DOI:
10.1039/D1NA00150G
(Paper)
Nanoscale Adv., 2021,
3, 4079-4088
A more effective catalysis of the CO2 fixation with aziridines: computational screening of metal-substituted HKUST-1†
Received
28th February 2021
, Accepted 3rd June 2021
First published on 3rd June 2021
Abstract
A vital issue for the fixation and conversion of CO2 into useful chemical products is to find effective catalysts. In this work, in order to develop more effective and diverse catalysts, we implemented the first computational screening study (at M06-2X//B3LYP level) on the cycloaddition of CO2 with aziridines under eighteen metal-substituted HKUST-1 MOFs and tetrabutylammonium bromide (TBAB) as a co-catalyst. For all considered catalytic systems, the ring-opening of aziridine is calculated to be the rate-determining step. Up to 11 M-HKUST-1 systems, i.e., Rh (31.87 kcal mol−1), Y (31.02), Sc (30.50), V (30.02), Tc (29.90), Cd (29.80), Ti (29.32), Mn (29.05), Zn (28.29), Fe (27.85) and Zr (25.09), possess lower ring-opening barrier heights than the original Cu-HKUST-1 (32.90), indicative of their superior catalytic ability to the original Cu-HKUST-1 in theory. With the lowest ring-opening barrier, Zr-HKUST-1 is strongly advocated for future synthetic and catalytic studies.
1. Introduction
Carbon dioxide (CO2), as the major waste gas from industrial and human activities, is considered one of the main greenhouse gases.1–5 On the other hand, carbon dioxide is a renewable, inexpensive, nontoxic and safe C1 feedstock.6–9 Therefore, currently, the effective utilization of CO2 has been quite a meaningful and challengeable task to meet the increasing energy demand and environmental sustainability. As one of the effective strategies for CO2 utilization, the cycloaddition of CO2 with aziridines to afford oxazolidinones stands out from many catalyzed reactions not only due to the satisfactory atom efficiency, but also due to the generation of high-value products.10–12 In fact, oxazolidinones, serving as the important intermediates and chiral auxiliaries in organic synthesis and anti-bacterial pharmaceuticals in medicinal chemistry, have an extensive application prospect.13–15 However, due to the chemical inertness and thermodynamic stability of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (bond enthalpy +805 kJ mol−1) of the CO2 molecule, efficient catalysts are required in CO2 conversion and utilization under mild conditions.16,17
O bond (bond enthalpy +805 kJ mol−1) of the CO2 molecule, efficient catalysts are required in CO2 conversion and utilization under mild conditions.16,17
During the past decades, various homogeneous catalysts (including alkali metal halide, tetraalkylammonium salt and α-amino acids) have been investigated and found to exhibit good catalytic activity.18–24 However, one usual drawback for such homogeneous processes is the difficulty for separating the catalyst and product. Alternatively, thanks to various attractive features of metal–organic frameworks (MOFs) such as the designable structures, adjustable functional groups, well-defined pores, high surface area, significant capability for gas-adsorption, and excellent recyclable catalytic performance,25–30 chemists have recently turned to exploring the possibility of various MOF-based catalysts for the cycloaddition of CO2 with aziridines.31–36 These laboratory studies have shown that as an efficient and heterogeneous catalyst, structurally diversified MOFs including [Cu30] nanocages ({[Cu2(BCP)(H2O)2]·3DMF}n, H4BCP = 5-(2,6-bis(4-carboxyphenyl)pyridin-4-yl)isophthalic acid),35 {[Zn(H2O)(C5H7NO4)]·H2O}n(ZnGlu),33 MMPF-10,34 [Zn24] nanocages,31 {[M2(XN)2(IPA)2]·2H2O}n(M = Co, Mn, Ni; XN = 4′-(4-pyridine)4,2′:2′,4′′-terpyridine, IPA = isophthalic acid)32 can be regarded as the optimal candidates for the catalytic reaction of CO2 with aziridines. Although the pursuit of novel, structurally diversified and high catalytic reactivity catalysts is a never-ending task, the frequently encountered synthetic challenge and difficulty in unraveling the reaction mechanism in the laboratory have greatly hindered the advancement of the catalytic chemistry of CO2 with aziridines by MOFs. In fact, the reported examples for MOF-catalyzed CO2 cycloaddition with aziridines have been very limited.31–36 Luckily, quantum chemical computational techniques have now evolved into a very powerful tool to tackle the above problems.37,38 The detailed mechanistic study could help deduce the key factors for an effective reaction pathway, with which we can screen out possible catalyst candidates from a number of experimentally untouched reactions.
It has proven to be quite applicable to design novel and powerful MOF-based catalysts simply by altering the metal centers, while retaining the rigid framework of a MOF.39–46 The recently reported [Cu30] nanocages35 contain the Cu-HKUST-1 building units. As is well known, the HKUST-1 (HKUST = Hong Kong University of Science and Technology) (also known as Cu-BTC or MOF-199, BTC = 1,3,5-benzenetricarboxylate) MOF possesses a number of attractive features, including designability and stability.47 Since the compound with [Cu30] nanocages was shown to perform best at a temperature of 100 °C,35 it should be quite desirable to find alternative catalysts that can allow milder reaction conditions (i.e., lower temperatures) for the sake of reducing the energy cost. In this work, we applied a metal-substitution strategy to M-HKUST-1 by considering a total of 18 transition metals (i.e., Cu, Ag, Mo, Cr, W, Nb, Ru, Rh, Y, Sc, V, Tc, Cd, Ti, Mn, Zn, Fe and Zr). Tetrabutylammonium bromide (TBAB) was used as the co-catalyst. The extensive computational study at the M06-2X//B3LYP level revealed that for all the considered systems, the ring-opening of aziridine is the rate-determining step. Thus, by taking the ring-opening barrier of Cu-HKUST-1 as a reference (32.90 kcal mol−1), the catalyst with better catalytic performance than the original Cu-HKUST-1 was screened out. This is the first computational screening for a Cu-HKUST-1-based catalyst for the cycloaddition of CO2 with aziridines.
2. Computational details
2.1. Models
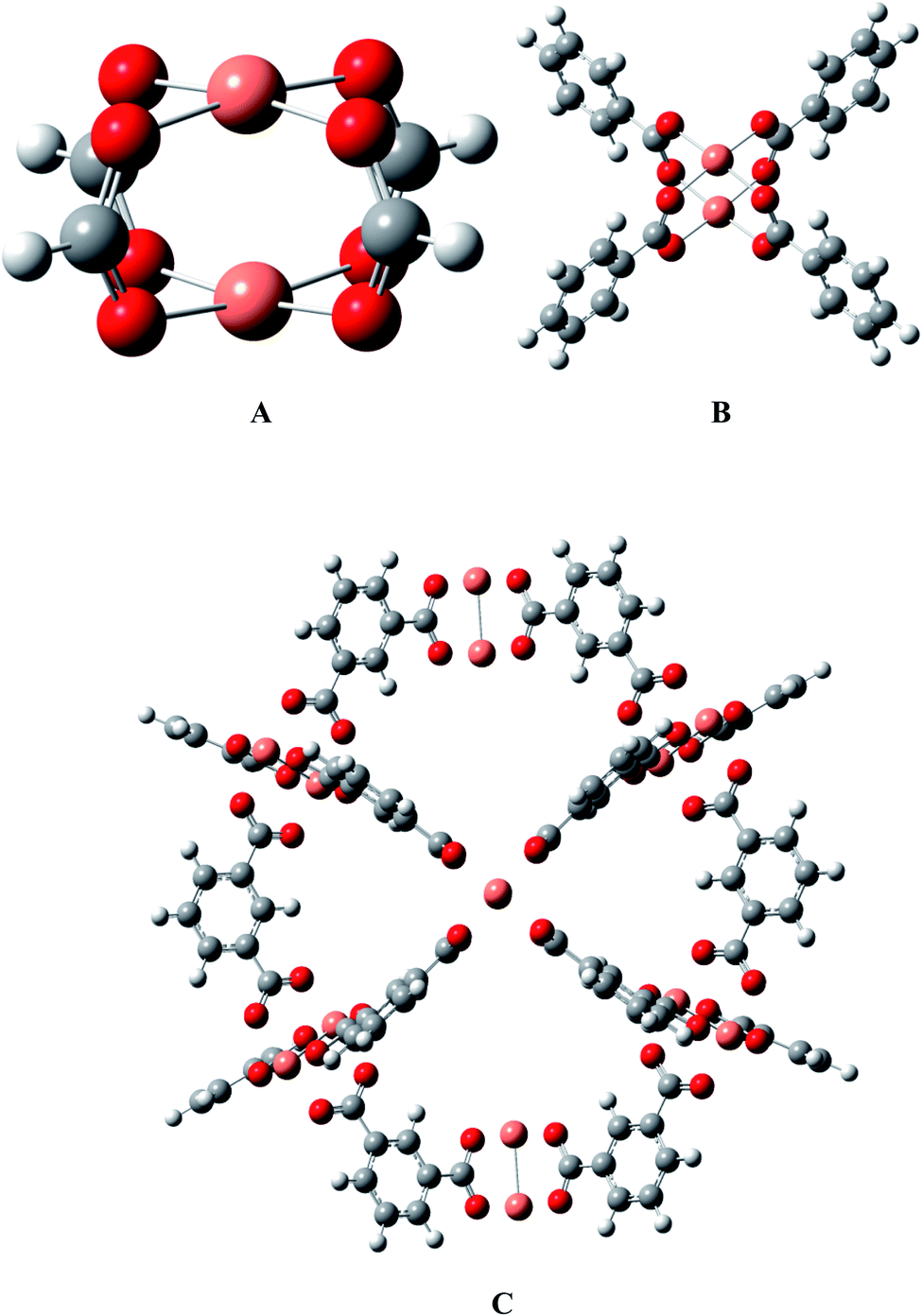
HKUST-1 is constructed from a binuclear copper paddlewheel unit connected with four benzene tricarboxylate ligands, which is deemed as a well-defined crystalline material with periodic structure, as shown in Fig. 1. According to the available research experiences, considering the calculation cost, the models could be cut from the periodic structure and saturated with protons so that the overall charge remains zero in the whole process of mechanistic exploration.48–53 The effectiveness of the Cu2(HCOO)4 model A has been validated in our group by two different models (see B and C in Fig. 1), i.e., a Cu2(C6H5COO)4 model B with four benzenecarboxylate ligands that can sufficiently describe the chemical environment of HKUST-1,36 and an ONIOM(QM/MM) supercage model with 408 atoms54 that can better present the surrounding atomic environment. The results showed that model A predicts the energetics of the key intermediates within several kcal mol−1 deviation of model B,36 and can capture the reaction mechanism from the supercage model C.54 Thus, it could be safe to apply model A to the present large-scale screening of M-HKUST-1 on the cycloaddition of CO2 with aziridines.
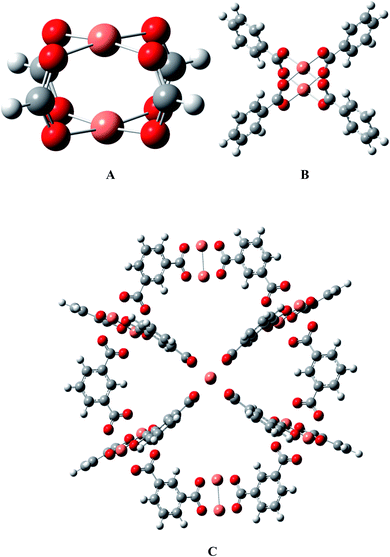 |
| | Fig. 1 Models for M-HKUST-1: (A) H-saturated cluster model, (B) benzenecarboxylate-saturated cluster model, and (C) supercage model. Color scheme: carbon-brown, hydrogen-white, oxygen-red, and M-pink (Cu, Ag, Mo, Cr, W, Nb, Ru, Rh, Y, Sc, V, Tc, Cd, Ti, Mn, Zn, Fe and Zr). | |
2.2. Computational methods
Calculations have been performed using density functional theory (DFT) on molecular models of HKUST-1. We carried out the B3LYP exchange–correlation functional and mixed basis sets for the geometries and frequencies of the complexes, intermediates, fragments and transition states,55 for which the 6-31+G(d) basis set was employed for C, O, H and N, while the LANL2DZ basis set was used to describe the transition metal atoms (Cu, Ag, Mo, Cr, W, Nb, Ru, Rh, Y, Sc, V, Tc, Cd, Ti, Mn, Zn, Fe and Zr) and bromine atom, respectively, as used in previous MOFs studies.56–59 Numerical integrations were executed using an ultrafine grid. In order to more accurately simulate the constraints imposed by the surrounding M-HKUST-1 structure, the terminal hydrogen atoms remained fixed in the framework, while the rest of the atoms were allowed to relax during the geometric optimization. Frequency analysis was conducted at the same level of theory to verify the stationary points to be the real minima. For each stationary point, the intrinsic reaction coordinate (IRC) analysis was undertaken to confirm that it connected the desired minimal reactant and product on the potential energy surface.60 Throughout the entire reaction pathways, we used the solvent Et2O to model this reaction and performed self-consistent reaction field calculations with the polarizable continuum model to consider the effects of the solvent, as was completed in previous similar investigations.39 Additionally, to obtain more reliable interaction energies, single-point energy calculations were conducted on the stationary points by using the M06-2X functional with a larger basis set, i.e., SDD for transition metal atoms and bromine atom, and 6-311++G(2df,p) for the other elements.61 The final energies were obtained from the Gibbs free energy corrections and single-point energies. Details of various cartesian coordinates and corresponding energies of the complexes, intermediates, transition states, reactants and products were supplied in the ESI.† All of the calculations were implemented in the Gaussian 09 program package.62
3. Results and discussion
3.1. Spin states of M-HKUST-1
In order to systematically consider the effects of the number of valence electrons and the number of periods in the metal element on the cycloaddition reaction of CO2 with aziridines, in principle, we considered a total of 20 transition metals of the first and second periods. It should be noted that among them, the transition metal structures of Co-HKUST-1, Ni-HKUST-1 and Pd-HKUST-1 are significantly different from that of Cu-HKUST-1, and have presented extreme difficulty in geometrical optimizations. Therefore, these three catalytic systems are not included in this work. In addition, it has been reported that W-HKUST-1 shows excellent catalytic activity for the cycloaddition reaction of CO2 and epoxide. Therefore, W-HKUST-1 was also taken into account in this paper, and there are 18 metal catalytic systems in total. Transition metals with various numbered d-electrons are known to bear varied spin states. Thus, it is necessary to consider the structures and energetics of M-HKUST-1 with different spin states, so as to obtain more reliable results. We considered a total of eight spin multiplicities ranging from 2n + 1 (n = 0–7), i.e., the singlet to 15-et state. The energies of M-HKUST-1 with different spin states are shown in Table S1 in the ESI.† The lowest energy spin state for these M-HKUST-1 is singlet for M = Mo, W, Sc, Zn, Zr, Y, Ag, Rh and Cd, triplet for M = Cu, Ru, Nb, Tc and Ti, septet for M = V, nonet for M = Fe and Cr, and undectet for M = Mn. It is reasonable that the entire calculation for the reaction channel over the metal exchanged M-HKUST-1 was analyzed on the lowest energy spin state of each M-HKUST-1, since for such thermally activated processes, the spin turnover (or intersystem crossing between different states) is usually unlikely to take place. Such a spin-selected methodology has been applied in previous studies.39,63
3.2. General reaction mechanism
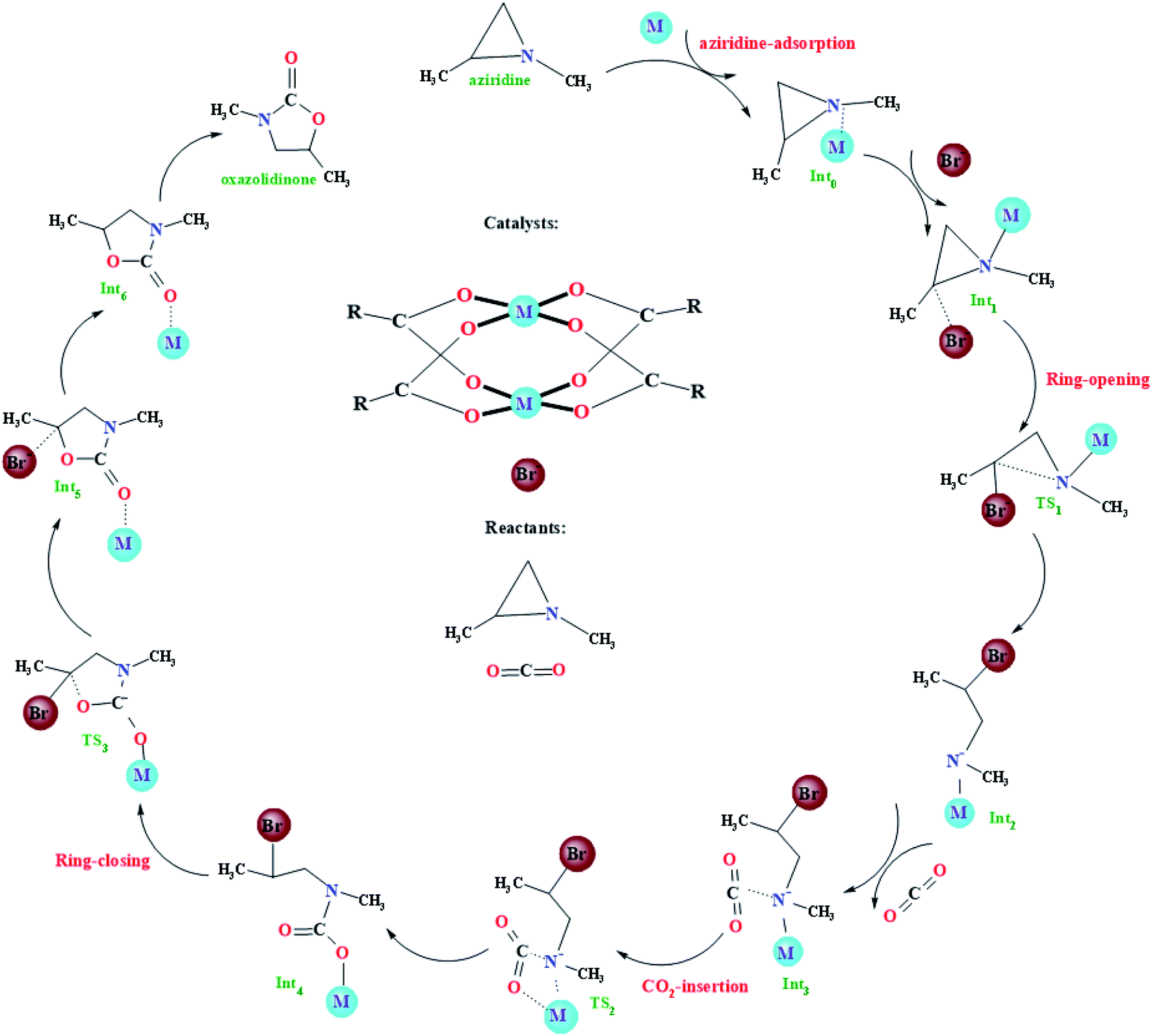
The very recent computational study on the model Cu-HKUST-1 catalyst showed that the CO2 cycloaddition with aziridines may experience four main steps: (i) adsorption of aziridines, (ii) ring-opening of aziridines, (iii) insertion of CO2 into the metal–nitrogen bond, and (iv) the formation of the oxazolidinones, as illustrated in Fig. 2. Such a general picture is consistent with other limited studies of the CO2 cycloaddition with aziridines using non-MOF catalysts.18–24 With regard to the four main steps mentioned above, some simple instructions were introduced. It is worth mentioning that metal atoms have many empty d orbitals, which can interact with Lewis bases and have the characteristics of Lewis acids. According to a large number of literature reports, for the metal–organic framework taken as an catalyst, its framework usually plays a supporting and auxiliary role, whereas the unsaturated metal sites play catalytic roles, namely Lewis acid sites.36,39,64–67 In the beginning, the catalytic reaction is initiated when coordination of the N atom of aziridine into the unsaturated metal site in M-HKUST-1 occurs, generating an initial coordinated intermediate Int0. Then, the C atom of Int0 is gradually approached by a bromide anion from the co-catalyst, finally forming a weak van der Waals trimolecular complex Int1. Meanwhile, the bromide anion makes a nucleophilic attack on the carbon atom of aziridine from the backside, leading to the ring opening of aziridine, resulting in a stable structure Int2via a transition state TS1. Subsequently, a CO2 molecule was joined into the system, bringing about a stable trimolecular complex Int3. Following the formation of Int3, the CO2 molecule is inserted into the obtained metal–N bond, generating a new intermediate Int4 through the transition state TS2. At last, the Int4 is transformed to the Int5via the transition state TS3. In this particular step, the M-HKUST-1-coordinated oxazolidinone product Int5 forms with the concomitant elimination of the Br− anion. Then, the dissociation of oxazolidinone leads to the recovery of the M-HKUST-1 catalyst.
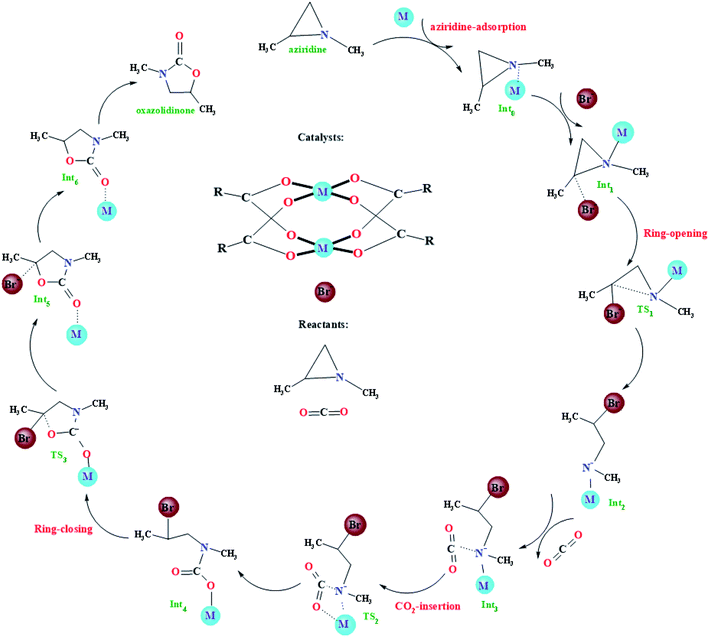 |
| | Fig. 2 Proposed reaction mechanism for the formation of oxazolidinone from CO2 and aziridines catalyzed by M-HKUST-1 and TBAB (M = Cu, Ag, Mo, Cr, W, Nb, Ru, Rh, Y, Sc, V, Tc, Cd, Ti, Mn, Zn, Fe and Zr). | |
3.3. Effect of metal centers in M-HKUST-1 catalysts
The open unsaturated Lewis-acidic sites (Cu-atoms) on the metal centers of the HKUST-1 are surely active in chemistry, and can activate the aziridines through the coordination with the N atoms from the aziridines. The nucleophilic attack of Br− to the aziridines can lead to ring-opening. In this work, we constructed a total of eighteen M-HKUST-1 catalysts for the catalysis of the cycloaddition reactions of the aziridines with CO2. It should be noted that only six M-HKUST-1 (M = Mo,68 Cr,69 Cu,47 Fe,70 Ru,71 and Zn72) have been synthesized experimentally. Moreover, five hypothetical M-HKUST-1 (M = Ti, W, Sc, Cd, V) have been devised by Hu et al.39 The remaining seven HKUST-1 (M = Nb, Zr, Tc, Ag, Mn, Rh and Y) were reported for the first time in this work.
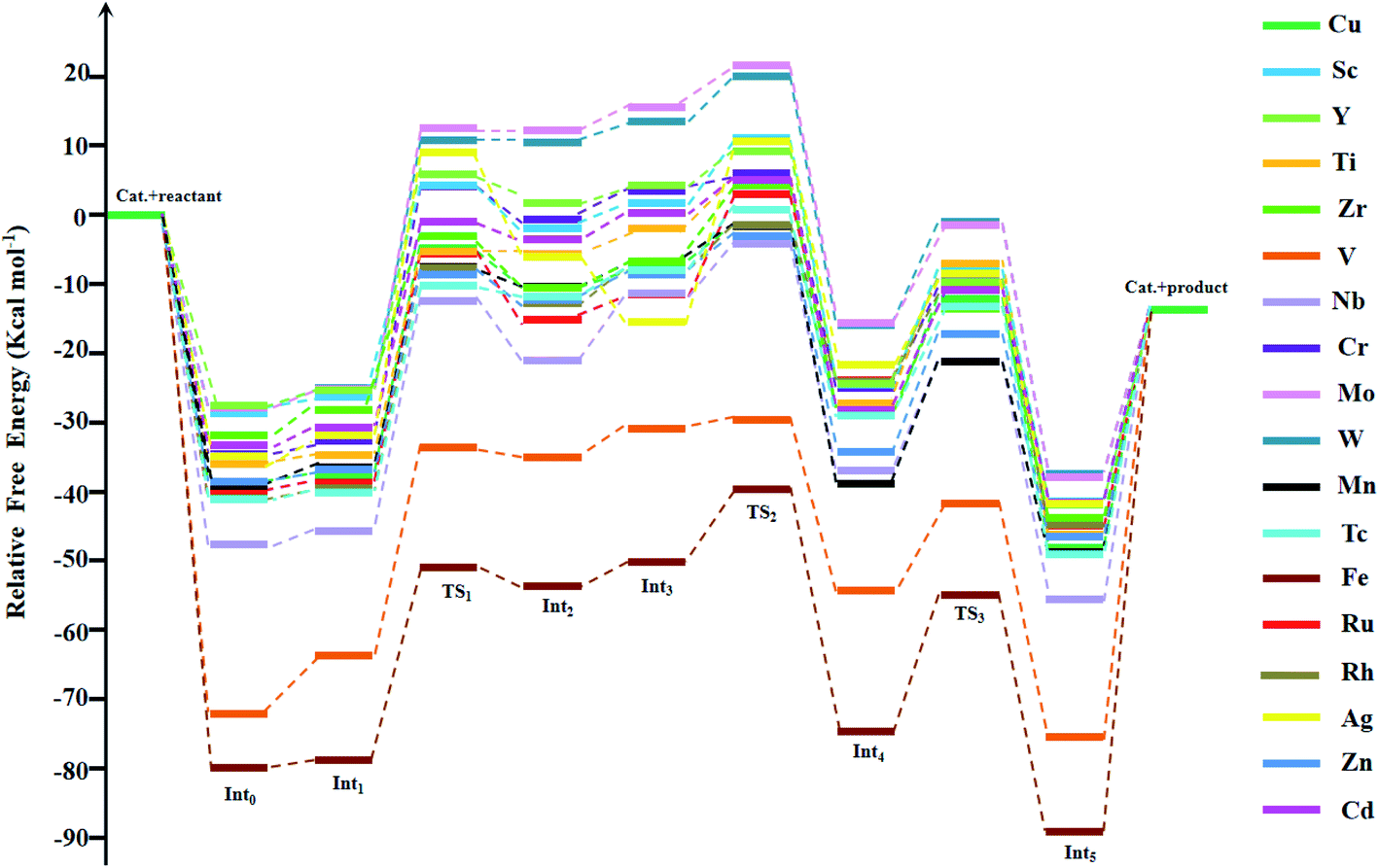
Experimentally, among the eighteen M-HKUST-1 catalytic systems for the cycloaddition of CO2 with aziridines, only M = Cu has been reported.35 Thus, we used Cu-HKUST-1 as a reference. The Gibbs free energy profiles of the eighteen cycloaddition reactions are shown in Fig. 3. The detailed relative Gibbs free energies for all stationary points along the reaction pathways of each M-HKUST-1 are listed in Table S2 of the ESI.† It should be pointed out that the remaining seventeen reactions were studied for the first time in this work. In the following, we will discuss and compare the reaction mechanism of M-HKUST-1 based on Fig. 3.
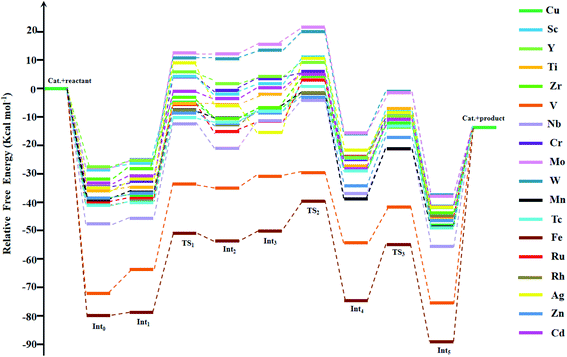 |
| | Fig. 3 Gibbs free energy profiles for stationary points along the reaction coordinates for CO2 fixation catalyzed by M-HKUST-1. | |
3.3.1. Aziridine adsorption.
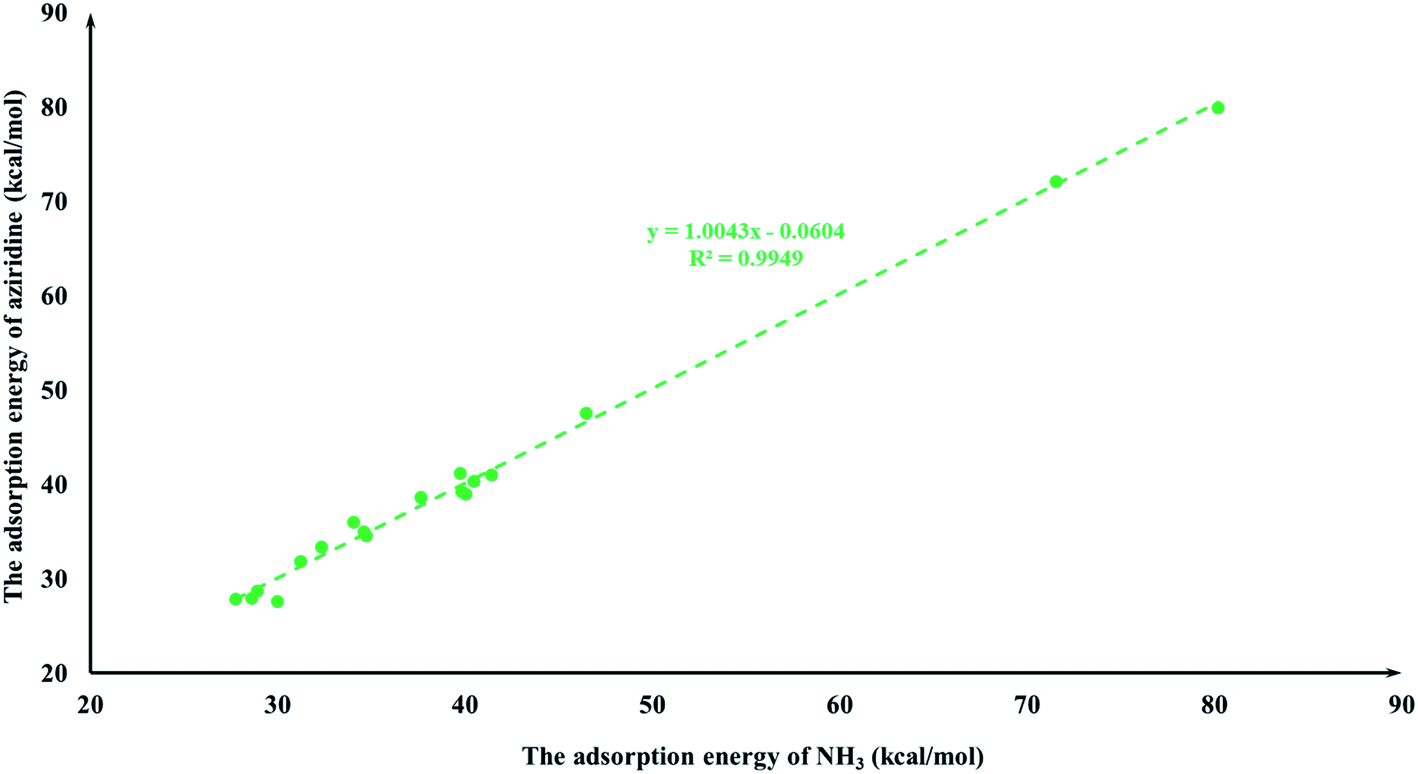
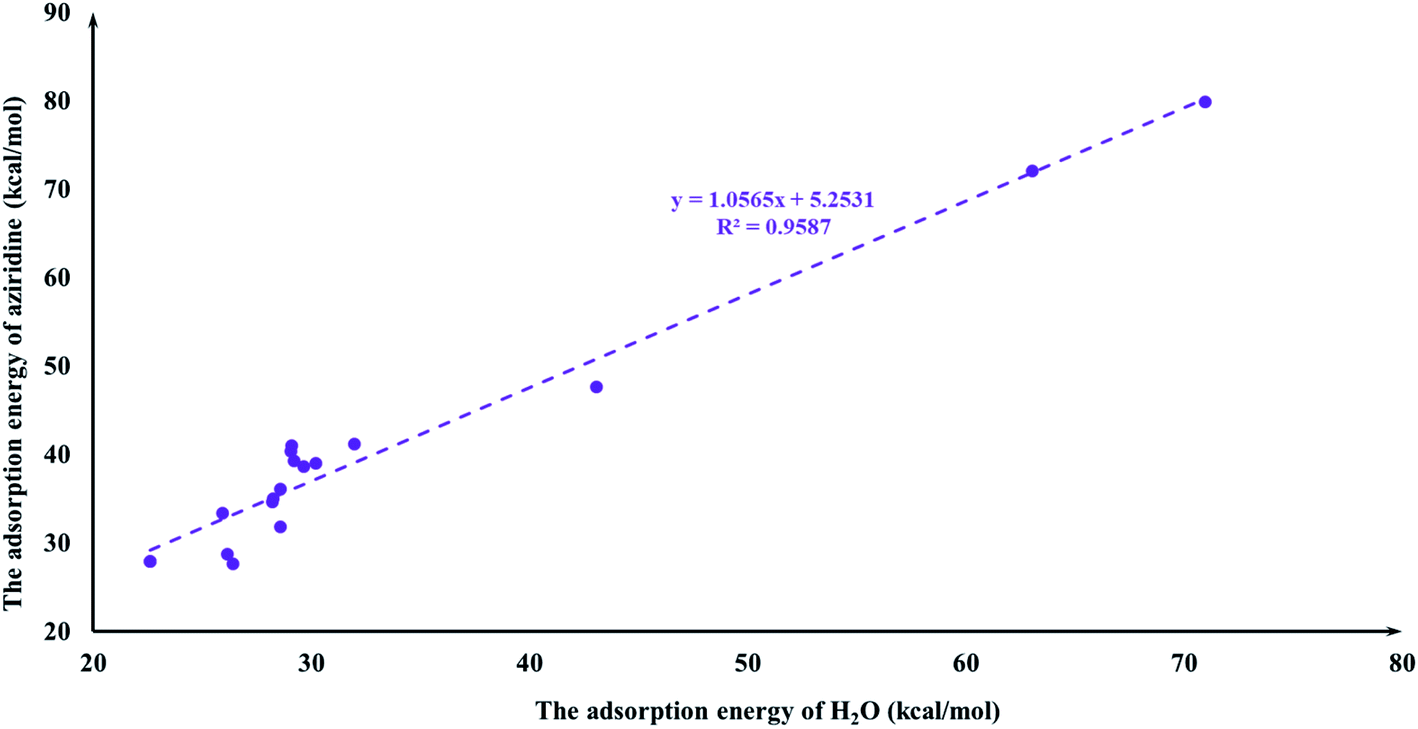
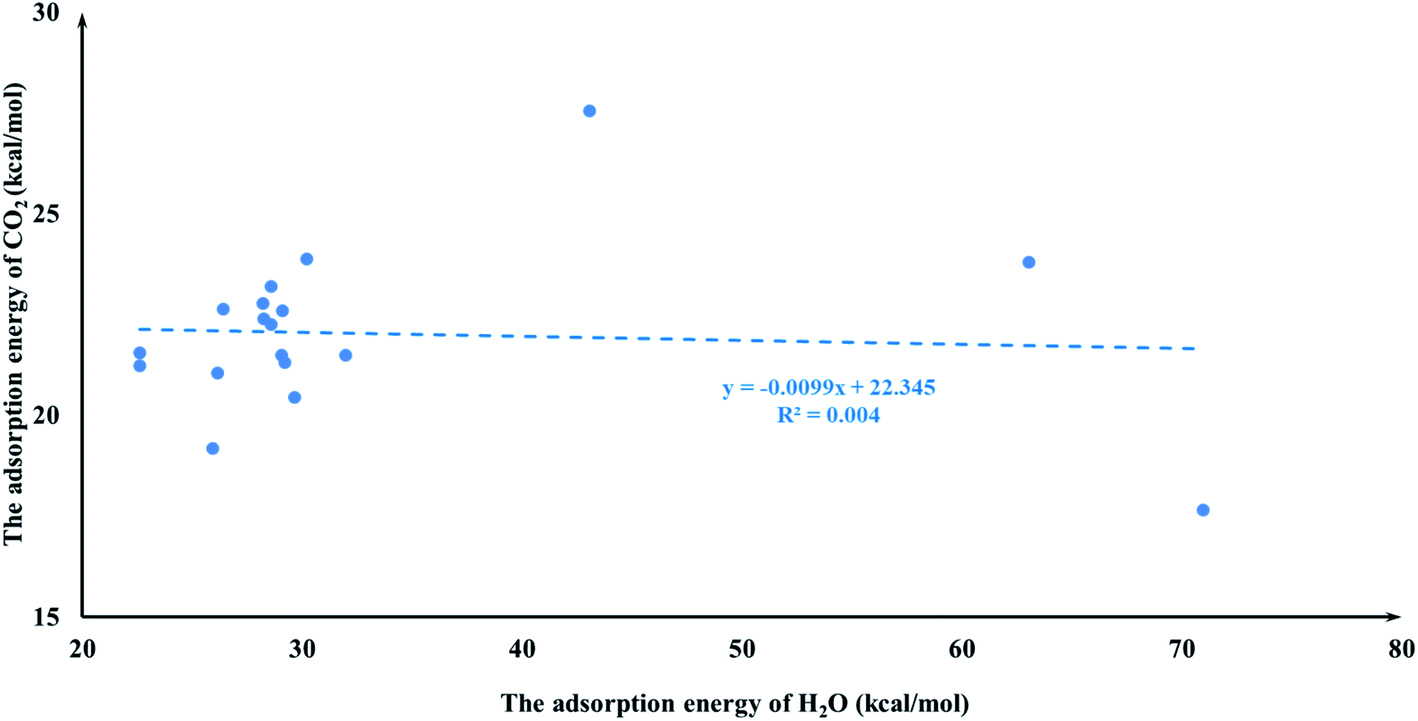
It is quite reasonable to consider aziridine adsorption as a starting point in regard to the CO2 cycloaddition reaction. Yet CO2 is a co-reagent in this system, and has the potential to get adsorbed to some extent on the Lewis acid active sites of the M-HKUST-1 with aziridine. For the title cycloaddition reactions, the two reagents, i.e., aziridine and CO2, contain the electron lone-pair (LP) at N and O-sites, respectively. The adsorption of both reagents to the catalyst should in principle be considered. We presented a correlation diagram of the adsorption energy of aziridine with respect to that of the probing molecule (see Fig. 4 and 6). NH3 and H2O were taken as probing molecules since both are key environmental gases, and can act as Lewis base models due to the inherent lone-pair electrons. Interestingly, the adsorption energy of aziridine with M-HKUST-1 is associated with an excellent linear correlation with that of NH3 (R2 = 0.9949) and H2O (R2 = 0.9587). Moreover, by comparison of the R2 values, we can find that the adsorption of aziridine to M-HKUST-1 is better correlated with NH3 than with H2O, which is ascribed to the closer similarity of aziridine to NH3 than to H2O. Thus, NH3 is a more suited probing molecule than H2O for the study of aziridine's adsorption. By sharp contrast and yet quite understandably, the adsorption of CO2 on M-HKUST-1 correlates rather poorly with that of NH3 and H2O (R2 is 0.0125 and 0.004, respectively) since the electronic features of CO2 (π-type) significantly differ from that of NH3 and H2O (σ-type) (Fig. 5 and 7). The detailed information about their adsorption energies is shown in Table S3 of the ESI.† Since CO2 itself is quite stable and versatile, it could act as a probing species for other π-type molecules.
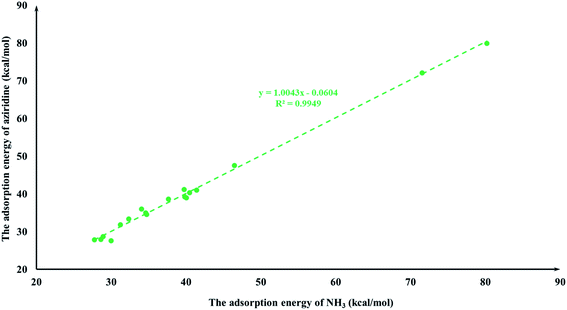 |
| | Fig. 4 The plot of the aziridine adsorption energies for M-HKUST-1 against the NH3 adsorption energy. | |
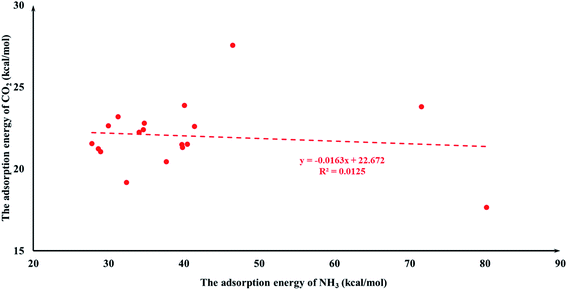 |
| | Fig. 5 The plot of CO2 adsorption energies for M-HKUST-1 against the NH3 adsorption energy. | |
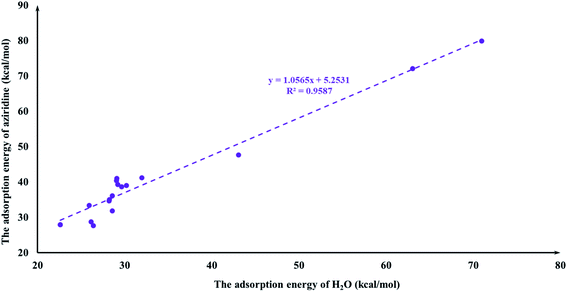 |
| | Fig. 6 The plot of the aziridine adsorption energies for M-HKUST-1 against the H2O adsorption energy. | |
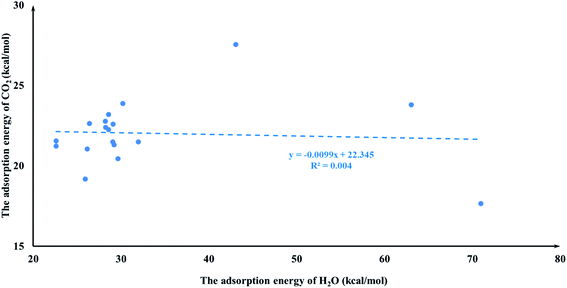 |
| | Fig. 7 The plot of the CO2 adsorption energies for M-HKUST-1 against the H2O adsorption energy. | |
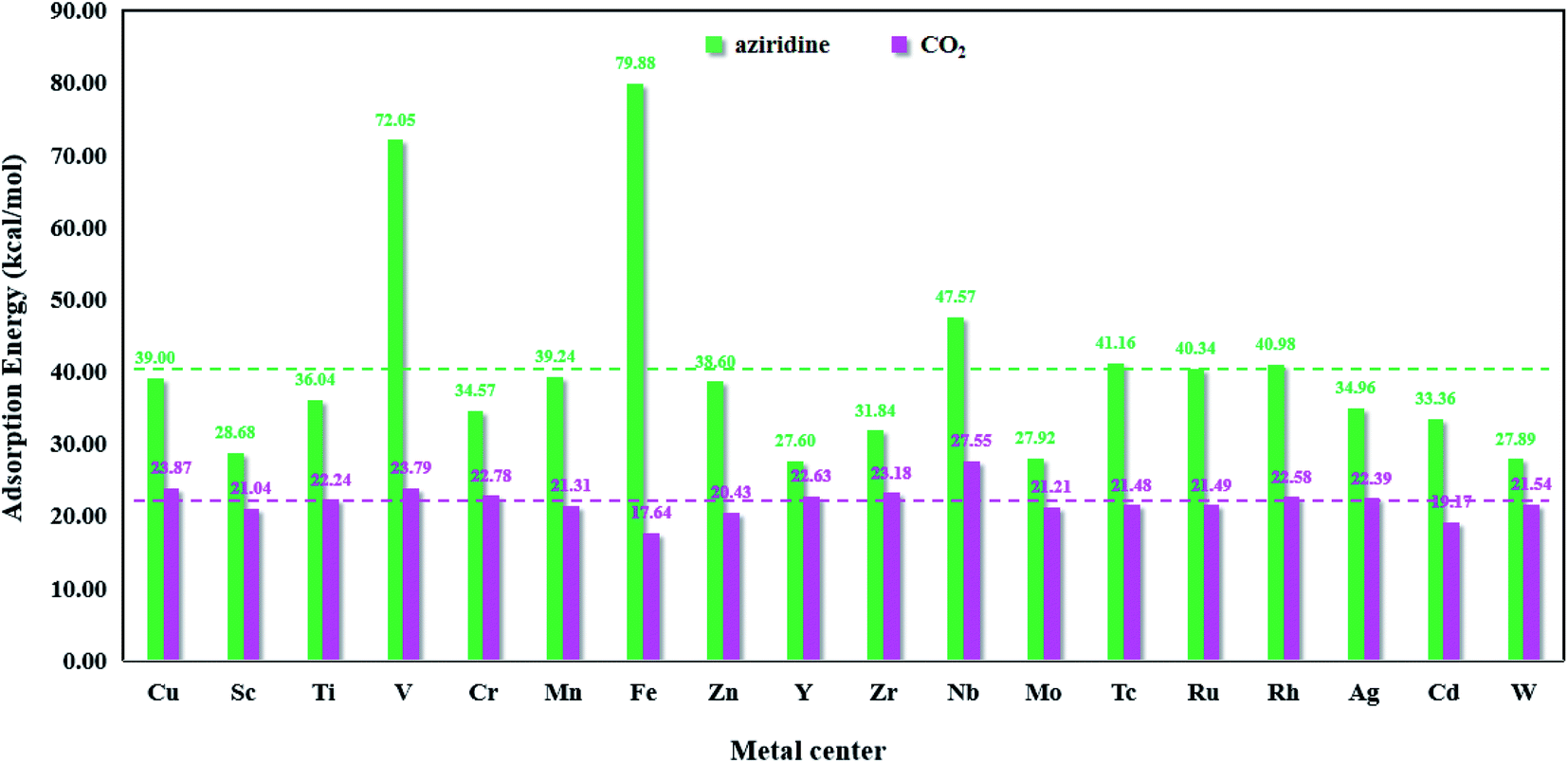
Fig. 8 presents the bar chart of aziridine and CO2 on M-HKUST-1. For the 18 M-HKUST-1 catalysts, the average adsorption values for aziridine and CO2 are shown by the red (22.02 kcal mol−1) and blue (40.09 kcal mol−1) dashed lines, respectively. It can be seen that the adsorption of CO2 does not vary much (17.64 to 27.55 kcal mol−1), whereas that of aziridine is altered dramatically (from 27.60 to 79.88 kcal mol−1) across different M-HKUST-1 systems. We can say that the adsorption of CO2 is much less sensitive to the exchanged metal-centers in M-HKUST-1 than that of aziridine. Fe-HKUST-1 and V-HKUST-1 are associated with the two largest adsorption energies, i.e., 79.88 and 72.05 kcal mol−1, respectively, possibly because Fe-HKUST-1 and V-HKUST-1 have the highest ability in attaching electrons from aziridine.
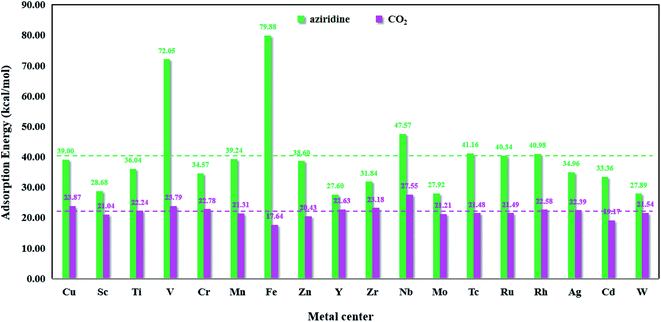 |
| | Fig. 8 The adsorption energies of aziridine and CO2 on M-HKUST-1. | |
3.3.2. Ring-opening.
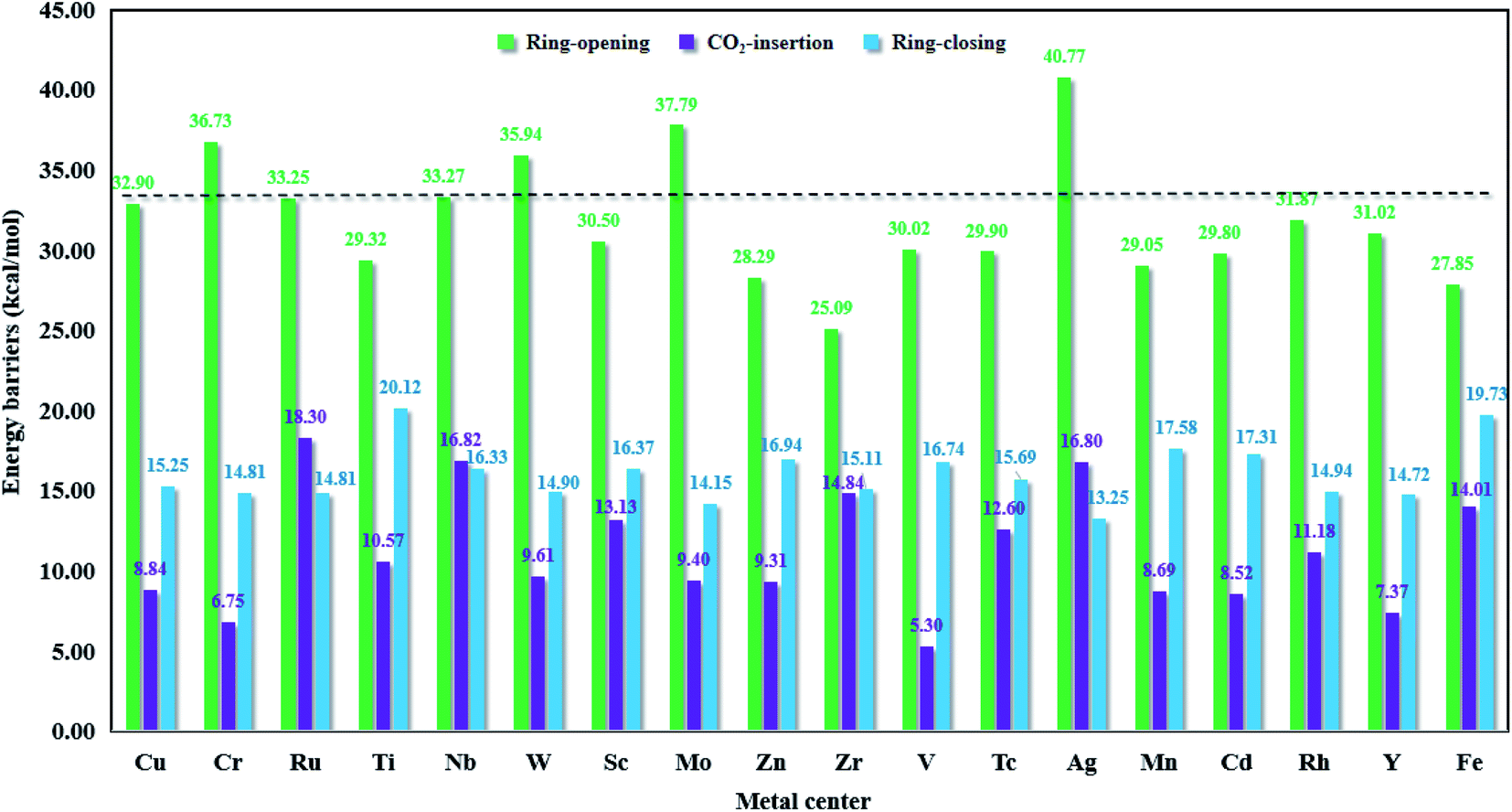
As shown in Fig. 9, we can see that for the whole cycloaddition reaction of CO2, the ring-opening (green bar) is the rate-determining step (i.e., the step with the highest activation energy), whose barrier is overwhelmingly higher than those of the CO2 insertion (purple bar) and ring-closing (blue bar) steps. For M = Ru, Nb and Ag, the barrier height of the CO2 insertion step is higher than that of the ring-closing step, whereas for the remaining metals, the ring-closing step has a higher barrier height than the CO2 insertion step. For simplicity, we would like to only discuss the ring-opening step.
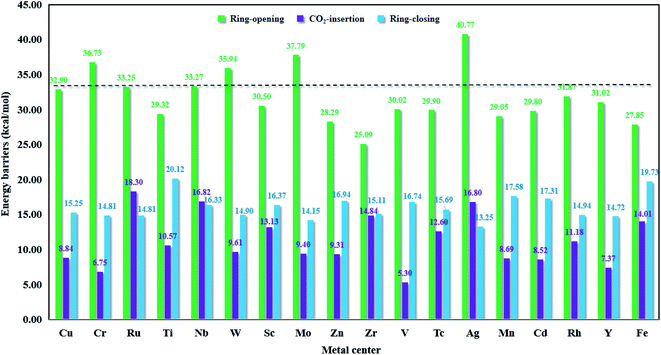 |
| | Fig. 9 The Gibbs free energy barriers in the key steps for M-HKUST-1 (M = Cu, Cr, Ru, Ti, Nb, W, Sc, Mo, Zn, Zr, V, Tc, Ag, Mn, Cd, Rh, Y and Fe). | |
The ring-opening energy barriers for different M-HKUST-1 systems increase as follows: Zr (25.09 kcal mol−1) < Fe (27.85) < Zn (28.29) < Mn (29.05) < Ti (29.32) < Cd (29.80) < Tc (29.90) < V (30.02) < Sc (30.50) < Y (31.02) < Rh (31.87) < Cu (32.09) < Ru (33.25) < Nb (33.27) < W (35.94) < Cr (36.73) < Mo (37.79) < Ag (40.77). It is of interest to compare the ring-opening activity of M-HKUST-1 (11 species were computationally studied with M = Mo, Cr, Ti, Cu, W, Sc, Ru, Zn, Cd, V and Fe)39 towards epoxide and aziridine. The ring-opening barrier of epoxide (by breaking C–O bond) is much lower than that of aziridine (by breaking C–N bond). This clearly contradicts with the prediction simply based on the bond dissociation energies of C–O (1076.63 kJ mol−1) and C–N (749.31 kJ mol−1).73 According to the general understanding, a stronger bond should be more difficult to break than a weaker bond, which would predict a more difficult ring-opening for epoxide than for aziridine. What is the cause? We are aware that both epoxide and aziridine are of the strongly ring-strained molecules. Regarding the rule of calculating ring-strain energy, we calculated cyclopropene, cyclopropane, cyclobutane, pure hydrogen-substituted epoxide and aziridine by using the homodesmotic reactions proposed by George et al.74 It should be noted that in the calculation process of the hydrocarbon ring-strain energies, in most cases, the contribution of the electron correlation and zero-point vibration energy cancel each other out.75,76 In order to keep consistent, the ring-strain energies were computed at the B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) level without including the zero-point vibration energy. As shown in Table S4 in the ESI,† one can see that the calculated ring-strain energies differ very little from those obtained in the literature (either experimental or theoretical),77,78 indicative of the suitability of the applied method for the ring-strain energy in this work. Notably, the ring-strain energy difference between the parent epoxide and aziridine (no substitution) is very close at 0.72 kcal mol−1 (the value is 0.4 kcal mol−1 in literature77,78). Upon methyl substitution, the ring-strain energy difference between epoxide and aziridine is increased to 1.47 kcal mol−1. This is understandable as methyl can better protect the tri-coordinate N than hydrogen, whereas the di-coordinate O-atom is naked. At the B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) level, the ring-strain energies of epoxide and aziridine were computed to be 25.15 and 23.68 kcal mol−1. This indicates a higher strain-induced activity for epoxide than for aziridine. Therefore, the larger ring-strain in epoxide counteracts the larger bond dissociation energy of C–O, totally resulting in a lower barrier height. The detailed information of the ring-strain energies can be shown in Table S4 in the ESI.†
3.4. Computational screening of M-HKUST-1 catalysts
In this section, in order to find more promising catalyst candidates, let us assess the catalytic activities of different M-HKUST-1 systems based on the involved rate-determining step barrier height. In Fig. 9, the energy barriers for the key step of each catalytic system obviously indicated that the metal centers of the M-HKUST-1/Br− catalysts have a major impact on the rate-determining energy barrier. We reasonably supposed that a higher-barrier rate-determining step might possess lower catalytic activity. In light of this assumption, we obtained the following catalytic activity sequence of the M-HKUST-1 series for CO2 cycloaddition with aziridines, i.e., Ag (40.77 kcal mol−1) > Mo (37.79) > Cr (36.73) > W (35.94) > Nb (33.27) > Ru (33.25) > Cu (32.90) > Rh (31.87) > Y (31.02) > Sc (30.50) > V (30.02) > Tc (29.90) > Cd (29.80) > Ti (29.32) > Mn (29.05) > Zn (28.29) > Fe (27.85) > Zr (25.09). We can find that M-HKUST-1 catalysts with M = Rh, Y, Sc, V, Tc, Cd, Ti, Mn, Zn, Fe and Zr metal centers possess superior catalytic activity to the parent Cu-HKUST-1 (32.90), which has been shown experimentally to have high catalytic activity in the CO2 chemical conversion. Through the computational screening, we obtained 11 M-HKUST-1 catalytic activities that were better than the original Cu-HKUST-1. For these 11 M-HKUST-1, we divided them into three cases. The first is that only the rate-determining step energy barrier (open loop energy barrier) is lower than the original Cu-HKUST-1 (i.e., Rh-HKUST-1, Sc-HKUST-1, Tc-HKUST-1, Ti-HKUST-1, Zn-HKUST-1 and Fe-HKUST-1). The second is that except for the energy barrier of the rate-determining step being lower than Cu-HKUST-1, one of the other two energy barriers is lower than Cu-HKUST-1, which are Zr-HKUST-1, V-HKUST-1, Cr-HKUST-1 and Mn-HKUST-1. The last case is that the three-step energy barrier is lower than Cu-HKUST-1 (i.e., only Y-HKUST-1). For each catalytic system, the energy barrier of the rate-determining step is much higher than that of the other two steps, so the rate-determining step plays a crucial role in the reaction rate of the whole reaction. In summary, Zr-HKUST-1 has the lowest rate-determining step energy barrier. For good catalysts, the adsorption energies usually are neither too strong nor too weak, indicating that a balanced adsorption energy energy is highly appealing. This is exactly what Zr-HKUST-1 does. Therefore, Zr-HKUST-1 has the best catalytic activity among the 18 selected catalytic systems, and can be synthesized in experiments in the future. We would like to note that the present study applied a cluster model. In future study, the large periodic model might reduce the adsorption energy. Moreover, in this work, our main task is to screen M-HKUST-1 with a certain catalytic potential, and we mainly consider the influence of the skeleton center (i.e., metal atoms) on the reaction energy barrier. The acid stability, alkali stability, thermal stability, light stability and hydrolysis stability of the catalyst will be studied in depth in future work. Up to now, six M-HKUST-1 (M = Mo,68 Cr,69 Cu,47 Fe,70 Ru,71 and Zn72) have been experimentally available. However, only the parent Cu-HKUST-1 has been used in the laboratory to catalyze the CO2 fixation by aziridines. Moreover, for the first time, eleven M-HKUST-1 (M = Rh, Y, Sc, V, Tc, Cd, Ti, Mn, Zn, Ag and Zr) were shown to exhibit better catalytic performance than Cu-HKUST-1.
4. Conclusions
Very recently, the MOF-based catalysts have been designed experimentally to convert CO2 to the high-valued oxazolidinones by the cycloaddition reactions with aziridines. To develop novel, structurally rich and catalytically better MOF-systems for the cycloaddition reactions of CO2 with aziridines, we carried out the first computational screening of a series of metal-substituted HKUST-1 (Ag, Mo, Cr, W, Nb, Ru, Rh, Y, Sc, V, Tc, Cd, Ti, Mn, Zn, Fe and Zr) MOFs based on the well-known Cu-HKUST-1 with the aid of tetrabutylammonium bromide (TBAB) as a co-catalyst. Using the rate-determining step (ring-opening) of each catalytic system as the screening criteria, we promisingly found that a total of 11 M-HKUST-1 systems, i.e., Rh (31.87 kcal mol−1), Y (31.02), Sc (30.50), V (30.02), Tc (29.90), Cd (29.80), Ti (29.32), Mn (29.05), Zn (28.29), Fe (27.85) and Zr (25.09), might possess superior catalytic ability to the parent Cu-HKUST-1 (32.90). Surely, Zr-HKUST-1 with the lowest ring-opening barrier strongly deserves to be tested in the near future.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2016YFB0701100), and the National Natural Science Foundation of China (No. 22073069 and 21773082). The authors would like show great attitude to the reviewers' insighful comments and suggestions to improve the manuscript.
References
- M. H. Beyzavi, C. J. Stephenson, Y. Liu, O. Karagiaridi, J. T. Hupp and O. K. Farha, Front. Energy Res., 2015, 2, 63 Search PubMed.
- P. García-Gutiérrez, R. M. Cuéllar-Franca, D. Reed, G. Dowson, P. Styring and A. Azapagic, Green Chem., 2019, 21, 4100–4114 Search PubMed.
- M. R. Allen, D. J. Frame, C. Huntingford, C. D. Jones, J. A. Lowe, M. Meinshausen and N. Meinshausen, Nature, 2009, 458, 1163–1166 Search PubMed.
- M. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 Search PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 Search PubMed.
- S. L. Hou, J. Dong, X. L. Jiang, Z. H. Jiao and B. Zhao, Angew. Chem., Int. Ed. Engl., 2019, 58, 577–581 Search PubMed.
- X. M. Kang, L. H. Yao, Z. H. Jiao and B. Zhao, Chem.–Asian J., 2019, 14, 3668–3674 Search PubMed.
- J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Today, 2009, 148, 221–231 Search PubMed.
- Y. Shi, S. Hou, X. Qiu and B. Zhao, Top. Curr. Chem., 2020, 378, 11 Search PubMed.
- H. L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304–1306 Search PubMed.
- Q. Yang, Q. Xu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed. Engl., 2016, 55, 3685–3689 Search PubMed.
- G. Zhang, H. Yang and H. Fei, ACS Catal., 2018, 8, 2519–2525 Search PubMed.
- T. A. Mukhtar and G. D. Wright, Chem. Rev., 2005, 105, 529–542 Search PubMed.
- L. Aurelio, R. T. Brownlee and A. B. Hughes, Chem. Rev., 2004, 104, 5823–5846 Search PubMed.
- M. R. Barbachyn and C. W. Ford, Angew. Chem., Int. Ed. Engl., 2003, 42, 2010–2023 Search PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 Search PubMed.
- T. Seki and A. Baiker, Chem. Rev., 2009, 109, 2409–2454 Search PubMed.
- Y. Du, Y. Wu, A.-H. Liu and L.-N. He, J. Org. Chem., 2008, 73, 4709–4712 Search PubMed.
- M. T. Hancock and A. R. Pinhas, Tetrahedron Lett., 2003, 44, 5457–5460 Search PubMed.
- H.-F. Jiang, J.-W. Ye, C.-R. Qi and L.-B. Huang, Tetrahedron Lett., 2010, 51, 928–932 Search PubMed.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Catal. Today, 2020, 356, 557–562 Search PubMed.
- A. Sudo, Y. Morioka, E. Koizumi, F. Sanda and T. Endo, Tetrahedron Lett., 2003, 44, 7889–7891 Search PubMed.
- A. Sudo, Y. Morioka, F. Sanda and T. Endo, Tetrahedron Lett., 2004, 45, 1363–1365 Search PubMed.
- P. Tascedda and E. Duñach, Chem. Commun., 2000, 449–450 Search PubMed.
- W. Y. Gao, M. Chrzanowski and S. Ma, Chem. Soc. Rev., 2014, 43, 5841–5866 Search PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2011, 112, 869–932 Search PubMed.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 Search PubMed.
- S. O. Odoh, C. J. Cramer, D. G. Truhlar and L. Gagliardi, Chem. Rev., 2015, 115, 6051–6111 Search PubMed.
- S. Yang, L. Peng, S. Bulut and W. L. Queen, Chemistry, 2019, 25, 2161–2178 Search PubMed.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 Search PubMed.
- C. S. Cao, Y. Shi, H. Xu and B. Zhao, Dalton Trans., 2018, 47, 4545–4553 Search PubMed.
- X.-M. Kang, Y. Shi, C.-S. Cao and B. Zhao, Sci. China: Chem., 2019, 62, 622–628 Search PubMed.
- A. C. Kathalikkattil, R. Roshan, J. Tharun, R. Babu, G. S. Jeong, D. W. Kim, S. J. Cho and D. W. Park, Chem. Commun., 2016, 52, 280–283 Search PubMed.
- X. Wang, W.-Y. Gao, Z. Niu, L. Wojtas, J. A. Perman, Y.-S. Chen, Z. Li, B. Aguila and S. Ma, Chem. Commun., 2018, 54, 1170–1173 Search PubMed.
- H. Xu, X. F. Liu, C. S. Cao, B. Zhao, P. Cheng and L. N. He, Adv. Sci., 2016, 3, 1600048 Search PubMed.
- T.-d. Hu and Y.-h. Ding, Organometallics, 2020, 39, 505–515 Search PubMed.
- L. Xu, G. Fang, J. Tao, Z. Ye, S. Xu and Z. Li, ACS Catal., 2018, 8, 11910–11925 Search PubMed.
- L. Xu, G. Fang, Y. Yu, Y. Ma, Z. Ye and Z. Li, Mol. Catal., 2019, 467, 1–8 Search PubMed.
- T.-d. Hu, Y. Jiang and Y.-h. Ding, J. Mater. Chem. A, 2019, 7, 14825–14834 Search PubMed.
- S. Ehrling, M. Mendt, I. Senkovska, J. D. Evans, V. Bon, P. Petkov, C. Ehrling, F. Walenszus, A. Pöppl and S. Kaskel, Chem. Mater., 2020, 32, 5670–5681 Search PubMed.
- A. Kirchon, P. Zhang, J. Li, E. A. Joseph, W. Chen and H. C. Zhou, ACS Appl. Mater. Interfaces, 2020, 12, 9292–9299 Search PubMed.
- P. Krokidas, S. Moncho, E. N. Brothers, M. Castier and I. G. Economou, Phys. Chem. Chem. Phys., 2018, 20, 4879–4892 Search PubMed.
- M. R. Momeni and C. J. Cramer, J. Phys. Chem. C, 2019, 123, 15157–15165 Search PubMed.
- M. A. Syzgantseva, C. P. Ireland, F. M. Ebrahim, B. Smit and O. A. Syzgantseva, J. Am. Chem. Soc., 2019, 141, 6271–6278 Search PubMed.
- A. S. Yasin, J. Li, N. Wu and T. Musho, Phys. Chem. Chem. Phys., 2016, 18, 12748–12754 Search PubMed.
- You, Y. Liu, J. D. Howe, D. Tang and D. S. Sholl, J. Phys. Chem. C, 2018, 122, 27486–27494 Search PubMed.
- A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 Search PubMed.
- S. Ketrat, T. Maihom, S. Wannakao, M. Probst, S. Nokbin and J. Limtrakul, Inorg. Chem., 2017, 56, 14005–14012 Search PubMed.
- N. Planas, A. L. Dzubak, R. Poloni, L. C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 Search PubMed.
- B. Supronowicz, A. Mavrandonakis and T. Heine, J. Phys. Chem. C, 2013, 117, 14570–14578 Search PubMed.
- P. Verma, X. Xu and D. G. Truhlar, J. Phys. Chem. C, 2013, 117, 12648–12660 Search PubMed.
- J. G. Vitillo, A. Bhan, C. J. Cramer, C. C. Lu and L. Gagliardi, ACS Catal., 2019, 9, 2870–2879 Search PubMed.
- K. Yu, K. Kiesling and J. R. Schmidt, J. Phys. Chem. C, 2012, 116, 20480–20488 Search PubMed.
- B. Supronowicz, A. Mavrandonakis and T. Heine, J. Phys. Chem. C, 2015, 119, 3024–3032 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 Search PubMed.
- D. Liu and C. Zhong, J. Phys. Chem. Lett., 2009, 1, 97–101 Search PubMed.
- T. Pham, K. A. Forrest, J. Eckert, P. A. Georgiev, A. Mullen, R. Luebke, A. J. Cairns, Y. Belmabkhout, J. F. Eubank, K. McLaughlin, W. Lohstroh, M. Eddaoudi and B. Space, J. Phys. Chem. C, 2013, 118, 439–456 Search PubMed.
- T. Pham, K. A. Forrest, P. Nugent, Y. Belmabkhout, R. Luebke, M. Eddaoudi, M. J. Zaworotko and B. Space, J. Phys. Chem. C, 2013, 117, 9340–9354 Search PubMed.
- Y. Wu, D. Liu, H. Chen, Y. Qian, H. Xi and Q. Xia, Chem. Eng. Sci., 2015, 123, 1–10 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4166 Search PubMed.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- B. D. Montejo-Valencia, Y. J. Pagán-Torres, M. M. Martínez-Iñesta and M. C. Curet-Arana, ACS Catal., 2017, 7, 6719–6728 Search PubMed.
- T.-d. Hu, Y.-w. Sun and Y.-h. Ding, J. CO2 Util., 2018, 28, 200–206 Search PubMed.
- L. Grajciar, A. D. Wiersum, P. L. Llewellyn, J.-S. Chang and P. Nachtigall, J. Phys. Chem. C, 2011, 115, 17925–17933 Search PubMed.
- J. Kim, S.-N. Kim, H.-G. Jang, G. Seo and W.-S. Ahn, Appl. Catal., A, 2013, 453, 175–180 Search PubMed.
- E. E. Macias, P. Ratnasamy and M. A. Carreon, Catal. Today, 2012, 198, 215–218 Search PubMed.
- M. Kramer, U. Schwarz and S. Kaskel, J. Mater. Chem., 2006, 16, 2245–2248 Search PubMed.
- L. J. Murray, M. Dinca, J. Yano, S. Chavan, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2010, 132, 7856–7857 Search PubMed.
- L. Xie, S. Liu, C. Gao, R. Cao, J. Cao, C. Sun and Z. Su, Inorg. Chem., 2007, 46, 7782–7788 Search PubMed.
- O. Kozachuk, K. Yusenko, H. Noei, Y. Wang, S. Walleck, T. Glaser and R. A. Fischer, Chem. Commun., 2011, 47, 8509–8511 Search PubMed.
- J. Lu, A. Mondal, B. Moulton and M. J. Zaworotko, Angew. Chem., Int. Ed., 2001, 40, 2113–2116 Search PubMed.
- B. Ruscic, D. Feller and K. A. Peterson, Theor. Chem. Acc., 2013, 133, 1415–1426 Search PubMed.
- P. George, M. Trachtman, C. W. Bock and A. M. Brett, Tetrahedron, 1976, 32, 317–323 Search PubMed.
- D. Barić and Z. B. Maksić, Theor. Chem. Acc., 2005, 114, 222–228 Search PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 1998, 120, 4450 Search PubMed.
-
E. V. Anslynand and D. E. Dougherty, Modern Physical Organic Chemistry, Edwards Brothers, Inc., 2006, p. 850 Search PubMed.
- H. T. Lacey, Ind. Eng. Chem., 1954, 46, 1827–1835 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00150g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab