
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHybrid chemoenzymatic heterogeneous catalyst prepared in one step from zeolite nanocrystals and enzyme–polyelectrolyte complexes†
Margot
Van der Verren
 ,
Valentin
Smeets
,
Aurélien
vander Straeten
,
Christine
Dupont-Gillain
and
Damien P.
Debecker
*
,
Valentin
Smeets
,
Aurélien
vander Straeten
,
Christine
Dupont-Gillain
and
Damien P.
Debecker
*
Institute of Condensed Matter and Nanosciences, UCLouvain, Place Louis Pasteur 1, 1348 Louvain-la-Neuve, Belgium. E-mail: damien.debecker@uclouvain.be
First published on 8th February 2021
Abstract
The combination of inorganic heterogeneous catalysts and enzymes, in so-called hybrid chemoenzymatic heterogeneous catalysts (HCEHCs), is an attractive strategy to effectively run chemoenzymatic reactions. Yet, the preparation of such bifunctional materials remains challenging because both the inorganic and the biological moieties must be integrated in the same solid, while preserving their intrinsic activity. Combining an enzyme and a zeolite, for example, is complicated because the pores of the zeolite are too small to accommodate the enzyme and a covalent anchorage on the surface is often ineffective. Herein, we developed a new pathway to prepare a nanostructured hybrid catalyst built from glucose oxidase and TS-1 zeolite. Such hybrid material can catalyse the in situ biocatalytic formation of H2O2, which is subsequently used by the zeolite to trigger the epoxidation of allylic alcohol. Starting from an enzymatic solution and a suspension of zeolite nanocrystals, the hybrid catalyst is obtained in one step, using a continuous spray drying method. While enzymes are expectedly unable to resist the conditions used in spray drying (temperature, shear stress, etc.), we leverage on the preparation of “enzyme–polyelectrolyte complexes” (EPCs) to increase the enzyme stability. Interestingly, the use of EPCs also prevents enzyme leaching and appears to stabilize the enzyme against pH changes. We show that the one-pot preparation by spray drying gives access to hybrid chemoenzymatic heterogeneous catalysts with unprecedented performance in the targeted chemoenzymatic reaction. The bifunctional catalyst performs much better than the two catalysts operating as separate entities. We anticipate that this strategy could be used as an adaptable method to prepare other types of multifunctional materials starting from a library of functional nanobuilding blocks and biomolecules.
Introduction
Targeting the development of more sustainable processes, researchers continuously explore new synthesis pathways for producing chemicals under milder conditions while limiting the formation of polluting and hazardous substances.1 In this context, catalysis is known to play a central role.2 As “the 9th principle of green chemistry”,3 catalysis allows inventing more efficient chemical routes for the production of chemicals, evolving towards intensified processes, and simplifying industrial workup procedures.4 While catalysis science is classically compartmented into three sub-fields – homogeneous, heterogeneous and enzymatic catalysis – it nowadays appears essential to harness the strengths of different types of catalysts to design greener chemical processes.5On the one hand, heterogeneous catalysts are considered as robust, with moderate intrinsic performance (selectivity, yield) but large scope of application. On the other hand, biocatalysis allows producing chemicals under mild conditions, with a low environmental impact, while achieving high (enantio)-selectivity and (enantio)-specificity.6 Microbial enzymes are currently widely used at the industrial scale. Aminopeptidases and lipases, for example, are routinely utilized in the food industry.7–9 Several recent reviews have highlighted the industrial interest of microbial enzymes and their various applications.7,10–12 However, the scope of reactions, substrates and conditions remains relatively limited when working with enzymes.13,14
Combining heterogeneous catalysis with biocatalysis is increasingly envisaged as a solution to leverage on the respective advantages of each partner to effectively run chemoenzymatic reactions.15–18 In most reported examples, an enzyme and a solid catalyst were used in conjunction, as two distinct entities. Advantageously, the enzyme can be immobilized on a carrier to facilitate recovery and reuse. Even more interestingly, the two catalytic species can be combined in a unique bifunctional solid18–21 by immobilizing the enzyme directly onto an active solid catalyst, to form a so-called “hybrid chemoenzymatic heterogeneous catalyst” (HCEHC).5,18,22–24 The latter materials are designed to host at least two catalytic active species – a chemical one and an enzymatic one – and to be easily separated from the reaction medium (and potentially reused).
However, working with such hybrid chemoenzymatic heterogeneous catalyst to run one-pot cascade reactions raises several issues. It is not always possible to establish a suitable operational window, because enzymes are active in mild conditions such as aqueous medium (exclusive of extreme pH values) and ambient temperature, while most inorganic catalysts require relatively harsh conditions (pressure and temperature) and sometimes organic solvents.25,26 The preparation of HCEHCs, itself represents an important challenge. The successful combination of an enzyme and an inorganic catalyst in a single solid requires to ensure an effective integration of both partners (e.g. by covalent attachment, adsorption, encapsulation, etc.) while preserving the essential properties that confer their activity. In particular, it is crucial to preserve the enzyme three-dimensional structure and to maintain access to active sites on the solid catalyst surface. Possible issues associated with the preparation of hybrid catalysts include pore plugging, enzyme leaching, enzyme denaturation, surface poisoning, etc. In fact, only few studies have investigated methods to synthesize true hybrid catalysts.18,27–30
A telling example is the combination of an enzyme with zeolite catalysts. These latter are ubiquitous in heterogeneous catalysis, including at the industrial scale for biomass upgrading,31 selective oxidation,32 organic synthesis,33etc. However, their crystalline structure and microporous texture make it complicated to envisage the coupling with an enzyme: the surface usually offers limited anchoring points for covalent attachment and the micropores are too small to host enzymes of a few nanometers. One of the scarce examples found in the literature is the combination of the biocatalytic production of hydrogen peroxide by glucose oxidase (GOx) with the subsequent oxidation of lower olefins to epoxides using titanosilicalite-1 zeolites (TS-1).22,27,34 TS-1 zeolites feature outstanding catalytic activity in epoxidation at moderate temperature and do not suffer from deactivation in aqueous medium.35–38 In 2010, Vennestrøm et al. proposed the one-pot chemoenzymatic epoxidation of allyl alcohol using the TS-1 zeolite as catalyst and H2O2 produced in situ by free glucose oxidase (GOx), as the oxidant.27,34 Interestingly, they also disclosed the first example of a true hybrid chemoenzymatic heterogeneous catalyst for this reaction, having covalently grafted GOx at the surface of TS-1 nanocrystals. However, only traces of epoxide were detected in this case, mostly due to the fact that only a low enzyme loading could be attained by covalent grafting. This can be directly associated with the fact that the TS-1 zeolite – featuring small micropores and a low density of surface hydroxyls – is intrinsically not well suited to host an enzyme.
To overcome these drawbacks, we have recently proposed a two-step strategy for the preparation of an efficient GOx/TS-1 hybrid catalyst.28 First, leveraging on a spray drying technique,39 hollow zeolite microspheres were designed, starting from a suspension of TS-1 nanocrystals. Second, the hollow zeolite microspheres were loaded with GOx and the latter was irreversibly trapped through the formation of aggregates. This is a change in paradigm with respect to the classic approach where the enzyme has to be immobilized onto the surface or into the pores of a solid. The GOx loading could be easily tuned and the enzyme also gained in stability. This original design allowed reaching relatively high levels of epoxide yield in the one-pot chemoenzymatic reaction.28
Moving further, it is appealing to envisage a more direct procedure to form a GOx/TS-1 hybrid catalyst directly in one step by spray drying of a precursors suspension containing both the zeolite nanocrystals and the enzyme. On the first hand, aerosol processing is well suited for the preparation of many types of inorganic heterogeneous catalysts.39–47 In fact, aerosol methods were already demonstrated to be successful and scalable to form TS-1 microspheres with a hierarchical porosity.48 On the second hand, spray drying is also widely used to dry proteins for food and pharmaceutical applications.49–52 However, in the case of enzymes, spray drying often leads to denaturation, due to the relatively harsh conditions applied during drying (high temperature, liquid/air interfacial stress, shear stress).53–56 Applying spray drying to process enzymes for biocatalysis as the targeted application is not straightforward because the fragile enzyme structure has to be maintained so that the catalytic activity is preserved.
To mitigate deactivation, enzyme stability has to be enhanced. Generally speaking, enzyme stability can be improved through immobilization on a support,57 through the formation of cross-linked enzyme aggregates (CLEAs)58 or through the creation of enzyme mutants by directed evolution.59–62 Another technique that has recently gained interest to stabilize proteins is their complexation with polyelectrolytes.63 The formation of protein–polyelectrolyte complexes is a self-assembly process known for more than a decade64,65 that is now used in drug formulation66–68 or in layer-by-layer deposition methods.63 The spontaneous assembly of enzyme–polyelectrolyte complexes (EPCs) is directed by electrostatic and van der Waals forces.69,70 The polyelectrolyte, a positively- or negatively-charged polymer, coils around the enzyme which features a pH-dependent surface charge.71 This versatile technique can be used to control and stabilize the enzyme activity.64,72,73 For example, Maruyama et al. and Izaki et al. have both shown, respectively with protein–polyelectrolyte complexes and antibody–polyelectrolyte complexes, that the formation of these complexes leads to a better protein stabilization against thermally-induced denaturation and mechanical stress.74,75 Souza et al. also highlighted a decrease in the denaturation rate of complexed β-galactoxidase compared to the free enzyme when exposed to low pH.76 These results prompted us to investigate the possible stabilization of GOx through the formation of EPCs, in the perspective of the preparation of a GOx/TS-1 hybrid catalyst by one-step spray drying.
In this work, a novel pathway is developed for the one-pot preparation of hybrid chemoenzymatic heterogeneous catalysts combining both an enzyme and an inorganic catalyst in one bifunctional solid (Scheme 1). More precisely, we aim to combine the GOx enzyme with TS-1 zeolite nanocrystals using spray drying as a rapid, scalable, and direct production method. To obtain an active chemo-biocatalyst, our strategy is to leverage on the stabilization effect of a polyelectrolyte, via the formation of EPCs. The obtained hybrid catalyst will be exploited for the in situ production of hydrogen peroxide and subsequent allyl alcohol epoxidation (Scheme 2).
 | ||
| Scheme 1 Schematic representation of the one-pot synthesis strategy. The EPCs are primarily self-assembled by mixing the PAH and GOx. The TS-1 colloids suspension and the silica precursors solution, mixed beforehand, are mixed with the EPCs suspension. | ||
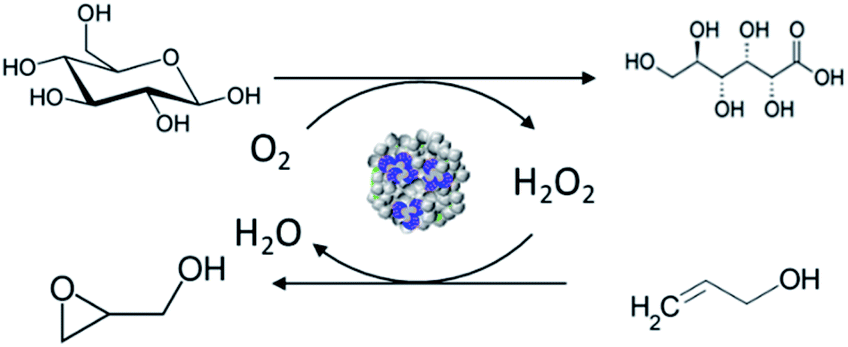 | ||
| Scheme 2 Representation of the oxidation of glucose to gluconic acid with the production of hydrogen peroxide used for the subsequent epoxidation of allyl alcohol to glycidol. | ||
Results and discussion
The results are presented and discussed in 3 distinct parts so that the two partners of the bifunctional hybrid catalyst are first described as separate entities, before being combined in the hybrid chemoenzymatic heterogeneous catalyst. Thus, the inorganic part of the catalyst is studied in the form of TS-1 nanocrystals and TS-1 microspheres (TS-1 and Aer_TS-1) tested in the epoxidation of allyl alcohol. Then the enzyme is analysed in its free (GOx) and complexed (EPCs) forms, and tested in the oxidation of glucose. Finally, the two catalysts are combined to form the hybrid materials. The enzyme is incorporated either in its free or in its complexed form in the hybrid chemoenzymatic heterogeneous catalysts (Hybrid_GOx and Hybrid_EPCs), and these are tested in the cascade reaction.Study of the inorganic catalyst (TS-1 and Aer_TS-1)
As prepared titanium silicalite (TS-1) zeolites can be described as nanocrystals with dimensions between 100 and 150 nm (Fig. S2A†), consistent with the size reported in the literature for such synthesis protocols.77,78 The microspheres obtained by the aggregation of these TS-1 nanocrystals via spray drying (Aer_TS-1) consist in spherical particles with a size ranging from 0.8 to 5 μm (Fig. 1A and B). Their shape results from the progressive drying of spherical aerosol droplets, leading to the formation spherical aggregates of TS-1 nanocrystals bound together by silica. Zooming in, the TS-1 nanocrystals are clearly visible (see also Fig. S2B†). Their size is equal to the size of the starting TS-1 nanocrystals.28 Interparticular voids are also observable between the aggregated nanocrystals with a size ranging from 40 to 65 nm. N2 adsorption–desorption isotherms of TS-1 and Aer_TS-1 are shown in Fig. 3 along with the pore size distribution (PSD) computed from adsorption. For both samples, the strong N2 uptake at low pressure accounts for the presence of micropores. While TS-1 is microporous, as evidenced by the type I isotherm (Vμ = 0.14 ± 0.01 cm3 g−1), interparticular voids also contribute to the total volume (Vp = 0.43 ± 0.01 cm3 g−1). According to the t-plot, the external specific surface area (i.e. excluding the surface developed by micropores) is 126 ± 7 m2 g−1. Similarly, Aer_TS-1 exhibits a micropore volume of 0.13 ± 0.01 cm3 g−1 showing that the microporous structure of the material is preserved. However, the total pore volume is slightly reduced (0.30 ± 0.01 cm3 g−1), possibly accounting for the fact that the packing (and therefore the interparticular voids) is more compact when the TS-1 is processed in the spray drier in the presence of the silica binder. The small hysteresis observed at high p/p0 is representative of a type IV isotherm and is associated with the presence of a low amount of large mesopores in the 50–80 nm range created during the spray drying process (see inset in Fig. 3). The external surface area is also decreased down to 82 ± 3 m2 g−1, possibly associated with the presence of the binder. The catalytic activity of TS-1 nanocrystals was evaluated in the reaction of allyl alcohol epoxidation with H2O2 as the oxidant. The final epoxide yield (see calculation in ESI†) reached 73% after 180 min, and no trace of glycerol was found (Fig. 3). This is consistent with the known excellent catalytic activity and selectivity of this zeolite for the epoxidation of light olefins in water.37,79,80 The level of activity is also in line with previously reported data.35,75 | ||
| Fig. 1 (A and B) SEM images of Aer_TS-1 at different magnifications. (C and D) SEM images of Hybrid_EPCs at different magnifications. | ||
When processed by spray drying, the obtained TS-1-based microspheres (here denoted Aer_TS-1) showed a slightly lower activity, with a final epoxide yield of 58% (Fig. 2). However, it should be noted that this material also contains 10 vol% of silica, used as a binder. Thus, normalizing the activity by the actual amount of TS-1 present in the sample, the epoxide production rate (calculated from the initial production of glycidol at 15 min) of TS-1 and Aer_TS-1 is respectively 26.6 and 25.0 mmol of glycidol per gram of TS-1 present in the material, showing that the intrinsic activity of the TS-1 nanocrystals is in fact maintained after spray drying. The selectivity toward glycidol is also fully preserved during the catalytic test. This result highlights that the active sites of each individual TS-1 crystals remain accessible in the Aer_TS-1 catalyst. It also indicates that the physico-chemical properties of TS-1 are not altered by the spray drying.
 | ||
| Fig. 2 N2 adsorption–desorption isotherms of TS-1, Aer_TS-1 and Hybrid_EPCs. In inset, the pore size distributions (PSDs) based on the BJH model are shown. | ||
 | ||
| Fig. 3 Kinetic data for the Ti-catalysed conversion of allyl alcohol into glycidol in H2O using aqueous solution of hydrogen peroxide as oxidant. Experimental conditions: T = 45 °C, [catalyst] = 5 g L−1, [H2O2] = 0.18 M, [allyl alcohol] = 0.9 M. The experiment has been repeated three times with TS-1 and two times with Hybrid_EPCs to evaluate the experimental error on these measurements (see error bars). | ||
Study of the enzyme (GOx and EPCs)
The enzyme specific activity was measured at 45 °C and at its optimal pH (i.e. pH 6)28 through the consumption of oxygen during the reaction. GOx exhibited a specific activity of 146 μmolO2 min−1 mgGOx−1 (Fig. 4). | ||
| Fig. 4 Comparison of the initial specific activity in GOx, EPCs and hybrid chemoenzymatic heterogeneous catalysts. Experimental conditions: T = 45 °C, [glucose] = 200 mM, PBS buffer, pH 6. The experiment has been repeated three times with Hybrid_EPCs to evaluate the experimental error (see error bars, relative error = 11.6%). | ||
In the perspective of spray drying, the complexation of GOx with poly(allylamine hydrochloride) (PAH) and its influence on the enzymatic activity was investigated. At pH 6, PAH is a positively-charged weak polyelectrolyte able to coil around the enzymes to make electrostatic interactions with the negative surface charges, thereby forming so-called enzyme–polyelectrolyte complexes (EPCs).65,71,81 Dynamic light scattering experiments were performed to measure the size of the EPCs and to determine the amount of polyelectrolyte that had to be added to the enzyme solution to ensure the complete complexation of GOx (i.e. no free GOx left in solution) (Fig. S1†). With 2.95 mg of PAH (corresponding to a +/− charge ratio of 6.6), no more signal of free GOx was detected, which means that all enzyme molecules were incorporated in the EPCs.
The specific enzymatic activity of the EPCs reached 95 μmolO2 min−1 mgGOx−1 (Fig. 4), thus lower than for free GOx. This decrease is tentatively explained by the structure of the complexes where PAH is coiled around the enzyme molecules and may hinder the accessibility of the active sites. Also, the formation of EPCs can induce conformational change in the enzyme structure and some active sites might be impaired.82 Finally, the polyelectrolyte creates a microenvironment in which the local conditions (such as the pH) might be slightly different from the bulk of the solution and affect the enzymatic activity.78,83,84 Indeed, the degree of ionization of PAH can affect the local pH around and inside the EPCs.83 In the conditions chosen in this work, there is an excess of positive charges (+/− charge ratio = 6.6) so that the complexes are surrounded by positive charges that will affect the charge distribution in water. To support this claim, the enzymatic activity was also measured at pH 7 to compare the results of GOx and EPCs (Fig. S3†). Unlike GOx – which was much less active at pH 7 – the specific activity of EPCs remained almost identical at pH 6 and pH 7. Spectrophotometry measurements confirmed that this effect was not induced by the release of GOx from the EPCs (Fig. S4†). These results tend to indicate that the formation of EPCs can result in a certain stabilization of the enzyme against pH changes, via modifications of the microenvironment around the enzymes. We suggest further systematic study on the effect of pH on the enzyme in its free and complexed forms could help supporting this hypothesis.
The possible stabilization of GOx in the EPCs against thermally-induced deactivation was also investigated by measuring the enzymatic activity of free GOx and EPCs after different times (1–5 h) of incubation at 45 °C (Fig. S5†). EPCs suffered from thermal deactivation similarly to free GOx after 5 h meaning that the formation of complexes did not protect the enzyme effectively against thermal denaturation.
Study of hybrid catalysts (Hybrid_GOx and Hybrid_EPCs)
First, a hybrid catalyst was synthesized in one-pot by the aerosol process with free GOx (Hybrid_GOx). The initial specific activity of GOx in Hybrid_GOx reached only 3 μmolO2 min−1 mgGOx−1 which corresponds to less than 2% of the initial specific activity of GOx (Fig. 4). Thus, an almost complete enzyme deactivation occurred upon spray drying, even though the inlet gas was set at a low temperature (50 °C) and the residence time in the drying chamber was short (∼3 s). As discussed in the literature,55 the important shear stress (as encountered in the nozzle of the spray drier) and the sudden transition from a liquid–solid interface to a gas–solid interface (that occurs during drying) may be the cause of this deactivation. It should be noted that the enzyme was efficiently trapped into the hybrid materials. A Bradford assay performed on the very pale yellow supernatant (Fig. 5A) obtained after 3 washings of the hybrid catalyst showed that only 1.8% of GOx had leached from this sample (Fig. 5B). However, when the washed catalyst was re-tested, it showed no enzymatic activity at all, suggesting that the only enzyme molecules responsible for the low level of activity reported in Fig. 4 were those that had leached out from the solid, subsequently acting as homogeneous biocatalysts. In other words, even though GOx was effectively trapped in Hybrid_GOx through spray drying, it was totally deactivated, and such one-step preparation starting from the free enzyme is ineffective. | ||
| Fig. 5 (A) Pictures of the GOx solution (26.32 mg mL−1), Hybrid_GOx and Hybrid_EPCs samples in suspension (100 mgcatalyst mL−1). Hybrid_GOx sample exhibits a very slightly yellow supernatant and a whitish powder attributed to the leaching of GOx in solution. Hybrid_EPCs sample shows a clear supernatant with no apparent leaching and a slightly yellow powder proving that GOx is inserted in the hybrid material. (B) Leaching of GOx from the hybrid material for Hybrid_GOx and Hybrid_EPCs after 24 h storage, measured by Bradford colorimetric assay (leaching = amount of GOx recovered in the supernatant(s)/amount of GOx introduced in the synthesis). | ||
To alleviate the loss of enzymatic activity, we investigated the possibility to protect the enzyme from the stress encountered during spray drying. Thus, a second type of hybrid catalyst (Hybrid_EPCs) was synthesized using GOx complexed with PAH (EPCs) instead of free GOx. The same spray drying conditions were applied. In SEM, this hybrid catalyst resembles Aer_TS-1: it consists in spherical aggregates of TS-1 with a size distribution ranging from 0.8 to 5 μm (Fig. 1C and D). It is not possible to highlight the presence of the EPCs in the SEM images, but we assume that they are intimately and randomly mixed with the TS-1 nanocrystals, as the aerosol droplets are dried very rapidly. The zeolite nanocrystals integrity is preserved in Hybrid_EPCs (Fig. S2†). This is further confirmed by N2 physisorption analyses (Fig. 3), since the micropore volume reached 0.12 cm3 g−1 and the external surface area was 125 m2 g−1, close to the value obtained for TS-1. Expectedly, the spray drying in the presence of the EPCs did not affect the textural properties of the TS-1 nanocrystals. The total pore volume is 0.33 cm3 g−1 which fits closely with the value obtained for Aer_TS-1. Pore size distribution (Fig. 3) shows smaller interparticular mesopores, in the 10–20 nm range, suggesting that the presence of the EPCs somewhat affects the way nanocrystals aggregate. Overall, looking at textural data, the two materials prepared by spray drying showed similar micropore volumes as in TS-1, indicating that the process did not affect the porosity inside nanocrystals. The decrease in total pore volume with respect to TS-1 indicates that the aggregation of the nanocrystals is more compact in spray dried materials, with a possible impact of the presence of the binder.
The specific enzymatic activity of Hybrid_EPCs reached 31 μmolO2 min−1 mgGOx−1, which has to be compared to the 95 μmolO2 min−1 mgGOx−1 reached by EPCs (Fig. 4). This corresponds to a residual activity of 33%. In other words, while the unprotected GOx suffered a complete deactivation upon spray drying, about one third of the enzymatic activity of the EPCs was preserved. It can be said that the use of complexes effectively contributed to the stabilization of the enzyme against deactivation during spray drying. This finding is consistent with several reports from the literature that indicate a beneficial effect of complexation against deactivation (vs. thermal, mechanical, or chemical stress).74,75 When measured at pH 7, the specific enzymatic activity of Hybrid_EPCs reached 29 μmolO2 min−1 mgGOx−1 which again suggests that EPCs are less sensitive to pH changes as compared to free GOx (Fig. S3†).
Hybrid_EPCs was subjected to a sequence of washing-centrifugation-Bradford assay and the amount of leached GOx in solution was measured in the supernatant. The latter supernatant was colorless (Fig. 5A) and the total amount of leached GOx after 3 washings corresponded to about 0.1% of the total enzyme loading (Fig. 5B). On the contrary, the pellet obtained after centrifugation remained yellowish. This result shows that the interaction between GOx and PAH is strong enough to maintain the enzymes inside the hybrid material and avoid leaching. It represents another advantage of complexing GOx before spray drying. The experimental loading of EPCs in Hybrid_EPCs was evaluated by thermogravimetric analysis (Fig. S6†). The mass loss corresponding to the decomposition of GOx and PAH occurred between 200 and 750 °C. The experimental loading obtained (53 mgorganic matter gcatalyst−1) roughly matched the theoretical one (50 mgorganic matter gcatalyst−1) showing that the spray drying process allows a precise control of the composition of the hybrid material.
The epoxidation activity of the TS-1 zeolite nanocrystals embedded in Hybrid_EPCs was measured to verify if the incorporation of EPCs affected the catalytic performance of the inorganic catalyst (Fig. 2). The epoxide yield reached 56% after 180 minutes. Initial specific activity reached 12.8 mmol h−1 gTS-1−1 for Hybrid_EPCs, which has to be compared to 25.0 mmol h−1 gTS-1−1 for Aer_TS-1. This drop may be attributed to the fact that complexes integrated in the hybrid catalyst hinder the access to the active site of the zeolite nanocrystals.
To validate our approach, the hybrid catalyst was tested in the cascade reaction, i.e. the epoxidation of allyl alcohol carried out in the absence of externally-added hydrogen peroxide, using the H2O2 produced in situ by the enzyme trapped in the hybrid catalyst (Fig. 6A). In the conditions used for the cascade reaction, a purely inorganic catalyst is totally inactive.28 Here, the detection of glycidol proved that the chemoenzymatic epoxidation was indeed catalysed by the HCEHC. The production of glycidol reached the value of 48.9 mM after 24 h, corresponding to an epoxide yield of 30% whereas the glucose conversion was 50%. The selectivity for glycidol was estimated at 66% (the balance corresponds to the epoxide ring opening leading to glycerol). To verify reproducibility, an independent experiment was carried out with a fresh Hybrid_EPCs sample and showed similar levels of activity (Fig. S7A†), indicating that the experimental error in this procedure accounts for approximately 10% in relative.
 | ||
Fig. 6 (A) One-pot chemoenzymatic epoxidation of allyl alcohol with Hybrid_EPCs. Experimental conditions: T = 45 °C, [allyl alcohol] = 0.9 M, [glucose] = 0.16 M, [Hybrid_EPCs] = 5 g L−1. (B) One-pot chemoenzymatic epoxidation of allyl alcohol with the two separate catalytic species Aer_TS-1 and EPCs. Experimental conditions: T = 45 °C, [allyl alcohol] = 0.9 M, [glucose] = 0.16 M, [Aer_TS-1] = 5 g L−1, [GOx–PAH complexes] = 0.048 g L−1.  hydrogen peroxide, ● glycidol, hydrogen peroxide, ● glycidol,  glucose, glucose,  total (glycidol + glycerol). total (glycidol + glycerol). | ||
The catalytic performance reported here is higher than the one obtained by a two-pot synthesis using hollow zeolite microparticles and enzyme encapsulation in the form of CLEAs (glycidol yield of 37.4 mM).28 Interestingly, the GOx in Hybrid_EPCs also showed a better resistance against deactivation during the cascade reaction. Indeed, the enzymes in Hybrid_EPCs were still active after 24 h of test, as attested by the continuous consumption of glucose. This contrasts with the rapid loss of enzymatic activity reported in the case of the hybrid catalyst based on CLEAs in hollow zeolite microspheres.28 In fact, GOx is known to suffer deactivation by the product (H2O2).85 In the case of Hybrid_EPCs, however, the EPCs are intimately mixed with TS-1 nanocrystals, and we surmise that hydrogen peroxide is rapidly consumed by the epoxidation reaction thereby mitigating enzyme deactivation. Moreover, the EPCs structure might bring an additional protection against the accumulation of hydrogen peroxide around GOx.
A Bradford assay was performed on the filtered medium after the 24 h test to estimate the leaching of GOx from Hybrid_EPCs. 3.5 mgGOx gcatalyst−1 had leached during the catalytic test accounting for 8% of the initial GOx loading. This may tentatively be explained by a progressive loss of the strong interactions that maintain GOx and PAH together in the complexes and in the hybrid material. Indeed, these electrostatic interactions in EPCs can be affected by a change in the ionic strength, which is increasing during the cascade test due to the production of gluconic acid and its concomitant neutralisation by sodium hydroxide. It was actually already demonstrated that the addition of low molecular weight electrolytes, such as sodium, had a screening effect on the intermolecular interactions ensuring EPCs stability.86
The same chemoenzymatic test was carried out with the two separate catalytic species composing the hybrid catalyst (i.e. EPCs and Aer_TS-1) (Fig. 6B). The amount of each catalyst was adapted to reach the same intrinsic activity for each catalytic species. The epoxide yield only reached 7% with a selectivity of 70%. The rapid stabilization of the glucose consumption rate indicates a fast deactivation of GOx in the EPCs, possibly provoked by the shear stress of the recirculation pump. In comparison, the embedded EPCs and TS-1 in hybrid microspheres appears to provide additional protection against the encountered stress. These results highlight again that the preparation of a bifunctional solid material bearing two catalytic species is advantageous over the use of two separate catalysts. Beyond the proof-of-concept presented here, we argue that further optimization can be pursued by playing on a series of relevant parameters of the preparation, such as the PAH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enzyme ratio, the TS-1:enzyme ratio, the relative amount of silica binder, etc.
:enzyme ratio, the TS-1:enzyme ratio, the relative amount of silica binder, etc.
Conclusions
In conclusion, we report a novel pathway for combining both an enzyme and a zeolite in one hybrid bifunctional material effective in a cascade chemoenzymatic reaction. We leverage on the spray drying process – as a rapid, direct, and continuous production method – to build TS-1 zeolites microspheres containing embedded glucose oxidase, in one step. The direct spray drying of the TS-1 suspension together with free GOx provokes the virtually complete deactivation of the enzyme. To obtain an active chemo-biocatalyst, we exploit the stabilization effect of a polyelectrolyte, via the formation of EPCs before spray drying. We report a strongly improved enzyme stability against the stress associated with spray drying, but also against leaching and against pH variations. The one-pot synthesized hybrid catalyst is active for the in situ production of hydrogen peroxide (by GOx) and the subsequent allyl alcohol epoxidation (by TS-1), reaching higher performance as compared to the systems already reported in the literature for this chemoenzymatic cascade reaction.We anticipate that this strategy can be transposed to other catalytic systems and multistep cascade reactions. In fact, the integration of EPCs in hybrid materials is favored by the fact that, while different enzymes may behave very differently upon assembly, the interactions of EPCs with other species are standardized in the reason of the presence of the polyelectrolyte shell that surrounds them. Thus, these results offer new perspectives, not only in the field of hybrid catalysts synthesis, but also in the broader field of multifunctional materials.
Experimental
Materials
Titanium isopropoxide (TiiP; ≥98%), allyl alcohol (≥99%; extra pure), D-(+)-glucose (ACS reagent, anhydrous) and glycidol (≥96%) were purchased from Acros Organics. Tetraethyl orthosilicate (TEOS; ≥98%), butan-1-ol (≥99.4%), hydrogen peroxide solution (H2O2; ∼30% w/w in H2O), poly(allylamine hydrochloride) (PAH, average molar mass 17500 g mol−1) and Bradford reagent were purchased from Sigma-Aldrich. Ethyl acetate (for gas chromatography ECD and FID) and tetrapropylammonium hydroxide (TPAOH; 40% in H2O) were purchased from Merck. Titanium(IV) sulfate (∼15%) was purchased from Fisher Scientific. Hydrochloric acid (HCl; ∼37%) and isopropyl alcohol (iPrOH) were respectively purchased from VWR Chemicals and VWR Life Sciences. Glucose oxidase (GOx) from Aspergillus niger was purchased from TCI. Distilled water was used for all synthesis and treatment processes.
TS-1 synthesis
The titanosilicalite-1 suspension was prepared via hydrothermal synthesis according to a protocol adapted from the literature.28,38,87 Solution A was prepared by solubilizing 0.55 g TiiP in 5 g iPrOH. 22.5 g TEOS were mixed with 16.51 g aqueous solution of TPAOH (40% w/w) and 9.84 g distilled water to form solution B. Both solutions were stirred for 5 min before solution A was added dropwise to solution B. The turbid mixture was stirred for another 15 min before a solution composed of 5.47 g TPAOH and 38.25 g distilled H2O was added. The resulting mixture was kept under stirring and heated at 75 °C for 3 h to evaporate the alcohol. Then, 30 mL distilled H2O were added and the mixture was transferred into a 70 mL Teflon-lined stainless-steel autoclave for 24 h at 160 °C. After rapid cooling, the resulting white precipitate was isolated by centrifugation and washed multiple times with distilled H2O until reaching neutral pH. The white solid was recovered and dried under vacuum overnight at 70 °C and calcined under static air at 550 °C for 5 h (heating rate of 5 °C min−1). The calcined powder was mixed with distilled water to obtain a 23% (w/w) colloidal suspension. The latter suspension was sonicated for 30 min and then kept under stirring at room temperature until use.Formation of GOx–PAH complexes (EPCs suspension)
The enzyme–polyelectrolyte complexes (EPCs) were formed by mixing 3.8 mL of GOx solution (26.32 mg mL−1) and 2.66 mL of PAH solution (4.22 mg mL−1), brought to pH 6.5 with TPAOH (40%) beforehand, to reach a (+)/(−) charge ratio = 6.6 (details on the charge ratio calculations can be found in Fig. S1†).Preparation of the hybrid catalysts in one pot by spray drying
A solution of silica precursor was synthesized by mixing 0.387 g TEOS, 5.993 g distilled water and 0.984 g HCl (0.012 M). It was kept under vigorous stirring overnight to allow the pre-hydrolysis of the silicon alkoxide. Then, 4.36 g of TS-1 suspension (23% w/w) was added. This corresponds to a TS-1:SiO2 volume ratio of 9:1, where silica acts as the binder. The mixture was brought to pH 6.5 by addition of TPAOH (40%) and then stirred for 5 min. Then 3.309 g of the EPCs solution (corresponding to 50.1 mg of GOx) was added and the mixture was stirred for another 5 min. Then, the mixture was processed in a flow of air in a Büchi Mini Spray Dryer B-290 with an air pressure of 4 bar in a glass reactor heated at 50 °C. This protocol was used to produce ∼1.1 g hybrid chemo-enzymatic heterogeneous catalyst in the form of a yellow powder which was denoted “Hybrid_EPCs” and had a nominal GOx loading of 45 mgGOx gcatalyst−1. This hybrid was stored at 4 °C.
The procedure was also applied, using a solution of free GOx (15.48 mg mL−1) instead of the EPCs solution, to obtain a hybrid chemo-enzymatic heterogeneous catalyst called “Hybrid_GOx” with the same nominal loading of 45 mgGOx gcatalyst−1. Another control material was prepared by replacing the enzyme solution by water to obtain a reference inorganic catalyst called “Aer_TS-1”.
Characterization
A Beckman Coulter DU800 Spectrophotometer was used to measure the turbidity of the suspension and thereby monitor the formation of EPCs. The effect of temperature and pH on their integrity was also investigated through spectrophotometry. 1 mL of GOx solution (26.32 mg mL−1) was put in a cuvette, increasing amounts of PAH solution (4.22 mg mL−1) were added and the absorbance was measured at 600 nm.Nitrogen physisorption analyses were carried out at −196 °C using a Tristar 3000 (Micromeritics, USA) instrument to determine de textural properties of the samples. Prior to analysis, the samples were degassed overnight under vacuum at 150 °C. The total pore volume was measured at p/p0 = 0.98 and the micropores volume and external surface area were determined via the t-plot. The pore size distribution was obtained from the adsorption part of the isotherm using the BJH method.
Scanning electron microscopy (SEM) analysis was performed using a JEOL 7600F microscope at 15.0 kV voltage. Samples were coated with a 15 nm layer of gold with a Sputter Metal 208 HR (Cressington) under vacuum.
Dynamic Light Scattering (DLS) measurements were performed using a Malvern CGS-3 equipped with a He–Ne laser at a wavelength of 633 nm and at a 90° scattering angle. The samples were analysed as prepared (in water). Experiments were performed at room temperature and 10 readouts were taken and averaged for each sample.
Thermogravimetric analysis (TGA) was carried out on a TGA/DSC 3+ (Mettler Toledo) thermogravimetric apparatus. Samples were dried overnight at 120 °C and measurements were performed with the following program of analysis: purge under dry nitrogen for 2 min at 25 °C, followed by a dwell at 25 °C in air for 10 min, and finally the temperature was increased from 25 °C to 900 °C (10 °C min−1) in a dry air flow (100 mL min−1).
A modified Bradford assay was used to evaluate the amount of enzyme in solution. A calibration was performed by mixing 0.5 mL of Bradford reagent and 0.5 mL of GOx solution ranging from 0 to 0.1 mg mL−1. Absorbance was measured at 450 nm and 590 nm using a Thermo Scientific Genesys 10s Vis Spectrophotometer and the value of A590 nm/A450 nm was plotted as a function of the enzyme amount.88,89 Leaching of the enzyme from the hybrid catalysts was evaluated using the same method. The hybrid catalysts were washed with distilled water and centrifuged to recover the supernatant and measure the enzyme concentration. Leaching tests were also performed on the filtered medium after the catalytic cascade test.
Biocatalytic production of H2O2, allyl alcohol epoxidation, and one-pot chemoenzymatic reaction
Enzymatic activity was measured based on the rate of oxygen consumption. O2 concentration in the reactor was monitored using an OXY-4 fiber optic oxygen meter connected to an optical oxygen sensor PreSens GmbH fixed on the inner side of the vessel. Reaction medium was prepared in a 50 mL volumetric flask with 500 μL pH buffer (phosphate buffer, 1 M) and 49.5 mL glucose solution (200 mM) and the resulting reaction medium was transferred to a 100 mL glass reactor equipped with a magnetic stirrer. pH was adjusted to 6.0 with HCl (0.2 M) and the medium was saturated with O2 by sparging oxygen (Air Liquide Alphagaz 1, purity 5.0) and kept at 45 °C. The specific enzymatic activity was approximated by the initial O2 consumption rate and normalized to the introduced amount of enzyme. The latter amount was chosen to determine the enzymatic activity taking into account the response time of the instrument. This test was performed at different pH and to evaluate the thermal stability of the enzyme after different incubation times. In all the tests (either with free GOx, EPCs or with the hybrid catalysts), the enzyme loading was set to about 4.5 mg mL−1.To evaluate the catalytic activity of the inorganic catalyst (TS-1), 9.152 g distilled H2O, 0.528 g allyl alcohol (0.9 M), 0.037 g butan-1-ol (used as internal standard) and 50 mg of catalyst (5 g L−1) were mixed in a two-necked glass round-bottomed reactor heated at 45 °C and equipped with a magnetic stirrer and a rubber septum. After 10 min, 180 μL aqueous H2O2 (0.18 M) were added with a syringe to initiate the reaction. This reactant is introduced as the limiting reagent in the reaction. Aliquots were collected at regular time intervals during the 3 h of analysis to monitor the formation of glycidol. Extraction was performed with ethyl acetate (10:90 v/v) to remove water and the samples were analysed with a CP-3800 Gas Chromatography Varian Chrompack equipped with a FID detector and a capillary column (BR-5, 30 m, 0.32 mm i.d., 1.0 μm film thickness). A calibration was performed beforehand for glycidol at concentration ranging from 0 to 100000 ppm.
The cascade chemoenzymatic epoxidation of allyl alcohol was studied in a 100 mL glass reactor equipped with a thermostatic bath and a magnetic stirrer. The reaction medium was prepared with 5.28 g allyl alcohol (0.9 M), 0.373 g butan-1-ol and 81.46 glucose solution (200 mM). The medium was saturated with oxygen by using a PDMS hollow fiber membrane fed with oxygen at 1 L h−1 rate. Once temperature was stabilized (45 °C), 11.9 mL of a suspension containing the hybrid catalyst (500 mg) was added. The pH of the reaction medium was maintained in the 5.5–6 range by addition of NaOH thanks to a TitroLine 7000 automatic titrator (Xylem Inc, Germany) to avoid acidification due to the production of gluconic acid. The reaction was monitored for 24 h by regular samplings. The amount of NaOH used for continuous titration was used to calculate the enzyme specific activity (via the production of gluconic acid). The conversion of allyl alcohol and the formation of glycidol was analysed through gas chromatography. The production of H2O2 was measured by colorimetric assay with 15% w/w titanium(IV) sulfate (Fischer Chemicals). The absorbance was measured at 405 nm using a Thermo Scientific Genesys 10s Vis Spectrophotometer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. Van der Verren is thankful to F.R.S. – FNRS for her PhD fellowship, under the project PDR-T.0058.19. V. Smeets and A. Vander Straeten are is thankful to F.R.S. – F.N.R.S. for their FRIA PhD grants. D. P. Debecker thanks the Francqui Foundation for the “Francqui Research Professor” chair. Prof. Patrick Gerin is gratefully acknowledged for providing access to the optical oxygen sensor. F. Devred is acknowledged for the technical and logistical support.Notes and references
- E. S. Beach, Z. Cui and P. T. Anastas, Energy Environ. Sci., 2009, 2, 1038 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2003, 77, 287 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Chem. Rev., 2020, 120, 7219 CrossRef.
- F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12 CrossRef.
- N. J. Turner, Trends Biotechnol., 2003, 21, 474 CrossRef CAS.
- R. Singh, M. Kumar, A. Mittal and P. K. Mehta, 3 Biotech., 2016, 6, 174 CrossRef.
- R. R. Singhania, A. K. Patel, L. Thomas, M. Goswami, B. S. Giri and A. Pandey, in Industrial Biorefineries & White Biotechnology, Elsevier, 2015, p. 473 Search PubMed.
- B. Andualema and A. Gessesse, Biotechnology, 2012, 11, 100 CrossRef CAS.
- S. Thapa, H. Li, J. OHair, S. Bhatti, F.-C. Chen, K. A. Nasr, T. Johnson and S. Zhou, Mol. Biotechnol., 2019, 61, 579 CrossRef CAS.
- N. Gurung, S. Ray, S. Bose and V. Rai, BioMed Res. Int., 2013, 329121, 1 Search PubMed.
- S. Raveendran, S. B. Ummalyma, A. K. Mathew, A. Madhavan, S. Rebello and A. Pandey, Food Technol. Biotechnol., 2018, 56, 16 CAS.
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859 CrossRef CAS.
- R. A. Sheldon and D. Brady, Chem. Commun., 2018, 54, 6088 RSC.
- W. Zhang, E. Fernández-Fueyo, Y. Ni, M. Van Schie, J. Gacs, R. Renirie, R. Wever, F. G. Mutti, D. Rother, M. Alcalde and F. Hollmann, Nat. Catal., 2018, 1, 55 CrossRef CAS.
- S. J. Freakley, S. Kochius, J. van Marwijk, C. Fenner, R. J. Lewis, K. Baldenius, S. S. Marais, D. Opperman, S. T. L. Harrison, M. Alcalde, M. S. Smit and G. J. Hutchings, Nat. Commun., 2019, 10, 4178 CrossRef.
- H. Huang, C. A. Denard, R. Alamillo, A. J. Crisci, Y. Miao, J. A. Dumesic, S. L. Scott and H. Zhao, ACS Catal., 2014, 4, 2165 CrossRef CAS.
- D. P. Debecker, V. Smeets, M. Van der Verren, H. Meerssman Arango, M. Kinnaer and F. Devred, Current Opinion in Green and Sustainable Chemistry, 2021, 28, 100437 CrossRef.
- X. Li, Y. Cao, K. Luo, Y. Sun, J. Xiong, L. Wang, Z. Liu, J. Ma, J. Ge, H. Xiao and R. N. Zare, Nat. Catal., 2019, 2, 718 CrossRef CAS.
- X. Li, X. Cao, J. Xiong and J. Ge, Small, 2019, 16, 1902751 CrossRef.
- A. Gimbernat, M. Guehl, M. Capron, N. Lopes Ferreira, R. Froidevaux, J.-S. Girardon, P. Dhulster, D. Delcroix and F. Dumeignil, ChemCatChem, 2017, 9, 2080 CrossRef CAS.
- F. Dumeignil, M. Guehi, A. Gimbernat, M. Capron, N. Lopez Ferreira, R. Froidevaux, J.-S. Girardon, R. Wojcieszak, P. Dhulster and D. Delcroix, Catal. Sci. Technol., 2018, 8, 5708 RSC.
- R. Ye, J. Zhao, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, Nat. Catal., 2018, 1, 318 CrossRef.
- Y. Wang, H. Ren and H. Zhao, Crit. Rev. Biochem. Mol. Biol., 2018, 53, 115 CrossRef CAS.
- J. M. Sperl, J. M. Carsten, J.-K. Guterl, P. Lommes and V. Sieber, ACS Catal., 2016, 6, 6329 CrossRef CAS.
- N. Balistreri, D. Gaboriau, C. Jolivalt and F. Launay, J. Mol. Catal. B: Enzym., 2016, 127, 26 CrossRef CAS.
- P. N. R. Vennestrøm, E. Taarning, C. H. Christensen, S. Pedersen, J.-D. Grunwaldt and J. M. Woodley, ChemCatChem, 2010, 2, 943 CrossRef.
- V. Smeets, W. Baaziz, O. Ersen, E. M. Gaigneaux, C. Boissière, C. Sanchez and D. P. Debecker, Chem. Sci., 2020, 11, 954 RSC.
- K. Engström, E. V. Johnston, O. Verho, K. P. J. Gustafson, M. Shakeri, C.-W. Tai and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2013, 52, 14006 CrossRef.
- X. Zhang, L. Jing, F. Chang, S. Chen, H. Yang and Q. Yang, Chem. Commun., 2017, 53, 7780 RSC.
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584 RSC.
- J. Přech, Catal. Rev., 2018, 60, 71 CrossRef.
- J.-J. Wang, Y.-Y. Chuang, H.-Y. Hsu and T.-C. Tsai, Catal. Today, 2017, 298, 109 CrossRef CAS.
- P. N. R. Vennestrøm, C. H. Christensen, S. Pedersen, J.-D. Grunwaldt and J. M. Woodley, ChemCatChem, 2010, 2, 249 CrossRef.
- G. N. Vayssilov, Catal. Rev., 1997, 39, 209 CrossRef CAS.
- R. S. Drago, S. C. Dias, J. M. McGilvray and A. L. M. L. Mateus, J. Phys. Chem. B, 1998, 102, 1508 CrossRef CAS.
- M. G. Clerici and P. Ingallina, J. Catal., 1993, 140, 71 CrossRef CAS.
- V. Smeets, E. M. Gaigneaux and D. P. Debecker, Microporous Mesoporous Mater., 2020, 293, 109801 CrossRef CAS.
- D. P. Debecker, S. L. Bras, C. Boissière, A. Chaumonnot and C. Sanchez, Chem. Soc. Rev., 2018, 47, 4112 RSC.
- N. Godard, A. Vivian, L. Fusaro, L. Cannavicci, C. Aprile and D. P. Debecker, ChemCatChem, 2017, 9, 2211 CrossRef CAS.
- S. Aghamohammadi and M. Haghighi, Appl. Clay Sci., 2019, 170, 70 CrossRef CAS.
- A. Kim, C. Sanchez, B. Haye, C. Boissière, C. Sassoye and D. P. Debecker, ACS Appl. Nano Mater., 2019, 2, 3220 CrossRef CAS.
- R. Li, Y. Yang, N. Sun and L. Kuai, Chem.–Eur. J., 2019, 25, 15586 CrossRef CAS.
- S. Ramesh and D. P. Debecker, Catal. Commun., 2017, 97, 102 CrossRef CAS.
- V. Smeets, C. Boissière, C. Sanchez, E. M. Gaigneaux, E. Peeters, B. F. Sels, M. Dusselier and D. P. Debecker, Chem. Mater., 2019, 31, 1610 CrossRef CAS.
- A. Zelcer, E. A. Franceschini, M. V. Lombardo, A. E. Lanterna and G. J. A. A. Soler-Illia, J. Sol-Gel Sci. Technol., 2020, 94, 195 CrossRef CAS.
- S. Maksasithorn, P. Praserthdam, K. Suriye and D. P. Debecker, Microporous Mesoporous Mater., 2015, 213, 125 CrossRef CAS.
- C. Niu, M. Liu, X. Gao, X. Ye, Y. Wen, Y. Liu and X. Wang, ACS Omega, 2019, 4, 4397 CrossRef CAS.
- A. Sosnik and K. P. Seremeta, Adv. Colloid Interface Sci., 2015, 223, 40 CrossRef CAS.
- C. Arpagaus, A. Collenberg, D. Ruetti, E. Assadpour and S. Mahdi Jafari, Int. J. Pharm., 2018, 546, 194 CrossRef CAS.
- H. Yoshii, F. Buche, N. Takeuchi, C. Terrol, M. Ohgawara and T. Furuta, J. Food Eng., 2008, 87, 34 CrossRef CAS.
- N. S. Mohtar, M. B. Abdul Rahman, S. Mustafa, M. S. Mohamad Ali and R. N. Z. R. A. Rahman, PeerJ, 2019, 7, e6880 CrossRef.
- V. Lauruengtana, V. Paramita, T. L. Neoh, T. Furuta and H. Yoshii, Jpn. J. Food Eng., 2009, 10, 79 CrossRef.
- A. Chinnaswamy, C. Rielly and A. Stapley, Drying Technol., 2007, 25, 799 CrossRef.
- Y.-F. Maa and C. C. Hsu, Biotechnol. Bioeng., 1997, 54, 503 CrossRef CAS.
- K. Samborska, D. Witrowa-Rajchert and A. Gonçalves, Drying Technol., 2005, 23, 941 CrossRef CAS.
- N. R. Mohamad, N. H. C. Marzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205 CrossRef CAS.
- R. A. Sheldon, R. Schoevaart and L. M. Van Langen, Biocatal. Biotransform., 2005, 23, 141 CrossRef CAS.
- A. A. Homaei, R. Sariri, F. Vianello and R. Stevanato, J. Chem. Biol., 2013, 6, 185 CrossRef.
- S. Nisha, A. Karthick and G. Nallathambi, Chem. Sci. Rev. Lett., 2012, 1, 148 Search PubMed.
- M. T. Reetz, Chem. Rec., 2016, 16, 2449 CrossRef CAS.
- J. L. Porter, R. A. Rusli and D. L. Ollis, ChemBioChem, 2016, 17, 197 CrossRef CAS.
- A. vander Straeten, A. Bratek-Skicki, L. Germain, C. D'Haese, P. Eloy, C.-A. Fustin and C. Dupont-Gillain, Nanoscale, 2017, 9, 17186 RSC.
- J. Xia and P. L. Dubin, in Macromolecular Complexes in Chemistry and Biology, ed. P. Dubin, J. Bock, R. Davis, D. N. Schulz and C. Thies, Springer Berlin Heidelberg, Berlin, Heidelberg, 1994, p. 247 Search PubMed.
- C. L. Cooper, P. L. Dubin, A. B. Kayitmazer and S. Turksen, Curr. Opin. Colloid Interface Sci., 2005, 10, 52 CrossRef CAS.
- X. Wan, T. Liu, J. Hu and S. Liu, Macromol. Rapid Commun., 2013, 34, 341 CrossRef CAS.
- T. Kurinomaru and K. Shiraki, Int. J. Biol. Macromol., 2017, 100, 11 CrossRef CAS.
- K. Shiraki, T. Kurinomaru and S. Tomita, Curr. Med. Chem., 2016, 23, 276 CrossRef CAS.
- L. E. Nita, A. P. Chiriac, E. Stoleru, A. Diaconu and N. Tudorachi, Des. Monomers Polym., 2016, 19, 596 CrossRef CAS.
- F. Caruso and C. Schüler, Langmuir, 2000, 16, 9595 CrossRef CAS.
- J. Choi and M. F. Rubner, Macromolecules, 2005, 38, 116 CrossRef CAS.
- N. I. Larionova, L. Y. Unksova, V. A. Mironov, I. Y. Sakharov, N. F. Kazanskaya and I. V. Berezin, Polym. Sci. U.S.S.R., 1981, 23, 1999 CrossRef.
- A. vander Straeten, A. Bratek-Skicki, A. M. Jonas, C.-A. Fustin and C. Dupont-Gillain, ACS Nano, 2018, 12, 8372 CrossRef CAS.
- T. Maruyama, S. Izaki, T. Kurinomaru, K. Handa, T. Kimoto and K. Shiraki, J. Biosci. Bioeng., 2015, 120, 720 CrossRef CAS.
- S. Izaki, T. Kurinomaru, K. Handa, T. Kimoto and K. Shiraki, J. Pharm. Sci., 2015, 104, 2457 CrossRef CAS.
- C. J. F. Souza, E. E. Garcia-Rojas and C. S. Favaro-Trindade, Food Hydrocolloids, 2018, 83, 88 CrossRef CAS.
- A. Thangaraj, M. J. Eapen, S. Sivasanker and P. Ratnasamy, Zeolites, 1992, 12, 943 CrossRef CAS.
- T. Zor and Z. Selinger, Anal. Biochem., 1996, 236, 302 CrossRef CAS.
- M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS.
- Y. Xue, Y. Wen, W. Huijuan, M. Liu, X. Huang, X. Ye, X. Wang and B. Li, RSC Adv., 2015, 5, 51563 RSC.
- G. Zhang, J. Sterte and B. J. Schoeman, Chem. Mater., 1997, 9, 210 CrossRef CAS.
- R. Murugavel and H. W. Roesky, Angew. Chem., Int. Ed. Engl., 1997, 36, 477–479 CrossRef CAS.
- M. G. Clerici, Kinet. Catal., 2015, 56, 450 CrossRef CAS.
- A. B. Kayitmazer, D. Seeman, B. B. Minsky, P. L. Dubin and Y. Xu, Soft Matter, 2013, 9, 2553 RSC.
- F. Cousin, J. Gummel, D. Ung and F. Boué, Langmuir, 2005, 21, 9675 CrossRef CAS.
- Y. Zhang, Q. Wang and H. Hess, ACS Catal., 2017, 7, 2047 CrossRef CAS.
- L. Goldstein, Y. Levin and E. Katchalski, Biochemistry, 1964, 3, 1913 CrossRef CAS.
- K. Kleppe, Biochemistry, 1966, 5, 139 CrossRef CAS.
- C. L. de Vasconcelos, P. M. Bezerril, D. E. S. dos Santos, T. N. C. Dantas, M. R. Pereira and J. L. C. Fonseca, Biomacromolecules, 2006, 7, 1245 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00834f |
| This journal is © The Royal Society of Chemistry 2021 |