
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize-tunable and stable cesium lead-bromide perovskite nanocubes with near-unity photoluminescence quantum yield†
Roberto
Grisorio
 *a,
Daniele
Conelli
a,
Elisabetta
Fanizza
bc,
Marinella
Striccoli
c,
Davide
Altamura
d,
Cinzia
Giannini
d,
Ignazio
Allegretta
e,
Roberto
Terzano
e,
Mihai
Irimia-Vladu
f,
Nicola
Margiotta
b and
Gian Paolo
Suranna
ag
*a,
Daniele
Conelli
a,
Elisabetta
Fanizza
bc,
Marinella
Striccoli
c,
Davide
Altamura
d,
Cinzia
Giannini
d,
Ignazio
Allegretta
e,
Roberto
Terzano
e,
Mihai
Irimia-Vladu
f,
Nicola
Margiotta
b and
Gian Paolo
Suranna
ag
aDipartimento di Ingegneria Civile, Ambientale, del Territorio, Edile e di Chimica (DICATECh), Politecnico di Bari, Via Orabona 4, 70125 Bari, Italy. E-mail: roberto.grisorio@poliba.it
bDipartimento di Chimica, Università degli Studi di Bari, “, A. Moro, ”, Via Orabona 4, 70126 Bari, Italy
cCNR, Istituto per i Processi Chimico Fisici, UOS Bari, Via Orabona 4, 70126 Bari, Italy
dCNR, Istituto di Cristallografia, via Amendola 122/O, Bari 70126, Italy
eDipartimento di Scienze del Suolo, della Pianta e degli Alimenti, Università degli Studi di Bari “Aldo Moro”, Via G. Amendola 165/A, 70126 Bari, Italy
fInstitute of Physical Chemistry, Linz Institute of Organic Solar Cells, Johannes Kepler University Linz, Altenberger Straße 69, 4040 Linz, Austria
gCNR NANOTEC, Istituto di Nanotecnologia, Via Monteroni, 73100 Lecce, Italy
First published on 11th May 2021
Abstract
Stable cesium lead bromide perovskite nanocrystals (NCs) showing a near-unity photoluminescence quantum yield (PLQY), narrow emission profile, and tunable fluorescence peak in the green region can be considered the ideal class of nanomaterials for optoelectronic applications. However, a general route for ensuring the desired features of the perovskite NCs is still missing. In this paper, we propose a synthetic protocol for obtaining near-unity PLQY perovskite nanocubes, ensuring their size control and, consequently, a narrow and intense emission through the modification of the reaction temperature and the suitable combination ratio of the perovskite constituting elements. The peculiarity of this protocol is represented by the dissolution of the lead precursor (PbBr2) as a consequence of the exclusive complexation with the bromide anions released by the in situ SN2 reaction between oleylamine (the only surfactant introduced in the reaction mixture) and 1-bromohexane. The obtained CsPbBr3 nanocubes exhibit variable size (ranging from 6.7 ± 0.7 nm to 15.2 ± 1.2 nm), PL maxima between 505 and 517 nm, and near-unity PLQY with a narrow emission profile (fwhm of 17–19 nm). Additionally, the NCs synthesized with this approach preserve their high PLQYs even after 90 days of storage under ambient conditions, thus displaying a remarkable optical stability. Through the rationalization of the obtained results, the proposed synthetic protocol provides a new ground for the direct preparation of differently structured perovskite NCs without resorting to any additional post-synthetic treatment for improving their emission efficiency and stability.
Introduction
Since the first report by Kovalenko and coworkers in 2015,1 all-inorganic cesium lead-halide perovskite nanocrystals (NCs) with the generic formula CsPbX3 (where X represents Cl, Br, I or a mixture thereof) have attracted an enormous interest from the scientific community.2–4 Lead-halide perovskite NCs combine high photoluminescence quantum yields (PLQYs) with narrow emission profiles and tunable fluorescence related to the halide composition5 or to the morphology.6 On the other hand, although lead-halide perovskite NCs can tolerate a relatively large concentration of defects without compromising their semiconducting properties,7 the achievement of near-unity PLQYs is severely undermined by halide vacancies8 prevalently placed at their surface.9–11In order to cope with this issue, it has recently been proposed, in the course of the synthesis, to separately combine the precursors of the three elements (Cs, Pb and X) comprising the CsPbX3 NCs, thus allowing to establish the proper halide-rich synthetic conditions,12 difficultly obtained by using the common approach based on the use of PbX2 salts as both lead and halide sources.13 However, differently from the usual post-synthetic treatments14–16 and the doping processes17,18 used to improve the emission efficiencies of the as-synthesized nanoparticles, rare are the reports of the systematic achievement of near-unity PLQY perovskite NCs directly from their synthesis.19
In the three-precursor synthetic approach, the choice of the appropriate halide source proved to be the key for directly obtaining perovskite NCs endowed with emission efficiencies approaching unity. Near-unity PLQY CsPbBr3 NCs were initially prepared by utilizing the bromide anions released by the reaction between an alternative halide source (benzoyl bromide) and the capping ligands.20,21 Subsequent efforts were directed toward the application of the three-precursor synthetic method to a compositional and morphological variety of perovskite NCs endowed with near-unity PLQY. Among these, noteworthy are (i) CsPbX3 perovskite nanocubes (X = Cl, Br, and I) prepared using the corresponding halide ammonium salts at high temperatures;22 (ii) APbBr3 perovskite nanocubes (A = cesium, methylammonium and formamidinium) obtained with N-bromosuccinimide;23 and (iii) CsPbBr3 nanorods synthesized using 1-bromohexane as the halide source.24
To date, the most popular hot-injection synthetic approaches required the presence of carboxylic acids as the capping ligands and also necessary to promote the dissolution of the lead precursors (as the corresponding carboxylates) in apolar solvents. Long-chain carboxylates, however, compete with bromide for the coordination to lead, causing the introduction of halide vacancies in perovskite NCs.25 The observed variability in generating defect-free (and consequently near-unity PLQY) perovskite NCs can actually be rationalized by considering the necessity of establishing the proper balance between the relative quantities of the independent perovskite constituting elements and the capping ligands.26 In addition, a general synthetic route for ensuring the size modulation of perovskite nanocubes while simultaneously preserving the achievement of near-unity PLQYs is still missing.
It was established that the factors which could determine the modulation of the NC dimensions are the cesium/lead molar ratio and the reaction temperature.27 However, it has been demonstrated that the modification of the former parameter under specific conditions could promote the formation of lead-poor perovskite phases28,29 or introduce a non-innocent additional quantity of aliphatic carboxylates required for dissolving the cesium precursor (typically Cs2CO3) in apolar solvents.30 At the same time, relatively low reaction temperatures (generally comprised between 90 and 140 °C) are known to induce an anisotropic growth of the forming nanoparticle with a dramatic effect on their optical properties.31,32
With these premises, we excogitated a new synthetic protocol avoiding carboxylic acids as dissolving agents of the proper lead precursor (PbBr2). The synthesis involves the coordination of bromide ions released by the reaction of oleylamine and 1-bromohexane with the formation of bromoplumbate species. We ascertained that the simultaneous presence of primary and secondary amines ensures the phase purity, preservation of the NC cuboidal-shape as well as the sufficient passivation of the nanomaterials obtained employing a wide range of reaction temperatures and cesium/lead molar ratios (Scheme 1). The size modulation of the perovskite nanocubes was accompanied with a size-dependent, near-unity QY and long-term stable emission. We are confident that our findings could fill a major gap in the synthetic methodologies for obtaining perovskite NCs, paving the way for deeper knowledge in this promising research field.
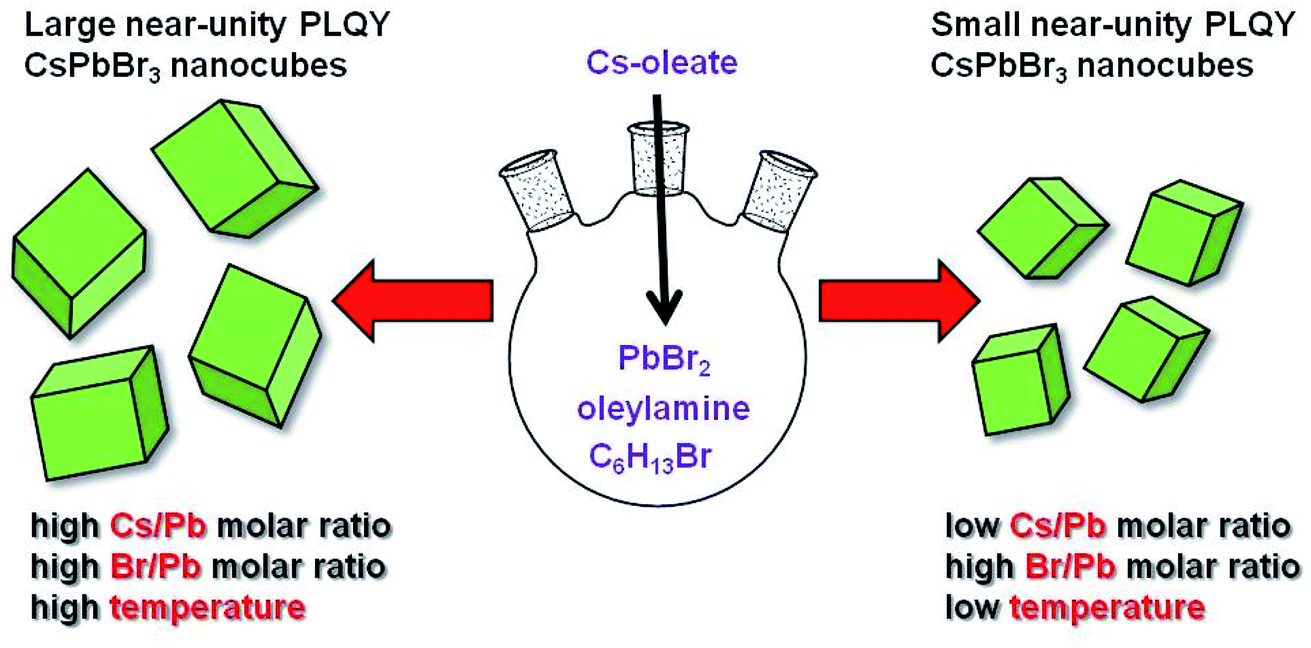 | ||
| Scheme 1 Schematic representation of the reaction conditions leading to obtaining near-unity PLQY perovskite nanocubes with variable sizes. | ||
Results and discussion
Synthesis and characterization
In order to find the suitable conditions for obtaining near-unity PLQY NCs, a first set of experiments was carried out by mixing PbBr2 and 1-bromohexane (1.0, 3.0 or 6.0 equivalents with respect to lead) in the presence of oleylamine (8.0 equivalents with respect to lead) as the surfactant and 1-octadecene (ODE) as a non-coordinating, high-boiling point solvent. The obtained mixtures were heated at 160 °C for 30 min before the hot-injection of the determined quantity of the cesium oleate solution previously prepared (Cs/Pb = 1/4). The resulting perovskite NCs were isolated by centrifugation and their optical properties were preliminarily studied (Fig. 1S†). Initially, we focused our attention on the emission efficiencies of the obtained NCs. We ascertained a remarkable increase of the PLQYs as a function of the corresponding C6H13Br/PbBr2 molar ratio used (Fig. 1A), reaching the highest value (∼100%) for the NCs synthesized in the presence of 6.0 equivalents of 1-bromohexane with respect to lead. These results suggest that the strong bromide excess under these peculiar reaction conditions is the sine qua non requirement for preventing the defect formation in our perovskite NCs.33,34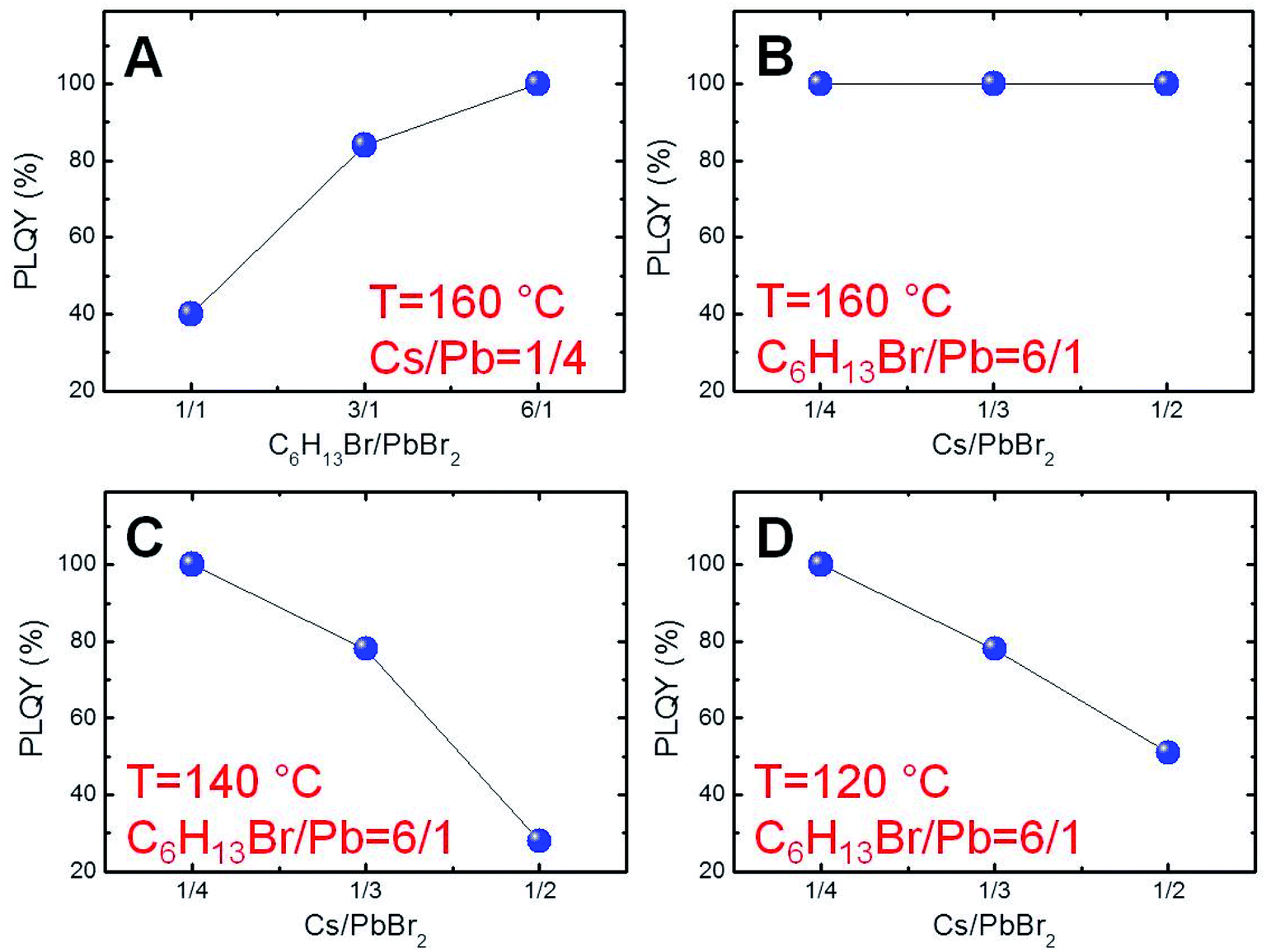 | ||
| Fig. 1 (A) Plot of perovskite NC PLQY as a function of the C6H13Br/PbBr2 molar ratio employed in the synthesis carried out at 160 °C with a Cs/Pb molar ratio of 1/4. (B) Plot of the perovskite NC PLQY with the Cs/Pb molar ratio employed in the synthesis carried out at 160 °C with C6H13Br/Pb = 6/1. Plot of the perovskite NC PLQY with a Cs/Pb molar ratio employed in the synthesis carried out at 140 °C (C) and 120 °C (D) with C6H13Br/Pb = 6/1. | ||
On these bases, we explored the possibility of adjusting the synthetic parameters for modulating the size of the perovskite nanoparticles without precluding the achievement of near-unity PLQY NCs. To this aim, we initially investigated the effect of Cs/Pb molar ratios on the emission efficiency of the resulting NCs under reaction conditions dictated by a strong bromide excess (C6H13Br/PbBr2 = 6.0 mol/mol) at 160 °C. Significantly, we recorded near-unity PLQYs also for the nanoparticles synthesized with higher Cs/Pb molar ratios (i.e., 1/3 or 1/2, as reported in Fig. 1B). Subsequently, in order to evaluate the effect of the reaction temperature, we repeated the same approach for the reactions carried out at 140 °C and 120 °C (Fig. 1C and D, respectively). In these cases, however, near-unity PLQYs were measured only for the lowest Cs/Pb molar ratio (1/4), thus obtaining five different experimental conditions ensuring near-unity PLQY perovskite NCs.
Furthermore, we deemed it worthwhile to investigate the emission properties of the obtained NCs as thin films to verify whether the high PLQY exhibited by these samples in solution was also preserved in the solid state. As a proof of concept, we measured the fluorescence of selected nanoparticles (T = 160 °C and Cs/Pb = 1/4) as neat films. As reported in Fig. 4S,† the PL maximum of the sample in the solid state was observed to be slightly red-shifted with respect to that recorded in solution, exhibiting a remarkable PLQY (80%) and a narrow emission profile (fwhm = 16 nm).
We gained insight into the morphology of the synthesized nanoparticles by transmission electron microscopy (TEM) measurements. Focusing on the near-unity PLQY NCs of the series, we evidenced the formation of cubic shapes independently of the reaction temperature and the Cs/Pb molar ratio (Fig. 2A–E). The NCs' edge lengths (measured by TEM) were 9.7 ± 1.4 nm (Cs/Pb = 1/4), 13.4 ± 1.4 nm (Cs/Pb = 1/3), and 15.2 ± 1.2 nm (Cs/Pb = 1/2) for the near-unity PLQY nanocubes synthesized at 160 °C. It can thus be concluded that the higher the Cs/Pb molar ratio, the larger the nanocubes formed at 160 °C and the narrower their size distribution (Fig. 3A). The same behaviour can also be deduced for the nanoparticles synthesized at 140 °C and 120 °C (Fig. 2S and 3S,† respectively), but if we consider only the NCs synthesized with the lowest Cs/Pb molar ratio, the measured edge lengths are 9.7 ± 1.4 nm (T = 160 °C), 8.6 ± 0.9 nm (T = 140 °C), and 6.7 ± 0.7 nm (T = 120 °C). Therefore, our synthetic protocol also allowed the size control of near-unity PLQY nanocubes through temperature modulation (Fig. 3B).
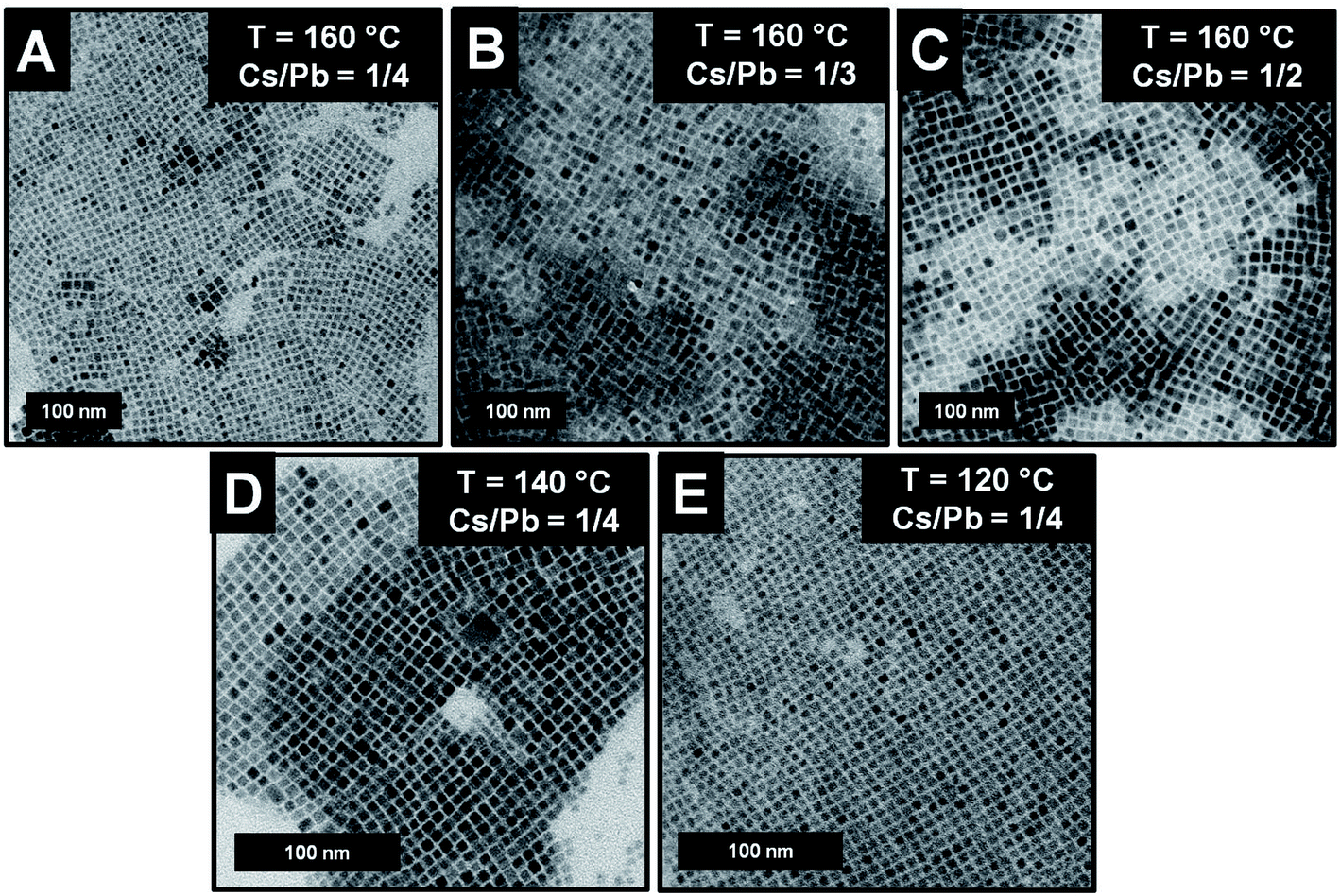 | ||
| Fig. 2 (A–E) TEM images of the NCs exhibiting a near-unity PLQY and obtained under different reaction conditions. | ||
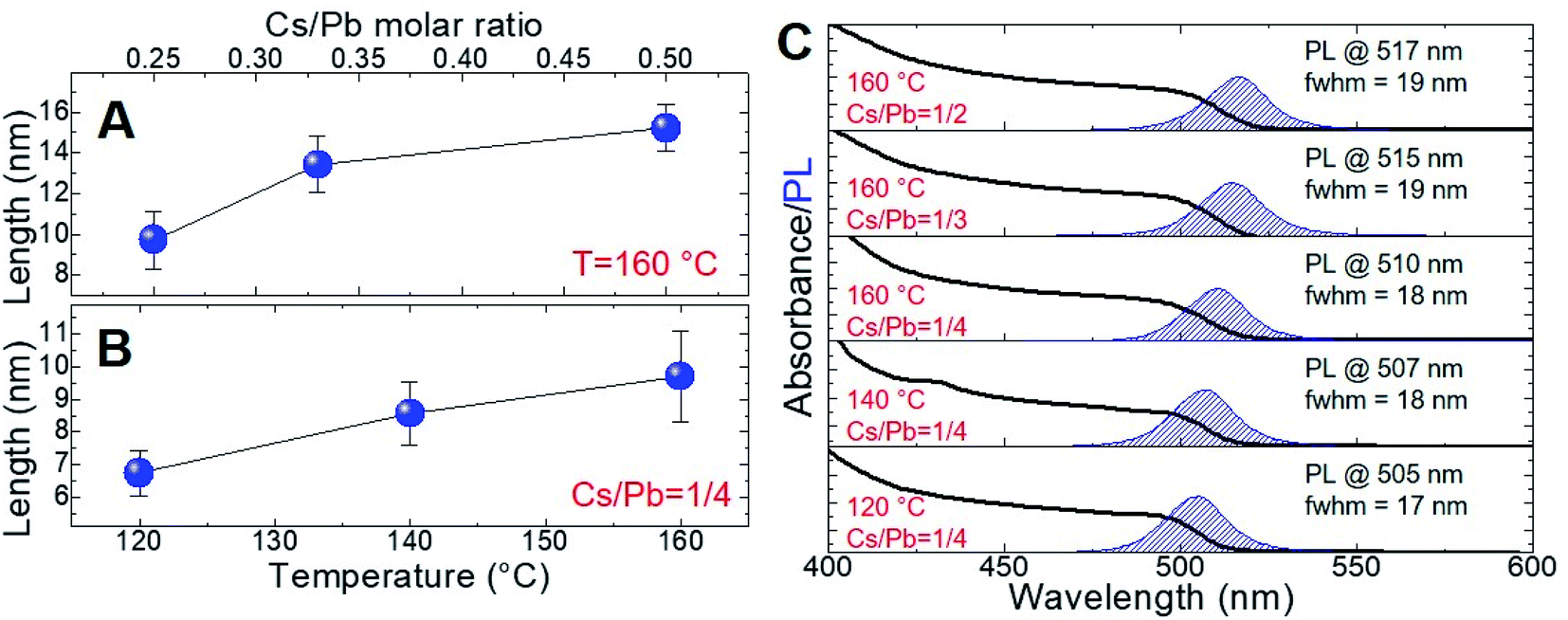 | ||
| Fig. 3 (A) Behaviour of the nanocube length (deduced by TEM measurements) of the NCs synthesized at 160 °C with different Cs/Pb molar ratios. (B) Behaviour of the nanocube length (deduced by TEM measurements) of the NCs synthesized with a Cs/Pb molar ratio of 1/4 at different temperatures. (C) UV-vis and PL spectra of the same nanocubes recorded in cyclohexane (only shown for the sample with near-unity PLQY). | ||
In addition, the size modulation of the near-unity PLQY nanocubes also determined the wavelength variation of the corresponding PL peak, as reported in Fig. 3C. For the reactions carried out at 160 °C, a red-shift of the PL maximum was observed (510 nm → 517 nm) upon the increase of the Cs/Pb molar ratio in the relevant NC synthesis. Conversely, the decrease of the reaction temperature caused a blue-shift of the corresponding PL maximum (510 nm → 505 nm) of the near-unity PLQY NCs synthesized with the same Cs/Pb molar ratio. Hence, the fluorescence behaviour of our near-unity PLQY NCs allowed to cover a considerable portion of the green region, preserving the excellent emission efficiencies (PLQY = 100%). Furthermore, the PL profiles were remarkably narrow for all near-unity PLQY NCs (fwhm = 17–19 nm), making them particularly suited for optoelectronic applications.35
Next, we investigated their elemental composition by field emission gun scanning electron microscopy (FEG-SEM) coupled with energy dispersive X-ray spectroscopy (EDX). The observed Cs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb:Br ratios were 1.2:1.0:3.0 (T = 160 °C and Cs/Pb = 1/4), 1.2:1.0:3.0 (T = 160 °C and Cs/Pb = 1/3), 1.2:1.0:3.1 (T = 160 °C and Cs/Pb = 1/2), 0.9:1.0:2.9 (T = 140 °C and Cs/Pb = 1/4), and 0.9:1.0:2.7 (T = 120 °C and Cs/Pb = 1/4). Therefore, the elemental compositions of our NCs with these sizes are not compatible with a bromide-rich surface,36 notwithstanding the excess of the halide source (1-bromohexane) used in the reaction mixture. By contrast, the observed values are in good agreement with the [CsPbBr3](PbBr2)k{CsBr}n structure consisting of a CsPbBr3 core exposing a PbBr2 inner shell (k) and residual monovalent cations (Cs+) and anions (Br−) of the original outer shell of CsBr (n).36 At the same time, the partial consumption of oleylamine in the SN2 reaction (vide infra) reduces the quantity of primary amines available in the reaction mixture for driving the transformation of CsPbBr3 to Cs4PbBr6 NCs through a dissolution and recrystallization process exclusively promoted by the primary amines.37,38
:Pb:Br ratios were 1.2:1.0:3.0 (T = 160 °C and Cs/Pb = 1/4), 1.2:1.0:3.0 (T = 160 °C and Cs/Pb = 1/3), 1.2:1.0:3.1 (T = 160 °C and Cs/Pb = 1/2), 0.9:1.0:2.9 (T = 140 °C and Cs/Pb = 1/4), and 0.9:1.0:2.7 (T = 120 °C and Cs/Pb = 1/4). Therefore, the elemental compositions of our NCs with these sizes are not compatible with a bromide-rich surface,36 notwithstanding the excess of the halide source (1-bromohexane) used in the reaction mixture. By contrast, the observed values are in good agreement with the [CsPbBr3](PbBr2)k{CsBr}n structure consisting of a CsPbBr3 core exposing a PbBr2 inner shell (k) and residual monovalent cations (Cs+) and anions (Br−) of the original outer shell of CsBr (n).36 At the same time, the partial consumption of oleylamine in the SN2 reaction (vide infra) reduces the quantity of primary amines available in the reaction mixture for driving the transformation of CsPbBr3 to Cs4PbBr6 NCs through a dissolution and recrystallization process exclusively promoted by the primary amines.37,38
Mechanistic insights
In order to rationalize the formation of near-unity PLQY NCs under these conditions, we gained insights into the process leading to the dissolution of the lead precursor by analyzing the absorption spectrum of the mixture composed by PbBr2, 1-bromohexane (6.0 equivalents with respect to lead) and oleylamine in 1-octadecene (ODE) immediately before the cesium introduction (Fig. 4A) for the NC assembly. The necessity of unveiling the details of this process is correlated to the fact that our synthetic protocol rules out the presence of carboxylic acids typically required for the dissolution of the lead precursors in apolar solvents in conventional preparations of all-inorganic perovskite NCs.39,40Fig. 4A shows that the UV-vis spectrum of the mixture obtained after the incubation period (necessary for the lead dissolution) is characterized by several absorption bands, which could be ascribed to bromoplumbate species.24,41 In the course of the incubation stage, it is reasonable to suppose that the bromoplumbate species are generated as a consequence of the reactions given below:| C18H35NH2 + C6H13Br → C18H35(C6H13)NH2Br | (1) |
| nC18H35(C6H13)NH2Br + PbBr2 → [C18H35(C6H13)NH2]nPbBr2+n | (2) |
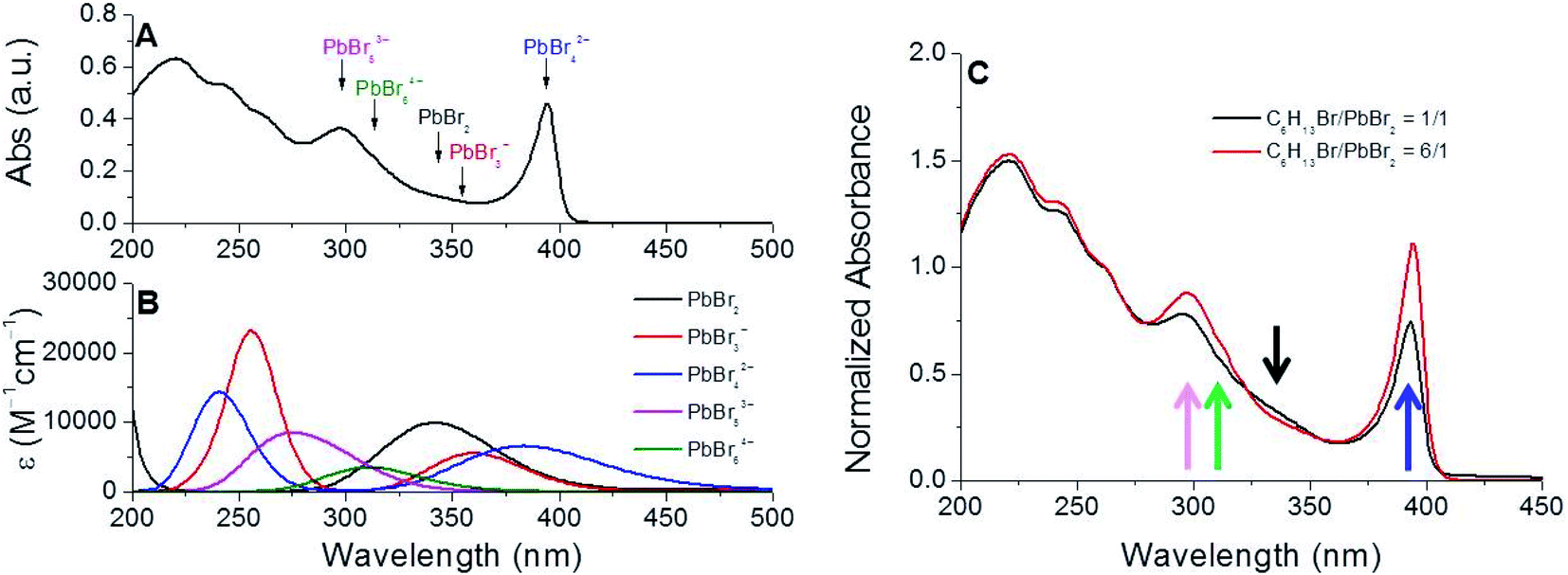 | ||
| Fig. 4 (A) UV-vis spectrum (cyclohexane) of an aliquot of the reaction mixture composed of PbBr2, 1-bromohexane (6.0 equiv. with respect to lead), and oleylamine (8.0 equiv. with respect to lead) in ODE at 160 °C for 30 min. (B) Calculated absorption spectra of the bromoplumbate species at the B3LYP/LANL2DZ level of theory including solvent effects (CPCM, cyclohexane). (C) Normalized (λ = 250 nm) absorption spectra of the reaction mixture at different C6H13Br/PbBr2 molar ratios, suggesting the relative increase of the concentration of highly coordinated bromoplumbate species (PbBr42−, PbBr53− and PbBr64−) with respect to PbBr3− and PbBr2. | ||
In this scenario, the bromide ions released by the SN2 reaction between oleylamine and 1-bromohexane42 should be available for the lead coordination of the PbBr2 precursor, thus generating the corresponding bromoplumbate species soluble in an apolar solvent owing to their organic countercations.
The association of each peak in this absorption spectrum with the plausible bromoplumbate species present in the reaction mixture was determined by simulating its vertical electronic transitions with time-dependent density functional theory (TDDFT) calculations. As shown in Fig. 4B, the theoretical absorption profiles of the plausible bromoplumbate species resulted to be strongly dependent on the coordination state of the lead atom. The accuracy of our theoretical calculations was validated by comparing the experimental value of the absorption maximum exhibited by the Cs4PbBr6 NCs (λmax = 314 nm in cyclohexane)43 with the simulated vertical transition attributable to the PbBr64− species (λmax = 312 nm in cyclohexane).
With this knowledge, we have been able to ascribe the narrow absorption band (λmax = 394 nm, Fig. 4A) of the reaction mixture to the PbBr42− species, showing a peculiar vertical electronic transition at λmax = 385 nm. Furthermore, the theoretical investigations revealed that higher coordinated bromoplumbate species (PbBr53−) are characterized by vertical transitions occurring at shorter wavelengths (λ = 275 nm) than PbBr3− and PbBr2 species, exhibiting calculated absorption transitions at λ = ∼350 nm. The UV-vis spectrum of the reaction mixture measured at the end of the incubation stage was characterized by structured absorption bands occurring in the wavelength range predicted by theoretical calculations. Consequently, we can reasonably consider the formation of all bromoplumbate species as the necessary event for the solubilization of the lead precursor. The distribution of the differently coordinated bromoplumbate species was influenced by the variation of the C6H13Br/PbBr2 molar ratio in the reaction mixture: the relative increase of signals attributable to the highly coordinated bromoplumbate species was in fact observed under conditions imposing a strong halide excess (Fig. 5C).
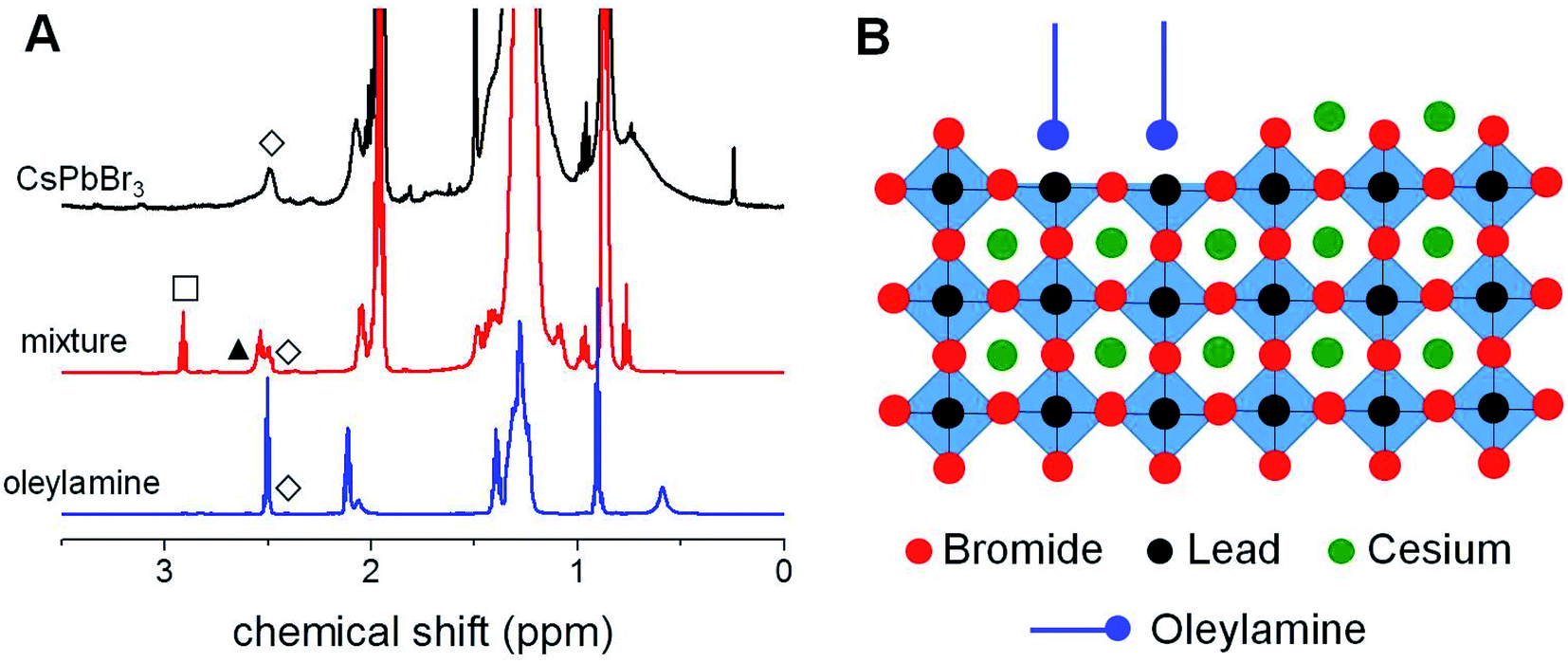 | ||
| Fig. 5 (A) Comparison between the 1H-NMR spectra (benzene-d6) of CsPbBr3 NCs, the reaction mixture constituted by PbBr2, oleylamine and 1-bromohexane in ODE at 160 °C for 30 min and pure oleylamine. The plot highlights the characteristic resonances of the secondary amine (–CH2NH–, ▲), oleylamine (–CH2NH2, ◊) and residual 1-bromohexane (–CH2Br, □). (B) Schematic representation of the NC surface. | ||
On the basis of these experimental evidences, the reaction pathway leading to the formation of CsPbBr3 NCs can be divided into two distinct stages in which the formation of the Pb–Br bonds anticipates the assembly of the tridimensional perovskite structure induced by the cesium intercalation between the formed PbBr64− octahedra. It is therefore reasonable to presume that, after the nucleation stage,44,45 the NC growth could proceed through the alternate addition of the different bromoplumbate species and cesium cations to the reactive nanoparticle facets. The eventual bromide surplus is removed from the surface by the action of the organic cations. The larger availability of highly coordinated bromoplumbate species involved in the crystallization process prevents the formation of halide vacancies in the final perovskite structure, thus explaining the achievement of near-unity PLQY NCs in the reactions characterized by high C6H13Br/PbBr2 molar ratios. However, the use of a large amount of the cesium precursor inevitably introduces a competitor of the bromide anions for the lead complexation (the oleate ion), which can be considered effectively responsible for the introduction of halide defects only in the perovskite structure synthesized at relatively low temperatures (i.e., 140 °C and 120 °C), as ascertained by the lower emission efficiencies of the corresponding NCs.
The selective formation of size-tunable nanocubes under the explored reaction conditions could be explained by the presence of secondary ammonium cations (generated by the SN2 reaction between oleylamine and 1-bromohexane) as the prevalent positively charged capping ligands, which, as demonstrated by Imran and coworkers, are not involved in the passivation of the forming nanoparticles, as described in eqn (1). During the addition of the bromoplumbate species and cesium in the growth of the forming nanoparticles, the larger steric hindrance of the secondary ammonium cations46 probably hampers the formation of a compact organic layer passivating the NC, which was proved to favor the anisotropic growth typically occurring at relatively low temperatures.47 On these bases, the size modulation of the nanocubes with the Cs/Pb molar ratio is made possible because all the lead species present in the reaction mixture are potentially reactive (as bromoplumbates) in the absence of carboxylic acids as the ligands. For the same reasons, by reducing the reaction temperature, the availability of reactive bromoplumbate species approaching the NC surface during its growth is kinetically limited, thus promoting the formation of smaller nanoparticles.
The NC ligand shell
In order to investigate the ligand shell of the synthetized NCs, we carried out the 1H-NMR characterization of the nanoparticles prepared at T = 160 °C with Cs/Pb = 1/4 as a representative sample of our series. As evident in its 1H-NMR spectrum shown in Fig. 5A, the presence of relatively broad proton signals in the aliphatic region can be observed, suggesting that these NCs are passivated by organic ligands and are also supported by the negative cross-peaks characterizing the corresponding NOESY spectrum reported in Fig. 5S.† To discriminate the nature of the ligands passivating our CsPbBr3 NCs, we compared the corresponding 1H-NMR spectrum with that of the mixture obtained before the injection of the cesium source, i.e. the solution obtained by mixing PbBr2, oleylamine and 1-bromohexane (6.0 equivalents with respect to lead) in ODE at 160 °C for the predetermined incubation time (30 min). The 1H-NMR characterization of the above-mentioned mixture (Fig. 5A) clearly evidenced the formation of an additional species (the corresponding secondary amine as described in eqn (1)) released by the SN2 reaction between 1-bromohexane and oleylamine. However, the NMR signals of the CsPbBr3 NCs appear to be superimposable with those of the pure oleylamine as clearly evident from the peculiar –CH2NH2 resonance (δ = 2.54 ppm). This observation leads to the conclusion that, in the NCs prepared with the proposed synthetic approach, the organic passivation involves L-type neutral species (i.e., oleylamine) rather than charged ones (i.e., the corresponding ammonium cations), whose resonances should be remarkably downfield shifted in the relevant 1H-NMR spectrum. Therefore, we can reasonably conclude that the undercoordinated lead atoms at the NC surface are passivated by oleylamine, as schematically represented in Fig. 5B. It is also possible the formation of hydrogen bonds between oleylamine and the bromine atoms at the surface.Long-term stability investigations on the perovskite NCs
We deemed it necessary to complete our investigation by studying the long-term colloidal stability of our near-unity PLQY perovskite nanocubes by storing the relevant colloidal dispersions under ambient conditions of humidity and illumination, while monitoring the evolution of their PLQY for 90 days. As shown in Fig. 6, all synthesized NCs retained most of the initial emission efficiency, showing remarkable residual PLQY (85–95%) after 3 months. However, the nanoparticles synthesized at 160 °C exhibited a higher resistance to the PLQY degradation (Fig. 6A–C) with respect to those prepared at lower temperatures (Fig. 6D and E) independently of the Cs/Pb molar ratio used in the synthesis.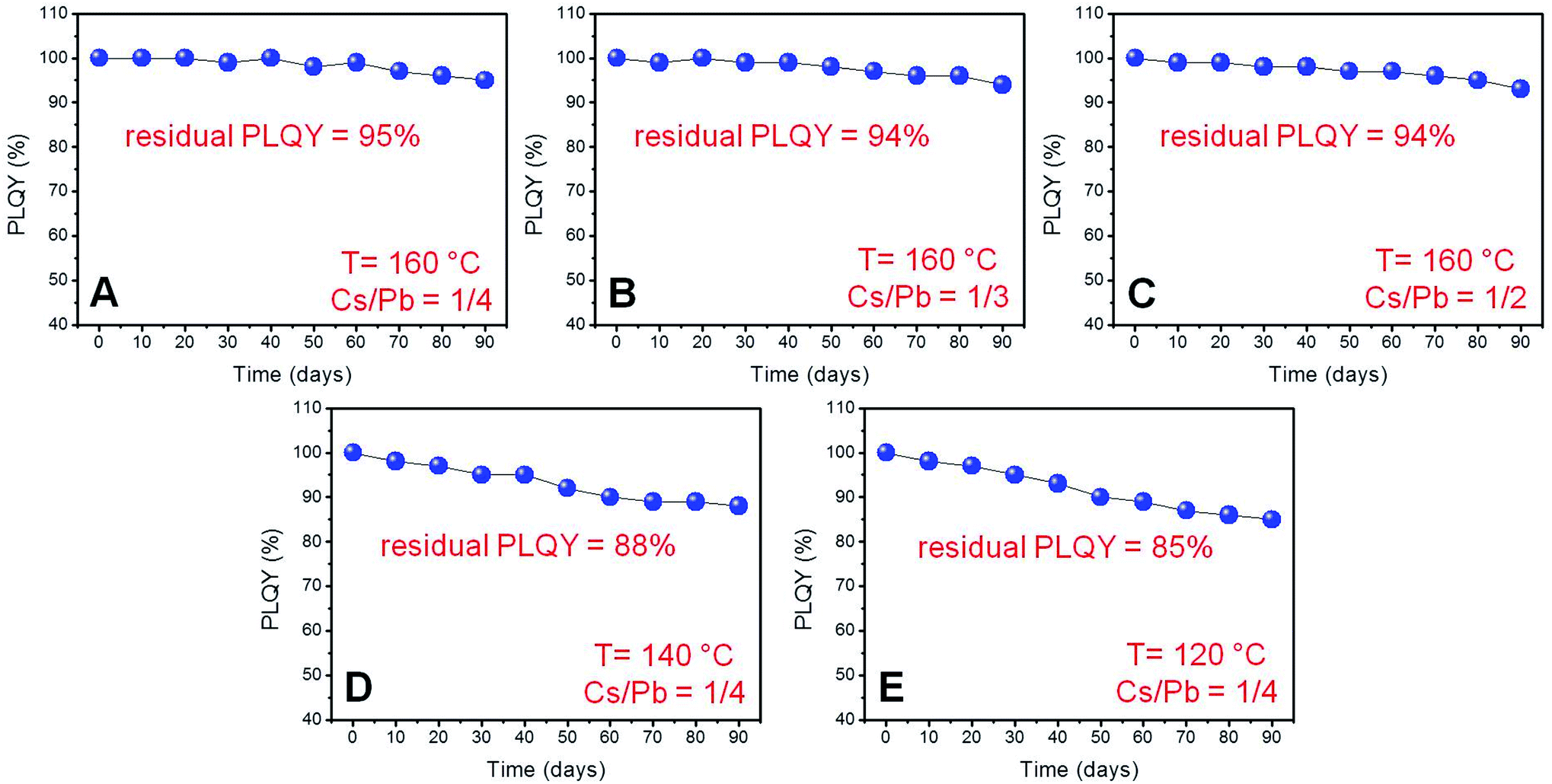 | ||
| Fig. 6 PLQY trend as a function of time (days) for solutions in cyclohexane of the samples (A–E) exhibiting near-unity PLQYs stored under ambient conditions. | ||
The remarkable optical and colloidal stability recorded for these NCs resulted in a noticeable contrast with our previous observations, revealing the rapid PLQY degradation of near-unity PLQY perovskite nanocubes synthesized in the presence of both oleic acid and oleylamine as the capping ligand.24 This behaviour was also observed in nanoparticles synthesized in the presence of oleic acid (an equivolumetric amount with respect to oleylamine) at 160 °C with a Cs/Pb molar ratio of 1/4, adopting the same synthetic protocol described in this study. Notwithstanding the relatively high initial PLQY (91%), the synthesized nanoparticles rapidly lose their high emission efficiencies (Fig. 6S†) if stored in cyclohexane under ambient conditions.
On these bases, we can argue that the factors contributing to the exceptional stability and optical properties of our near-unity PLQY CsPbBr3 NCs (synthesized in the presence of oleylamine as the only surfactant) cannot be rationalized solely considering that they were obtained in the presence of an uncommonly high Pb/Br precursor ratio, as suggested by the current literature.48 In fact, the EDX measurements (vide supra) on our samples suggested that these nanoparticles cannot properly be considered as bromide-rich perovskite NCs, inevitably introducing other reasons for explaining their stability.
First, it should be taken into account that the residual presence of oleylamine can provide the suitable passivation of the NC surfaces, conferring the colloidal integrity to the relevant nanoparticles. In fact, it was theoretically demonstrated and experimentally confirmed that secondary amines weakly interact with the NC surface,24,38 while the reaction conditions proposed in this study lead to the formation of PbBr2-terminating surfaces, which could be effectively passivated by the primary amines, such as oleylamine.49 A second factor which should be scrutinized in this context is the absence (in the dissolution process of the lead precursor) of carboxylates, which are competitors of bromides for the lead coordination at the NC surface.50 It is well-known, in fact, that carboxylates are easily removed along with cesium ions from the surface of perovskite NCs, causing, if not properly replaced by equally efficient neutral passivating species, the structural destabilization of the nanoparticle and the downgrade of its emission efficiency.51
Structural investigations
Obtaining near-unity PLQY CsPbBr3 NCs under different reaction conditions (in terms of temperature and element ratio) offers the possibility to establish a link between the crystal structure and the optical properties of the obtained particles. Therefore, XRD measurements were performed to reveal the CsPbBr3 crystal phase of the obtained near-unity PLQY nanomaterials. A whole-pattern fitting was performed (on the 2θ scan profiles in Fig. 7S and 8S†) based on the Rietveld approach, by using the Rietveld program package QUANTO.52 It is well-known that the definite assessment of the crystalline phase of CsPbBr3 NCs is hampered mainly by the intrinsic peak broadening due to the small crystalline domain size, basically overlapping peak positions of the typical CsPbBr3 crystalline phases (orthorhombic, cubic, and tetragonal), and the altered relative peak intensities due to NC anisotropy and/or preferred orientations.9,16,53 Nevertheless, meaningful clues to the main crystalline phase in the samples can be obtained based on the goodness of fit (GoF) results of the XRD whole profile fits for CsPbBr3 orthorhombic and cubic/tetragonal structures, as well as directly from the intensity ratio between the two main diffraction peaks at ∼15° and 30° (2θ). Whole profile fitting was applied to the detector (2θ) scan profiles (Fig. 7S and 8S†), featuring more complete data despite the altered relative peak intensities due to preferred orientations; on the other hand, the intensity ratio was evaluated on the peak areas in the coupled (θ/2θ) scans (reported in Fig. 7 after background subtraction).9,16 Both methods feature advantages and drawbacks: the former is affected by altered intensity ratios due to preferred orientations and the specific structure models used in the fitting procedure, as well as by data quality and background interpolation (see e.g. profile fitting for the sample at T = 160 °C and Cs/Pb = 1/2), also affected by scattering contributions from surface amorphous components (due to the relatively small incidence angle); the latter is mainly affected by possible sample inhomogeneity (the illuminated area decreases with increasing q) and transparency effects (X-ray penetration depth increases with increasing q), leading to a decrease in the intensity of the higher angle peak and hence of the ratio between the two peak intensities; finally, both methods are in general affected by experimental errors within the structure solution process leading to the final crystallographic information file (.cif) or to the powder diffraction file (.pdf) taken as references. The combination of both methods is therefore expected to produce more reliable results, provided that their application and results are suitably considered.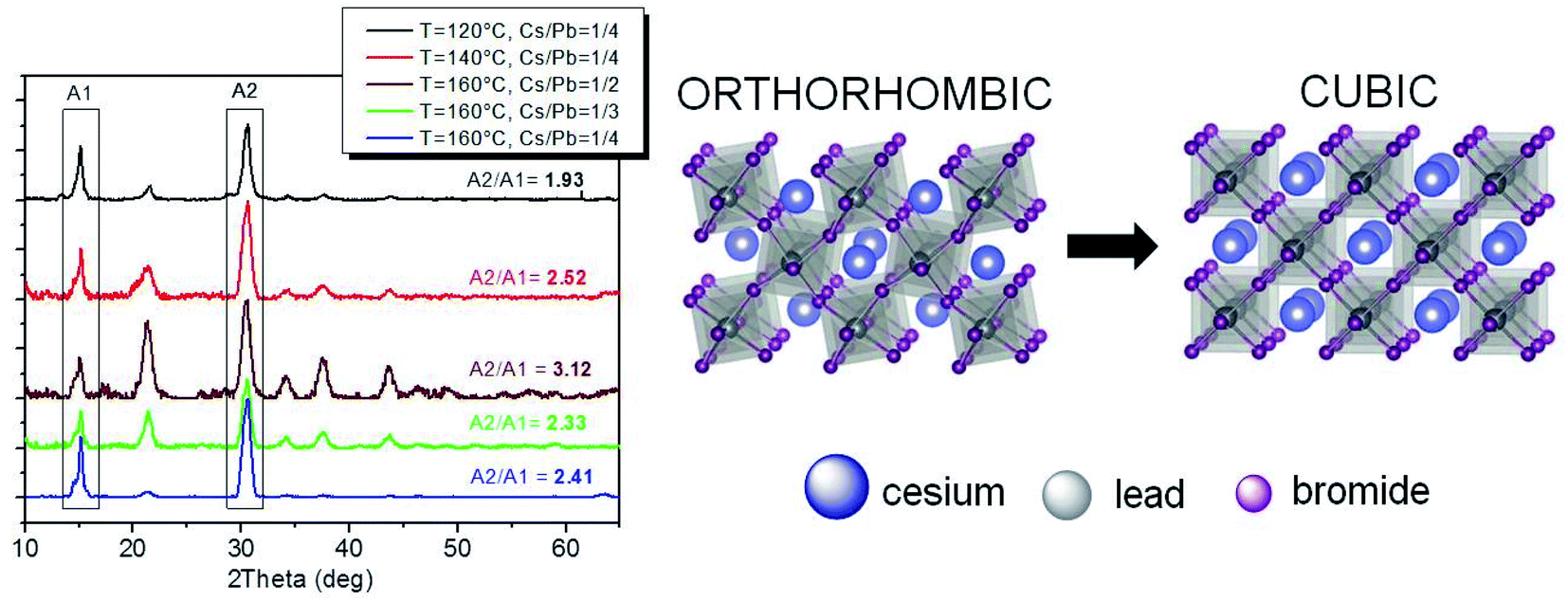 | ||
| Fig. 7 (left) XRD experimental patterns (θ/2θ scan and background subtracted) showing a higher preferred orientation for samples at 120 °C and 160 °C with Cs/Pb = 1/4 (black and blue curves, respectively). (right) A schematic representation of the orthorhombic and cubic structures of CsPbBr3 differing for the tilting of PbBr64− octahedra with respect to each other. | ||
In the hypothesis of similar and uniform thickness of the deposited NC films, transparency effects will similarly affect all samples, so that the intensity ratios will follow the same trend predicted by the relevant (.cif) files for the different crystal symmetries, although with values differing by a scale factor dependent on experimental conditions (in particular on the film thickness). This was actually found for the whole set of samples. In fact, as shown in Fig. 7, a lower intensity ratio (1.9) was obtained for NCs obtained at T = 120 °C, which is compatible with the orthorhombic phase (1.8 ratio),9,16 whereas larger intensity ratios are compatible with the cubic (2.5 for NCs obtained at T = 140 °C and 2.4 and 2.3 for T = 160 °C with Cs/Pb = 1/4 and 1/3, respectively) or the tetragonal16 (T = 160 °C with Cs/Pb = 1/2) phase. Such a trend is similar to that expected based on the analysis of the (.cif) files, with an intensity ratio close to 9.0 for Pbnm and Pnma (ICSD-97851 and COD-4510745, respectively) and to 11.3 and 11.5 for the cubic and tetragonal phase, respectively (COD-1533063 for Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and ICSD-109295 for P4mm). The intensity ratios derived from the powder diffraction file (.pdf)9,16 and from the (.cif) files are thus related by a scale factor (∼5.0), which can be accounted for if a suitable film thickness is assumed for the samples, and the attenuation of the X-ray beam traveling across the CsPbBr3 nanocrystalline film in the θ/2θ scans is calculated based on the Lambert–Beer law54 for the two main diffraction orders. If the Bragg angle corresponding to the first peak (lower angle) is assumed to allow film irradiation through its whole thickness down to the interface with the substrate; a film thickness of ∼1.3 μm can be estimated, which is definitely plausible, given a CsPbBr3 density of 4.75 g cm−3.
m and ICSD-109295 for P4mm). The intensity ratios derived from the powder diffraction file (.pdf)9,16 and from the (.cif) files are thus related by a scale factor (∼5.0), which can be accounted for if a suitable film thickness is assumed for the samples, and the attenuation of the X-ray beam traveling across the CsPbBr3 nanocrystalline film in the θ/2θ scans is calculated based on the Lambert–Beer law54 for the two main diffraction orders. If the Bragg angle corresponding to the first peak (lower angle) is assumed to allow film irradiation through its whole thickness down to the interface with the substrate; a film thickness of ∼1.3 μm can be estimated, which is definitely plausible, given a CsPbBr3 density of 4.75 g cm−3.
Unexpectedly, such results were confirmed by the GoF values obtained from the whole XRD profile fits only for samples prepared at T = 120 °C and (with notable tiny differences) at T = 160 °C with Cs/Pb = 1/3 (Fig. 7S†); on the contrary, for samples prepared at 140 °C and 160 °C (with Cs/Pb = 1/2 or 1/4), the orthorhombic structure was found to have a higher or similar probability (lower or similar GoF) with respect to the cubic one. However, the low temperature (from room temperature on) phases are expected to be monoclinic and orthorhombic, whereas the high temperature phases are expected to be tetragonal and cubic.55,56
Such discrepancy between the two approaches is ascribed to the overall relatively high GoF values with limited capability to discriminate among different structures based on small variations, possibly due to low data quality. Indeed, for the samples prepared at T = 140 °C or at T = 160 °C with Cs/Pb = 1/4, basically identical GoF values were obtained (Fig. 8S†) for the orthorhombic (Pbnm) and cubic (Pmm) structures, which testifies the insufficient capacity of the whole profile fitting to discriminate such tiny structural differences in a non-random population of NCs. Moreover, similar GoF values were also obtained based on the tetragonal structure (P4mm, not shown). For this reason, the fitting results obtained for the sample prepared at T = 160 °C with Cs/Pb = 1/2 (Fig. 8S†), indicating an opposite trend with respect to the intensity ratio approach, were not considered as reliable either. As a consequence, the intensity ratio between the two main diffraction orders in the θ/2θ scan is confirmed as a practical and suitable approach to discriminate tiny differences between crystalline phases mainly related to lattice distortions. From the experimental point of view, the accuracy of this method would be as good as thickness uniformity achieved in sample preparation. Vice versa, the film thickness could be in general derived from the peak intensity ratios if the crystalline structure is known.
We can therefore conclude that, under our specific reaction conditions leading to near-unity PLQY NCs, the crystalline phase of the corresponding samples is mainly influenced by the reaction temperature. Specifically, relatively low reaction temperatures (≤140 °C) promoted the formation of the orthorhombic crystalline phase, while relatively high reaction temperatures (160 °C) induced the formation of the cubic crystalline phase. Nevertheless, both phases are compatible with the achievement of near-unity PLQYs.
Conclusions
In this work we have proposed a synthetic protocol with wide applicability for obtaining near-unity PLQY perovskite nanocubes. In this method, the dissolution of the lead precursor (PbBr2) occurs in the incubation stage of the process and results from the complexation by the bromide anions released by the in situ SN2 reaction between oleylamine (the only surfactant introduced in the reaction mixture) and 1-bromohexane with the formation of an additional ligand (the corresponding secondary amine). The control over the nanocube size was achieved through the modulation of the reaction temperature and the proper combination of the perovskite constituting elements. The obtained CsPbBr3 nanocubes exhibited PL maxima in the range of 505–517 nm and near-unity PLQY with a narrow emission profile (fwhm = 17–19 nm). The pre-formation of highly coordinated bromoplumbate species occurring before the cesium injection was evaluated as the key factor for determining the cubic shape of the resulting NCs under different reaction conditions, while preserving their near-unity PLQY. This peculiarity is due to the selective involvement of the scarcely passivating secondary ammonium cations (the countercations of the bromoplumbate species formed during the incubation stage) in the nucleation and growth of the forming nanoparticles. Conversely, the colloidal stability of the corresponding nanoparticles was conferred by the simultaneous presence of oleylamine acting as the L-type ligand of the PbBr2-terminating surfaces characterizing these nanocubes. In fact, the NCs synthesized by our protocol were remarkably stable, preserving their high PLQYs even after 90 days of storage under ambient conditions. Therefore, the proposed synthetic protocol brings about the direct preparation of differently structured perovskite NCs without the need to improve the emission efficiency and stability by additional post-synthetic treatment.Experimental section
Chemicals
Cesium carbonate (Cs2CO3, 99.9% metals basis, Alfa Aesar), lead bromide (PbBr2, 99.999% metals basis, Aldrich), 1-bromohexane (98%, Aldrich), oleylamine (technical grade 70%, Aldrich), oleic acid (technical grade 90%, Aldrich), 1-octadecene (ODE, technical grade 90%, Aldrich), and cyclohexane (Aldrich, HPLC grade).Synthesis of near-unity PLQY CsPbBr3 NCs
The syntheses were carried out under a nitrogen atmosphere using standard Schlenk techniques. The cesium-oleate solution was prepared as follows: Cs2CO3 (0.407 g, 1.25 mmol), oleic acid (1.55 mL, 4.91 mmol) and ODE (20 mL) were mixed in a 50 mL Schlenk tube and kept in a vacuum for 1 hour at 120 °C. After the complete solubilization of the salt, the tube was filled with nitrogen and kept at 120 °C before the injection. To a 50 mL Schlenk tube, ODE (7.5 mL), lead bromide (0.138 g, 0.38 mmol), oleylamine (1.0 mL) and 1-bromohexane (2.28 mmol, 0.320 mL) were added and the mixture was heated at a specified temperature (160, 140 or 120 °C). After complete solubilization of the reactants (5 min), the Cs-oleate solution (0.8, 1.6 or 2.4 mL) was injected. Subsequently, the reaction mixture was rapidly cooled in an ice bath and the crude mixture was centrifuged at 4000 rpm for 40 min. The supernatant solution, containing the unreacted precursors was discarded and the obtained precipitate was dispersed in cyclohexane.Optical characterization
UV-vis absorption spectra were collected using a Jasco V670 spectrometer operated in the transmission mode. Steady-state photoluminescence (PL) spectra from solutions were recorded on a Varian Cary Eclipse instrument. The quantum yields were determined by using fluorescein as the standard, according to standard procedures.57Transmission electron microscopy (TEM) characterization
TEM micrographs were acquired using a JEOL JEM1011 microscope, operating at an accelerating voltage of 100 kV. The instrument was equipped with a tungsten electron source and a high-resolution CCD camera. Samples for TEM analysis were prepared by dipping the carbon-coated copper grid into the NC solution diluted with anhydrous n-hexane. Statistical size analysis (NC average size and standard deviation) was performed by using a freeware image analysis application (AxioVision); a size of not less than one hundred nanoparticles was measured for each sample.Field emission gun scanning electron microscopy coupled with energy dispersive X-ray spectroscopy
FEG-SEM-EDX analysis was performed using a Zeiss Sigma 300VP electron microscope equipped with an Oxford C-MaxN SDD detector with an active area of 20 mm2. Perovskite NCs were suspended in cyclohexane and deposited on aluminum stubs. Analysis was performed using a working distance of 7.5 mm, an acceleration voltage of 15 kV and a magnification of 1000×. The accuracy of the analysis was checked using the MAC (Micro-Analysis Consultants Ltd) reference materials.X-ray diffraction
XRD data were collected both in the coupled sample-detector (θ/2θ) scan mode and in detector (2θ) scan mode with 5° incidence angle, by using a Bruker D8 Discover equipped with a Cu source (Kα line), a Göbel mirror, and a scintillation point detector. Background subtraction in the θ/2θ scans was performed by using the program QUALX.58Computational details
All stationary points were optimized at the density functional theory (DFT) level with the hybrid B3LYP functional as implemented in the Gaussian 09 software package (Rev. D.01).59 Geometry optimizations were computed using the LANL2DZ (Pb and Br) basis sets. Time-dependent DFT (TD-DFT) analysis was carried out including the solvent effects (CPCM, cyclohexane).Nuclear magnetic resonance
1H-NMR spectra were recorded on a Agilent 500 MHz instrument. All chemical shifts were referred to the non-deuterated benzene residue signal at 7.16 ppm. The NOESY spectra were acquired using standard pulse sequences; mixing time was set to 300 ms.Author contributions
R. Grisorio: conceptualization, investigation, methodology, supervision, writing – original draft, writing – review & editing; D. Conelli: investigation; E. Fanizza: investigation; M. Striccoli: investigation, methodology; D. Altamura: investigation, writing – original draft; C. Giannini: methodology; I. Allegretta: investigation; R. Terzano: methodology; M. Irimia-Vladu: validation; N. Margiotta: investigation; G. P. Suranna: conceptualization, writing – review & editing, funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. G. acknowledges REFIN project NANO-3D (COD. 55FF6B6F) funded by Regione Puglia.References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- Q. Zhang and Y. Yin, ACS Cent. Sci., 2018, 4, 668–679 CrossRef CAS PubMed.
- D. Yang, M. Cao, Q. Zhong, P. Li, X. Zhang and Q. Zhang, J. Mater. Chem. C, 2019, 7, 757–789 RSC.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- C. Zhang, J. Chen, S. Wang, L. Kong, S. W. Lewis, X. Yang, A. L. Rogach and G. Jia, Adv. Mater., 2020, 32, 2002736 CrossRef CAS PubMed.
- J. Kang and L. Wang, J. Phys. Chem. Lett., 2017, 8, 489–493 CrossRef CAS PubMed.
- Y. Wu, X. Li and H. Zeng, ACS Energy Lett., 2019, 4, 673–681 CrossRef CAS.
- R. Grisorio, M. E. Di Clemente, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, M. Irimia-Vladu and G. P. Suranna, Nanoscale, 2019, 11, 986–999 RSC.
- Y. Chen, S. R. Smock, A. H. Flintgruber, F. A. Perras, R. L. Brutchey and A. J. Rossini, J. Am. Chem. Soc., 2020, 142, 6117–6127 CrossRef CAS PubMed.
- D. Yang, X. Li and H. Zeng, Adv. Mater. Interfaces, 2018, 5, 1701662 CrossRef.
- P. Liu, W. Chen, W. Wang, B. Xu, D. Wu, J. Hao, W. Cao, F. Fang, Y. Li, Y. Zeng, R. Pan, S. Chen, W. Cao, X. W. Sun and K. Wang, Chem. Mater., 2017, 29, 5168–5173 CrossRef CAS.
- R. Grisorio, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, V. Vergaro, G. Ciccarella, N. Margiotta and G. P. Suranna, Nanoscale, 2020, 12, 623–637 RSC.
- N. Mondal, A. De and A. Samanta, ACS Energy Lett., 2019, 4, 32–39 CrossRef CAS.
- F. D. Stasio, S. Christodoulou, N. Huo and G. Konstantatos, Chem. Mater., 2017, 29, 7663–7667 CrossRef CAS.
- E. Fanizza, F. Cascella, D. Altamura, C. Giannini, A. Panniello, L. Triggiani, F. Panzarea, N. Depalo, R. Grisorio, G. P. Suranna, A. Agostiano, M. L. Curri and M. Striccoli, Nano Res., 2019, 12, 1155–1166 CrossRef CAS.
- Y. Dong, T. Qiao, D. Kim, D. Parobek, D. Rossi and D. H. Son, Nano Lett., 2018, 18, 3716–3722 CrossRef CAS PubMed.
- S. Seth, T. Ahmed, A. De and A. Samanta, ACS Energy Lett., 2019, 4, 1610–1618 CrossRef CAS.
- Y. Zhang, T. D. Siegler, C. J. Thomas, M. K. Abney, T. Shah, A. De Gorostiza, R. M. Greene and B. A. Korgel, Chem. Mater., 2020, 32, 5410–5423 CrossRef CAS.
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS PubMed.
- B. Zhang, L. Goldoni, J. Zito, Z. Dang, G. Almeida, F. Zaccaria, J. de Wit, I. Infante, L. De Trizio and L. Manna, Chem. Mater., 2019, 31, 9140–9147 CrossRef CAS.
- A. Dutta, R. K. Behera, P. Pal, S. Baitalik and N. Pradhan, Angew. Chem., Int. Ed., 2019, 58, 5552–5556 CrossRef CAS PubMed.
- S. Paul and A. Samanta, ACS Energy Lett., 2020, 5, 64–69 CrossRef CAS.
- R. Grisorio, D. Conelli, R. Giannelli, E. Fanizza, M. Striccoli, D. Altamura, C. Giannini, I. Allegretta, R. Terzano and G. P. Suranna, Nanoscale, 2020, 12, 17053–17063 RSC.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L.-W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS PubMed.
- A. Dutta, S. K. Dutta, S. Das Adhikari and N. Pradhan, ACS Energy Lett., 2018, 3, 329–334 CrossRef CAS.
- A. Feng, X. Jiang, X. Zhang, X. Zheng, W. Zheng, O. F. Mohammed, Z. Chen and O. M. Bakr, Chem. Mater., 2020, 32, 7602–7617 CrossRef CAS.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS PubMed.
- R. Grisorio, E. Fanizza, M. Striccoli, D. Altamura, C. Giannini, I. Allegretta, R. Terzano and G. P. Suranna, ChemNanoMat, 2020, 6, 356–361 CrossRef CAS.
- C. Lu, M. W. Wright, X. Ma, H. Li, D. S. Itanze, J. A. Carter, C. A. Hewitt, G. L. Donati, D. L. Carroll, P. M. Lundin and S. M. Geyer, Chem. Mater., 2019, 31, 62–67 CrossRef CAS.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS PubMed.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS PubMed.
- X. Zheng, Y. Hou, H.-T. Sun, O. F. Mohammed, E. H. Sargent and O. M. Bakr, J. Phys. Chem. Lett., 2019, 10, 2629–2640 CrossRef CAS PubMed.
- J. Pradhan, P. Moitra, Umesh, B. Das, P. Mondal, G. S. Kumar, U. K. Ghorai, S. Acharya and S. Bhattacharya, Chem. Mater., 2020, 32, 7159–7171 CrossRef CAS.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- M. I. Bodnarchuk, S. C. Boehme, S. ten Brinck, C. Bernasconi, Y. Shynkarenko, F. Krieg, R. Widmer, B. Aeschlimann, D. Günther, M. V. Kovalenko and I. Infante, ACS Energy Lett., 2019, 4, 63–74 CrossRef CAS PubMed.
- T. Udayabhaskararao, L. Houben, H. Cohen, M. Menahem, I. Pinkas, L. Avram, T. Wolf, A. Teitelboim, M. Leskes, O. Yaffe, D. Oron and M. A. Kazes, Chem. Mater., 2018, 30, 84–93 CrossRef CAS.
- M. Imran, P. Ijaz, D. Baranov, L. Goldoni, U. Petralanda, Q. Akkerman, A. L. Abdelhady, M. Prato, P. Bianchini, I. Infante and L. Manna, Nano Lett., 2018, 18, 7822–7831 CrossRef CAS PubMed.
- S. K. Balakrishnan and P. V. Kamat, Chem. Mater., 2018, 30, 74–78 CrossRef CAS.
- J. Hui, Y. Jiang, Ö. Ö. Gökçinar, J. Tang, Q. Yu, M. Zhang and K. Yu, Chem. Mater., 2020, 32, 4574–4583 CrossRef CAS.
- A. Ray, D. Maggioni, D. Baranov, Z. Dang, M. Prato, Q. A. Akkerman, L. Goldoni, E. Caneva, L. Manna and A. L. Abdelhady, Chem. Mater., 2019, 31, 7761–7769 CrossRef CAS PubMed.
- J.-R. Wen, B. J. Roman, F. A. Rodriguez Ortiz, N. Mireles Villegas, N. Porcellino and M. Sheldon, Chem. Mater., 2019, 31, 8551–8557 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- M. Koolyk, D. Amgar, S. Aharon and L. Etgar, Nanoscale, 2016, 8, 6403–6409 RSC.
- T. Udayabhaskararao, M. Kazes, L. Houben, H. Lin and D. Oron, Chem. Mater., 2017, 29, 1302–1308 CrossRef CAS.
- S. Aharon, M. Wierzbowska and L. Etgar, ACS Energy Lett., 2018, 3, 1387–1393 CrossRef CAS.
- V. M. Burlakov, Y. Hassan, M. Danaie, H. J. Snaith and A. Goriely, J. Phys. Chem. Lett., 2020, 11, 6535–6543 CrossRef CAS PubMed.
- J. Y. Woo, Y. Kim, J. Bae, T. G. Kim, J. W. Kim, D. C. Lee and S. Jeong, Chem. Mater., 2017, 29, 7088–7092 CrossRef CAS.
- Q. Zhong, M. Cao, Y. Xu, P. Li, Y. Zhang, H. Hu, D. Yang, Y. Xu, L. Wang, Y. Li, X. Zhang and Q. Zhang, Nano Lett., 2019, 19, 4151–4157 CrossRef CAS PubMed.
- S. R. Smock, T. J. Williams and R. L. Brutchey, Angew. Chem., Int. Ed., 2018, 57, 11711–11715 CrossRef CAS PubMed.
- M. Imran, P. Ijaz, L. Goldoni, D. Maggioni, U. Petralanda, M. Prato, G. Almeida, I. Infante and L. Manna, ACS Energy Lett., 2019, 4, 819–824 CrossRef CAS.
- A. Altomare, M. C. Burla, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Rizzi, J. Appl. Crystallogr., 2001, 34, 392–397 CrossRef CAS.
- P. Cottingham and R. L. Brutchey, Chem. Commun., 2016, 52, 5246–5249 RSC.
- J. Als-Nielsen and D. McMorrow, Elements of Modern X-ray Physics, John Wiley and Sons, Ltd., NY, 2011 Search PubMed.
- C. K. Møller, Nature, 1958, 182, 1436 CrossRef.
- M. Rodová, J. Brožek, K. Knížek and K. Nitsch, J. Therm. Anal. Calorim., 2003, 71, 667–673 CrossRef.
- M. Grabolle, M. Spieles, V. Lesnyak, N. Gaponik, A. Eychmüller and U. Resch-Genger, Anal. Chem., 2009, 81, 6285–6294 CrossRef CAS.
- A. Altomare, N. Corriero, C. Cuocci, A. Falcicchio, A. Moliterni and R. Rizzi, J. Appl. Crystallogr., 2015, 48, 598–603 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional spectroscopic and structural investigations. See DOI: 10.1039/d1na00142f |
| This journal is © The Royal Society of Chemistry 2021 |