
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagneto-mechanical treatment of human glioblastoma cells with engineered iron oxide powder microparticles for triggering apoptosis†
C.
Thébault
 ab,
M.
Marmiesse
a,
C.
Naud
a,
K.
Pernet-Gallay
c,
E.
Billiet
a,
H.
Joisten
ad,
B.
Dieny
a,
M.
Carrière
*b,
Y.
Hou
*b and
R.
Morel
*a
ab,
M.
Marmiesse
a,
C.
Naud
a,
K.
Pernet-Gallay
c,
E.
Billiet
a,
H.
Joisten
ad,
B.
Dieny
a,
M.
Carrière
*b,
Y.
Hou
*b and
R.
Morel
*a
aUniv. Grenoble Alpes, CEA, CNRS, Spintec, 38000 Grenoble, France. E-mail: robert.morel@cea.fr
bUniv. Grenoble Alpes, CEA, CNRS, IRIG-SyMMES, 38000 Grenoble, France. E-mail: marie.carriere@cea.fr; yanxia.hou-broutin@cea.fr
cUniv. Grenoble Alpes, Inserm, U1216, CHU Grenoble Alpes, CEA, Grenoble Institut Neurosciences, Grenoble, France
dUniv. Grenoble Alpes, CEA, LETI, 38000 Grenoble, France
First published on 28th September 2021
Abstract
In nanomedicine, treatments based on physical mechanisms are more and more investigated and are promising alternatives for challenging tumor therapy. One of these approaches, called magneto-mechanical treatment, consists in triggering cell death via the vibration of anisotropic magnetic particles, under a low frequency magnetic field. In this work, we introduce a new type of easily accessible magnetic microparticles (MMPs) and study the influence of their surface functionalization on their ability to induce such an effect, and its mechanism. We prepared anisotropic magnetite microparticles by liquid-phase ball milling of a magnetite powder. These particles are completely different from the often-used SPIONs: they are micron-size, ferromagnetic, with a closed-flux magnetic structure reminiscent of that of vortex particles. The magnetic particles were covered with a silica shell, and grafted with PEGylated ligands with various physicochemical properties. We investigated both bare and coated particles' in vitro cytotoxicity, and compared their efficiency to induce U87-MG human glioblastoma cell apoptosis under a low frequency rotating magnetic field (RMF). Our results indicated that (1) the magneto-mechanical treatment with bare MMPs induces a rapid decrease in cell viability whereas the effect is slower with PEGylated particles; (2) the number of apoptotic cells after magneto-mechanical treatment is higher with PEGylated particles; (3) a lower frequency of RMF (down to 2 Hz) favors the apoptosis. These results highlight a difference in the cell death mechanism according to the properties of particles used – the rapid cell death observed with the bare MMPs indicates a death pathway via necrosis, while PEGylated particles seem to favor apoptosis.
Introduction
Magnetic nanoparticles are widely studied and used for different biomedical and bioengineering applications, including hyperthermia, magnetic resonance imaging, drug delivery, and biosensing.1 More recently, the use of magnetic microparticles (MMPs) for biomedical applications has attracted an increasing interest. One promising example is the destruction of cancer cells induced by low-frequency magneto-mechanical vibration of particles, by the application of an external magnetic field.2,3 Such a research field opens perspectives for a new and effective therapeutic approach.4 The interest in the modulation of the cell physiology using mechanical stimulation also stems from the recent recognition of the importance of mechanotransduction in the mechanisms associated with tumor progression, besides the classical molecular pathways governing cancer.5From a material design point of view, the magnetic properties that are required for this application differ from those of the widely used superparamagnetic iron oxide nanoparticles (SPIONs).6 First, their vibration must result in a mechanical stress sufficient to induce a response of the cell. Some examples show either the opening of mechanosensitive ion channels, for which a stress in the range of 1–10 kPa is required,7 or damage to the cell membrane, for which a higher stress is required (10–15 kPa).8 Other cell responses are reported in the review by Golovin et al.9 The magnetic torque experienced by a particle under the application of an external magnetic field is proportional to its magnetization. With micron size particles, it can reach a few tens of fN m, which will locally generate stresses of a few tens of kPa.10
In addition to this size requirement, the particles should have a low magnetization remanence to avoid agglomeration caused by their magnetostatic interactions. They should also have good biocompatibility, i.e., no intrinsic cell toxicity and good colloidal stability under physiological conditions. Finally, they should allow surface functionalization to enable specific interactions with the cells, for instance to ensure specific cell-targeting.
Most often these particles are thin discs with a diameter close to a micron and a thickness of a few tens of nanometers, whose magnetic properties result from appropriate choice of size, shape, and multilayered composition. The most commonly used structures are synthetic antiferromagnets with planar or perpendicular magnetizations, and vortex microparticles.11 These particles are essentially made of ferromagnetic transition metals (Fe, Co, Ni), with other non-magnetic elements (Ru, B, Pt, Ta) to provide them the desired properties. They are produced by top-down approaches including lithography, etching and/or lift-off techniques, most often in a clean room facility. Consequently, the main disadvantage compared to SPIONs is the higher cost of fabrication, their low production yield, as well as their lack of biocompatibility.
To address these drawbacks and insure that the particles can be biocompatible, an alternative solution is to use easily accessible micron-size iron oxide particles. The SPIONs that are widely used for biomedical applications are in a size range of a few nanometer in diameter at which they are superparamagnetic. The particles we discuss here are in the normal ferrimagnetic state. One important feature making them suitable for biomedical application is that they consist of magnetite grains with a closed-flux magnetic structure.12,13 This contributes to the low magnetic remanence, which is one of the requirement to ensure good dispersion of the particles in solutions.14
It must however be kept in mind that the use of magnetic particles for biomedical applications involves many pitfalls, as evidenced by the difficulties encountered with SPIONs, notably in the development of particles for drug delivery and imaging. These difficulties have been described and discussed in many review papers to which the reader is referred.15–17
One example of below-expectation result is with drug delivery where, despite interesting results often obtained with SPIONs in in vitro experiment, the delivery efficiency with intravenous injection is as low as 0.7%. Many factors may contribute to this, among which is the fact that tumor targeting often relies on the enhanced permeability and retention (EPR) effect, which in some cases may not be as efficient as expected. Even if the targeting efficiency can be improved by specific surface functionalization, the protein-corona that rapidly covers the particles significantly modify their physico-chemical properties in an adverse way.18,19 Micron-size magnetic particles differ from SPIONs regarding these two aspects. For one thing, they are not intended for targeted-delivery using the EPR effect (their diameter is above the reported endothelial gap size20) and their administration is done locally, at the tumor site, by injection.4 Although reports of in vivo experiment are scarce, it appears that even many days after injection the particles remain mostly close to the injection site, meaning that they can be magnetically activated over long period.
The cell–particles interaction is also different according to the particle size. Small particles enter the cell via clathrin-mediated endocytosis, and cells can internalize thousands of SPIONs that accumulate in lysosomal cavities, where their decay can induce dysfunction.21 Lysosome internalization is nevertheless not necessarily detrimental, as the magneto-mechanical stimulation of lysosome-entrapped SPIONs has been reported to induce cancer cell death, either by lysosomal membrane permeabilization22–24 or by damaging the cytoskeleton.25
Regarding larger particles, caveolae-mediated internalization has been reported for latex spheres up to, but not larger than 500 nm.26 Apart from the endocytosis mechanism, one observation is that, contrary to small particles, no delivery to the lysosomes is observed for the large ones. Internalization of magnetic particles and nanowires with length well above 1 μm has also been reported, although no mechanism for internalization has been identified.27–29
In this paper, we report on the development of a new type of magnetic microparticles, obtained via ball milling of magnetite powder, and their use for triggering apoptosis of cancer cells. Such a simpler manufacturing process allows the production of large quantities of particles in an easy way and at low cost. The average particle size and magnetic properties are optimized to be suitable for the destruction of cancer cells. In addition, to ensure their long-term stability in physiological conditions, the magnetic microparticles are covered by a silica shell. One advantage using silica as a coating material lies in its compatibility with various surface chemistry for the introduction of diverse surface properties such as charge and hydrophobicity.30 It has been reported that surface charge and surface hydrophobicity are among the main influencing factors for cytotoxicity in addition to the material, size, shape, and composition of particles.31 Herein, the silica shell is further functionalized with diverse poly(ethylene glycol) (PEG) ligands with different charge and length to study their effect on the efficiency for cell apoptosis induction. PEGylated coatings are highly biocompatible with excellent anti-fouling property and high steric hindrance to stabilize particles. They are often used to prolong vascular circulation of nanoparticles like SPIONs.1 In this study, the effect of surface coating charge and thickness, duration and frequency of the applied magnetic field on their efficiency for apoptosis induction was investigated.
Results and discussion
Magnetic microparticle synthesis and optimization by ball milling
MMP were obtained by ball milling of magnetite powder. To characterize the size of these anisotropic particles, the Feret and MinFeret diameters were measured from SEM pictures of initial powder and after 1 h, 2 h, 4 h or 12 h of ball milling (some examples in Fig. 1a–c). Fig. 1d shows the size distribution of the initial powder compared to the size distribution of MMP after 4 h of ball milling.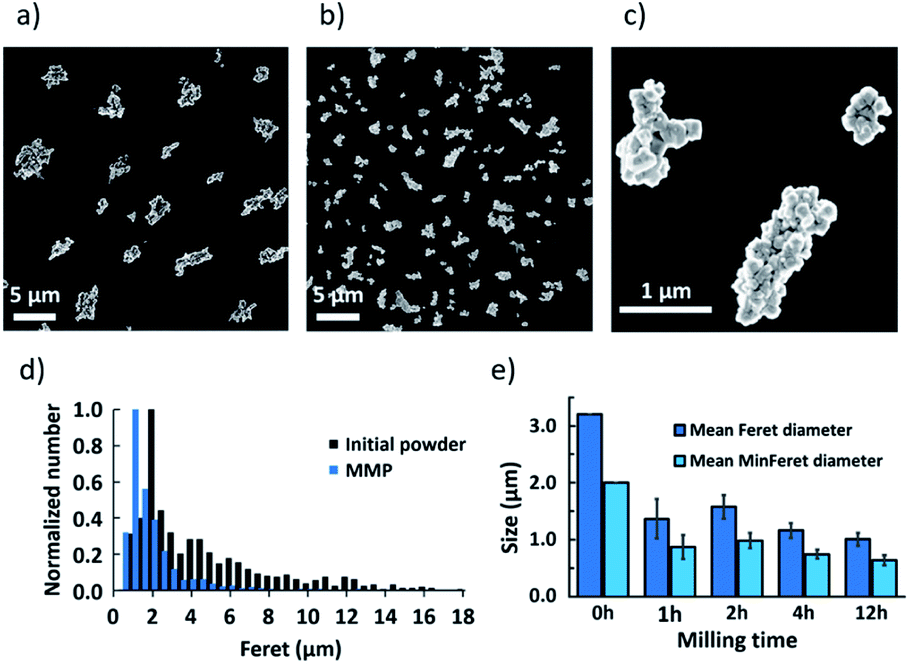 | ||
| Fig. 1 SEM images of the initial powder (a) and particles after 4 h of ball milling (b and c). (d) Feret diameter distribution for the initial powder and for particles after 4 h of ball milling (MMP). (e) Mean Feret and Mean MinFeret diameters for the initial powder and after 1, 2, 4 and 12 h of ball milling, calculated from the lognormal size distribution fitting. | ||
When fitting the MMP size distribution using a lognormal law, after 1 h, 2 h, 4 h or 12 h of ball milling, the Mean Feret diameter of the initial powder that was measured at 3.2 μm decreased to 1.4 μm, 1.6 μm, 1.2 μm and 1.0 μm, respectively (Fig. 1e). Accordingly, the Mean MinFeret diameter initially at 2.0 μm was reduced to 0.9 μm, 1.0 μm, 0.7 μm and 0.6 μm, respectively.
In particular, 4 h-ball milling process resulted in the formation of magnetite particles, composed of sintered smaller crystallites, with an average size close to one micron that is well suited for our purpose (Fig. 1b and c). Therefore, in this study, the particles obtained after 4 h of ball milling, which we hereafter denote MMP, were used for the further experiments. All these results confirm that ball milling is a very efficient, simple, rapid and cost-effective process for preparation of large quantities of anisotropic magnetite microparticles.
Characterization of the magnetic microparticles
First, bare MMP were characterized to obtain their XRD profile and magnetization curve. The XRD measurements with both initial powder and MMP matched with the expected Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m(227) space group for Fe3O4 (PDF 01-080-6402 (ref. 32)), with a slight peak deformation indicating a small structural inhomogeneity (Fig. 2a). On the other hand, no contribution from maghemite or other iron oxide phase could be identified before or after milling. The lattice parameters for the initial powder and for MMP were, respectively, a = 8.392(5) Å and a = 8.391(5) Å. These values are very close to the reported lattice parameter for bulk magnetite (a = 8.3967(3) Å).33 The crystallite sizes before and after milling were, respectively, 80 nm and 98 nm. Given the peak deformation, the difference is not significant.
m(227) space group for Fe3O4 (PDF 01-080-6402 (ref. 32)), with a slight peak deformation indicating a small structural inhomogeneity (Fig. 2a). On the other hand, no contribution from maghemite or other iron oxide phase could be identified before or after milling. The lattice parameters for the initial powder and for MMP were, respectively, a = 8.392(5) Å and a = 8.391(5) Å. These values are very close to the reported lattice parameter for bulk magnetite (a = 8.3967(3) Å).33 The crystallite sizes before and after milling were, respectively, 80 nm and 98 nm. Given the peak deformation, the difference is not significant.
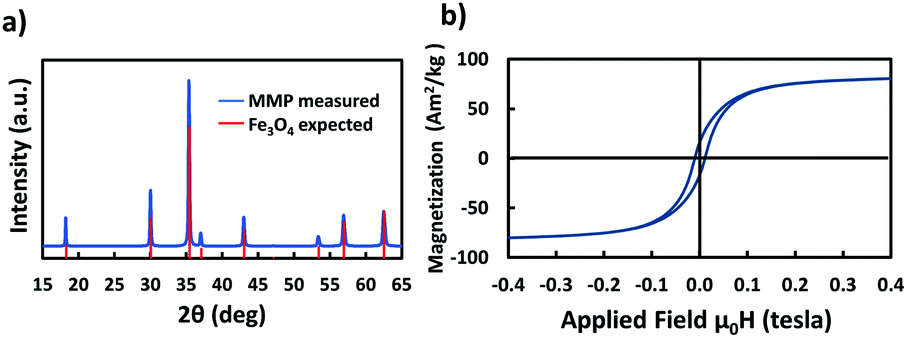 | ||
| Fig. 2 (a) X-ray diffraction spectra and (b) magnetization curve of MMP obtained after 4 h of ball milling. | ||
The saturation mass magnetization of the MMP was measured at 83 A m2 kg−1 (Fig. 2b). This is lower than the bulk value (92 A m2 kg−1),34 but is typical for magnetite particles.8 The remanent magnetization was 16 A m2 kg−1, and the coercivity 10.6 mT; both values are again typical for particles close to the single domain – multi domain critical size.35 Afterwards, MMP were first covered by a silica shell and then functionalized with diverse PEG ligands following the procedures given in Fig. 3. The functionalized particles were characterized by measuring their zeta potential, which is related to their net surface charge. In this study, in order to evaluate the effect of surface coating charge and thickness on the magneto-mechanical treatment efficacy, different functionalization of the MMP were performed using diverse PEG with distinct terminal group (–OH, –NH2, –COOH) and length (the detailed step-by-step functionalization of MMP is described hereafter in the Materials and methods section). To simplify notation, particles were labeled as detailed in Table 1.
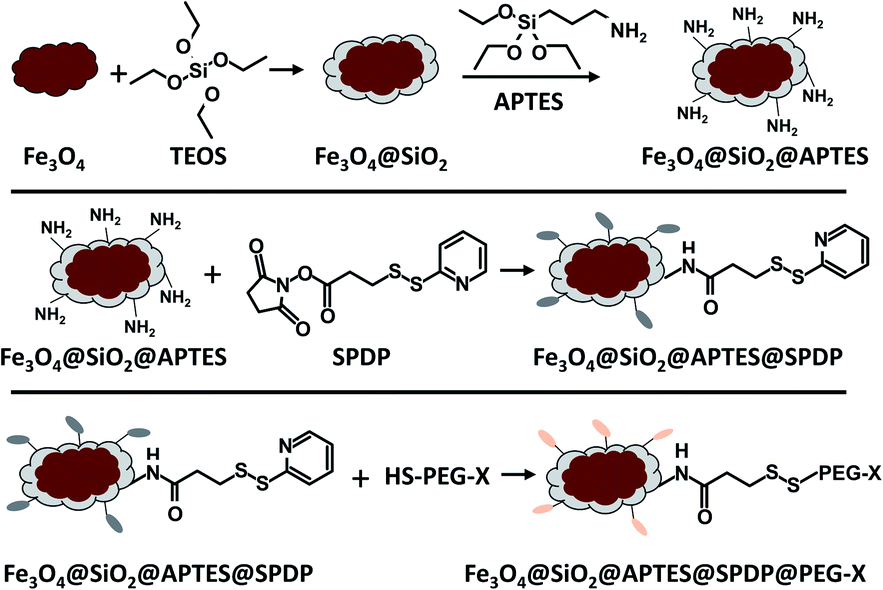 | ||
| Fig. 3 Schematic illustration of the surface functionalization of magnetite particles. | ||
| Bare magnetite particles after 4 h of milling | MMP |
|---|---|
| Fe3O4@SiO2 | MMP@SiO2 |
| Fe3O4@SiO2@APTES | MMP@APTES |
| Fe3O4@SiO2@APTES@SPDP@(CH2)11PEG280NH2 | MMP@PEG280NH2 |
| Fe3O4@SiO2@APTES@SPDP@(CH2)11PEG280CH2COOH | MMP@PEG280COOH |
| Fe3O4@SiO2@APTES@SPDP@(CH2)6PEG150OH | MMP@PEG150OH |
| Fe3O4@SiO2@APTES@SPDP@PEG2000OH | MMP@PEG2000OH |
The MMP have a negative zeta potential of −50 mV. After the formation of the silica shell, as expected, zeta potential of MMP@SiO2 shifted to −36 mV. At the end, four different PEGs were grafted onto the magnetic particles, allowing to obtain PEGylated particles MMP@PEG280NH2, MMP@PEG280COOH, MMP@PEG150OH and MMP@PEG2000OH. The zeta potential of these particles were −10 mV, −38 mV, −24 mV and −22 mV, respectively (Table 2). These results show that the functionalized particles have differential surface charge and confirm the successful immobilization of PEG on the particles. It is generally admitted that a zeta potential above 30 mV or below −30 mV is indicative of good NP dispersion property, while small zeta potential values indicate a tendency to aggregation. In our case, as will be shown hereafter, the PEGylated particles show the best in vitro dispersion while bare particles, with the highest zeta potential, tend to aggregate in more complex milieu. Possible cause for the aggregation is either the magnetic dipolar interaction or the formation of protein corona on the particle surface, which in the case of PEGylated particles is reduced due to the steric repulsion and anti-fouling ability.
| Zeta potential (mV) | |
|---|---|
| MMP | −50.0 ± 2.3 |
| MMP@SiO2 | −36.2 ± 0.9 |
| MMP@PEG280NH2 | −10.1 ± 1.5 |
| MMP@PEG280COOH | −37.7 ± 1.7 |
| MMP@PEG150OH | −24.0 ± 1.1 |
| MMP@PEG2000OH | −22.4 ± 1.9 |
Evaluation of toxicity of bare and functionalized magnetic microparticles
First, the intrinsic toxicity of the particles after a 24 h incubation with glioblastoma cells without the application of an external magnetic field was evaluated by two different tests: the WST-1 assay, which measures the cellular metabolic activity and the LDH leakage assay, which evaluates the cell membrane integrity by determining the amount of LDH enzyme released into the culture medium. Toxicity was assessed for the six different types of particles with various concentrations from 3 mg L−1 up to 0.2 g L−1 (or even up to 1 g L−1 for MMP and MMP@SiO2) (supporting info Fig. S1†). It was found that the concentration of particles has no significant effect on the toxicity. Therefore, as examples, Fig. 4 shows the metabolic activity and the LDH leakage of glioblastoma cells 24 h after incubation with 50 mg L−1 (except otherwise indicated) for all the tested particles. Both tests indicate very limited toxicity with metabolic activity above 90% and non-significant LDH leakage, except for the MMP@PEG280NH2 and MMP@PEG280COOH. Indeed, it was reported that the charge of particles plays a significant role in their toxicity.36 Generally speaking, the charged particles are more toxic than neutral ones.31 It has been demonstrated that coating inorganic nanoparticles with PEG-NH2 increases their intracellular accumulation,37 and this may be because of the positive charge of these particles, which promotes a better interaction of the inorganic particle with the negatively-charged cell membrane. This would explain the slightly higher cytotoxicity of MMP@PEG280NH2, which could potentially result from adhesion to the cell membrane leading to intracellular accumulation. Although it is still modest, these particles with relatively higher toxicity have been discarded for the following in vitro magneto-mechanical experiments. Also, based on previous results obtained with the same magneto-mechanical treatment of cancer cells, but with different magnetic particles, the particle concentration was fixed at 50 mg L−1.3,4 At this concentration, the MMP do not induce any significant cell mortality. Still, it should be kept in mind that this does not necessarily imply that they do not affect the cells, since the activation of some cell signaling has already been reported at sub-lethal concentrations of iron oxide nanoparticles.21 | ||
| Fig. 4 Metabolic activity and LDH leakage of U87-MG cells after a 24 h incubation with 50 mg L−1 (unless otherwise indicated) of magnetic microparticles with different functionalization. The LDH leakage for samples with no reported result was not significant. | ||
Efficacy of the magneto-mechanical treatment to promote cell death via apoptosis
The effects of the magneto-mechanical treatment on the metabolic activity and membrane permeability, with respect to the duration and frequency of the applied magnetic field, are then investigated and reported in Fig. 5 and 6, respectively. The study was performed using four different types of particles for comparison, including bare MMP, MMP@SiO2, MMP@C6PEG150OH and MMP@PEG2000OH.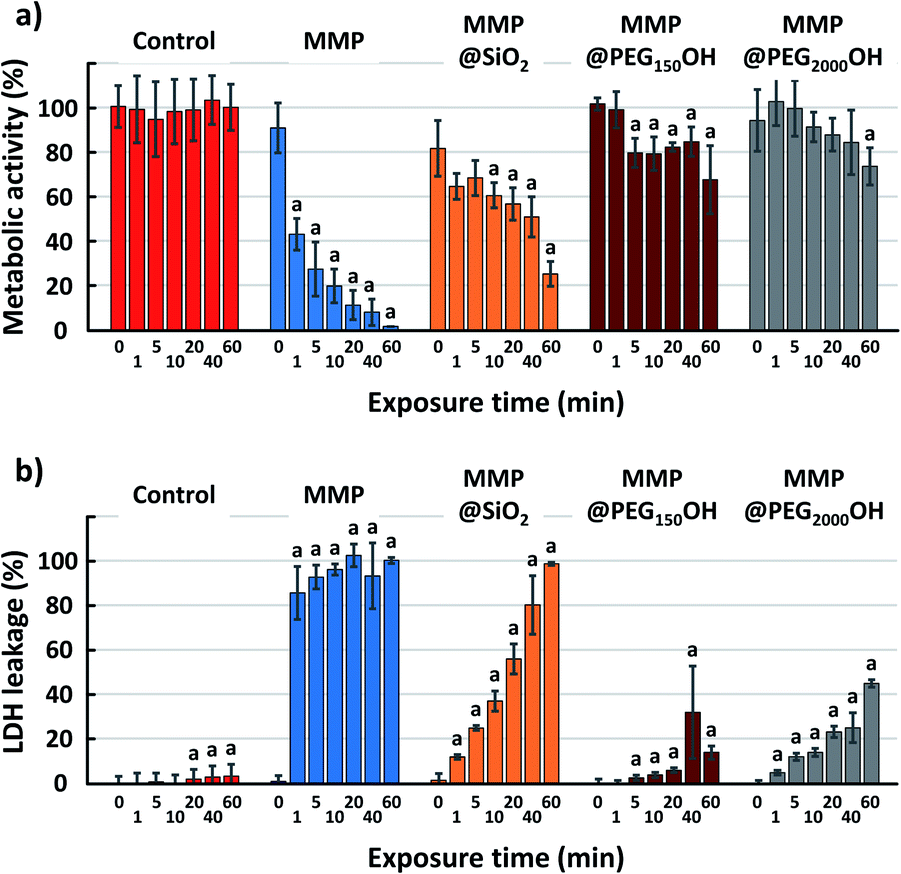 | ||
| Fig. 5 (a) Metabolic activity and (b) LDH leakage of U87-MG cells 18 h after magneto-mechanical treatment at 20 Hz during 0, 1, 5, 10, 20, 40 or 60 min. One-way ANOVA for each group and then Student t-test. (a) p < 0.05 for Student t-test compared with the point at 0 min for the same group. | ||
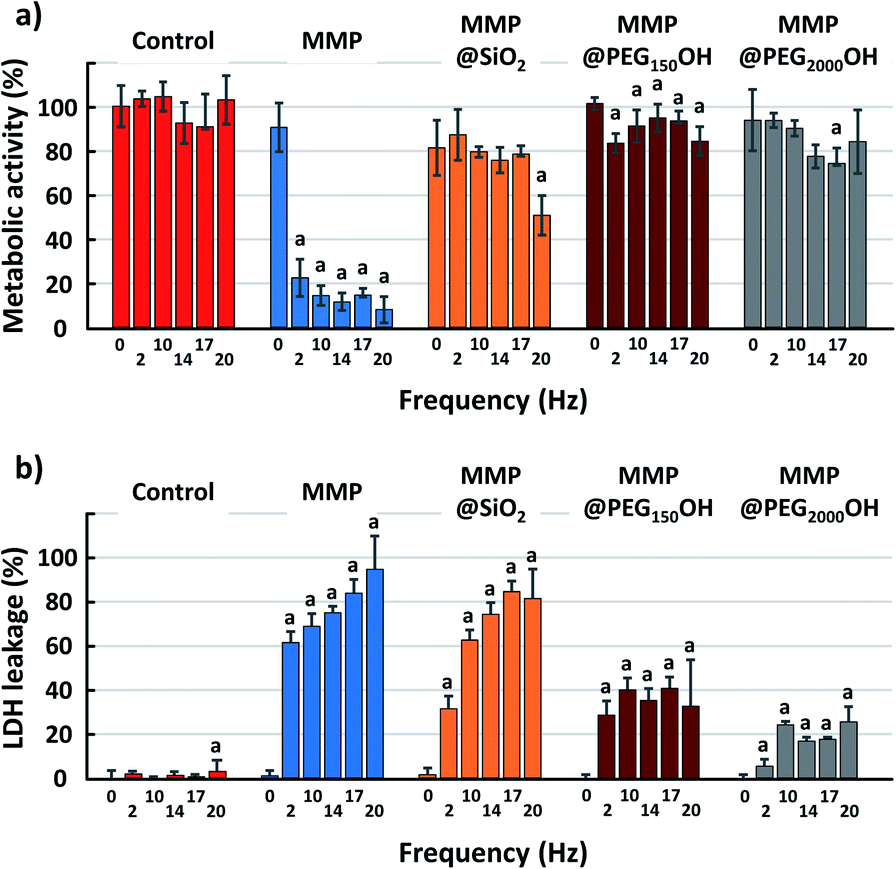 | ||
| Fig. 6 (a) Metabolic activity and (b) LDH leakage of U87-MG cells 18 h after magneto-mechanical treatment during 40 min at 2, 10, 14, 17 or 20 Hz. One-way ANOVA for each group and then Student t-test. (a) p < 0.05 for Student t-test compared with the point at 0 Hz for the same group. | ||
A large decrease in metabolic activity is also observed with MMP@SiO2, but it is more gradual, with a significant decrease after 10 min and a value of 25% for metabolic activity after 60 min. On the other hand, the treatment with PEGylated MMP shows less pronounced decreases in metabolic activity with a value at 67% (MMP@C6PEG150OH) and 73% (MMP@PEG2000OH) even after 60 min.
Such discrepancy between the results of LDH and WST-1 assay are unexpected. Both assays are used classically as markers of cell viability, and it is generally accepted that cells with damaged membranes are not viable anymore. Consequently, cells showing intense LDH release would also show decreased metabolic activity via the WST-1 assay, while the opposite is not necessarily true (cells can show temporary reduced metabolic activity but remain viable, with no release of LDH). It suggests that cells could show transient membrane breakage due to the magneto-mechanical treatment, with no impact on their metabolism, and could stay alive.
Evaluation of the ratio of apoptotic cells
The previous results show that the MMP and MMP@SiO2 seem to be the most effective in triggering cell death. Nevertheless, a closer look at the induced apoptotic response from the cells indicates that the picture is different in these two cases. For this, we choose to compare the change in the total number of cells in each culture well, and the change in the number of apoptotic cells, after 40 min magneto-mechanical treatment using different frequencies (Fig. 7).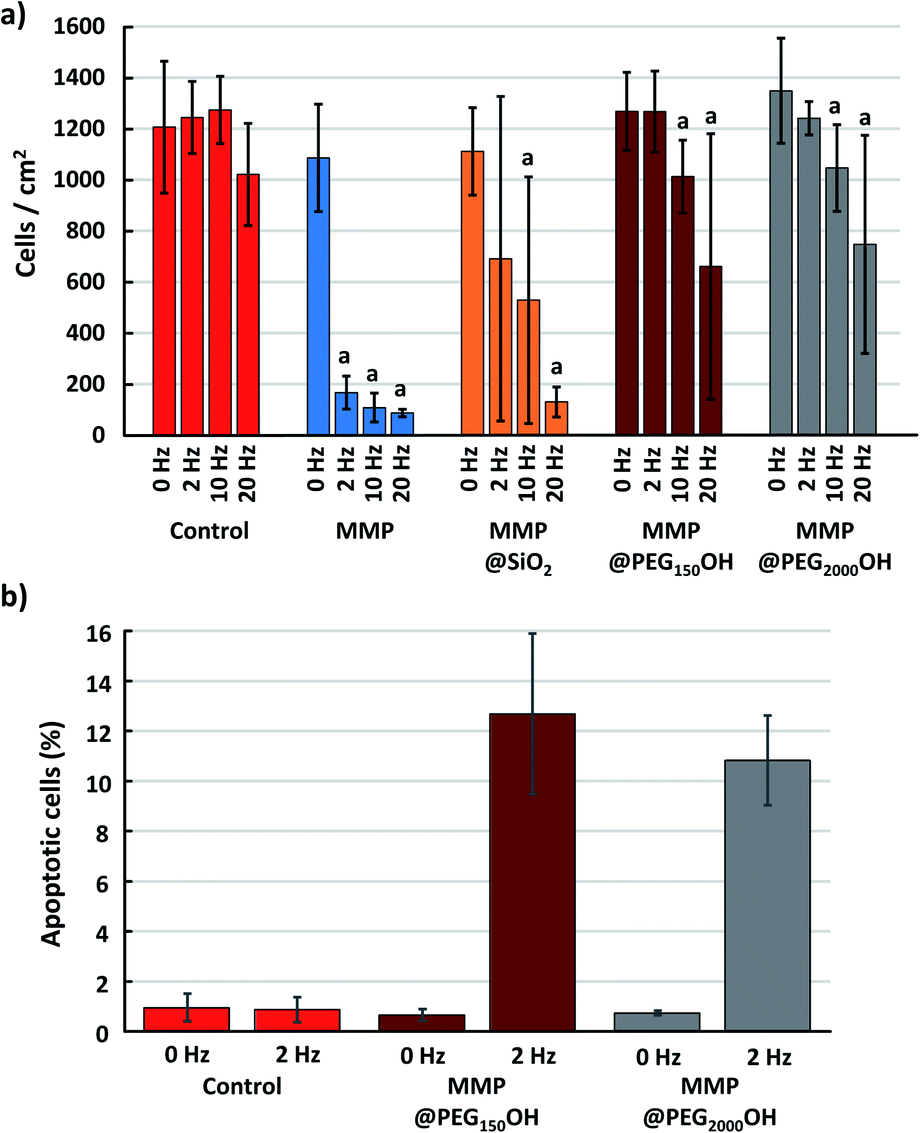 | ||
| Fig. 7 (a) Number of cells per mm2 measured 5 h after magneto-mechanical treatment during 40 min at 0, 2, 10 or 20 Hz. One-way ANOVA for each group and then Student t-test. (a) p < 0.05 for Student t-test compared with the point at 0 Hz for the same group. (b) Ratio of apoptotic cells relative to the number of cells per mm2, for 0 Hz and 2 Hz. | ||
As already observed when analyzing the evolution of cell metabolic activity and LDH leakage, application of the magneto-mechanical treatment to cells exposed to MMPs leads to the death of 90% of the cells, whatever the frequency of the applied magnetic field. Cell death is less intense when the cells are exposed to MMP@SiO2, compared to bare MMP. Moreover, MMP PEGylation greatly reduces cell death, which is negligible when a 2 Hz magnetic field is applied on cells exposed to MMP@PEG150OH and MMP@PEG2000OH (Fig. 7a). Focusing on this condition, while most of the cells remain viable when treated with MMP@PEG150OH and MMP@PEG2000OH, 13% and 11% of them, respectively, are apoptotic after application of the 2 Hz magneto-mechanical treatment (Fig. 7b). This level of apoptotic cells is comparable to what is generally observed upon magneto-mechanical treatments using more complex particles produced by a top-down approach.4 This shows that the MMP used here, obtained from the simple processing of easily accessible material, and surface modified using classical techniques, are a promising alternative to those complex particles for future magneto-mechanical treatments.
Cell interaction with the magnetic microparticles
To delve into the mechanisms of the observed effects, the next question is whether the magnetic microparticles are simply adsorbed on the surface of cells, or whether they are internalized inside the cells. If adsorbed on the cell membrane, the induction of apoptosis could result from opening of mechanoreceptors of the plasma membrane, leading to the influx of some ions such as calcium inside the cells, which would trigger apoptosis. Transmission electron microscopic observation of cells exposed to MMP, MMP@PEG150OH or MMP@PEG2000OH submitted to 2 Hz magneto-mechanical treatment shows that whatever their surface modification, magnetic microparticles both get adsorbed on the cell plasma membrane (e.g., in Fig. 8C, *) and internalized inside cells (Fig. 8, arrows). This cellular adsorption and internalization shows that even negatively charged particles coated with long PEG chains can interact with negatively charged cell membranes. After the magneto-mechanical treatment, evidence of cell death is observed (Fig. 8D shows a dead cell, where organelles have been released in the extracellular compartment and are interacting with MMP@PEG2000OH, surrounded by two live cells – upper and lower third of the image – with intact nucleus and mitochondria). Magnetic microparticles concentrate in the region of the fragmented dead cell; this may be due to release of intracellular components that locally modify the chemical characteristics of the milieu, triggering the particle agglomeration and/or interaction with dead cell fragments.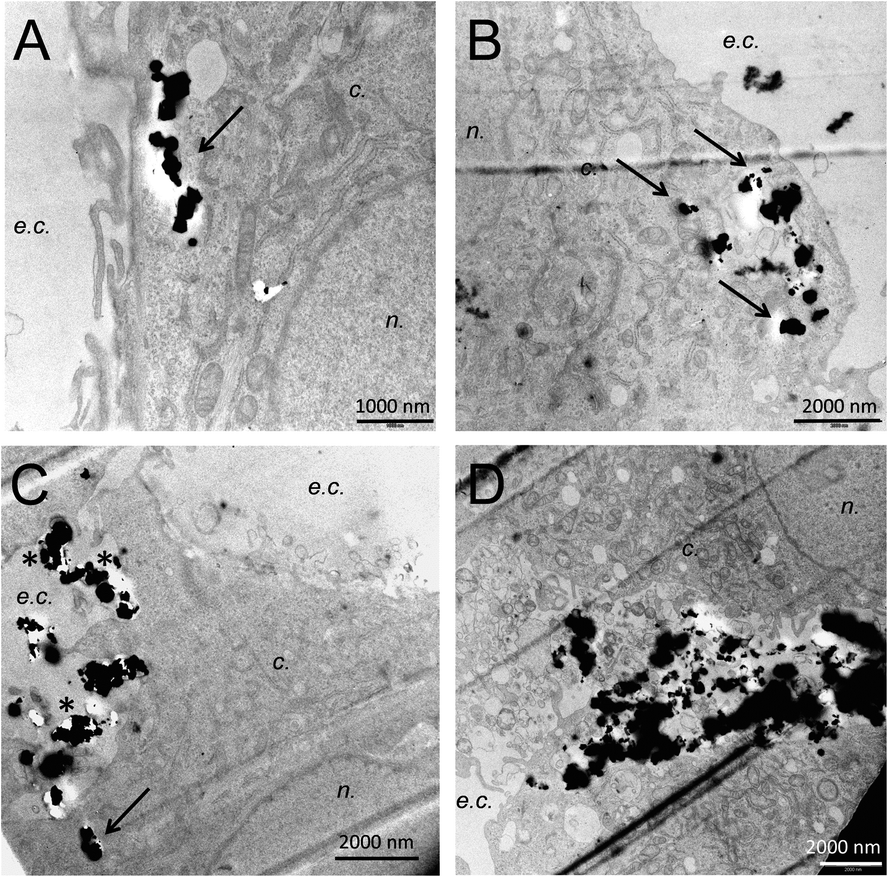 | ||
| Fig. 8 Microparticles interaction with cell membrane and accumulation into the cell cytoplasm. Cells are exposed to 50 μg mL−1 of MMP (A and B), MMP@PEG2000OH (C and D) and submitted to a magneto-mechanical treatment at 2 Hz for 40 min (D); n.: nucleus, c.: cytoplasm, e.c.: extracellular compartment. | ||
Materials and methods
Materials
Magnetite powder (iron (II, III) oxide, Fe3O4, particle size <5 μm), tetraethyl orthosilicate (TEOS), (3-aminopropyl)triethoxysilane (APTES), LDH and WST-1 kits were all purchased from Sigma-Aldrich. DMEM + Glutamax, succinimidyl 3-(2-pyridyldithio)propionate (SPDP), Hoechst 33342 and CellEvent Caspase-3/7 Green Detection Reagent were purchased from Thermo Fisher. PEGs (HS-PEG-X), including HS-C6PEG150-OH, HS-C11PEG280-NH2, and HS-C11PEG280-OCH2COOH were purchased from Prochimia Surfaces. The PEG HS-PEG2000-OH was purchased from Creative PEGWorks. Glioblastoma cells U87-MG were purchased from ATCC.Solvents (ethanol, isopropanol, and dimethyl sulfoxide (DMSO)), ammonium hydroxide solution (28% NH3 in H2O), phosphate buffered saline tablet (PBS, pH 7.4) were all supplied by Sigma-Aldrich. MilliQ ultrapure water (18.2 MΩ cm) was used in all experiments.
Particle fabrication by ball milling
0.5 g of magnetite powder was added to the 50 mL zirconia-coated grinding jars with 15 mL of isopropanol and 10 zirconia grinding balls, with 10 mm diameter. The planetary ball milling (Retsch-PM100) was performed for 1, 2, 4 or 12 h at 600 rpm with a 10 s pause every 10 min and with a change of the rotation direction. The obtained magnetite microparticles were washed twice with isopropanol by attracting the particles in the bottom of a tube with a magnet followed by supernatant removal. MMP were then dried, weighted and dispersed in isopropanol at 30 g L−1.Particle functionalization
The functionalization path is illustrated in Fig. 3 and detailed below.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethanol and ultrapure water and 2% v/v of APTES was added.42 The suspension was maintained at 50 °C for 24 h while agitating. The obtained Fe3O4@SiO2@APTES particles were carefully washed with ethanol and then with ultrapure water before being dispersed in PBS at 1 g L−1 and stored at 4 °C.
:1 mixture of ethanol and ultrapure water and 2% v/v of APTES was added.42 The suspension was maintained at 50 °C for 24 h while agitating. The obtained Fe3O4@SiO2@APTES particles were carefully washed with ethanol and then with ultrapure water before being dispersed in PBS at 1 g L−1 and stored at 4 °C.
Particle characterization
Cell culture
U87-MG glioblastoma cells were cultured in DMEM Glutamax medium supplemented with 10% of fetal bovine serum, 100 μM of streptomycin, and 100 units per mL of penicillin. They were maintained at 37 °C in a 5% CO2-humidified atmosphere and passaged twice a week using 0.05% Trypsin-EDTA.Toxicity assays
For cytotoxicity assays, cells were seeded in 96 well plates at a density of 50000 cells per well in 100 μL of culture medium. 24 h after cell seeding, MMP were washed and dispersed in the culture medium at concentrations ranging from 3 mg L−1 to 1 g L−1 and 100 μL of each MMP suspension was applied to cells (6 wells per condition, n = 6). 0.1% Triton-X-100 was used as positive control. After 24 h of exposure, cell viability was assessed using LDH and WST-1 assays.
For the LDH leakage assay, 50 μL of the supernatant of each well was transferred to a clean 96 well plate. LDH assay substrate, cofactor and dye were mixed as recommended by the supplier, added to each well and incubated at room temperature in the dark for 5 min. The reaction was stopped by the introduction of 10 μL of 1 N HCl per well. The absorbance (Abs) was recorded at 490 nm with subtraction of the baseline signal at 690 nm. The percentage of LDH leakage was calculated as:
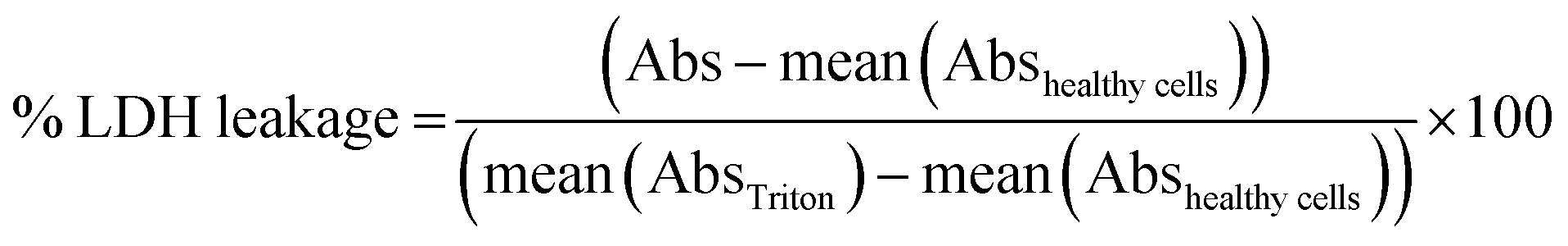
For the WST-1 assay, after the sampling exposure medium for the LDH assay, the remaining supernatant was replaced by a 10% WST-1 solution prepared in cell culture medium and incubated for 1 h at 37 °C. Interference of nanoparticles with the readout of cytotoxicity assays has been largely described in the literature. To avoid such bias, the MMP were allowed to settle down at the bottom of the plate and 50 μL of the supernatant was transferred to a clean 96 well plate. The ability of metabolically-active cells to cleave WST-1 to formazan was evaluated via absorbance measurement at 450 nm and subtraction of the baseline signal at 650 nm. The percentage of viability was calculated as:
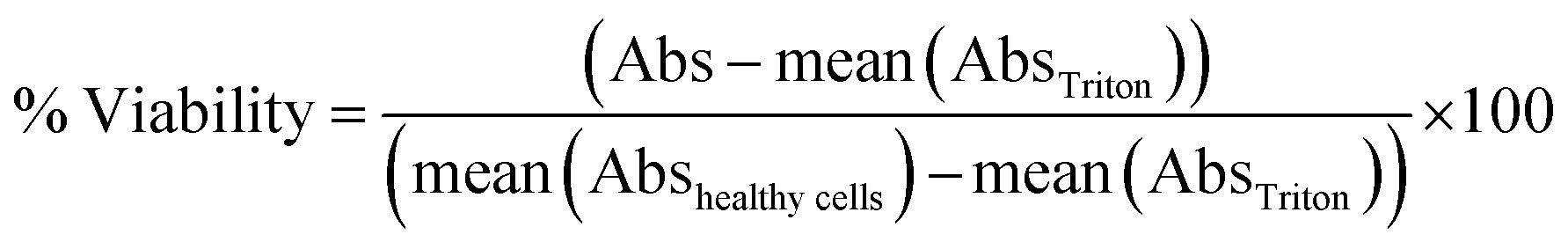
Magneto-mechanical treatment
Cells were seeded in 8 well LABTEK with 50000 cells per well in 200 μL of culture medium and incubated for 24 h at 37 °C in a 5% CO2-humidified atmosphere. MMP were washed and dispersed in the culture medium at 50 mg L−1 and 200 μL of this MMP suspension was applied to cells. After 4 h of incubation, the cells were placed inside a rotating Halbach cylinder, with a magnetic field of 0.6 T at its center. In one set of experiments, cells were treated with a RMF at 20 Hz for a duration of 1, 5, 10, 20, 40 and 60 min, respectively. In another set of experiments, cells were treated with the RMF for a fixed duration of 40 min with varied frequency of 2, 10, 14, 17 and 20 Hz, respectively.
LDH and WST-1 tests were performed 18 h after treatment as previously described.
Viabilities and LDH leakage were compared with the ones of cells treated with the same conditions of RMF but without the presence of magnetic particles. Cells exposed to Triton at 0.1% were used as positive control.
Evaluation of the efficacy of the magneto-mechanical treatment
The efficacy of the magneto-mechanical treatment was evaluated by measuring both the cell viability and the proportion of apoptotic cells after application of the magnetic field to the MMP. The viability was assessed via LDH leakage and WST-1 assays, 18 h after the magneto-mechanical treatment, using 0.1% Triton as positive control as described in the toxicity test section. The proportion of apoptotic cells was quantified via fluorescence microscopy observation after staining with the CellEvent Caspase 3/7 assay kit (Invitrogen) and 0.3 mg L−1 of Hoechst 33342, 5 h after the mechano-magnetic treatment. After 30 min of incubation at 37 °C, cells were observed by fluorescence microscopy. The ImageJ software (National Institutes of Health, Bethesda) was then used to count cells for determining the total number of cells (i.e. the number of nuclei, stained blue by Hoechst 33342) and the number of apoptotic cells (stained green by the CellEvent marker).Transmission electron microscopy (TEM)
For TEM imaging, after exposure to MMP and magneto-mechanical treatment, cells were rinsed with PBS, fixed in 2% glutaraldehyde prepared in cacodylate buffer then post-fixed using 1% osmium tetroxide. They were then dehydrated through a graded series of ethanol and embedded in Epon resin. Ultra-thin sections were cut and stained with 1% uranyl acetate. Sections were observed on a JEOL 1200EX TEM operating at 80 kV (Grenoble Institut des Neurosciences, Grenoble, France).Conclusions
We prepared anisotropic magnetite microparticles by liquid-phase ball milling of a magnetite powder, with magnetic properties such that a magnetic field induces mechanical vibration. After coating the particles with silica (MMP@SiO2), different PEGylated molecules were grafted to the surface thanks to a 3-step functionalization (MMP@C6PEG150OH, MMP@PEG2000OH, MMP@C11PEG280NH2, MMP@C11PEG280COOH).The particles showed very low in vitro intrinsic cytotoxicity on human glioblastoma U87-MG cells, while significant cell death was observed under RMF. To optimize the magneto-mechanical treatment efficacy, the metabolic activity and the LDH leakage were measured while varying the RMF frequency (2–20 Hz) and exposure time (1–60 min). Our results indicate that (1) the magneto-mechanical treatment with bare MMP induced a rapid decrease in cell viability whereas the effect was slower with PEGylated particles; (2) the number of apoptotic cells after magneto-mechanical treatment was higher with PEGylated particles; (3) a lower RMF frequency (down to 2 Hz) favored apoptosis. These results highlight a difference in the cell death mechanism according to the type of particle used – the rapid cell death observed with MMP favoring necrosis, while PEGylated particles rather induce apoptosis.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work has received support from the European Union through the H2020 ERA-Net EuroNanoMed II project Nanoviber. The authors would like to thank Christine Saint-Pierre for the zeta potential measurement.References
- S. M. Dadfar, K. Roemhild, N. I. Drude, S. von Stillfried, R. Knüchel, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2019, 138, 302–325 CrossRef CAS PubMed.
- D. H. Kim, E. A. Rozhkova, I. V. Ulasov, S. D. Bader, T. Rajh, M. S. Lesniak and V. Novosad, Nat. Mater., 2010, 9, 165–171 CrossRef CAS PubMed.
- S. Leulmi, X. Chauchet, M. Morcrette, G. Ortiz, H. Joisten, P. Sabon, T. Livache, Y. Hou, M. Carrière, S. Lequien and B. Dieny, Nanoscale, 2015, 7, 15904–15914 RSC.
- C. Naud, C. Thébault, M. Carrière, Y. Hou, R. Morel, F. Berger, B. Diény and H. Joisten, Nanoscale Adv., 2020, 2, 3632–3655 RSC.
- F. Broders-Bondon, T. H. N. Ho-Bouldoires, M. E. Fernandez-Sanchez and E. Farge, J. Cell Biol., 2018, 217, 1571–1587 CrossRef CAS PubMed.
- Q. A. Pankhurst, N. T. K. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- V. S. Markin and F. Sachs, Phys. Biol., 2004, 1, 110–124 CrossRef CAS PubMed.
- D. Gonzalez-Rodriguez, L. Guillou, F. Cornat, J. Lafaurie-Janvore, A. Babataheri, E. de Langre, A. I. Barakat and J. Husson, Biophys. J., 2016, 111, 2711–2721 CrossRef CAS PubMed.
- Y. I. Golovin, S. L. Gribanovsky, D. Y. Golovin, N. L. Klyachko, A. G. Majouga, А. M. Master, M. Sokolsky and A. V. Kabanov, J. Controlled Release, 2015, 219, 43–60 CrossRef CAS PubMed.
- S. Leulmi, H. Joisten, T. Dietsch, C. Iss, M. Morcrette, S. Auffret, P. Sabon and B. Dieny, Appl. Phys. Lett., 2013, 103, 132412 CrossRef.
- M. Goiriena-Goikoetxea, D. Muñoz, I. Orue, M. L. Fernández-Gubieda, J. Bokor, A. Muela and A. García-Arribas, Appl. Phys. Rev., 2020, 7, 011306 CAS.
- A. R. Muxworthy, D. J. Dunlop and W. Williams, J. Geophys. Res.: Solid Earth, 2003, 108, 2281 Search PubMed.
- T. P. Almeida, A. R. Muxworthy, A. Kovács, W. Williams, L. Nagy, P. Ó. Conbhuí, C. Frandsen, R. Supakulopas and R. E. Dunin-Borkowski, Geophys. Res. Lett., 2016, 43, 8426–8434 CrossRef PubMed.
- H. Joisten, T. Courcier, P. Balint, P. Sabon, J. Faure-Vincent, S. Auffret and B. Dieny, Appl. Phys. Lett., 2010, 97, 253112 CrossRef.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- M. Torrice, ACS Cent. Sci., 2016, 2, 434–437 CrossRef CAS PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- A. Frtús, B. Smolková, M. Uzhytchak, M. Lunova, M. Jirsa, Š. Kubinová, A. Dejneka and O. Lunova, J. Controlled Release, 2020, 328, 59–77 CrossRef PubMed.
- B. Kharazian, N. L. Hadipour and M. R. Ejtehadi, Int. J. Biochem. Cell Biol., 2016, 75, 162–174 CrossRef CAS PubMed.
- F. Yuan, M. Dellian, D. Fukumura, M. Leunig, D. A. Berk, V. P. Torchilin and R. K. Jain, Cancer Res., 1995, 55, 3752–3756 CAS.
- M. Uzhytchak, B. Smolková, M. Lunova, M. Jirsa, A. Frtús, Š. Kubinová, A. Dejneka and O. Lunov, Cells, 2020, 9, 1015 CrossRef CAS PubMed.
- E. Zhang, M. F. Kircher, X. M. Koch, L. Eliasson, S. N. Goldberg and E. Renstro, ACS Nano, 2014, 8, 3192–3201 CrossRef CAS PubMed.
- O. Lunov, M. Uzhytchak, B. Smolková, M. Lunova, M. Jirsa, N. M. Dempsey, A. L. Dias, M. Bonfim, M. Hof, P. Jurkiewicz, Y. Petrenko, Š. Kubinová and A. Dejneka, Cancers, 2019, 11, 1873 CrossRef CAS PubMed.
- P. Clerc, P. Jeanjean, N. Hallali, M. Gougeon, B. Pipy, J. Carrey, D. Fourmy and V. Gigoux, J. Controlled Release, 2018, 270, 120–134 CrossRef CAS PubMed.
- A. M. Master, P. N. Williams, N. Pothayee, N. Pothayee, R. Zhang, H. M. Vishwasrao, Y. I. Golovin, J. S. Riffle, M. Sokolsky and A. V. Kabanov, Sci. Rep., 2016, 6, 33560 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- M. Contreras, R. Sougrat, A. Zaher, T. Ravasi and J. Kosel, Int. J. Nanomed., 2015, 10, 2141–2153 CrossRef CAS PubMed.
- M. M. Song, W. J. Song, H. Bi, J. Wang, W. L. Wua, J. Sun and M. Yu, Biomaterials, 2010, 31, 1509–1517 CrossRef CAS PubMed.
- I. Papageorgiou, C. Brown, R. Schins, S. Singh, R. Newson, S. Davis, J. Fisher, E. Ingham and C. P. Case, Biomaterials, 2007, 28, 2946–2958 CrossRef CAS PubMed.
- K. Turcheniuk, A. V. Tarasevych, V. P. Kukhar, R. Boukherroub and S. Szunerits, Nanoscale, 2013, 5, 10729–10752 RSC.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- S. Gates-Rector and T. Blanton, Powder Diffr., 2019, 34, 352–360 CrossRef CAS.
- F. Bosi, U. Halenius and H. Skogby, Am. Mineral., 2009, 94, 181–189 CrossRef CAS.
- B. D. Cullity and C. D. Graham, Introduction to Magnetic Materials, 2nd edn, 2008 Search PubMed.
- Q. Li, C. W. Kartikowati, S. Horie, T. Ogi, T. Iwaki and K. Okuyama, Sci. Rep., 2017, 7, 9894 CrossRef PubMed.
- N. M. Schaeublin, L. K. Braydich-Stolle, A. M. Schrand, J. M. Miller, J. Hutchison, J. J. Schlager and S. M. Hussain, Nanoscale, 2011, 3, 410–420 RSC.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- A. Aliyandi, I. S. Zuhorn and A. Salvati, Front. Bioeng. Biotechnol., 2020, 8, 599454 CrossRef PubMed.
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailänder and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed.
- W. Stober, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- P. Riani, M. A. Lucchini, S. Thea, M. Alloisio, G. Bertoni and F. Canepa, Inorg. Chem., 2014, 53, 9166–9173 CrossRef CAS PubMed.
- S. Villa, P. Riani, F. Locardi and F. Canepa, Materials, 2016, 9, 826 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00461a |
| This journal is © The Royal Society of Chemistry 2021 |