
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology
Lukas
Krasny
 and
Paul H.
Huang
*
and
Paul H.
Huang
*
Division of Molecular Pathology, The Institute of Cancer Research, 237 Fulham Road, London, SW3 6JB, UK. E-mail: paul.huang@icr.ac.uk
First published on 9th October 2020
Abstract
Data-independent acquisition mass spectrometry (DIA-MS) is a next generation proteomic methodology that generates permanent digital proteome maps offering highly reproducible retrospective analysis of cellular and tissue specimens. The adoption of this technology has ushered a new wave of oncology studies across a wide range of applications including its use in molecular classification, oncogenic pathway analysis, drug and biomarker discovery and unravelling mechanisms of therapy response and resistance. In this review, we provide an overview of the experimental workflows commonly used in DIA-MS, including its current strengths and limitations versus conventional data-dependent acquisition mass spectrometry (DDA-MS). We further summarise a number of key studies to illustrate the power of this technology when applied to different facets of oncology. Finally we offer a perspective of the latest innovations in DIA-MS technology and machine learning–based algorithms necessary for driving the development of high-throughput, in-depth and reproducible proteomic assays that are compatible with clinical diagnostic workflows, which will ultimately enable the delivery of precision cancer medicine to achieve optimal patient outcomes.
 Lukas Krasny | Dr Lukas Krasny received his Master's degree followed by PhD in Biochemistry from the University of Chemical Technology in Prague (Czech Republic). In 2015, he joined the Molecular and Systems Oncology Team at the Institute of Cancer Research in London (UK) where he focused mainly on analysis of proteins in extracellular matrix by MS-based tools and proteomic characterisation of soft tissue sarcomas. He explored the potential of DIA mass spectrometry for analysis of matrisomal proteins and recently published a mouse reference spectral library for proteomic applications of SWATH/DIA-MS. |
Introduction
Rapid advances in the development of Omics technologies (e.g. genomics, transcriptomics, proteomics, metabolomics and glycomics) in the past two decades has significantly broadened our understanding of cancer biology. For instance, the comprehensive molecular characterisation of tumours has enabled better classification of different cancer types and improved the speed and accuracy of disease diagnosis while the discovery of new oncogenes and tumour suppressors has led to novel drug targets and more effective treatment strategies.1,2 Due to democratisation of cancer genomics as a result of the introduction of standardised platforms and decreasing costs, next generation DNA and RNA sequencing has been rapidly adopted as the method of choice for molecular characterisation of tumours by the cancer research community.3 To date, hundreds of gene aberrations have been identified as tumour drivers or suppressors and genomic profiles of hundreds of thousands of tumour specimens have been analysed across more than 20 cancer types.2,4,5In contrast to the cancer genome, there is a significant gap in our knowledge of the cancer proteome. Proteins, as downstream effector molecules of the genetic code, reflect the phenotypic consequence of the cancer genome and allows one to link the relatively static genetic information with the dynamic proteomic landscape within the cell. Furthermore, given that the majority of druggable targets in tumour cells are proteins, a global overview of the cancer proteome may reveal new options for drug discovery and development. Recognising this gap, there has been significant investment in recent years in the large-scale characterisation of the tumour proteome led largely by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) of the National Cancer Institute.6 These studies have provided publicly available proteogenomic datasets for several cancer types such as breast cancer, ovarian cancer and colon cancer with ongoing studies in other cancer types.7–9
Since the discovery of soft ionization techniques such as matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI), mass spectrometry (MS) has become an unrivalled analytical tool for the identification, characterization and quantification of proteins and their post-translational modifications. In particular, the combination of liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) has provided a sensitive high-throughput platform enabling analysis of several thousand proteins from an individual sample. In oncology, proteomic analysis by LC-MS/MS has been widely used in multiple applications such as biomarker discovery, drug screens and personalized medicine. Most of these applications use conventional data-dependent acquisition (DDA) or targeted methods such as single or multiple reaction monitoring (SRM/MRM) which have been comprehensively reviewed elsewhere.10–12 In this review, we focus on the use of data-independent acquisition (DIA) (also known as sequential window acquisition of all theoretical mass spectra (SWATH-MS))13 and provide an overview of specific applications in cancer proteomics to inform molecular classification, biomarker discovery and the identification of new drug targets. This review will focus on DIA-MS applications in tissue and cell line analysis, and readers who are interested in the use of this technology in liquid biopsies and plasma proteomics are referred to these excellent reviews on the topic.14–16 We further present the latest innovations in DIA-MS that will push the boundaries of this technology and accelerate its implementation in precision cancer medicine.
Principles and workflow of DDA versus DIA mass spectrometry
Conventional DDA-MS involves the scanning of all precursor peptide ions during the survey scan (MS1) which is followed by the selection of a predefined number of precursor ions for subsequent fragmentation (MS2). This sequential selection and fragmentation approach provides detailed peptide sequencing information about precursor ions (Fig. 1A). Technological advances in MS instrumentation has not only led to faster scanning speeds but also increased sensitivity. These developments have resulted in the development of a next generation proteomic strategy known as DIA-MS or SWATH-MS which provides better reproducibility and sensitivity when compared to conventional DDA-MS.13,17–19 In contrast to DDA-MS, DIA-MS is based on the fragmentation of all precursor ions identified in a MS1 survey scan where fragment ions are accumulated in a fixed number of wide isolation windows that span the entire mass-to-charge ratio (m/z) range (Fig. 1B).13 In this fashion, rather than only acquiring fragmentation data from a predefined set of selected precursor ions as is the case in DDA-MS, all detected precursor ions within a survey scan are fragmented. The minimum instrument requirements for DIA-MS experiments are mass spectrometers capable of high-resolution MS/MS spectra acquisition at fast scan speeds. Modern and commonly used mass spectrometers with quadrupole time-of-flight (QTOF) or hybrid quadrupole Orbitrap mass analyzers comply with these requirements, enabling straightforward adoption with minimal adaptation for use in DIA-MS.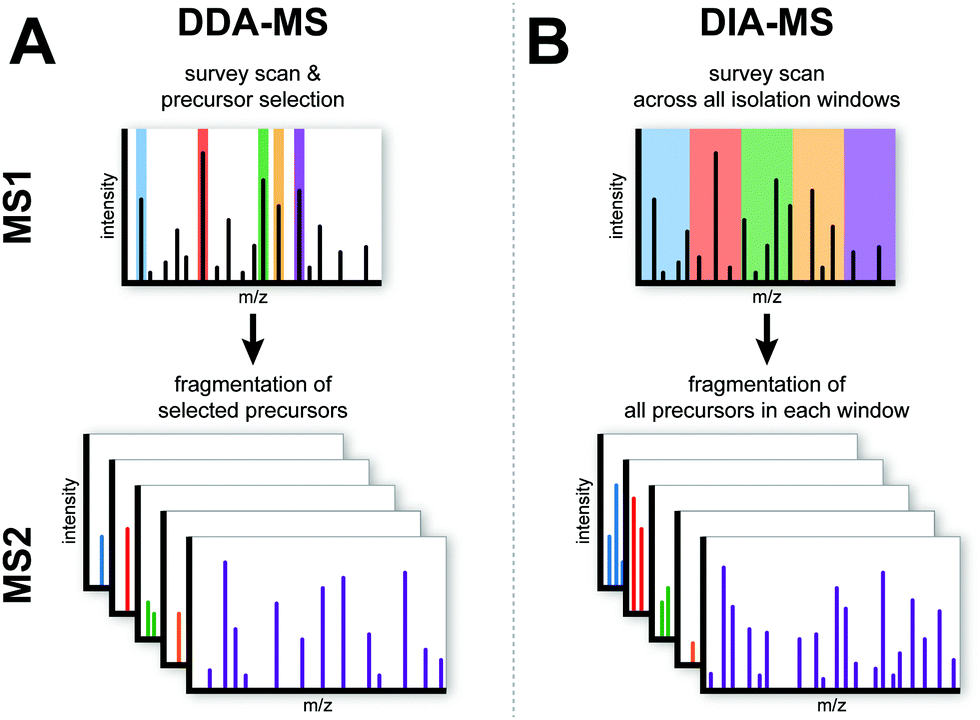 | ||
| Fig. 1 Schematic overview of the DDA-MS and DIA-MS. In DDA-MS, the top n most abundant precursor ions are selected based on the survey scan (MS1) and selected ions are fragmented in MS2. In DIA-MS, the survey scan provides snapshot of the precursor ions (MS1). Pre-defined wide isolation windows cover the whole MS1 m/z range and all precursor ions within each isolation window are fragmented in MS2. | ||
Typical sample processing workflows for label-free DDA-MS analysis (Fig. 2A) often include the steps of protein extraction, digestion, data acquisition and data processing (indicated by solid arrows in Fig. 2A). To increase the depth of proteomic analysis, off-line fractionation such as SDS-PAGE or liquid chromatography are often used. However, such pre-fractional steps will increase total sample amount requirements for the experiment. In DIA-MS, the sample processing and data acquisition steps are identical to single-shot DDA-MS (Fig. 2B). However, because all precursor ions in a survey scan are fragmented (Fig. 1), there is a need to incorporate post-acquisition in silico data processing steps to deconvolute the resulting complex fragment ion spectra which involves interrogating MS data with spectral libraries (Fig. 2B). A spectral library is a database which contains mass spectrometric and chromatographic parameters such as precursor and fragment m/z value, fragment type, charge and elution time for each individual peptide in the analysed sample.13,20 These study-specific spectral libraries are conventionally generated by extensive DDA-based proteomic characterization of the same samples prior to analysis by DIA-MS (Fig. 2B).21–23 However, study-specific libraries can vary between laboratories due to the lack of consistency in DDA experiments and spectral library generation. This can result in wide variations in the number and type of proteins identified and quantified between studies. As a result of the extensive number of DDA-MS experiments required to generate study-specific spectral libraries, there are also cost and time implications to consider which may decrease the attractiveness of DIA-MS. More recently, the generation of comprehensive spectral libraries as a community resource have been employed as an alternative solution. To date, comprehensive reference libraries have been generated for number of organisms including human,24 mouse,25,26 zebrafish,27 fruit fly,28 yeast,29 and various bacteria.30–32 Most of these libraries are publicly available in repositories such as SWATHAtlas.org for community use. These comprehensive reference libraries remove the need to generate study-specific libraries for each DIA-MS experiment, thus increasing inter-laboratory reproducibility while economising sample requirements and MS instrument time. This high inter-laboratory reproducibility was demonstrated by Collins et al. who undertook a multi-laboratory assessment of HEK293 cell lysates in 11 laboratories across the world and showed a very high median inter-laboratory Pearson correlation coefficient of 0.94 in the quantification of 4,077 proteins.33
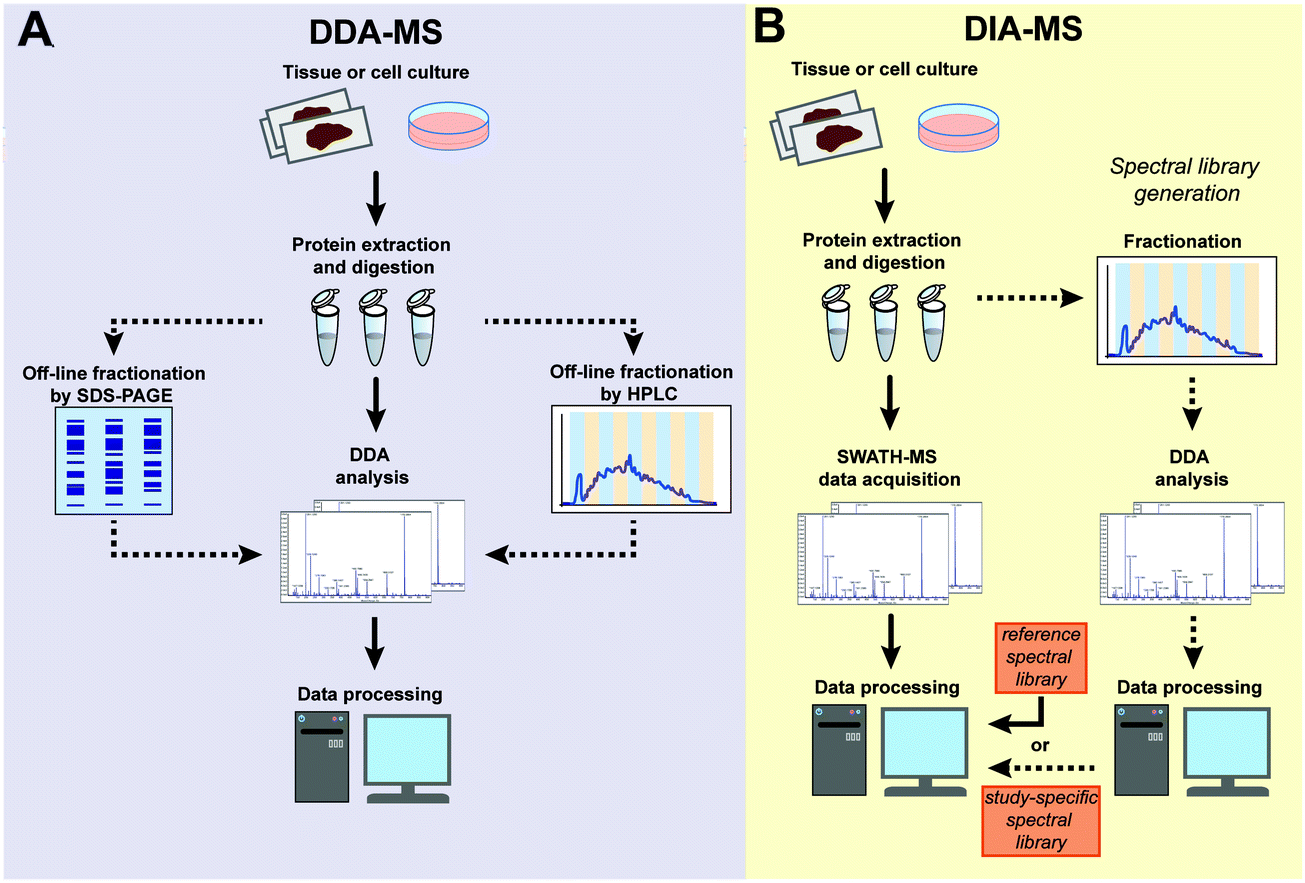 | ||
| Fig. 2 Schematic workflow of the label-free DDA-MS and DIA-MS experiments. (A) In DDA-MS, extracted proteins are digested and either directly analysed by single-shot DDA-MS (solid arrows) or subjected to off-line fractionation (dotted arrows) prior to DDA-MS analysis of individual fractions. The acquired data is searched against a database of known protein sequences and further processed by software tools. (B) In DIA-MS analysis, extracted proteins are digested and directly analysed by single-shot analysis. The complex spectra generated is processed using either reference spectral library (solid arrow) or a study-specific library that is generated from the same samples by a parallel DDA-MS analysis (dotted arrow). | ||
Strengths and limitations of DIA-MS
A major strength of DIA-MS is the exceptional reproducibility in protein identification across multiple experiments (Fig. 3). In DDA-MS, the stochastic nature of the automated precursor ion selection in the survey scan prior to fragmentation leads to a well-documented inability of this method to reproducibly identify the same set of proteins across technical replicate experiments.18,19,34,35 This lack of consistency in precursor ion fragmentation results in a large number of missing values in large-scale experiments involving multiple samples which significantly impacts the level of reproducibility necessary for contemporary biological experiments. DIA-MS overcomes this challenge by the cyclic acquisition of fragment ions for all precursor ions in the survey scan thereby significantly improving reproducibility in protein identification between technical replicate experiments. For instance, Bruderer et al. reported that in an MS analysis comprising of 24 samples, DIA-MS resulted in only 1.6% missing values across all samples compared to 51% missing values in DDA-MS.19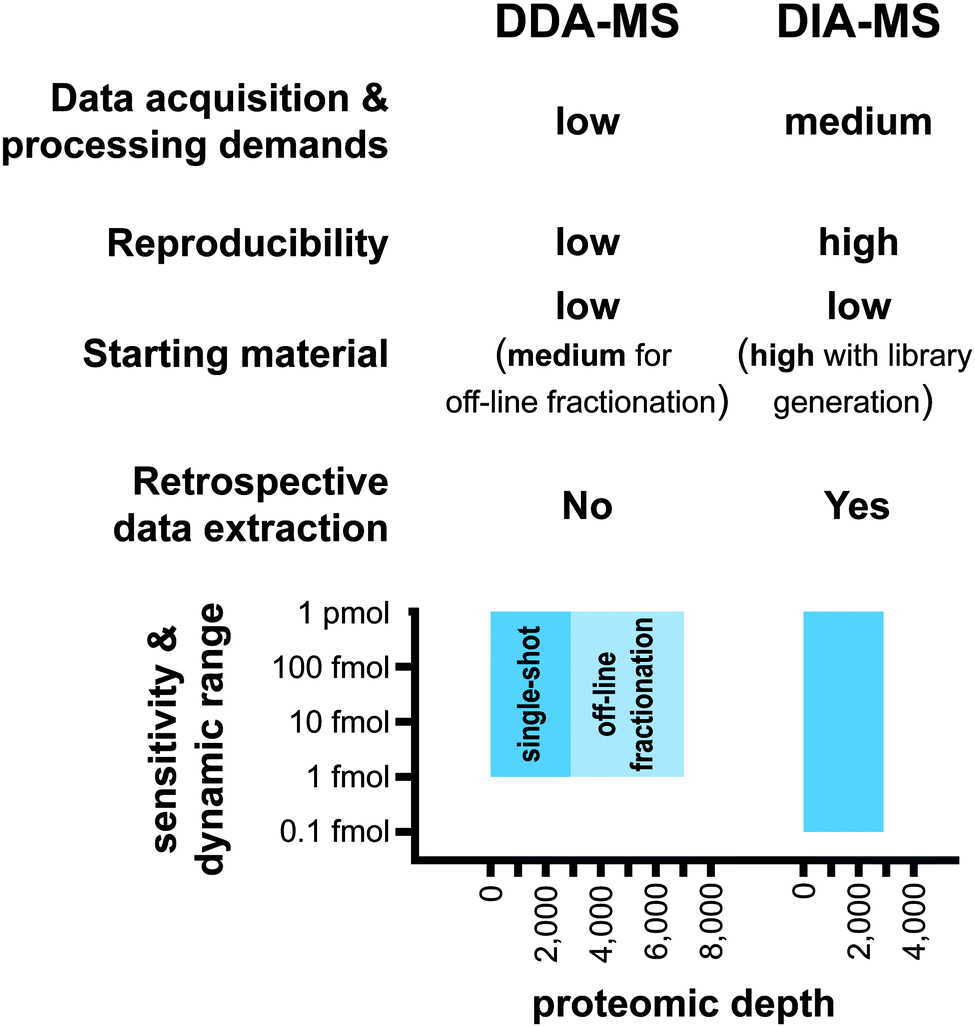 | ||
| Fig. 3 Advantages and limitations of DDA-MS in comparison to DIA-MS. | ||
Both methods typically quantify similar number of proteins (∼3000–5000) in a single shot analysis.17 Based on the published reports, it has been shown that the limit of detection (LOD) of the DIA-MS is ∼100 amol and its dynamic quantification range spans over 4–5 orders of magnitude13,33 (Fig. 3). A comparison of DDA-MS and DIA-MS performed by Gillet et al. showed that DDA-MS failed to identify reference peptides spiked into a yeast lysate background at 2–10 fold higher concentration than the LOD of DIA-MS.13 Furthermore, an up to 10-fold gain in the sensitivity of DIA-MS was reported when compared to label-free workflows based on extraction of precursor ion trace from MS1 scans.13,33 These analyses suggest that the sensitivity of DIA-MS is superior to DDA-MS although a direct head-to-head comparison of the sensitivity of these two methodologies has yet to be performed.
The nature of the LC-MS/MS analysis is based on the destructive sampling of the analyte eluted from the LC column into the MS instrument. Therefore, once the sample has been injected into the LC-MS/MS system and the data acquired, it cannot be regenerated. Given the stochastic nature of DDA-MS and the missing values resulting from this technique, it is challenging to undertake comprehensive retrospective analysis of the acquired mass spectra. Retrospective signal extraction from DDA-MS data is therefore only available for precursor ions with acquired fragmentation spectra. In contrast, DIA-MS fragments all detected precursor ions in a sample which opens new possibilities for retrospective analysis. The acquired digitized proteome files can be reprocessed with different spectral libraries and provide reliable quantitative information for new sets of queries including post-translational modifications.36,37 As a result, DIA-MS proteomic data can become an invaluable repository for the community for subsequent analyses without the need of additional data acquisition.
One major limitation of DIA-MS is the need to generate spectral libraries for data processing (Fig. 3). In situations where a comprehensive reference spectral library is not available for use or if the study involves analysis of a sub-proteome (e.g. specific subcellular compartments or post-translational modifications) that is underrepresented in reference spectral libraries, there will be a need to generate study-specific libraries. As discussed above, building a new study-specific spectral library for DIA-MS involves significantly higher starting sample amounts, instrument time and costs. This barrier may have important implications particularly where sample availability is limiting such as in the case of tissue biopsies or in rare diseases.
Applications of DIA-MS in cancer proteomics
Since the first publication of DIA-MS in 2012,13 the use of this method in cancer proteomics has been steadily increasing. In 2019 alone, DIA-MS was employed in 42 published studies across a range of cancer types to analyse a variety of different types of biological material. In the following section, we provide a broad overview demonstrating the versatility and utility of DIA-MS in key cancer proteomics applications including molecular characterisation and classification, evaluating treatment response and biomarker discovery (Fig. 4 and Table 1).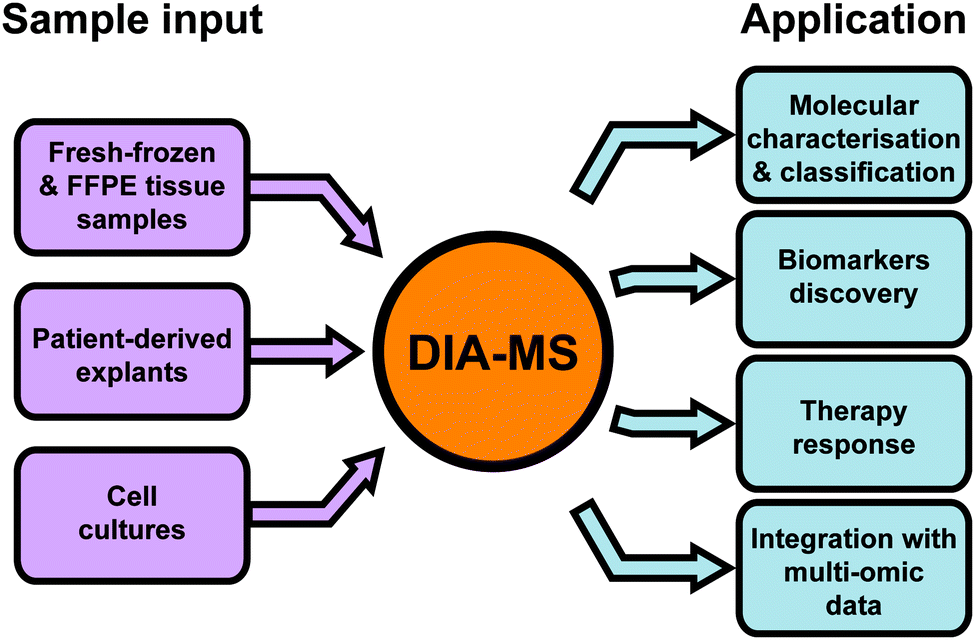 | ||
| Fig. 4 Common applications of DIA-MS in oncology. FFPE – formalin-fixed paraffin-embedded. | ||
| Cancer type | Study | Ref. | Study design | Number and type of samples | Proteome coverage | Key findings |
|---|---|---|---|---|---|---|
| Breast cancer | Bouchal et al. 2019 | 22 | Quantitative profiling of global proteome in biopsy samples from 4 breast cancer subtypes | 96 fresh frozen needle biopsies from 4 breast cancer subtypes: Luminal A (n = 48), Luminal B (n = 24), Her2-enriched (n = 8), triple-negative (n = 16) | 2842 proteins | • NF-KB pathway upregulated in luminal subtypes, VEGF pathway upregulated in Her2+ subtypes. |
| • Decision tree classifier developed based on expression of ERBB2, INPP4B and CDK1 with correct identification rate of 84% when applied on the original dataset | ||||||
| Prostate cancer | Liu et al. 2014 | 45 | Quantitative profiling of N-glycoproteins in tissue samples from prostate cancer patients | 75 fresh-frozen tissue specimens; normal (n = 10) tissue, non-aggressive (n = 24), aggressive (n = 16) and metastatic (n = 25) prostate cancer | 897 N-glycoproteins | • NAAA and PTK7 identified as potential markers for stratification of high- and low-risk prostate cancer. |
| Keam et al. 2018 | 57 | Quantitative profiling of global proteome in tumour and matched adjacent tissue samples pre- and post-radiotherapy | Fresh-frozen (n = 4) and FFPE (n = 16) biopsies taken pre- and post-radiotherapy from 8 patients | 4665 proteins in fresh frozen samples | • Wound healing, extracellular remodelling and acute inflammatory response pathways were enriched in the samples after radiation therapy | |
| 3974 proteins in FFPE sample | ||||||
| Nguyen et al. 2018 | 58 | Quantitative proteomic profiling of prostate cancer patient-derived explants treated with HSP90 inhibitors | 46 patient-derived explant tumours; discovery study (n = 16), validation (n = 30) | 4095 proteins in discovery cohort | • mRNA translation, ribosome function and RNA metabolism pathways were found downregulated and TCA metabolism upregulated after treatment with HSP90 inhibitors. | |
| 5450 proteins in validation cohort | • 9 proteins are universally decreased after inhibition of HSP90. | |||||
| • TRFC and TIMP1 identified as candidate drug response markers for treatment of prostate cancer by AUY922 | ||||||
| Latonen et al. 2019 | 66 | Multi-omic analysis of fresh frozen tissue samples by genomics, trascriptomics and proteomics | 38 fresh frozen tissue specimens; BPH (n = 10), treatment naïve PC (n = 17) and CRPC (n = 11) | 3394 proteins | • A panel of 95 miRNA identified as an important mechanism of gene expression regulation in prostate cancer. | |
| • Decreased expression of miR-22 and miR-205 related to upregulation of MDH2 in CRPC compared to PC | ||||||
| Kidney cancer | Guo et al. 2015 | 41 | Quantitative profiling of global proteome in 9 tumour and matched tissue biopsies | Fresh frozen tumour and matched adjacent tissue biopsy specimens from 9 patients with ccRCC (n = 6), pRCC (n = 2) and chRCC (n = 1) | 2375 proteins | • Proof-of-principle study demonstrating utility of DIA-MS for molecular characterization and biomarker identification in cancer research. |
| • A set of 21 known diagnostic markers of kidney cancer identified in the dataset including AMACR, VIM and GSTA1. | ||||||
| Lymphoma | Schwarzfischer et al. 2017 | 69 | Metabolomic analysis of cell lysates and tissue samples by GC-MS, LC-MS and NMR spectroscopy combined with quantitative analysis of global proteome by DIA-MS | 24 lymphoma cell lines (BL: n = 6, DLBCL: n = 18), fresh-frozen (n = 11) and FFPE (n = 13) tissue specimens | 3041 proteins in cell lines | • Higher intra- and extracelullar level of pyruvic acid in DLBCL compared to BL. |
| 2938 proteins in fresh-frozen tissues | • Upregulation of proteins involved in non-oxidative phosphorylation and one-carbon metabolism in BL identified as a result of metabolic reprogramming | |||||
| 1442 proteins in FFPE tissues. | ||||||
| Liver cancer | Gao et al. 2017 | 42 | Quantitative profiling of global proteome in 14 pairs of tumour and non-tumour tissue samples by DIA-MS | 28 fresh-frozen specimens; tumour (n = 14) and adjacent normal tissue (n = 14) | 4216 proteins | • Significant upregulation of spliceosome pathway and downregulation of 37 metabolic pathways in HCC compared to adjacent normal tissue. |
| • Expression of 9 proteins validated by IHC on separate cohort of 6 pairs of samples | ||||||
| Zhu et al. 2019 | 43 | Quantitative profiling of global proteome in 19 pairs of tumour and non-tumour tissue samples by DIA-MS | 38 fresh-frozen specimens; tumour (n = 19) and adjacent normal tissue (n = 19) | 2579 proteins | • MCM7, proteins from HSP family and mitochondrial ribosomal proteins found upregulated in HCC samples compared to adjacent normal tissues. | |
| • Upregulation of MCM7 validated by IHC on separate cohort | ||||||
| Other | Guo et al 2019 | 60 | Global proteomic profiling of the NCI-60 cancer cell lines | 60 cell lines included in NCI-60 panel | 3171 proteins | • Drug response prediction based on DIA-MS data outperforms prediction based on DDA-MS data. |
| • DIA-MS data can be integrated with mutational and transcriptomic data to obtain optimal predictive power for drug response simulations | ||||||
| Mehnert et al. 2020 | 70 | Multi-layered proteomic analysis of Dyrk2 mutant cell lines | 6 HEK293 mutant cell lines; HEK293 wild type | 5138 proteins in | • Individual mutations of Dyrk2 cause mutation-specific reorganization of the protein–protein interactions network and changes in phosphoproteomic profile. | |
| 2888 phospho-peptides | • Subset of the mutations modulate Cancer Driver Proteins suggesting that these mutations are associated with cancer progression. | |||||
Molecular characterization of tumour specimens for defining biological pathways, subtype classification and biomarker discovery
While the traditional classification of tumours based primarily on histopathological assessment has played a critical role in diagnosis and clinical management of disease, the increasing use of molecular and Omics based approaches have provided unprecedented insights into the underlying biology of cancer and facilitated new classification systems based on molecular alterations.38–40 In line with this, recent advances in MS technologies have driven new opportunities for deep proteomic profiling of clinical cohorts for the refinement of current cancer classification systems as well as revealing important disease-specific biological pathways. There is also a high demand for robust cancer markers for early and reliable tumour diagnostics, selection of appropriate treatments or prediction of patient outcomes. In this regard, DIA-MS has been employed in the proteomic characterization of multiple cancer types including breast, kidney, liver and prostate cancer,22,41–45 a selection of which are reviewed in this section.The first reported application of DIA-MS in cancer proteomics was published by Guo et al. who analysed biopsy samples obtained from kidney cancer patients.41 In this pioneering work, the authors presented a novel approach of combining pressure cycling technology (PCT) for sample preparation with DIA-MS data acquisition as a rapid proteomic pipeline for the analysis of human tissue specimens. Given that DIA-MS generates profiles comprising all fragment ions in a sample, this methodology results in a permanent digital proteome map for each individual patient which can be routinely interrogated for the identification and quantification of proteins of interest. In this proof-of-principle experiment, the authors analysed tumour and matched adjacent tissue samples from 9 patients in three different subtypes of renal cell carcinoma (RCC); clear cell RCC (ccRCC), papillary RCC (pRCC) and chromophobe RCC (chRCC).41 Overall 2375 proteins were quantified by PCT-DIA-MS across all 18 samples, including 21 proteins such as alpha-methylacyl-CoA racemase (AMACR), vimentin (VIM) and glutathion-S-transferase A1 (GSTA1) which are currently used as diagnostic or prognostic biomarkers in kidney cancer. Unsupervised clustering of the whole proteomic dataset clearly separated pRCC from ccRCC suggesting that proteomic profiling is an effective means for molecular classification of this disease. In particular, the authors showed by MS that AMACR, an established diagnostic biomarker used in immunohistochemistry for distinguishing pRCC and ccRCC,46 was 13 times higher in pRCC samples in comparison to ccRCC, validating the methodology. Conversely, VIM and GSTA1, were significantly increased in ccRCC which is in accordance with previously published literature.41,46 The comparison of the ccRCC tumours versus adjacent non-tumour regions identified 296 upregulated and 317 downregulated proteins in the tumour tissue including protein kinases, transcription factors and other proteins involved in biological processes such as apoptosis, immune response or in signalling. Taken together, this work showed for the first time that DIA-MS can be applied to the analysis of human tissue biopsies in order to generate digital proteome maps that are useful for molecular classification and identification of tumour-relevant biomarkers.
Breast cancer can be molecularly classified into five intrinsic subtypes (luminal A, luminal B consisting of Luminal B and Luminal B-like, Her2 enriched, normal-like and triple-negative).38,47 There have been several published MS-based studies focused on profiling the proteomic landscape of these molecular subtypes using conventional DDA approaches.7,48–50 DIA-MS has only recently been employed by Bouchal et al. to profile 96 breast cancer needle biopsies across four of the breast cancer subtypes (48 × Luminal A, 24 × Luminal B comprising 16 × Luminal B and 8 × Luminal B-like, 8 × Her2-enriched, 16 × triple-negative).22 In total, 2842 proteins were quantified across all samples and analysis of this data led to the identification of biological pathways which are enriched in each individual subtype. For instance, the authors showed that the nuclear factor kappa-B (NF-κB) pathway was upregulated in the luminal subtypes while an enrichment of vascular endothelial growth factor (VEGF) pathway components was found in Her2-positive subtypes (Luminal B-like, Her2-enriched). Subsequent statistical analysis of the subtype-specific proteomic maps resulted in the construction of a decision tree for subtype classification based on the expression levels of three proteins – receptor tyrosine-protein kinase erbB-2 (ERBB2) or Her2, inositol polyphosphate 4-phosphatase (INPP4B) and cyclin-dependent kinase 1 (CDK1). This decision tree correctly classified 84% samples from the original cohort of 96 samples into the appropriate molecular subtype. As an orthogonal validation, the authors extended the protein-based decision tree to evaluate the gene expression levels of ERBB2, INPP4B and CDK1 in published microarray and RNASeq datasets from 883 and 1078 breast cancer patients respectively, which confirmed the association of expression levels of these three genes with individual breast cancer subtypes.
Hepatocellular carcinoma (HCC) represents ∼90% of all liver cancers and due to the asymptomatic manifestation in the early stages, patients often present with advanced disease.51,52 The availability of curative therapy consisting of liver resection and transplantation for patients with early stage HCC increases the importance of identifying biomarkers for early detection.42,52 DIA-MS has been used in a small number of studies to characterise the biology of this disease and identify new protein-based diagnostic biomarkers of HCC.42–44 For instance, Gao et al. performed a comparative proteomic analysis on 14 matched pairs of HCC tumour and adjacent non-tumour tissue resections.42 In total, the authors quantified 4216 proteins and identified 191 upregulated and 147 downregulated proteins in tumour compared to adjacent normal tissue. Gene ontology and KEGG pathway enrichment analysis revealed a significant upregulation of the spliceosome pathway in HCC as well as a downregulation of 37 metabolic pathways including the metabolism of glycine, serine and sarcosine, metabolism of retinol and biosynthesis of antibiotics.42 Based on these observations, the authors selected 9 proteins for further validation by immunoblotting in an independent set of 6 matched HCC pairs which showed expression levels changes which were consistent with the DIA-MS data. In another study, Zhu et al. analysed 19 matched pairs of HCC and adjacent tissue samples and quantified 2579 proteins by DIA-MS with 541 differentially expressed proteins between HCC and adjacent tissue.43 A number of proteins from the heat-shock proteins (HSP) family as well as mitochondrial ribosomal proteins were found to be upregulated in tumour samples compared to the adjacent tissue. The authors focused on the DNA replication licensing factor MCM7 (MCM7), which was found by DIA-MS to be upregulated in tumour specimens, and further validated this observation by IHC in an additional series of three tumour and adjacent matched tissue specimens. The authors also separated HCC samples into two groups based on the serum alpha-fetoprotein (AFP) levels, which is an FDA approved serum marker to indicate risk for liver cancer and for early detection of HCC. A comparison of adjacent normal tissue and tumour regions in HCC cases with high levels of serum AFP (>20 ng ml−1) identified 419 upregulated and 192 downregulated proteins in the tumour specimens. Conversely, no significantly altered proteins were found in the cases with low serum AFP when tumour specimens were compared to adjacent normal tissue. While hypothesis generating in nature, these studies suggest that complex metabolic reprogramming may play a role in HCC and that there are protein alterations that are specific in high risk (high serum AFP) HCC that could potentially be developed as early detection biomarkers. These findings open new opportunities in drug development for therapy and biomarker validation in this difficult-to-treat disease.
One interesting area where DIA-MS has shown some success in biomarker discovery is in glycoproteomic analysis of tissue specimens. The glycoproteome is comprised of all N- and O-glycosylated proteins present in tissue and is thought to be more amenable to biomarker discovery due to their accessibility as cell surface or secreted proteins.53 In one example, Liu et al. characterised the N-glycoproteome in prostate cancer by utilising a combination of solid phase deglycosylation of peptides and DIA-MS.45 To achieve this, they developed a novel spectral library optimised for the human N-glycoproteome generated from multiple DDA-MS sources. In this study, the authors analysed 75 tissue specimens including 10 normal prostate samples, 40 prostate cancer samples and 25 metastatic prostate cancer samples. The aim of the study was to identify protein biomarkers associated with aggressive prostate cancer. Based on the histopathological staging of the tumours (using Gleason score), the authors further divided the prostate cancer specimens into two groups, namely non-aggressive (NAG, Gleason score = 6) and aggressive (AG, Gleason score = 7–9) prostate cancer. Overall 2188 N-glycosites were identified across all 4 pooled sample groups (normal, NAG, AG and metastatic) that enabled quantification of 897 distinct N-glycoproteins. Fifty glycoproteins were found to be significantly altered between NAG and AG which included the glycoproteins N-acylethanolamine-hydrolyzing acid amidase (NAAA) and protein tyrosine kinase 7 (PTK7) which was significantly decreased and increased in AG respectively.45 These proteins were further evaluated by IHC analysis in tissue microarrays (TMA) on an expanded cohort of 56 prostate cancer cases which showed that a combined panel of these two proteins was able to discriminate between AG and NAG. These data suggest that the NAAA and PTK7 glycoproteins may be candidate markers for staging of low-risk versus high-risk prostate cancer. However, given the relatively small single centre cohort used in this study, validation in larger multi-centre independent cohorts is required to further validate their clinical utility as robust biomarkers.
These exemplar studies demonstrate the utility of DIA-MS in the acquisition of biologically relevant protein profiles from small starting sample amounts such as biopsies. These profiles not only aid in the classification of the tumour samples into molecular and histological subtypes, they also shed light on the specific biological pathways that operate within individual cancer types which may be ultimately be useful for downstream functional investigation, drug discovery and biomarker development.
Unravelling mechanisms of therapy response
Therapy resistance remains one of the key challenges in cancer management today. The inherent intratumoural heterogeneity and plasticity in cancers results in the evolution of a wide spectrum of distinct and often unpredictable resistance mechanisms when patients receive chemo- and radio-therapy. Given that the majority of druggable targets in cells are proteins, proteomics remains a pivotal technology for characterising the molecular reprogramming that occurs upon drug treatment, especially in instances where the mechanisms of drug action are unknown.54–56 In addition, knowledge of the activation or suppression of individual biological pathways initiated by treatment may shed light on heterogeneity in the patient responses commonly seen in the clinic. The ability of DIA-MS to comprehensively analyse many different cell lines and tissue specimens reproducibly without missing data has immense potential for interrogating mechanisms of drug action or resistance in large cohorts. Linking this information with other clinic-pathological characteristics or underlying genetic information may result in the identification of predictive biomarkers of therapy response and help design new treatment strategies. Several key examples of DIA-MS applications in the study of cancer treatment response and resistance mechanisms are summarised in this section.57–60The NCI-60 panel comprises of 60 cancer cell lines from nine distinct tissue types. This panel is a preclinical workhorse for the cancer community and has been subjected to in-depth molecular (genomic and transcriptional) and pharmacological (over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 chemical compounds) profiling. Guo et al. employed DIA-MS to analyse the proteomic landscape of the NCI-60 panel and identified 3171 proteins across all cell lines.60 The authors then used univariate and multivariate regression analysis to evaluate drug response predictions of 224 pharmacological compounds either based on the DIA-MS data alone or integrated with genomic and transcriptional features. Interrogating existing data available in CellMiner, they showed that the proteomic data contributed to a higher percentage of drug response prediction features (12%) that those derived from DNA mutations (2%) and RNA transcripts (6%). They further showed that the response of 49 screened drugs were best predicted by DIA-MS data while response to 83 compounds had optimal predictive power when combining DIA-MS data with transcript and mutational data. Notably, the authors found that the protein expression levels of multiple ATP-binding cassette family transporters were strongly associated with response to cancer drugs across several classes, including alkylating agents, histone deacetylase inhibitors and kinase inhibitors. This result underscores the importance of this family of transporters as a putative mechanism of drug response and their use as candidate biomarkers for optimisation of cancer therapy. The authors further demonstrated that the predictive power of the regression models based on DIA-MS data was generally higher compared to the models using DDA data61 due to the better quantitative accuracy and data consistency of the DIA-MS dataset. This study highlights the role that DIA-MS can play important role in the burgeoning field of pharmacoproteomics where protein level measurements not only enable deep insights into mechanisms of drug action but may also lead to predictive biomarkers of therapy response.
000 chemical compounds) profiling. Guo et al. employed DIA-MS to analyse the proteomic landscape of the NCI-60 panel and identified 3171 proteins across all cell lines.60 The authors then used univariate and multivariate regression analysis to evaluate drug response predictions of 224 pharmacological compounds either based on the DIA-MS data alone or integrated with genomic and transcriptional features. Interrogating existing data available in CellMiner, they showed that the proteomic data contributed to a higher percentage of drug response prediction features (12%) that those derived from DNA mutations (2%) and RNA transcripts (6%). They further showed that the response of 49 screened drugs were best predicted by DIA-MS data while response to 83 compounds had optimal predictive power when combining DIA-MS data with transcript and mutational data. Notably, the authors found that the protein expression levels of multiple ATP-binding cassette family transporters were strongly associated with response to cancer drugs across several classes, including alkylating agents, histone deacetylase inhibitors and kinase inhibitors. This result underscores the importance of this family of transporters as a putative mechanism of drug response and their use as candidate biomarkers for optimisation of cancer therapy. The authors further demonstrated that the predictive power of the regression models based on DIA-MS data was generally higher compared to the models using DDA data61 due to the better quantitative accuracy and data consistency of the DIA-MS dataset. This study highlights the role that DIA-MS can play important role in the burgeoning field of pharmacoproteomics where protein level measurements not only enable deep insights into mechanisms of drug action but may also lead to predictive biomarkers of therapy response.
Commercial immortalised cell lines such as those in the NCI-60 panel have been subjected to decades of cell culture and thus may not retain many of the molecular features present in the tumours from which they were originally derived. In recent years, there has been a push towards the development of patient-derived models for preclinical cancer research. These models encompass patient-derived xenografts, organoids or tumour explants and are thought to better recapitulate the human disease.62,63 DIA-MS has been used as a characterisation tool to profile such models to identify clinical response mechanisms of drug action. One example is the study undertaken by Nguyen et al., who employed prostate cancer patient-derived explants obtained from men undergoing radical prostatectomy to study tumour-specific response to treatment with heat shock protein 90 (HSP90) inhibitors 17-AAG and AUY922.58 The use of fresh tumour specimens from different patients was important in modelling the heterogeneity inherent in prostate cancer and highlight any conserved mechanisms of treatment response found across all patients. Proteomic analysis identified a consistent downregulation of 44 proteins involved in pathways associated with mRNA translation, ribosome function and RNA metabolism. Conversely, 54 proteins were found to be increased with drug treatment with an enrichment of tricarboxylic acid metabolism components. Despite the heterogeneity amongst the 46 cases examined, the authors were remarkably able to identify 9 proteins that were universally downregulated by AUY922 treatment, including two proteins from the HIF-1 pathway, transferrin receptor protein 1 (TRFC) and metalloproteinase inhibitor 1 (TIMP1), which could serve as candidate markers of drug response. This study provides proof-of-principle evidence for the use of DIA-MS profiling in patient-derived models and brings the field one step closer to implementing this next generation proteomic strategy in precision cancer medicine.
Another interesting area of research is the design of window of opportunity studies to better understand mechanisms of therapy response and resistance.64 Such studies involve the sampling of tumour tissue prior to and after the treatment of interest for thorough pharmacodynamic assessment. In addition to chemotherapy and surgery, radiotherapy is the mainstay local treatment in a wide array of different cancer types including prostate cancer. To investigate the major cellular pathways that are regulated following the use of radiotherapy, Keam et al. performed DIA-MS based proteomic profiling of matched tissue biopsies collected at pre-treatment and 14 days post brachytherapy from 8 prostate cancer patients.57 The authors found that out of >5000 proteins identified, 24 proteins and 3 proteins were consistently up- or down-regulated post radiation respectively in all patients. The authors also identified a number of upregulated pathways in the post-radiation samples including wound healing, extracellular matrix remodelling and acute inflammatory response. These biological processes are consistent with tissue deposition and remodelling associated with radiation response. One of the limitations of this study is that it is descriptive in nature and lacks any clinical response and patient outcome data which restricts the ability to define proteins associated with brachytherapy response. Nonetheless, the identification of a number of candidate proteins which are universally regulated as a result of radiotherapy provides a useful resource for future studies elucidating their mechanistic role in radiotherapy response and resistance.
Collectively, the aforementioned examples demonstrate that DIA-MS is a useful tool for the investigation of how therapeutic interventions impact the proteomic landscape in cell lines, patient-derived models and human tissue and thus refines our current understanding of treatment responses at the molecular level. Such correlative studies can aid in revealing putative mechanisms of drug resistance and identify novel response markers to both chemotherapy and radiotherapy for subsequent functional and clinical evaluation.
Multi-omic and integrative analysis
With the dramatic decrease in the cost of genomic and transcriptomic profiling over the past decade, there has been a push towards integrative multi-omic analysis as a means to provide a holistic view of the molecular landscape of cancer. The underlying basis for this integrative approach is the idea that multiple orthogonal measurements of the same tumour specimens may reveal new findings that are likely to be masked due to the inherent limitations of any single type of Omic measurement. This is a burgeoning field for DIA-MS and here we provide some recent examples of how this methodology has been integrated with other Omics modalities in cancer research.Castration resistant prostate cancer (CRPC) is a chemoresistant form of prostate cancer that is unresponsive to androgen-deprivation therapy.65 Currently there are no alternative treatment options available for CRPC patients.66,67 To study the genomic, transcriptomic and proteomic changes during different stages of prostate cancer disease progression, Latonen et al. undertook an integrative multi-omic study of 11 tumour specimens from CRPC patients and compared them to profiles obtained from 17 untreated prostate cancer (PC) and 10 benign prostate hyperplasia (BPH) tissue specimens.66 Using DIA-MS, the authors quantified 3394 proteins across all samples and identified 382 and 728 differentially expressed proteins between CRPC and PC samples and PC and BPH samples, respectively. A comparison of the acquired proteomic dataset with the copy number and transcriptomic data obtained from the same specimens revealed a poor correlation between genomic, transcriptomic and proteomic measures. The authors hypothesized that this discrepancy may be due to alterations in the levels of cellular microRNA (miRNA) which can either directly lead to the degradation of mRNA targets or block the protein translation process by binding to mRNA and forming mRNA/miRNA complexes. Such complexes may alter levels of the expressed protein without affecting the overall mRNA levels of the coding gene.68 To test this hypothesis, the authors undertook miRNA sequencing and identified 95 differentially expressed miRNAs between PC and CRPC samples and these miRNAs have the potential to target almost 500 genes. From this list of potential gene targets, only 24% were differentially expressed between PC and CRPC at the mRNA level, while 45% were differentially expressed at the protein level supporting the concept that miRNAs may decrease protein levels but not the corresponding mRNA levels of the same gene target. To validate this, the authors focused on miR-22 and miR-493 that were differentially expressed between PC and CRPC and transfected them into PC-3 prostate cancer cells. The mRNA levels of the miRNA targets Endonuclease domain containing 1 (ENDOD1) and Golgi membrane protein 1 (GOLM1) were significantly decreased in the transfected cells while miRNA targets KH-type splicing regulatory protein (KHRSP1) and dynamin 1-like protein (DNML1) showed no change on the mRNA level but displayed decreased protein expression levels. In a second example, the authors identified two miRNAs (miR-22 and miR-205) with the potential to target malate dehydrogenase (MDH2). DIA-MS and RT-qPCR analysis of PC-3 cells transfected with these miRNAs revealed a decrease in MDH2 protein levels but no change in MDH2 mRNA levels. This comprehensive study demonstrates capability of DIA-MS to reveal novel insights into the regulation of gene expression in therapy resistant prostate cancer when integrated as part of multi-omic investigation.
In another example, Schwarzfischer et al. performed an integrative metabolomic and proteomic analysis of two forms of high-grade non-Hodgkin lymphomas, Burkitt's lymphoma (BL) and Diffuse large B-cell lymphoma (DLBCL).69 Metabolomic analysis of 24 lymphoma cell lines (6 BL and 18 DLBCL) identified increased intracellular levels of pyruvic acid in DLBCL compared to BL as well as higher secretion of pyruvate by DLBCL cell lines. Higher levels of pyruvate were also detected in 6 DLBCL cryopreserved tumour tissue samples when compared to 5 BL tumours. Pyruvate is a key intermediate energy metabolism and a central intersection for a number of vital metabolic pathways. To test whether the difference in pyruvate levels observed in the metabolic studies is reflected by alterations in proteins involved in specific metabolic pathways, the authors performed proteomic analysis of 11 lymphoma cell lines (5 × BL and 6 × DLBCL), 11 fresh-frozen and 13 formalin-fixed paraffin-embedded (FFPE) tissue samples. DIA-MS analysis of the lymphoma cell lines revealed a downregulation of proteins involved in pyruvate metabolism, glycolysis and oxidative phosphorylation pathways in BL compared to DLBCL. For instance, key glycolytic enzymes such as hexokinase (HXK1) and phosphoglycerate kinase (PGK1) were significantly downregulated in BL. In contrast, an upregulation of lactate dehydrogenase (LDH1), phosphoglycerate dehydrogenase (PHGDH) and phosphoserine aminotransferase (PSAT1) in BL suggests that the metabolism of glucose using non-oxidative phosphorylation and the one carbon metabolic pathway may be the predominant processes operating in this disease. The differences in expression levels of the key enzymes described above in BL and DLBCL were further confirmed by proteomic analysis of the fresh-frozen and FFPE tissue samples. This study underscores the important complementary role that DIA-MS has in the interpretation of metabolomics data and highlights the power of this integrative approach in revealing new insights into the complex metabolic reprogramming underlying the development of non-Hodgkin lymphoma.
Recent studies employing integration of orthogonal MS strategies to sample different facets of tumour biology have also been promising. For instance, Mehnert et al. developed a multi-layered proteomic approach to study effects of different mutations of Dual specificity tyrosine-phosphorylation-regulated kinase 2 (Dyrk2) on protein topology, protein–protein interactions (PPI) and global proteomic and phosphoproteomic profiles.70 Through interactions with the EDVP E3 ubiquitin ligase complex, Dyrk2 plays a key role in cell cycle and apoptosis and has been identified as both a putative tumour suppressor and oncogene.71,72 Based on published data, the authors generated a series of cancer-associated Dyrk2 mutants which were expressed in HEK293 cells. Analysis of the PPI networks by affinity purification-mass spectrometry (AP-MS) identified mutation-specific reorganization of the Dyrk2 PPI network in truncated and catalytically inactive mutants of this protein. MS-based quantitative crosslinking analysis revealed topological changes in the Dyrk2 structure as well as a decrease in Dyrk2 phosphorylation status particularly in the truncated and catalytically inactive mutants. To explore the broader effects of Dyrk2 mutations on the proteome, the authors employed DIA-MS for proteomic and phosphoproteomic analysis of the HEK293 mutant cell lines. When combined with the PPI AP-MS data, this workflow showed that a subset of Dyrk2 mutants modulated multiple proteins annotated as Cancer Driver Proteins in Cancer Gene Census catalogue, suggesting that these Dyrk2 cancer-associated mutations have the potential to contribute to cancer progression. This study highlights the power of combining orthogonal MS-based strategies with DIA-MS to deliver multi-scale molecular information to dissect the functional roles of oncogenes and tumour suppressors.
These examples provide proof-of-principle that DIA-MS can be an integral part of proteogenomic or metaboproteomic analysis of tissue samples and cell lines and we anticipate the use of such comprehensive integrative studies will continue to grow and ultimately become a routine toolkit in cancer research.
Looking ahead: harnessing the latest innovations in DIA-MS technology
Despite the clear promise of DIA-MS, as with all other technologies, further innovations in instrumentation and informatics will be key to pushing the boundaries of improved sensitivity and throughput. In this section, we delve into some of the latest innovations in DIA-MS technology which are likely to have a direct impact on future applications in oncology.New developments in data acquisition and MS instrumentation
Some notable technological advances in data acquisition and MS instrumentation include the development of scanning quadrupole isolation (SONAR, scanningSWATH), high field asymmetric waveform ion mobility spectrometry (FAIMS) and parallel accumulation – serial fragmentation (diaPASEF).73–79 Scanning quadrupole isolation is a novel method where fixed precursor isolation windows are replaced by a single isolation window that periodically slides through the entire MS1 range.73,74 The main advantage of this approach is the improvement in acquisition speed as the sliding scan can be completed more rapidly than the conventional method of scanning through fixed isolation windows. This set-up allows for the use of shorter LC gradients and higher flow rates resulting in an up to 3 times increased overall sample throughput.74 FAIMS is an ion selection and separation device that can be used as an interface between the ion source and orifice of the mass spectrometer.76 FAIMS uses a combination of oscillating high and low electric fields to focus the ions generated by the source which results in a reduction of chemical noise (neutral particles) and the removal of interfering ions. In this manner, FAIMS further improves the sensitivity, robustness and reproducibility of DIA-MS quantification.75 Rapid improvements in DIA-MS sensitivity has also been achieved by diaPASEF. This method is enabled by the development of trapped ion mobility spectrometry (TIMS).77,79,80 TIMS technology allows the separation of ions in the gas phase based on their size and shape and in diaPASEF, two TIMS regions in the mass spectrometer are employed. The first TIMS region (TIMS1) is used for the accumulation of the precursor ions which are later released into the second TIMS region (TIMS2) to be separated by their size and shape prior to release and fragmentation. In parallel to the ion separation in TIMS2, TIMS1 is accumulating a new set of precursor ions, which rapidly improves the ion sampling efficiency from ∼3% used in conventional DIA-MS to nearly 100%. This brings about a dramatic increase in sensitivity as demonstrated by Meier at al. who identified more than 4000 proteins from as little as 10 ng of input protein extract from HeLa cells.79 In addition, diaPASEF offers 10 times faster sequencing speed which is advantageous for rapid MS analysis and increased throughput.78,79 Collectively, these advances in data acquisition and instrumentation dramatically improve the speed and sensitivity of DIA-MS analysis leading to enhanced proteomic depth and sample throughput critical for large-scale biological studies.Advances in informatics strategies for DIA-MS data processing
As indicated in earlier sections, one of the main drawbacks of the DIA-MS is the need to generate spectral libraries from DDA-MS experiments prior to undertaking any investigation. To address this limitation, there has been a strong interest in the development of algorithms that generate in silico spectral libraries which dispense with the requirement for experimentally-derived libraries.81–84 These tools can be divided into two categories. The first category is spectrum-centric algorithms (DIA-Umpire, Group-DIA) which use the intensity profiles of co-eluting fragments and precursors from a DIA-MS experiment to generate “pseudo-MS/MS” spectra that can then be searched against a database of in silico digested protein sequences in a similar manner as conventional DDA-MS workflows.81,82 The second category is peptide-centric algorithms (PECAN, DIA-NN) where in silico spectral libraries are simulated from protein sequence databases and subsequently queried against DIA-MS datasets.83,84 Recent advances in computational modelling based on deep learning-based methods have further improved the quality of in silico libraries for peptide-centric data processing. For instance, deep neural networks have been used in tools such as Prosit or DeepDIA to train model spectral libraries based on large DDA datasets, which lead to highly accurate predictions of peptide fragmentation patterns and chromatographic retention times.85,86 It has been shown that these two categories of data processing tools result in the reliable identification and quantification of thousands of proteins. For instance, a comparison of the spectrum-centric algorithm DIA-Umpire with conventional processing tools demonstrated good agreement in protein quantification (R2 > 0.76) with a slight ∼10–15% decrease in protein and peptide identification.87 It should be noted that these in silico approaches are not mutually exclusive from experimentally-derived spectral libraries and can be readily used to augment existing spectral libraries to further increase the depth of protein coverage in retrospective analysis of DIA-MS datasets.Another limitation of DIA-MS is that the complex mass spectra arising from this methodology is compounded when a short chromatographic separation is applied in order to increase sample throughput. The reason for this increased complexity is due to the lower number of data points during acquisition in combination with very high number of co-eluting peptides. The resulting complex spectra poses significant challenges for deconvolution with conventional data processing platforms. To address this challenge, machine learning algorithms have been exploited to distinguish real signals from interfering background.19,84 A very recent innovation in this area is the development of the DIA-NN algorithm which uses deep neural networks to improve proteome coverage in DIA-MS data analysis.84 Demichev et al., compared the performance of DIA-NN to conventional platforms such as Spectronaut, Skyline and OpenSWATH. In a 30 minute DIA-MS experiment, DIA-NN identified more precursors than Spectronaut and Skyline at the same false discovery rate (FDR) threshold, while OpenSWATH failed to process the data. Moreover, DIA-NN identified more precursors in a 30 minute experiment compared to Skyline and OpenSWATH in 60 minute experiment using the same FDR threshold. Such novel approaches could enable a step-change in the translation of DIA-MS into the clinical setting where fast and reliable analysis may be necessary for applications in personalised cancer medicine.
Conclusion
Omic technologies are rapidly changing the conventional way of cancer classification, biomarker discovery and drug development. As a next-generation proteomic method, DIA-MS can reproducibly quantify thousands of proteins from a single tissue section or biopsy opening new frontiers in large-scale oncology studies with minimal sample requirements. Such studies can offer the statistical power to identify proteomic signatures for molecular subtypes and provide a more comprehensive description of the underlying cancer biology. In addition, DIA-MS can both complement and be integrated with genomic, transcriptomic or metabolomic analyses. The results of such integrative multi-omic studies may be imperative for the development of more accurate predictive and prognostic biomarkers in oncology where any one analyte is likely to be insufficient to fully describe the complexity inherent in cancer development and progression. We anticipate that new developments in DIA-MS technology and machine learning–based algorithms will usher a new era of rapid, in-depth and reproducible proteomic measurements that are compatible with clinical diagnostic workflows and will ultimately facilitate the delivery of precision cancer medicine to achieve better patient outcomes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Our laboratory is supported by grants from the Institute of Cancer Research, Cancer Research UK and Breast Cancer Now.References
- F. Sanchez-Vega, M. Mina, J. Armenia, W. K. Chatila, A. Luna and K. C. La, et al., Oncogenic Signaling Pathways in The Cancer Genome Atlas, Cell, 2018, 173(2), 321–337 CrossRef CAS.
- P. J. Campbell, G. Getz, J. O. Korbel, J. M. Stuart, J. L. Jennings and L. D. Stein, et al., Pan-cancer analysis of whole genomes, Nature, 2020, 578(7793), 82–93 CrossRef.
- M. F. Berger and E. R. Mardis, The emerging clinical relevance of genomics in cancer medicine, Nat. Rev. Clin. Oncol., 2018, 15(6), 353–365 CrossRef CAS.
- J. N. Weinstein, E. A. Collisson, G. B. Mills, K. R. M. Shaw, B. A. Ozenberger and K. Ellrott, et al., The Cancer Genome Atlas Pan-Cancer analysis project, Nat. Genet., 2013, 45(10), 1113–1120 CrossRef.
- M. H. Bailey, C. Tokheim, E. Porta-Pardo, S. Sengupta, D. Bertrand and A. Weerasinghe, et al., Comprehensive Characterization of Cancer Driver Genes and Mutations, Cell, 2018, 173(2), 371–385 CrossRef CAS.
- M. J. Ellis, M. Gillette, S. A. Carr, A. G. Paulovich, R. D. Smith and K. K. Rodland, et al., Connecting Genomic Alterations to Cancer Biology with Proteomics: The NCI Clinical Proteomic Tumor Analysis Consortium, Cancer Discovery, 2013, 3(10), 1108–1112 CrossRef CAS.
- P. Mertins, D. R. Mani, K. V. Ruggles, M. A. Gillette, K. R. Clauser and P. Wang, et al., Proteogenomics connects somatic mutations to signalling in breast cancer, Nature, 2016, 534(7605), 55–62 CrossRef CAS.
- B. Zhang, J. Wang, X. Wang, J. Zhu, Q. Liu and Z. Shi, et al., Proteogenomic characterization of human colon and rectal cancer, Nature, 2014, 513(7518), 382–387 CrossRef CAS.
- H. Zhang, T. Liu, Z. Zhang, S. H. Payne, B. Zhang and J. E. McDermott, et al., Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer, Cell, 2016, 166(3), 755–765 CrossRef CAS.
- J. F. Timms, O. J. Hale and R. Cramer, Advances in mass spectrometry-based cancer research and analysis: from cancer proteomics to clinical diagnostics, Expert Rev. Proteomics, 2016, 13(6), 593–607 CrossRef CAS.
- W. C. Cho, Mass spectrometry-based proteomics in cancer research, Expert Rev. Proteomics, 2017, 14(9), 725–727 CrossRef CAS.
- B. Zhang, J. R. Whiteaker, A. N. Hoofnagle, G. S. Baird, K. D. Rodland and A. G. Paulovich, Clinical potential of mass spectrometry-based proteogenomics, Nat. Rev. Clin. Oncol., 2019, 16(4), 256–268 CrossRef.
- L. C. Gillet, P. Navarro, S. Tate, H. Rost, N. Selevsek and L. Reiter, et al., Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis, Mol. Cell. Proteomics, 2012, 11(6), O111.016717 CrossRef.
- M. Pernemalm and J. Lehtio, Mass spectrometry-based plasma proteomics: state of the art and future outlook, Expert Rev. Proteomics, 2014, 11(4), 431–448 CrossRef CAS.
- P. Ghodasara, P. Sadowski, N. Satake, S. Kopp and P. C. Mills, Clinical veterinary proteomics: Techniques and approaches to decipher the animal plasma proteome, Vet. J., 2017, 230, 6–12 CrossRef CAS.
- D. Pascovici, J. X. Wu, M. J. McKay, C. Joseph, Z. Noor and K. Kamath, et al., Clinically Relevant Post-Translational Modification Analyses-Maturing Workflows and Bioinformatics Tools, Int. J. Mol. Sci., 2019, 20(1), 16 CrossRef.
- C. Ludwig, L. Gillet, G. Rosenberger, S. Amon, B. Collins and R. Aebersold, Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial, Mol. Syst. Biol., 2018, 14(8), e8126 CrossRef.
- K. Barkovits, S. Pacharra, K. Pfeiffer, S. Steinbach, M. Eisenacher and K. Marcus, et al., Reproducibility, Specificity and Accuracy of Relative Quantification Using Spectral Library-based Data-independent Acquisition, Mol. Cell. Proteomics, 2020, 19, 181–197 CrossRef CAS.
- R. Bruderer, O. M. Bernhardt, T. Gandhi, S. M. Miladinovic, L. Y. Cheng and S. Messner, et al., Extending the Limits of Quantitative Proteome Profiling with Data-Independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues, Mol. Cell. Proteomics, 2015, 14(5), 1400–1410 CrossRef CAS.
- O. T. Schubert, L. C. Gillet, B. C. Collins, P. Navarro, G. Rosenberger and W. E. Wolski, et al., Building high-quality assay libraries for targeted analysis of SWATH MS data, Nat. Protoc., 2015, 10(3), 426–441 CrossRef CAS.
- L. M. von Ziegler, N. Selevsek, R. Y. Tweedie-Cullen, E. Kremer and I. M. Mansuy, Subregion-Specific Proteomic Signature in the Hippocampus for Recognition Processes in Adult Mice, Cell Rep., 2018, 22(12), 3362–3374 CrossRef CAS.
- P. Bouchal, O. T. Schubert, J. Faktor, L. Capkova, H. Imrichova and K. Zoufalova, et al., Breast Cancer Classification Based on Proteotypes Obtained by SWATH Mass Spectrometry, Cell Rep., 2019, 28(3), 832–843 CrossRef CAS.
- S. Arya, D. Wiatrek-Moumoulidis, S. A. Synowsky, S. L. Shirran, C. H. Botting and S. J. Powis, et al., Quantitative proteomic changes in LPS-activated monocyte-derived dendritic cells: A SWATH-MS study, Sci. Rep., 2019, 9(1), 4343 CrossRef.
- G. Rosenberger, C. C. Koh, T. N. Guo, H. L. Rost, P. Kouvonen and B. Collins, et al., A repository of assays to quantify 10,000 human proteins by SWATH-MS, Sci. Data, 2014, 1, 140031 CrossRef CAS.
- C. Q. Zhong, J. F. Wu, X. F. Qiu, X. Chen, C. C. Xie and J. H. Han, Generation of a murine SWATH-MS spectral library to quantify more than 11,000 proteins, Sci. Data, 2020, 7(1), 104 CrossRef CAS.
- L. Krasny, P. Bland, J. Burns, N. C. Lima, P. T. Harrison and L. Pacini, et al., A mouse SWATH-MS reference spectral library enables deconvolution of species-specific proteomic alterations in human tumour xenografts, Dis. Models Mech., 2020, 13(7), dmm044586 CrossRef CAS.
- P. Blattmann, V. Stutz, G. Lizzo, J. Richard, P. Gut and R. Aebersold, Generation of a zebrafish SWATH-MS spectral library to quantify 10,000 proteins, Sci. Data, 2019, 6, 190011 CrossRef CAS.
- B. Fabre, D. Korona, C. I. Mata, H. T. Parsons, M. J. Deery and M. L. A. T. M. Hertog, et al., Spectral Libraries for SWATH-MS Assays for Drosophila melanogaster and Solanum lycopersicum, Proteomics, 2017, 17(21), 1700216 CrossRef.
- P. Picotti, M. Clement-Ziza, H. Lam, D. S. Campbell, A. Schmidt and E. W. Deutsch, et al., A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis, Nature, 2013, 494(7436), 266–270 CrossRef CAS.
- S. Michalik, M. Depke, A. Murr, M. G. Salazar, U. Kusebauch and Z. Sun, et al., A global Staphylococcus aureus proteome resource applied to the in vivo characterization of hostpathogen interactions, Sci. Rep., 2017, 7(1), 9718 CrossRef.
- D. B. Muller, O. T. Schubert, H. Rost, R. Aebersold and J. A. Vorholt, Systems-level Proteomics of Two Ubiquitous Leaf Commensals Reveals Complementary Adaptive Traits for Phyllosphere Colonization, Mol. Cell. Proteomics, 2016, 15(10), 3256–3269 CrossRef CAS.
- O. T. Schubert, C. Ludwig, M. Kogadeeva, M. Zimmermann, G. Rosenberger and M. Gengenbacher, et al., Absolute Proteome Composition and Dynamics during Dormancy and Resuscitation of Mycobacterium tuberculosis, Cell Host Microbe, 2015, 18(1), 96–108 CrossRef CAS.
- B. C. Collins, C. L. Hunter, Y. S. Liu, B. Schilling, G. Rosenberger and S. L. Bader, et al., Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry, Nat. Commun., 2017, 8(1), 291 CrossRef.
- D. L. Tabb, L. Vega-Montoto, P. A. Rudnick, A. M. Variyath, A. J. L. Ham and D. M. Bunk, et al., Repeatability and Reproducibility in Proteomic Identifications by Liquid Chromatography-Tandem Mass Spectrometry, J. Proteome Res., 2010, 9(2), 761–776 CrossRef CAS.
- A. Michalski, J. Cox and M. Mann, More than 100,000 Detectable Peptide Species Elute in Single Shotgun Proteomics Runs but the Majority is Inaccessible to Data-Dependent LC-MS/MS, J. Proteome Res., 2011, 10(4), 1785–1793 CrossRef CAS.
- Z. L. Ye, Y. Mao, H. Clausen and S. Y. Vakhrushev, Glyco-DIA: a method for quantitative O-glycoproteomics with in silico-boosted glycopeptide libraries, Nat. Methods, 2019, 16(9), 902–910 CrossRef CAS.
- L. Krasny, P. Bland, N. Kogata, P. Wai, B. A. Howard and R. C. Natrajan, et al., SWATH mass spectrometry as a tool for quantitative profiling of the matrisome, J. Proteomics, 2018, 189, 11–22 CrossRef CAS.
- N. Eliyatkin, E. Yalcin, B. Zengel, S. Aktas and E. Vardar, Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to A New Age, and A New Way, J. Breast Health, 2015, 11(2), 59–66 CrossRef.
- J. J. Berman, Tumor classification: molecular analysis meets Aristotle, BMC Cancer, 2004, 4, 10 CrossRef.
- S. Zhu, W. Yu, X. Yang, C. Wu and F. Cheng, Traditional Classification and Novel Subtyping Systems for Bladder Cancer, Front. Oncol., 2020, 10, 102 CrossRef.
- T. N. Guo, P. Kouvonen, C. C. Koh, L. C. Gillet, W. E. Wolski and H. L. Rost, et al., Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps, Nat. Med., 2015, 21(4), 407–413 CrossRef CAS.
- Y. Y. Gao, X. Z. Wang, Z. H. Sang, Z. C. Li, F. Liu and J. Mao, et al., Quantitative proteomics by SWATH-MS reveals sophisticated metabolic reprogramming in hepatocellular carcinoma tissues, Sci. Rep., 2017, 7, 45913 CrossRef CAS.
- Y. Zhu, J. Zhu, C. Lu, Q. S. Zhang, W. Xie and P. Sun, et al., Identification of Protein Abundance Changes in Hepatocellular Carcinoma Tissues Using PCT-SWATH, Proteomics Clin. Appl., 2019, 13(1), e1700179 CrossRef.
- T. Sajic, R. Ciuffa, V. Lemos, P. Xu, V. Leone and C. Li, et al., A new class of protein biomarkers based on subcellular distribution: application to a mouse liver cancer model, Sci. Rep., 2019, 9(1), 6913 CrossRef.
- Y. S. Liu, J. Chen, A. Sethi, Q. K. Li, L. J. Chen and B. Collins, et al., Glycoproteomic Analysis of Prostate Cancer Tissues by SWATH Mass Spectrometry Discovers N-acylethanolamine Acid Amidase and Protein Tyrosine Kinase 7 as Signatures for Tumor Aggressiveness, Mol. Cell. Proteomics, 2014, 13(7), 1753–1768 CrossRef CAS.
- F. Algaba, H. Akaza, A. Lopez-Beltran, G. Martignoni, H. Moch and R. Montironi, et al., Current Pathology Keys of Renal Cell Carcinoma, Eur. Urol., 2011, 60(4), 634–643 CrossRef.
- C. M. Perou, T. Sorlie, M. B. Eisen, M. van de Rijn, S. S. Jeffrey and C. A. Rees, et al., Molecular portraits of human breast tumours, Nature, 2000, 406(6797), 747–752 CrossRef CAS.
- H. J. Johansson, F. Socciarelli, N. M. Vacanti, M. H. Haugen, Y. Zhu and I. Siavelis, et al., Breast cancer quantitative proteome and proteogenomic landscape, Nat. Commun., 2019, 10(1), 1600 CrossRef.
- S. Tyanova, R. Albrechtsen, P. Kronqvist, J. Cox, M. Mann and T. Geiger, Proteomic maps of breast cancer subtypes, Nat. Commun., 2016, 7, 10259 CrossRef CAS.
- J. J. Kennedy, S. E. Abbatiello, K. Kim, P. Yan, J. R. Whiteaker and C. W. Lin, et al., Demonstrating the feasibility of large-scale development of standardized assays to quantify human proteins, Nat. Methods, 2014, 11(2), 149–155 CrossRef CAS.
- J. M. Llovet, J. Zucman-Rossi, E. Pikarsky, B. Sangro, M. Schwartz and M. Sherman, et al., Hepatocellular carcinoma, Nat. Rev. Dis. Primers, 2016, 2, 16018 CrossRef.
- J. M. Llovet, R. Lencioni, A. M. Di Bisceglie, P. R. Gaile, J. F. Dufour and T. F. Greten, et al., EASL-EORTC Clinical Practice Guidelines: Management of hepatocellular carcinoma, J. Hepatol., 2012, 56(4), 908–943 CrossRef.
- A. Kirwan, M. Utratna, M. E. O’Dwyer, L. Joshi and M. Kilcoyne, Glycosylation-Based Serum Biomarkers for Cancer Diagnostics and Prognostics, BioMed Res. Int., 2015, 490531 Search PubMed.
- M. Frantzi, A. Latosinska and H. Mischak, Proteomics in Drug Development: The Dawn of a New Era?, Proteomics Clin. Appl., 2019, 13(2), 1800087 CrossRef CAS.
- Y. An, L. Zhou, Z. Huang, E. C. Nice, H. Zhang and C. Huang, Molecular insights into cancer drug resistance from a proteomics perspective, Expert Rev. Proteomics, 2019, 16(5), 413–429 CrossRef CAS.
- G. Roti and K. Stegmaier, Genetic and proteomic approaches to identify cancer drug targets, Br. J. Cancer, 2012, 106(2), 254–261 CrossRef CAS.
- S. P. Keam, T. Gulati, C. Gamell, F. Caramia, C. Huang and R. B. Schittenhelm, et al., Exploring the oncoproteomic response of human prostate cancer to therapeutic radiation using data-independent acquisition (DIA) mass spectrometry, Prostate, 2018, 78(8), 563–575 CrossRef CAS.
- E. V. Nguyen, M. M. Centenera, M. Moldovan, R. Das, S. Irani and A. D. Vincent, et al., Identification of Novel Response and Predictive Biomarkers to Hsp90 Inhibitors Through Proteomic Profiling of Patient-derived Prostate Tumor Explants, Mol. Cell. Proteomics, 2018, 17(8), 1470–1486 CrossRef CAS.
- M. Garrido-Rodriguez, I. Ortea, M. A. Calzado, E. Munoz and V. Garcia, SWATH proteomic profiling of prostate cancer cells identifies NUSAP1 as a potential molecular target for Galiellalactone, J. Proteomics, 2019, 193, 217–229 CrossRef CAS.
- T. N. Guo, A. Luna, V. N. Rajapakse, C. C. Koh, Z. C. Wu and W. Liu, et al., Quantitative Proteome Landscape of the NCI-60 Cancer Cell Lines, iScience, 2019, 21, 664–680 CrossRef CAS.
- A. M. Gholami, H. Hahne, Z. X. Wu, F. J. Auer, C. Meng and M. Wilhelm, et al., Global Proteome Analysis of the NCI-60 Cell Line Panel, Cell Rep., 2013, 4(3), 609–620 CrossRef CAS.
- M. Bleijs, M. van de Wetering, H. Clevers and J. Drost, Xenograft and organoid model systems in cancer research, EMBO J., 2019, 38(15), e101654 CrossRef.
- G. P. Risbridger, R. Toivanen and R. A. Taylor, Preclinical Models of Prostate Cancer: Patient-Derived Xenografts, Organoids, and Other Explant Models, Cold Spring Harbor Perspect. Med., 2018, 8(8), a030536 CrossRef.
- S. Schmitz, F. Duhoux and J.-P. Machiels, Window of opportunity studies: Do they fulfil our expectations?, Cancer Treat. Rev., 2016, 43, 50–57 CrossRef.
- S. J. Hotte and F. Saad, Current management of castrate-resistant prostate cancer, Curr. Oncol., 2010, 17, S72–S79 Search PubMed.
- L. Latonen, E. Afyounian, A. Jylha, J. Nattinen, U. Aapola and M. Annala, et al., Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression, Nat. Commun., 2018, 9(1), 1176 CrossRef.
- Y. N. S. Wong, R. Ferraldeschi, G. Attard and J. de Bono, Evolution of androgen receptor targeted therapy for advanced prostate cancer, Nat. Rev. Clin. Oncol., 2014, 11(6), 365–376 CrossRef CAS.
- L. He and G. J. Hannon, Micrornas: Small RNAs with a big role in gene regulation, Nat. Rev. Genet., 2004, 5(7), 522–531 CrossRef CAS.
- P. Schwarzfischer, J. Reinders, K. Dettmer, K. Kleo, L. Dimitrova and M. Hummel, et al., Comprehensive Metaboproteomics of Burkitt's and Diffuse Large B-Cell Lymphoma Cell Lines and Primary Tumor Tissues Reveals Distinct Differences in Pyruvate Content and Metabolism, J. Proteome Res., 2017, 16(3), 1105–1120 CrossRef CAS.
- M. Mehnert, R. Ciuffa, F. Frommelt, F. Uliana, A. van Drogen and K. Ruminski, et al., Multi-layered proteomic analyses decode compositional and functional effects of cancer mutations on kinase complexes, Nat. Commun., 2020, 11(1), 3563 CrossRef CAS.
- R. Mimoto, N. Taira, H. Takahashi, T. Yamaguchi, M. Okabe and K. Uchida, et al., DYRK2 controls the epithelial-mesenchymal transition in breast cancer by degrading Snail, Cancer Lett., 2013, 339(2), 214–225 CrossRef CAS.
- P. Stephens, S. Edkins, H. Davies, C. Greenman, C. Cox and C. Hunter, et al., A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer, Nat. Genet., 2005, 37(6), 590–592 CrossRef CAS.
- M. A. Moseley, C. J. Hughes, P. R. Juvvadi, E. J. Soderblom, S. Lennon and S. R. Perkins, et al., Scanning Quadrupole Data-Independent Acquisition, Part A: Qualitative and Quantitative Characterization, J. Proteome Res., 2018, 17(2), 770–779 CrossRef CAS.
- C. Messner, V. Demichev, N. Bloomfield, G. Ivosev, F. Wasim and A. Zelezniak, et al., Scanning SWATH enables ultra-fast proteomics using high-flow chromatography and minute-scale gradients, bioRxiv, 2019, 656793, DOI:10.1101/656793.
- D. B. Bekker-Jensen, A. Martinez-Val, S. Steigerwald, P. Ruther, K. L. Fort and T. N. Arrey, et al., A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients, Mol. Cell. Proteomics, 2020, 19(4), 716–729 CrossRef CAS.
- A. S. Hebert, S. Prasad, M. W. Belford, D. J. Bailey, G. C. McAlister and S. E. Abbatiello, et al., Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer, Anal. Chem., 2018, 90(15), 9529–9537 CrossRef CAS.
- F. Meier, S. Beck, N. Grassl, M. Lubeck, M. A. Park and O. Raether, et al., Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device, J. Proteome Res., 2015, 14(12), 5378–5387 CrossRef CAS.
- F. Meier, A.-D. Brunner, S. Koch, H. Koch, M. Lubeck and M. Krause, et al., Online Parallel Accumulation Serial Fragmentation (PASEF) with a Novel Trapped on Mobility Mass Spectrometer, Mol. Cell. Proteomics, 2018, 17(12), 2534–2545 CrossRef CAS.
- F. Meier, A.-D. Brunner, M. Frank, A. Ha, I. Bludau and E. Voytik, et al., Parallel accumulation – serial fragmentation combined with data-independent acquisition (diaPASEF): Bottom-up proteomics with near optimal ion usage, bioRxiv, 2020, 656207, DOI:10.1101/656207.
- F. A. Fernandez-Lima, D. A. Kaplan and M. A. Park, Note: Integration of trapped ion mobility spectrometry with mass spectrometry, Rev. Sci. Instrum., 2011, 82(12), 126106 CrossRef CAS.
- C. C. Tsou, D. Avtonomov, B. Larsen, M. Tucholska, H. Choi and A. C. Gingras, et al., DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics, Nat. Methods, 2015, 12(3), 258–264 CrossRef CAS.
- Y. Y. Li, C. Q. Zhong, X. Z. Xu, S. W. Cai, X. R. Wu and Y. Y. Zhang, et al., Group-DIA: analyzing multiple data-independent acquisition mass spectrometry data files, Nat. Methods, 2015, 12(12), 1105–1106 CrossRef CAS.
- Y. S. Ting, J. D. Egertson, J. G. Bollinger, B. C. Searle, S. H. Payne and W. S. Noble, et al., PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data, Nat. Methods, 2017, 14(9), 903–908 CrossRef CAS.
- V. Demichev, C. B. Messner, S. I. Vernardis, K. S. Lilley and M. Ralser, DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput, Nat. Methods, 2020, 17(1), 41–44 CrossRef CAS.
- Y. Yang, X. Liu, C. Shen, Y. Lin, P. Yang and L. Qiao, In silico spectral libraries by deep learning facilitate data-independent acquisition proteomics, Nat. Commun., 2020, 11(1), 146 CrossRef CAS.
- S. Gessulat, T. Schmidt, D. P. Zolg, P. Samaras, K. Schnatbaum and J. Zerweck, et al., Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning, Nat. Methods, 2019, 16(6), 509–518 CrossRef CAS.
- P. Navarro, J. Kuharev, L. C. Gillett, O. M. Bernhardt, B. MacLean and H. L. Rost, et al., A multicenter study benchmarks software tools for label-free proteome quantification, Nat. Biotechnol., 2016, 34(11), 1130–1136 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |