DOI:
10.1039/D0NA00831A
(Paper)
Nanoscale Adv., 2021,
3, 593-603
High-performance Bi2O3-NC anodes through constructing carbon shells and oxygen vacancies for flexible battery-supercapacitor hybrid devices†
Received
8th October 2020
, Accepted 30th November 2020
First published on 9th December 2020
Abstract
Battery-supercapacitor hybrid (BSH) devices generally provide both high energy density and power density, but usually suffer from the serious electrochemical kinetics mismatch of cathodes and anodes mainly due to complex faradaic reactions of the unmatched battery-type electrodes used for charge storage, which inevitably degrade the rate capability and power density. To solve this, we propose a facile and efficient strategy of constructing carbon shells and oxygen vacancies. Oxygen-deficient Bi2O3 nanoflakes stabilized by N-doped carbon and supported on graphite fibers (GF@Bi2O3–NCs) were prepared to improve specific capacity, rate capability and cycling stability. The N/S-codoped carbon aerogels supported on graphite fibers (GF@NS-CAGs) provided a high capacitance of 312 F g−1 at 1 A g−1, which was mainly attributed to the microporous structure and high active N content. The flexible quasi-solid-state BSH device based on the GF@Bi2O3-NC anode and the GF@NS-CAG cathode with a stable voltage window of 2.3 V could deliver a remarkable capacity of 103 mA h g−1, an energy density of 118 W h kg−1 and capacity retention of 95.7% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, reflecting that this was a highly-efficient approach to develop high-performance flexible energy storage devices.
000 cycles, reflecting that this was a highly-efficient approach to develop high-performance flexible energy storage devices.
1. Introduction
The rigorous environmental pollution problems brought by the sustained consumption of non-renewable energy have stimulated the rapid development of renewable energy resources. Efficient and clean electrochemical energy storage technologies, such as supercapacitors and batteries, have been playing a significant role in the utilization and development of renewable energy.1 Battery-type electrode materials rely on strong Faraday reactions to store charges,2 while capacitive electrode materials are based on three charge storage mechanisms: surface-controlled electric double layer capacitance (EDLC), surface-controlled redox pseudocapacitance and diffusion-controlled intercalation pseudocapacitance.3 Battery-supercapacitor hybrid (BSH) devices, a type of asymmetric supercapacitors, are typically composed of a high-capacity battery-type electrode, such as LiMn2O4,4 Bi2O3,5–8 Fe3O4,9 Ni12P5,10 Ni-Co,11 Fe3C,12 BiFeO3,13 and Bi2MoO6,14 and a high-rate capacitive electrode, such as carbon nanomaterials,15–17 conducting polymers,17,18 Nb2O5,19 MoS2,20 MXenes,21 and LaMnO3.22 Though BSH devices have emerged as promising and highly efficient energy storage devices with both high energy density and power density, they usually suffer from the serious mismatch of electrochemical kinetics between the cathodes and anodes mainly due to complex faradaic reactions of the unmatched battery-type electrodes used for charge storage, which inevitably degrade the rate capability and power density.23
By taking full advantage of the different potential windows of the cathodes and anodes, BSH devices can extend their operating voltage windows and break the overpotential barrier usually found in symmetric supercapacitors, and this can be attributed to the thermodynamic decomposition of the electrolytes.24 Bi2O3 anodes have previously been reported to exhibit the reversible redox reaction of Bi3+, which is regarded as a “quasi-conversion reaction” and can suppress the hydrogen evolution reaction of aqueous electrolytes.5 However, Bi2O3-based BSH devices, such as Bi2O3//AC,6 Bi2O3–Ni–F//graphite7 and Bi2O3//MnO2,8 have been reported to provide poor specific capacitance (<100 F g−1), low energy density (<36 W h kg−1) and bad cyclic stability (<85% for 4000 cycles). The bad rate capability and low power density are mainly attributed to the multielectron faradaic reactions of the unmatched Bi2O3 electrodes used for charge storage, which reflect relatively slower electrochemical kinetics than those of capacitive electrodes with high rates. To solve this problem, constructing a composite structure has been considered as an effective strategy, which results in the formation of Bi2O3/carbon composite electrodes, such as Bi2O3/activated carbon,25,26 Bi2O3/carbon quantum dot,27,28 and Bi2O3/graphene.28 For instance, a graphene/carbon dot encapsulation structure of the Bi2O3 electrode supported on carbon nanotube films has been reported to not only improve conductivity but also inhibit loss of capacity, which result in an asymmetric supercapacitor with the energy density of 98.2 W h kg−1 and capacity retention of 80.1% after 8000 cycles.28 Furthermore, the introduction of oxygen vacancies into the crystal lattice of Bi2O3 has been considered as another effective route for the improvement of ion diffusion kinetics,29 which is essential for reasonably matching the battery-type electrodes with the capacitive electrodes in BSH devices. It has been previously confirmed that oxygen vacancies play an important role in the enhancement of electronic conductivity30,31 and anion-based intercalation pseudocapacitance.22 With oxygen-deficient Bi2O3, the assembled BSH device could provide an energy density of 86.6 W h kg−1 and capacity retention of 85% after 10000 cycles;29 but it seems to be not enough.
Carbon shell protection is usually used to improve the rate performance and cyclic stability of metallic oxides for electrochemical energy storage.9 However, the necessary carbonization of precursors cannot only form carbon shells on the surface of metallic oxides as expected, but also inevitably produce oxygen vacancies in the crystal lattices of metallic oxides, which tend to be neglected in carbon-stabilized metallic oxides. Herein, we propose a novel BSH device based on oxygen-deficient Bi2O3 nanoflakes that are stabilized by N-doped carbon and supported by graphite fibers (GF@Bi2O3–NCs) as the battery-type anode, N/S-codoped carbon aerogels supported by graphite fibers (GF@NS-CAGs) as the capacitive cathode, and PVA/LiCl/H2O gel as the electrolyte. The oxygen-deficient Bi2O3-NC nanoflakes were prepared through the carbonization of the electrochemically-deposited Bi2O3 nanoflake precursors previously immersed in a melamine solution. The N-doped carbon shells as the outer layer of the Bi2O3-NC nanoflakes effectively improved specific capacity, rate capability and cycling stability. The GF@NS-CAG cathode was obtained by the carbonization of the GF@PSS-PPy hydrogel precursors. With the GF@Bi2O3-NC anode and the GF@NS-CAG cathode, flexible quasi-solid-state BSH devices with different mass loadings were assembled in a two-electrode configuration. The electrochemical kinetics was analyzed in order to further understand the energy storage mechanism of the BSH devices.
2. Experimental
2.1 Fabrication of GF@Bi2O3-NC
2.4 g bismuth(III) nitrate pentahydrate (0.068 mol) was dissolved in a mixed solution consisting of 48 mL acetone and 24 mL ethylene glycol. The electrochemical deposition (ED) system was composed of a GF electrode as the working electrode, a platinum foil as the counter electrode and the as-prepared mixture as the electrolyte. The ED process was conducted at 1 V for 20 min on an electrochemical workstation (CHI 760E). This product, denoted as GF@Bi2O3, was successively dried at 80 °C for 24 h, immersed in a melamine solution (0.01 M) for 10 minutes, and carbonized at 1000 °C for 30 min at the heating rate of 5 °C min−1 in a tube furnace under an N2 flow. After cooling down to room temperature, the final product was obtained and denoted as GF@Bi2O3-NC.
2.2 Fabrication of GF@NS-CAGs
350 μL polystyrene sulfonate (PSS) aqueous solution (1 mg mL−1) and 4 mL isopropanol solution containing 420 μL pyrrole (Py) monomer (6 mmol) were mixed together with the immersed GF electrodes. Then, 1.37 g ammonium persulfate (6 mmol) was dissolved in 2 mL of 1 M H2SO4 solution and successively added into the PSS-pyrrole mixed solution. After keeping for 2 h, the resultant product (denoted as GF@PSS-PPy) was taken out with a pair of tweezers and washed with ethanol and deionized water thoroughly to remove impurities. The wet GF@PSS-PPy hydrogels were frozen in the refrigerator at −15 °C overnight and dried in a freeze-drying machine (Bilon FD-1A-50) for two days. Then, the freeze-dried samples were heated at 600 °C, 800 °C, and 1000 °C for 2 h at the heating rate of 5 °C min−1 in a tube furnace under an N2 flow, resulting in GF@NS-CAG-600, GF@NS-CAG-800, and GF@NS-CAG-1000-1, respectively. As a comparative experiment, the as-prepared GF@PSS-PPy hydrogels were immersed into a melamine solution (0.01 M) for 10 minutes, subsequently freeze-dried and carbonized at 1000 °C according to the above-mentioned carbonization condition, resulting in GF@NS-CAG-1000-2.
2.3 Assembly of flexible BSH devices
The PVA/LiCl/H2O gel electrolyte was prepared as follows: 0.42 g LiCl (0.01 mol) and 1 g PVA were added into 10 mL deionized water, heated at 90 °C and kept at 90 °C under stirring until the mixture became clear. For the assembly of the flexible BSH devices, the charge balance of the two electrodes must essentially follow the theoretical relationship q+ = q− based on the mass loading of active materials in the anodes and cathodes. The as-prepared GF@NS-CAG-1000-2 cathode (2.4 mg cm−2) and GF@Bi2O3-NC anode (1.4 mg cm−2) were cut into rectangular strips and pressed on carbon papers at 1 MPa. Then, the PVA/LiCl/H2O gel electrolyte was slowly poured on the two electrodes and air-dried at room temperature to evaporate excess water. The two electrodes were pressed together and packed with parafilm to form a flexible BSH device denoted as BSH-1 (3.8 mg cm−2). The BSH-2 (8.4 mg cm−2) and BSH-3 (11.6 mg cm−2) devices were based on GF@NS-CAG-1000-2 (6.0 mg cm−2) and GF@Bi2O3-NC (2.4 mg cm−2), and GF@NS-CAG-1000-2 (7.2 mg cm−2) and GF@Bi2O3-NC (4.4 mg cm−2), respectively.
2.4 Characterization
The morphologies of the samples were observed by field-emission scanning electron microscopy (FE-SEM, Carl Zeiss Ultra 55) and transmission electron microscopy (TEM, JEM-2100). X-ray photoelectron spectrometry (XPS) was carried out on a Thermo Scientific ESCALAB 250Xi X-ray photoelectron spectrometer using monochromated Al Kα X-ray beams as the excitation source (1486.6 eV). The X-ray diffraction (XRD) patterns were collected on a Rigaku D/Max-cA X-ray diffractometer with a Ni filter and Cu–Kα radiation (30 kV, 30 mA). The nitrogen adsorption–desorption isotherms of the NS-CAG samples vacuum-degassed at 200 °C for 12 h were measured at 77 K on an ASAP2460. The specific surface area was measured by the Brunauer–Emmett–Teller (BET) method with the meso- and micro-pore size distributions calculated by the Barrett–Joyner–Halenda (BJH) model and the density functional theory (DFT) model, respectively. Raman scattering was performed on a Renishaw inVia Reflex Raman spectrometer using a 10 mW laser with an excitation wavelength of 532 nm.
2.5 Electrochemical testing
The electrochemical performances were measured on an electrochemical workstation (CHI 760E). For the three-electrode tests, the cyclic voltammetry (CV) and galvanostatic charge/discharge (GCD) tests were conducted in 1 M LiCl. The gravimetric specific capacitance (Cs) and specific capacity (Qs) based on the mass of active materials for single electrodes were calculated from the GCD curves as follows:| | 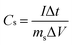 | (1) |
| | 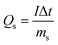 | (2) |
where I is the discharge current, Δt is the discharge time, ms is the mass of active materials in one electrode, and ΔV is the voltage window.
For the BSH devices, the specific capacitance (Ccell) and specific capacity (Qcell) based on the mass of active materials in both electrodes were calculated from the GCD and CV curves22 as follows:
| | 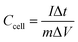 | (3) |
| | 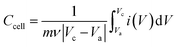 | (4) |
| | 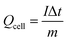 | (5) |
where
m is the mass of active materials in both electrodes,
ν is the scan rate,
Va and
Vc are the anodic and cathodic potentials, respectively, and
i(
V) is the current response at potential
V. The gravimetric energy density (
E) and power density (
P) were calculated by using the equations:
| | 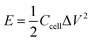 | (6) |
| | 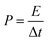 | (7) |
The electrochemical impedance spectroscopy (EIS) measurements were performed at open-circuit voltages ranging from 105 to 10−2 Hz.
3. Results and discussion
Based on the GF@Bi2O3-NC anode and the GF@NS-CAG-1000-2 cathode, a flexible BSH device was assembled in a two-electrode configuration, as shown in Fig. 1. The GF@Bi2O3-NC anode was fabricated through the ED process of Bi2O3 on the surface of GFs and the carbonization process of melamine adsorbed on Bi2O3, which resulted in the NC layer on the surface of Bi2O3. The GF@NS-CAG-1000-2 cathode was prepared by the polymerization of Py and the carbonization of the PPy/melamine composite. The morphologies and structures of GF@Bi2O3-NC and GF@NS-CAG-1000-2 were obtained by the following characterization methods and compared with those of other relevant samples.
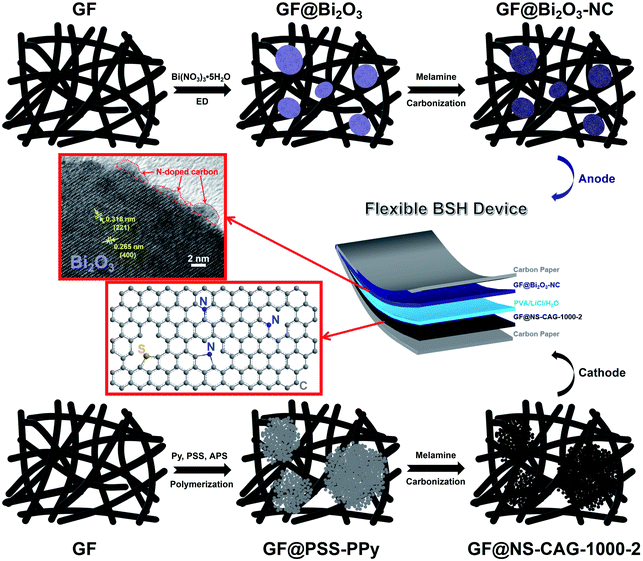 |
| | Fig. 1 Schematic illustration of the assembly of a flexible BSH device based on the GF@Bi2O3-NC anode and the GF@NS-CAG-1000-2 cathode. | |
The SEM image of GF@Bi2O3 showed abundant nanoplates assembled into an amorphous bulk (Fig. 2a). The Bi2O3 particles showed diameters ranging from several micrometers to one dozen micrometers, and the average diameter was about 8.52 μm (Fig. S1a†). After coated with the NC layer, the obtained Bi2O3-NC particles reflected a sphere-like morphology with a relatively smooth surface (Fig. 2b and S1b†). As seen in the inset of Fig. 2b, many nanoplates could be clearly observed in a broken sphere with a uniform distribution of C, N, O, and Bi both inside and outside Bi2O3-NC (Fig. 2c). The in situ growth of Bi2O3 on the surface of GFs ensured the intimate contact of Bi2O3-NC with GFs. The low- and high-magnification TEM images of Bi2O3 and Bi2O3-NC showed a nanoplate morphology (Fig. S2a† and 2d) with average lattice fringe spacings of about 0.32 nm and 0.26 nm ascribed to the lattice planes (221) and (400), respectively (Fig. S2b† and 2e). An amorphous carbon layer could be observed, indicating the existence of N-doped carbon. The corresponding selected-area electron diffraction (SAED) patterns of Bi2O3 and Bi2O3-NC reflected their good crystal structure (Fig. S2c† and 2f), which was further demonstrated by XRD. In Fig. 2g, the XRD spectrum of Bi2O3-NC shows the 2θ values of 24.9°, 27.8°, 31.1°, 32.8°, 47.0°, 53.3°, 55.4°, 57.4°, and 62.2° ascribed to the lattice planes (310), (221), (002), (400), (440), (512), (621), (442), and (711), respectively, which well-indexed to α-Bi2O3 (JCPDS 74-1374).28 The XPS spectra were analyzed to determine the atomic composition of Bi2O3 (Fig. S3†) and Bi2O3-NC (Fig. 2h–k), and the fitting results are listed in Table S1.† The Bi 4f spectra of Bi2O3 and Bi2O3-NC were composed of two Gaussian peaks at the binding energies of 158.3 eV (Bi 4f7/2) and 163.6 eV (Bi 4f5/2), demonstrating the existence of Bi3+.5 The O 1s spectra were composed of two characteristic peaks at 529.2 eV (O2−) and 530.8 eV (O vacancy).29 It is worth mentioning that Bi2O3-NC showed a higher content of O vacancy (64.4%) than that in Bi2O3 (52.5%), indicating that more oxygen vacancies were introduced into the crystal lattice of Bi2O3 in the carbonation process. In addition, Bi2O3-NC exhibited the C 1s fitted peaks at 284.6 eV (sp2 C), 285.2 eV (C–O/C–N), 286.1 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/CN), and 288.6 eV (O–CO)32–34 and the fitted N 1s peaks ascribed to pyridinic N (N-6) at 396.9 eV, pyrrolic N (N-5) at 398.9 eV, and graphitic N (N-Q) at 402.7 eV,32–34 further confirming the existence of N-doped carbon, which is consistent with the EDS and TEM results. The weight ratio of the N-doped carbon shells in the sample of Bi2O3-NC was determined to be 13.2% by XPS analysis.
O/CN), and 288.6 eV (O–CO)32–34 and the fitted N 1s peaks ascribed to pyridinic N (N-6) at 396.9 eV, pyrrolic N (N-5) at 398.9 eV, and graphitic N (N-Q) at 402.7 eV,32–34 further confirming the existence of N-doped carbon, which is consistent with the EDS and TEM results. The weight ratio of the N-doped carbon shells in the sample of Bi2O3-NC was determined to be 13.2% by XPS analysis.
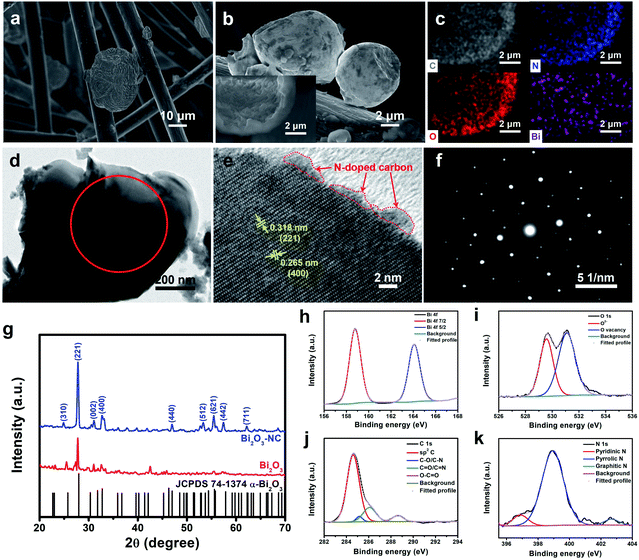 |
| | Fig. 2 The SEM images of (a) GF@Bi2O3 and (b) GF@Bi2O3-NC (inset: the SEM image of a broken sphere). (c) EDS C, N, O and Bi elemental mappings in Bi2O3-NC. The (d) low- and (e) high-magnification TEM images of Bi2O3-NC with (f) the corresponding SAED pattern. (g) The XRD spectra of Bi2O3 and Bi2O3-NC. The XPS (h) Bi 4f, (i) O 1s, (j) C 1s and (k) N 1s spectra of Bi2O3-NC. | |
The morphologies and structures of the obtained NS-CAGs were characterized, as shown in Fig. 3. Compared with NS-CAG-600, NS-CAG-800 and NS-CAG-1000-1 (Fig. S4† and 3a), NS-CAG-1000-2 showed more obvious macroporous frameworks composed of abundant nanoparticles (Fig. 3b and c), which reflected the uniform distribution of C, O, N and S on the surface (Fig. S5†). There was no doubt that their morphologies were derived from the PSS-PPy hydrogel precursors, which have a 3D porous structure with a nanoparticle morphology and uniform distribution of C, O, N and S on the surface (Fig. S6†). The TEM images of NS-CAG-1000-2 showed irregular nanoparticles (Fig. 3d) with plenty of micropores (Fig. 3e) and a lattice fringe spacing of 0.384 nm (Fig. 3f), which is slightly higher than the theoretical value of 0.34 nm for graphite.35,36 The XPS spectra of NS-CAG-1000-1 and NS-CAG-1000-2 were analyzed to determine the atomic composition, and the fitting results of C 1s, N 1s and S 2p are listed in Table S2.† As shown in Fig. 3g, the C 1s peaks of NS-CAG-1000-2 were composed of four Gaussian peaks with the binding energies of 284.6 eV (sp2 C), 285.1 eV (C–O/C–N/C–S), 286.7 eV (CO/CN) and 290.0 eV (O–CO).32–34 In Fig. 3h, the fitted N 1s peaks of NS-CAG-1000-2 were ascribed to pyridinic N (N-6) at 397.4 eV, pyrrolic N (N-5) at 400.1 eV and graphitic N (N-Q) at 401.3 eV.33,34 Compared with NS-CAG-1000-1, NS-CAG-1000-2 exhibited an improved N content (from 1.48% to 2.58%), with a higher active N (N-6 and N-5) content (71.6%) and a lower N-Q content (28.4%) due to the addition of N-rich melamine during the fabrication of the PSS-PPy hydrogel precursors. In Fig. 3i, the S 2p fitted peaks of NS-CAG-1000-2 belong to C–S–C (S 2p3/2) at 163.1 eV, C–S–C (S 2p1/2) at 164.2 eV and C–SOx–C at 166.3 eV, thus confirming S doping.33,34 The Raman spectra of the NS-CAGs showed a D band and a G band at 1360 cm−1 and 1580 cm−1 (Fig. S7†), reflecting the intrinsic characteristic of carbon materials.35,36 As a result, NS-CAG-1000-1 showed a lower peak intensity ratio of the D band and G band (ID/IG) than those of NS-CAG-600 and NS-CAG-800, indicating its higher graphitization degree due to the higher carbonization temperature, which favored improving conductivity.16,17 Compared with NS-CAG-1000-1, NS-CAG-1000-2 exhibited a higher ID/IG value mainly due to more defects, which were considered as active N sites, as confirmed by the previously-mentioned XPS results. The pore structures of the obtained NS-CAGs were analyzed by the nitrogen adsorption and desorption isotherms (Fig. 3j) and the DFT-based average pore width distributions (Fig. 3k), and the results were listed in Table S3.† At low pressure, the adsorption capacity increased obviously, reflecting the abundance of micropores,17 which was well consistent with the results obtained in Fig. 3e. Compared with the other samples, NS-CAG-800 showed the largest SSA (737 m2 g−1), as measured by the BET method, and the maximum micropore volume (0.244 cm3 g−1) and surface area (482 m2 g−1) calculated by the t-plot method. Compared with NS-CAG-1000-1, NS-CAG-1000-2 exhibited a lower SSA (436 m2 g−1), but the pore radius (9.09 nm) obtained by the BJH method and the pore width (0.733 nm) based on the DFT model were higher, which could facilitate electrolyte ion diffusion, thereby boosting the supercapacitive performances.
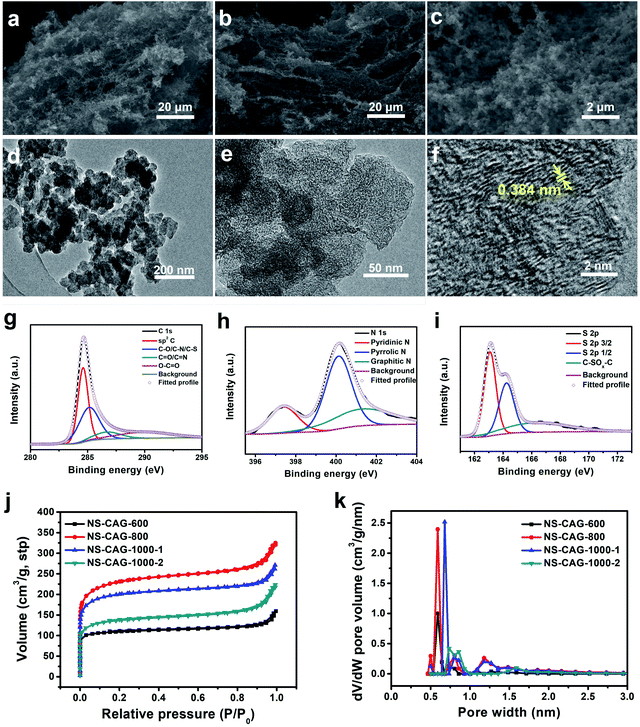 |
| | Fig. 3 The SEM images of (a) NS-CAG-1000-1 and (b) NS-CAG-1000-2. (c) A high-magnification SEM image of NS-CAG-1000-2. The (d) low-, (e) medium- and (f) high-resolution TEM images of NS-CAG-1000-2. The XPS (g) C 1s, (h) N 1s and (i) S 2p spectra of NS-CAG-1000-2. (j) The nitrogen adsorption and desorption isotherms and (k) the DFT-based average pore width distributions of NS-CAG-600, NS-CAG-800, NS-CAG-1000-1 and NS-CAG-1000-2. | |
The electrochemical performances of the GF@Bi2O3 and GF@Bi2O3-NC electrodes were evaluated by three-electrode tests in 1 M LiCl from −1.3 V to 0.3 V. The CV and GCD curves of the GF electrode (Fig. S8†) reflected its poor capacitive performance (1.9 F g−1 at 1 A g−1), which could be negligible in the energy storage performances of the GF@Bi2O3-NC and GF@NS-CAG electrodes. In Fig. 4a, the CV curves show two pairs of reversible redox peaks probably attributed to a “quasi-conversion reaction” mechanism, which can be described as follows:5
| | | Bi2O3 + H2O + 2e− ↔ Bi2O2 + 2OH− | (8) |
| | | Bi2O2 + 2H2O +4e− ↔ 2Bi + 4OH− | (9) |
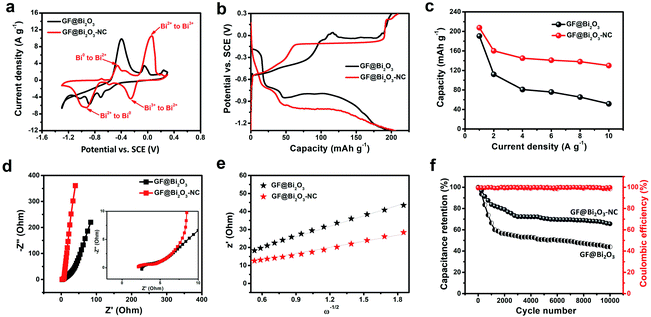 |
| | Fig. 4 Electrochemical performances of the GF@Bi2O3 and GF@Bi2O3-NC electrodes. (a) CV curves at 10 mV s−1. (b) GCD curves at 1 A g−1. (c) Specific capacity. (d) Nyquist plots (inset: magnification of the high-frequency region). (e) Plots of Z′ against ω−1/2 for the calculation of the Li+ diffusion coefficient. (f) Cycling stability and coulombic efficiency obtained by consecutive GCD tests at 5 A g−1 for 10000 cycles. | |
Compared with Bi2O3, Bi2O3-NC showed higher intensities of the anodic and cathodic peaks at high potentials, which could be ascribed to the reversible reaction of Bi2O3 ↔ Bi2O2. This reversible reaction might lead to an enhanced Li+ intercalation pseudocapacitance, which was beneficial to improve the capacitive-controlled contribution to total charge storage. Additionally, Bi2O3-NC showed a smaller potential difference between the anodic and cathodic peaks (Bi3+ ↔ Bi2+) than that of original Bi2O3, indicating that oxygen-deficient Bi2O3 exhibited higher reversibility of the redox reaction (Bi2O3 ↔ Bi2O2) mainly due to its higher conductivity resulting from the carbon shells and oxygen vacancies. The GCD curves of the GF@Bi2O3 and GF@Bi2O3-NC electrodes were compared at 1 A g−1 (Fig. 4b), and the GF@Bi2O3-NC electrode had a smaller voltage drop (0.038 V) than that of the GF@Bi2O3 electrode (0.096 V) mainly because of the NC layer and oxygen vacancies previously confirmed by the TEM and XPS analysis, which could improve the electrical conductivity of Bi2O3. Based on the discharge curves of the GCD tests (Fig. S9†), the GF@Bi2O3-NC electrode provided a higher specific capacity of 208 mA h g−1 (at 2.9C) and a better rate capability of 67.3% (from 2.9C to 29C) than those of the GF@Bi2O3 electrode (Fig. 4c). The Nyquist plots of the GF@Bi2O3-NC electrode showed a transition from a vertical curve to a −45° line, which was more inclined to high-frequency regions than that of the GF@Bi2O3 electrode, indicating fast electrolyte ion diffusion (Fig. 4d). The Li+ diffusion coefficient (D) was calculated from the slope of the line in the Nyquist plots according to the following equations:37,38
| | | Zre = Rs + Rct + σωω−1/2 | (10) |
| | 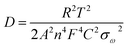 | (11) |
where
Zre is the real part of the
Z plots,
Rs is the internal resistance,
Rct is the charge transfer resistance,
ω is the angular frequency,
σω is the Warburg factor,
R is the gas constant,
T is the absolute temperature,
A is the surface area of the electrode,
n is the number of transferred electrons,
F is the Faraday's constant, and
C is the Li
+ concentration. As a result, the GF@Bi
2O
3-NC electrode showed a lower slope (
σω) of
Z′ against
ω−1/2 than that of the GF@Bi
2O
3 electrode (
Fig. 4e), indicating a higher Li
+ diffusion coefficient (6.32 × 10
−12 cm
2 s
−1). The long-term cycling stability and coulombic efficiency were evaluated by consecutive GCD tests at 5 A g
−1 for 10
000 cycles (
Fig. 4f). Compared with the GF@Bi
2O
3 electrode, the GF@Bi
2O
3-NC electrode showed higher capacitance retention (65.9%) at the end of the cycling test and an excellent coulombic efficiency with no significant variation during the cycling test, indicating its excellent electrochemical stability. The capacitance retentions of GF@Bi
2O
3 and GF@Bi
2O
3-NC exhibited a rapid decline in capacity during the first 1000 cycles, especially GF@Bi
2O
3. After the cycling stability tests, Bi
2O
3 and Bi
2O
3-NC were further characterized by SEM measurements (Fig. S10
†). The samples of Bi
2O
3 and Bi
2O
3-NC showed the morphology of broken monolithic blocks probably due to severe lattice expansion caused by Li
+ intercalation/de-intercalation during the charge/discharge process. This volume expansion could probably destroy the electrode structure and lead to the significant loss of active materials (Bi
2O
3 and Bi
2O
3-NC), resulting in a rapid decline in capacity during the first 1000 cycles. In conclusion, compared with the GF@Bi
2O
3 electrode, the GF@Bi
2O
3-NC electrode possessed better energy storage performance, including voltage drop, specific capacity, rate capability, cycling stability and Li
+ diffusion coefficient, indicating that it was more suitable as a battery-type anode for high-performance BSH devices.
The electrochemical performances of the GF@NS-CAG electrodes were evaluated by three-electrode tests in 1 M LiCl from 0 V to 1 V. The CV and GCD curves of the GF@NS-CAG electrodes showed a rectangle-like shape (Fig. 5a) and a triangle-like shape (Fig. 5b), respectively, indicating that the capacitive response depended on EDLC. The GF@NS-CAG-1000-2 electrode exhibited a pair of unimpressive reversible humps related to pseudocapacitance produced by heteroatom doping.17,39 The gravimetric capacitance of the GF@NS-CAG electrodes was calculated based on the discharging curves at different current densities (Fig. S11†). As a result, the GF@NS-CAG-1000-2 electrode exhibited a much higher gravimetric capacitance of 312 F g−1 at 1 A g−1 than those of the other GF@NS-CAG electrodes (Fig. 5c), which could be mainly attributed to its higher active N content and the larger mesopores and micropores, as confirmed by the XPS and BET measurements. Even when the current density was increased to 10 A g−1, it retained a specific capacitance of 244 F g−1, revealing an excellent rate capability of 78.2%. The Nyquist plots were obtained to ascertain the electrolyte ion diffusion (Fig. 5d). The characteristic frequency at the phase angle of −45° reflected the transition from resistive behavior to capacitive behavior, which was figuratively called “knee point”.40 At the knee point, the frequency of the GF@NS-CAG-1000-2 electrode (317 Hz) was much higher than those of the other GF@NS-CAG electrodes, indicating faster electrolyte ion diffusion probably due to the existence of larger mesopores and micropores. All these results indicated that the GF@NS-CAG-1000-2 electrode with the highest supercapacitive performance among the tested GF@NS-CAG electrodes could be compatibly used as a capacitive cathode for high-performance BSH devices.
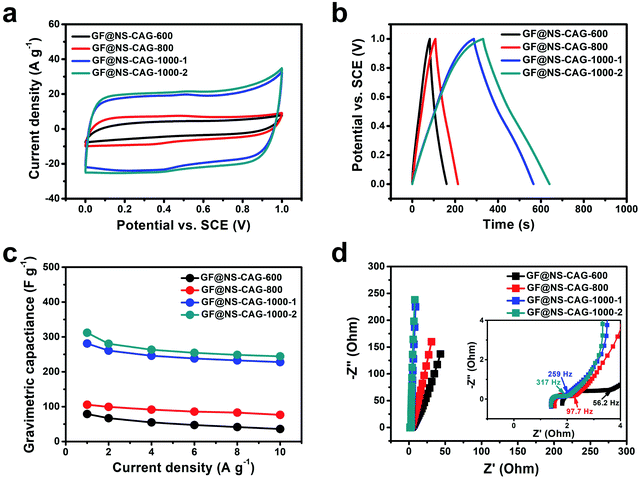 |
| | Fig. 5 Electrochemical performances of the GF@NS-CAG electrodes. (a) CV curves at 20 mV s−1. (b) GCD curves at 1 A g−1. (c) Gravimetric capacitance. (d) Nyquist plots (inset: magnification of the high-frequency region). | |
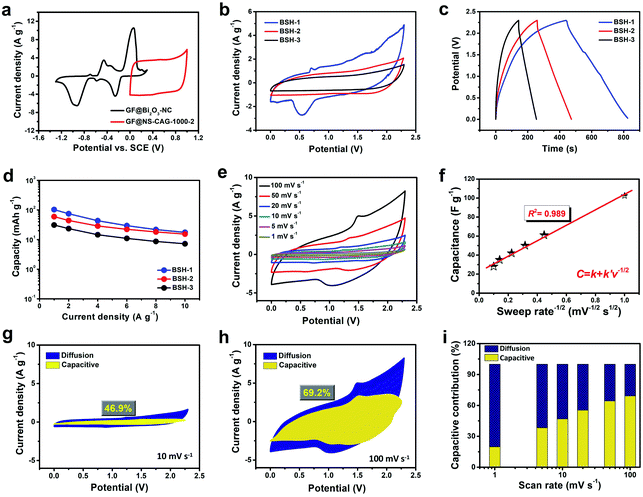
Based on the GF@Bi2O3-NC anode and the GF@NS-CAG-1000-2 cathode, flexible quasi-solid-state BSH devices with different mass loadings were assembled in a two-electrode configuration and denoted as BSH-1 (3.8 mg cm−2), BSH-2 (8.4 mg cm−2) and BSH-3 (11.6 mg cm−2). In Fig. 6a, the CV curves of the GF@Bi2O3-NC anode and the GF@NS-CAG-1000-2 cathode showed the voltage range of −1.3–0.3 V and 0–1 V respectively, reflecting a stable operating voltage window of 0–2.3 V for the assembled BSH devices. As a result, BSH-1 showed a pair of relatively obvious redox peaks (Fig. 6b) and longer discharging time (Fig. 6c) compared with those of BSH-2 and BSH-3, indicating its higher specific capacity. There were no apparent redox peaks in the CV curves of BSH-2 and BSH-3 with high mass loadings mainly because of their larger interior resistance and slower electrolyte ion diffusion than those of BSH-1 with a low mass loading. According to the GCD curves of these BSH devices (Fig. S12†), the capacities were calculated and compared (Fig. 6d), and the highest specific capacity of 103 mA h g−1 at 1 A g−1 was obtained for BSH-1. To further understand the energy storage mechanism, we analyzed the electrochemical kinetics of BSH-1 with the capacitive- and diffusion-controlled capacities separated by Dunn's method.19,24,41,42 The relationship between the current (i) and the scan rate (v) is expressed by the following equation:
where
a and
b are adjustable values. The linear dependence of specific capacitance
versus v−1/2 was employed to determine the
b value, according to the CV curves (
Fig. 6e). The linear correlation coefficient of the fitted curve was obtained as 0.989 (
Fig. 6f), reflecting a
b value near 1 and that the energy storage behavior was dominated by the capacitive-controlled contribution. At a certain potential,
i can be expressed as the combination of capacitive-controlled contribution (
k1v) and diffusion-controlled contribution (
k2v1/2):
9,24,42
 |
| | Fig. 6 Electrochemical evaluation of the BSH devices. (a) CV curves of the GF@Bi2O3-NC anode and the GF@NS-CAG-1000-2 cathode at 10 mV s−1. (b) CV curves at 10 mV s−1. (c) GCD curves at 1 A g−1. (d) Specific capacity. (e) CV curves of BSH-1 at different scan rates. (f) Specific capacitance versus v−1/2. Contributions of the capacitive- and diffusion-controlled capacities to total charge storage at (g) 10 mV s−1 and (h) 100 mV s−1. (i) Normalized contribution ratio of capacitive-controlled capacity at different scan rates. | |
The normalized capacitive- and diffusion-controlled contributions to total charge storage at different scan rates were shown in Fig. 6g, h, and S13,† and summarized in Fig. 6i. At 10 mV s−1, the capacitive-controlled contribution (illustrated as the yellow area) was 46.9% of the total charge storage compared with the diffusion-controlled contribution marked as the blue area. When the scan rate was increased to 100 mV s−1, 69.2% of the total charge storage was contributed by capacitive behavior, indicating that capacitive-controlled contribution was predominant in the energy storage behavior mainly attributed to the pseudocapacitance of the GF@NS-CAG-1000-2 cathode and the Li+ intercalation capacity of the GF@Bi2O3-NC anode. In the crystal lattice of Bi2O3, the oxygen vacancies confirmed by XPS analysis serve as the centers of positive charge for the construction of built-in electric fields, which can accelerate Li+ transport in the crystal lattice, thus enhancing the Li+ intercalation capacity.43 The rational utilization of the GF@Bi2O3-NC anode instead of the GF@Bi2O3 anode can effectively improve the ion diffusion kinetics of the unmatched Bi2O3 electrodes and facilitate the enhancement of rate capability and cycling stability.
EIS measurements of the BSH devices were performed to ascertain the interior resistance and electrolyte ion diffusion. The high-frequency region in the Nyquist plots (Fig. 7a) was fitted to a Randles equivalent circuit (Fig. S14†), resulting in the equivalent circuit parameters listed in Table S4.† Compared with BSH-2 and BSH-3, BSH-1 showed a smaller Rs value of 1.72 Ω and a shorter Warburg region (confirmed by a higher Wo-P value), indicating faster electrolyte ion diffusion.17 The Ragone plots of the BSH devices were compared with those of the previously-reported symmetric and asymmetric devices based on Bi2O3, as shown in Fig. 7b and listed in Table S5.† BSH-1 provided an amazing energy density of 118 W h kg−1 at the power density of 1.15 kW kg−1, which was much higher than those of the reported symmetric device based on CQD-Bi2O3 (88 W h kg−1)27 and asymmetric devices, such as LiMn2O4//Bi2O3 (78 W h kg−1),5 Bi2O3//AC (35.4 W h kg−1),6 Bi2O3–Ni–F//graphite (11 W h kg−1),7 Bi2O3//MnO2 (11.3 W h kg−1),8 CNT@NiCo2O4//CNT@Bi2O3-G (98.2 W h kg−1),28 N-GNTs@OV-Bi2O3 NSAs//N-GNTs@CoNi2S4 NP (86.6 W h kg−1),29 and NiO//Bi2O3@C (34.3 W h kg−1).44 Additionally, BSH-1 also delivered an ultrahigh power density of 11.5 kW kg−1 with the energy density maintained at 20 W h kg−1. The cycling stability was evaluated by consecutive GCD tests at the different current densities, such as 2, 4, 6, 8, 10 A g−1, for 10000 cycles in total (Fig. 7c). BSH-1 showed the capacitance retention of 95.7% at the end of the cycling test, which was better than those of the reported asymmetric devices, such as LiMn2O4//Bi2O3 (60%, 50 cycles),5 Bi2O3//AC (72%, 1000 cycles),6 Bi2O3–Ni–F//graphite (83%, 2000 cycles),7 Bi2O3//MnO2 (85%, 4000 cycles),8 CNT@NiCo2O4//CNT@Bi2O3-G (80.1%, 8000 cycles),28 N-GNTs@OV-Bi2O3 NSAs//N-GNTs@CoNi2S4 NP (85%, 10000 cycles),29 and NiO//Bi2O3@C (83%, 1000 cycles).44 Moreover, BSH-3 with a high mass loading also exhibited satisfactory capacitance retention of 92.1% and excellent coulombic efficiency without significant variation during the cycling test, revealing its huge potential in commercial applications. For practical application, it is important to evaluate the capacitance stability of flexible energy storage devices under bent states. The CV curves of BSH-1 at 10 mV s−1 displayed slight deformation at different bending angles from 0° to 135° (Fig. S15†), demonstrating good capacitance stability under bent states. The capacitance stability of the BSH-1 device reflected the stable electrochemical performance of the GF@Bi2O3-NC electrode under different bending angles. The CV curves of two BSH-1 devices connected in series showed that the potential window extended from 2.3 V for a single device to 4.6 V for the tandem device (Fig. S16a†). The GCD curves of two BSH-1 devices connected in series (Fig. 7d) and parallel (Fig. S16b†) confirmed that the capacity of each BSH device was well-maintained. Additionally, the tandem device based on two BSH-1 devices could power a red LED array (Fig. 7e), indicating its great potential in high-performance energy storage devices.
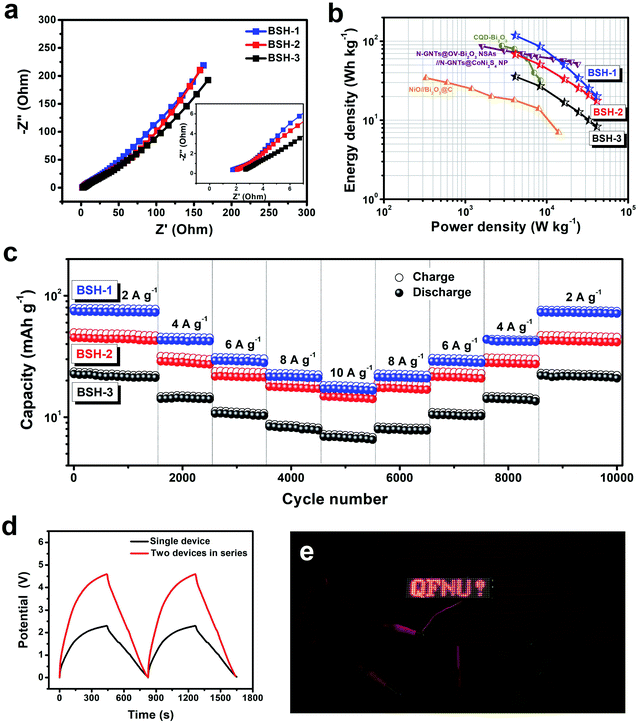 |
| | Fig. 7 (a) Nyquist plots (inset: magnification of the high-frequency region). (b) Ragone plots of our devices compared with previously-reported symmetric and asymmetric devices based on Bi2O3. (c) Cycling stability obtained by consecutive GCD tests at different current densities for 10000 cycles in total. (d) GCD curves of two BSH-1 devices connected in series at 1 A g−1. (e) A photograph of a red LED array powered by two BSH-1 devices connected in series. | |
4. Conclusions
Flexible BSH devices with different mass loadings were facilely assembled by using a GF@Bi2O3-NC anode, a GF@NS-CAG-1000-2 cathode, and the PVA/LiCl/H2O gel electrolyte. The oxygen-deficient Bi2O3-NC nanoflakes prepared by the carbonization of the Bi2O3/melamine precursors were confirmed to improve the voltage drop, specific capacity, rate capability, cycling stability and Li+ diffusion coefficient. Additionally, the GF@NS-CAG-1000-2 cathode obtained by the carbonization of the GF@PSS-PPy hydrogel precursors provided a high gravimetric capacitance of 312 F g−1 at 1 A g−1 mainly attributed to its microporous structure and high active N content. The flexible quasi-solid-state BSH device based on the GF@Bi2O3-NC anode and the GF@NS-CAG-1000-2 cathode, showed the stable voltage window of 2.3 V and delivered a specific capacity of 103 mA h g−1 (at 1 A g−1), an energy density of 118 W h kg−1 (at 1.15 kW kg−1) and capacity retention of 95.7% (after 10000 cycles), indicating that this was a facile and highly-efficient approach to exploit high-performance energy storage devices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The financial support from National Natural Science Foundation of China (22005174) is gratefully acknowledged. The authors would like to thank Shiyanjia Lab (http://www.shiyanjia.com) for the TEM measurements.
References
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2020, 5, 5–19 CrossRef.
- K. Suzuki, K. Kim, S. Taminato, M. Hirayama and R. Kanno, J. Power Sources, 2013, 226, 340–345 CrossRef CAS.
- W. Zuo, W. Zhu, D. Zhao, Y. Sun, Y. Li, J. Liu and X. W. Lou, Energy Environ. Sci., 2016, 9, 2881–2891 RSC.
- S. T. Senthilkumara, R. K. Selvana, M. Ulaganathanb and J. S. Meloc, Electrochim. Acta, 2014, 115, 518–524 CrossRef.
- N. M. Shinde, Q. Xia, J. Yun, S. Singh, R. S. Mane and K. Kim, Dalton Trans., 2017, 46, 6601–6611 RSC.
- H. H. Xu, X. L. Hu, H. L. Yang, Y. M. Sun, C. C. Hu and Y. H. Huang, Adv. Energy Mater., 2015, 5, 1401882 CrossRef.
- R. Li, Y. Wang, C. Zhou, C. Wang, X. Ba, Y. Li, X. Huang and J. Liu, Adv. Funct. Mater., 2015, 25, 5384–5394 CrossRef CAS.
- Y. Gan, C. Wang, X. Chen, P. Liang, H. Wan, X. Liu, Q. Tan, H. Wu, H. Rao, H. Wang, J. Zhang, Y. Wang, P. A. van Aken and H. Wang, Chem. Eng. J., 2020, 392, 123661 CrossRef CAS.
- C. Wang, Z. Song, H. Wan, X. Chen, Q. Tan, Y. Gan, P. Liang, J. Zhang, H. Wang, Y. Wang, X. Peng, P. A. van Aken and H. Wang, Chem. Eng. J., 2020, 400, 125955 CrossRef CAS.
- Q. Tan, X. Chen, H. Wan, B. Zhang, X. Liu, L. Li, C. Wang, Y. Gan, P. Liang, Y. Wang, J. Zhang, H. Wang, L. Miao, J. Jiang, P. A. van Aken and H. Wang, J. Power Sources, 2020, 448, 227403 CrossRef CAS.
- C.-J. Ma, Y. Chen, C. Zhu, Q. Chen, W.-L. Song, S. Jiao, H. Chen and D. Fang, J. Mater. Chem. A, 2019, 7, 12176–12190 RSC.
- F. Wu, X. Wang, W. zheng, H. Gao, C. Hao and C. Ge, Electrochim. Acta, 2017, 245, 685–695 CrossRef CAS.
- T. Lin, I.-W. Chen, F. Liu, C. Yang, H. Bi, F. Xu and F. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS.
- C. Yang, N. Hu, W. Wang and B. Cao, J. Power Sources, 2018, 399, 115–124 CrossRef CAS.
- Q. Jia, C. Yang, Q. Pan, Y. Xin, F. Xu, W. Qi, H. Wei, S. Yang, C. Zhou, N. Hu and B. Cao, Chem. Eng. J., 2020, 383, 123153 CrossRef CAS.
- X. Chu, H. Huang, H. Zhang, H. Zhang, B. Gu, H. Su, F. Liu, Y. Han, Z. Wang, N. Chen, C. Yan, W. Deng, W. Deng and W. Yang, Electrochim. Acta, 2019, 301, 136–144 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS.
- J. T. Mefford, W. G. Hardin, S. Dai, K. P. Johnston and K. J. Stevenson, Nat. Mater., 2014, 13, 726–732 CrossRef CAS.
- W. Zuo, R. Li, C. Zhou, Y. Li, J. Xia and J. Liu, Adv. Sci., 2017, 4, 1600539 CrossRef.
- Y. Shao, M. F. El-Kady, J. Sun, Y. Li, Q. Zhang, M. Zhu, H. Wang, B. Dunn and R. B. Kaner, Chem. Rev., 2018, 118, 9233–9280 CrossRef CAS.
- J. Li, Q. Wu and G. Zan, Eur. J. Inorg. Chem., 2015, 5751–5756 CrossRef CAS.
- S. X. Wang, C. C. Jin and W. J. Qian, J. Alloys Compd., 2014, 615, 12–17 CrossRef CAS.
- A. Prasath, M. Athika, E. Duraisamy, A. S. Sharma, V. S. Devi and P. Elumalai, ACS Omega, 2019, 4, 4943–4954 CrossRef CAS.
- W. Wang, Y. Xiao, X. Li, Q. Cheng and G. Wang, Chem. Eng. J., 2019, 371, 327–336 CrossRef CAS.
- J. Zhao, Z. Li, T. Shen, X. Yuan, G. Qiu, Q. Jiang, Y. Lin, G. Song, A. Meng and Q. Li, J. Mater. Chem. A, 2019, 7, 7918–7931 RSC.
- X. Lu, Y. Zeng, M. Yu, T. Zhai, C. Liang, S. Xie, M.-S. Balogun and Y. Tong, Adv. Mater., 2014, 26, 3148–3155 CrossRef CAS.
- S. Yang, Y. Liu, Y. Hao, X. Yang, W. A. Goddard III, X. L. Zhang and B. Cao, Adv. Sci., 2018, 5, 1700659 CrossRef.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092–7102 CrossRef CAS.
- T. Tsubota, K. Takenaka, N. Murakami and T. Ohno, J. Power Sources, 2011, 196, 10455–10460 CrossRef CAS.
- L. Sun, H. Zhou, L. Li, Y. Yao, H. Qu, C. Zhang, S. Liu and Y. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 26088–26095 CrossRef CAS.
- L. Zhang, C. Yang, N. Hu, Z. Yang, H. Wei, C. Chen, L. Wei, Z. J. Xu and Y. Zhang, Nano Energy, 2016, 26, 668–676 CrossRef CAS.
- J. Yang, X. Zhou, D. Wu, X. Zhao and Z. Zhou, Adv. Mater., 2017, 29, 1604108 CrossRef.
- W. Zuo, C. Xie, P. Xu, Y. Li and J. Liu, Adv. Mater., 2017, 29, 1703463 CrossRef.
- Y. Huang, M. Xie, Z. Wang, Y. Jiang, Y. Yao, S. Li, Z. Li, L. Li, F. Wu and R. Chen, Small, 2018, 14, 1801246 CrossRef.
- F. Béguin, K. Szostak, G. Lota and E. Frackowiak, Adv. Mater., 2005, 17, 2380–2384 CrossRef.
- Y. Shao, M. F. El-Kady, C. W. Lin, G. Zhu, K. L. Marsh, J. Y. Hwang, Q. Zhang, Y. Li, H. Wang and R. B. Kaner, Adv. Mater., 2016, 28, 6719–6726 CrossRef CAS.
- J. Li, L. An, H. Li, J. Sun, C. Shuck, X. Wang, Y. Shao, Y. Li, Q. Zhang and H. Wang, Nano Energy, 2019, 63, 103848 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- Y. Zheng, T. Zhou, X. Zhao, W. K. Pang, H. Gao, S. Li, Z. Zhou, H. Liu and Z. Guo, Adv. Mater., 2017, 1700396 CrossRef.
- D. Ba, Y. Li, Y. Sun, Z. Guo and J. Liu, Sci. China Mater., 2019, 62, 487–496 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00831a |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Qi
Jia
,
Qianqian
Pan
,
Wentao
Qi
,
Rui
Ling
and
Bingqiang
Cao
*
,
Qi
Jia
,
Qianqian
Pan
,
Wentao
Qi
,
Rui
Ling
and
Bingqiang
Cao
*














