
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of CO2 activation on the structure, composition, and performance of Sb/C nanohybrid lithium/sodium-ion battery anodes†
Suzhe
Liang
 ab,
Ya-Jun
Cheng
*ac,
Xiaoyan
Wang
a,
Zhuijun
Xu
ad,
Liujia
Ma
ae,
Hewei
Xu
a,
Qing
Ji
af,
Xiuxia
Zuo
a,
Peter
Müller-Buschbaum
bg and
Yonggao
Xia
*ah
ab,
Ya-Jun
Cheng
*ac,
Xiaoyan
Wang
a,
Zhuijun
Xu
ad,
Liujia
Ma
ae,
Hewei
Xu
a,
Qing
Ji
af,
Xiuxia
Zuo
a,
Peter
Müller-Buschbaum
bg and
Yonggao
Xia
*ah
aNingbo Institute of Materials Technology & Engineering, Chinese Academy of Sciences, 1219 Zhongguan West Rd, Ningbo, Zhejiang Province 315201, P. R. China. E-mail: chengyj@nimte.ac.cn; xiayg@nimte.ac.cn
bPhysik-Department, Lehrstuhl für Funktionelle Materialien, Technische Universität München, James-Franck-Str. 1, 85748, Garching, Germany
cDepartment of Materials, University of Oxford, Parks Rd, OX1 3PH, Oxford, UK
dUniversity of Chinese Academy of Sciences, 19A Yuquan Rd, Shijingshan District, Beijing, 100049, P. R. China
eState Key Laboratory of Separation Membranes and Membrane Processes, Tianjin Polytechnic University, Tianjin, 300387, P. R. China
fThe University of Nottingham Ningbo China, 199 Taikang East Rd, Ningbo, Zhejiang Province 315100, P. R. China
gHeinz Maier-Leibnitz Zentrum (MLZ), Technische Universität München, Lichtenbergstr. 1, 85748, Garching, Germany
hCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, 19A Yuquan Rd, Shijingshan District, Beijing 100049, P. R. China
First published on 28th January 2021
Abstract
Antimony (Sb) has been regarded as one of the most promising anode materials for both lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs) and attracted much attention in recent years. Alleviating the volumetric effect of Sb during charge and discharge processes is the key point to promote Sb-based anodes to practical applications. Carbon dioxide (CO2) activation is applied to improve the rate performance of the Sb/C nanohybrid anodes caused by the limited diffusion of Li/Na ions in excessive carbon components. Based on the reaction between CO2 and carbon, CO2 activation can not only reduce the excess carbon content of the Sb/C nanohybrid but also create abundant mesopores inside the carbon matrix, leading to enhanced rate performance. Additionally, CO2 activation is also a fast and facile method, which is perfectly suitable for the fabrication system we proposed. As a result, after CO2 activation, the average capacity of the Sb/C nanohybrid LIB anode is increased by about 18 times (from 9 mA h g−1 to 160 mA h g−1) at a current density of 3300 mA g−1. Moreover, the application of the CO2-activated Sb/C nanohybrid as a SIB anode is also demonstrated, showing good electrochemical performance.
Introduction
Due to its high theoretical capacity (660 mA h g−1), appropriate reaction potential (0.8–0.9 V vs. Li/Li+, 0.5–0.8 V vs. Na/Na+) and abundant reserves in Earth's crust, antimony (Sb) is regarded as a promising alternative anode material for both lithium-ion batteries (LIBs) and sodium-ion batteries (SIBs).1–12 However, the volume expansion of Sb during lithiation/sodiation leads to electrode pulverization and quick capacity fading, which has become one of the major impediments for practical applications of the Sb-based anodes.8 In order to address this issue, several effective strategies have been proposed including decreasing the particle size, introducing a buffer matrix and creating a void space inside the anode material.13–18 At present, utilization of multiple strategies has gradually been a mainstream concept to improve the electrochemical performance of Sb-based anodes.19–25 For example, Sb nanoparticles encapsulated in 1-D N-doped porous carbon were fabricated by an in situ nanoconfined replacement reaction.26 Sb nanoparticles with a size of 10–20 nm were uniformly encapsulated in the 1-D N-doped porous carbon scaffolds. This Sb/C composite exhibited a high reversible capacity of 556 mA h g−1 at 200 mA g−1 after 100 cycles for LIBs and a reversible capacity of 401 mA h g−1 at 100 mA g−1 after 100 cycles for SIBs. One-dimensional yolk–shell Sb@Ti–O–P nanostructures were prepared by reducing core–shell Sb2O3@TiO2 nanorods with NaH2PO2.20 It delivered a capacity of about 760 mA h g−1 after 200 cycles at 500 mA g−1 for SIBs, with a capacity retention of about 94%. However, most of the existing synthesis methods require careful structure control and are difficult to scale up.27–30 Thus, it is necessary to develop new strategies to synthesize Sb-based anodes in a facile and scalable way.In our previous studies, we have developed new concepts to synthesize various nanohybrid anodes for LIBs in facile scalable ways.31–41 The precursors or active materials are first homogeneously dissolved in a methacrylate resin monomer solution with the addition of initiators. Through photo or thermal polymerization, the structural units of precursors are integrated into the cross-linking network of dimethacrylate polymers. After calcination in an inert atmosphere, the thermosetting methacrylate polymers are transformed into a carbon matrix, while the precursors are in situ converted to active components assisted by the carbothermic reaction at the same time. During the calcination process, the nucleation, growth, and agglomeration of the in situ-formed nanoparticles are significantly inhibited within the in situ-formed carbon matrix because of the cross-linking structure of the polymerized difunctional methacrylates. As a result, tiny nanoparticles are in situ formed and homogeneously embedded in the continuous micrometer-sized carbon matrix, which can effectively alleviate the absolute volume changes of active materials during charging/discharging processes.
According to this unique strategy, a hierarchical Sb/C nanohybrid anode material was successfully synthesized and good electrochemical performance was achieved.37 Particularly, by applying this Sb/C nanohybrid as the anode of LIBs, a reversible specific capacity of 362 mA h g−1 was exhibited after 300 cycles at 66 mA g−1, corresponding to the capacity retention of 79%. However, due to the excessive amount of compact carbon matrix within the Sb/C nanohybrid, the diffusion capability of lithium ions was limited, leading to compromised rate performance. To solve this problem, carbon dioxide (CO2) activation is in this work applied to improve the rate performance of Sb/C hybrid anodes.42,43 Due to the reaction between carbon and carbon dioxide at high temperatures (as shown by eqn (1)), CO2 activation can not only reduce the carbon content of the Sb/C composite but also create more porous structures within the carbon matrix.42–46
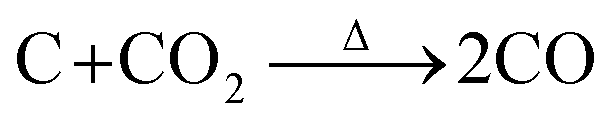 | (1) |
The activation temperature is selected as 950 °C to ensure the sufficient reaction between carbon and CO2, according to the previous report.47–50 Besides, considering the low melting point of Sb, we chose a relatively short activation time of 30 min in order to decrease the Sb loss as low as possible during the activation process. The pristine Sb/C nanohybrid anode material is first synthesized according to our previous work, as shown in Scheme 1. The liquid antimony(III) n-butoxide is used as the precursor of Sb, which is mixed well with the methacrylate resin solution. After photopolymerization, the as-obtained polymer is calcined in an argon/hydrogen (Ar/H2) atmosphere, then activated under CO2 conditions. Combining the advantages of both in situ synthesis and CO2 activation, the final Sb/C nanohybrid anode exhibits a significantly enhanced electrochemical performance. On the one hand, Sb nanoparticles are homogeneously embedded in the carbon matrix after calcination in an Ar/H2 atmosphere, which is beneficial for the stable cyclability of the Sb-based anode. On the other hand, the CO2 activation can partially eliminate the carbon component of the as-synthesized Sb/C nanohybrid anode and create more porous structures within the carbon matrix simultaneously. In this work, we aim to establish a mechanism of CO2 activation treatment on the Sb/C composite electrode. The impacts of carbon dioxide activation treatment on the structure, composition, and electrochemical performance of the Sb/C electrode are investigated comprehensively.
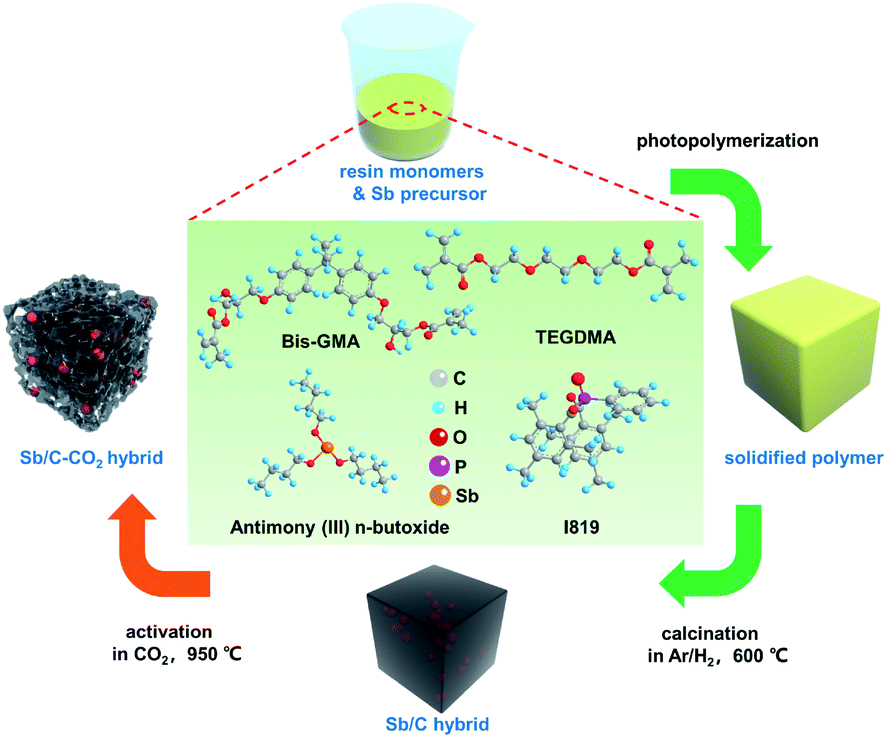 | ||
| Scheme 1 Schematic illustration of the preparation process of the Sb-based nanohybrids without CO2 activation (Sb/C hybrid) and with CO2 activation (Sb/C–CO2 hybrid). | ||
Experimental section
Materials
Antimony(III) n-butoxide (Sb (OC4H9)3, 99+%) was purchased from Alfa Aesar Co., Ltd., China. Bisphenol A glycidyl dimethacrylate (Bis-GMA, analytical reagent grade) and trimethylene glycol dimethacrylate (TEGDMA, 95%) were purchased from Aladdin Reagent Co., Ltd., China. The photoinitiator, phenyl-bis-(2,4,6-trimethyl benzoyl)-phosphine oxide (Irgacure I819), was purchased from Sigma-Aldrich Co., Ltd., China. Conductive carbon Super-P was purchased from SCM Chem. Shanghai, China, and poly(vinylidene fluoride) (PVDF) was donated by Solvay. All chemicals were directly used as received without further purification.Sample preparation and characterization
Except for the carbon dioxide (CO2) activation process, the typical sample preparation procedure was similar to our previously reported work.37 The resin solvent was obtained by mixing Bis-GMA and TEGDMA with a mass ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. Then, I819, as a photoinitiator was dissolved in the resin mixture with a mass ratio of 2%, forming the photoactive B/T solution. Thereafter, various amounts of antimony(III) n-butoxide (0 g, 0.25 g, 0.5 g, and 1 g) were added to every single B/T solution (2 g), respectively. After 30 min of stirring, all solutions were transferred into silicone rubber molds (1.0 cm × 4.5 cm × 0.4 cm) clamped by two pieces of glass slides and the photo-polymerization process was implemented with a visible light-curing unit (Huge G01D05; blue light: 9 W; emission wavelength range: 430–510 nm). Illumination was done for 2 minutes on each side. The as-obtained solid samples were cut into small particulate powders by a grinder, where the approximate diameter of final powders ranged from 0.1 mm to 1.0 mm. All pulverized samples were put into boat crucibles and transferred into a tube furnace for the calcination process in an argon/hydrogen atmosphere (Ar/H2 95:5 by volume). The calcination process started from room temperature and reached 600 °C with a ramp rate of 5 °C min−1, then this temperature was kept for 10 h. CO2 activation was immediately carried out following the calcination step. The Ar/H2 gas was quickly switched to CO2 when the calcination time was up to 10 h, and the temperature was increased rapidly with a ramp rate of 10 °C min−1 from 600 °C to 950 °C. The CO2 activation procedure lasted for 30 min. The atmosphere was then switched back to Ar/H2 again, followed by natural cooling to room temperature. As a contrast, samples with the same feeding ratio were synthesized under the same calcination conditions without CO2 activation. The pure carbon samples with or without activation were prepared through the same protocol without the addition of antimony(III) n-butoxide. Totally, eight samples were synthesized and denoted Carbon, Carbon–CO2, Sb/C-1/8, Sb/C-1/8-CO2, Sb/C-1/4, Sb/C-1/4-CO2, Sb/C-1/2, and Sb/C-1/2-CO2, respectively. The details of the synthesis conditions are shown in Table 1. These samples were further ball-milled with a planetary ball miller (XQM-0.2, Changsha TianChuang Powder Technology Co., Ltd.) for 100 min with the speed of 1000 rpm. A pause of 10 min was set after each 15 min of ball milling. The parameters of the jar and beads were identical to those employed in our previous work.31,32
:3. Then, I819, as a photoinitiator was dissolved in the resin mixture with a mass ratio of 2%, forming the photoactive B/T solution. Thereafter, various amounts of antimony(III) n-butoxide (0 g, 0.25 g, 0.5 g, and 1 g) were added to every single B/T solution (2 g), respectively. After 30 min of stirring, all solutions were transferred into silicone rubber molds (1.0 cm × 4.5 cm × 0.4 cm) clamped by two pieces of glass slides and the photo-polymerization process was implemented with a visible light-curing unit (Huge G01D05; blue light: 9 W; emission wavelength range: 430–510 nm). Illumination was done for 2 minutes on each side. The as-obtained solid samples were cut into small particulate powders by a grinder, where the approximate diameter of final powders ranged from 0.1 mm to 1.0 mm. All pulverized samples were put into boat crucibles and transferred into a tube furnace for the calcination process in an argon/hydrogen atmosphere (Ar/H2 95:5 by volume). The calcination process started from room temperature and reached 600 °C with a ramp rate of 5 °C min−1, then this temperature was kept for 10 h. CO2 activation was immediately carried out following the calcination step. The Ar/H2 gas was quickly switched to CO2 when the calcination time was up to 10 h, and the temperature was increased rapidly with a ramp rate of 10 °C min−1 from 600 °C to 950 °C. The CO2 activation procedure lasted for 30 min. The atmosphere was then switched back to Ar/H2 again, followed by natural cooling to room temperature. As a contrast, samples with the same feeding ratio were synthesized under the same calcination conditions without CO2 activation. The pure carbon samples with or without activation were prepared through the same protocol without the addition of antimony(III) n-butoxide. Totally, eight samples were synthesized and denoted Carbon, Carbon–CO2, Sb/C-1/8, Sb/C-1/8-CO2, Sb/C-1/4, Sb/C-1/4-CO2, Sb/C-1/2, and Sb/C-1/2-CO2, respectively. The details of the synthesis conditions are shown in Table 1. These samples were further ball-milled with a planetary ball miller (XQM-0.2, Changsha TianChuang Powder Technology Co., Ltd.) for 100 min with the speed of 1000 rpm. A pause of 10 min was set after each 15 min of ball milling. The parameters of the jar and beads were identical to those employed in our previous work.31,32
| Sample | Sb(OC4H9)3/BT resin | Activation temperature | Activation time |
|---|---|---|---|
| Carbon | 0 g/2 g | None | None |
| Carbon–CO2 | 0 g/2 g | 950 °C | 30 min |
| Sb/C-1/8 | 0.25 g/2 g | None | None |
| Sb/C-1/8-CO2 | 0.25 g/2 g | 950 °C | 30 min |
| Sb/C-1/4 | 0.5 g/2 g | None | None |
| Sb/C-1/4-CO2 | 0.5 g/2 g | 950 °C | 30 min |
| Sb/C-1/2 | 1 g/2 g | None | None |
| Sb/C-1/2-CO2 | 1 g/2 g | 950 °C | 30 min |
The morphology of the samples was measured by field emission scanning electron microscopy (FESEM, Hitachi S4800, Japan), transmission electron microscopy (TEM, Tecnai F20, America FEI, USA), high-resolution transmission electron microscopy (HRTEM, Tecnai F20, America FEI, USA), and energy-dispersive X-ray spectroscopy (EDX, FEI QUANTA 250 FEG, USA). The crystalline phases of Sb and graphitic structures of Sb/C nanohybrids were measured by X-ray diffraction (XRD, Bruker AXS D8 Advance, Germany) and Raman spectroscopy (Renishaw, inVia Reflex, UK), respectively. The Fourier-transform infrared (FTIR) spectra were recorded on a Nicolet 6700 infrared spectrometer (Thermo, USA). The spectra (4000–500 cm−1) were recorded with a resolution of 0.9 cm−1 and 32 scans per sample. The carbon contents of Sb/C nanohybrids were estimated from the results of thermogravimetric analysis (TGA, Mettler Toledo, Switzerland) and accurately measured with an organic elemental analyzer (CHNSO, Elementar, Germany). The surface areas and porosity of all samples were measured by the Brunauer–Emmett–Teller (BET) method and the Barrett–Joyner–Halenda (BJH) method, respectively, via an accelerated surface area and porosimetry system (2020M, USA).
Electrochemical measurements
The electrochemical performance of all samples was measured using standard 2032-type coin half-cells with lithium/sodium foil as counter electrodes. The as-prepared active material, Super-P (conductive agent) and PVDF (binder) were dissolved in N-methyl pyrrolidone (NMP) to prepare a slurry mixture, according to a mass ratio of 8:1:1. The electrodes were fabricated using a scraper to spread the slurry mixture onto a piece of copper foil, followed by drying at 80 °C in a vacuum for 12 h. The whole electrodes were cut into circular pieces with a diameter of 13 mm. The mass loading of the active material was controlled to be from 1.3 mg cm−2 to 1.7 mg cm−2. The electrodes were further dried in an oven at 80 °C for 4 h before assembling the cells. As to the lithium-ion half-cells, the electrolyte was purchased from Dongguan Shanshan Battery Material Co., Ltd., wherein 1.0 M LiPF6 was dissolved in a solvent mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:2 v/v). The separator was purchased from Celgard, LLC (C210, PP/PE/PP, 16 μm). In terms of sodium-ion half-cells, the electrolyte was formulated by Suzhou Fosai New Material Co., Ltd., where 1.0 M NaPF6 was dissolved in a solvent mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) (1:1 v/v) with 5% of fluoroethylene carbonate (FEC). The separator was purchased from GE Whatman, LLC (1825-257 glass microfiber filters, 257 mm).
The cycling performance and rate performance of all cells were tested using a multichannel land battery test system. For lithium-ion half-cells, the cyclic voltammetry measurements were carried out at a current density of 66 mA g−1 in the voltage range of 3.0–0.005 V (vs. Li/Li+) for certain cycles. The rate performance was measured at the current density sequence of 66 mA g−1, 132 mA g−1, 330 mA g−1, 660 mA g−1, 1320 mA g−1, 3300 mA g−1 and 66 mA g−1 with a voltage range between 3.0 V and 0.005 V (vs. Li/Li+) (five cycles at each current density). As to sodium-ion half-cells, the voltage range of the test was changed between 2.5 V and 0.01 V (vs. Na/Na+), and other parameters were identical to the performance tests of lithium-ion half-cells. The specific capacity was calculated based on the active material only. The lithiation/delithiation (sodiation/desodiation) processes were indexed as the discharge and charge processes, respectively. Cyclic voltammetry tests were performed using a 1470E potentiostatic/galvanostatic analyzer (Solartron Analytical, UK) at a scanning rate of 0.01 mV s−1 with the voltage range between 0.005 V and 3.0 V.
Results and discussion
Structure and composition
In this work, eight samples are synthesized and denoted carbon, carbon–CO2, Sb/C-1/8, Sb/C-1/8-CO2, Sb/C-1/4, Sb/C-1/4-CO2, Sb/C-1/2, Sb/C-1/2-CO2, respectively. The details of synthesis conditions are shown in Table 1. The morphology and structure of all samples are investigated by TEM and HRTEM measurements. As shown in Fig. 1, the carbon matrix of Sb/C nanohybrids incorporated with different amounts of Sb is mainly amorphous. However, the graphitized carbon can also be observed in HRTEM, as displayed in Fig. S1.† The distribution of graphitized carbon in the carbon matrix is sparse, and not all carbon particles contain the graphitized carbon. This phenomenon indicates that both amorphous carbon and graphitized carbon exist in the carbon matrix, and the amount of amorphous carbon is dominant. Regarding the pure carbon samples, no obvious difference is observed with or without CO2 activation. In terms of Sb/C nanohybrids, all samples display a similar structure where pentagonal Sb nanoparticles are embedded in the carbon matrix homogeneously. Lattices of Sb nanoparticles are clearly displayed by the HRTEM images, corresponding to the (012) crystal facet of synthesized Sb (JCPDS no. 35-0732). As shown in the TEM images of Sb/C nanohybrids [Fig. 1(e, g, i, k, m and o)], two different types of Sb crystals are distributed in the carbon matrix, Sb nanoparticles and Sb nanorods. The size range of Sb nanoparticles is relatively broad. The Sb nanoparticles are classified into smaller Sb nanoparticles (diameter < 100 nm) and larger Sb nanoparticles (diameter > 100 nm), and detailed size statistics of two types of Sb crystals is presented in Table S1.† For the non-activated samples, the average size of Sb nanoparticles increases with increasing mass ratio of antimony(III) n-butoxide against the B/T resin. The amount of B/T resin is kept constant (2 g) in the sample synthesis. Therefore, more antimony(III) n-butoxide leads to agglomeration of Sb nanoparticles in the later calcination process. Moreover, it is found that the Sb particle size increased after carbon dioxide activation with respect to each Sb/C nanohybrid. The size increase is mainly induced by the high temperature (950 °C) applied in the activation procedure. This temperature is much higher than the melting point of Sb (630 °C), where flow and coalescence of molten Sb would cause an enlargement of the particle size.6 | ||
| Fig. 1 TEM and HRTEM images of carbon (a, b), carbon–CO2 (c, d), Sb/C-1/8 (e, f), Sb/C-1/8-CO2 (g, h), Sb/C-1/4 (i, j), Sb/C-1/4-CO2 (k, l), Sb/C-1/2 (m, n) and Sb/C-1/2-CO2 (o, p). | ||
The surface morphologies on a larger scale of all samples are probed by SEM measurements, as presented in Fig. S2.† In general, featureless particles with a broad size ranging from 100 nm to 2 μm are observed in all samples. Neither different feed ratios of antimony(III) n-butoxide and B/T resin nor running with or without CO2 activation can influence the apparent morphologies of these samples. This irregular morphology is mainly attributed to crushed powder after ball milling. The powders are mainly in the form of micrometer-size structure feature. It is difficult to identify the Sb species by SEM because the Sb nanoparticles are well embedded in the continuous carbon matrix. TEM and HRTEM measurements are carried out to further probe the internal structures and morphologies of the Sb/C nanohybrids. Additionally, the spatial element distribution in a large area is characterized by EDX images, as shown in Fig. S3.† It is clearly observed that the Sb species is homogeneously dispersed in the carbon matrix over a large size scale area of around 10 μm × 10 μm for all Sb/C nanohybrid samples.
The XRD patterns and Raman spectra of all samples are presented in Fig. S4 and S5,† respectively. The crystalline information of both the Sb component and carbon component is studied comprehensively. The rhombohedral-phase Sb in all Sb/C nanohybrids can be clearly distinguished by the Sb characteristic diffraction peaks (JCPDS no. 35-0732). Based on the Raman spectra results, it is demonstrated that both amorphous and graphitic carbon domains exist in the carbon matrix of Sb/C nanohybrids. The integration ratio of ID/IG can be affected by the composition of Sb/C nanohybrids and the CO2 activation. Besides, the FTIR spectra of carbon and carbon–CO2 samples are shown in Fig. S6.† The detailed discussion of XRD patterns, Raman spectra, and FTIR spectra can be found in the ESI.†
The effect of CO2 activation on carbon content reduction is investigated by thermogravimetric analysis (TGA) under an air atmosphere. As presented in Fig. 2, a major mass loss occurs above 400 °C, which is attributed to the combustion of carbon. After 490 °C, the rates of mass losses (reflected by the slope of curves) are significantly reduced, which is mainly ascribed to the oxidation of Sb. Compared to the Sb/C nanohybrids, the initial mass-loss temperature of pure carbon samples is increased, which reaches about 500 °C. Good thermal conductivity of metallic Sb nanoparticles promotes heat transfer to the surrounding carbon matrix, leading to a faster mass loss and a lower combustion temperature compared to those of the pure carbon sample. According to the TGA curves, the content of metallic Sb in the Sb/C hybrids is calculated based on eqn (2) by assuming that the final product of the Sb/C nanohybrids is Sb2O4.51
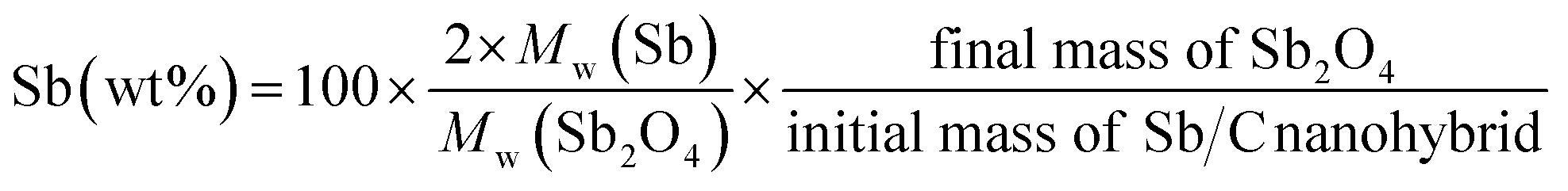 | (2) |
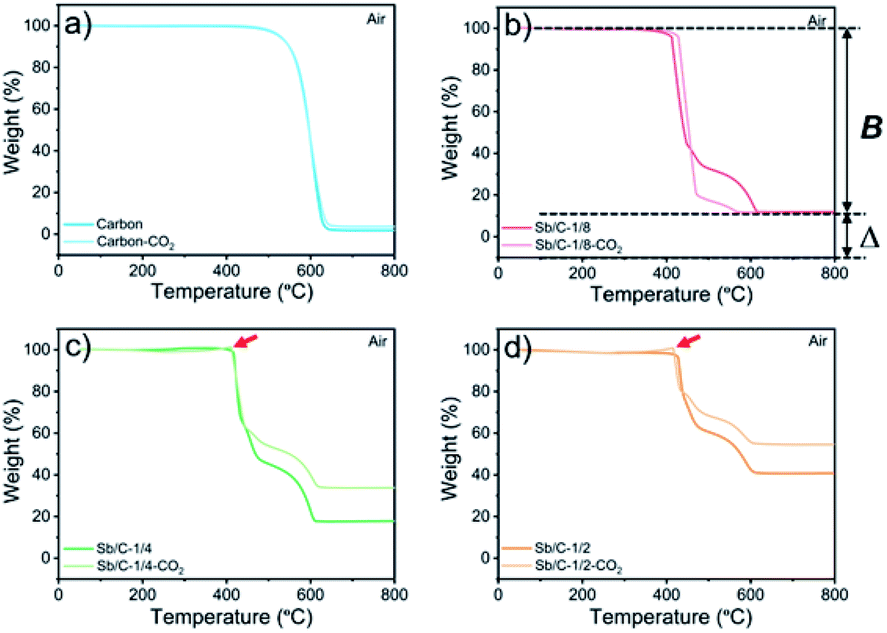 | ||
| Fig. 2 TGA profiles of pure carbon and Sb/C nanohybrids tested in an air atmosphere: carbon and carbon–CO2 (a), Sb/C-1/8 and Sb/C-1/8-CO2 (b), Sb/C-1/4 and Sb/C-1/4-CO2 (c), and Sb/C-1/2 and Sb/C-1/2-CO2 (d). B refers to the percentage of mass loss from both carbon combustion and Sb evaporation under air conditions (50–800 °C). Δ refers to the weight percentage of the remaining material after the heating process under air conditions (50–800 °C). | ||
M w denotes the molecular weight. The carbon contents of the Sb/C nanohybrids can be further calculated based on the Sb contents. The carbon contents of the Sb/C-1/8, Sb/C-1/8-CO2, Sb/C-1/4, Sb/C-1/4-CO2, Sb/C-1/2 and Sb/C-1/2-CO2 samples are derived to be 90.5 wt%, 90.7 wt%, 85.9 wt%, 78.1 wt%, 80.4 wt% and 52.3 wt%, respectively. In terms of Sb/C-1/4 and Sb/C-1/2 samples before and after activation (Sb/C-1/4 vs. Sb/C-1/4-CO2 and Sb/C-1/2 vs. Sb/C-1/2-CO2), the carbon contents of activated samples are lower compared to those of the corresponding pristine samples. This confirms the assumption that the CO2 activation treatment can reduce the carbon content within the Sb/C nanohybrids. However, the carbon contents of the Sb/C-1/8 sample before and after activation are almost equal (Fig. 2(b)). It originates from evaporation of Sb at high temperatures because of its relatively low melting point. The TGA test contains three origins of mass change: mass loss from combustion of carbon, mass loss from Sb evaporation, and mass increase from Sb oxidation. Therefore, the above carbon content calculation equation does not reflect the carbon content of Sb/C nanohybrid accurately. Based on the three mass change mechanisms during the TGA test, it is reasonable to conclude that the identical mass before and after activation, as displayed in Fig. 2(b), is just a coincidence. To prove this speculation, TGA measurements under a nitrogen atmosphere are performed with all Sb/C nanohybrid samples, where the results are shown in Fig. 3.
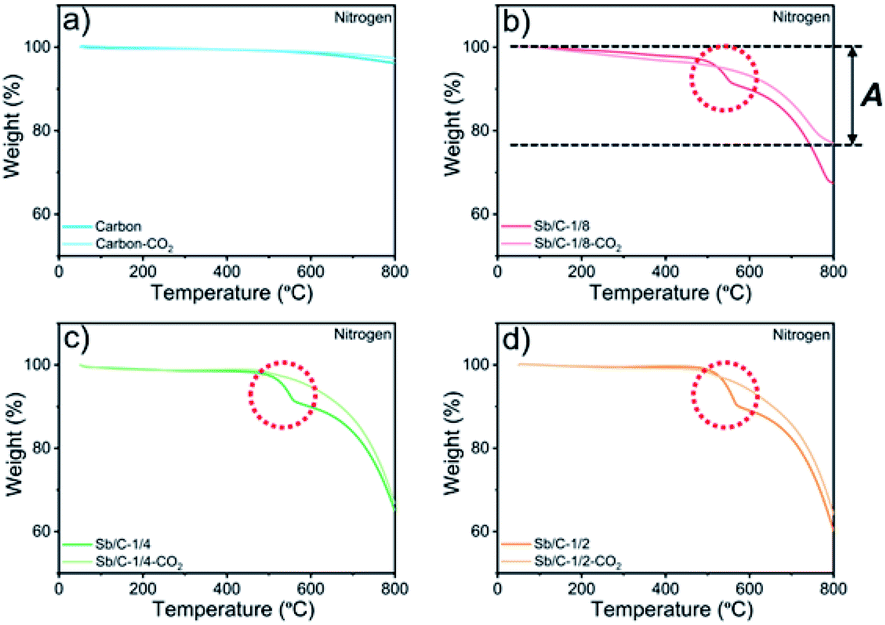 | ||
| Fig. 3 TGA profiles of pure carbon and Sb/C nanohybrids tested under a nitrogen atmosphere: carbon and carbon–CO2 (a), Sb/C-1/8 and Sb/C-1/8-CO2 (b), Sb/C-1/4 and Sb/C-1/4-CO2 (c), Sb/C-1/2 and Sb/C-1/2-CO2 (d). A refers to the percentage of mass loss from Sb evaporation under N2 conditions (50–800 °C). | ||
The results of TGA tests under a N2 atmosphere demonstrate that all Sb/C samples display mass losses from 50 °C to 800 °C. Therefore, it confirms the existence of Sb evaporation during the TGA measurement due to the high temperature. Moreover, it also indicates that the carbon contents of Sb/C nanohybrids calculated using eqn (1) deviates from the actual values. Herein, based on the TGA results under both air and N2 atmospheres, a modified method is proposed to estimate the carbon content in the Sb/C nanohybrid in a more precise way. The percentage of mass loss from Sb evaporation under N2 conditions (50–800 °C) is denoted A; the percentage of mass loss from both carbon combustion and Sb evaporation under air conditions (50–800 °C) is denoted B; the percentage of the amount of oxidized Sb under air conditions (50–800 °C) is denoted C (C = Δ × 2Mw(Sb)/Mw(Sb2O4)); the estimated carbon content of the Sb/C nanohybrid is denoted D. The values of A can be calculated from the results of Fig. 3; while the values of B and C can be derived from the results in Fig. 2, as displayed in the corresponding figures (with the Sb/C-1/8-CO2 sample as an example). There is an assumption that the mass loss due to Sb evaporation in N2 (50–800 °C) is equal to the Sb evaporation in air (50–800 °C). According to eqn (3), the values of D can be calculated (all numbers in this arithmetic are percentages).
| D = B[1 − A(1 − C)] | (3) |
All calculation results are presented in Table 2. According to the abovementioned assumption, A(1 − C) means the percentage of mass loss of Sb evaporation in an air atmosphere. Thus, B[1 − A(1 − C)] refers to the carbon content of the Sb/C nanohybrid. To verify the rationality of this assumption and arithmetic, the carbon content of the Sb/C nanohybrids is further measured using an organic elemental analyzer. The results are shown in Table 2 as well, which are marked as E. The differences between the D values and E values are less than 4%, which could be regarded as allowable measurement errors of the organic elemental analyzer. The differences could be mainly attributed to the different highest testing temperatures of the thermogravimetric analyzer (800 °C) and the organic elemental analyzer (1200 °C) used in this work. Therefore, the rationality of this modified calculation method for carbon content estimation is verified. As the data presented in Table 2, after activation, the carbon content of the Sb/C-1/4-CO2 and Sb/C-1/2-CO2 samples decreased compared to that of their pristine counterparts. However, the carbon content of Sb/C-1/8-CO2 sample is higher than that of Sb/C-1/8 sample, which seems contradictory to the effect of CO2 activation. As discussed above, Sb can evaporate at high temperatures. With CO2 activation at 950 °C, not only the carbon matrix can be etched by CO2 but also the Sb evaporates due to the high temperature.
| Sample | A | B | C | D | E |
|---|---|---|---|---|---|
| Sb/C-1/8 | 32.5% | 88.0% | 9.5% | 62.1% | 62.9% |
| Sb/C-1/8-CO2 | 23.0% | 88.8% | 9.3% | 70.3% | 70.3% |
| Sb/C-1/4 | 35.0% | 82.2% | 14.1% | 57.5% | 54.6% |
| Sb/C-1/4-CO2 | 33.3% | 64.8% | 27.9% | 49.2% | 51.5% |
| Sb/C-1/2 | 39.7% | 59.3% | 32.2% | 43.3% | 39.8% |
| Sb/C-1/2-CO2 | 35.8% | 45.5% | 43.2% | 36.2% | 37.6% |
In addition, there are also some interesting phenomena in the TGA results that are worth discussing. First, as shown in Fig. 2(b)–(d), the rates of mass loss are reduced after 490 °C, but the slopes of the TGA curves are different among those samples. In Fig. 2(b), in the range from 450 °C to 600 °C, the slope of the light-red curve (Sb/C-1/8-CO2 sample) is smaller than that of the red curve (Sb/C-1/8 sample), which is mainly ascribed to a lower Sb content in the Sb/C-1/8-CO2 sample (Table 1). Thus, the mass increment from Sb oxidation of the Sb/C-1/8-CO2 sample is not as distinct as that of the Sb/C-1/8 sample. In the other two control groups, the rates of mass loss of Sb/C-1/4 and Sb/C-1/4-CO2 and of Sb/C-1/2 and Sb/C-1/2-CO2 samples are almost the same. This is mainly attributed to the similar metallic Sb contents of the two samples in the same control group. However, when the temperature is higher than 500 °C, the mass loss rates of Sb/C nanohybrids become faster. Sb begins to evaporate at such high temperatures, leading to an increment in the mass loss rates of samples. Besides, in Fig. 2(c) and (d), there are small sharp angles in the curve of the activated sample, which are marked by red arrows. The reason is that the temperature of Sb oxidation is a little lower than that of the carbon combustion. During the TGA measurement, Sb is oxidized slowly at first, then carbon starts to decompose in large quantities when the temperature reaches its decomposition point. Because of a sudden decrease in mass, the small sharp angle appears in the TGA curve. Moreover, due to high Sb contents in the Sb/C-1/4-CO2 and Sb/C-1/2-CO2 samples as well as the porous carbon matrix after CO2 activation, the small sharp angles exist only in the TGA curves of these two samples. Furthermore, in Fig. 3(b)–(d), it is noticeable that there is a steep slope in each TGA curve of the non-activated samples marked by red dash circles; while there is no comparable phenomenon observed in the corresponding activated samples. It is assumed that this steep slope is derived from an accelerated mass loss process of Sb in samples without activation. For the Sb/C nanohybrids, some Sb species exist on the surface of the carbon matrix. These surface-existing Sb species are more susceptible to high temperatures during the TGA measurement and more easily evaporate than the Sb species embedded in the carbon matrix. Regarding the activated samples, the surface-existing Sb species evaporate away after the high temperature CO2 activation. The mass loss processes are more gradual when a secondary high temperature treatment is applied during the TGA measurement under a nitrogen atmosphere. To confirm this hypothesis, the three pristine Sb/C nanohybrids are studied by TGA twice under a N2 atmosphere. As shown in Fig. S7,† all the first-time TGA curves (dashed lines) display the steep slopes which are consistent with the results in Fig. 3. However, the slopes disappear in the second-time TGA curve (solid lines), because the surface-existing Sb species have already evaporated in the first-time TGA measurement process.
All samples are further tested by nitrogen absorption–desorption measurements to determine the porosity of the samples before and after carbon dioxide activation. As shown in Fig. S8,† all samples exhibit type IV absorption–desorption isotherm with an H3 hysteresis loop. It indicates the existence of mesoporous structures in all the Sb/C nanohybrids.52–54 Compared to the pristine Sb/C nanohybrids, all activated samples exhibit higher Brunauer–Emmett–Teller (BET) specific areas. Besides, the hysteresis loops of activation samples are more obvious than those of the pristine samples, manifesting the existence of more porous structures. Therefore, it confirms that the CO2 activation creates mesoporous structures within the Sb/C nanohybrids due to partial etching of the carbon matrix. Detailed data including the BET specific area and pore volume (based on sample mass) of all eight samples are summarized in Table 3. The solid Sb nanoparticles cannot make significant contribution to the porous structure and specific surface area. Therefore, the increased specific surface area mainly originates from the carbon matrix and the interface between the carbon matrix and Sb nanoparticles. The BET specific area and pore volume of all samples calculated against the carbon mass are also displayed in Table 3, where the carbon mass is taken from the data listed in Table 2 (E). The specific surface area and pore volume against the carbon mass better reflect the actual porosity of the Sb/C nanohybrids. Therefore, the BET specific area and pore volume addressed later refer to the values calculated against the carbon mass. The BET specific areas of the pristine samples increase gradually along with increasing amount of Sb, which is mainly due to more interfaces generated between the carbon matrix and Sb nanoparticles. However, the BET specific area of the Sb/C-1/2 sample declines slightly compared to that of the Sb/C-1/4 sample. For the Sb/C-1/2 sample, the agglomeration of Sb nanoparticles is more pervasive than that of the Sb/C-1/4 sample, which leads to a reduced specific surface area originating from the interface between the carbon matrix and Sb nanoparticles. Compared with the pristine counterparts, the extent of specific surface area increase is 82.4%, 48.8%, and 66.7% with respect to the Sb/C-1/8-CO2, Sb/C-1/4-CO2 and Sb/C-1/2-CO2 samples. However, the pure carbon sample exhibits a less specific surface area increase after carbon dioxide activation compared to those of the Sb/C nanohybrids. It is ascribed to the absence of interfaces between the carbon matrix and Sb nanoparticles in the pure carbon samples. The pore volumes of all samples are increased after CO2 activation. The change in the pore volumes regarding different Sb/C nanohybrids before and after carbon dioxide activation follows the trend of the specific surface area.
| Sample | BET SA based on SM (m2g−1) | BET SA based on CM (m2g−1) | PS (nm) | PV based on SM (cm3 g−1) | PV based on CM (cm3 g−1) |
|---|---|---|---|---|---|
| Carbon | 350.4 | 350.4 | 11.1 | 0.19 | 0.19 |
| Carbon–CO2 | 391.6 | 391.4 | 11.0 | 0.20 | 0.20 |
| Sb/C-1/8 | 237.5 | 377.6 | 11.1 | 0.14 | 0.22 |
| Sb/C-1/8-CO2 | 484.1 | 688.6 | 11.0 | 0.27 | 0.38 |
| Sb/C-1/4 | 255.0 | 467.0 | 11.1 | 0.16 | 0.29 |
| Sb/C-1/4-CO2 | 357.8 | 694.7 | 11.0 | 0.20 | 0.39 |
| Sb/C-1/2 | 180.1 | 452.5 | 11.2 | 0.11 | 0.28 |
| Sb/C-1/2-CO2 | 283.7 | 754.5 | 11.1 | 0.18 | 0.48 |
The Barrett–Joyner–Halenda (BJH) pore size distribution curves (adsorption) of Sb/C nanohybrids exhibit uniform pore size distribution profiles (Fig. S9†). According to the pore size displayed in Table 3, all samples have a similar pore size distribution. The diameter of the mesopores is around 11 nm. The mesopores are supposed to exist within the carbon matrix and at the interface between the carbon matrix and Sb nanoparticles. All activated samples display more intensive characteristic peaks located around 11 nm compared to the pristine counterparts. In addition, the pore volumes of the activated samples are also higher than those of the corresponding non-activated samples. This implies that CO2 activation can create more mesopores within the Sb/C nanohybrids. In summary, the nitrogen adsorption and desorption tests prove that the CO2 activation treatment effectively increases the specific surface area due to the formation of additional mesopores within the Sb/C nanohybrids.
Electrochemical performance
The electrochemical performance of the Sb/C nanohybrids as LIB anodes are investigated systematically. The cyclic voltammetry (CV) curves of the pure carbon and Sb/C nanohybrid samples are displayed in Fig. S10.† The discharge/charge curves of each sample for the 1st, 2nd, 50th and 100th cycle under a constant current density of 66 mA g−1 are presented in Fig. S11,† and the detailed data are summarized in Table S2.† The related discussion can be found in the ESI.† The cycling and rate performance of all samples are displayed in Fig. 4 and 5, and the detailed data are presented in Table S3.† The current density of the cycling tests is set to 66 mA g−1. The current densities of the rate performance tests ranged from 66 mA g−1 to 3300 mA g−1, and the cells are tested for five cycles at each current density. As shown in Fig. 4, the cycling performance changing trend of these samples is consistent with the results of discharge/charge curves. Due to the lower amount of amorphous carbon in the activated samples, the reversible capacities slightly decrease compared to the pristine counterparts. With the increase of Sb content, the volumetric effect becomes drastic gradually, leading to compromised cycling stability. The capacity retentions after 200 cycles of the carbon, carbon–CO2, Sb/C-1/8, Sb/C-1/8-CO2, Sb/C-1/4, Sb/C-1/4-CO2, Sb/C-1/2 and Sb/C-1/2-CO2 samples are 94%, 98%, 84%, 90%, 24%, 26%, 19% and 38%, respectively. Because of the more porous structure created by CO2 activation, the activated samples exhibit better cycling stability than their counterparts. Generally, the samples with a higher Sb content display a worse capacity retention, which is roughly consistent with the above regulation.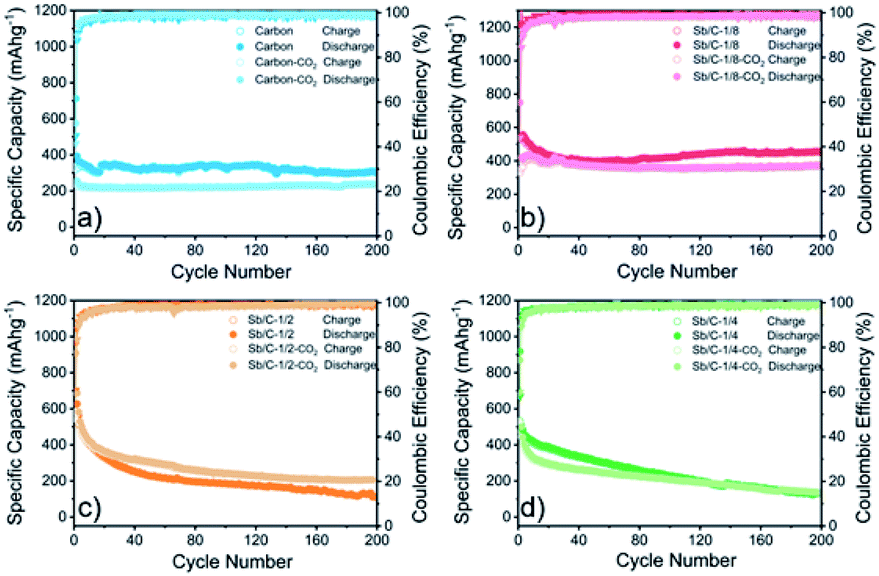 | ||
| Fig. 4 Cycling performance of pure carbon and Sb/C nanohybrid anodes in LIBs measured at 66 mA g−1: carbon and carbon–CO2 (a), Sb/C-1/8 and Sb/C-1/8-CO2 (b), Sb/C-1/4 and Sb/C-1/4-CO2 (c), Sb/C-1/2 and Sb/C-1/2-CO2 (d). | ||
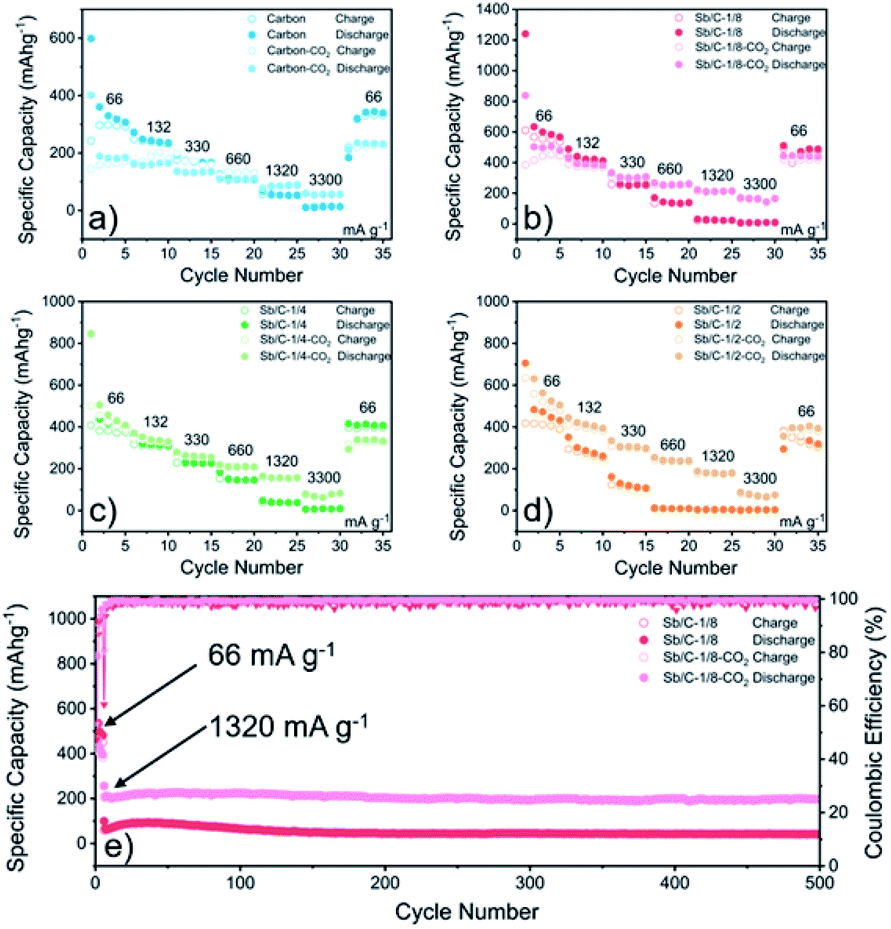 | ||
| Fig. 5 Rate performance of pure carbon and Sb/C nanohybrid anodes in LIBs measured at 66 mA g−1, 132 mA g−1, 330 mA g−1, 660 mA g−1, 1320 mA g−1, and 3300 mA g−1, respectively: carbon and carbon–CO2 (a), Sb/C-1/8 and Sb/C-1/8-CO2 (b), Sb/C-1/4 and Sb/C-1/4-CO2 (c), Sb/C-1/2 and Sb/C-1/2-CO2 (d). Cycling performance of (e) Sb/C-1/8 and Sb/C-1/8-CO2 in LIBs measured at 66 mA g−1 for the first five cycles and at 1320 mA g−1 for the following cycles. | ||
As to the rate performance, when the current density is not larger than 132 mA g−1, the specific capacities of the pristine samples are higher than those of corresponding activated samples. When the current density is above 132 mA g−1, the activated samples exhibit a better rate performance than the corresponding pristine samples. Under the current densities of 660 mA g−1, 1320 mA g−1 and 3300 mA g−1, the average reversible capacities of the Sb/C-1/8-CO2 sample are 254 mA h g−1, 210 mA h g−1, and 160 mA h g−1, respectively. However, at the same current densities, the pristine Sb/C-1/8 sample can only retain average reversible capacities of 137 mA h g−1, 23 mA h g−1, and 9 mA h g−1, respectively. The rate performance of other samples treated with CO2 activation also exhibit similar advantages over the corresponding pristine Sb/C nanohybrids. At high current densities, the average reversible capacities of activated samples are significantly increased compared to those of the non-activated counterparts. Especially, when the current densities are as high as 1320 mA g−1 and 3300 mA g−1, the average reversible capacities of the activated samples are 1 or 2 orders of magnitude higher than those of the pristine counterparts. For example, at a current density of 1320 mA g−1, the average reversible capacities of Sb/C-1/4-CO2 and Sb/C-1/2-CO2 samples are 156 mA h g−1 and 177 mA h g−1, respectively. However, the average capacities of the pristine Sb/C-1/4 and Sb/C-1/2 samples are merely 38 mA h g−1 and 5 mA h g−1, respectively. When the current density continues to increase to 3300 mA g−1, the average reversible specific capacities of Sb/C-1/4-CO2 and Sb/C-1/2-CO2 samples are both 73 mA h g−1; while the capacities of Sb/C-1/4 and Sb/C-1/2 samples are only 7 mA h g−1 and 4 mA h g−1, respectively. The specific capacities of all samples can almost recover to the values when the current density is restored to 66 mA g−1. Fig. S12(c, d)† presents the rate curves of Sb/C-1/8 and Sb/C-1/8-CO2 samples, respectively. The superior rate characteristic of the Sb/C-1/8-CO2 sample is clearly demonstrated. The capacity proportion that originated from the plateau region of the Sb/C-1/8 sample declines more rapidly than that of the Sb/C-1/8-CO2 sample as the current density increases. It suggests that the kinetics of lithium ion insertion is weakened within the Sb/C-1/8 sample, where the lithium ions cannot be absorbed by the active sites.55 After CO2 activation, the high specific surface area and abundant mesopores within the Sb/C-1/8-CO2 sample facilitate lithium ion transportation, leading to enhanced rate performance. A similar phenomenon is also observed in other control groups, as shown in Fig. S12.† The excellent rate performance of the activated samples under a high current density is mainly attributed to their high specific surface area and abundant mesoporous structures in the carbon matrix, as illustrated in Scheme 2. Without CO2 activation, the excessive carbon content of Sb/C nanohybrids limits fast diffusion and transportation of Li ions to access the active sites (Sb nanoparticles). In contrast, the porous structure created by CO2 activation can significantly improve the electrochemical kinetics of the lithiation/delithiation process, enlarge the contact area between the electrolyte and active material, and greatly reduce the transport distance of the charge carriers. With carbon dioxide activation, the rate performance of Sb/C nanohybrids is significantly enhanced with a tiny sacrifice of the cycling performance.
 | ||
| Scheme 2 Schematic illustration of the effect of CO2 activation on the rate performance of Sb/C nanohybrid anode. | ||
Due to the most distinct rate performance improvement, the Sb/C-1/8-CO2 sample is further selected to carry out long-cycle test at a high current density, while the Sb/C-1/8 sample is also tested to act as a contrast. The cells are first cycled for five cycles at 66 mA g−1, followed by cycling at 1320 mA g−1 until 500 cycles are complete. As shown in Fig. 5(e), during the first five cycles, the average reversible capacity of the Sb/C-1/8-CO2 sample is slightly lower than that of the Sb/C-1/8 sample, which is consistent with the result shown in Fig. 5(b). However, when the current density increases to 1320 mA g−1, the activated sample exhibits higher capacities immediately. During the long-cycle test at 1320 mA g−1, both samples present good cycling stability. The reversible specific capacity of the Sb/C-1/8-CO2 sample remains 197 mA h g−1 after 500 cycles with a capacity retention of 96% against the capacity of the sixth cycle. However, the Sb/C-1/8 sample can only maintain a reversible capacity of 43 mA h g−1 after 500 cycles, corresponding to the capacity retention of 72% against the value of the sixth cycle.
To further understand the origin of the enhanced rate performance brought by the CO2 activation, the lithium-storage kinetics are studied based on CV measurements. Fig. 6(a) and (b) show the CV curves of Sb/C-1/8 and Sb/C-1/8-CO2 samples at scan rates from 0.1 mV s−1 to 1.0 mV s−1. The results of CV curves display similar shapes with broad cathodic and anodic peaks. The peak current (Ip) increases with increasing scan rate (v). In Fig. 6(c), the apparent diffusion coefficient of lithium ions (DLi+) can be estimated from the relationship between the peak current (Ip) of cathodic peaks (C1 and C2) and the square root of scan rate (v1/2). On the basis of CV data and eqn (4)55,56
| Ip = 2.69 × 105An3/2C0DLi+1/2v1/2 | (4) |
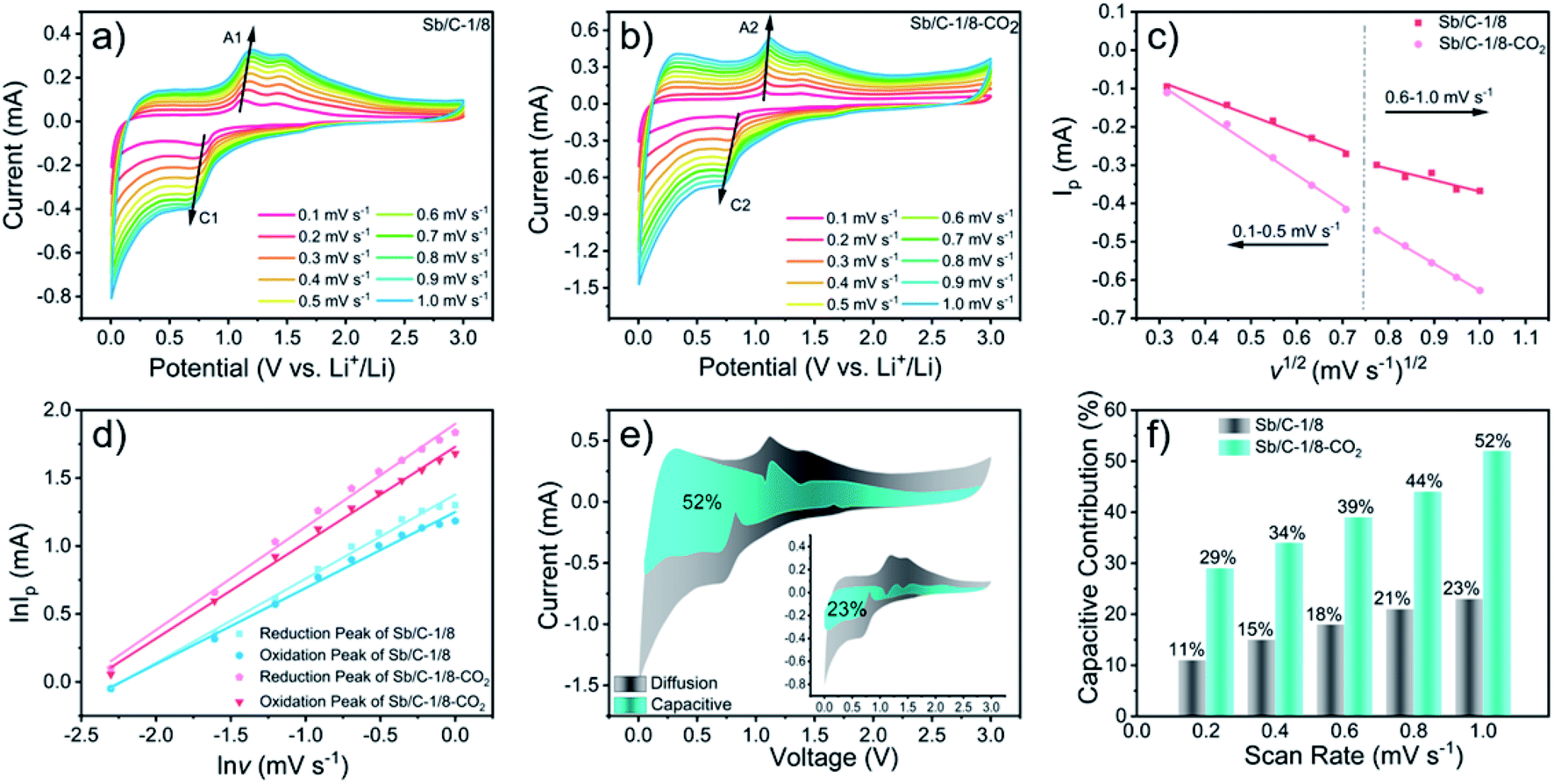 | ||
| Fig. 6 CV curves of Sb/C-1/8 (a) and Sb/C-1/8-CO2 (b) samples measured at various sweep rates (from 0.1 to 1.0 mV s−1). Relationship (c) between the peak current (Ip) and the square root of scan rate (ν1/2) of the cathodic peaks of both samples. Linear relationship (d) between log Ip (logarithm peak currents) and log v (logarithm scan rate) of cathodic and anodic peaks in the scan rate region (from 0.1 to 1.0 mV s−1). Cyclic voltammogram (e) of Sb/C-1/8-CO2 and Sb/C-1/8 (inset) samples with the capacitive contribution at a scan rate of 1.0 mV s−1. Normalized contribution ratio (f) of specific capacities of Sb/C-1/8-CO2 and Sb/C-1/8 samples at different scan rates. | ||
Generally, the relationship between the measured current (i) and the scan rate (v) can be expressed as eqn (5)57
| i = avb | (5) |
| i(V) = k1v1/2 + k2v | (6) |
By changing eqn (6) to
| i(V)/v1/2 = k1 + k2v1/2 | (7) |
The cycling and rate performances of Sb/C-1/8 and Sb/C-1/8-CO2 samples in SIBs are also measured, and the results are displayed in Fig. 7. The cycling performance of the activated sample is clearly better than that of the non-activated one. The initial specific charge and discharge capacities of the Sb/C-1/8 sample are 248 mA h g−1 and 987 mA h g−1, respectively; while those of the Sb/C-1/8-CO2 sample are 106 mA h g−1 and 422 mA h g−1, respectively. The difference between initial capacities of the two samples is similar to their exhibitions in LIBs. The lower initial capacities of the activated sample could be attributed to the lower amount of amorphous carbon inside the carbon matrix. Additionally, the larger diameter of the sodium ion could magnify such effect, leading to more obvious difference between the initial capacities of non-activated and activated samples in SIBs than those in LIBs. After 500 cycles, the reversible specific capacity of the Sb/C-1/8 sample is 27 mA h g−1 with a capacity retention of 11%; while the capacity of the Sb/C-1/8-CO2 sample is 120 mA h g−1 with a capacity retention of 80% (calculated by the stabilized value after initial several cycles). Thus, the cyclic stability of the Sb/C-1/8-CO2 sample is much better than that of the Sb/C-1/8 sample. The porous structures of the activated sample provide more buffering space for alleviating the volume expansion of Sb upon sodiation. In regard to the rate performance, the Sb/C-1/8-CO2 sample exhibits a better performance. When the current density increases to 330 mA g−1, the reversible specific capacity of the Sb/C-1/8 sample is almost zero. The Sb/C-1/8-CO2 sample can retain reversible capacities of 85 mA h g−1, 60 mA h g−1 and 35 mA h g−1 at 330 mA g−1, 660 mA g−1, and 1320 mA g−1, respectively. The drastically enhanced rate performance of the activated sample originates from the high specific surface area and abundant mesoporous structures.
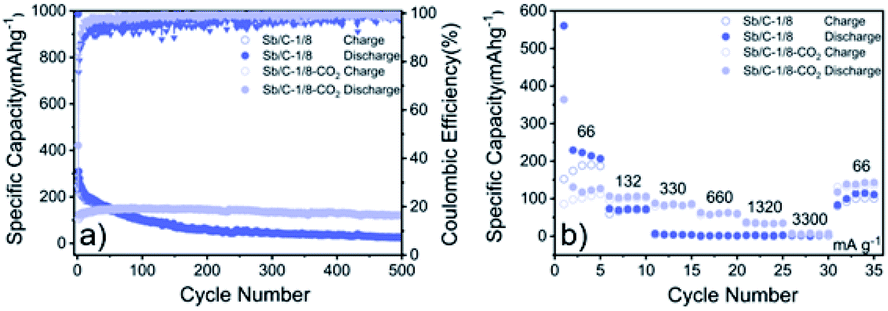 | ||
| Fig. 7 Cycling (a) and rate (b) performances of Sb/C-1/8 and Sb/C-1/8-CO2 samples in SIBs. Cycling tests are measured at 66 mA g−1, and rate tests are measured from 66 mA g−1 to 3300 mA g−1. | ||
Conclusions
In summary, a new concept for improving the rate performance of Sb/C nanohybrid anodes is developed in this work. Sb/C nanohybrids are first synthesized using dimethacrylate monomers as a solvent and a carbon source, followed by calcination under an inert atmosphere and CO2 activation treatment. A series of samples with different contents of Sb have been synthesized and characterized systematically. According to the results of TEM and EDX, Sb nanoparticles are embedded in the carbon matrix homogeneously. The CO2 activation treatment removes the disordered carbon more than the graphitic carbon structure, as suggested by Raman spectroscopy. Thus, CO2 activation reduces the carbon content, modifies the microstructure of the carbon matrix, and creates mesopores within the carbon matrix as well. The specific surface area and specific pore volumes are increased due to carbon dioxide activation, which has been confirmed by the nitrogen absorption–desorption measurements. Particularly, based on the TGA test results, a new calculation method for carbon content estimation of the Sb/C nanohybrid is proposed in this work, where the rationality and accuracy are confirmed by the elemental analysis. In terms of electrochemical performance, on the basis of maintaining original cycling stability, the rate performance of Sb/C nanohybrid anodes are enhanced significantly after CO2 activation. Particularly, the Sb/C-1/8-CO2 sample can maintain an average capacity of 160 mA h g−1 at a high current density of 3300 mA g−1, whereas the control sample (Sb/C-1/8) could only gain a capacitance of 9 mA h g−1 under the same testing conditions. The increased specific surface area and more mesoporous structures created by CO2 activation effectively shorten the diffusion distances for charge carries, leading to improved rate performance. Through a series of CV tests at different scanning rates, the apparent diffusion coefficient of Li ions and contributions of the capacitive capacities are studied. The results demonstrate that the CO2 activation process plays a key role in improving the rate performance of the Sb/C nanohybrid anodes. Besides, the long cycling performance under high current densities of the Sb/C nanohybrid anodes are also studied in this work. The activated Sb/C nanohybrid anodes still exhibit distinct advantages over the corresponding pristine samples. When applied as the anodes of SIBs, the activated Sb/C nanohybrid retains a reversible capacity of 120 mA h g−1 after 500 cycles, corresponding to a capacity retention over 80%. Such performance is significantly higher than that of the non-activated Sb/C nanohybrid SIB anode. The knowledge generated in this work provides a fundamental understanding on the CO2 activation process. Based on the comprehensive studies in this work, the impacts of CO2 activation treatment on the structure, composition, and performance of the Sb/C composite electrode are clearly revealed. It is applicable not only to the specific Sb/C system but also to many various functional materials in principle. It offers a powerful tool to tune the microstructure and performance of the carbon compositing-based materials in a green facile scalable way, where a tedious post-treatment process is totally circumvented. It is instructive to the advanced materials design and synthesis in various fields where carbon compositing plays an important role, such as energy storage, energy conversion, and catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Key R&D Program of China (Grant No. 2016YFB0100100), the National Natural Science Foundation of China (51702335 and 21773279), the Zhejiang Non-profit Technology Applied Research Program (LGG19B010001), the Ningbo Municipal Natural Science Foundation (2018A610084), the CAS-EU S&T Cooperation Partner Program (174433KYSB20150013), and Key Laboratory of Bio-based Polymeric Materials of Zhejiang Province. S. L. acknowledges the China Scholarship Council (CSC) for funding and Ms Zhuoyi Feng for the hand-drawn style TOC. P. M.-B. acknowledges funding from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via International Research Training Group 2022 Alberta/Technical University of Munich International Graduate School for Environmentally Responsible Functional Materials (ATUMS) and by TUM.battery.References
- W.-J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- Z. Hou and A. Li, J. Mater. Sci. Technol., 2009, 20, 743–745 Search PubMed.
- M. N. Obrovac and V. L. Chevrier, Chem. Rev., 2014, 114, 11444–11502 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS.
- S. Y. Hong, Y. Kim, Y. Park, A. Choi, N.-S. Choi and K. T. Lee, Energy Environ. Sci., 2013, 6, 2067–2081 RSC.
- N. Wang, Z. Bai, Y. Qian and J. Yang, Adv. Mater., 2016, 28, 4126–4133 CrossRef CAS.
- W. M. Stanley, Chem. Rev., 2004, 10, 4271–4302 Search PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS.
- Z. Liu, X.-Y. Yu, X. W. (David) Lou and U. Paik, Energy Environ. Sci., 2016, 9, 2314–2318 RSC.
- L. Fan and X. Li, Nano Energy, 2018, 53, 630–642 CrossRef CAS.
- X. Liu, Y. Hao, J. Shu, H. M. K. Sari, L. Lin, H. Kou, J. Li, W. Liu, B. Yan, D. Li, J. Zhang and X. Li, Nano Energy, 2019, 57, 414–423 CrossRef CAS.
- D. Xiong, X. Li, Z. Bai and S. Lu, Small, 2018, 14, 1703419 CrossRef.
- J. L. Gómez-Cámer, C. Villevieille and P. Novák, J. Mater. Chem. A, 2013, 1, 13011–13016 RSC.
- Y. N. Ko and Y. C. Kang, Chem. Commun., 2014, 50, 12322–12324 RSC.
- W. Luo, S. Lorger, B. Wang, C. Bommier and X. Ji, Chem. Commun., 2014, 50, 5435–5437 RSC.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, Energy Environ. Sci., 2014, 7, 323–328 RSC.
- H. Hou, M. Jing, Y. Yang, Y. Zhu, L. Fang, W. Song, C. Pan, X. Yang and X. Ji, ACS Appl. Mater. Interfaces, 2014, 6, 16189–16196 CrossRef CAS.
- H. Kim and J. Cho, Chem. Mater., 2008, 20, 1679–1681 CrossRef CAS.
- J. Liu, L. Yu, C. Wu, Y. Wen, K. Yin, F.-K. Chiang, R. Hu, J. Liu, L. Sun, L. Gu, J. Maier, Y. Yu and M. Zhu, Nano Lett., 2017, 17, 2034–2042 CrossRef CAS.
- N. Wang, Z. Bai, Y. Qian and J. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 447–454 CrossRef CAS.
- M. Wang, Z. Yang, J. Wang, W. Li, L. Gu and Y. Yu, Small, 2015, 11, 5381–5387 CrossRef CAS.
- Z. Yi, Q. Han, P. Zan, Y. Wu, Y. Cheng and L. Wang, J. Power Sources, 2016, 331, 16–21 CrossRef CAS.
- Z. Liu, X.-Y. Yu, X. W. (David) Lou and U. Paik, Energy Environ. Sci., 2016, 9, 2314–2318 RSC.
- X. Xu, Z. Dou, E. Gu, L. Si, X. Zhou and J. Bao, J. Mater. Chem. A, 2017, 5, 13411–13420 RSC.
- J. Song, P. Yan, L. Luo, X. Qi, X. Rong, J. Zheng, B. Xiao, S. Feng, C. Wang, Y.-S. Hu, Y. Lin, V. L. Sprenkle and X. Li, Nano Energy, 2017, 40, 504–511 CrossRef CAS.
- Q. Yang, J. Zhou, G. Zhang, C. Guo, M. Li, Y. Zhu and Y. Qian, J. Mater. Chem. A, 2017, 5, 12144–12148 RSC.
- S. Qiu, X. Wu, L. Xiao, X. Ai, H. Yang and Y. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 1337–1343 CrossRef CAS.
- W. Luo, P. Zhang, X. Wang, Q. Li, Y. Dong, J. Hua, L. Zhou and L. Mai, J. Power Sources, 2016, 304, 340–345 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS.
- H. Lv, S. Qiu, G. Lu, Y. Fu, X. Li, C. Hu and J. Liu, Electrochim. Acta, 2015, 151, 214–221 CrossRef CAS.
- X. Wang, J.-Q. Meng, M. Wang, Y. Xiao, R. Liu, Y. Xia, Y. Yao, E. Metwalli, Q. Zhang, B. Qiu, Z. Liu, J. Pan, L.-D. Sun, C.-H. Yan, P. Müller-Buschbaum and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2015, 7, 24247–24255 CrossRef CAS.
- Y. Xiao, X. Wang, Y. Xia, Y. Yao, E. Metwalli, Q. Zhang, R. Liu, B. Qiu, M. Rasool, Z. Liu, J.-Q. Meng, L.-D. Sun, C.-H. Yan, P. Müller-Buschbaum and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2014, 6, 18461–18468 CrossRef CAS.
- X. Wang, Y.-J. Cheng, Q. Ji, S. Liang, L. Ma, Z. Xu, X. Zuo, J.-Q. Meng, J. Zhu, P. Müller-Buschbaum and Y. Xia, Energy Technol., 2020, 6(8), 2000034 CrossRef.
- X. Wang, L. Ma, Q. Ji, J.-Q. Meng, S. Liang, Z. Xu, M. Wang, X. Zuo, Y. Xiao, J. Zhu, Y. Xia, P. Müller-Buschbaum and Y.-J. Cheng, Adv. Mater. Interfaces, 2019, 6, 1900335 CrossRef.
- X. Wang, D. Zhao, C. Wang, Y. Xia, W. Jiang, S. Xia, S. Yin, X. Zuo, E. Metwalli, Y. Xiao, Z. Sun, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, Chem.–Asian J., 2019, 14, 1557–1569 CrossRef CAS.
- L. Zheng, X. Wang, Y. Xia, S. Xia, E. Metwalli, B. Qiu, Q. Ji, S. Yin, S. Xie, K. Fang, S. Liang, M. Wang, X. Zuo, Y. Xiao, Z. Liu, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2018, 10, 2591–2602 CrossRef CAS.
- S. Liang, X. Wang, Y. Xia, S. Xia, E. Metwalli, B. Qiu, Q. Ji, S. Yin, S. Xie, K. Fang, L. Zheng, M.-M. Wang, X. Zuo, R. Li, Z. Liu, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, Acta Metall. Sin., 2018, 31, 910–922 CrossRef CAS.
- S. Yin, D. Zhao, Q. Ji, Y. Xia, S. Xia, X. Wang, M. Wang, J. Ban, Y. Zhang, E. Metwalli, X. Wang, Y. Xiao, X. Zuo, S. Xie, K. Fang, S. Liang, L. Zheng, B. Qiu, Z. Yang, Y. Lin, L. Chen, C. Wang, Z. Liu, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, ACS Nano, 2018, 12, 861–875 CrossRef CAS.
- M. Wang, Y. Xia, X. Wang, Y. Xiao, R. Liu, Q. Wu, B. Qiu, E. Metwalli, S. Xia, Y. Yao, G. Chen, Y. Liu, Z. Liu, J.-Q. Meng, Z. Yang, L.-D. Sun, C.-H. Yan, P. Müller-Buschbaum, J. Pan and Y.-J. Cheng, ACS Appl. Mater. Interfaces, 2016, 8, 13982–13992 CrossRef CAS.
- X. Zuo, X. Wang, Y. Xia, S. Yin, Q. Ji, Z. Yang, M. Wang, X. Zheng, B. Qiu, Z. Liu, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, J. Power Sources, 2019, 412, 93–104 CrossRef CAS.
- X. Wang, Y. Xia, X. Zuo, S. J. Schaper, S. Yin, Q. Ji, S. Liang, Z. Yang, S. Xia, Y. Xiao, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, Ceram. Int., 2019, 45, 14327–14337 CrossRef CAS.
- L. Chang, Z. Fu, M. Liu, L. Yuan, J. Wei, Y. wei He, X. Liu and C. Wang, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2014, 29, 213–218 CrossRef CAS.
- S.-Z. Zeng, Y. Yao, X. Zeng, Q. He, X. Zheng, S. Chen, W. Tu and J. Zou, J. Power Sources, 2017, 357, 11–18 CrossRef CAS.
- K. Xia, Q. Gao, C. Wu, S. Song and M. Ruan, Carbon, 2007, 45, 1989–1996 CrossRef CAS.
- M. Kunowsky, Á. Linares-Solano, A. Garcia-Gomez, V. Barranco, J. M. Rojo and J. D. Carruthers, Int. J. Appl. Ceram. Technol., 2015, 12, E127–E132 CrossRef CAS.
- H. D. Pham, V. H. Pham, T. V. Cuong, T.-D. Nguyen-Phan, J. S. Chung, E. W. Shin and S. Kim, Chem. Commun., 2011, 47, 9672–9674 RSC.
- K. Xia, Q. Gao, C. Wu, S. Song and M. Ruan, Carbon, 2007, 45, 1989–1996 CrossRef CAS.
- K. Xia, Q. Gao, S. Song, C. Wu, J. Jiang, J. Hu and L. Gao, Int. J. Hydrogen Energy, 2008, 33, 116–123 CrossRef CAS.
- K. Xia, Q. Gao, J. Jiang and J. Hu, Carbon, 2008, 46, 1718–1726 CrossRef CAS.
- L. Chang, Z. Fu, M. Liu, L. Yuan, J. Wei, Y. wei He, X. Liu and C. Wang, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2014, 29, 213–218 CrossRef CAS.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, Energy Environ. Sci., 2014, 7, 323–328 RSC.
- Z. Yi, Q. Han, Y. Cheng, Y. Wu and L. Wang, Chem. Commun., 2016, 52, 7691–7694 RSC.
- Y. Xia, Z. Xiao, X. Dou, H. Huang, X. Lu, R. Yan, Y. Gan, W. Zhu, J. Tu, W. Zhang and X. Tao, ACS Nano, 2013, 7, 7083–7092 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- P. Lu, Y. Sun, H. Xiang, X. Liang and Y. Yu, Adv. Energy Mater., 2018, 8, 1702434 CrossRef.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- H. Zou, X. Liang, X. Feng and H. Xiang, ACS Appl. Mater. Interfaces, 2016, 8, 21407–21416 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS.
- L. Yang, X. Li, S. He, G. Du, X. Yu, J. Liu, Q. Gao, R. Hu and M. Zhu, J. Mater. Chem. A, 2016, 4, 10842–10849 RSC.
- L. C. Yang, W. Sun, Z. W. Zhong, J. W. Liu, Q. S. Gao, R. Z. Hu and M. Zhu, J. Power Sources, 2016, 306, 78–84 CrossRef CAS.
- S. Li, J. Qiu, C. Lai, M. Ling, H. Zhao and S. Zhang, Nano Energy, 2015, 12, 224–230 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- D. Chao, C. Zhu, P. Yang, X. Xia, J. Liu, J. Wang, X. Fan, S. V. Savilov, J. Lin, H. J. Fan and Z. X. Shen, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- D. Chao, P. Liang, Z. Chen, L. Bai, H. Shen, X. Liu, X. Xia, Y. Zhao, S. V. Savilov, J. Lin and Z. X. Shen, ACS Nano, 2016, 10, 10211–10219 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1na00008j |
| This journal is © The Royal Society of Chemistry 2021 |