
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning protein adsorption on graphene surfaces via laser-induced oxidation†
Efstratios D.
Sitsanidis
 a,
Johanna
Schirmer
a,
Aku
Lampinen
a,
Kamila K.
Mentel
a,
Vesa-Matti
Hiltunen
b,
Visa
Ruokolainen
c,
Andreas
Johansson
ab,
Pasi
Myllyperkiö
a,
Maija
Nissinen
a and
Mika
Pettersson
*a
a,
Johanna
Schirmer
a,
Aku
Lampinen
a,
Kamila K.
Mentel
a,
Vesa-Matti
Hiltunen
b,
Visa
Ruokolainen
c,
Andreas
Johansson
ab,
Pasi
Myllyperkiö
a,
Maija
Nissinen
a and
Mika
Pettersson
*a
aDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, P. O. Box 35, FI-40014 JYU, Finland. E-mail: mika.j.pettersson@jyu.fi
bDepartment of Physics, Nanoscience Center, University of Jyväskylä, P. O. Box 35, FI-40014 JYU, Finland
cDepartment of Biological and Environmental Sciences, Nanoscience Center, University of Jyväskylä, P. O. Box 35, FI-40014 JYU, Finland
First published on 22nd February 2021
Abstract
An approach for controlled protein immobilization on laser-induced two-photon (2P) oxidation patterned graphene oxide (GO) surfaces is described. Selected proteins, horseradish peroxidase (HRP) and biotinylated bovine serum albumin (b-BSA) were successfully immobilized on oxidized graphene surfaces, via non-covalent interactions, by immersion of graphene-coated microchips in the protein solution. The effects of laser pulse energy, irradiation time, protein concentration and duration of incubation on the topography of immobilized proteins and consequent defects upon the lattice of graphene were systemically studied by atomic force microscopy (AFM) and Raman spectroscopy. AFM and fluorescence microscopy confirmed the selective aggregation of protein molecules towards the irradiated areas. In addition, the attachment of b-BSA was detected by a reaction with fluorescently labelled avidin-fluorescein isothiocyanate (Av-FITC). In contrast to chemically oxidized graphene, laser-induced oxidation introduces the capability for localization on oxidized areas and tunability of the levels of oxidation, resulting in controlled guidance of proteins by light over graphene surfaces and progressing towards graphene microchips suitable for biomedical applications.
Introduction
Graphene is a carbon allotrope, consisting of a monolayer of sp2 hybridized carbon atoms bound together in a honeycomb lattice. Due to its mechanical, physicochemical, optical, electronic and biological properties, this two-dimensional (2D) nanomaterial has attracted much attention in the biomedical field. Indeed, functionalized graphene-based nanostructures have been already reported for tissue engineering, cancer therapy, drug delivery, regenerative medicine, imaging, cytotoxicity and biosensing applications.1–5Graphene and its derivatives (graphene oxide, GO, reduced graphene oxide, rGO) can be prepared in a scalable and cost-effective manner. They demonstrate a strong antimicrobial activity and their amphiphilic nature allows conjugation with biomolecules. At the same time, a high specific surface area results in the enhancement of cell adhesion, proliferation and in certain cases, cell differentiation.6–8 To this extent, graphene-based implantable materials have already led to advanced therapeutic approaches by fixing or replacing tissues and organs damaged by accident or disease. Such sophisticated bionic devices exploit the synergy of biology with electronics (bioelectronics) and focus on restoring vision, treating spinal cord injuries and ameliorating neurodegenerative diseases.9 Certain devices (neural interfaces) which have been already clinically developed, consist of cortical electrodes, retina implants, spinal cord and vagal nerve stimulators, deep brain stimulation (DBS) and cochlear implants.10
In terms of biocompatibility, the response of the human body towards injectable or implantable graphene-based materials is not yet fully understood. As different physicochemical properties may generate different interactions with biological systems, all implantable devices need to meet specific key requirements such as minimal inflammation, an adequate signal-to-noise ratio and mechanical compliance towards the affected tissues.10,11 Any chemical contamination during their formulation process can cause adverse biological reactions therefore, to minimize potential toxic effects, an effective sterilization and depyrogenation procedure should be followed. In addition, their stability and functionality need to be assessed over time.12 Although the phenomenon of biocompatibility is complex and its mechanisms not fully known, for those materials exposed to the human body, it is associated with the adsorption of proteins on their surface.13
While working towards bioinspired surfaces, our study aims at the preparation of biocompatible graphene-based microchips, to induce graphene–cell interactions, as potential implantable devices. Here, we describe the selective functionalization of laser-induced GO patterns on graphene surfaces by protein adsorption via non-covalent interactions.
Our protein models were HRP enzyme and b-BSA. HRP was previously immobilized on GO sheets in aqueous suspensions by Zhang et al.14 Based on their findings, certain pH values favoured either electrostatic interactions or hydrogen bonding, when the pH was below or above the enzyme's isoelectric point (pI = 7.2), respectively. According to the authors, although physical adsorption resulted in conformational changes of the HRP molecule, its observed catalytic activity was mainly determined by the type of interactions with the GO sheets. In addition, loading the enzyme on GO sheets did not affect its activity, provided that the surface of the sheets was not crowded, thus allowing free diffusion of the HRP substrate and the respective product molecules.
BSA is the most abundant protein found in the blood plasma, while its adsorption over the surface of implantable materials passivates the reaction of the human body.13 Huang et al. reported that incubation of GO with 10% fetal bovine serum reduced significantly its cytotoxicity.15 Furthermore, in silico studies based on BSA's adsorption over graphene, showed that free adsorption occurred with little structural rearrangements.16,17 In contrast, forced adsorption simulations led to orientations capable of preserving the structural properties of the majority of the protein's binding sites. Herein, we used the biotinylated derivative (b-BSA) due to its high affinity towards avidin to detect its immobilization by the reaction with the fluorescently labelled Av-FITC.
In our case, the immobilization of proteins is based on non-covalent interactions by immersion of the laser-patterned microchips into protein solutions. In contrast to covalent binding, supramolecular forces maintain the structure and the properties of graphene. Physical adsorption is caused by weak interactions among graphene and protein molecules, such as H-bonding, van der Waals forces, hydrophobic, electrostatic and aromatic–aromatic (π–π stacking) interactions. It is of note that such forces depend on the morphology and hydrophobicity of the GO surface. Additionally, physical adsorption is mainly hydrophobic in nature due to the sp2 hybridization of carbon atoms within the honeycomb lattice, and it depends on both the electron density and geometry of the protein molecules. By adapting their shape, proteins orientate their aromatic residues accordingly, resulting in stronger π–π stacking with graphene.18,19
In this study, the focus is on the topography of the protein molecules rather than their functionality, therefore the catalytic activity of the immobilized HRP enzyme was not assessed. A range of different conditions for the irradiation process and adsorption protocols were used to introduce tunability in protein functionalization and investigate any consequently generated defects upon graphene. The methodology presented here can be used to selectively localize various proteins on graphene, which is highly beneficial for the development of bioinspired graphene devices suitable, for example, in virus detection,20 electrochemical catalysis21 and cell/tissue adhesion.
Results and discussion
Raman and AFM assessment of oxidized graphene and protein immobilization
We used a set of squared silicon chips (7 mm × 7 mm) consisting of a 300 nm upper layer of SiO2 and a monolayer of graphene on top (5 mm × 5 mm). A reference metal grid was prepared on the surface of graphene, to ease exposure of the oxidation patterns by laser-induced 2P oxidation.22–24 Details of the microchip preparation and its full characterization are described in our previous studies.25 Graphene was irradiated using a range of laser pulse energies (5 to 30 pJ) and duration of irradiation from 0.2 up to 1.5 s per spot, to assess its effect on the level of oxidation and optimal conditions for protein adhesion. The matrices of squares with sizes ∼2 × 2 μm2 were patterned by step-by-step irradiation with steps of 0.1 μm and laser spot diameter of ∼0.6 μm (Fig. 1). | ||
| Fig. 1 Graphene microchip and depiction of the matrix of squares after irradiation. (A) Schematic representation and (B) optical images of a Si/SiO2 microchip bearing a monolayer of graphene on top with given dimensions of the pattern grid; (C) AFM height sensor image of the irradiated area (matrix of squares), using a range of laser pulse energies and duration of irradiation; the field of view (FOV) is 22 × 22 μm2. | ||
When comparing the height distribution profiles of the irradiated graphene squares before any treatment with protein solutions, it was evident that an increase of the laser pulse energy (from 5 to 30 pJ) for the same irradiation time (1.5 s) resulted in higher height profiles for the corresponding graphene spots (Fig. 2-D; irradiated area within the red frame).
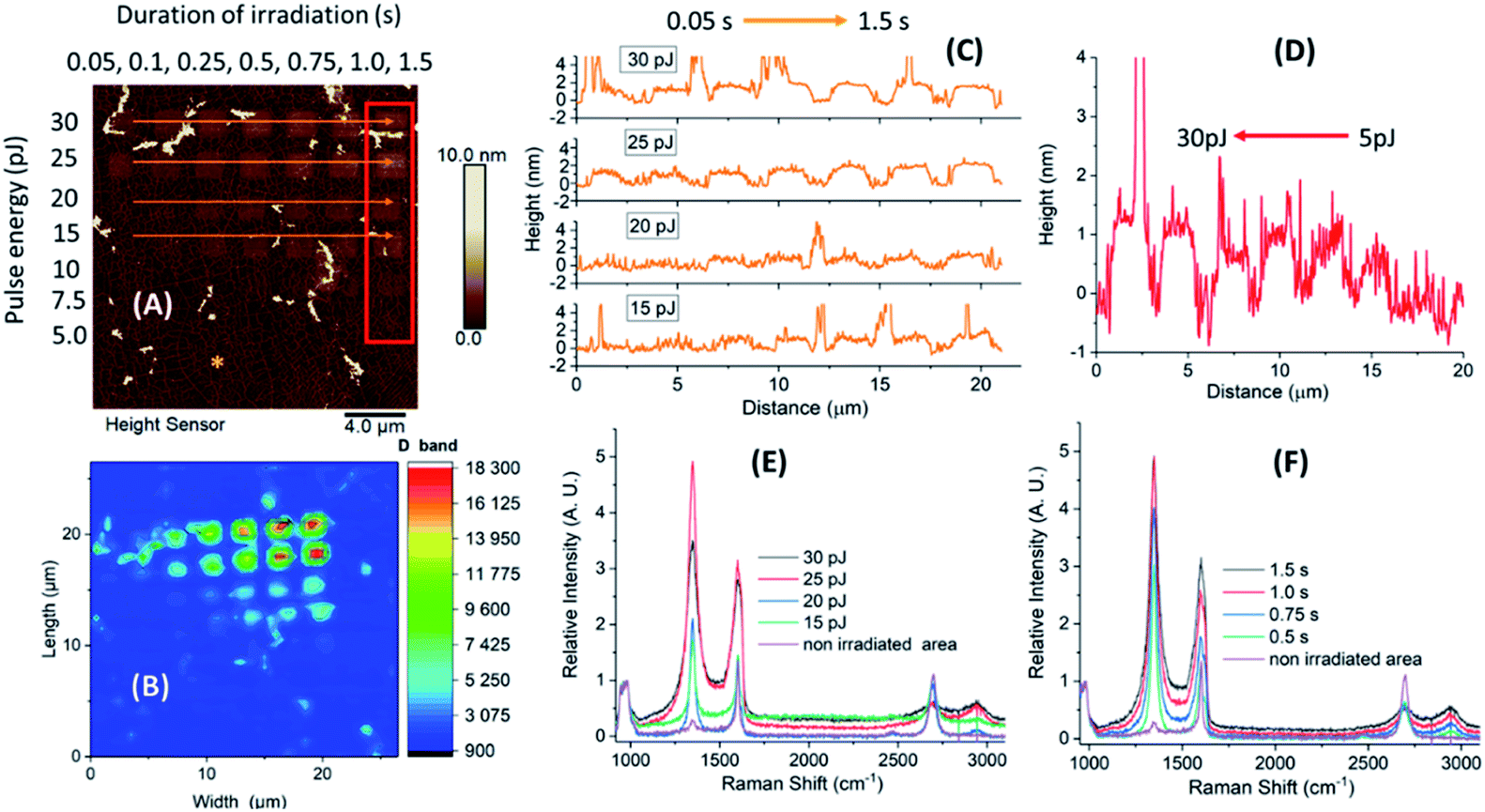 | ||
| Fig. 2 Evaluation of laser induced 2P oxidation of a microchip prior to the treatment with protein solutions. (A) AFM height sensor image of the irradiated microchip with designated oxidized areas (red frame and orange arrows); (B) Raman map showing integrated intensity in the D band area at ∼1350 cm−1. Height distribution profiles of the designated areas; (C) increased duration of irradiation (0.05–1.5 s, orange arrows); (D) increased laser pulse energy (5–30 pJ, red frame). Raman spectra of the designated irradiated and non-irradiated (denoted by the asterisk) spots, normalized with respect to the Si band intensities; (E) increased pulse energy at a constant irradiation of 1.5 s; (F) increased duration of irradiation at a constant pulse energy of 25 pJ. | ||
On the other hand, when increasing the irradiation time (from 0.05 to 1.5 s), no significant increase in height was noticed for higher pulse energies (30 and 25 pJ). In contrast, for lower energies (20 and 15 pJ), the difference in heights was more profound (Fig. 2-C; irradiated areas marked by the orange arrows). The observed differences in height indicate a different level of graphene oxidation achieved by laser-induced oxidation, as stated in our previous studies.22 In addition, Raman measurements of the oxidized samples showed that the intensity of the D band and interestingly that of G were increasing by irradiation (in all times ID > IG). The G band frequency was slightly shifted towards higher wavenumbers (blue-shifted) which denotes a signature of oxidized graphene.
The effect of laser oxidation on HRP immobilization
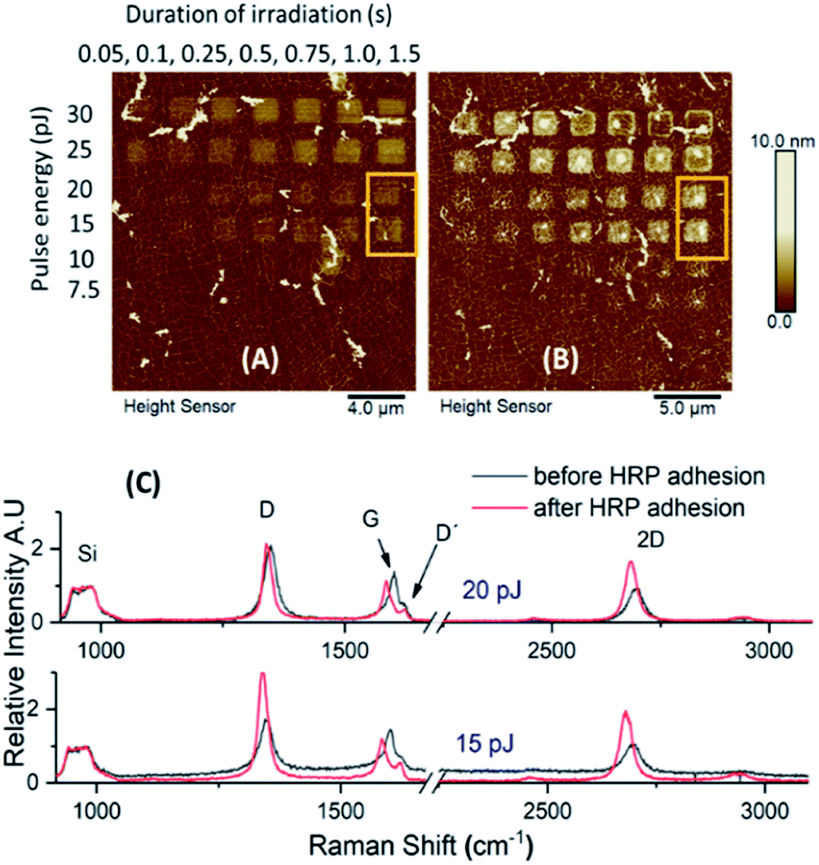
A combination of different laser pulse energies and irradiation times allowed us to assess the effects of different levels of graphene oxidation on protein adhesion and consequently defects upon the graphitic lattice, induced by protein immobilization. A microchip sample was therefore incubated in HRP solution for one hour, and AFM images and Raman spectra were recorded prior and after incubation.The topographic height profiles of the irradiated squares verified an increase in height from 2 nm to almost ∼7 nm after HRP adhesion (Fig. 3). A dense aggregation of enzyme molecules was observed in the middle and over the edges of each irradiated square. For such a short incubation time (1 h) there were no significant differences among the observed height profiles for the squares irradiated by intense pulse energies (30 and 25 pJ). For moderate pulse energies (20 and 15 pJ), however, HRP adhesion was denser when irradiation time had been longer. This suggests that both the irradiation conditions and the incubation time may affect protein adhesion, vide infra.
 | ||
| Fig. 3 Evaluation of laser pulse energy and duration of irradiation upon protein adhesion. AFM height sensor images of the microchip, before (A) and after (B) HRP adhesion; (C) topographic height profiles of a designated area (yellow frame) before and after functionalization; (D) HRP aggregation towards the middle and over the edges of the squares, irradiated by laser pulse energy of 25 pJ at different durations (red arrows and corresponding height profiles). | ||
In contrast to the height profiles, the Raman spectra appeared both qualitatively and quantitatively different regarding the given irradiation conditions (Fig. S1–S3 and Tables S1–S3†). As anticipated, the intensity of the G and 2D bands were prominent for all non-irradiated graphene areas. In contrast, for the oxidized areas, both D and G peaks were more intense compared to the 2D vibrational mode. It is of note that the position, intensity and full width at half maximum (FWHM) of the 2D and D + D′ peaks were changing depending on the used irradiation conditions.
Furthermore, less intense peaks of the D′ at ∼1620 cm−1 and G* at ∼2460 cm−1 were also observed. The D′ band appeared as a shoulder of the G peak in the spectra of graphene squares irradiated under moderate conditions (15 and 20 pJ/irradiation time over 0.5 s). For the regions treated with an increased laser pulse energy and irradiation time (25 and 30 pJ/irradiation over 1.5 s), both peaks merged into a broader band (Fig. S1–S3†). On the other hand, for the non-irradiated graphene regions, the G* peak was observed at a very low intensity.
The D or disorder-induced band usually ranges at ∼1250–1450 cm−1 and is indicative of lattice defects or appears near the edges of graphene. It originates from an intervalley process by the transverse optical phonons and associates to the breathing oscillation of the six-membered ring due to the presence of structural defects. The G or graphitic vibrational mode appears at ∼1580 cm−1 and is due to the in-plane motion of the sp2 hybridized carbon atoms (bond stretching). The less intense D′ peak usually appears as a shoulder of the G band and is associated with lattice defects. The G* band can be attributed to two different time-order phonon processes, a combination of transverse optical and longitudinal acoustic phonons.26 The 2D vibrational mode, an overtone of the D band, appears near 2700 cm−1 and is a second-order, two phonon, double resonance process. The D + D′ located near 2900 cm−1 is a combination of defect activated phonons related to the D and D′ vibrations.27–30
For all studied cases (irradiated and non-irradiated graphene), the Raman bands were shifted towards lower wavenumbers after HRP adhesion. The observed downshift was quantitatively different for each peak. One of our main findings is that a significant downshift was evident for areas irradiated under less intense conditions (laser pulse energy of 15 and 20 pJ for a duration of 1.5 s), especially for the G and 2D bands (Table 1 and Fig. 4). When more intense irradiation was applied, however, not any notable shift was observed for the G band in contrast to the 2D peak, which appeared notably shifted after functionalization. In addition, the shift of the D band was smaller in areas treated under intense pulse energy, following the same pattern as above (Fig. S1–S3†).
| Conditions | Red shift (cm−1) | ||
|---|---|---|---|
| D | G | 2D | |
| Increased pulse energy (pJ)/constant irradiation time of 1.5 s | |||
| 30 | 4.2 | 0.8 | 8.8 |
| 25 | 5.3 | 1.0 | 9.7 |
| 20 | 6.1 | 13.6 | 11.2 |
| 15 | 7.4 | 14.0 | 14.4 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Increased irradiation time (s)/constant pulse energy of 25 pJ | |||
| 1.5 | 4.7 | 1.1 | 4.3 |
| 1.0 | 5.2 | 2.3 | 8.3 |
| 0.75 | 6.5 | 4.9 | 12.8 |
| 0.5 | 7.4 | 12.8 | 13.5 |
|
|||
| Non-irradiated spots | |||
| Spot 1 | 4.2 | 3.7 | 6.5 |
| Spot 2 | 4.6 | 4.2 | 5.2 |
| Spot 3 | 3.2 | 2.4 | 2.8 |
| Spot 4 | 3.7 | 5.0 | 7.2 |
| Spot 5 | 2.5 | 3.9 | 4.3 |
 | ||
| Fig. 4 Observed Raman shifts. AFM height sensor images of the microchip, depicting the designated areas before (A) and after (B) HRP adhesion. (C) Raman spectra recorded prior and after HRP adhesion. Moderate irradiation conditions: laser pulse energy of 15 and 20 pJ and duration of irradiation of 1.5 s; all spectra are normalized with respect to the Si band intensities. | ||
For more intense irradiation conditions (laser pulse energy of 25 and 30 pJ/irradiation time > 1.0 s), our observations indicate that the defects on the graphitic lattice, caused by laser-induced oxidation, generated a strong Raman fingerprint. Therefore, the weak supramolecular interactions, induced by HRP adhesion, when added to the overall number of defects were not strong enough to cause significant changes at the spectroscopic features of the densely deformed graphene areas and thus causing a smaller downshift. Furthermore, for any specific irradiated spot on the lattice, the Raman vibrational modes originate by different phonon processes and therefore respond to a different extent towards the applied supramolecular forces.
Similar shifts caused by strain on the graphitic lattice have already been reported.31,32 In a previous case, the deposition of gold nanoparticles (GNPs) on graphene resulted in the whole Raman spectrum to downshift.33 More specifically, the shift of the 2D peak (∼6 cm−1) was more significant than that of the G band (∼3 cm−1). In the attempt to explain the observed downshift, a model of local strain was proposed, generated by the charge transfer among GNPs and graphene. Regarding our sample, the observed downshift of the G and 2D bands could be associated to both strain and doping effects due to charge transfer from the protein to graphene. Based on Ferrari et al., a downshift for both bands would indicate a decrease of positive doping.34,35 In general, for strained or doped graphene, both the G and 2D bands are shifted. Depending on its origin, such shifting is slightly different, which makes it possible to differentiate their contributions. We, therefore, calculated the level of strain and doping from the obtained Raman data (Fig. 5, S4 and S5†)36–39 following a methodology described by Lee et al.36
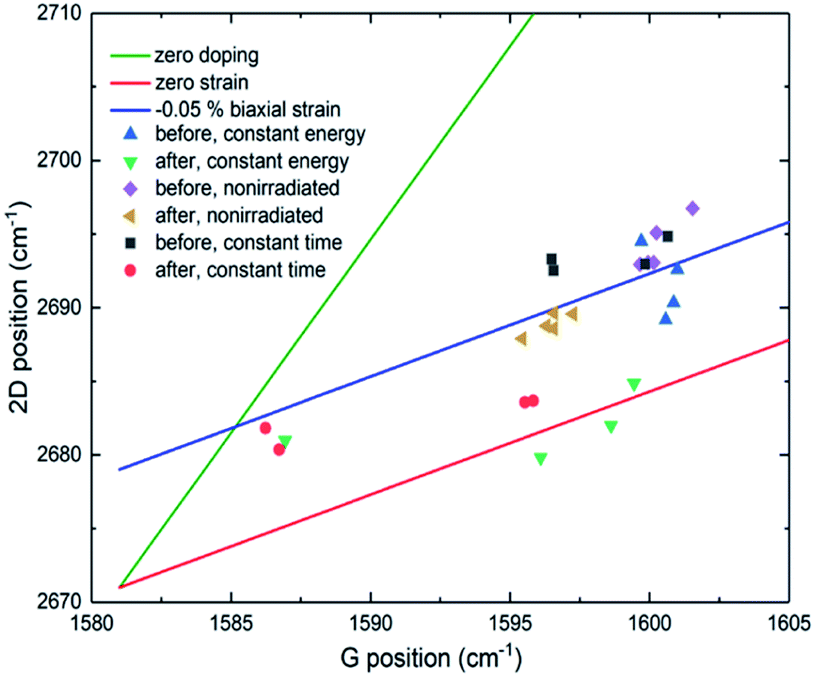 | ||
| Fig. 5 2D vs. G Raman band position plot. The green line shows the effect of pure strain without doping, and the red line the effect of doping without strain. The blue line shows a case of changing doping with constant −0.05% biaxial strain. Symbols show measurement points in different places on the sample. The blue triangle represents measurements from spots irradiated by constant 25 pJ pulse energy before HRP adhesion and green triangles after HRP adhesion. Black squares represent measurements from spots irradiated by different pulse energies (30–15 pJ) with a constant 1.5 s exposure time before functionalization and red circles after functionalization. Purple diamonds refer to non-irradiated graphene before functionalization and yellow triangles after. | ||
Non-irradiated graphene is initially p-type doped (about 0.35 eV) because of annealing enhanced graphene/SiO2 substrate conformity, which is the case for all our graphene samples on SiO2 substrate. Laser-induced oxidation does not seem to change the doping substantially, and the change is in different directions at the different irradiated spots as can be seen by the Fermi level plots (black squares, Fig. S4 and S5†). HRP adhesion (red circles, Fig. S4 and S5†) does not change the doping significantly for most of the patterns. However, in those squares where it does (15 and 20 pJ laser pulse energy; Fig. S4-A†/irradiation over 0.5 s; Fig. S5-A†), the change is large, and the doping is almost neutralized. It changes from ∼0.35 eV to ∼0.05 eV but still remains p-type.
According to the applied experimental conditions, the HRP molecules were positively charged as we adjusted the pH of the phosphate buffer solution slightly below the pI (7.2) of the enzyme. Our intention was to induce coulombic forces between the protein molecules and GO. Albeit a dense aggregation of HRP was observed by AFM upon the irradiated areas, the enzyme interacted with the whole graphitic surface. Indeed, the non-irradiated spot 4 (pristine graphene, Fig. S3†), although positioned far from any irradiated areas, showed a similar downshift pattern to the irradiated areas after the functionalization process. However, the magnitude of the shift was smaller. The differences in shifting among the irradiated and non-irradiated regions of graphene can only suggest differences in the type of supramolecular interactions (electrostatic forces or hydrogen bonding) between the protein molecules and the graphitic lattice.
For the non-irradiated spots and those oxidized under moderate conditions, a sharp 2D band was present before and after HRP adhesion. For regions bearing denser deformities, however, the sharp 2D band was significantly broadened. According to Ferreira et al., an increase of the lattice defects leads to an increase in the intensity of the defect activated bands. In contrast, for denser deformities, the 2D band is suppressed as the honeycomb graphene network is disturbed.40
This explains the increase of the FWHM of the 2D peak and other defect activated bands (D and D + D′) towards regions oxidized under more intense conditions. Therefore, for irradiation time of 1.5 s and pulse energy ranging from 15 pJ to 30 pJ, the FWHM of the D and 2D bands was increased by 56 and 170 cm−1 accordingly, while the D + D′ vibrational mode was visible only at regions bearing denser deformities. Similarly, by increasing the irradiation time from 0.5 s to 1.5 s under constant pulse energy of 25 pJ, the FWHM of the D, 2D and D + D′ bands increased by 18, 99 and 25 cm−1 respectively.
The effect of HRP immobilization on GO was further assessed by comparison of the ID/IG ratio before and after the functionalization of the microchip (Fig. 6). Significant changes in the ratio values were noticed only over the areas of graphene oxidized under moderate conditions. For a constant irradiation time (1.5 s), the highest difference in the ID/IG ratio was spotted at areas treated with the lowest laser pulse energy (15 pJ). The ratio increased from 1.29 to 2.16, whereas minor changes were observed as the pulse energy increased. For those regions treated with constant pulse energy (25 pJ) over different irradiation times, the alterations in the ID/IG ratio were significant at 0.5 s of irradiation.
 | ||
| Fig. 6 I D/IG ratios prior and after incubation of the microchip in HRP solution. AFM height sensor image of the microchip, before (A) and after (B) HRP adhesion with designated areas. The blue spot is non-irradiated graphene. (C) Ratio values correspond to graphene squares (red frame) irradiated at a constant duration of 1.5 s over a range of pulse energies (top plot) and those squares (yellow frame) irradiated at a constant pulse energy of 25 pJ over a range of time durations (bottom plot). | ||
Differences in the ID/IG ratio related to differences in doping have been previously reported. In their work, Ferrari et al. describe how the intensity of the D band decreases as doping increases.35 In our case, the same observation applies; the intensity of the D band is increased when doping is decreased, which explains the increase in ID/IG ratio. Such a result denotes that although HRP adhesion introduces defects upon the graphene monolayer, these are noticeable at a different extent, depending on the initial deformities achieved by laser-induced 2P oxidation. In other words, the weak supramolecular interactions between the enzyme and GO surface are notably visible when the deformities over the lattice of graphene are less dense.
The effect of incubation cycles on HRP immobilization
A second microchip was successively incubated in HRP solution for 1, 2 and 3 h, respectively, (totally 6 h of incubation) to investigate potential concentration and sample handling effects on protein immobilization. The enzymatic solution was diluted to a quarter of the initial concentration, AFM height sensor images were captured after each incubation cycle, and Raman spectra were recorded prior any treatment and after functionalization (Fig. 7).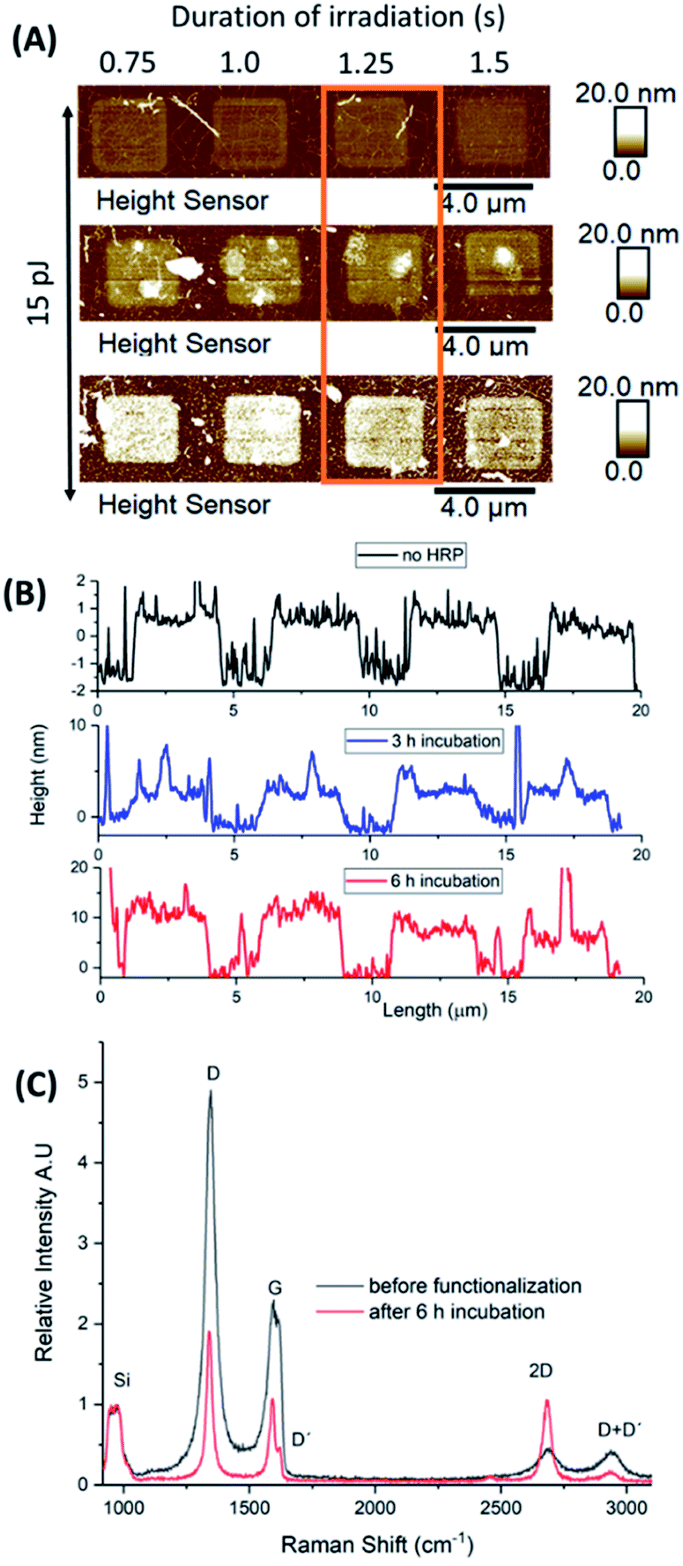 | ||
| Fig. 7 Incubation studies in HRP solution. (A) AFM height sensor images of a selected area at different cycles of incubation. Graphene was irradiated by laser pulse energy of 15 pJ at different time durations; (B) topographic height profiles of the designated area after 3 and 6 h of incubation; (C) Raman spectra of a selected irradiated square (orange frame) before and after functionalization. All spectra are normalized to the Si bands intensity. | ||
For a selected area of graphene, irradiated by 15 pJ for different lengths of times, the 2P oxidation process increased the height of the irradiated squares up to 1–1.5 nm in a homogeneous manner. After two cycles of incubation (totally 3 h of treatment), the designated squares increased their heights due to protein adhesion. Based on the topographic height profiles, a denser accumulation of the enzyme molecules was observed at the centre and over the edges of each square. A third incubation cycle for 3 more hours (totally 6 h of treatment) resulted in heights up to 15 nm. It is of note that for an increased irradiation time (1.5 s) the same aggregation pattern was observed. For shorter irradiation times (0.75–1.25 s), however, HRP enzymes immobilized upon the surface of the squares almost evenly.
To ensure that the observed changes in height and the topography of HRP enzyme are not related to non-specific adsorption of the buffer solutes, we compared the height profile of a non-irradiated area of the microchip after each incubation cycle. Indeed, no changes in height were present after 6 h of treatment, indicating the absence of HRP molecules upon pristine graphene (Fig. S6†). In addition, although scanning electron microscopy (SEM) images of the microchip showed the deposition of crystals on its surface, their shape was rather distinct and could not be related to immobilized HRP (Fig. S6-D†).
Further to this, we generated histograms corresponding to the topographic height profiles of the irradiated area (Fig. 7-A) by using the depth function of Nanoscope analysis software (Fig. S7-C†). The x axis of each histogram represents the measured heights from the AFM height sensor channel versus the percentage of data points (y axis) related to the corresponding heights. Before treatment, two distinct peaks are visible (black histogram). By comparison to the AFM height sensor image (Fig. 7-A and B) it is evident that the peak at ∼0 nm corresponds to non-irradiated graphene while the peak at ∼0.5 nm to oxidized graphene. After each incubation cycle, both peaks were broadened (blue and red histograms) while a notable shift was observed for those peaks related to irradiated graphene. Indeed, shifting denotes an increase of height (x axis values) by increasing the time of incubation from 3 h (4 nm) to 6 h (10 nm). Prior treatment, two distinct height levels (x axis values) are observed and almost all data points (y axis) fall within this range. The broadening of the peaks after each incubation cycle, suggests the preference of HRP molecules to immobilize upon irradiated graphene resulting in a rougher surface with variations in height. The shifting towards increased heights (x axis) supports the increased deposition of HRP molecules.
To relate the successive incubation cycles with the formation of multiple HRP layers upon irradiated graphene (measured heights, Fig. 7-B) we took into account the dimensions and hydrophobic–hydrophilic distribution of the enzyme itself (Protein Data Bank ID, 1HCH).41 Assuming that the GO–HRP interaction is mostly electrostatic then one could expect that regions with a higher amount of hydrophilic amino-acids will be more prone to adsorption. According to the measured height profiles (Fig. 3-C, S8-A†) the nominal height of immobilized HRP in a single layer should be around 3 nm (±0.6 nm owing to the deformation-induced upon AFM indentation). Based on our findings, after 3 h of incubation 1–2 layers of the enzyme seem to have been deposited (1 nm + 3 nm) while after 6 h, 3–4 layers (Fig. S8†).
After 6 h of incubation, the Raman spectra showed a significant decrease in the intensity of the D and G bands (ID > IG in both cases). Also, the initial broad G band appeared rather sharp after 6 h of treatment, bearing the D′ peak as a shoulder, while the 2D signal increased its intensity almost to that of the G band (Fig. 7-C). The ID/IG ratio values, prior and after HRP adhesion were 2.28 and 1.85, respectively, which suggests an increase in doping after 6 h of treatment. Finally, a downshift was also observed which was qualitatively different for all peaks (D: 3.5 cm−1, G: 5.7 cm−1, 2D: no shift observed, D + D′: 5.6 cm−1) showing the presence of strain and/or doping effects due to charge transfer over the graphitic lattice.
In contrast to shorter incubation times (1 h, first microchip), additional immersion cycles in HRP solution (second microchip) increased further the height of the irradiated squares, indicating a denser aggregation of proteins. The differences in Raman spectroscopy between the two microchips suggest that the initial incubation cycles favoured interactions among the protein molecules and graphene. Indeed, the induced supramolecular forces had an immediate structural effect on the graphitic lattice, which was reflected by the outstanding vibrational responses (i.e. downshifting) of the Raman peaks (first microchip). Depending on the type of interactions (i.e. electrostatic or hydrogen bonding), graphene is affected either by charge transfer and/or mechanical deformations, leading to different phonon processes. A denser accumulation of enzyme molecules through additional incubation cycles (second microchip), favours intermolecular interactions, causing minor effects on the graphitic lattice. This hypothesis is justified by the smaller downshifting before and after functionalization. In addition, the moderate changes in the Raman spectra can be attributed to the reduced concentration of HRP used during the treatment of the second microchip.
Detection of b-BSA/Av-FITC attachment by fluorescence microscopy
In our attempt to visualize the topography/selective aggregation of protein molecules over irradiated graphene, a third microchip was initially treated with a b-BSA solution and reacted thereafter with fluorescently labelled Av-FITC, before imaging by fluorescence confocal microscopy. Using ImageJ software, we analysed the obtained red, green, blue (RGB) colour image and generated the given grayscale plot profiles of the selected areas of the microchip (Fig. 8). The surface plot displays the intensity of light which reflects from the chosen areas, designated by the dashed lines. It defines the brightness of the pixels along the drawn lines and relates to the number of immobilized protein molecules. The denser the protein aggregation, the higher the grayscale value is. | ||
| Fig. 8 Evaluation of the b-BSA/Av-FITC topographic immobilization by fluorescence microscopy. (A) Fluorescence microscopy image of the microchip after treatment with given oxidation parameters; (B) grayscale surface plot profiles corresponding to constant laser pulse energies of 30 and 10 pJ; (C) grayscale surface plot profiles corresponding to constant irradiation times of 1.5 and 0.2 s. | ||
The black and yellow horizontal lines correspond to graphene treated by a constant pulse energy of 30 and 10 pJ respectively, over a range of irradiation times (0.2–1.5 s). The red and blue vertical lines represent graphene irradiated for 1.5 and 0.2 s respectively, at a range of laser pulse energies (30–10 pJ). From the plot, it is evident that for intense oxidation conditions (higher pulse energy and longer irradiation time), b-BSA adhered on graphene almost evenly (black and red plots). On the other hand, for moderate oxidation conditions, the intensity varies. The intensity is reaching the highest values on the grayscale when higher pulse energies (30 and 25 pJ) are used over short irradiation times (blue plot) and when longer irradiation is applied (1.5 to 0.8 s) at lower energies (yellow plot).
As the protein carries a net negative charge at the used pH value (7.3), it was expected to adhere less upon areas bearing a high surface density of oxygen-containing groups due to electrostatic repulsion. The experimental results, however, can be attributed to the induced coulombic forces between the negatively charged GO and the positively charged amino acids of b-BSA (lysine, histidine).42 This also explains the poor adhesion of protein molecules over graphene areas irradiated under moderate conditions hence bearing a lower surface density of oxygen functional groups.
The functionalization of graphene surfaces by proteins expands the potential of the microchips towards applications in biomedicine, such as in biosensors, drug delivery systems and immunological assays. In comparison to the existing patterning technologies,43,44 laser-induced oxidation introduces tunability on protein adhesion by controlling the levels of oxidation. In other words, proteins can be guided over graphene surfaces by light. Sequential irradiation of different areas followed by the immobilization of proteins can be used to functionalize chips with multiple proteins, localized in separate regions. Since the selective immobilization of proteins is based on non-covalent interactions, our approach does not require the addition of chemicals or the use of chemical masking techniques resulting in a facile and free from chemical contaminants patterning method.
Conclusions
We studied protein adhesion upon laser-induced GO patterns. Our key observations are: (i) by modification of the irradiation conditions we introduced tunability of the levels of graphene oxidation and hence induced the selective immobilization of protein molecules; (ii) the type of protein, its concentration, incubation time and pH value of the buffer solution have an immediate effect on physical adsorption; (iii) any consequent deformities upon the graphitic lattice due to protein adhesion are notably visible by Raman spectroscopy over areas oxidized under moderate conditions; (iv) we facilitated a Raman spectroscopic fingerprint of protein adhesion and observed the topography of protein immobilization by AFM and fluorescence microscopy.Working towards bioinspired graphene surfaces, we can control the topography of protein immobilization by laser-induced oxidation in contrast to chemically oxidized graphene. Our methodology has a high potential for advancing the development of bioelectronic devices and sensors, for example, via the selective immobilization of antibodies on active sensor areas.
Materials and methods
Material
Horseradish peroxidase, HRP (type VI, ≥250 units per mg solid), biotinylated Bovine Serum Albumin, b-BSA (A8549) and avidin-fluorescein isothiocyanate, Av-FITC from egg white (A2901) were purchased from Sigma Aldrich.Preparation of protein solutions
HRP solutions were prepared in phosphate-buffered saline, PBS (0.0754 M Na2HPO4·2H2O, 0.0246 M NaH2PO4·2H2O, pH 7.1) with final concentrations of 2.5 units per mL or 10 μg mL−1 and 0.625 units per mL or 2.5 μg mL−1. b-BSA and Av-FITC solutions were prepared in PBS (137 mmol L−1 NaCl, 2.7 mmol L−1 KCl, 10 mmol L−1 Na2HPO4, 2.0 mmol L−1 KH2PO4, pH 7.3 pH) at final concentrations of 10 μg mL−1 and 80 μg mL−1 respectively.Immobilization of HRP enzyme
The microchip was immersed in HRP solution (2.5 units per mL) for 1 h at 0 °C. The graphene surface was then extensively washed with PBS solution (pH 7.1) and MilliQ water before dried under the stream of N2 gas. A lower concentration of the enzyme was used (0.625 units per mL) to assess concentration and immersion timing effects on HRP adsorption. The microchip was therefore immersed into HRP solution for 1, 2 and 3 h respectively. After each immersion cycle, washings were performed using PBS solution (pH 7.1) and MilliQ water followed by drying.Immobilization of b-BSA and binding with Av-FITC
The microchip was immersed in b-BSA solution (10 μg mL−1) for 24 h at r.t. The graphene surface was washed with a PBS solution (pH 7.3) and MilliQ water to remove unbound proteins. The chip was then dried under a stream of N2 gas before it was immersed for a second time into an Av-FITC solution (80 μg mL−1) for 1 h at r.t. Excess of Av-FITC was removed by PBS solution and water washings followed by drying.Instruments
Oxidized patterns were curved on graphene monolayer by irradiation with 515 nm focused femtosecond laser in the ambient atmosphere (pulse duration 250 fs, repetition rate: 600 kHz). A home-built Raman setup was used for mapping and spectra acquisition, as previously described.22,45 The laser power of 0.250 mW and 532 nm of excitation was utilized, the exposure time was set at 10 s and mapping was conducted in a 25 × 25 μm area. AFM imaging was carried on a Bruker Dimension Icon atomic force microscope, using Peak Force Tapping mode. ScanAsyst-Air probes from Bruker were used during imaging with the peak force set to 2.0 nN. All AFM images were processed with NanoScope Analysis 1.9 software. Images of the height channel were smoothed with a Gaussian low-pass filter to suppress noise to generate 3D structures. Fluorescence imaging was performed on a Nikon A1R laser scanning confocal microscope. Images were acquired by using argon laser excitation at 488 nm and a 512/30 emission filter for detection. All images were processed with Fiji2 software.AFM and Raman analysis
The height distribution profile of the functionalized surfaces was evaluated by PeakForce tapping (PFT) mode AFM.46 The irradiated graphene surfaces were visualized by Raman mapping of the integrated intensity of the D band, while Raman spectra were recorded for all samples at selected spots (irradiated and non-irradiated). By comparison of the vibrational response of the graphene surfaces before and after protein adhesion, we attempted to facilitate a spectroscopic fingerprint of the functionalized areas. Both first (1050–1750 cm−1) and second (2400–3100 cm−1) order scattering spectral regions were evaluated prior and after protein functionalization to assess potential defects on the lattice of graphene. Our study focused on features such as the position, intensity and displacement of the recorded bands, the ID/IG ratios and the FWHM of the vibrational modes which were quantified by the Lorentzian function fitting procedure.To calculate the level of strain and doping, the sensitivity factors we used were (Δω2D/ΔωG)holen = 0.7 and (Δω2D/ΔωG)biaxialε = 2.63. Doping was estimated simply by n = (EF/(ħνF))/π, where n is carrier concentration, EF Fermi energy and νF Fermi velocity. The shift of the G band frequency is given by ΔωG = EF × 42 cm−1/eV.37 The effect of strain on the G band frequency was estimated with a sensitivity factor of −69.1 cm−1/%, which is an average value derived from previous studies.36–39 The point of zero doping and strain we used in calculations was 1581 cm−1 (G band frequency) and 2671 cm−1 (2D band frequency) (Fig. 5, S4 and S5†).
Author contributions
E. D. S. and J. S. contributed equally to this work. E. D. S. wrote the manuscript and with J. S. functionalized the microchips, performed Raman mapping/spectroscopy measurements, conducted AFM imaging and data analysis. A. L. conceived and performed preliminary studies on HRP adhesion. K. K. M. oxidized and characterized the microchips. V. M. H. calculated the level of strain and doping. V. R. performed fluorescence imaging. A. J. evaluated the fabrication of the microchips. P. M. designed and developed the laser setups. M. N. supervised the protein adhesion work. M. P. planned the experiments, coordinated and supervised the work. All of the authors discussed the results, commented on the manuscript and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Jane and Aatos Erkko Foundation for supporting the current work and the Academy of Finland for funding the postdoctoral program of the Nanoscience Center and for supporting the current project (decision no. 318905). V. M. H. is grateful to Finnish Cultural Foundation for his financial support.References
- S. Y. Wu, S. S. A. An and J. Hulme, Int. J. Nanomed., 2015, 10, 9–24 CrossRef CAS.
- W. Cheng, K. Ping and C. Teck, Biomaterials, 2018, 155, 236–250 CrossRef.
- Z. Gu, S. Zhu, L. Yan, F. Zhao and Y. Zhao, Adv. Mater., 2019, 31, e1800662 CrossRef.
- J. A. Jaleel, S. Sruthi and K. Pramod, J Control Release, 2017, 255, 218–230 CrossRef CAS.
- T. A. Tabish, Biochem. Biophys. Res. Commun., 2018, 504, 686–689 CrossRef CAS.
- M. Wierzbicki, S. Jaworski, M. Kutwin, M. Grodzik, B. Strojny, N. Kurantowicz, K. Zdunek, R. Chodun, A. Chwalibog and E. Sawosz, Int. J. Nanomed., 2017, 12, 7241–7254 CrossRef CAS.
- S. Liu, T. H. Zeng, M. Hofmann, E. Burcombe, J. Wei and R. Jiang, ACS Nano, 2011, 9, 6971–6980 CrossRef.
- J. Vlček, L. Lapčík, M. Havrdová, K. Poláková, B. Lapčíková, T. Opletal, J. P. Froning and M. Otyepka, Nanoscale, 2019, 11, 3222–3228 RSC.
- D. Kireev and A. Offenhäusser, 2D Mater., 2018, 5, 042004 CrossRef CAS.
- K. Kostarelos, M. Vincent, C. Hebert and J. A. Garrido, Adv. Mater., 2017, 29, 1700909 CrossRef.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- C. J. Bullock and C. Bussy, Adv. Mater. Interfaces, 2019, 6, 1900229 CrossRef.
- M. Seredych, L. Mikhalovska, S. Mikhalovsky and Y. Gogotsi, C, 2018, 4, 3 Search PubMed.
- J. Zhang, F. Zhang, H. Yang, X. Huang, H. Liu and J. Zhang, Langmuir, 2010, 26, 6083–6085 CrossRef CAS.
- W. Hu, C. Peng, M. Lv, X. Li, Y. Zhang, N. Chen, C. Fan and Q. Huang, ACS Nano, 2011, 5, 3693–3700 CrossRef CAS.
- J. G. Vilhena, P. Rubio-Pereda, P. Vellosillo, P. A. Serena and R. Pérez, Langmuir, 2016, 32, 1742–1755 CrossRef CAS.
- P. Rubio-Pereda, J. G. Vilhena, N. Takeuchi, P. A. Serena and R. Pérez, J. Chem. Phys., 2017, 146, 214704 CrossRef.
- G. Faccio, Sensors, 2018, 18, 1204 CrossRef.
- M. Simsikova and T. Sikola, J. Nanomed. Res., 2017, 5, 00109 Search PubMed.
- E. Vermisoglou, D. Panáček, K. Jayaramulu, M. Pykal, I. Frébort, M. Kolář, M. Hajdúch, R. Zbořil and M. Otyepka, Biosens. Bioelectron., 2020, 166, 112436 CrossRef CAS.
- H. Seelajaroen, A. Bakandritsos, M. Otyepka, R. Zbořil and N. S. Sariciftci, ACS Appl. Mater. Interfaces, 2020, 12, 250–259 CrossRef CAS.
- J. Aumanen, A. Johansson, J. Koivistoinen, P. Myllyperkiö and M. Pettersson, Nanoscale, 2015, 7, 2851–2855 RSC.
- J. Koivistoinen, L. Sládková, J. Aumanen, P. Koskinen, K. Roberts, A. Johansson, P. Myllyperkiö and M. Pettersson, J. Phys. Chem. C, 2016, 120, 22330–22341 CrossRef CAS.
- A. Johansson, H. C. Tsai, J. Aumanen, J. Koivistoinen, P. Myllyperkiö, Y. Z. Hung, M. C. Chuang, C. H. Chen, W. Y. Woon and M. Pettersson, Carbon, 2017, 115, 77–82 CrossRef CAS.
- A. Johansson, P. Myllyperkiö, P. Koskinen, J. Aumanen, J. Koivistoinen, H. C. Tsai, C. H. Chen, L. Y. Chang, V. M. Hiltunen, J. J. Manninen, W. Y. Woon and M. Pettersson, Nano Lett., 2017, 17, 6469–6474 CrossRef CAS.
- J. Zhu, R. Rao, A. M. Rao and R. Podila, Recent Pat. Mater. Sci., 2018, 11, 24–32 CrossRef CAS.
- K. Tsirka, A. Katsiki, N. Chalmpes, D. Gournis and A. S. Paipetis, Front. Mater., 2018, 5, 37 CrossRef.
- S. Eigler, C. Dotzer and A. Hirsch, Carbon, 2012, 50, 3666–3673 CrossRef CAS.
- A. Kaniyoor and S. Ramaprabhu, AIP Adv., 2012, 2, 032183 CrossRef.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- E. Del Corro, M. Taravillo and V. G. Baonza, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 033407 CrossRef.
- F. Ding, H. Ji, Y. Chen, A. Herklotz, K. Dörr, Y. Mei, A. Rastelli and O. G. Schmidt, Nano Lett., 2010, 10, 3453–3458 CrossRef CAS.
- X. Zheng, W. Chen, G. Wang, Y. Yu, S. Qin, J. Fang, F. Wang and X. A. Zhang, AIP Adv., 2015, 5, 057133 CrossRef.
- A. Das, S. Pisana, B. Chakraborty, S. Piscanec, S. K. Saha, U. V. Waghmare, K. S. Novoselov, H. R. Krishnamurthy, A. K. Geim, A. C. Ferrari and A. K. Sood, Nat. Nanotechnol., 2008, 3, 210–215 CrossRef CAS.
- M. Bruna, A. K. Ott, M. Ijäs, D. Yoon, U. Sassi and A. C. Ferrari, ACS Nano, 2014, 8, 7432–7441 CrossRef CAS.
- J. E. Lee, G. Ahn, J. Shim, Y. S. Lee and S. Ryu, Nat. Commun., 2012, 3, 1024 CrossRef.
- C. F. Chen, C. H. Park, B. W. Boudouris, J. Horng, B. Geng, C. Girit, A. Zettl, M. F. Crommie, R. A. Segalman, S. G. Louie and F. Wang, Nature, 2011, 471, 617–620 CrossRef CAS.
- J. Zabel, R. R. Nair, A. Ott, T. Georgiou, A. K. Geim, K. S. Novoselov and C. Casiraghi, Nano Lett., 2012, 12, 617–621 CrossRef CAS.
- T. M. G. Mohiuddin, A. Lombardo, R. R. Nair, A. Bonetti, G. Savini, R. Jalil, N. Bonini, D. M. Basko, C. Galiotis, N. Marzari, K. S. Novoselov, A. K. Geim and A. C. Ferrari, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 205433 CrossRef.
- E. H. Martins Ferreira, M. V. O. Moutinho, F. Stavale, M. M. Lucchese, R. B. Capaz, C. A. Achete and A. Jorio, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125429 CrossRef.
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463–468 CrossRef CAS.
- A. Hasan, G. Waibhaw and L. M. Pandey, Langmuir, 2018, 34, 8178–8194 CrossRef CAS.
- H. Zhao, Y. Lee, M. Han, B. K. Sharma, X. Chen, J. H. Ahn and J. A. Rogers, Nano Today, 2020, 30, 100825 CrossRef CAS.
- N. Vigneswaran, F. Samsuri, B. Ranganathan and Padmapriya, Procedia Eng., 2014, 97, 1387–1398 CrossRef CAS.
- M. Pettersson, J. Rintala, O. Herranen, A. Johansson and M. Ahlskog, J. Phys. Chem. C, 2009, 113, 15398–15404 CrossRef.
- K. Xu, W. Sun, Y. Shao, F. Wei, X. Zhang, W. Wang and P. Li, Nanotechnol. Rev., 2018, 7, 605–621 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Raman spectra, Fermi level plots, compressive strain plots. See DOI: 10.1039/d0na01028f |
| This journal is © The Royal Society of Chemistry 2021 |