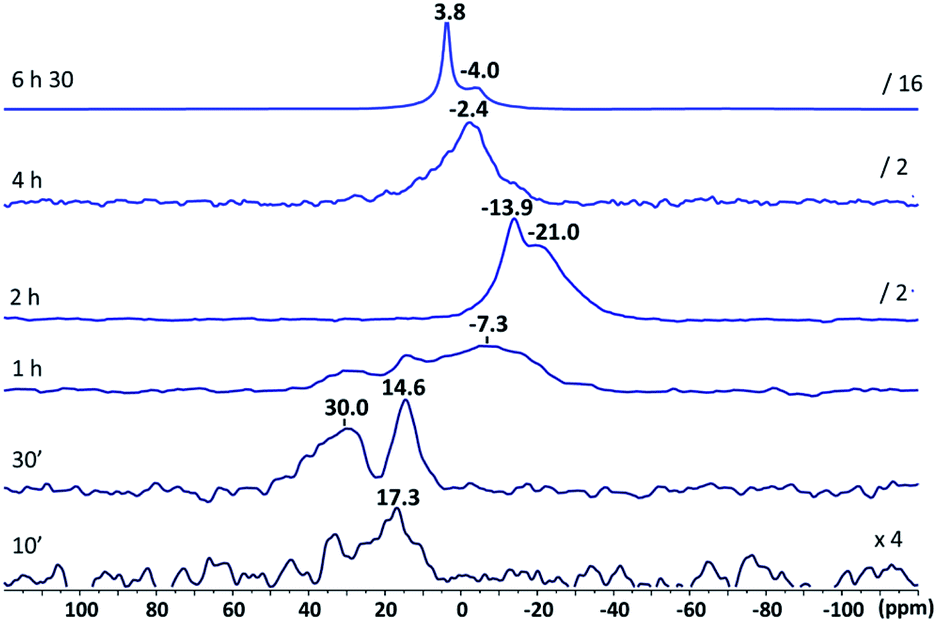 | ||
| Fig. 3 17O NMR spectra (298 K, toluene-d8) of [ZnCy2] mixed with 2 eq. of C12-NH2 after different hydrolysis reaction times. | ||
After 1 h, the new broad resonance observed at −7.3 ppm can be assigned to various small-sized [Znx+yOx(OH)2y] clusters, nuclei of future ZnO NCs. Even if the molecular structure of these clusters has not been specifically determined in this work (crystallization of the cluster with such a long alkyl chain length is very tricky), a structure with a central Zn2(μ4-O)2 core encapsulated by the parent RZn[N(CH2)11CH3] moieties as the ones already reported in the literature for more bulky primary amines can be considered.89,90 The evolution of the 17O signals over time confirms this hypothesis: after 2 h, signals at −13.9 and −21.0 ppm remained the ones observed for ZnO NCs,87 suggesting the formation of small dispersible ZnO NCs at this early stage of the reaction. The disappearance of these signals at longer times is due to the formation of large ZnO NCs that no longer give observable liquid-state NMR signals. The signal around 0.0 ppm is characteristic of water molecules interacting with the ZnO NC surface, in fast exchange between the solution and the surface of ZnO.87
Overall, the detailed multinuclear NMR analysis established that the hydrolysis reaction takes precedence over any oligomerization process that occurs with primary amine surfactants in the absence of water. These results suggest that the difference in morphology is not chiefly due to significant differences at the early stage of the molecular state between primary and secondary amines. This prompted us to examine, in more detail, the interaction and surface dynamics of various alkyl amines on the ZnO NCs.
Amine interaction with ZnO NCs and surface dynamics
The interaction of amines on the surface of ZnO NCs can be studied by solid state NMR.87,91 In brief, while DP is not selective and shows all the species on the NC surface regardless of their dynamics, CP and INEPT sequences evidence selectively rigid and mobile species, respectively. Fig. 4 shows the comparison of the 13C CP, DP and INEPT MAS NMR spectra of ZnO NCs stabilized by either C12-NH2 (ZnO@C12-NH2, Fig. 4 left) or C12-NH(CH3) (ZnO@C12-NH(CH3), Fig. 4 right). First of all, INEPT spectra exhibit similar central CH2 signals for both ZnO@C12-NH(CH3) and ZnO@C12-NH2 stating that the mobile amine possesses the same conformation regardless of their structure. Second, ZnO@C12-NH(CH3) exhibits similar 13C spectra regardless of the polarization mode which suggests that C12-NH(CH3) possesses the same conformation regardless of its dynamics. This is in contrast with the ZnO@C12-NH2 spectra whose CP and INEPT spectra are very different stating that mobile and rigid C12-NH2 possess different conformations. Moreover, sharper CP MAS signals are clearly observed for ZnO@C12-NH(CH3) NCs than for ZnO@C12-NH2. This important result points out to an increase in the chain mobility of the rigid C12-NH(CH3) compared to the rigid C12-NH2 at the ZnO surface and suggests weaker interactions of the C12-NH(CH3) with the ZnO surface than the ones of C12-NH2. Importantly, a CP J-resolved experiment performed on ZnO@C12-NH2 evidenced a bent conformation of C12-NH2 at the ZnO surface induced by hydrogen bonds.91 Such bent conformation is not observed for C12-NH(CH3) highlighting the absence of hydrogen bonds in ZnO@C12-NH(CH3).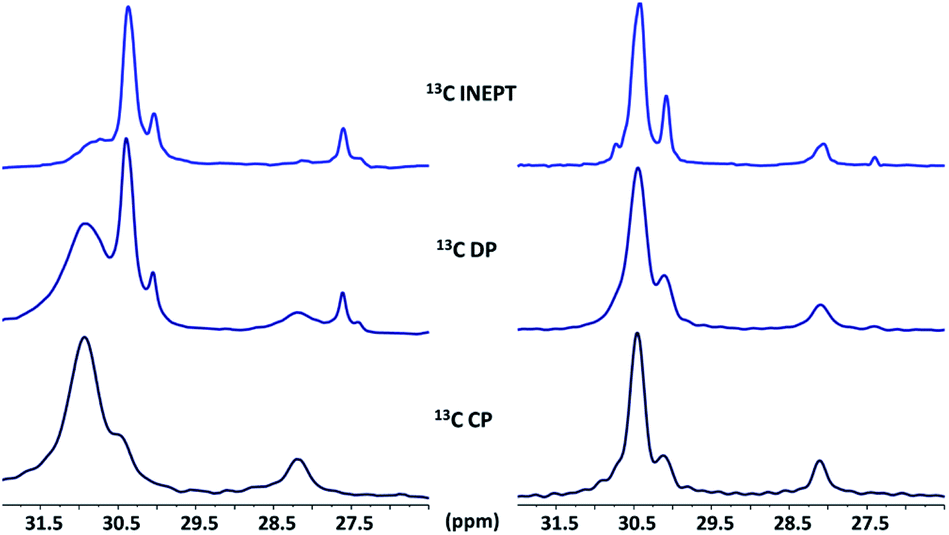 | ||
| Fig. 4 13C CP, DP and INEPT MAS NMR spectra (295 K) of ZnO stabilized with 0.2 eq. of C12-NH2 (left), and with 0.2 eq. of C12-NH(CH3) (right). | ||
Finally, a transferred NOE experiment86 was performed on ZnO NCs in solution with 1 equiv. of C12-NH2 and 10 equiv. of C12-NH(CH3) (Fig. S20†). Despite the large excess of secondary amine introduced, the typical primary amine 1H α-CH2 signal at 2.54 ppm is strongly broadened and showed an intense transferred NOE correlation, which evidences that the presence of a large 10-fold excess of secondary amine compared to the corresponding primary amine does not lead to any sensible decrease of the quantity of primary amine at the ZnO surface. This clearly confirms the higher affinity of the primary amine for ZnO.
From a geometrical perspective (Scheme 1), multiple H-bonding interactions reduce the mobility of primary amines at the surface of the NC. This additional network mainly localizes along the lateral faces of the NCs (left),91,92 and is directly responsible for the 1D anisotropic growth of ZnO NCs. Conversely, the secondary amine surfactant does not form such a hydrogen bonding network, or forms to a much limited extent (right). It consequently remains mobile over the entire surface of the NCs, and a formal 3D isotropic growth is expected from such a situation. In the last system, in ZnO aggregates formed in the presence of tertiary amine C12-N(CH3)2, the amine much weakly interacts with the surface of NCs, and it does not participate to segregate the particles or to orientate any particular growth. Accordingly, no NOE-Tr signal is observed (Fig. S21†).86 The precursor colloids formed in solution are not sufficiently stabilized and therefore aggregate.
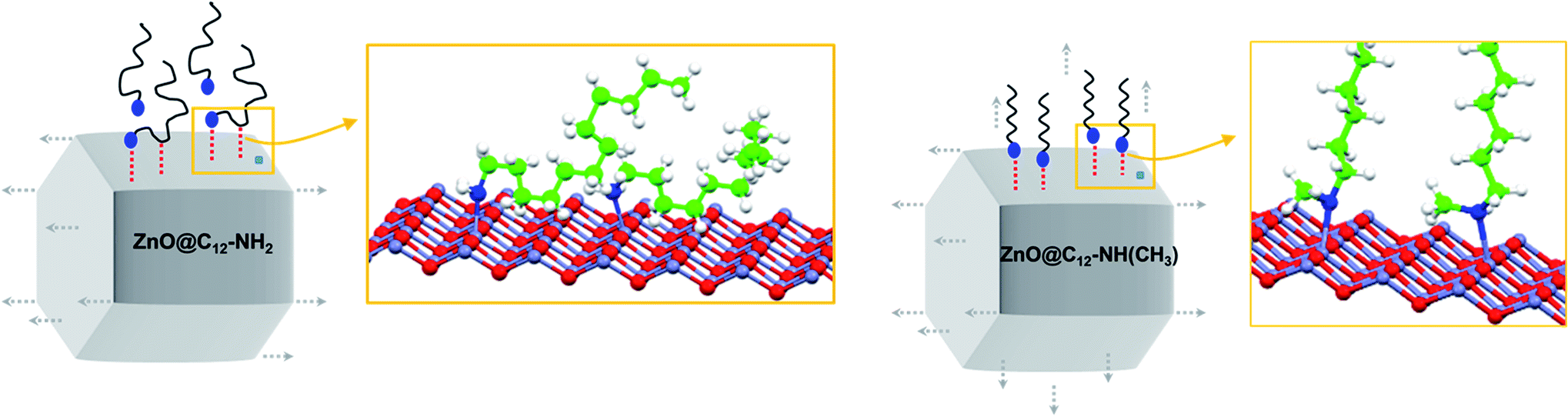 | ||
| Scheme 1 Overview of the proposed model for either the anisotropic or isotropic growth of ZnO depending on the structure of the amines which are localized on the lateral faces of the NCs, namely primary or secondary amine, respectively. H-bonds formed by primary amine reduced their mobility while secondary amine remains mobile at the surface of the NC. | ||
Experimental
Materials and reagents
All starting compounds in this work were stored in a MBraun glove box with an argon system. The zinc precursor [Zn(C6H11)2] (denoted as [ZnCy2]) was purchased from Nanomeps (http://www.nanomeps.fr). Secondary amine ligands (N-methyldodecylamine, C12-NH(CH3)) and tertiary amine (N,N-dimethyldodecylamine, C12-N(CH3)2) were all purchased from Alfa Aesar. A primary amine ligand (dodecylamine, C12-NH2) was purchased from Aldrich and stored in a glove box and used without any further purifications. THF was obtained from an MBraun purificator. The residual water content of the solvent was systematically measured by Karl Fischer coulometric titration by using Metrohm equipment. All mixing processes were performed in a glove box. Hydrolysis steps were performed outside such a glove box and performed with a standard vacuum line under an argon atmosphere.NMR spectroscopy
Liquid state NMR: 1D and 2D 1H NMR experiments were performed on a Bruker Avance 600 NEO spectrometer that was equipped with a 5 mm triple resonance inverse Z-gradient probe. Toluene-d8 was used as solvent. All diffusion measurements were conducted by using the stimulated echo pulse sequence with bipolar gradient pulses. 1D 17O NMR experiments were performed with a recovery delay of 0.2 s. Ka measurements were performed from 1H NMR titration by following chemical shift evolution of [ZnCy2] resonances. The concentration of [ZnCy2] was kept constant in the presence of increasing amine concentrations (with concentration ratios from 0 to about 15 equivalents). Ka was extracted from fitting the evolution of chemical shifts with eqn (4)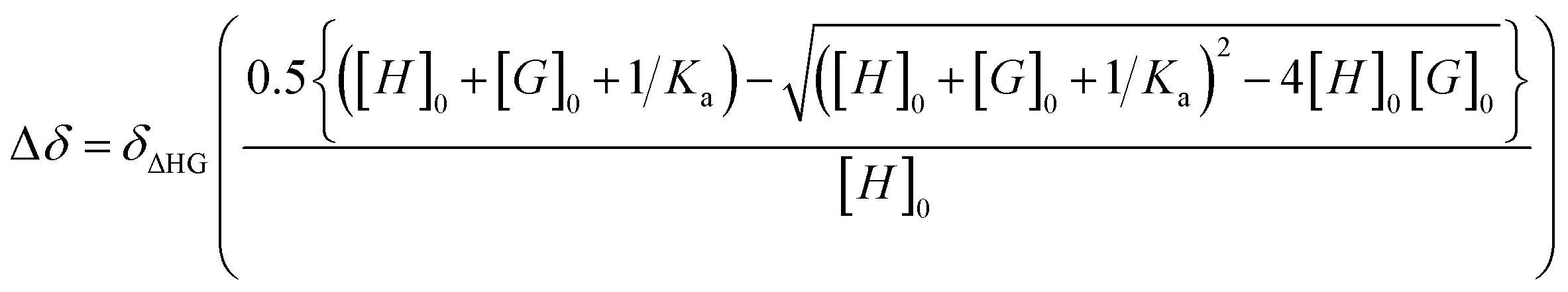 | (4) |
Solid-state NMR experiments were performed on a Bruker Avance III HD 400 spectrometer. Samples were packed into 4 mm zirconia rotors inside a glove box. The rotors were spun at 5 kHz at 293 K. 13C MAS with cross-polarization (CP) was performed with a recycle delay of 2 s and a contact time of 2 ms. 13C MAS with Insensitive Nuclei Enhanced by Polarization Transfer (INEPT) were performed with a recycle delay of 3 s.
1H and 13C chemical shifts are relative to TMS and 17O is referenced to water.
Transmission electron microscopy
Samples for TEM analysis were prepared by the slow evaporation of droplets of colloidal solution deposited on carbon-supported copper grids. The samples were carefully dried overnight under a pressure of 5 × 10−5 mbar by using a BOC Edward turbo molecular pump. The TEM experiments were performed on a JEOL JEM1011 electron microscope operating at 100 kV with a resolution point of 0.45 nm. Image J software was used manually or with a macro in order to extract width and length of nano-objects in various micrographs. The macro is based on the PSA macro, available at http://www.code.-google.com/p/psa-macro. Overlapping nanoparticles are automatically rejected for the statistical analysis. Nanoparticle size-distribution histograms were generated by using magnified TEM images. For each sample, the size-distribution of the particles was determined by measuring a minimum of 200 particles with image J software. It was generally analyzed using 2D plots.Synthesis
Colloids of [ZnCy2]/2C12-NH(CH3)([ZnCy2]: 57.9 mg, 0.25 mmol; C12-NH(CH3): 99.69 mg, 0.5 mmol), [ZnCy2]/2C12-N(CH3)2([ZnCy2]: 57.9 mg, 0.25 mmol; C12-N(CH3)2: 106.7 mg, 0.5 mmol) in d8-toluene were prepared the same as the [ZnCy2]/C12-NH2 colloid.
Computational details
Geometries were fully optimized at the PBE-D3/DGDZVP level of calculation using Gaussian09.93 Vibrational analysis was performed at the same level as the geometry optimization. Solvent effects of octylamine (ε = 3.1)94 were included using the polarizable continuum model (PCM) implemented in Gaussian09. The influence of the length of the alkyl chain of the model amine of C12-NH2 was studied. No significant difference was found in the results using either hexylamine (H2NC6H13) or octylamine (H2NC8H17). The former was therefore selected in order to reduce the computational cost. However, octylamine was used as the solvent in PCM calculations, as its required dielectric constant was known.Electron Localization Function (ELF)95,96 topological analysis and Quantum Theory of Atoms in Molecules (QTAIM)97,98 analysis were performed with the TopMod package.99 ELF maps were plotted using the Molekel program.100 QTAIM analysis was also performed with the AIMAll software.101
Conclusions
In a crucial attempt toward progress in the understanding and control of the construction of functional nano-objects at the molecular scale, the present work aims at elucidating the inherent complexity of solution organometallic mild synthesis of ZnO NCs. We focused on some of the most widely used surfactants for nanoparticle growth, fatty alkylamines. By keeping the same aliphatic chain length and varying the structure of the amine (primary, secondary, or tertiary) the complementary NMR and DFT analyses allowed us to study the interaction of these surfactants with the metallic precursor of the ZnO NCs along the synthetic process. Regardless of the amine structure, a 1:1 adduct is originally formed. However, while its association constant is consistent for related primary and secondary amines, it is one order of magnitude smaller for the corresponding tertiary amine. The difference between primary and secondary amine surfactants comes from the propensity of primary amines to form zinc-amido oligomers, a reaction which is impeded in the case of secondary amine because of higher steric hindrance.
However, the reaction rate of such oligomerization was proved to be much slower than the competitive hydrolysis reaction generating the NCs, and has a negligible influence over NC structuring. We thus demonstrated that the difference in morphology between ZnO NCs observed as a function of the amine structure arises from a strong difference in their dynamics at the surface of the growing NCs. Multiple H-bonding interactions at the ZnO surface for primary amines, identified by 13C MAS NMR, leads to reduced mobility of these amines compared to secondary amines, which remain mobile at the surface of the NCs in all the dimensions of space.
Our approach in the present study could clearly be extended to a great number of metal and semi-conducting nanoparticles stabilized in solution by surfactants such as thiols, carboxylic acids, phosphines, carbenes, etc. This would provide fundamental knowledge towards a rationalized vision of the mild and controlled synthesis of NCs in solution. Most importantly, we have shown herein that the efficient and very widespread use of primary amines in the synthesis of nanoparticles via solution protocols is attributable to weak interaction hydrogen bonds with strong influence on the surface of the growing nanoparticle and final morphology and the structure of the end hybrid material.
Author contributions
The manuscript was written through contributions of all authors and all authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by both the Centre National de la Recherche Scientifique (CNRS) and the China Scholarship Council (CSC). V. Collière is gratefully acknowledge for TEM measurements. The theoretical studies were performed using HPC resources from CALMIP (Grant 2020 [16028]]) and from GENCI-[CINES/IDRIS] (Grant 2020 [0805008]).References
- Z. Li, R. Wang, J. Xue, X. Xing, C.-C. Yu, T. Huang, J. Chu, K.-L. Wang, C. Dong, Z. Wei, Y. Zhao, Z.-K. Wang and Y. Yang, J. Am. Chem. Soc., 2019, 141, 17610–17616 CrossRef CAS PubMed.
- H. Li and W. Zhang, Chem. Rev., 2020, 120, 9835–9950 CrossRef CAS PubMed.
- Y. Peng, B. Lu, F. Wu, F. Zhang, J. E. Lu, X. Kang, Y. Ping and S. Chen, J. Am. Chem. Soc., 2018, 140, 15290–15299 CrossRef CAS PubMed.
- N. Nasiri, R. Bo, L. Fu and A. Tricoli, Nanoscale, 2017, 9, 2059 RSC.
- M. H. Tran, T. Park and J. Hur, Appl. Surf. Sci., 2021, 539, 148222 CrossRef.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- A. Pinkas, N. Waiskopf, S. Gigi, T. Naor, A. Layani and U. Banin, Nanoscale, 2021, 13, 7152 RSC.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- H. Ma, W. Ma, J.-F. Chen, X.-Y. Liu, Y.-Y. Peng, Z.-Y. Yang, H. Tian and Y.-T. Long, J. Am. Chem. Soc., 2018, 140, 5272–5279 CrossRef CAS PubMed.
- Y. Hu, H.-R. Zhang, L. Dong, M.-R. Xu, L. Zhang, W.-P. Ding, J.-Q. Zhang, J. Lin, Y.-J. Zhang, B.-S. Qiu, P.-F. Wei and L.-P. Wen, Nanoscale, 2019, 11, 11789 RSC.
- O. Lupan, V. Postica, M. Hoppe, N. Wolff, O. Polonskyi, T. Pauporté, B. Viana, O. Majérus, L. Kienle, F. Faupel and R. Adelung, Nanoscale, 2018, 10, 14107 RSC.
- H. Hu, L. Sun, Y. Gao, T. Wang, Y. Huang, C. Lv, Y.-F. Zhang, Q. Huang, X. Chen and H.-X. Wu, J. Hazard. Mater., 2020, 387, 121670 CrossRef CAS PubMed.
- K. Vyavhare, R. B. Timmons, A. Erdemir, B. L. Edwards and P. B. Aswath, Langmuir, 2021, 37, 1743–1759 CrossRef CAS PubMed.
- F. Gonell, C. M. Sánchez-Sánchez, V. Vivier, C. Méthivier, C. Laberty-Robert and D. Portehault, Chem. Mater., 2020, 32, 4241–4247 CrossRef CAS.
- Q. Xiong, S. Huang, J. Du, X. Tang, F. Zeng, Z. Liu, Z. Zhang, T. Shi, J. Yang, D. Wu, H. Lin, Z. Luo and Y. Leng, Adv. Opt. Mater., 2020, 8, 2000977 CrossRef CAS.
- M. Yan, S. M. Collins, P. A. Midgley and J. I. Feldblyum, Chem. Mater., 2020, 32, 2379–2388 CrossRef CAS.
- G. Gouget, D. Bregiroux, R. Grosjean, D. Montero, S. Maier, F. Gascoin, C. Sanchez and D. Portehault, Chem. Mater., 2021, 33, 2099–2109 CrossRef CAS.
- J. Ye, Y. Li, A. A. Medjahed, S. Pouget, D. Aldakov, Y. Liu and P. Reiss, Small, 2021, 17, 2005671 CrossRef CAS PubMed.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed.
- S. Pradhan, D. Bhujel, B. Gu-rung, D. Sharma, S. Basel, S. Rasaily, S. Thapa, S. Borthakur, W. L. Ling, L. Saikia, P. Reiss, A. Pariyar and S. Tamang, Nanoscale Adv., 2021, 3, 1464 RSC.
- N. Moghaddam, C. Dabard, M. Dufour, H. Po, X. Xu, T. Pons, E. Lhuillier and S. Ithurria, J. Am. Chem. Soc., 2021, 143, 1863–1872 CrossRef CAS PubMed.
- Y. Prado, J.-L. Qu, C. Gréboval, C. Dabard, P. Rastogi, A. Chu, A. Khalili, X. Z. Xu, C. Delerue, S. Ithurria and E. Lhuillier, Chem. Mater., 2021, 33, 2054–2061 CrossRef CAS.
- P. Krupiński, A. Grala, M. Wolska-Pietkiewicz, W. Danowski, I. Justyniak and J. Lewiński, ACS Sustainable Chem. Eng., 2021, 9, 1540–1549 CrossRef.
- M. Siemer, G. Tomaschun, T. Klüner, P. Christopher and K. Al-Shamery, ACS Appl. Mater. Interfaces, 2020, 12, 27765–27776 CrossRef CAS PubMed.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, Chem. Rev., 2019, 119, 4819–4880 CrossRef CAS PubMed.
- S. Busatto, M. de Ruiter, J. T. B. H. Jastrzebski, W. Albrecht, V. Pinchetti, S. Brovelli, S. Bals, M.-E. Moret and C. d. M. Donega, ACS Nano, 2020, 14, 13146–13160 CrossRef CAS PubMed.
- L.-M. Lacroix, A. Meffre, C. Gatel, P.-F. Fazzini, S. Lachaize, M. Respaud and B. Chaudret, ChemPlusChem, 2019, 84, 302–306 CrossRef CAS PubMed.
- N. Liakakos, B. Cormary, X. Li, P. Lecante, M. Respaud, L. Maron, A. Falqui, A. Genovese, L. Vendier, S. Koinis, B. Chaudret and K. Soulantica, J. Am. Chem. Soc., 2012, 134, 17922–17931 CrossRef CAS PubMed.
- F. Wu, Z. Zhang, Z. Zhu, M. Li, W. Lu, M. Chen, E. Xu, L. Wang and Y. Jiang, CrystEngComm, 2018, 20, 4492–4498 RSC.
- M. A. Ben Aissa, B. Tremblay, A. Andrieux-Ledier, E. Maisonhaute, N. Raouafi and A. Courty, Nanoscale, 2015, 7, 3189–3195 RSC.
- S. C. Padmanabhan, J. McGrath, M. Bardosova and M. E. Pemble, J. Mater. Chem., 2012, 22, 11978–11987 RSC.
- J. Cure, Y. Coppel, T. Dammak, P. F. Fazzini, A. Mlayah, B. Chaudret and P. Fau, Langmuir, 2015, 31, 1362–1367 CrossRef CAS PubMed.
- Y. Zhou, F. Wang and W. E. Buhro, Chem. Mater., 2020, 32, 8350–8360 CrossRef CAS.
- F. Li, Y. Xie, Y. Hu, M. Long, Y. Zhang, J. Xu, M. Qin, X. Lu and M. Liu, ACS Energy Lett., 2020, 5, 1422–1429 CrossRef CAS.
- Y.-H. Kim, Y. Zhai, E. A. Gaulding, S. N. Habisreutinger, T. Moot, B. A. Rosales, H. Lu, A. Hazarika, R. Brunecky, L. M. Wheeler, J. J. Berry, M. C. Beard and J. M. Luther, ACS Nano, 2020, 14, 8816–8825 CrossRef CAS PubMed.
- C. Garnero, M. Lepesant, C. Garcia-Marcelot, Y. Shin, C. Meny, P. Fuger, B. Warot-Fonrose, R. Arenal, G. Viau, K. Soulantica, P. Fau, P. Poveda, L.-M. Lacroix and B. Chaudret, Nano Lett., 2019, 19, 1379–1386 CrossRef CAS PubMed.
- S. Tamang, C. Lincheneau, Y. Hermans, S. Jeong and P. Reiss, Chem. Mater., 2016, 28, 2491–2506 CrossRef CAS.
- B. Fritzinger, R. K. Capek, K. Lambert, J. C. Martins and Z. Hens, J. Am. Chem. Soc., 2010, 132, 10195–10201 CrossRef CAS PubMed.
- B. Cormary, T. Li, N. Liakakos, L. Peres, P.-F. Fazzini, T. Blon, M. Respaud, A. J. Kropf, B. Chaudret, J. T. Miller, E. A. Mader and K. Soulantica, J. Am. Chem. Soc., 2016, 138, 8422–8431 CrossRef CAS PubMed.
- S. Mourdikoudis and L. M. Liz-Marzan, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- K. Lee, Y. Huang and J. F. Corrigan, Chem. Commun., 2019, 55, 11466 RSC.
- B. A. Tappan, M. K. Horton and R. L. Brutchey, Chem. Mater., 2020, 32, 2935–2945 CrossRef CAS.
- R. G. Ellis, J. W. Turnley, D. J. Rokke, J. P. Fields, E. H. Alruqobah, S. D. Deshmukh, K. Kisslinger and R. Agrawal, Chem. Mater., 2020, 32, 5091–5103 CrossRef CAS.
- R. M. Freire, J. Rojas-Nunez, A. L. Elias-Arriaga, K. Fujisawa, L. Troncoso, J. C. Denardin and S. E. Baltazar, Inorg. Chem. Front., 2020, 7, 4902 RSC.
- K. Tabatabaei, H. R. Sully, Z. Ju, K. Hellier, H. Lu, C. J. Perez, K. A. Newton, R. L. Brutchey, F. Bridges, S. A. Carter and S. M. Kauzlarich, ACS Nano, 2021, 15, 1685–1700 CrossRef CAS PubMed.
- G. Gouget, M. Pellerin, R. Al Rahal Al Orabi, L. Pautrot-D'Alençon, T. Le Mercier and C. B. Murray, J. Am. Chem. Soc., 2021, 143, 3300–3305 CrossRef CAS PubMed.
- A. Zakhtser, A. Naitabdi, R. Benbalagh, F. Rochet, C. Salzemann, C. Petit and S. Giorgio, ACS Nano, 2021, 15, 4018–4033 CrossRef CAS PubMed.
- R. Dören, B. Leibauer, M. A. Lange, E. Schechtel, L. Prädel, M. Panthöfer, M. Mondeshki and W. Tremel, Nanoscale, 2021, 13, 8146 RSC.
- G. M. Leteba, Y.-C. Wang, T. J. A. Slater, R. Cai, C. Byrne, C. P. Race, D. R. G. Mitchell, P. B. J. Levecque, N. P. Young, S. M. Holmes, A. Walton, A. I. Kirkland, S. J. Haigh and C. I. Lang, Nano Lett., 2021, 21, 3989–3996 CrossRef CAS PubMed.
- Y.-H. Kim, Y. Zhai, E. A. Gaulding, S. N. Habisreutinger, T. Moot, B. A. Rosales, H. Lu, A. Hazarika, R. Brunecky, L. M. Wheeler, J. J. Berry, M. C. Beard and J. M. Luther, ACS Nano, 2020, 14, 8816–8825 CrossRef CAS PubMed.
- O. J. Ashton, A. R. Marshall, J. H. Warby, B. Wenger and H. J. Snaith, Chem. Mater., 2020, 32, 7172–7180 CrossRef CAS.
- L. Chen, H. Hu, Y. Chen, Y. Li, J. Gao and G. Li, Chem.–Eur. J., 2021, 27, 1057–1065 CrossRef CAS PubMed.
- B. M. Sperry and C. K. Luscombe, Chem. Mater., 2021, 33, 136–145 CrossRef CAS.
- C. K. Ng, W. Yin, H. Li and J. J. Jasieniak, Nanoscale, 2020, 12, 4859–4867 RSC.
- M. Liu, Y.-Y. Wang, Y. Liu and F.-L. Jiang, J. Phys. Chem. C, 2020, 124, 4613–4625 CrossRef CAS.
- F. J. Garcia-Garcia, R. Klee, P. Lavela, M. R. D. Bomio and J. L. Tirado, ChemElectroChem, 2020, 7, 3528–3534 CrossRef CAS.
- M. L. Kahn, A. Glaria, C. Pages, M. Monge, L. Saint Macary, A. Maisonnat and B. Chaudret, J. Mater. Chem., 2009, 19, 4044–4060 RSC.
- V. Ischenko, S. Polarz, D. Grote, V. Stavarache, K. Fink and M. Driess, Adv. Funct. Mater., 2005, 15, 1945–1954 CrossRef CAS.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 12871–12934 RSC.
- M. Muller, S. Hermes, K. Kaehler, M. W. E. van den Berg, M. Muhler and R. A. Fischer, Chem. Mater., 2008, 4576–4587 CrossRef.
- B. Fritzinger, I. Moreels, P. Lommens, R. Koole, Z. Hens and J. C. Martins, J. Am. Chem. Soc., 2009, 131, 3024–3032 CrossRef CAS PubMed.
- D. Lee, M. Wolska-Pietkiewicz, S. Badoni, A. Grala, J. Lewinski and G. De Paepe, Angew. Chem., Int. Ed., 2019, 58, 17163–17168 CrossRef CAS PubMed.
- C. H. Hung and W. T. Whang, J. Mater. Chem., 2005, 15, 267–274 RSC.
- Z. H. Zhang, M. Lu, H. R. Xu and W. S. Chin, Chem.–Eur. J., 2007, 13, 632–638 CrossRef CAS PubMed.
- Z. Zhao, Z. Zheng, C. Roux, C. Delmas, J. D. Marty, M. L. Kahn and C. Mingotaud, Chem.–Eur. J., 2016, 22, 12424–12429 CrossRef CAS PubMed.
- H. Ding, J. Yan, Z. Wang, G. Xie, C. Mahoney, R. Ferebee, M. Zhong, W. F. M. Daniel, J. Pietrasik, S. S. Sheiko, C. J. Bettinger, M. R. Bockstaller and K. Matyjaszewski, Polymer, 2016, 107, 492e502 CrossRef.
- X. Han, S. Huang, Y. Wang and D. Shi, Mater. Sci. Eng., C, 2016, 64, 87–92 CrossRef CAS PubMed.
- C. Sun and D. Xue, J. Phys. Chem. C, 2013, 117, 5505–5511 CrossRef CAS.
- M. L. M. Napi, A. F. A. Noorden, M. Loong, P. Tan, H. Jamaluddin, F. Abd Hamid, M. K. Ahmad, U. Hashim, M. R. Ahmad and S. M. Sultan, J. Electrochem. Soc., 2020, 167, 137501 CrossRef CAS.
- V. Amendola, D. Amans, Y. Ishikawa, N. Koshizaki, S. Scirè, G. Compagnini, S. Reichenberger and S. Barcikowski, Chem.–Eur. J., 2020, 26, 9206–9242 CrossRef CAS PubMed.
- W. Lee, J.-S. Yoon, Y.-M. Kim and Y.-M. Sung, J. Mater. Chem. C, 2019, 7, 12019 RSC.
- S. Murali, P. K. Dammala, B. Rani, R. Santhosh, C. Jadhao and N. K. Sahu, J. Alloys Compd., 2020, 844, 156149 CrossRef CAS.
- Z. Zheng, R. Butynska, C. Valverde Serrano, J.-D. Marty, C. Mingotaud and M. L. Kahn, Chem.–Eur. J., 2016, 22, 15614–15618 CrossRef PubMed.
- Z. Zhao, Y. Coppel, J. Fitremann, P. Fau, C. Roux, C. Lepetit, P. Lecante, J.-D. Marty, C. Mingotaud and M. L. Kahn, Chem. Mater., 2018, 30, 8959–8967 CrossRef CAS.
- M. L. Kahn, M. Monge, V. Collière, F. Senocq, A. Maisonnat and B. Chaudret, Adv. Funct. Mater., 2005, 15, 458–468 CrossRef CAS.
- S. Saliba, M. L. Kahn, J.-D. Marty, Y. Coppel, C. Mingotaud and B. Chaudret, J. Mater. Chem., 2011, 21, 6821–6823 RSC.
- S. Saliba, Y. Coppel, C. Mingotaud, J.-D. Marty and M. L. Kahn, Chem.–Eur. J., 2012, 18, 8084–8091 CrossRef CAS PubMed.
- S. Saliba, Y. Coppel, M.-F. Achard, C. Mingotaud, J.-D. Marty and M. L. Kahn, Angew. Chem., Int. Ed., 2011, 50, 12032–12035 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- E. Espinosa, I. Alkorta, I. Rozas, J. Elguero and E. Molins, Chem. Phys. Lett., 2001, 336, 457–461 CrossRef CAS.
- E int = −1/2Vbcp and Eint (kcal mol−1) = −313.754 × Vbcp (au).
- C. Lepetit and M. L. Kahn, Res. Chem. Intermed., 2021, 47, 377–395 CrossRef CAS.
- R. J. Ouellette and J. David Rawn, Organic Chemistry Study Guide, 2015, pp. 465–494 Search PubMed.
- W. Tiznado, E. Chamorro, R. Contreras and P. Fuentealba, J. Phys. Chem., 2005, 109, 3220–3224 CrossRef CAS PubMed.
- Y. Coppel, G. Spataro, C. Pages, B. Chaudret, A. Maisonnat and M. L. Kahn, Chem.–Eur. J., 2012, 18, 5384–5393 CrossRef CAS PubMed.
- G. Spataro, Y. Champouret, P. Florian, Y. Coppel and M. L. Kahn, Phys. Chem. Chem. Phys., 2018, 20, 12413–12421 RSC.
- Y. Champouret, Y. Coppel and M. L. Kahn, J. Am. Chem. Soc., 2016, 138, 16322–16328 CrossRef CAS PubMed.
- D. Prochowicza, K. Sokołowski and J. Lewinski, Coord. Chem. Rev., 2014, 270–271, 112–126 CrossRef.
- J. A. Garden and S. D. Pike, Dalton Trans., 2018, 47, 3638 RSC.
- M. L. Kahn, T. Cardinal, B. Bousquet, M. Monge, V. Jubera and B. Chaudret, ChemPhysChem, 2006, 7, 2392–2397 CrossRef CAS PubMed.
- G. Spataro, Y. Champouret, Y. Coppel and M. L. Kahn, ChemPhysChem, 2020, 21, 2454–2459 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P.-M. Chassaing, F. Demangeot, V. Paillard, A. Zwick, N. Combe, C. Pagès, M. L. Kahn, A. Maisonnat and B. Chaudret, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 153306 CrossRef.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- B. Silvi and A. Savin, Nature, 1994, 371, 683–686 CrossRef CAS.
- R. F. W. Bader, in Atoms In Molecules, Clarendon Press, Oxford, UK, 1990 Search PubMed.
- R. F. W. Bader and H. Essen, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
- Molekel 4.3 from CSCS, http://www.cscs.ch/molekel/ Search PubMed.
- T. A. Keith, AIMAll (Version 17.11.04), TK Gristmill Software, Overland Park KS, USA, http://www.aim.tkgristmill.com Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional NMR measurements and calculation details are provided as a supplementary material. See DOI: 10.1039/d1na00566a |
| This journal is © The Royal Society of Chemistry 2021 |