
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreservation of the donor–acceptor character of a carbazole–phenalenone dyad upon adsorption on Pt(111)†
René
Ebeling
 ab,
Narendra P.
Arasu
cd,
Lisa
Bensch
e,
Bertram
Schulze Lammers
a,
Bernhard
Mayer
e,
Thomas J. J.
Müller
e,
Héctor
Vázquez
*c and
Silvia
Karthäuser
*a
ab,
Narendra P.
Arasu
cd,
Lisa
Bensch
e,
Bertram
Schulze Lammers
a,
Bernhard
Mayer
e,
Thomas J. J.
Müller
e,
Héctor
Vázquez
*c and
Silvia
Karthäuser
*a
aPeter Grünberg Institut (PGI-7), JARA-FIT, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: s.karthaeuser@fz-juelich.de
bFaculty 1 – Mathematics, Computer Science and Natural Science, RWTH Aachen University, 52056 Aachen, Germany
cInstitute of Physics, Academy of Sciences of the Czech Republic, CZ-162 00 Prague 6, Czech Republic. E-mail: vazquez@fzu.cz
dDepartment of Condensed Matter Physics, Faculty of Mathematics and Physics, Charles University, CZ-12116 Prague 2, Czech Republic
eInstitut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine Universität Düsseldorf, Universitätsstrasse 1, 40225 Düsseldorf, Germany
First published on 8th December 2020
Abstract
Donor–acceptor molecules are a subject of great attention due to their immense potential in molecular electronics and photovoltaics. Despite numerous extensive studies demonstrating their functionality in solution, the donor–acceptor character is usually lost upon adsorption on a conducting substrate. Here the concept of breaking the conjugation between the donor and acceptor unit by insertion of a bridge is used. Furthermore, the bridge introduces a kink into the dyad and thus, reduces the possibility of hybridization with the substrate. A donor–bridge–acceptor dyad composed of carbazole and phenalenone units joined through a flexible bridge is synthesized and deposited on a Pt(111) surface. Its electronic properties are investigated with a combination of low temperature scanning tunneling microscope measurements and density functional theory simulations. Two preferential adsorption configurations are identified, in which individual molecules form strong bonds to the substrate and to a Pt adatom. Differential conductance measurements and atomistic simulations evidence the preservation of a reduced donor–acceptor character upon adsorption of the molecule, where this reduction is ascribed to the strong molecule-metal hybridization. Our results highlight the changes in donor–acceptor character of the dyad induced by the substrate and provide guidelines for the use of donor–bridge–acceptor molecules as functional units in solid-state devices.
1. Introduction
An increased interest and manifold activities in the research field of donor–acceptor dyads are motivated by their electronic and optoelectronic properties which render possible to use them as memory devices, sensors, organic solar cells1 or organic light emitting diodes.2 Donor–acceptor dyads enable intramolecular electron transfer induced by thermal, electrical or optical stimulation and often show fluorescence or solvatochromism.3–7 However, the application of donor–acceptor dyads in advanced devices requires a careful molecular design regarding the efficiency of electron transfer and the lifetime of the charge separated state. Several factors have been suggested to enhance the quantum yields and lifetimes in the charge separated states, such as spin restriction rules, conformational gating, and orientation effects. These concepts have successfully been applied to develop highly efficient dyads with long-lived charge separated states showing that the bridge mediating the charge transfer in donor–bridge–acceptor systems has a pronounced effect upon the charge transfer kinetics.6–9 However, usually these studies are performed on molecules in solution.To date, only a few studies exist which characterize groups or single donor–acceptor dyads deposited on solid surfaces by scanning probe methods that have the potential to reveal the intrinsic molecular properties with sub-nanometer resolution. It was found, that the theoretically predicted spatial localization of the frontier orbitals in donor–acceptor dyads, based on ab initio modeling of isolated molecules, could be observed experimentally if the dyads were electronically decoupled from the metallic substrate by an insulating layer.10,11 On the other hand, the direct adsorption on metallic substrates or the formation of chemical contacts to metal atoms, which is a fundamental requirement for the application in functional devices, is expected to change the intrinsic molecular properties.12,13 Therefore the preservation of the donor–acceptor character after adsorption of a dyad and forming a chemical contact to a metallic electrode remains a challenge.
Recently, we studied a non-planar naphthalene diimide (NDI) derivative in contact with a Pt(111) electrode using low-temperature ultra-high vacuum scanning tunneling microscopy (LT-UHV-STM) at the single molecule level.14 Here, a phenyl group is forced to adopt an out-of-plane coordination by a methylene bridge, which links it to the NDI acceptor. Thus, the phenyl group is decoupled from the substrate as well as from the NDI acceptor. However, the inspection of dyads composed of donors and acceptors, both with an extended π-electron system, indicates that larger donor groups cannot easily be forced to exhibit an out-of-plane coordination. Therefore, in this work, we investigate an especially designed non-planar donor–acceptor dyad with a bridging unit. The employed dyad is composed of a carbazole and a phenalenone derivative used as donor and acceptor, respectively. Hereby, the 9-hydroxy-1H-phenalen-1-one (HP) acceptor shows attractive opportunities like the formation of a stable radical anion, interesting photophysical characteristics and it is employed as a ligand in multiple stable metal complexes.15,16 Recently 5-(hetero)aryl substituted conjugated 9-HP dyads were successfully synthesized in a modular fashion and their ensemble electronic ground and excited state properties were studied in detail.17 However, here the delocalized donor and acceptor π-systems of the studied 5-[4-(9H-carbazol-9-yl)benzyl]-9-hydroxy-1H-phenalen-1-one (CBHP) are separated electronically by an aliphatic methylene bridge which introduces a twist angle between acceptor and donor and breaks the conjugation. In the following, single CBHP molecules will be analyzed on the Pt(111) surface by high-resolution STM in order to reveal their exact conformation and thus, their adsorption behavior. By analyzing the spatially resolved topographic and electronic features of this donor–bridge–acceptor dyad in comparison to Density Functional Theory (DFT) calculations we reveal two on-surface geometries characterized by the lateral extension of the donor and acceptor π-electron systems. We will show that adsorption on metallic surfaces induces a reduction, though not loss, of intrinsic molecular properties including the donor–acceptor character.
2. Results and discussion
2.1 Chemical synthesis of CBHP
The title compound CBHP was synthesized in a five-step sequence starting from carbazole (1) (Scheme 1). First, carbazole (1) was coupled with p-bromobenzaldehyde (2) under Ullmann conditions18 to give 4-(9H-carbazol-9-yl)benzaldehyde (3) in 60% yield. The aldehyde was transformed to the benzyl alcohol 4 by sodium borohydride reduction in 83%, which was in turn reacted with PBr3 to give benzyl bromide 5 in 99% yield. The benzyl bromide 5 was then transformed into the organozinc bromide–lithium chloride 6 by a literature known protocol,19,20 which was directly submitted to Negishi coupling with 5-bromo-9-hydroxy-1H-phenalen-1-one (7) to give the dyad CBHP in 72% yield over two steps.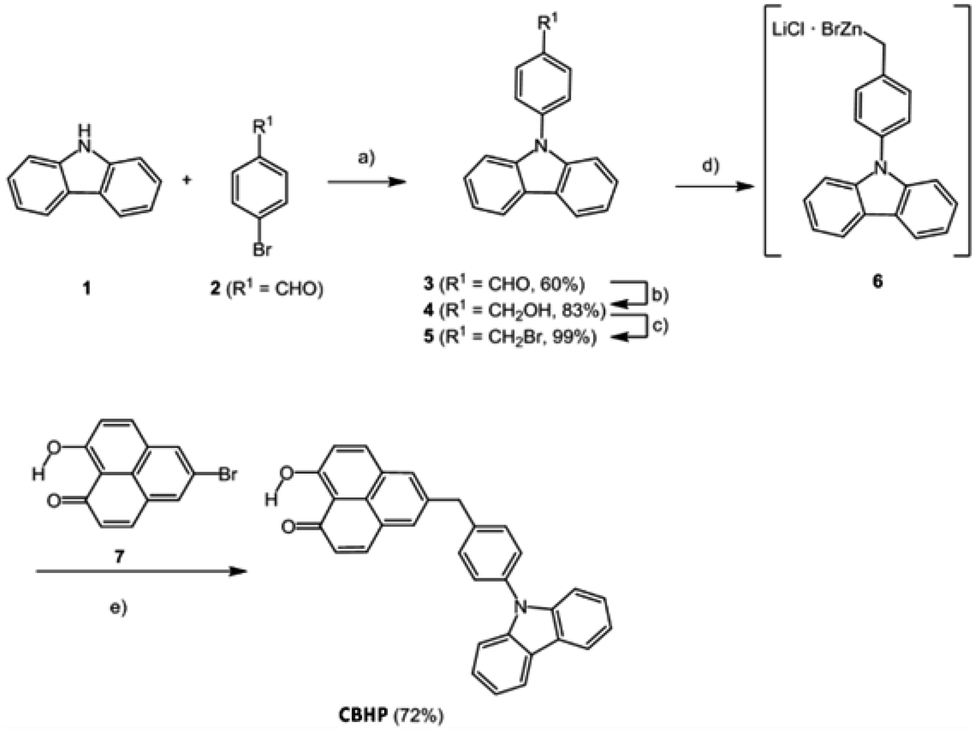 | ||
| Scheme 1 Five-step synthesis of CBHP. (a) Cu (0.70 equiv.), K2CO3 (2.2 equiv.), [18]-crown-6 (0.14 equiv.), 1,2-dichlorobenzene, 185 °C, 3d, 60%. (b) NaBH4 (1.20 equiv.), MeOH, 20 °C, 3 h, 83%. (c) PBr3 (1.00 equiv.), THF, 20 °C, 23 h, 99%. (d) LiCl (1.12 equiv.), Zn (1.52 equiv.), 1,2-dibromoethane (0.35 equiv.), Me3SiCl (0.35 equiv.), THF, 20 °C, 18 h. (e) 10 mol% Pd(OAc)2, 10 mol% SPhos (2-dicyclohexylphosphano-2′,6′-dimethoxybiphenyl), THF, 65 °C, 24 h, 72%. | ||
The fluorescence of the dyad is minimal and can be detected with a fluorescence quantum yield Φf < 0.01 at 461 nm. Moreover, in the electronic ground state, as investigated by cyclic voltammetry, an electrochemically reversible one-electron oxidation is found at E00/+1 = 1290 mV, as well as an electrochemically reversible one-electron reduction at E00/−1 = −1160 mV. From these redox potentials, an E00 transition λexc = 453 nm, and a mean donor–acceptor distance rDA = 0.82 nm, a Gibbs free enthalpy of photoinduced electron transfer ΔGPET = −0.487 eV can be estimated by the Rehm–Weller approximation.21
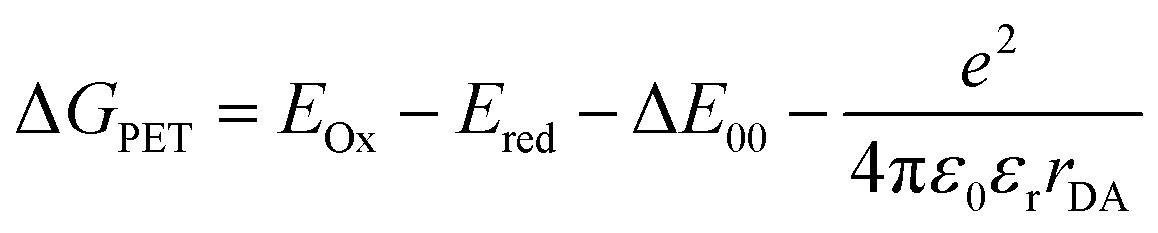 | (1) |
2.2 Structure and topography of CBHP
Sublimation of CBHP from powder onto a clean Pt(111) surface leads to reproducible surface coverages controlled by duration and temperature. Using LT-UHV-STM we could identify single intact CBHP molecules in two different recurring structures, which we denote ConA and ConB, as well as a few molecular fragments. High resolution images of CBHP in both configurations are given in Fig. 1a and b. A large-scale image of a prototypical Pt(111) surface covered with CBHP is given in Fig. S1 (ESI†). The CBHP molecules exhibit no directionality with respect to the principal directions in the Pt(111) plane, which reflects the adsorption behavior of a non-planar, unsymmetrical molecule with three freely rotatable single bonds in the gas phase. Nevertheless, by means of the adsorption process the multiple degrees of freedom of the gas phase molecule (see red arrows in Fig. 2b) are reduced and the prochiral center located at the methylene linker becomes a chiral center in combination with the surface. This is reflected by the existence of the adsorption conformations ConA and ConB also in their mirror forms, that is, ConA′ and ConB′, like shown exemplarily for ConA′ in Fig. S1 (ESI†). Moreover, due to the on-surface reduced rotational degrees of freedom of the single bonds to the methylene linker and the need to provide enough space for the phenylene hydrogen atoms, the kink between the donor and the acceptor group only holds values from 140° to 160° deduced from 20 molecules adsorbed on Pt(111). However, the existence of this range of values points to multiple, nearly energetically equivalent rotational conformers of CBHP on Pt(111).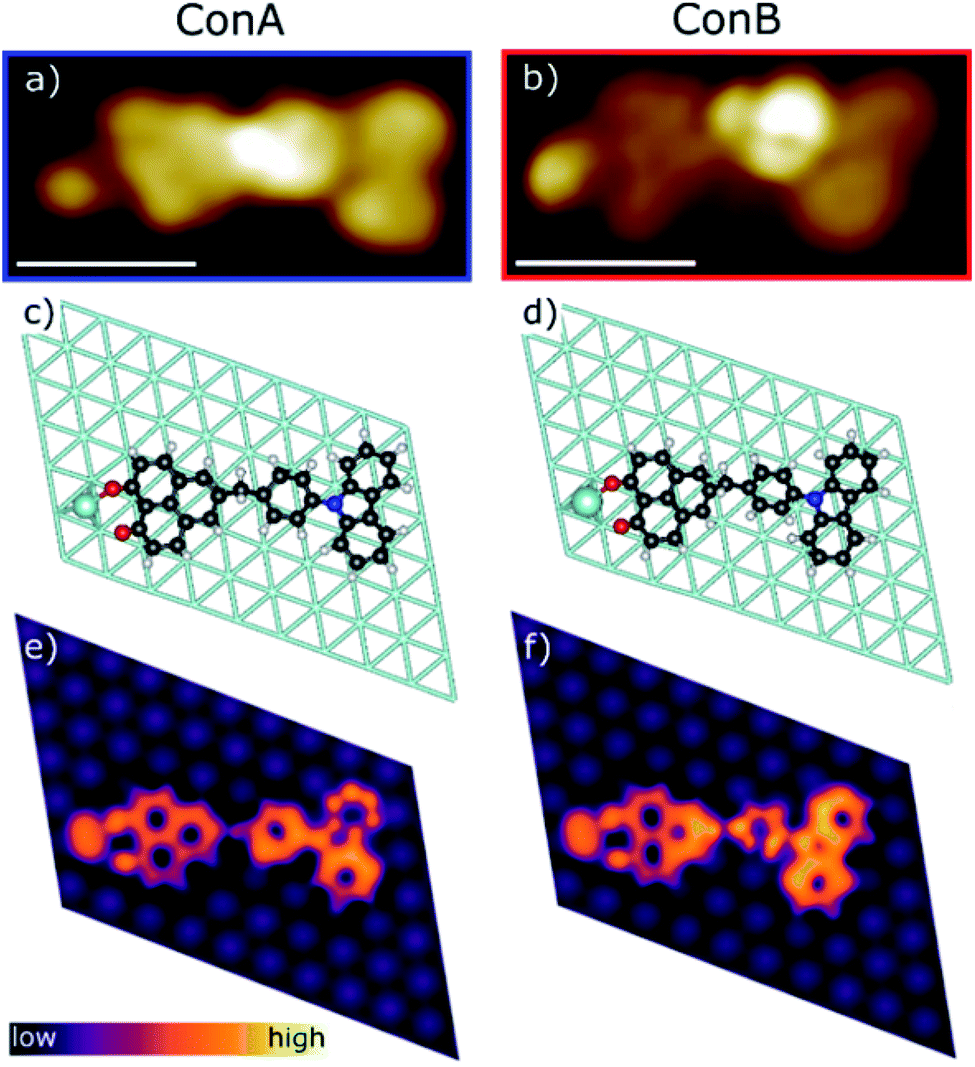 | ||
| Fig. 1 Comparison of the two adsorption configurations of CBHP, ConA and ConB. (a and b) High resolution STM images (Ubias = 50 mV, Iset = 200 pA, scale bar = 1 nm). (c and d) Geometries of the CBHP–Pt complex on the Pt(111) surface corresponding to local minima from DFT calculations, where the Pt adatom has been highlighted from the substrate. (e and f) Simulated constant-current STM images around EF. | ||
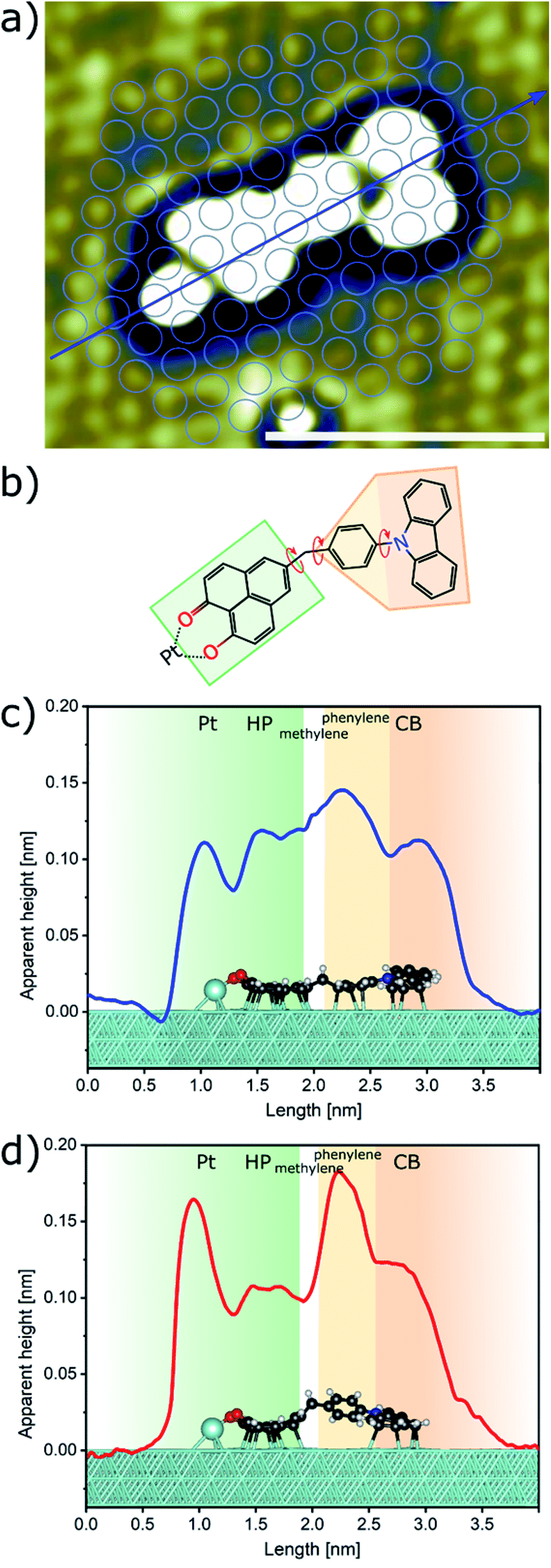 | ||
| Fig. 2 Detailed on-surface geometry of CBHP. (a) High resolution STM image of ConA filtered as described in ESI, Fig. S5† (Ubias = 50 mV, Iset = 200 pA, scale bar = 2 nm). (b) Chemical structure of CBHP–Pt. The hydroxyphenalenone acceptor (HP) is marked by a green and the carbazolyl phenyl donor (CB) by an orange contour. A methylene linker is connecting donor and acceptor while introducing a kink to CBHP. Bonds with a rotational degree of freedom in the gas phase molecule are indicated by red arrows. (c and d) Averaged height profiles along the long molecular axis for ConA (blue) and ConB (red) as indicated exemplarily in (a) for ConA. The corresponding calculated CBHP–Pt complex geometries on a 3-layer Pt substrate are shown below the respective height profiles. | ||
The two recurring adsorption configurations of the CBHP molecules (Fig. 1) exhibit the same general shape. They are identified as an elongated structure of 2.3 nm, a value which is in accordance with the dimension expected from the chemical structure given in Scheme 1. Three of the four main parts of the STM images can be identified by comparison of the orbital resolved structure with the chemical structure of CBHP. For both configurations we find from left to right in Fig. 1a and b, the rather flat HP acceptor, the brighter bridge unit (methylene and phenylene groups) which is the source for the conformational flexibility in the molecule, and the almost planar CB donor group. While the acceptor exhibits a width of only 0.9 nm perpendicular to the long molecular axis, this width amounts to 1.0 nm for the donor part (for details see ESI Fig. S2 and S3†). However, the single separated bright spot to the left of the acceptor cannot be explained in this way. Its position indicates an interaction with the HP group, but it is clearly not an orbital feature of CBHP. Indeed, each CBHP molecule observed on the surface shows this spot next to HP. Since HP is known to form multiple stable complex structures with metal atoms,15,16 we hypothesize that deprotonation of the hydroxyl group and subsequent complexation leading to a CBHP–Pt complex takes place during adsorption of CBHP on the Pt(111) surface. Calculations for both conformers indicate that the adsorbed CBHP–Pt complex plus an individual H atom is more stable than the adsorbed CBHP plus an individual Pt atom by approximately 3.3 eV, thus supporting the formation of CBHP–Pt complexes. The Pt atoms might originate from adatoms diffusing on the surface or even substrate atoms extracted from the surface, since Pt(111) surfaces tend to reconstruct under the influence of adsorbed molecules.22,23
In order to verify this CBHP–Pt complex formation hypothesis during adsorption and to evaluate the CBHP/Pt(111) interface interactions, we performed thorough DFT calculations including van der Waals interactions on this molecular system. These calculations lead to a variety of adsorption geometries of CBHP or CBHP–Pt. Simulations showed that the strong C–Pt interaction and the large conformational flexibility of the molecule influenced the CBHP and CBHP–Pt geometries on the substrate. The calculated average height of C atoms above the Pt surface layer is 2.9 Å on the donor unit and 2.2 Å on the acceptor for ConA. For ConB these heights are 2.6 Å and 2.3 Å, respectively. These values are characteristic of strong metal-molecule chemical bonds.24,25 Surface Pt atoms are also found to be displaced as a consequence of bonding to the adsorbate.26,27 In our case, Pt atoms bonded to the molecule can be lifted up to 0.3 Å. The O–Pt adatom bond length is 2.2 Å in both configurations, ConA and ConB, which is a typical size for a donor–acceptor bond.28 Both, C–Pt and O–Pt distances are close to previously computed values.29,30 We obtained a considerable conformity between the measured STM images of ConA and ConB and the calculated constant-current STM images of the CBHP–Pt complex corresponding to the two most stable local energy minima (Fig. 1e and f). The aforementioned general shape of CBHP is reproduced in length and width and the four distinguishable parts of the CBHP–Pt complex are distinctly and visibly identified. The experimentally obtained appearance of the carbazole moiety, consisting of 2 large spots with small depressions in their center, and of the planar phenalenone moiety match in detail the calculated STM images. Even the differences in the general structures between ConA and ConB are distinguishable in the simulated STM images. The central region shows the brightest spot located at the position of the phenylene ring in the case of ConB (Fig. 1b) while it is located at the position of the methylene linker for ConA (Fig. 1a) indicating a rotation of the bridge between acceptor and donor part. This rotation of ConB relative to ConA is represented in the simulated STM images as well. Thus, the calculated on-surface configurations are representatives of the experimentally obtained conformations adopted by the CBHP–Pt complexes.
Additional simulations of the CBHP molecule without the Pt adatom were conducted and are given in ESI Fig. S4.† While in the experimental STM images one bright, distinct spot is observed on the edge of the HP acceptor, two smaller spots appear in the simulated constant-current image for the CBHP molecule, which can be assigned to the two oxygen atoms of HP. Thus, DFT calculations strongly suggest the formation of the CBHP–Pt complexes, as presented in Fig. 1b–e, and we exclude the adsorption of single CBHP molecules without the formation of a Pt-complex on Pt(111).
A high-resolution image of CBHP–Pt with an atomically resolved Pt(111) surface is given in Fig. 2a, that enables the determination of the exact position of this molecule with respect to the substrate and shows the position of the averaged apparent height profile indicated by an arrow. In Fig. 2c and d the apparent height profiles through the molecule are overlaid with side views of the calculated on-surface geometries for ConA and ConB on three Pt layers. While the bridging phenylene group in ConA is only slightly rotated from an orientation parallel to the substrate, the one in ConB is rather upright. This rotation causes the methylene linker to be lifted from the surface in ConB as well. In consequence, this leads to an increased apparent height of the whole center of CBHP–Pt in ConB, consistent with the calculated geometries.
Based on these theoretically deduced insights the brightest features observed in the STM image of ConB (Fig. 1b) are interpreted as a phenylene ring standing obliquely to the substrate. This phenylene ring is represented by the two spots which differ by approximately 30 pm in their apparent height. They originate from the two lobes of the π-orbital (+π and −π) located above and below the phenylene ring which are separated by the atomic nodal plane located in the plane of the ring. A phenylene ring with an orientation exactly perpendicular to the substrate would be imaged as two equivalent spots related to the two parts of its π-orbital, as shown in a previous work.14
Different from the case of ConB, the slight asymmetry in the orbital appearance of the phenylene ring in ConA (Fig. 1a) can be attributed to the deviation from a parallel position to the substrate. The brighter side of the phenylene ring in ConA is found always at the inner side of the kink. This corresponds to a slight lifting of the inner side of the phenylene ring from the surface, owing to the steric hindrance introduced by the ring hydrogen atoms approaching the ones of HP and CB. ConB shows the rotation of the phenylene ring in the same direction but to a much higher extent. Here, always the outer side of the phenylene ring appears brighter, since in this configuration the geometry of the oblique π-orbital is recorded, as discussed above. Thus, the dominant differences in the appearance of ConA and ConB are mainly due to geometric effects. This has been verified by a series of high resolution STM images taken for ConA revealing no clear voltage dependent changes in the range between −2 V and 2 V (Fig. S6, ESI†). The simulated STM images in Fig. 1e and f show the differences in electronic and geometric properties of both conformers, while the latter are clearly visible in the side views of the calculated geometries (Fig. 2c and d).
While CBHP–Pt in ConB is chemisorbed to the Pt(111) surface through the HP acceptor and CB donor, all HP, the central part and CB units are chemisorbed in the case of ConA. The binding energy of the adsorbed CBHP molecule is −12.59 eV (ConA) and −11.10 eV (ConB). As a reference, values of 1.3–1.9 eV were measured and calculated previously for benzene on Pt(111),31,32 approximately 6–10 times smaller than the calculated binding energies of ConA and ConB. Given that the total number of five- and six-membered rings in the CBHP molecule is seven (ignoring N and O heteroatoms), the calculated binding energies are consistent and indicative of a strong interaction with the substrate. The binding of C atoms to the Pt(111) substrate drives the lateral position of the CB and HP units on the surface and induces strain along the molecule in order to enable the binding of acceptor and donor to adequate positions of the Pt(111) surface, leading to the lifted molecule center in ConB and resulting in a smaller angle between HP and CB in experiment and theory. The difference in binding energy between both configurations, ConA and ConB, of 1.49 eV suggests that in thermal equilibrium only dyads in conformation ConA should be found on the Pt(111) surface. However, the observation of dyads in ConB in a considerable amount points clearly to a kinetically trapping during the deposition process.
2.3 Molecular orbitals in gas phase and on surface
The influence of the adsorption of CBHP–Pt on Pt(111) on the electronic structure is studied using voltage dependent STM images as well as scanning tunneling spectroscopy (STS) and is compared to the Local Density Of States (LDOS) from the DFT calculations. STM measurements exhibit a small voltage dependence and are given in ESI Fig. S6† for ConA. These STM images reveal that throughout the whole investigated voltage range (−2 V to +2 V) the unoccupied states appear brighter than the occupied states. In addition, the phenylene group becomes the brightest feature for voltages of more than 1 V, while otherwise the position of the methylene linker is the most intense spot of the molecule.A full set of STS measurements was obtained for CBHP–Pt in configuration ConA, while this was not possible for ConB. CBHP–Pt in configuration ConB changed their appearance if voltage sweeps were applied indicating a considerable strain, as discussed above, so that voltage sweeps cause a strain release and thereby a change of the molecular conformation or even the decomposition of the molecule. Fig. 3a shows typical STS curves for ConA measured above four different positions of CBHP in comparison to the clean Pt(111) surface (gray background). Differential conductance spectra (dI/dV) were taken on the center of the acceptor, the benzyl group and both spots of the CB donor. Distinct differences are observed for the four positions. When the tip is positioned above the inner side of the donor (green curve) only small differential conductance values are obtained compared to the other parts of the molecule. However, this is the only tip position showing distinct features in the unoccupied part of the spectrum, at 0.9 eV (lowest unoccupied molecular orbital, LUMO) and 2.0 eV (LUMO+1). The dI/dV curve taken above the central part (purple) starts to rise at 1.0 eV, while on the other hand, the orange curve (outer part of the donor) begins to rise slowly not before 1.5 eV. Remarkably, the differential conductance characteristic of the HP acceptor (black curve) shows no distinct molecular features over the whole examined voltage range, which we attribute to a strong interaction with the Pt(111) surface. Occupied molecular orbital states can be identified by STS above the center of the molecule (purple curve) and the outer donor part (orange curve). The purple curve shows a first shoulder around −1.0 eV (highest occupied molecular orbital, HOMO) with a main peak at −1.9 eV while the orange curve has a feature at −2.1 eV. This is in accordance with occupied delocalized states over the CB donor inclusive the phenylene ring indicating the preservation of the donor character on Pt(111) in ConA. The thereof obtained HOMO–LUMO gap of CBHP adsorbed on Pt(111) amounts to 1.9 eV compared to 2.45 eV obtained from cyclic voltammetry investigations performed on CBHP in solution described above. This points to a reduction of the HOMO–LUMO gap during adsorption.
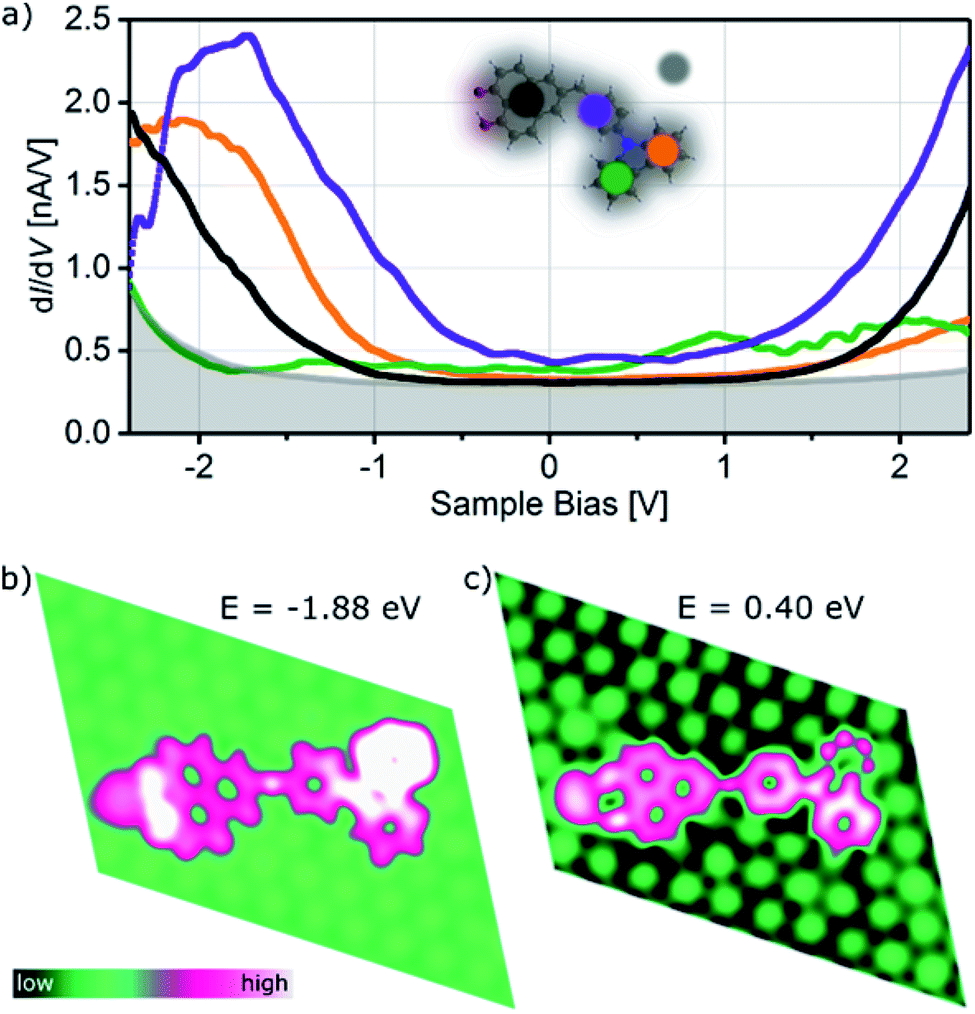 | ||
| Fig. 3 STS spectra and calculated LDOS of CBHP in ConA. (a) Differential conductance (set-point: Iset = 3 nA, Vmod = 20 mV) obtained over four positions of ConA as indicated in the inset by colored circles: HP acceptor (black), central part (purple) and two spots over the donor CB (green and orange), Pt(111) substrate (grey background). (b) Calculated LDOS of ConA illustrating the spatial localization over the dyad of occupied and unoccupied electronic states. | ||
Calculations for ConA capture some of the trends seen in the measured dI/dV curves for different tip positions, as shown in Fig. 3b. At energies below the Fermi level the calculated LDOS shows electronic states at −1.88 eV predominantly on the O atoms of the HP–Pt unit, the outer donor part and the central part (black, orange and purple lines in Fig. 3a, respectively). The calculated LDOS of the adsorbed ConA, for energies above the Fermi level, at 0.40 eV (Fig. 3c), exhibits bright LDOS features over the central phenylene ring and the inner CB unit and smaller features over the outer donor ring, in similarity with the purple, green, and orange curves of Fig. 3a for unoccupied states. Overall, spectral features of the adsorbed dyad reveal a high degree of hybridization with the Pt substrate leading to molecular states being significantly broadened in energy and distorted with respect to the isolated molecule. The calculated Projected Density Of States (PDOS) of ConA is given in Fig. S7 (ESI†) as well as LDOS images of ConA at additional positive and negative energies.
To investigate the preservation of the donor–acceptor character upon adsorption, electronic structure calculations were performed where the binding to the Pt adatom and then to the substrate were considered sequentially by calculating the isolated CBHP molecule, the isolated CBHP–Pt complex, and the CBHP–Pt complex adsorbed on the Pt(111) surface. Throughout, the positions of all atoms were kept frozen at the values of the metal-molecule junction, thus allowing us to disentangle electronic from geometric effects and analyze the former ones alone. Fig. 4 shows the results of this analysis for ConB.
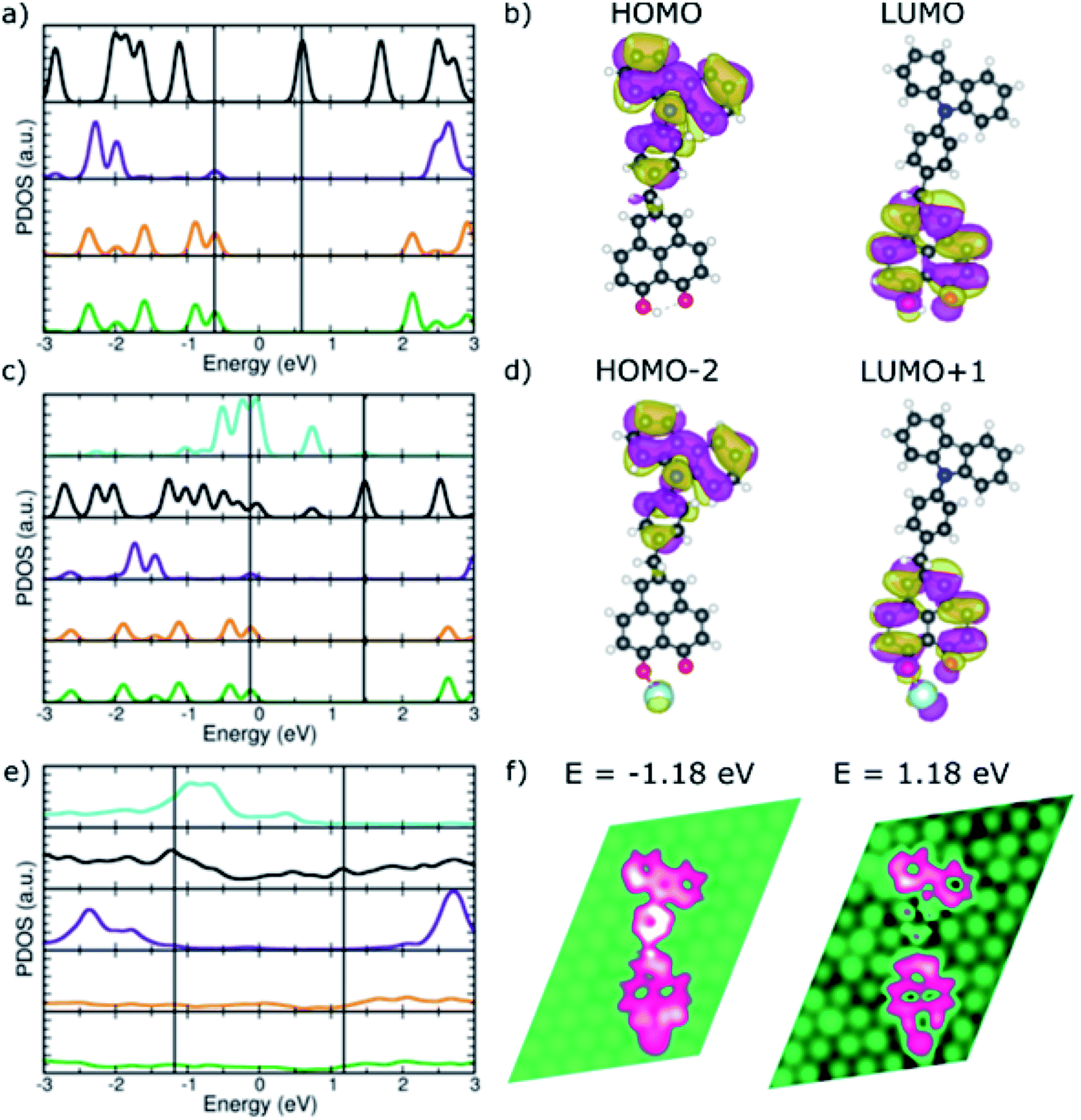 | ||
| Fig. 4 PDOS of the donor–acceptor dyad in ConB. (a) CBHP in the gas phase. (c) CBHP–Pt in the gas phase. (e) CBHP–Pt adsorbed on Pt(111) (color code as in Fig. 3a, in addition Pt-adatom in turquoise). (b and d) Relevant frontier molecular orbitals marked by black lines in (a) and (c). (f) LDOS plots at selected energies marked by black lines in (e). | ||
The CBHP molecule in the gas phase (Fig. 4a) exhibits discrete molecular orbitals shown by well-separated PDOS peaks. The DOS projected over different donor and acceptor subunits are represented in different colors, like defined in Fig. 3. The calculated HOMO–LUMO gap of the molecule in the ConB geometry is 1.23 eV, close to the value of 1.37 eV in the ConA geometry and 1.40 eV in the fully relaxed isolated molecule geometry. These gap values, calculated with the vdW-DF exchange–correlation functional, are smaller than with other functionals which feature a more accurate description of electron–electron interaction (Fig. S8, ESI†) but which cannot be applied to the metal-molecule interface. The donor–acceptor character of the isolated dyad in geometry ConB can be seen in Fig. 4a, where PDOS peaks projected over the inner and outer donor regions are predominant at negative energies (green and orange lines), while the HP acceptor states abound in the unoccupied part of the spectrum. Bridge states are found mostly far from the gap for the unoccupied states.
Fig. 4b illustrates the donor–acceptor character of the isolated molecule by plotting selected molecular orbitals, whose energies are indicated in panel (a) with vertical lines. The HOMO is localized over the donor rings and the bridge phenylene and methylene units. In contrast, the LUMO is delocalized on the HP acceptor unit but has no significant amplitude on the phenylene bridge group or the CB acceptor. This is consistent with the donor–acceptor character of the CBHP dyad, which is clearly manifested in the gas phase, even in the somewhat constrained geometry ConB.
Fig. 4c shows the calculated PDOS after bond formation between a Pt atom (turquoise PDOS) and one of the HP oxygen atoms of CBHP, while keeping the geometry frozen to that of the ConB/Pt(111) interface. Additional features appear near the HOMO–LUMO gap due to the mixing of molecular states located at the HP acceptor with the states of this Pt atom. Several states of the complex have appreciable Pt PDOS contribution (turquoise line). In particular, the buildup slightly below the Fermi level leads to the edge of the Pt band in the slab. Many wave functions of the CBHP–Pt complex are delocalized over both species. However, the gas-phase orbitals selected in panel (b) still retain their characteristic nodal patterns (Fig. 4d) even though their ordering in the spectrum has changed as they are now HOMO−2 and LUMO+1, respectively. Thus, despite the mixing and rearranging introduced by the states of the Pt atom, the CBHP–Pt complex largely preserves the donor–acceptor characteristics of the isolated dyad (see ESI, Fig. S9 and S10†).
This situation, however, is significantly changed when the complex is adsorbed on the Pt(111) surface. Fig. 4e shows the calculated DOS projected onto the different donor and acceptor regions when CBHP–Pt is adsorbed on Pt(111). Molecular states are significantly broadened due to the strong hybridization with the Pt substrate. This is expected from the multiple bond formation established between C atoms of the CB and HP units and the Pt surface, as seen in the junction geometries (Fig. 2c and d). The spectral features of the molecule have significant amplitudes over a wide energy range around the Fermi level. CB donor features are present below the Fermi level, but also above it. Similarly, features derived from the HP acceptor are present in unoccupied energies, however, they also peak below the Fermi level. The donor–acceptor character is thus clearly reduced upon adsorption.
Over a wide range of energies above and below the Fermi level, calculated LDOS features are spread to some extent over donor, bridge, and acceptor units. We found no energies at which the distinctive localization and nodal patterns of panels (b) and (d) were preserved. Fig. 4f shows the calculated LDOS at two energies above and below the Fermi level. Electronic states at −1.18 eV involve dominantly the bridge and donor parts of the molecule, but also the Pt adatom bound to the HP unit and the HP unit. At 1.18 eV, there is no significant LDOS on the bridge group, however, it is spread over the HP acceptor including the Pt adatom and a part of the donor unit simultaneously. LDOS plots at additional energies are given in ESI Fig. S11† and exhibit similar characteristics. Thus, the donor–acceptor character of the CBHP dyad is significantly modified due to the hybridization and chemical bonds with the Pt(111) substrate.
2.4 Donor–acceptor character of CBHP upon adsorption on Pt(111)
The changes described previously can be analyzed in terms of the calculated electrostatic potential of the isolated CBHP molecule, isolated CBHP–Pt complex, and adsorbed CBHP–Pt complex. The results are plotted in Fig. 5 for both conformations. The plots show the evolution of the electrostatic potential along the sequential binding to the Pt adatom and then onto the Pt(111) substrate, while excluding effects due to changes in geometry. Electrostatic potential values (in mV) are projected on an isocontour of the electron density and are shown as a color scale.33 High values of the electrostatic potential (blue) are related to a reduced electron density. Low electrostatic potential values (red) occur in electronegative regions and correspond to accumulation of electron density. The comparison between both conformers in Fig. 5 shows a similar pattern, indicating that the differences in geometry between ConA and ConB do not play an important role in determining the electrostatic distribution upon adsorption. The electrostatic potential of the isolated CBHP molecule (Fig. 5a and d) shows positive values near the peripheral H atoms, while negative values are found near the O atoms of the HP acceptor and, to a lesser extent, in the CB donor rings. Consequently, the planar carbon system forming the acceptor shows a lower electron density (green/blue) than the carbon system of the donor (yellow/green) confirming the donor–acceptor character of CBHP for both configurations in the gas phase. Binding of the complex to the Pt adatom (Fig. 5b and e) introduces changes in the O–Pt region but the potential in other parts of the molecule remains almost unchanged, thus the donor–acceptor character is maintained. This is consistent with the findings shown in Fig. 4, which indicate the introduction of Pt-related states but otherwise the preservation of the character of isolated molecular orbitals. Adsorption onto the Pt(111) substrate, however, leads to an increase of the electrostatic potential throughout the molecule. Negative values are only retained near the HP O atoms in both conformations, the center of ConB and on the donor rings of ConA and ConB which are detached from the substrate (see Fig. 2c). The Pt substrate has negative values. These findings reflect electron transfer from the molecule to the Pt substrate, resulting in the changes of the potential described. Therefore, the donor–acceptor character seems to be significantly changed but can be nevertheless identified at the surface.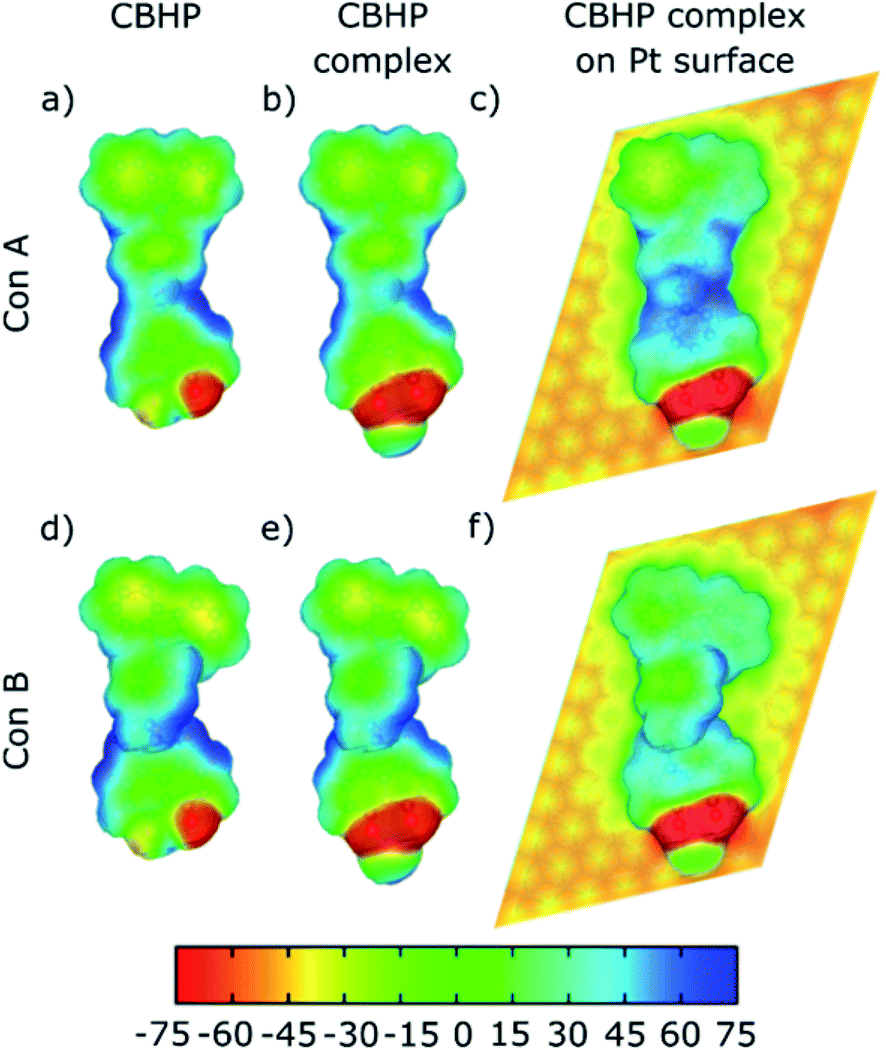 | ||
| Fig. 5 Calculated electrostatic potential in mV plotted over an electron density contour of 10−3 e Å−3 for ConA (top panels) and ConB (bottom panels). (a and d) Gas-phase CBHP. (b and e) CBHP–Pt complex. (c and f) CBHP–Pt on Pt(111). | ||
To analyze the charge transfer in detail, we calculated the electron density difference upon adsorption. We computed the electron density of the junction minus the sum of the individual CBHP–Pt complex and the Pt(111) substrate. As before, the position of all atoms was kept fixed to their values at the interface throughout. Fig. 6 shows the results for ConA (panels a–c) and ConB (panels d–f). Positive (negative) values correspond to electron accumulation (depletion) and are shown in blue (yellow). For both ConA and ConB, electrons are transferred from the molecule to the substrate, mostly to the surface Pt layer. This follows from the formation of multiple molecule–substrate bonds involving many C atoms. The charge transfer between detached parts of the molecule and the Pt surface is minimal, like indicated by the real-space representation of the electron density difference (Fig. 6). For ConA, this applies to the outer donor ring, which is lifted above the inner one. In the case of the more twisted ConB, this corresponds to the bridge unit formed by the methylene group and one phenylene ring. Notably, electron density is transferred to the Pt surface from both donor and acceptor units, which changes the donor–acceptor character of the dyad upon adsorption.
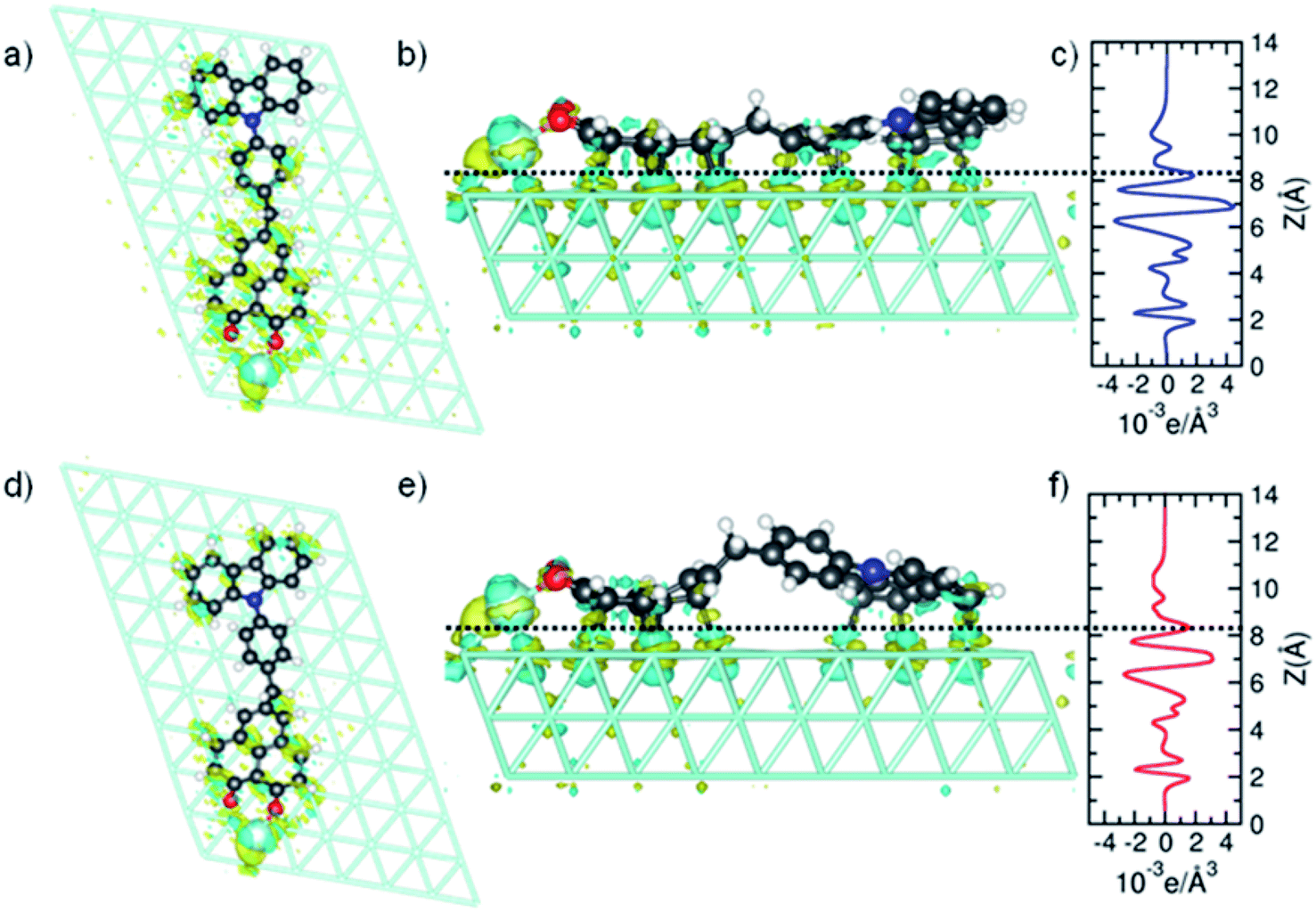 | ||
| Fig. 6 Calculated electron density difference upon adsorption of ConA (top panels) and ConB (bottom panels). (a, b, d and e) Top and side views of the real-space difference. (c and f) Plane-averaged electron density difference. | ||
Fig. 6c and f plot the plane-averaged electron density difference as a function of vertical distance for ConA and ConB. Both profiles exhibit oscillations and negative values in the molecular region and positive values below the Pt surface layer. The dashed horizontal lines indicate the mid height of the shortest C–Pt bond and are taken as a guide to separate the CBHP–Pt and Pt(111) subsystems. Integration of the charge difference above and below this dashed line gives a measure of the transferred charge. A value of 0.18e transferred from CBHP–Pt to the substrate is obtained for both ConA and ConB, which is in the size range found for other molecules.34,35 However, for ConB only the CB and HP groups are involved, whereas in ConA the bridge also participates in the charge transfer. By cleaving the CBHP–Pt complex into separate donor, bridge, and acceptor–Pt units (all appropriately H-saturated), the charge transfer of each constituent can be computed. As expected, in both conformers electron transfer to Pt(111) from the CB donor unit is higher than from the HP–Pt acceptor (0.10–0.13 electrons compared to 0.06 electrons, respectively). This is consistent with the high work function of 5.8 eV for the Pt (111) substrate,36 which possesses a higher acceptor character than either CB or HP. However, the analysis of Fig. 5 shows that the donor–acceptor character of the dyad does not disappear completely when adsorbed on Pt(111), although it is reduced.
These investigations indicate that the non-planarity of CBHP is the key factor, which enables the preservation of the donor–acceptor character upon adsorption on a metallic surface and in principle qualifies this kind of dyads to be employed in advanced devices. Further molecular engineering, like the introduction of bulky side groups, should lead to an increased decoupling of molecular parts from the surface und thus, should be advantageous for the preservation of the intrinsic molecular properties. In addition, by tuning the energy level alignment of dyad and metal, undesired charge redistributions upon adsorption should be diminished and a significant on-surface donor–acceptor character should be possible.
3. Conclusions
In conclusion, a novel donor–bridge–acceptor dyad, CBHP, was synthesized, deposited on a Pt(111) surface, and studied by a combination of LT-UHV-STM measurements and DFT simulations. The geometry and electronic properties were investigated using STM and differential conductance measurements. Two recurring conformations, ConA and ConB, were identified in these measurements by comparison to the chemical structure. A bright spot close to the acceptor side on all STM images was ascribed to a Pt adatom. STS of ConA above different positions of the molecule revealed donor-related peaks below the Fermi level and acceptor-derived states at higher energies. DFT-based simulations were performed for two representative stable structures obtained from a broad sampling of junction configurations. The stronger bound was adsorbed nearly flat on Pt(111) (ConA) while the other conformation (ConB) exhibited a slightly twisted geometry with the phenylene bridge lifted from the substrate. Calculations rationalize the experimental findings and the nature of the donor–acceptor character of the adsorbed CBHP dyad. The donor–acceptor properties are significantly reduced upon adsorption. This is due to the strong hybridization between adsorbed molecular units and substrate orbitals, which substantially broadens and intermixes donor and acceptor states, as well as to the high work function of the Pt substrate, which acts as an acceptor to both CB and HP constituents. Nevertheless, calculated electrostatic potential and electron density difference indicate that the donor–acceptor character of this non-planar dyad is partially retained even after the formation of strong chemical contacts to the metal substrate. Thus, we conclude that despite the substantial changes of intrinsic molecular properties upon chemical adsorption the principle applicability of donor–bridge–acceptor molecules in functional devices is feasible.4. Materials and methods
4.1 Chemical synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and crystallization from hexane/ethyl acetate furnished 4-(9H-carbazol-9-yl)benzaldehyde (3) (814 mg, 60%) as colorless crystals, Rf = 0.32 (n-hexane/ethyl acetate 10:1), mp 158 °C (156–158 °C as given in ref. 37).
:1) and crystallization from hexane/ethyl acetate furnished 4-(9H-carbazol-9-yl)benzaldehyde (3) (814 mg, 60%) as colorless crystals, Rf = 0.32 (n-hexane/ethyl acetate 10:1), mp 158 °C (156–158 °C as given in ref. 37).
1H NMR (600 MHz, CDCl3): δ 7.34 (d, 3J = 7.9 Hz, 2H), 7.46–7.43 (m, 2H), 7.50 (s, 2H), 7.81–7.79 (m, 2H), 8.17–8.14 (m, 4H), 10.12 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 109.9 (CH), 120.7 (CH), 121.0 (CH), 124.1 (Cquat), 126.4 (CH), 127.0 (CH), 131.5 (CH), 134.8 (Cquat), 140.2 (Cquat), 143.6 (Cquat), 191.1 (CH). MS (EI) (70 eV, m/z (%)): 272 (20), 271 (M+, 100), 243 (5), 242 ([M − CHO]+, 24), 241 (28), 240 (5), 120 (18). IR: ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) = 2824 (w), 2733 (w), 1701 (m), 1595 (m), 1512 (w), 1479 (w), 1451 (m), 1362 (m), 1337 (m), 1317 (w), 1300 (w), 1227 (m), 1209 (w), 1182 (w), 1161 (m), 1125 (w), 1103 (w), 916 (w), 845 (w), 829 (w), 750 (s), 714 (m) cm−1. Anal. calcd for C19H13NO (271.1): C 84.11, H 4.83, N 5.16. Found: C 84.11, H 5.10, N 4.99.
= 2824 (w), 2733 (w), 1701 (m), 1595 (m), 1512 (w), 1479 (w), 1451 (m), 1362 (m), 1337 (m), 1317 (w), 1300 (w), 1227 (m), 1209 (w), 1182 (w), 1161 (m), 1125 (w), 1103 (w), 916 (w), 845 (w), 829 (w), 750 (s), 714 (m) cm−1. Anal. calcd for C19H13NO (271.1): C 84.11, H 4.83, N 5.16. Found: C 84.11, H 5.10, N 4.99.
:1 → 4:1 → 2:1) to give [4-(9H-carbazol-9-yl)phenyl]methanol (4) (592 mg, 83%) as a colorless solid, Rf = 0.22 (n-hexane/ethyl acetate 4:1), mp 123 °C.
1H NMR (600 MHz, CDCl3): δ 1.83 (s, 1H), 4.84 (s, 2H), 7.29 (ddd, JH–H = 7.9 Hz, 5.7 Hz, 2.4 Hz, 2H), 7.45–7.38 (m, 4H), 7.64–7.54 (m, 4H), 8.15 (ddd, JH–H = 7.8 Hz, 7.8 Hz, 1.0 Hz, 2H). 13C NMR (151 MHz, CDCl3): δ 65.1 (CH2), 109.9 (CH), 120.1 (CH), 120.5 (CH), 123.5 (Cquat), 126.1 (CH), 127.4 (CH), 128.6 (CH), 137.2 (Cquat), 140.2 (Cquat), 141.0 (Cquat). MS (EI) (70 eV, m/z (%)): 274 (14), 273 (M+, 68), 272 (6), 271 (14), 256 ([M − OH]+, 18), 254 (21), 253 (7), 252 (25), 242 ([M − CH2OH]+, 8), 241 (12), 223 (8), 217 (8), 199 (12), 189 (10), 188 (89), 187 (90), 185 (16), 179 (10), 171 (42), 169 (43), 167 ([M − C7H7O]+, 10), 159 (17), 157 (23), 152 ([M − C7H7NO]+, 23), 149 (11), 108 (10), 107 (97), 91 (13), 90 (C6H4N+, 18), 89 (17), 79 (100), 78 (36), 77 (C6H4+, 96), 76 (12), 75 (13), 63 (10), 53 (13), 51 (25), 50 (20). IR: = 3233 (w), 1701 (w), 1607 (w), 1595 (w), 1518 (m), 1477 (w), 1451 (s), 1362 (w), 1335 (w), 1314 (w), 1229 (s), 1182 (w), 1171 (w), 1011 (s), 914 (w), 843 (m), 806 (w), 750 (s), 725 (s), 700 (w), 629 (m) cm−1. Anal. calcd for C19H23NO (281.2): C 83.49, H 5.53, N 5.12. Found: C 83.69, H 5.45, N 5.05.
:1) to give 9-[4-(bromomethyl)phenyl]-9H-carbazole (5) (2.32 g, 99%) a colorless solid, Rf = 0.80 (n-hexane/ethyl acetate 4:1), mp 100 °C.
1H NMR (600 MHz, CDCl3): δ 4.62 (s, 2H), 7.33–7.29 (m, 2H), 7.45–7.40 (m, 4H), 7.57 (d, 3J = 8.4 Hz, 2H), 7.64 (d, 3J = 8.4 Hz, 2H), 8.16 (d, 3J = 7.8 Hz, 1H). 13C NMR (151 MHz, CDCl3): δ 32.9 (CH2), 109.9 (CH), 120.3 (CH), 120.5 (CH), 123.6 (Cquat), 126.1 (CH), 127.4 (CH), 130.7 (CH), 136.9 (Cquat), 137.9 (Cquat), 140.8 (Cquat). MS (EI) (70 eV, m/z (%)): 337 (M(81Br)+, 15), 335 (M(79Br)+, 13), 257 (23), 256 ([M − Br]+, 100), 254 (16), 241 (6), 128 (19), 127 (12), 52 (6). IR: = 3061 (w), 3046 (w), 1593 (w), 1514 (m), 1478 (w), 1451 (s), 1418 (w), 1366 (w), 1335 (w), 1308 (m), 1287 (w), 1229 (s), 1206 (m), 1182 (m), 1169 (w), 1144 (w), 1119 (w), 1096 (w), 1020 (w), 995 (w), 916 (w), 831 (w), 812 (w), 747 (s), 721 (s), 702 (m), 644 (w), 629 (w) cm−1. Anal. calcd for C19H14BrN (335.0): C 67.87, H 4.20, N 4.17. Found: C 67.64, H 4.28, N 4.01.
In a second oven dried nitrogen-flushed screw-cap Schlenk tube with magnetic stir bar were placed 5-bromo-9-hydroxy-1H-phenalen-1-one (7) (69 mg, 0.250 mmol) and dry THF (2.00 mL) under nitrogen. To this solution palladium(II)acetate (6 mg, 25 μmol), and SPhos (21 mg, 50 μmol) were added. The decanted solution of the first Schlenk tube was placed in a syringe and by syringe pump this solution was added dropwise over 1.5 h to the stirred solution in the second Schlenk tube, which was warmed to 65 °C. Stirring was continued at 65 °C for 22.5 h. Then, after cooling to room temp the reaction was ended by addition of dichloromethane (5.00 mL), deionized water (10.0 mL), and 37% aqueous hydrochloric acid (2.00 mL). The aqueous layer was extracted with dichloromethane (3 × 10 mL) and the combined organic phases were dried (anhydrous magnesium sulfate), filtered and the solvents were removed in vacuo. The residue was purified by flash chromatography on reversed phase RP-18 (MeCN:H2O 1:1 → MeCN) to furnish 5-[4-(9H-carbazol-9-yl)benzyl]-9-hydroxy-1H-phenalen-1-one (CBHP) (82 mg, 72%) as a yellow solid, mp 259 °C.
1H NMR (600 MHz, CDCl3): δ 4.36 (s, 2H), 7.19 (d, 3J = 9.3 Hz, 2H), 7.30–7.27 (m, 2H), 7.42–7.36 (m, 4H), 7.47 (d, 3J = 8.1 Hz, 2H), 7.52 (d, 3J = 8.2 Hz, 2H), 7.94 (s, 2H), 8.09 (d, 3J = 9.3 Hz, 2H), 8.14 (d, 3J = 7.7 Hz, 2H), 15.89 (s, 1H). 13C NMR (151 MHz, CDCl3): δ 41.4 (CH2), 109.8 (CH), 111.2 (Cquat), 120.1 (CH), 120.5 (CH), 123.5 (Cquat), 124.1 (CH), 125.8 (Cquat), 126.0 (CH), 126.1 (Cquat), 127.5 (CH), 130.4 (CH), 133.4 (CH), 136.3 (Cquat), 136.8 (Cquat), 139.9 (Cquat), 140.95 (CH), 140.97 (Cquat), 179.1 (Cquat). MS (EI) (70 eV, m/z (%)): 453 (7), 452 (35), 451 (M+, 100), 285 (8), 284 (6), 256 (12), 255 (8), 254 (9), 241 (5), 239 (7), 226 (21), 211 (9). IR: = 3043 (w), 2963 (w), 2920 (w), 2361 (w), 1939 (w), 1632 (s), 1595 (s), 1558 (m), 1516 (s), 1477 (m), 1452 (s), 1418 (w), 1366 (m), 1335 (m), 1314 (w), 1267 (w), 1233 (s), 1184 (w), 1165 (m), 1144 (w), 1105 (m), 1022 (w), 930 (w), 914 (w), 893 (m), 833 (m), 812 (m), 746 (s), 723 (s), 692 (m), 638 (m) cm−1. Anal. calcd for C32H21NO2·1/5H2O (451.2 + 3.6): C 84.45, H 4.74, N 3.08. Gef.: C 84.39, H 4.82, N 3.02. UV/Vis (MeCN): λmax [nm] (ε [L mol−1 cm−1]): 239 (73400), 260 (29300), 285 (19900), 292 (21500), 353 (21600), 421 (7600), 446 (8200). Fluorescence (MeCN, λexc = 420 nm): λmax = 461 nm (Φf < 0.01), Stokes shift: Δ = 800 cm−1. E00/−1 = −1160 mV, E00/+1 = 1290 mV.
4.2 SPM characterization
All low temperature scanning tunneling microscopy and spectroscopy experiments were performed with custom-made electrochemically etched tungsten tips using a commercial Createc STM (Germany) at 4.2 K. The LT-STM is operated under ultra-high vacuum (UHV) with a base pressure of 1 × 10−10 mbar. It is composed of three parts, i.e., a load-lock, a preparation chamber, and a measurement chamber. Constant-current mode was used to record all STM images which are plotted after applying plane correction and optional noise filtering by a slight Fourier-filter using SPIP. Apparent heights are obtained from averaged height profiles in the corresponding images. The given length of molecules is the full width at half maxima obtained from the respective apparent height profiles. The precise current and voltage set-points are given in the caption of the respective STM images. STS experiments were performed with the feedback-loop switched off and the differential tunneling current was recorded using lock-in detection while the bias voltage was modulated. For each adsorption configuration several molecules were investigated on the Pt(111) surface. The differential conductance (dI/dV) spectra are usually averaged over five curves and their measurement parameters are given directly in the respective captions of the STS measurements.The substrate used in this work was a polished Pt(111) single crystal (purity 99.999%) purchased from MaTeck (Germany). Remaining surface artifacts were removed by repeated cycles of Ne+-ion sputtering (1 kV, 30 min, 2.5 μA), optional baking (600 °C, 30 min., 1 × 10−6 mbar, O2 partial pressure) and subsequent annealing (900 °C, 10 min). The resulting Pt(111) surface was investigated by STM and LEED to confirm its cleanliness. The preparation cycles were repeated until the Pt(111) surface showed large clean terraces with a diameter up to several 100 nm and sharp spots with a hexagonal symmetry in the LEED patterns.
After outgassing of the CBHP molecules at 200 °C for at least 1 hour they were deposited onto the Pt(111) substrate using a Knudsen cell. Suitable evaporation temperatures were found with the help of TGA and DTA measurements (Fig. S12, ESI†). A submonolayer coverage was obtained by sublimation at 180 °C for 10 minutes with a background pressure in the 10−10 mbar range and the Pt(111) surface at room temperature (RT). Thereafter the sample was kept at RT for 105 s before it was cooled to 4.2 K for the STM and STS characterization.
4.3 Atomistic simulations
The structure and electronic properties of the metal/molecule interface were studied using the DFT-based code SIESTA.38 A local-orbital pseudo-atomic basis was used. Molecular (substrate) atoms were described using double-(single-)zeta plus polarization bases. Exchange–correlation, including non-local dispersion interactions, was described with the vdW-DF functional of Dion et al. in the implementation of Román-Pérez and Soler.39,40 The unit cell in the calculations consisted of the CBHP–Pt complex and three Pt(111) layers, each consisting of 70 atoms. Almost 50 initial geometries, each with different lateral positions and orientations with respect to the substrate, were screened. The positions of the CBHP–Pt complex and of the surface layer Pt atoms were optimized until the forces acting on them were smaller than 0.02 eV Å−1. A real-space grid of 250 Ry was used. Reciprocal space was sampled using a single point for geometry optimization calculations, and a 5 × 5 Monkhorst–Pack grid to compute PDOS and LDOS. The LDOS at selected energies was obtained by integrating the electron density over 100 meV around that energy. Constant-current images were calculated from the LDOS within the Tersoff–Hamann approximation using a 0.25 Å Gaussian broadening to simulate the experimental broadening.41,42Author contributions
S. K. and T. J. J. M. planned the experiments. L. B. synthesized and analyzed the molecular components and B. M. contributed to it. R. E. performed the STM/STS experiments and processed the data. B. S. L. contributed to the STM measurements. N. P. A. performed and H. V. planned the DFT calculations. The manuscript was written by R. E. with contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge financial support from the Volkswagen-Stiftung through the ‘Optically Controlled Spin Logic’ project and the Czech Science Agency GAČR (grant number 19-23702S). Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures.References
- A. B. Ricks, K. E. Brown, M. Wenninger, S. D. Karlen, Y. A. Berlin, D. T. Co and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 4581–4588 CrossRef CAS.
- Y. Olivier, M. Moral, L. Muccioli and J.-C. Sancho-García, J. Mater. Chem. C, 2017, 5, 5718–5729 RSC.
- T. J. J. Müller and U. H. Bunz, Functional Organic Materials: Syntheses, Strategies and Applications, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- S. Bay, T. Villnow, G. Ryseck, V. Rai-Constapel, P. Gilch and T. J. J. Müller, ChemPlusChem, 2013, 78, 137–141 CrossRef CAS.
- L. Levi and T. J. J. Müller, Chem. Soc. Rev., 2016, 45, 2825–2846 RSC.
- T. Benelli, M. Tomasulo and F. M. Raymo, in Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH, Weinheim, Germany, 2011, pp. 697–717 Search PubMed.
- H. Kawauchi, S. Suzuki, M. Kozaki, K. Okada, D.-M. S. Islam, Y. Araki, O. Ito and K.-i. Yamanaka, J. Phys. Chem. A, 2008, 112, 5878–5884 CrossRef CAS PubMed.
- O. S. Wenger, Chem. Soc. Rev., 2011, 40, 3538–3550 RSC.
- M. Kuss-Petermann and O. S. Wenger, Angew. Chem., Int. Ed., 2016, 55, 815–819 CrossRef CAS.
- C. Tao, J. Sun, X. Zhang, R. Yamachika, D. Wegner, Y. Bahri, G. Samsonidze, M. L. Cohen, S. G. Louie, T. D. Tilley, R. A. Segalman and M. F. Crommie, Nano Lett., 2009, 9, 3963–3967 CrossRef CAS PubMed.
- B. Schuler, S.-X. Liu, Y. Geng, S. Decurtins, G. Meyer and L. Gross, Nano Lett., 2014, 14, 3342–3346 CrossRef CAS PubMed.
- A. Hinaut, T. Meier, R. Pawlak, S. Feund, R. Jöhr, S. Kawai, T. Glatzel, S. Decurtins, K. Müllen, A. Narita, S.-X. Liu and E. Meyer, Nanoscale, 2018, 10, 1337–1344 RSC.
- T. Meier, R. Pawlak, S. Kawai, Y. Geng, X. Liu, S. Decurtins, P. Hapala, A. Baratoff, S.-X. Liu, P. Jelínek, E. Meyer and T. Glatzel, ACS Nano, 2017, 11, 8413–8420 CrossRef CAS.
- R. Ebeling, S. Tsukamoto, E. Dirksen, V. Caciuc, T. J. J. Müller, N. Atodiresei and S. Karthäuser, J. Phys. Chem. C, 2017, 121, 26916–26924 CrossRef CAS.
- R. C. Haddon, R. Rayford and A. M. Hirani, J. Org. Chem., 1981, 46, 4587–4588 CrossRef CAS.
- X. Chi, M. E. Itkis, K. Kirschbaum, A. A. Pinkerton, R. T. Oakley, A. W. Cordes and R. C. Haddon, J. Am. Chem. Soc., 2001, 123, 4041–4048 CrossRef CAS PubMed.
- L. Bensch, I. Gruber, C. Janiak and T. J. J. Müller, Chem.–Eur. J., 2017, 23, 10551–10558 CrossRef CAS PubMed.
- K. Guzow, M. Czerwinska, A. Ceszlak, M. Kozarzewska, M. Szabelski, C. Czaplewski, A. Lukaszewicz, A. A. Kubicki and W. Wiczk, Photochem. Photobiol. Sci., 2013, 12, 284–297 CrossRef CAS PubMed.
- G. Manolikakes, C. Muñoz Hernandez, M. A. Schade, A. Metzger and P. Knochel, J. Org. Chem., 2008, 73, 8422–8436 CrossRef CAS PubMed.
- S. Xu, H. Kamada, E. H. Kim, A. Oda and E.-i. Negishi, in Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Bräse and M. Oestreich, Wiley-VCH, Weinheim, Germany, 2014, pp. 133–278 Search PubMed.
- A. Weller, Z. Phys. Chem., 1982, 133, 93–98 CrossRef CAS.
- R. Felici, M. Pedio, F. Borgatti, S. Iannotta, M. Capozi, G. Ciullo and A. Stierle, Nat. Mater., 2005, 4, 688–692 CrossRef CAS PubMed.
- M. Huang, Phys. Chem. Chem. Phys., 2012, 14, 4959–4963 RSC.
- M. Kiguchi, O. Tal, S. Wohlthat, F. Pauly, M. Krieger, D. Djukic, J. C. Cuevas and J. M. van Ruitenbeek, Phys. Rev. Lett., 2008, 101, 046801 CrossRef CAS PubMed.
- R. Otero, A. L. Vázquez de Parga and J. M. Gallego, Surf. Sci. Rep., 2017, 72, 105–145 CrossRef CAS.
- A. L. Pinardi, G. Biddau, K. van De Ruit, G. Otero-Irurueta, S. Gardonio, S. Lizzit, R. Schennach, C. F. Flipse, M. F. López, J. Méndez, R. Pérez and J. A. Martín-Gago, Nanotechnology, 2014, 25, 385602 CrossRef PubMed.
- A. Ugolotti, S. S. Harivyasi, A. Baby, M. Dominguez, A. L. Pinardi, M. F. López, J. A. Martín-Gago, G. Fratesi, L. Floreano and G. P. Brivio, J. Phys. Chem. C, 2017, 121, 22797–22805 CrossRef CAS.
- T. Jacob, R. P. Muller and W. A. Goddard, III, J. Phys. Chem. B, 2003, 107, 9465–9476 CrossRef CAS.
- G. Papoian, J. K. Norskov and R. Hoffmann, J. Am. Chem. Soc., 2000, 122, 4129–4144 CrossRef CAS.
- Z. Gu and P. B. Balbuena, J. Phys. Chem. C, 2007, 111, 9877–9883 CrossRef CAS.
- H. Ihm, H. M. Ajo, J. M. Gottfried, P. Bera and C. T. Campbell, J. Phys. Chem. B, 2004, 108, 14627–14633 CrossRef CAS.
- W. Liu, J. Carrasco, B. Santra, A. Michaelides, M. Scheffler and A. Tkatchenko, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 245405 CrossRef.
- V. P. Gupta, Principles and Applications of Quantum Chemistry, Academic Press, London, UK, 2016 Search PubMed.
- X. Jia and W. An, J. Phys. Chem. C, 2018, 122, 21897–21909 CrossRef CAS.
- L. Vitali, G. Levita, R. Ohmann, A. Comisso, A. De Vita and K. Kern, Nat. Mater., 2010, 9, 320–323 CrossRef CAS PubMed.
- P. C. Rusu and G. Brocks, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 073414 CrossRef.
- X. Yang, R. Lu, F. Gai, P. Xue and Y. Zhan, Chem. Commun., 2010, 46, 1088–1090 RSC.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- G. Román-Pérez and J. M. Soler, Phys. Rev. Lett., 2009, 103, 096102 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- J. Tersoff and D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 805 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00925c |
| This journal is © The Royal Society of Chemistry 2021 |