
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative epitope analysis reveals drastic 63% reduced immuno-affinity and 60% enhanced transmissibility for SARS-CoV-2 variants†
Giulio Brunetti ,
Annalisa De Pastina and
Martin Hegner*
,
Annalisa De Pastina and
Martin Hegner*
Center for Research on Adaptive Nanostructures and Nanodevices (CRANN), School of Physics, Trinity College Dublin (TCD), D02 Dublin, Ireland
First published on 23rd September 2021
Abstract
SARS-CoV-2 is the cause of a global pandemic that has led to more than 4 million deaths, continues to spread and holds the world in a tight grip. The virus has developed substantial mutations that undermine the efficacy of current vaccines and monoclonal antibody therapies. Semi-quantitative immuno – and neutralization assays are unable to provide direct quantitative insights about the minute variations of emerging mutants. Here, we develop a quantitative assay that enables synchronous screening of emerging variant epitopes with single amino acid resolution. We report on specific label-free quantitative nanomechanical analysis of pseudovirus spike interaction with ACE2 receptors. Within minutes, we can characterize the B.1.1.7 variant transmissibility due to its 63% increased binding, and measure a 60% reduced efficacy of antibodies towards B.1.351 and P.1 variants. Our technology can assist vaccine development studies, with focus on comparing protection patterns and novel vaccine candidates and tracking of immunity over time.
Introduction
As of 2020, there are 45 different known species of coronavirus, divided into four families: alpha (α), beta (β), delta (δ) and gamma (γ), classified based on their protein sequence. Mostly zoonotic, only α and β families developed the ability to transfer from animals to humans, thus becoming human pathogens.1 So far, there are seven recorded human coronaviruses (hCoV): five of these belong to the β family and include the severe acute respiratory syndrome coronavirus (SARS-CoV), the Middle East respiratory syndrome coronavirus (MERS-CoV), the SARS-CoV hCoV-HKU1, the hCoV-OC43 and the most recently discovered severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The remaining two, belonging to the α family, include hCoV-NL63 and hCoV-229E, mainly cause mild respiratory or gastrointestinal symptoms and are responsible for up to 30% of human common colds.2 Conversely, hCoV such as MERS, SARS and SARS-CoV-2 represent serious threats for humans due to their correlation with severe health conditions.In particular, SARS-CoV-2 is responsible for COVID-19 (Coronavirus disease 2019), which caused more than 180 million cases and 4 million deaths worldwide as of June 2021.3 COVID-19 pandemic is the third recorded outbreak of a coronavirus in the last two decades, preceded by SARS in 2002 and MERS in 2012 with a case fatality ratio (CFR) of 10% and 34%, respectively.2 COVID-19 is less deadly than SARS and MERS, with a CFR below 3%, but more infectious. In less than two months the number of confirmed COVID-19 cases surpassed the amount that SARS reached over several months.4
Through cryo-electron microscopy, this novel β-coronavirus appears as an approximately 100 nm spherical particle with a lipid bilayer surrounding a highly dense viroplasm.5 A 30 Kb positive single strand RNA encodes for four main structural proteins within its 3′ terminus genome region: nucleocapsid (N), envelope (E), membrane (M) and spike (S).2 The S protein is a 180–200 KDa homotrimeric structure that protrudes with its N-terminus domain from the viral surface with a substantial angular mobility.6,7 This enhanced mobility compensates for the moderate density distribution of the S trimers on the surface of each individual virion, up to 10 times lower than the one reported for influenza virus.5 Furthermore, SARS-CoV-2 exhibits an affinity between S protein and the human angiotensin converting enzyme 2 (ACE2) in the low nM range, about 15-fold higher compared to SARS-CoV.8,9 As expected from a class I viral fusion protein, the S protein presents a S1 receptor binding unit and a S2 membrane fusion unit, able to be activated from the invaded host cell proteases.10 This binding-activation process forces the S protein to undergo a structural rearrangement from a prefusion to a postfusion form, both highly conserved among coronavirus.5 As already known for SARS-CoV, SARS-CoV-2 uses a specific domain of its subunit S1 (domain B or receptor binding domain RBD) to bind to ACE2.8 A hinge point has been discovered between the N-terminal domain (NTD) and the receptor binding motif (RBM), portion of the RBD.5,6 This generates two conformations of the prefusion form, an open state and a closed state, with high or low exposure of the RBM, respectively.
Continuous evolution of SARS-CoV-2 has been reported since its initial appearance, and several sub-clades of SARS-CoV-2 have been identified only in 2020,11,12 mainly generated by single point polymorphisms of the wild type (WT) originated in Wuhan 2019. Considering the haplotype D614G as dominant initial mutation of Wuhan 2019, the SARS-CoV-2 B.1.1.7, SARS-CoV-2 B.1.351 and SARS-CoV-2 P.1 variants emerged in different geographical areas, such as South East England, South Africa and Brazil, respectively.11,13–15 The three new variants show 8 to 9 additional S protein mutations with respect to D614G strain and are commonly named after their geographic origin. High transmission rate and potential inefficacy of developed antibody-based treatments and vaccines are the major concerns associated to the newly identified variants.13,14
Several studies based on semi-quantitative assays such as enzyme linked immunosorbent assay (ELISA) and antibody neutralizations focus on specific RBM mutations, namely N501Y (characterizing B.1.1.7, B.1.351 and P.1 variants) and E484K (characterizing B.1.351 and P.1 variants).13,14,16 Indeed, recent findings highlighted how the immunodominant position of the mutations in the RBM, particularly for E484K, is responsible for substantial loss of antibodies neutralization activity in both convalescent plasma and vaccine sera.14
New semi-quantitative immunoassays targeting the rapid detection of SARS-CoV-2 use different detection mechanisms (e.g. electrochemical with polymerase chain reaction (PCR) amplification17 or local plasmonic with labelled secondary detection steps18), aim at reducing cost19 and enhance multiplexing.18,20 Reported assays can detect the virus RNA or developed antibodies, but are not able to provide direct quantitative insights about the minute variations of emerging mutants.
Results
In this work, we focus on label-free quantitative nanomechanical assays able to perform a single-step evaluation of a sensor array of the evolving humoral immune response in complex biological matrices (see Fig. 1). The piezo-actuated sensors are calibrated and detected in situ,21–25 thus enabling direct large scale multiplexing,26 while the inherent differential read-out with integrated controls reduces false positive results.23,27 The analysis relays on congruent mechanical properties of the array of sensors where the individual resonance frequencies were within <0.5% range. The time resolution of the array measurement across 18 sensors is in the order of 5 s, thus quick changes can be tracked in real-time. Synchronised differential readout with in situ reference probes is mandatory, otherwise evaluation of the biospecific signals are hampered owing to convolution with external environmental factors. Such analysis eliminates thermal drifts, flow-induced features and unspecific interactions as present in serum samples.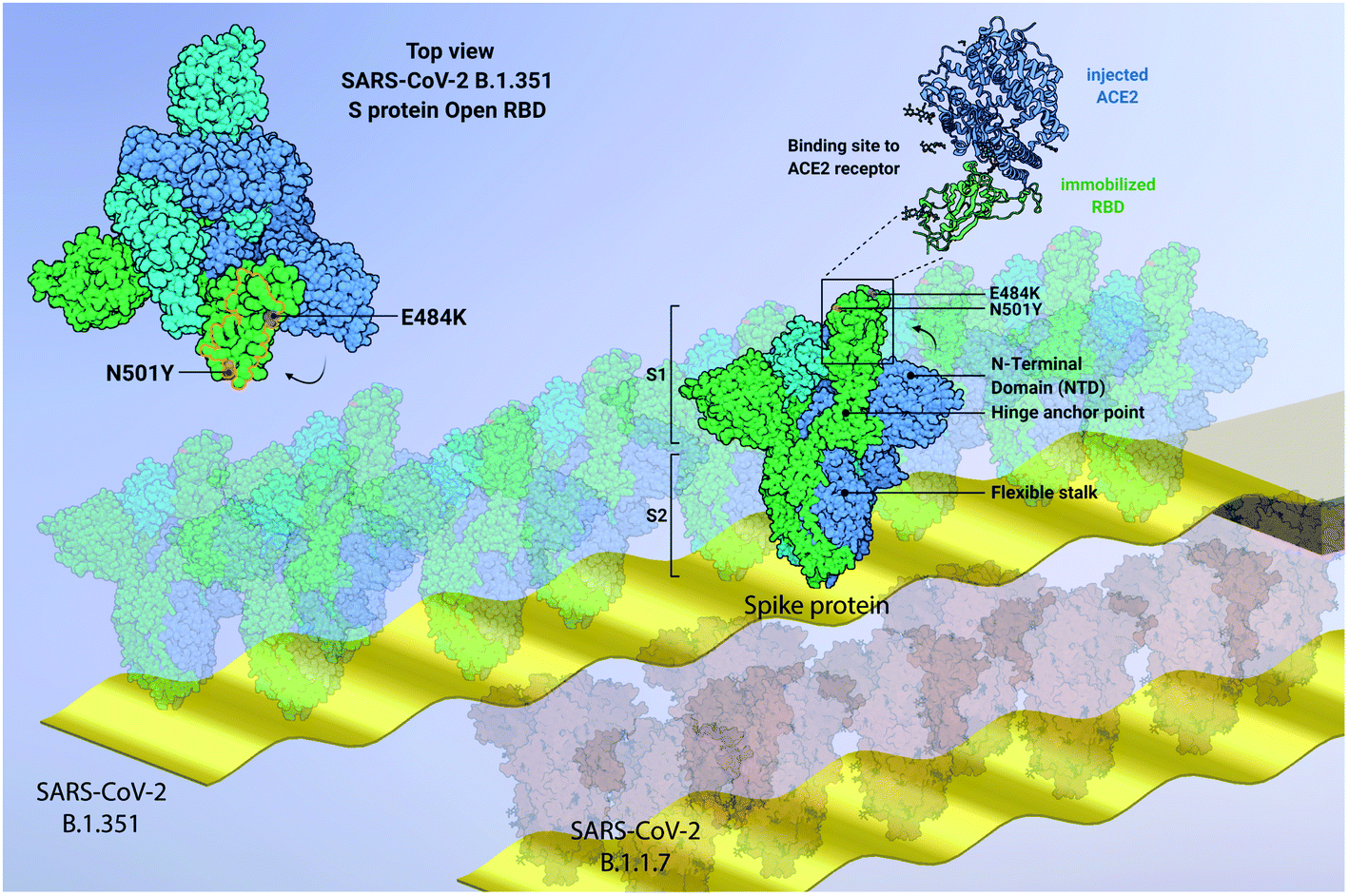 | ||
| Fig. 1 Graphical representation of developed quantitative nanomechanical assay. 18 microcantilevers are specifically functionalized to quantify the affinity between SARS-CoV-2 proteins (variants SARS-CoV-2 B.1.351, SARS-CoV-2 B.1.1.7 and SARS-CoV-2 P.1, and Wuhan WT) and ACE2 receptors. Each microresonator in the array is functionalized with an oriented variant-specific RBD monolayer, immobilized via Fc-tag/protein G or His-tag/NTA binding pattern (see ESI Fig. S1†). The microresonators are actively driven through piezo actuation and oscillate at specific resonance frequencies. The interaction between RBD and ACE2 is measured nanomechanically. The ACE2 binding on the RBD causes a resonance frequency shift that is proportional to the binding efficiency of each variant (see text and Fig. 5). In the centre, a 180–200 kDa SARS-CoV-2 spike homotrimer protruding from the bilayer viral membrane, with a highly flexible stalk, is represented. One S protein is shown in its open conformation (green), as indicated by the RBD projecting out of the proximate NTD and the other two S subunits (cyan and blue). The zoomed-in inset highlights the interaction between the injected ACE2 and the immobilized RBD (blue and green, respectively). On the top left, the top view of SARS-CoV-2 B.1.351 spike protein shows its homotrimeric S protein structure and the open configuration of one RBD (green). The pivotal positions of E484K (purple) and N501Y (orange) mutations are highlighted, both embedded into the ACE2 binding region RBM (delimited by orange line). The size of biomolecules and the amplitude of oscillation are exaggerated to effectively convey the concept. Image created from PDB database (7DDN) with http://biorender.com. | ||
We recently demonstrated a direct single-step label-free quantitative immunoassay in serum, investigating malaria vaccines.23 Our method exhibits 1 pg sensitivity, on par with the gold-standard multi-step ELISA, performs faster (5 s time resolution) and enables simultaneous mass uptake studies of multiple targets, due to epitope-specific recognition.23,28
The experimental workflow is indicated in Fig. 2. Sensors within the array are individually functionalized with a random receptor pattern across the 18 sensors (Fig. 2A and B, see also ESI Fig. S1†). The sensors are actuated at higher modes and their mechanics follows the harmonic oscillator model in a liquid environment.23,24,27,29 The minimum detectable mass is given if one takes the minimum measurable frequency change. Evaluating only individual sensors would lead to an overestimation of mechanical changes due to specific interactions, also due to variations in the hydrodynamic environment. The sensors are exposed synchronously to the same sample and multiples of individual functionalizations allow for averaging of the specific mechanical responses. That spread in individual sensor responses with the same receptors could stem from variability in the molecular grafting of the receptors and their local interactions with the ligands. Upon injection of the serum samples at time 0 minutes the sensor oscillations are slowing down due to binding of molecules to the interface and due to fluid dynamic changes (see Fig. 2C and D). Differential analysis further eliminates all unspecific effects as described above and will result in an absolute mass evaluation (see Fig. 3B).
 | ||
| Fig. 2 Experimental workflow. Sensors within the array are functionalized with DSU/Protein-G/Fc-tag protein monolayers (assembly A). Depending on the designed experiment, Fc-tagged proteins (i.e. RBD, S1, S2) are subsequently immobilized on each cantilever due to 1 h incubation via glass microcapillaries approach (B). (C) shows the frequency signal of each resonator in the array upon injection of the target molecules in serum. The individual responses of resonators functionalized with the same receptors are averaged and plot in (D), and the corresponding mass uptakes are shown in Fig. 3B. (D) highlights the increased binding recognition of the injected monoclonal antibodies towards RBD, with respect to S1 and S2. For an overview of the functionalization platform used in (B), see Fig. S1 of ref. 27. | ||
 | ||
| Fig. 3 Quantitative affinity analysis of SARS-CoV-2 S proteins: signal development over time (top) and 15 minutes after sample injection (bottom). (A, D), (B, E): SARS-CoV-2 RBD, SARS-CoV-2 S1 and SARS-CoV-2 S2 are immobilized in their native configuration on individual cantilevers, to have at least 4 sensors with the same functionalization distributed across the 18 resonator array. Each monolayer is binding in an oriented manner via protein G/Fc-tag recognition (see ESI Fig. S1†). 20 μl samples of 1.5 ng μl−1 ACE2 (A and D) or 0.3 ng μl−1 anti-SARS-CoV-2 RBD mAbs (B and E) diluted in human serum are injected into the analytical microfluidic chamber the resonators. A differential mass uptake is detected via higher oscillation mode analysis (see Methods) due to the specific interaction and binding of the injected ACE2 (A and D) or mAbs (B and E) with the immobilized SARS-CoV-2 proteins S1/S2/RBD. In (A and B), the average signal of resonators functionalized with SARS-CoV-2 RBD (n = 6, purple line) and the average signal of resonators functionalized with SARS-CoV-2 S1 (n = 6, orange line) are compared with resonators functionalized with SARS-CoV-2 S2 (n = 6), used as in situ reference. (C and F) 18 resonator array is functionalized with three pandemic-related hCoV anti-RBD mAbs (MERS, SARS-CoV and SARS-CoV-2) and a control immunoglobulin IgG. 20 μl of 1.5 ng μl−1 SARS-Cov-2 RBD WT in human serum samples are injected. In a single experiment, the average signals of resonators functionalized with anti-SARS-CoV RBD mAbs (n = 4, dark blue) and anti-SARS-CoV-2 RBD mAbs (n = 6, cyan) are compared with resonators functionalized with anti-MERS RBD mAbs (n = 4), used as reference. Also, the average signals of resonators functionalized with anti-SARS-CoV RBD mAbs (n = 4, dark red) and anti-SARS-CoV-2 RBD mAbs (n = 6, pink) are compared with resonators functionalized with control IgG (n = 4), used as a second reference. In (A–C) the light blue background indicates stabilization in buffer (PBS), the dark pink represents serum sample injection, and the light pink denotes signal stabilization in serum (no flow). In (A–C) the signal is tracked for 20 minutes and the raw data are shown, meanwhile in (D–F) the average after 15 minutes from sample injection is plotted with its Standard Deviation (SD) calculated over 6 min around t = 15 min (for details on bio-functionalisation see ESI Fig. S1†). | ||
SARS-CoV-2 wild type epitope specific mapping and hCov screening
We present an in situ quantitative analysis of the specific interaction between a set of neutralizing monoclonal antibodies (mAbs) generated against SARS-CoV-2 WT RBD, and individually modified RBDs with specific single point mutations. In particular, we study the N501Y and E484K mutations in order to recreate the surface topography of each variant under examination. We analyze variants transmissibility and compare their RBD interaction with human ACE2 receptors, characterizing the binding capacity of an exposed or occluded RBD towards ACE2 receptors. In addition, we carry out a quantitative analysis among different species of coronavirus, studying the interaction of SARS-CoV-2 WT RBD with three pandemic-related hCoV mAbs (MERS, SARS-CoV and SARS-CoV-2) in a competitive environment.Current vaccines are developed based on monomeric SARS-CoV-2 S protein (containing the RBD WT subunit), since the S domain induces higher IgG and IgA polyclonal antibody levels than the RBD alone.30 However, the spread of SARS-CoV-2 strands characterized by antigenic-dominant mutations of the S protein, led to antigenic alteration and immune-impairment for antibodies therapies and S protein-based vaccines. Two single point mutations, namely N501Y and E484K, have been used in this study to target the three variants commonly known as UK (B.1.1.7), South African (B.1.351) and Brazilian (P.1) variants. N501Y is shared between the three variants, while E484K is only found in B.1.351 and P.1. We show how each RBM mutation alters the ability of the corresponding variant to be recognized and neutralized by RBD-specific mAbs. To perform this analysis, the RBD of each variant was immobilized on individual cantilevers distributed across an 18 resonator array. Fig. 3A and D shows more than a 3-fold higher affinity between RBD and ACE2, compared to the one between S1 and ACE2. This result highlights how the steric hindrance of the native embedded configuration of RBD into S1 reduces the binding capacity of the whole S1 protein. Indeed, S glycoprotein trimers are usually found in partially opened states (thus exposing the RBM) in case of highly pathogenic human coronavirus, while they remain largely closed in human coronavirus associated with common colds.8 Furthermore, the RBD open state represents a structure targeted by many reported effective neutralizing antibodies.14
In Fig. 3B and E we analyze the specific binding of mAbs against the SARS-CoV-2 WT RBD. We measure a clear enhanced binding towards the open RBD receptor subunit, whereas binding to the occluded RBM in S1 is attenuated. The virus S2 subunit is adopted as internal reference, as it mimics a coronavirus-related structure but does not trigger relevant immune recognition on our sensor assay.
To further investigate the immunological binding pattern of SARS-CoV-2, a comparison between three pandemic related hCoV mAbs (MERS, SARS-CoV and SARS-CoV-2) has been carried out, as shown in Fig. 3C and F. Upon injection of SARS-CoV-2 WT RBD we observe a pronounced recognition on specific mAbs targeting SARS-CoV and SARS-CoV-2, compared with MERS. Therefore, the WT RBD constitutes an antigenic target for both SARS-CoV and SARS-CoV-2 specific mAbs, but eludes the recognition by immobilized MERS mAbs. This highlights a crosstalk between SARS-CoV-2 mAbs and SARS-CoV mAbs towards SARS-CoV-2 RBD, as expected from ref. 8. We also implement IgG antibodies as additional experimental control, to further confirm that our assay is capable to unambiguously distinguish the specific recognition of WT RBD binding to SARS-CoV and SARS-CoV-2 antibodies in serum. To investigate the immune-evading effect and the transmissibility of the variants, we studied their RBDs interaction with WT mAbs and ACE2, respectively. This allowed us to quantify not only the effect of each mutation on the enhanced transmissibility of the relative variant, but also the variant-specific neutralization loss (mAbs binding ratio between specific variant and WT).
To evaluate in situ neutralization of the spike RBD interaction with ACE2 receptors we performed an experiment that allowed us to quantify the blockage of the receptor binding domain with ACE2 receptors.31 Due to the native off-rate of the interaction we cross-linked the receptor to some of the RBD sensors within the array. This enabled subsequent interaction measurements with soluble ACE2 in the diagnostic chamber. A clear neutralization of the viral spike protein open domain was recorded (see Fig. 4).
 | ||
| Fig. 4 Quantitative affinity analysis of wild type (WT) SARS-CoV-2 RBD neutralization with ACE2: signal development over time (A) and 15 minutes after sample injection (B). SARS-CoV-2 RBD, neutralized SARS-CoV-2 RBD (RBD neutr.) and SARS-CoV-2 S2 are immobilized on 18 resonator arrays in their native configuration on individual cantilevers (n = 6, each). To obtain neutralized RBD, ACE2 has been used as neutralization agent in combination with a dimethyl pimelimidate (DMP) cross-linker. The whole sensor array is exposed to DMP. For the neutralized RBD, this prevents dissociation of the receptor–ligand pair. Each monolayer is binding with a controlled orientation due to protein G/Fc-tag recognition (see Fig. S1†) and 20 μl of 3 ng μl−1 ACE2 solution in PBS is injected. The average signal of sensors functionalized with RBD (n = 6) is compared both with resonators functionalized with neutralized RBD (n = 6, purple), and with S2 (n = 6, orange). The differential mass uptake due to the binding interaction of immobilized spike components (RBD, neutralized RBD and S2) with ACE2 is shown. 180 pg are detected between the WT and the neutralized RBD, indicating that ACE2, in combination with DMP, has been able to neutralize the binding capacity of WT RBD in its native conformation. S2 is used as a standard internal control and a mass uptake of 100 pg is recorded, confirming the binding capacity of the unblocked RBD. We recognize a reduced mass uptake due to the crosslinker modification of the global protein signature on all sensors with respect to Fig. 3A. In A the time evolution of the signal is tracked for 20 minutes, meanwhile in B the average signal after 15 minutes from sample injection is plot with its Standard Deviation (SD) calculated over 10 min around t = 15 min. The light blue background is referring to stabilization time in buffer (PBS), dark pink is referring to serum sample injection and light pink to signal stabilization in PBS (no flow). | ||
Immuno evasion and transmissibility quantification with single amino acid polymorphism resolution
We performed a comparison between N501Y RBD (UK variant B.1.1.7), E484K RBD (South African B.1.351 and Brazilian P.1 variants) and WT RBD (Wuhan 2019).11 Crucial topographic information can be extrapolated from the epitope-specific comparison in Fig. 5A and B, showing that N501Y and E484K respectively exhibit 30% and 60% mAbs neutralization loss, compared with mAbs recognition by WT RBD in Fig. 3B (350 pg). This can be explained via the remodeling of the binding footprint between neutralizing mAbs and variants RBD: as recently reported,15 the specific location of the mutation E484K (Fig. 1) alters the RBD local charge from a negative glutamic amino acid to a positive lysine amino acid. | ||
| Fig. 5 Quantitative analysis of SARS-CoV-2 variants B.1.1.7, B.1.351 and P.1 mAbs and ACE2 recognition. A-D, SARS-CoV-2 B.1.1.7 RBD, SARS-CoV-2 B.1.351 RBD and WT SARS-CoV-2 RBD are immobilized on 18 (A and B) and 15 (C and D) resonator arrays in their native configuration on individual cantilevers, to have at least 4 sensors with the same functionalization distributed across the array. Each variant-specific RBD monolayer is immobilized with a controlled orientation due to NTA/His-tag recognition (see supplementary Fig. S1†). 20 μl of 1.5 ng μl−1 anti-SARS-CoV-2 RBD mAbs (A and B) and 0.3 ng μl−1 ACE2 (C and D) solutions in human serum are injected into the microfluidic chamber. The average signal of resonators functionalized with WT SARS-CoV-2 RBD (n = 6) is compared both with resonators functionalized with SARS-CoV-2 B.1.351 RBD (n = 6, E484K mutation, purple), and with SARS-CoV-2 B.1.1.7 (n = 6, N501Y mutation, orange). The differential mass uptakes due to the binding interaction of immobilized variant-specific SARS-CoV-2 RBD with anti-WT SARS-CoV-2 RBD mAbs (A and B) or ACE2 (C and D) are shown. In (A and C) the time evolution of the signal is tracked for 20 minutes and the raw data are shown, meanwhile in (B and D) the average signal 15 minutes after sample injection is plotted with its Standard Deviation (SD) calculated over 6 min around t = 15 min. In (A and C) the light blue background indicates stabilization in buffer (PBS), the dark pink represents serum sample injection, and the light pink denotes signal stabilization in serum (no flow). | ||
In Fig. 5 (panel C and D), we present the impact of N501Y and E484K mutations on the RBD ability to bind ACE2. The single point mutation N501Y located in the RBM region enhances the formation of hydrogen bonds with ACE2 receptors,14,15 therefore an alteration in the binding efficiency is expected. The main concern being that increased affinity is alarmingly correlated with an enhanced transmissibility of the relative virus strand. As shown in Fig. 5C and D, N501Y is drastically enhancing the B.1.1.7 RBD recognition towards ACE2 by 63% with respect to WT RBD (460 pg, Fig. 3A), by binding additional 290 pg, while a 25% increase is found for E484K. These results confirm the most recent findings related to the new SARS-CoV-2 variants and add a remarkable quantitative information via cantilever-based single-step assay for binding affinity analysis within minutes.
Discussion
The presented nanomechanical assay should be envisioned to be complementary to other antibodies investigation techniques for therapy and diagnostics, and for fine tuning of new vaccine targets. The capability to provide epitope-specific information, with single amino acid resolution, can support phase 1–3 of vaccine efficacy tests, reveal potential weaknesses and clarify protection against mutants.32 For instance, old vaccines coding for SARS-CoV-2 RBD can directly be compared with. Furthermore, quantifying binding interactions will allow to directly monitor infection immunity over time. In addition, as previously presented by the authors, this nanotechnological assay also permits simultaneous coagulation diagnostics.24 The inherent plasma viscoelastic blood status can be directly analyzed in parallel to the binding interaction, thus providing outstanding support in guiding different vaccination therapies.In conclusion, the presented method corroborates recent COVID-19 findings and provides a unique fast single-step label-free quantitative tool for studying the specific interaction between virus and cellular receptors or antibodies. Our measurements within minutes match the statistical analysis of the transmissibility of for instance the UK variant α – B.1.1.7 that can only be gained by analysing the development of the disease proliferation within a population within weeks. The emergence of new variants demands for the development of novel strategies that can improve and speed up the public health guidance process. PCR, ELISA and lateral flow immuno assays (LFA) are currently used worldwide as standard techniques for COVID-19 detection. The presented method with single amino acid resolution demonstrates to surpass the semi-quantitative ELISA assay with reduced number of steps and consumables.23 Furthermore, it is faster than PCR, ELISA and shows within the same timing higher sensitivity compared to LFA.
The unique data herein shown provide novel quantitative insights on the influence of specific single point RBD mutations in relation to immune-response and receptor binding affinity. The appearance of these new variants for SARS-CoV-2 are widely considered a starting point of an antigenic drift. At present, there is a substantial amount of coronavirus variants under investigation, including the recent δ – B.1.617, first identified in India, which surpassed the effects of N501Y and E484K in infectiousness and is now classified as a variant of concern. If this trend is confirmed, accumulation of new mutations occurring in immune-strategic locations can lead the virus to a faster spread, evading the current prophylaxis and vaccine-based immunization.
As a final remark, even the most advanced technology cannot keep us one step ahead of the next variant appearing worldwide. Only enhanced caution and mitigation measures can stop the virus transmission as quickly as possible and avoid a mutation-chase over new evolved variants of SARS-CoV-2.
Materials and methods
Measuring device
Microfabricated silicon arrays of 18 cantilevers used in this study (400 μm long, 70 μm wide and 2.3 μm thick, fabricated in Tyndall national institute, Cork, Ireland) have a spring constant down to 0.4 N m−1 and a mass of approximately 160 ng. They are mounted into a microfluidic measurement chamber, micromachined in polyether ether ketone (PEEK) and previously described in ref. 22. The chamber serves to mechanically clamp the chip and to immerse it in a 6 μl microfluidic volume, for in-liquid measurements. Automated syringes pumps (Kent Scientific Corporation, Lee Company), allow exchange of nl to ml liquid volumes via a software-controlled system of solenoid valves (ASCO Valve Inc.).22 In order to ensure temperature stability and insulation, the whole setup (including fluidic pumps, pipes and valves) is enclosed in a thermally insulated box (stable internal temperature with 0.02 °C precision).27 Dynamic mode operation is enabled via a custom-built piezo-ceramic stack actuator, placed in a pocket underneath the chip, isolated from the fluidic volume by a 200 μm-thick PEEK membrane.22 Mechanical signals are detected via optical beam deflection readout.22 For a complete description of the experimental device refer to our previous works.22,23,27Data acquisition
The resonance frequency are acquired by phase-locked-loop (PLL) frequency tracking and then converted to mass adsorption via differential read-out ref. 27. A proportional-integral-derivative (PID) controlled PLL is implemented via an in-house developed LabVIEW (National Instruments) code, directly interfaced with the experimental hardware, and able to track more than 4 modes of 18 sensors in parallel over several hours. For a complete description of the acquisition apparatus refer to ref. 27. For each experiment, up to 4 resonant modes and 18 cantilevers are measured, resulting in 72 parallel PID controls. If the system is ideally unperturbed, the frequency that locks the phase (resonant frequency) would remain constant, but upon perturbation (e.g. mass adsorption generated by protein–protein recognition) the frequency would shift accordingly and is recorded. The limit of detection of mass bound to the sensors is around 1 pg in serum with a signal acquisition time resolution of 5 seconds.23 The differential data analysis enables direct quantitative extraction of the newly bound mass based on specific interaction of the ligands to their receptors. An average shift in resonance frequency in the measuring sensors is directly correlated to the added mass and is simultaneously compared to the frequency behaviour of reference sensors on the same array. For further information on the in situ-calibration of such measurements please consult ref. 23, 24 and 27.Functionalization
Each cantilever used in the present study is coated with 3 nm of Titanium and 23/33 nm Gold (top and bottom side, respectively) via e-beam metal evaporation (Temescal FC-2000, Scotech). The exposed gold layer allows (i) to optimize the optical detection by maximizing the reflectivity of each cantilever surface and (ii) offers an anchoring platform for the thiol groups of the functionalization molecules. Two approaches are used to immobilize specific probe proteins on each cantilever in an oriented manner (see Fig. 2 and ESI Fig. S1†). (a) DSU/Protein-G/Fc-tag protein monolayers assembly: the gold coated array is immersed in 1 mM dithiobis(1-succinimidyl undecanoate) (DSU) in 1,4-dioxane for 1 h and then washed with dioxane, ethanol and PBS. This step creates a NHS-terminated self-assemble monolayer (SAM) that covalently binds to primary amine groups of proteins. The chip is immersed in 0.1 mg ml−1 recombinant protein G solution in PBS for 1 h. To avoid any subsequent unspecific binding the array is quenched in 0.1 mM ethanolamine solution overnight. Depending on the designed experiment, Fc-tagged proteins or monoclonal antibodies (mAbs) are subsequently immobilized on each cantilever due to 1 h incubation of 10 ng ml−1 proteins or mAbs solutions via glass microcapillaries approach (see Fig. 2B). (b) NTA-SAM/Ni2+/His-tag monolayers creation: the gold coated array is first immersed in 0.2 mM NTA-SAM solution in ethanol overnight and then incubated in 40 mM l−1 NiSO4 for 1 h in order to create a nickel-chelated nitrilotriacetate (NTA) self-assembled monolayer. 10 μg ml−1 experiment related his-tag proteins are then immobilized through complexation to the central Ni2+ on the NTA monolayer with 1 h incubation via glass microcapillaries approach. Both methods allow the array to be placed directly in the microfluidic measurement chamber or to be stored in buffer for several days.Biological samples preparation
40592-V02H SARS-CoV-2 (2019-nCoV) Spike RBD-Fc Recombinant Protein, 40592-V08B SARS-CoV-2 (2019-nCoV) Spike RBD-His, 40590-V02H SARS-CoV-2 (2019-nCoV) Spike S2 ECD-fc, 40592-V08H82 SARS-CoV-2 (2019-nCoV) Spike RBD(N501Y)-His, 40592-V08H84 SARS-CoV-2 (2019-nCoV) Spike RBD(E484K)-His, 40591-V02H SARS-CoV-2 (2019-nCoV) Spike S1-Fc, 10108-H08H ACE2 Protein, Human-His, 40592-MM57 SARS-CoV-2 (2019-nCoV) Spike Neutralizing Antibody (Mouse mAb), 40069-MM23 MERS-CoV Spike Protein S1 Antibody, Mouse mAb, 40150-D002 SARS-CoV-2 (2019-nCoV) Spike Antibody Chimeric mAb were purchased from SinoBiological (China). They were first reconstituted in 500 μl of nanopure H2O and then diluted in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 human serum (Sigma Aldrich, Ireland) to perform the experiments. Dithiobis(succinimidyl undecanoate) (DSU)33 was purchased from Dojindo Molecular Technologies, Japan. Phosphate buffered saline (PBS), 10 mM, pH 7.4 and 1,4-Dioxane (99.8%) and NTA-SAM were purchased from Sigma Aldrich, Ireland.
:50 human serum (Sigma Aldrich, Ireland) to perform the experiments. Dithiobis(succinimidyl undecanoate) (DSU)33 was purchased from Dojindo Molecular Technologies, Japan. Phosphate buffered saline (PBS), 10 mM, pH 7.4 and 1,4-Dioxane (99.8%) and NTA-SAM were purchased from Sigma Aldrich, Ireland.
Funding
The work was supported by Science Foundation Ireland under the IvP scheme SFI/09IN/1B2623 and SFI/15/IA/3023 and the CSET scheme SFI/10/CSET/B1821.Author contributions
G. B., A. D. P. and M. H. conceived the study, G. B. designed the protocols, performed the experiments and analyzed the data. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Dr Stefania Magnano and Dr Marilena Karavyraki (Trinity Biomedical Sciences Institute, TCD) for fruitful discussion and Patrick Murphy (Mechanical Workshop, TCD) for the manufacture of the temperature stabilized enclosure and the liquid chambers.References
- Z.-W. Ye, S. Yuan, K.-S. Yuen, S.-Y. Fung, C.-P. Chan and D.-Y. Jin, Int. J. Biol. Sci., 2020, 16, 1686–1697 CrossRef CAS PubMed.
- Z. Zhu, X. Lian, X. Su, W. Wu, G. A. Marraro and Y. Zeng, Respir. Res., 2020, 21, 224 CrossRef CAS PubMed.
- M. Roser, H. Ritchie, E. Ortiz-Ospina and J. Hasell, Coronavirus Pandemic (COVID-19), https://ourworldindata.org/coronavirus, 2020 Search PubMed.
- E. Callaway, D. Cyranoski, S. Mallapaty, E. Stoye and J. Tollefson, Nature, 2020, 579, 482–483 CrossRef CAS PubMed.
- Z. Ke, J. Oton, K. Qu, M. Cortese, V. Zila, L. McKeane, T. Nakane, J. Zivanov, C. J. Neufeldt, B. Cerikan, J. M. Lu, J. Peukes, X. Xiong, H.-G. Kräusslich, S. H. W. Scheres, R. Bartenschlager and J. A. G. Briggs, Nature, 2020, 588, 498–502 CrossRef CAS PubMed.
- R. A. Römer, N. S. Römer and A. K. Wallis, Sci. Rep., 2021, 11, 4257 CrossRef PubMed.
- Y. Huang, C. Yang, X.-f. Xu, W. Xu and S.-w. Liu, Acta Pharmacol. Sin., 2020, 41, 1141–1149 CrossRef PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 181, 281–292.e286 CrossRef CAS PubMed.
- I. Khatri, F. J. T. Staal and J. J. M. van Dongen, Front. Immunol., 2020, 11, 570018 CrossRef CAS PubMed.
- B. J. Bosch, R. van der Zee, C. A. M. de Haan and P. J. M. Rottier, J. Virol., 2003, 77, 8801–8811 CrossRef CAS PubMed.
- B. Korber, W. M. Fischer, S. Gnanakaran, H. Yoon, J. Theiler, W. Abfalterer, N. Hengartner, E. E. Giorgi, T. Bhattacharya, B. Foley, K. M. Hastie, M. D. Parker, D. G. Partridge, C. M. Evans, T. M. Freeman, T. I. de Silva, A. Angyal, R. L. Brown, L. Carrilero, L. R. Green, D. C. Groves, K. J. Johnson, A. J. Keeley, B. B. Lindsey, P. J. Parsons, M. Raza, S. Rowland-Jones, N. Smith, R. M. Tucker, D. Wang, M. D. Wyles, C. McDanal, L. G. Perez, H. Tang, A. Moon-Walker, S. P. Whelan, C. C. LaBranche, E. O. Saphire and D. C. Montefiori, Cell, 2020, 182, 812–827.e819 CrossRef CAS PubMed.
- D. Mercatelli and F. M. Giorgi, Frontiers in Microbiology, 2020, 11, 1800 CrossRef PubMed.
- O. T. R. Toovey, K. N. Harvey, P. W. Bird and J. W.-T. W.-T. Tang, J. Infect., 2021, 82, e23–e24 CrossRef CAS PubMed.
- P. Wang, M. S. Nair, L. Liu, S. Iketani, Y. Luo, Y. Guo, M. Wang, J. Yu, B. Zhang, P. D. Kwong, B. S. Graham, J. R. Mascola, J. Y. Chang, M. T. Yin, M. Sobieszczyk, C. A. Kyratsous, L. Shapiro, Z. Sheng, Y. Huang and D. D. Ho, Nature, 2021, 593, 130–135 CrossRef CAS PubMed.
- C. K. Wibmer, F. Ayres, T. Hermanus, M. Madzivhandila, P. Kgagudi, B. Oosthuysen, B. E. Lambson, T. de Oliveira, M. Vermeulen, K. van der Berg, T. Rossouw, M. Boswell, V. Ueckermann, S. Meiring, A. von Gottberg, C. Cohen, L. Morris, J. N. Bhiman and P. L. Moore, Nat. Med., 2021, 27, 622–625 CrossRef CAS PubMed.
- P. Wang, M. S. Nair, L. Liu, S. Iketani, Y. Luo, Y. Guo, M. Wang, J. Yu, B. Zhang and P. D. Kwong, Nature, 2021, 10, 130–135 CrossRef PubMed.
- T. Chaibun, J. Puenpa, T. Ngamdee, N. Boonapatcharoen, P. Athamanolap, A. P. O'Mullane, S. Vongpunsawad, Y. Poovorawan, S. Y. Lee and B. Lertanantawong, Nat. Commun., 2021, 12, 802 CrossRef CAS PubMed.
- N. C. Cady, N. Tokranova, A. Minor, N. Nikvand, K. Strle, W. T. Lee, W. Page, E. Guignon, A. Pilar and G. N. Gibson, Biosens. Bioelectron., 2021, 171, 112679 CrossRef CAS PubMed.
- A. Yakoh, U. Pimpitak, S. Rengpipat, N. Hirankarn, O. Chailapakul and S. Chaiyo, Biosens. Bioelectron., 2021, 176, 112912 CrossRef CAS PubMed.
- Z. Swank, G. Michielin, H. M. Yip, P. Cohen, D. O. Andrey, N. Vuilleumier, L. Kaiser, I. Eckerle, B. Meyer and S. J. Maerkl, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2025289118 CrossRef CAS PubMed.
- T. Braun, M. K. Ghatkesar, N. Backmann, W. Grange, P. Boulanger, L. Letellier, H.-P. Lang, A. Bietsch, C. Gerber and M. Hegner, Nat. Nanotechnol., 2009, 4, 179 CrossRef CAS PubMed.
- M. Walther, P. M. Fleming, F. Padovani and M. Hegner, EPJ Techniques and Instrumentation, 2015, 2, 7 CrossRef.
- G. Brunetti, F. Padovani, A. De Pastina, C. Rotella, A. Monahan, S. L. Hoffman, S. A. Jongo, S. Abdulla, G. Corradin, G. Pluschke, C. Daubenberger and M. Hegner, Nanoscale, 2021, 13, 2338–2349 RSC.
- F. Padovani, J. Duffy and M. Hegner, Anal. Chem., 2016, 89, 751–758 CrossRef PubMed.
- F. Padovani, J. Duffy and M. Hegner, Nanoscale, 2017, 9, 17939–17947 RSC.
- J. L. Arlett, E. B. Myers and M. L. Roukes, Nat. Nanotechnol., 2011, 6, 203–215 CrossRef CAS PubMed.
- A. De Pastina, F. Padovani, G. Brunetti, C. Rotella, F. Niosi, V. Usov and M. Hegner, Rev. Sci. Instrum., 2021, 92 Search PubMed.
- G. Brunetti, A. De Pastina and M. Hegner, Appl. Cell Biol., 2021, 9, 29–32 Search PubMed.
- M. K. Ghatkesar, T. Braun, V. Barwich, J.-P. Ramseyer, C. Gerber, M. Hegner and H. P. Lang, Appl. Phys. Lett., 2008, 92, 043106 CrossRef.
- C. B. Creech, S. C. Walker and R. J. Samuels, JAMA, 2021, 325, 1318–1320 CrossRef CAS PubMed.
- C. Lei, K. Qian, T. Li, S. Zhang, W. Fu, M. Ding and S. Hu, Nat. Commun., 2020, 11, 2070 CrossRef CAS PubMed.
- R. W. Sanders and M. D. de Jong, Lancet, 2021, 397, 1326–1327 CrossRef CAS.
- P. Wagner, M. Hegner, P. Kernen, F. Zaugg and G. Semenza, Biophys. J., 1996, 70, 2052–2066 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00554e |
| This journal is © The Royal Society of Chemistry 2021 |