
Cleaning the molecular machinery of cells via proteostasis, proteolysis and endocytosis selectively, effectively, and precisely: intracellular self-defense and cellular perturbations†
Rajiv
Kumar
 *a,
Bhupender S.
Chhikara
b,
Kiran
Gulia
c and
Mitrabasu
Chhillar
d
*a,
Bhupender S.
Chhikara
b,
Kiran
Gulia
c and
Mitrabasu
Chhillar
d
aNIET, National Institute of Medical Science, India. E-mail: chemistry_rajiv@hotmail.com
bDepartment of Chemistry, Aditi Mahavidyalaya, University of Delhi, Delhi, 110027, India
cMaterials and Manufacturing, School of Engineering, University of Wolverhampton, England TF29NN, UK
dInstitute of Nuclear Medicine and Allied Sciences (INMAS), Brig. S. K. Mazumdar Marg, Delhi, 110054, India
First published on 2nd November 2020
Abstract
Network coordinates of cellular processes (proteostasis, proteolysis, and endocytosis), and molecular chaperones are the key complements in the cell machinery and processes. Specifically, cellular pathways are responsible for the conformational maintenance, cellular concentration, interactions, protein synthesis, disposal of misfolded proteins, localization, folding, and degradation. The failure of cellular processes and pathways disturbs structural proteins and the nucleation of amyloids. These mishaps further initiate amyloid polymorphism, transmissibility, co-aggregation of pathogenic proteins in tissues and cells, prion strains, and mechanisms and pathways for toxicity. Consequently, these conditions favor and lead to the formation of elongated amyloid fibrils consisting of many-stranded β-sheets (N,N-terminus and C,C-terminus), and abnormal fibrous, extracellular, proteinaceous deposits. Finally, these β-sheets deposit, and cells fail to degrade them effectively. The essential torsion angles (φ, ψ, and ω) define the conformation of proteins and their architecture. Cells initiate several transformations and pathways during the regulation of protein homeostasis based on the requirements for the functioning of the cell, which are governed by ATP-dependent proteases. In this process, the kinetics of the molding/folding phenomenon is disturbed, and subsequently, it is dominated by cross-domain misfolding intermediates; however, simultaneously, it is opposed by small stretching forces, which naturally exist in the cell. The ubiquitin/proteasome system deals with damaged proteins, which are not refolded by the chaperone-type machinery. Ubiquitin-protein ligases (E3-Ub) participate in all the cellular activity initiated and governed by molecular chaperones to stabilize the cellular proteome and participate in the degradation phenomenon implemented for damaged proteins. Optical tweezers, a single-resolution based technique, disclose the folding pathway of linear chain proteins, which is how they convert themselves into a three-dimensional architecture. Further, DNA–protein conjugation analysis is performed to obtain folding energies as single-molecule kinetic and thermodynamic data.
 Rajiv Kumar | Dr Rajiv Kumar (Group Head) is a Scientist in the field of cell science, molecular omics, nanoscience and nanotechnology and acquired his MSc in Chemistry with honors (Institute Topper) from CCS University, and PhD in derivatives of chlorins, which are relatives of porphyrins from the University of Delhi, India. He has extensive research expertise in multiple discoveries in the area of chemical transformations, nano–bio interfaces, nanomedicines, computational nanotechnologies-interfaces, computational chemistry, apoptotic, mitochondrial and DNA damage pathway, cell cleaning, proteostasis, and proteolysis, signaling pathways of cell machinery, modeling of mechanical forces, conformational changes, molecular modeling, macromolecules, antibiotic drugs, spectroscopy, and biomarkers. He has contributed as a co-author for the nano/micro-series books (“Nanotechnology and Nanomaterials in the Treatment of Life-threatening Diseases”), and is a reviewer for almost 70 journals of repute published worldwide. |
 Bhupender S. Chhikara | Dr B. S. Chhikara is an organic and medicinal chemist with a PhD in Biomedical Sciences from the University of Delhi and Institute of Nuclear Medicine and Allied Science (DRDO), New Delhi. He did his postdoctoral research at the Indian Institute of Science, Bangalore, India, and the University of Rhode Island, Kingston, RI, USA. He has wide research experience with publications in the field of radiopharmaceuticals, organic synthesis, medicinal chemistry, catalysis, green chemistry, nanomedicine of carbon nanotubes and lipids, gene delivery, anti-cancer, and anti-HIV drugs development. His current research interests are in the development of new drug molecules using science at the interface of chemistry, biology, and nanotechnology. |
 Kiran Gulia | Dr Kiran Gulia is a Senior Academic: Lecturer and Scientist in Advanced Manufacturing and Materials, in the School of Engineering, University of Wolverhampton, Birmingham, UK. Her research is focused on advanced manufacturing and materials. Her core interest is in nanoscience. She obtained her PhD in Lasers and Optoelectronics from Heriot Watt University, Edinburgh Scotland. Her interest in the innovation and development of tiny machines, surefooted micro/nanorobots for biomedical claims is emerging. These engineered nanoscale drones and utensils will be capable of crossing biological barriers. These nanorobots can be utilized for tracking and drug delivery to cells. |
 Mitrabasu Chhillar | Dr Mitra Basu completed his MBBS and MD in Medicine from Pt BDS PGIMS, India, and has an MBA in Healthcare from the prestigious FMS, Delhi University, India. He is a trained Six Sigma Professional and a Gold Commander in the Strategic Management of CBRN Emergencies from Ryton, UK. He has more than 40 publications in the field of medicine, toxicology, management of healthcare facilities, and CBRN Defense in various journals of repute. He spearheaded a nationwide program on emerging coronary risk factors and remained head of the division of CBRN Defense at the Institute of Nuclear Medicine and Allied Sciences, Delhi, India. |
Introduction
Cells are capable of controlling and maintaining the equilibrium of numerous pathways (folding, biochemical transformation, and synthesis of proteins, and degradation of misfolded proteins) necessary for sustainability and proper functioning. Among them, protein misfolding and aggregation are not natural processes, which are caused by irregular, unnatural, and unwanted environmental factors. Consequently, the number of diseases triggered by protein misfolding and aggregation is increasing.1 Some mechanisms (cytoplasmic quality control, molecular chaperones, and ubiquitin–proteasome system) assist with protein folding and enable the deterioration of misfolded polypeptides.2 Genetic mutations, translational faults, irregular protein alterations, thermal or oxidative stress, and imperfect complex growth initiate protein misfolding. Therefore, this allows pathological conditions (inappropriate degradation, dominant-negative mutations, essential modifications, mislocalization, novel toxic utilities, and amyloid build-up) to develop.3 Consequently, these intricacies in the cellular environment prohibit the natural functioning of cells and entrap functional proteins.4 Since normal proteins cannot achieve their correct conformations, misfolding is initiated. Proteins adopt analogous folding, which favors the realization of their inherent configuration. Proteins contain a hydrophobic core cluster for the formation of β-hairpin, which is a perfectly folded assembly. On the other hand, the misfolding of proteins produces amyloid fibrils, which have a non-native β-turn configuration. The abovementioned irregular activities disturb the packing of the hydrophobic core cluster and initiate the formation of a misfolded assembly. However, competing (misfolded) structures can be initiated by various cellular pathways, mechanisms, phenomena (transcription, translation, signal transduction, genomic mutations, toxic compounds, environmental stress, different types of cellular stress, nutrient deprivation, hostile environments, dominant-negative mutations, trafficking defects, irregular post-translational alterations, high degradation percentage, off-pathway folding, oxidative stress, reactive oxygen species, oxygen radicals, oxygenic energy transduction, Lewis acids/base regulators, oxidants, metabolism, mitochondria-to-nucleus signaling pathway, proteostasis–ubiquitin proteasomal and lysosomal autophagy systems, mitochondrial electron transport chain, cell necrosis, endothelial apoptosis, angiogenesis, proteasomal degradation heat, and heavy metal ions) and other specific factors.5 The above-mentioned pathways, mechanisms, and phenomena influence the thermodynamic and kinetic parameters of proteins.The folding and misfolding of proteins can be dynamically described by free energy studies, where previous results indicate that the folding and misfolding of β-hairpins are analogous processes. The selection of the protein pathways to be followed depends on the competition between the construction of flexible turn conformations and cross-strand hydrophobic interface/interaction. During the endoplasmic reticulum-associated degradation (ERAD) pathway, molecular chaperones and other associated factors identify an approach, which are the components for retro-translocation in the cytoplasm.6 During these processes, these proteins are destroyed by the ubiquitin–proteasome mechanism.7 There are three main types of non-native conformations, namely stable misfolded, unstable misfolded, and aggregation-prone, which have three different effects, specifically functional shortage, dominant-negative possessions, and toxic cellular impact, respectively. Protein misfolding pathologies mainly result in intra- or extra-cellular aggregation of proteins during diseases, displaying a gain-of-function pathology.
Ca2+ homeostasis is controlled by the ER, where a higher concentration of Ca2+ is suitable for the proper folding of proteins. Furthermore, computational studies on protein–surface interactions, and ER, mitochondrial, nuclear, and ribosomal PQCPs showed that processes of protein folding are dynamic. The setup of chaperones contributes to folding by ATP hydrolysis to identify wide-open hydrophobic amino acid extended layers.8 These other pathways and mechanisms also require further investigations for a greater understating of proteins. The failure of cell cleaning mechanisms for misfolded proteins in cells is characteristically responsible for various complications and diseases (protein conformational disorders, trafficking defects, mitochondria-to-nucleus signaling pathway, cardiovascular diseases, neuronal dysfunction, endothelial dysfunction, aqueous-deficient dry eyes, cataracts, proteasome activity, Pick's disease, ubiquitination in the pathogenesis of human diseases, cystic fibrosis, dialysis-related amyloidosis, Alzheimer's disease, neurodegenerative disorders, transmissible spongiform, serpin-deficiency disorders, cancer, hemolytic anemia, Huntington's disease, amyotrophic lateral sclerosis, encephalopathies, Parkinson's disease, and age-related molecular degeneration9).10 The specific aim of “Cell Cleaning: Proteostasis and Proteolysis”, which involves determining how to stop cells from undergoing denaturing activities,11 has been made possible by computational and theoretical advances, thus revealing some of the folding machineries at the atomic scale.
Elucidating the molecular mechanism of diseases, and subsequently designing and developing the required therapies with new methodologies12,13 must be the main goal. Besides, there are protein quality control mechanisms (cytoplasmic quality control, molecular chaperones, and ubiquitin–proteasome system) that control protein homeostasis and provide protection during acute inflammation and stress conditions, and this must be considered specifically.14 The failures of these mechanisms and uneven homeostasis originate from diseases and aging. Herein, we present the most important cellular processes and their mechanisms at the molecular and cellular levels. Therefore, this review article addresses the requirement to determine the pathways for the perturbations in cellular proteostasis, proteolysis, and endocytosis by revealing the urgency for repairing and cleaning of the molecular machinery of cells selectively, effectively, and precisely.
Protein quality control pathways: molecular chaperone-mediated autophagy
Inconsistency, dysfunctionalities, and errors in the folding of the proteins occur because of transcriptional, translational (Fig. 1), mutational, and oxidative stress interrupt natural cellular processes.15 Molecular chaperones detect these folded proteins through hydrophobic patches and help them gain their native states accordingly. If it does not occur, then proteolytic processes16 and mechanisms (ubiquitin–proteasome, autophagic vacuolar, and lysosomal) can easily differentiate between folded and misfolded proteins, target misfolded proteins, and eradicate them through a natural cell cleaning process.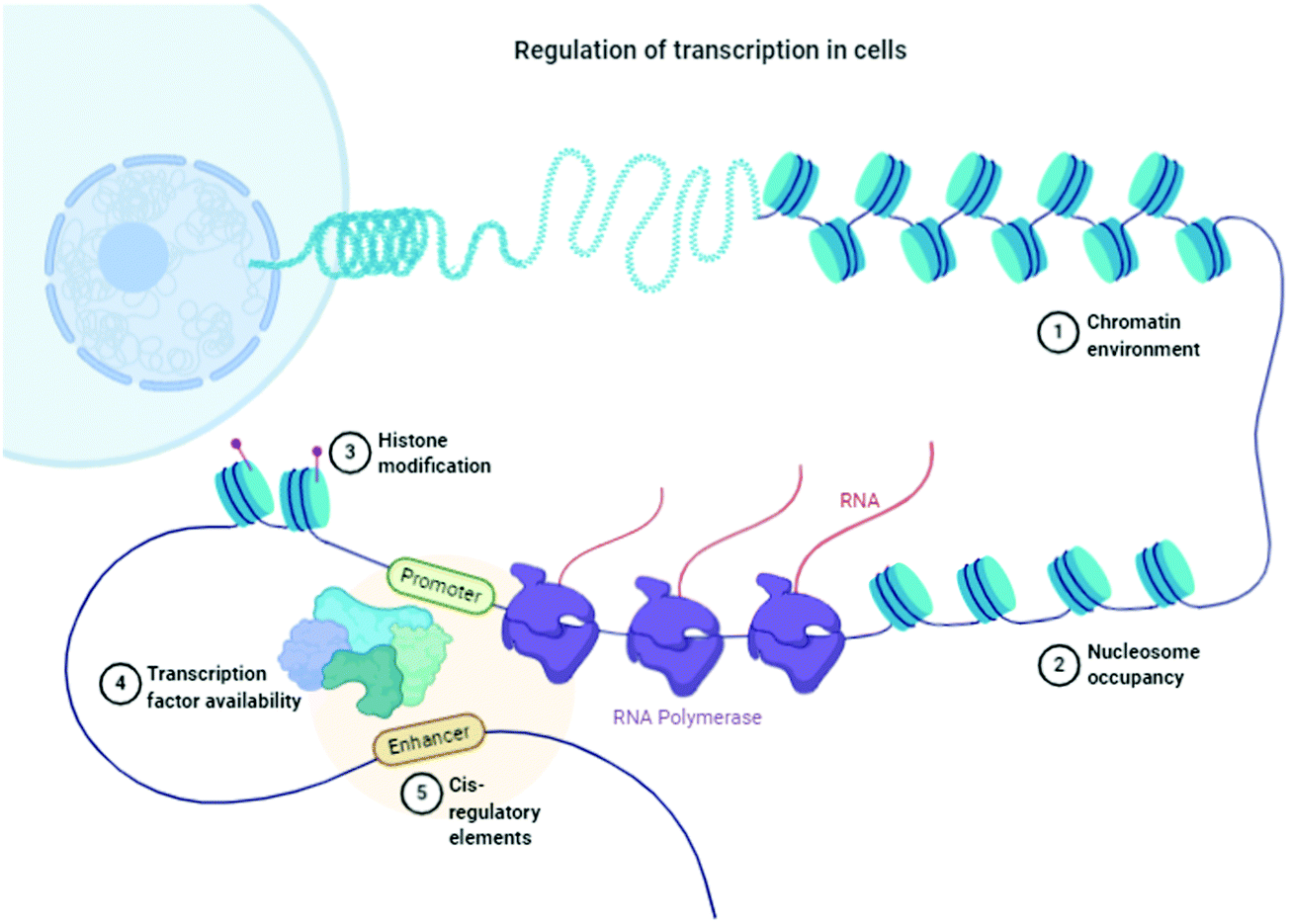 | ||
| Fig. 1 Schematic of the regulation of transcription in cells. “Adapted and created with permission from [http://biorender.com]. Same is referred and acknowledged as per instructions”. | ||
There is always an urgent need for quick cell cleaning via protein homeostasis (proteostasis), especially for neuron cells that have long extensions of cellular space. If the appropriate cleaning in these cells does not occur, cell heath will deteriorate. This complication is one of the main causes for the pathogenesis of several diseases. In this section, the α-Syn assemblies and autophagy-lysosome pathway are additionally included to expose their interlinked phenomenon. The key to this discussion is to explore the cellular processes to understand how the degradation of α-Syn aggregates occurs. To expose these mechanisms and activities, protein homeostasis, protein quality control checkpoints, the autophagy-lysosome pathway, and molecular chaperone functioning must be reexamined.17 Finally, the remaining misfolded proteins are eliminated by Golgi quality control. Macroautophagy involves one more intracellular system for the bulk degradation of decomposed self-components, as illustrated in Fig. 2.
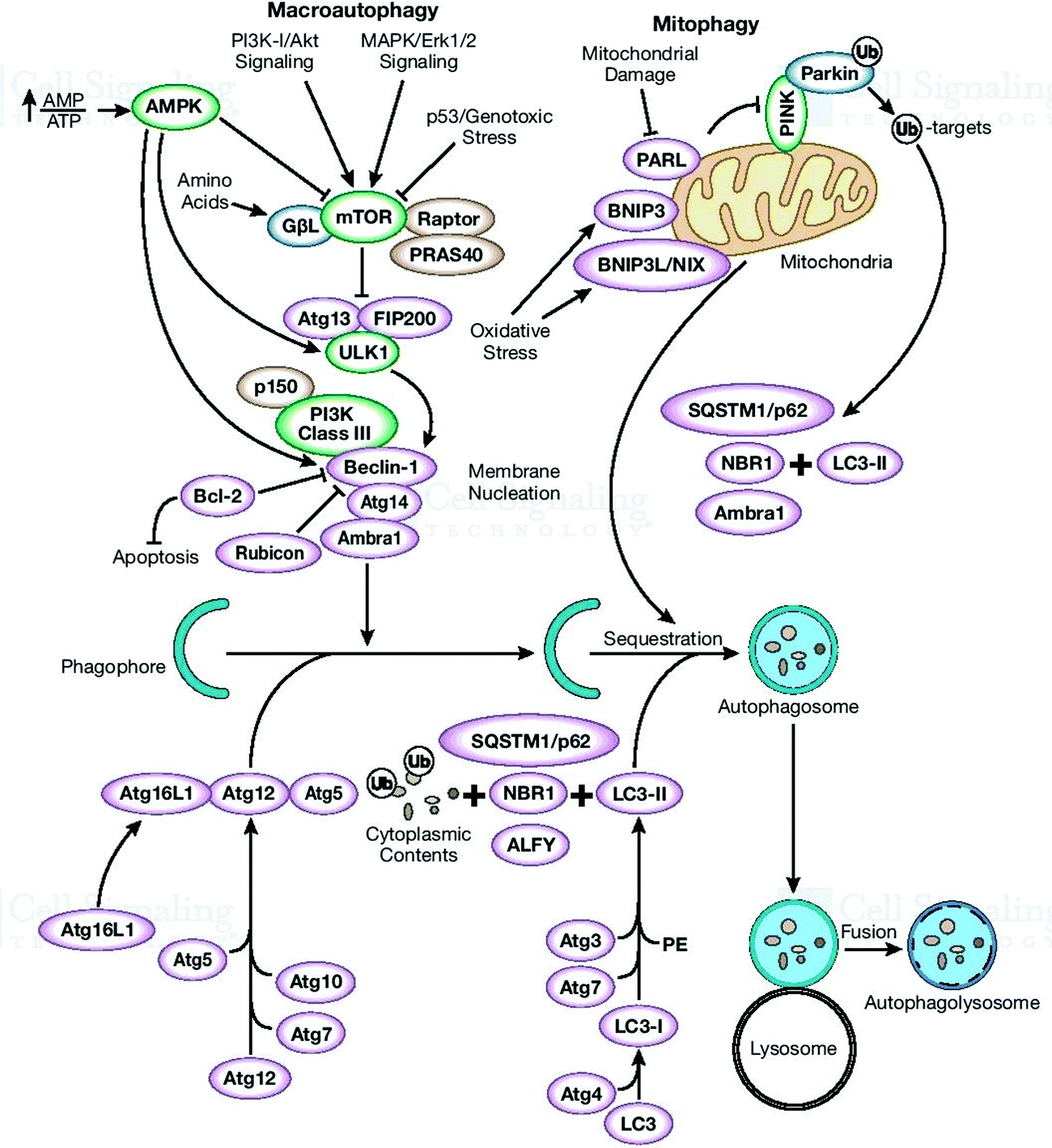 | ||
| Fig. 2 Illustrations of macroautophagy and autophagosome-lysosomal degradation or damaged organelles. ULK1- and ULK2-formed autophagy-related (Atg) gene product (mAtg13) and the scaffold protein FIP200 (an ortholog of yeast Atg17). “Adapted with permission from [http://www.cellsignal.com] and the same is acknowledged”. | ||
Autophagosomes carry out the autophagy process to damage the cytoplasmic contents (damaged organelles, mitochondria, lysosomes, and protein aggregates) and invade bacteria and harmful substances when the proteasomal degradation pathway fails to deal with them.18 It has also been highlighted in the literature that autophagy pathways exclusively eliminates unsafe substances according to the requirements of the cell, and also fulfil the needs of cells by providing back-up energy and building blocks during starvation. If the abovementioned harmful substances and damaged organelles remain in the cell, subsequently they play a crucial role in propagating neurodegeneration diseases.19 Therefore, the underlying molecular mechanisms of autophagy redefine it as a cellular defense mechanism. In the cytoplasm, the nuclear ubiquitin–proteasome system, which is a cytoplasmic autophagic route that can be macro, micro, or chaperone-associated autophagy (Fig. 3), and molecular chaperones deal with misfolded proteins to contribute to the necessity of cell cleaning by detecting, targeting, and repairing damaged and mislocalized proteins existing in the cellular environment.20 To achieve a perfect methodology for convoluted cell cleaning, theoretical, experimental and practical expertise need to be applied.
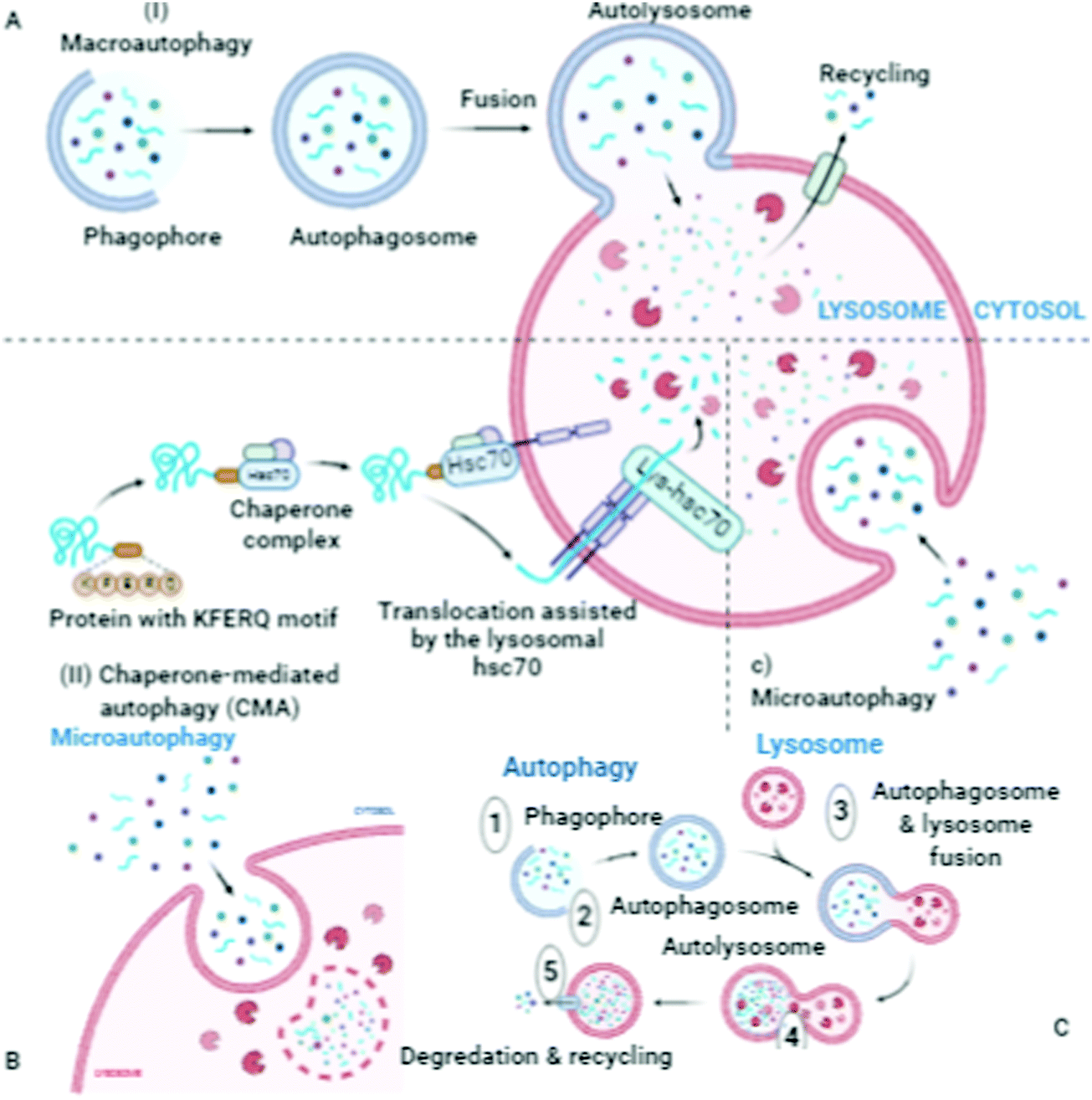 | ||
| Fig. 3 Schematic of macroautophagy, an intracellular system for the bulk degradation of self-components. Autophagosomes, autolysosomes and phagophores carry out the autophagy process to damage the cytoplasmic contents (damaged organelles, mitochondria, lysosomes, and protein aggregates), chaperone-mediated autophagy, microautophagy, autophagy, degradation, and recycling. “Adapted and created with permission from [http://biorender.com]. Same is referred and acknowledged as per instructions”. | ||
Knowledge of cytoplasmic protein quality control (CPQC) and ribosomal protein quality control pathways (PQCPs) must be implemented for developing new strategies to survive the failure of the protein quality control system to maintain the function and integrity of the cellular system.21 These are the main objectives of the goal to search for new therapeutic methodologies for curing protein conformational disorders (PCDs). The networks of cellular proteins require continuous surveillance to investigate the proper functioning of proteostasis, and it is necessary to determine declines during these processes. Reviewing the ubiquitin–proteasome system (UP), trafficking defects, mitochondria-to-nucleus signaling pathway, neuronal dysfunction, endothelial dysfunction, and causes responsible for the pathogenesis of diseases (for example in the case of the eyes: aqueous-deficient dry eye diseases, age-related macular degeneration, cataracts, and Pick's disease) will be helpful to overcome the challenges of cell cleaning.22 Cellular toxicity is also introduced by protein misfolding, which is a threat to the fitness and viability of cells. Molecular chaperones, which play a defensive role, can impound irregular configured proteins to disturb their toxic interfaces.23 Health and aging play a crucial role in oxygen sensing, mediating calcium homeostasis, neurodegenerative diseases, cell growth, heme biosynthesis, and cell death.24 The mitochondrial respiratory chain drives protons transversely onto the ion-impermeable interior membrane having a gradient-dependent (ΔμH) electrical potential (Δψ) and concentration ratio (ΔpH), corresponding to the Nernst equation, which can be defined as ΔμH = zFΔψ + RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln[H+]I/[H+]O. These sequences play a key role in the transportation and dispersal of Ca2+.25 Accordingly, the buffering capacity delivered by weak acids and the actively respiring mitochondria support an electrical gradient through the internal membrane.
ln[H+]I/[H+]O. These sequences play a key role in the transportation and dispersal of Ca2+.25 Accordingly, the buffering capacity delivered by weak acids and the actively respiring mitochondria support an electrical gradient through the internal membrane.
This indicates a tough thermodynamic process, which favors of the build-up of cations. The Ca2+ mitochondrial circulation bypasses the internal mitochondrial membrane via the extrusion of Ca2+ through two major routes, where one route is driven by Na+/Ca2+ exchange, and the other involves H+–Ca2+ exchange. In the matrix, Ca2+ excites the movement of three Ca2+-sensitive dehydrogenases in the pathway of the Krebs cycle. The gradient bypasses the internal membrane, which is crucial to maintain the mitochondrial state. Thus, this sequence occurs and is influenced by the movement of the mitochondrial respiratory chain.26 Analysis of protein conformational disorders and potentially identified protein clearance mechanisms (autophagy-lysosome or autophagic-lysosomal), exposed the necessity of healthier cellular processes to achieve an extended lifespan and protect organisms from associated disorders and age-associated diseases (Fig. 4).27
 | ||
| Fig. 4 Schematic of the theoretical, experimental, practical expertise, and knowledge of cytoplasmic protein quality control (CPQC) and ribosomal protein quality control pathways (PQCPs). New strategies, failure of the protein quality control system and maintaining the function and integrity of the cellular system. “Adapted with permission from [http://www.cellsignal.com] and the same is acknowledged”. | ||
To achieve appropriate cell cleaning processes and pathways, some unknown mechanisms and hidden pathways have been highlighted as follows: (1) metal–protein interface/interaction comprising metal-, ligand-, and enzyme-bridge complexes, defined as Lewis acids/base regulators. (2) Principal types of machinery maintaining proteostasis–ubiquitin proteasomal and lysosomal autophagy systems. (3) Cell survival responses to environmental stress, oxidants, metabolism, and signal transduction. (4) How cells adapt to proteasome assembly under stress conditions. (5) Boosting cellular resilience to protein misfolding, and ER-governed Ca2+ homeostasis. (6) Identifying strategies for proteasome activation by natural or chemical compounds. (7) Endothelial apoptosis, angiogenesis, and proteasomal degradation. (8) Serine-protease inhibitors, and (9) ring-finger domain, with the goal to tailor strategies to dismantle unassailable fortress of the cellular system, which requires significant efforts to be accomplished.
Dihedral angles (φ and Ψ) vs. torsion angles (ω): conformational analysis
Protein disorders and conformational scattering are linked, and thus quantitative and experimental analysis can evaluate the impact of the conformational changes. Further, computational analysis and bioinformatic evidence may confirm the results. This scientific evidence can be utilized to determine unfolded conditions of peptides and proteins. For the quantitative analysis of the intrinsic disorders of proteins, their structural configuration and conformational entropy are highly important features. This analysis can easily differentiate globular and disordered proteins. For the backbone torsions (φ and Ψ) of three-dimensional proteins, minor inaccuracies can only be predicted by computational analysis; however, this results in major errors.28 The dihedral angles (φ and ψ) of a protein sequence can be determined by potential energy and torque moment analysis. The β-hairpin, with two stands interlinked with the folding characteristics of proteins, shows a two-state transition. Further analysis established that its thermodynamic stability and fast folding kinetics are also interconnected. To avoid kinetic traps, the co-translational folding and C-terminal residue interactions must be evaluated. The rare codons that affect protein assembling are crucial for folding. However, how do proteins go from non-native (fully unfolded) structures to native (folded) structures or how distinguish folded and out-of-register misfolded β-hairpin assemblies (N,N-terminus and C,C-terminus), and how their hydrophobic side-chains remain above/below both hairpins remain unresolved fundamental questions.29 The β-strand has a predisposition to self-associate to form β-sheets, which are composed of several β-strands bound together by hydrogen bonds between the backbone atoms. Successive planes define the φ and Ψ angles of the O, N, C and H atoms, which exist at a similar level, and the conformation of a chain of η amino acids replicated 2η parameters, an energetically stable state.30 The high content of hydrophobic residues in the amino acid chains plays a crucial role in preventing misfolding. Specifically, they stabilize the inherent β-hairpin assembly. However, defective proteins may be unable to acquire the requisite post-translational modifications and assume their native conformations. Proteins are involved in many mechanisms including signal sequence cleavage, disulfide-bond formation/isomerization, N-glycosylation, and glycosylphosphatidylinositol (GPI)-anchoring of existing proteins. These transformations govern the various co- and post-translational alterations and modifications of proteins (Fig. 5).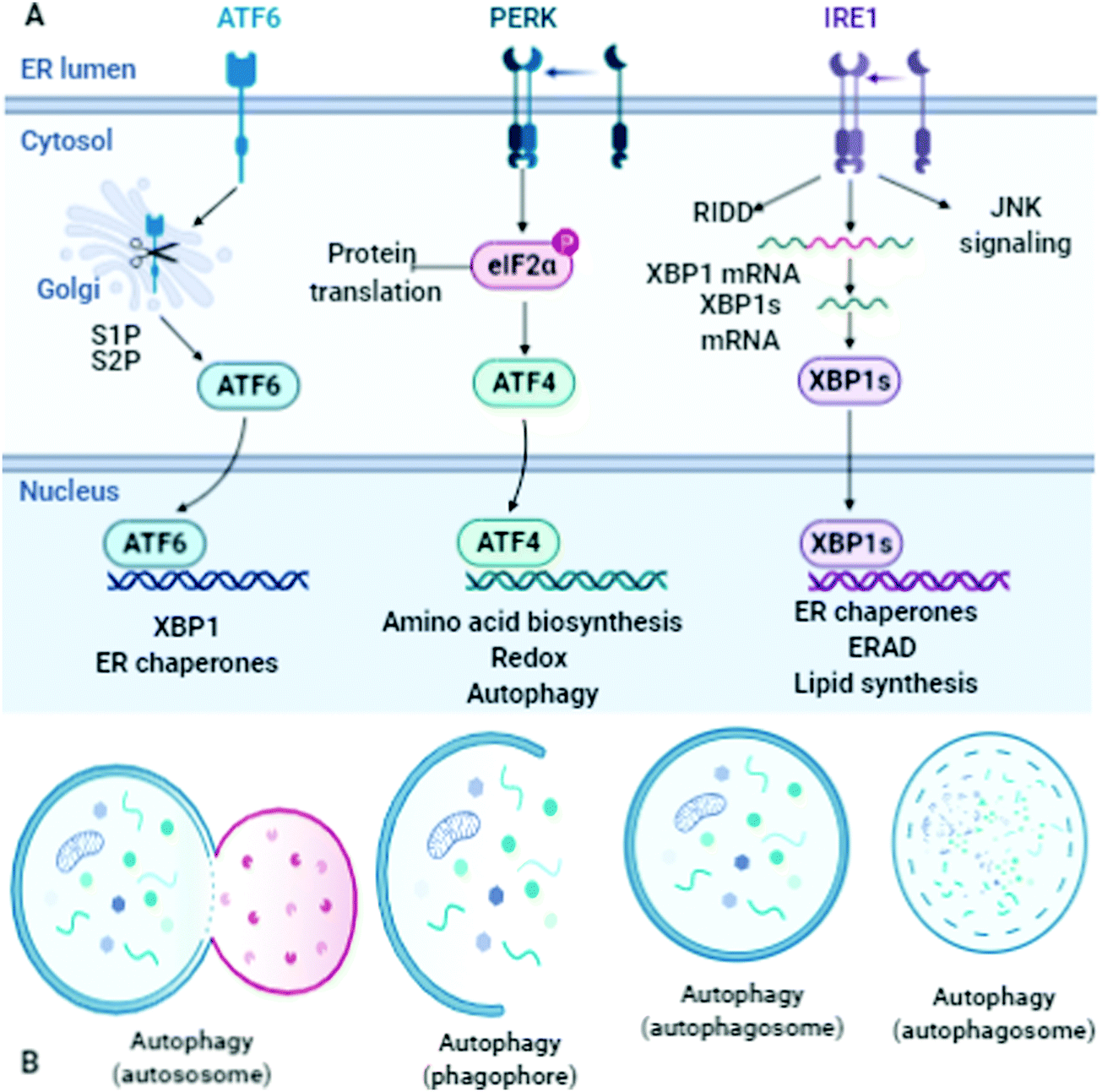 | ||
| Fig. 5 Schematic of the structure and molecular mechanism of stress signaling, and unfolded protein response signaling. Defective proteins and requisite post-translational modifications. Co- and post-translational modifications, the lumen of the ER, signal sequence cleavage, Golgi body, protein translation, autosome, phagophore, and autophagosomes. “Adapted and created with permission from [http://biorender.com]. Same is referred and acknowledged as per instructions”. Thanks also for this template, which is adapted and reproduced from Emma Madden PhD Candidate, NUI Galway. | ||
To elucidate the link between protein-misfolding/folding and chemical instability, there is a need to emphasize the bond formation (oxidation) and bond cleavage (hydrolysis) processes.31 Specifically, the interpretation of the denatured activity of cells with ongoing chemical conversions (reactions and mechanism, reaction intermediates, pathways of oxidation and reduction reactions, nucleophilic/electrophilic, molecular processes of chemical transformations, reaction discovery, pathways of enzymatic mechanisms, chemoinformatic processing of enzymatic transformations, reconstruction of metabolic pathways, computer-aided validation for proton and electron-driven reactions in protein-metallic catalysis, and computational analysis) must be strategically applied to elaborate chemical variability.
Generally, the oxidation of proteins is involved in various chemical transformations (metal ion-catalyzed), and if not, photochemical transformations occur via high energy γ-radiation or organic additives. There are many transformations (protein/peptide synthesis) initiated and governed by metal catalysis. Furthermore, there are many factors (purification, bulk storage, and dosage formula) necessary for successful metal-catalyzed oxidation. Amyloids, which are the final state of aggregated proteins, represent the nature of metal-catalyzed oxidation. The conformation of polypeptides is distinguished by the dihedral angles φ and Ψ. The values of a single set of φ and Ψ are sufficient to differentiate folded and misfolded types of proteins.32 Therefore, the dihedral angles are the key parameters to be highlighted for enhancing the understanding of proteins. A shift in residues leads to misfolding in the β-hairpin conformation, which is affected by the configuration of the hydrogen bonds present in the skeleton of a particular molecule. The underlined aspect of this interpretation is that the dihedral angles are not only the calculated features of particular macromolecules, but also define other characteristics. For example, they maintain the equilibrium conformation, which is a key factor to restructure the folded and misfolded states. The values of the φ and ψ angles can be transformed into reaction coordinates. It is well known that these parameters demonstrate the two-dimensional free energy landscape. However, the question, how does β-hairpin misfolding occur, remains unanswered. There are only three torsion angles, which are φ, ψ, and ω. Their mathematical values define the conformation of the skeleton.33 Further, these torsion angles are related to the angle of rotation around the formed bonds. The N–Cα bond angle is known as the phi (φ) angle. The rotation around the Cα–C bind is the angle denoted as psi (ψ). Furthermore, the rotation for the C–N bond gives the angle called omega (ω). There is a possibility that at one position or location, any structure or residue of the secondary configuration has similar values of the dihedral angle (φ and ψ) existing within identified regions. These landscapes indicate the existence of secondary assemblies having very well defined dihedral angles. Specifically, there two sub-types, which are as follows (i) right-handed α-helix and left-handed α-helix and (ii) β-strand. The α-helix is the main component of the protein states and is solely responsible for their spiral appearance. These arrangements are further balanced by the parallel hydrogen bonds formed between the skeleton atoms of successive residues of the protein.
Pathological conditions: chaperones, ATP hydrolysis, and mitochondrial Ca2+ uptake
Interference in proteostasis results from pathogenesis and initiates aging and the possibilities for the propagation of degenerative diseases. Thus, understanding the mechanisms of chaperone and co-chaperones in both situations (regulating natural folding and pathological conditions where misfolding of proteins occurs) is the key factor for the interpretation of occurring sequences. Generally, chaperones are assisted by the bind-and-release cycle because they are ATPases, and the necessary energy may be produced by ATP hydrolysis.34 Unrestricted heat shock chaperones take part in protein folding at the expense of ATP. Molecular chaperones (Hsp60s, Hsp70s, Hsp90s, and sHsps) govern folding intermediates, aggregation, and act as continuous hurdles for misfolding processes.35 Thus, these chaperones are directly involved in the propagation of diseases and amyloid disorders. One of the varieties of these chaperones, Hsp70, exists in two different states, i.e. low/high affinity ATP states, which also control folding catalysis. Therefore, ATP binding and hydrolysis are the key components for the proper functioning of co-chaperones, and chaperone-associated and interlinked pathways (Fig. 6). Thus, it is a challenging task to develop theoretical and computational methods for protein–surface examination. Accordingly, we identify the applicability of different modeling and simulation techniques to address this issue. Quantum mechanics is based on atom/molecules and is utilized for the analysis of all-atom molecular mechanics by calculating free energy with a coarse-grained methodology. Ca2+ activates metabolic enzymes by stimulating energy-consuming routes in the cytosol during the processes of contraction and secretion simultaneously. Meanwhile, mitochondrial dehydrogenases stimulate the associated routes. These types of mechanisms are considered as the aerobic metabolism process, which occurs to complete the energy requirements of active cells. Therefore, the route of mitochondrial Ca2+ acceptance has been identified for proper understanding.36 The pathological conditions governed by the mitochondria indicate an alteration in the Ca2+ signals and can be induced by oxidative stress. These uncontrolled transformations change the performance of these organelles, and it is evident that these unwanted activities can alter their structure. Finally, in some cases, the cells undergo apoptosis. The functioning of the mitochondria is influenced by these degradations, wherein some of the β-cells neighboring the secretory insulin granules act to stop proper functioning.37 These factors play a crucial role in the metabolism–secretion coupling.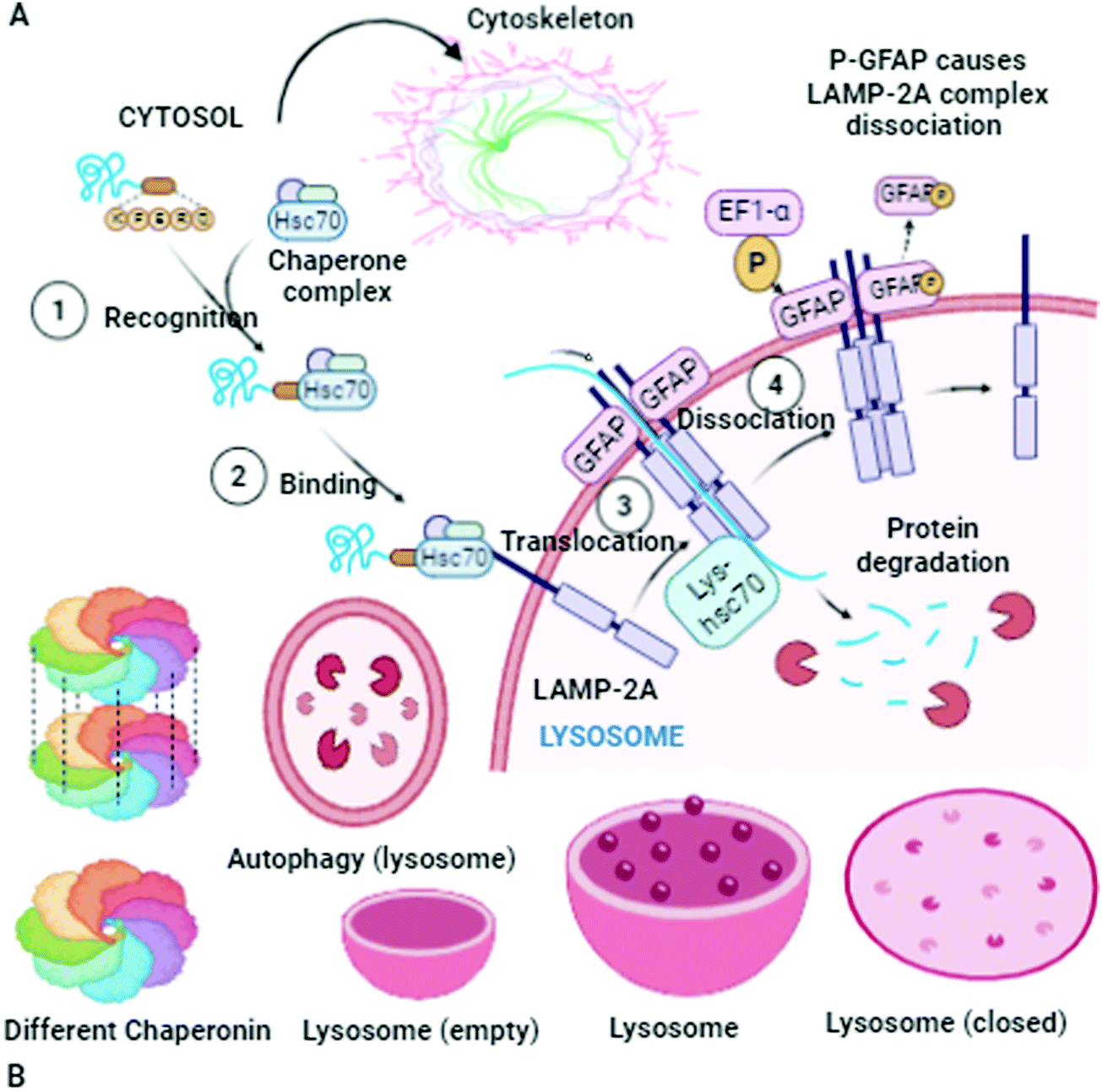 | ||
| Fig. 6 Schematic of the chaperone-assisted and bind-and-release cycles due to ATPases and ATP hydrolysis. The heat shock chaperones and protein folding. These mechanisms to consume ATP. Molecular chaperones (Hsp60s, Hsp70s, Hsp90s, and sHsps) govern folding intermediates, aggregation and act as hurdles for misfolding processes. Propagation of diseases and amyloid disorders. “Adapted and created with permission from [http://biorender.com]. Same is referred and acknowledged as per instructions”. | ||
In all cell activities and transformations, ATP supplies the required energy for the imitation of insulin granules and priming of exocytosis. The phenomenon of insulin exocytosis occurs when the level of glycolysis in β-cells is amplified, and consequently, the percentage of blood glucose becomes high. These transformations require plenty energy, which is supplied by ATP, and thus, the production of ATP increases. One more important transformation involved is known as the glucose-stimulation route. In this process, β-cells secrete insulin, which further initiates various signaling pathways.38,39 The Ca2+ signal originating from the mitochondrial activation plays a role to end these chain events. Overall, these transformations and pathways govern the uptake of glucose by β-cells and the process of insulin exocytosis.
Mitochondrial Ca2+ homeostasis: decoding of extracellular stimuli
The outputs of mitochondrial biogenesis can be utilized for pharmacological benefits. Therefore, mitochondrial Ca2+ homeostasis must be considered as a potential therapeutic to treat degenerative diseases occurring in mitochondria for enhancing the lifestyle of patients. Analysis of mitohormesis and intracellular signaling, can be used to develop new methodologies towards new pharmacological targets and natural therapies.40 Mitochondria play a crucial role in governing numerous pathways and chemical transformations. Signal transduction is one of the routes utilized by mitochondria for decoding. Fundamental intercellular actions such as energy production route and apoptosis pathways are triggered by extracellular stimuli. Thus, modifications and alterations in mitochondrial calcium Ca2+ homeostasis are the key mechanism through which cells are involved in various functions and processes.41 Any perturbation in mitochondrial Ca2+ homeostasis can lead to pathological conditions. This also depends on the functions of the cell. Mitochondrial impairment causes various neurodegenerative and neuroinflammatory disorders.42 Thus, to recognize these complications, the analysis and interpretation of the Ca2+ homeostasis mechanism definitely will be a crucial factor to identify these complications. With the help of molecular dynamics simulations, it is quite possible to investigate the biomolecular assembly and the associated features of the dynamics occurring at the atomic level. This methodology is also capable to expose the complicated molecular processes occurring during protein conformations and other mechanisms and pathways, for example the enzyme-reaction mechanisms and ligand binding43–45 routes occurring during protein folding and denaturation. Therefore, by employing computer and theoretical innovations, it is possible to shed light on the routes and the pathways occurring during protein folding. The same can be evident from conducting related experiments for elucidating the folding machineries. These techniques can be further utilized for the detection of other atomic details.46 By employing a multidisciplinary approach, it may be possible to break the boundaries existing among various fields, i.e. biochemistry, chemistry, medicine, and physics. These new themes display potential for a successful outcome. Investigation of the landscape of the energy of the folding process indicates intermediate constitutes and their nature. A proposed 3D model will reveal the nature of the barriers and help out to overcome them. These investigations can further help to understand the mechanism of amyloidogenic diseases. These efforts can bring hope to find remedies for these diseases. The outputs and procedures can be utilized to expose hidden routes and paths of hemodialysis-related amyloidosis. These pathological conditions are among the main causes that initiate aggregation in the sheet protein 2-microglobulin and protein fibrils.47Finally, there are various proteins such as the Bcl-2 family proteins that control the process of mitochondrial Ca2+ homeostasis for proper regularization of cell activity by controlling the permeability of the mitochondrial membrane. These specific proteins have various features and can intervene in the various routes responsible for apoptosis.25 The decoding tactics of extracellular stimuli can further be useful. There are possibilities to develop anti-apoptotic interventions, which can identify the features of the Bcl-2 protein family required for the possible development of anti-ischemic therapy. These possibilities can be achieved by aiming to generate anti-apoptotic functions or initiate loss in the pro-apoptotic function. If a therapeutic that can inhibit the permeability of the mitochondrial outer membrane is available, it can be recommended to stop apoptosis. These strategies can be successful as a technique that can control and alter the features of the Bcl-2 family proteins. This can also be achieved by shielding the myocardium during any ischemia or reperfusion injury.48 These efforts may be fruitful for gaining the desired therapeutic for the clinical trials shortly. The main features of the formation of perfectly folded β-hairpin assembly involve three steps as follows: (i) turn formation, (ii) hydrophobic core packing, and (iii) assembling of H-bonds.49 A comparative analysis of the structural features of misfolded/correctly folded proteins reveals that there are three main differences. They have different turn configurations, ineffective filling of cross-strand hydrophobic side-chains, and altered hydrogen bonding existing in their backbone. These features are present between antiparallel strands, which are initiated by the shifting of the related residue. It is quite complicated to determine the folding/misfolding pathway of the mechanisms of β-hairpin.50 Also, it is quite challenging to analyze a misfolded assembly by comparing the native folded structure of β-hairpin with the folded structure from β-hairpin redesign experiments. The data analysis of the topologies and measurement of the hydrophobic residues of the amino acids of these two states can be employed for this interpretation. A low percentage of hydrophobic residues in amino acids is an indication of the possibility of misfolding. However, if this percentage is high, then it improves the firmness of the native β-hairpin assembly.
Mitochondria-based pharmacological strategies: decoding of oxidative insults
The processes of signal transduction are supported by mitochondria because they are capable of decoding numerous and different categories of signals of extracellular stimuli into action (intracellular actions, energy production, and cell death). If the process of mitochondrial calcium (Ca2+) homeostasis occurs, it will further initiate several other pathological conditions. Recently, it was identified that some proteins are involved in mitochondrial calcium homeostasis, and here, is the source of this discussion, which will be helpful in the development of mitochondria-based pharmacological strategies.51 Therefore, failure of the mitochondrion activities initiates metabolic and nervous disorders. However, the pathways of mitochondrial biogenesis, its dynamics, and mitophagy have the potential to remedy these imbalances in the functioning of the mitochondrion. Any therapeutic strategy for the regulation of mitochondrion activities will be a crucial innovation for drug discovery for the treatment of rare diseases. Mitochondrial oxidative phosphorylation and glycolytic processes are influenced by disorders in oxidative metabolism, further enhancing neurodegenerative processes. Again, the initiated imbalance between the inflammatory cytokines and cellular energy makes it worse.52 Thus, the urgency for repairing and cleaning the mitochondrial machinery of the cell must be well addressed to remove the perturbations in cellular proteostasis, proteolysis, and endocytosis. However, the computational predictions of protein conformational errors, accumulation of errors in displacement deteriorated cell environmental conditions, coding failure in the mitochondria-to-nucleus signaling pathway (trafficking defects, mitochondria-to-nucleus signaling pathway, neuronal dysfunction, endothelial dysfunction, macular degeneration, proteasome activity, and ubiquitination) and several other similar hidden pathways have never been experimentally investigated, and therefore, these mechanisms will be the foremost area to highlight.53This review article emphasizes the specific and aforementioned aim of determine how to stop the cell from undergoing denaturing activities, and if these activities persist, how to stop them to clean the cell. There is an urgency to tailor ground-breaking strategies for cell cleaning, with the foremost ambition to prevent the product of cell necrosis and apoptotic bodies from the cellular environment for cells to recuperate their natural operability. In these circumstances, organs lose their proper functioning because of these degradation activities, which further initiate a lot of deteriorating disorders. Especially, these degeneration activities promote apoptosis in non-proliferating cells. ATP synthesis occurring in the mitochondria is related to the generation of reactive oxygen species. The formation of superoxide (O2−) also observed in the respiratory chain pathway.54 Subsequently, the superoxide initiates unwanted oxidants (peroxynitrite and hydrogen peroxide) and fast-responding hydroxyl radicals. The molecular mechanism of the route of apoptosis identified and highlighted the impact of the mitochondria in decoding processes. Finally, these transformations identify the oxidative insults. Meanwhile, the proteins released are catalyzed by the presence of cytochrome c, which acts as a cofactor of effector caspases.
Ubiquitin proteasome pathway, DNA repair, and endocytosis
The cell defense network consists of the genome and proteome, and any degradation in their related pathways or components further triggers various pathogenic states and aging. Thus, the blueprint of the pathways of genome and proteome damage must be highlighted for further research. The analysis and interpretation of induced DNA damage must be urgently performed. This is because these phenomena play a key role and act as a deciding factor in shaping the cellular landscape. These observations must be employed to develop strategies for the innovation of therapeutic modalities. Why do the molecular pathways force cell aging, pathogenesis, and apoptosis? How does proteostasis interlink with translation and oxidative damage? Also, how do the pathways of protein folding, molecular chaperone functioning, and protein quality control deal with irreparably damaged proteins? All these undiscovered occurrences are interlinked with each other, and thus need to be investigated and exposed. A few of them are performed to improve cellular functioning.55 One more important set of cellular processes is the control of the chaperon activities, how does it happen to save healthy proteins, which are required and in demand by cells during the impactful action of molecular chaperons. An imbalance between the cellular process of the generation of good proteins and damaged proteins initiates apoptosis. It was reported that if there are more chaperones or proteasomes in the cell, this is good for longevity. The rate of the accumulation of cellular damage and the normal proteostasis of cells is directly correlated with aging. Therefore, there is an urgent need to develop technologies for repairing damaged DNA56,57 since DNA repair pathways are directly responsible for genome stability.58 Quality proteins are necessary to repair DNA, and this is the main concern to be highlighted as the necessity of cell cleaning. Interestingly, naturally, some proteins exist in the cellular environment and have the potential to avoid misfolding, degradation, and aggregation and can be used to repair DNA damage for proper regulation. It has been established that the molecular chaperones play a crucial role in repairing damaged DNA. During pathological conditions, the DNA repair machinery compensates for defects in protein quality control and vice versa.59 It is widely accepted that the genome controls cellular perturbations and performs the necessary maintenance. On the other hand, the reassembled proteome repairs the genome and maintains proper cellular functioning. Damage at the structural and genomic levels occur due to the oxidative proteome. Genome mutations influence the reactive oxygen species, oxidative DNA damage,60 and irretrievable oxidative proteome destruction.61 These pathways directly affect the efficacy and precision of cellular processes, consequently resulting in degeneracy. The unfolded protein response influences the transcription of genes and participates in DNA repair and cell-cycle regulation. Therefore, it is very helpful in the development of therapeutic modalities for the repair of damaged DNA.62 Uncontrolled pathways initiate abnormal protein deposition, and the inability of the protein quality control, proteostasis, to deal with it may cause several diseases. Therefore, protein oxidation and damaged DNA are directly responsible for protein destruction. It can be assumed that DNA and DNA-binding proteins affect gene quality. Thus, to prevent and control DNA damage,63 all cells have kinase-based signaling networks64 as safeguards, and which are a critical factor for apoptotic machinery (Fig. 7). Genes participate in these cellular processes by transcriptional activation.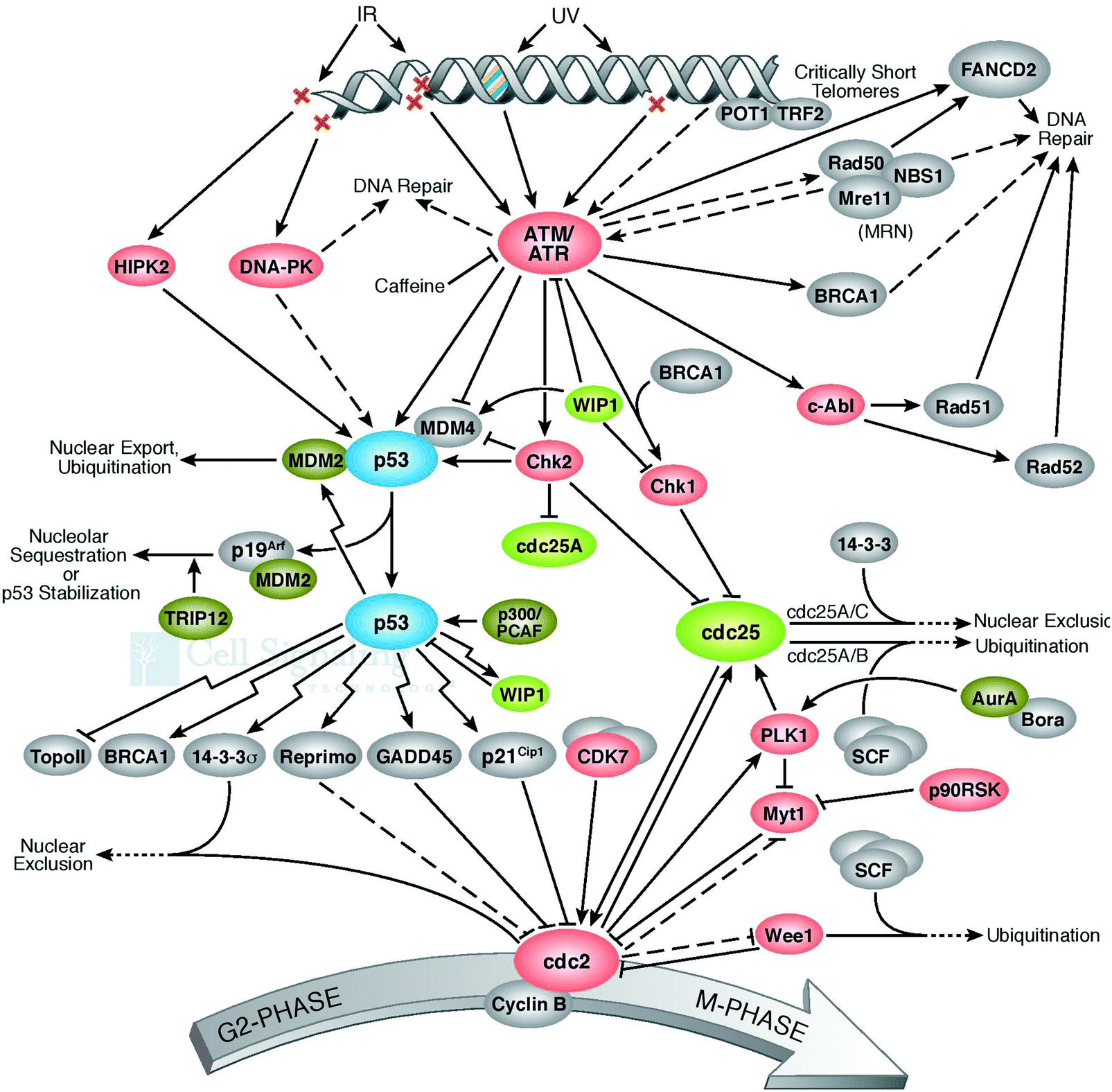 | ||
| Fig. 7 Schematic of the repair of damaged DNA. DNA repaired pathways and genome stability. Proteins needed to repair the DNA. Molecular chaperones and repairing DNA. “Adapted with permission from [http://www.cellsignal.com] and the same is acknowledged”. | ||
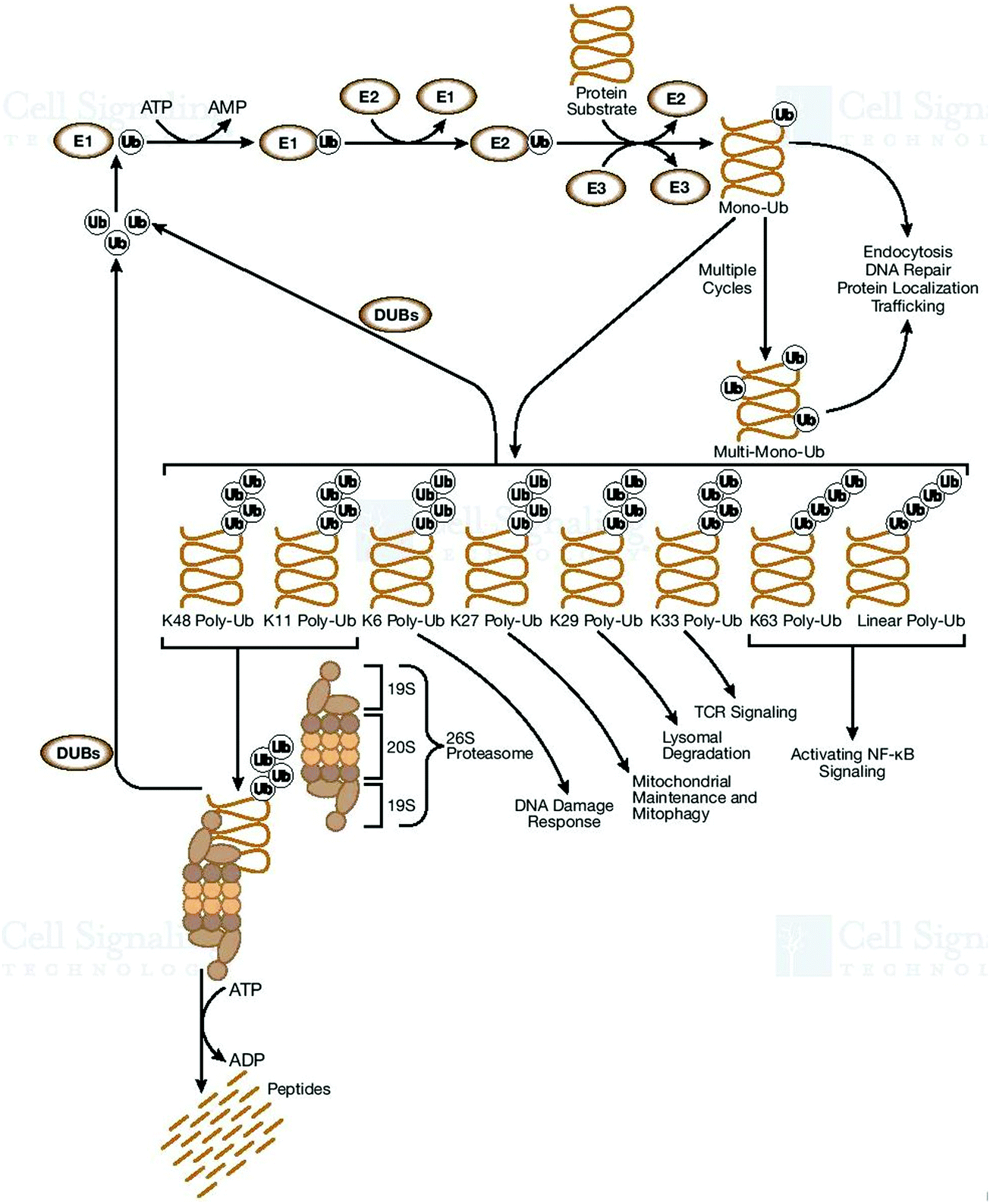
Ubiquitylation regulates a dynamic cellular process for stability and cellular homeostasis. Ubiquitin signaling trigger the ubiquitin-conjugation machinery for proper surveillance.65 Monoubiquitination is associated with numerous cellular functions (endocytic trafficking and DNA repair) and trafficking cargoes, it highlights the correctness of the endocytic route, receptor endocytosis, and degradation, and acts as Ub-receptors in the endosome. By being passive and not depending on specific molecular components, these principles reveal the general mechanisms (adaptation and rearrangement) of a significant portion of the cell membrane area.66 Furthermore, mutations also cause misfolding, which are increased by the non-functional forms of proteins. This proves that many pathological conditions are fundamentally rooted in protein-folding problems. Thus, all cells must have the ability to resolve them on a priority basis to maintain their functioning and integrity. The cellular proteome is a complex microcosm of structural and regulatory networks required for continuous surveillance and is modified to meet the dynamic needs of the cell.67 Genetic alterations, ranging from chromosome imbalance to oncogene initiation, influence the rate, fidelity, and ability of cells to perform protein biogenesis.
Any imbalance in the functioning of the proteome often initiates the degradation of cellular functioning and environment. Since the ubiquitin pathways participate in protein alteration and modification and regulate DNA and endocytosis,68 any imbalance or aggregation of misfolded proteins initiates various types of neurodegenerative diseases. Therefore, the ubiquitin–proteasome mechanism alters and destroys a variety of proteins that participate in definite signals, or ‘degrons’.
Serine-protease inhibitors and ring-finger domain
The folding patterns of proteins adopt more stable thermodynamic states. The serine protease inhibitors modify several biological mechanisms (blood clotting and inflammation) and are highly capable of altering their configuration. The ability of conformational conversion allows them to trap protease. Thus, to study the conformational patterns and adoption of serpin, α1-antitrypsin will be further prolific considering its ability to check the pathogenic states.69 During protein folding and the related enzymatic activities, the pro-peptide does not participate and is removed intramolecularly by auto-processing. Thus, there is a need to address and rectify the possibilities of how an intramolecular chaperone defines the role of the pro-peptide in protein folding.70 Serine protease inhibitors are involved in conformation modifications desirable for biological function, but are there any other structural features (buried polar groups, cavities, and over-packing of side-chains) to oppose proper folding? Thus, the processes of the folding of serpin polypeptides can be disrupted, resulting in an inactive conformation. Some amino acid subunits are responsible for clear unfavorable interactions, stabilize substitutions of α1-antitrypsin, and also enhance thermostability.71 Serpins also defend immune cells from the attack of cytotoxic proteases. It is well known that the serpin mechanism is highly stable during disease-associated mutations. This highlights their importance in maintaining the balance and health of the inhibitor conformation. Conformational stability and changes persisting due to the difference in higher free energy states and lower free energy states support their performance in physiological functioning.Sometimes, these conformational changes influence the free energy to inhibit target proteases by removing the protease. For example, during blood coagulation, some varieties of serpin regulate thrombin and other proteases, and thus with any dysregulation, their misfolding leads to thrombosis and other diseases. The serpins have an N-terminus and C-terminus with disulfide bonds, and their bonding patterns govern their metastable states.72 For effective and efficient folding, N- and C-bonded glycans support the molecular chaperones.73 Thus, analysis of these processes contribute to determining disease phenotypes. The proteases and their natural protein inhibitor ratio are a crucial factor in a balanced biological system. There are numerous small folds in protein inhibitors having disulfide bonding, which govern oxidative folding. For the prediction or analysis of the folding patterns of the two-domain protease inhibitors, it is easy to expose their oxidative folding pathways. The intermediates of the protease inhibitors and their molecular mechanisms reveal the oxidative folding of the disulfide-rich proteins. These interpretations will give researchers insight into the functioning of N-glycans, physiological exchanges, and biochemical transformations.74 The role of the N-glycosylation mechanisms in folding (Fig. 8) and their extracellular expression must be highlighted.
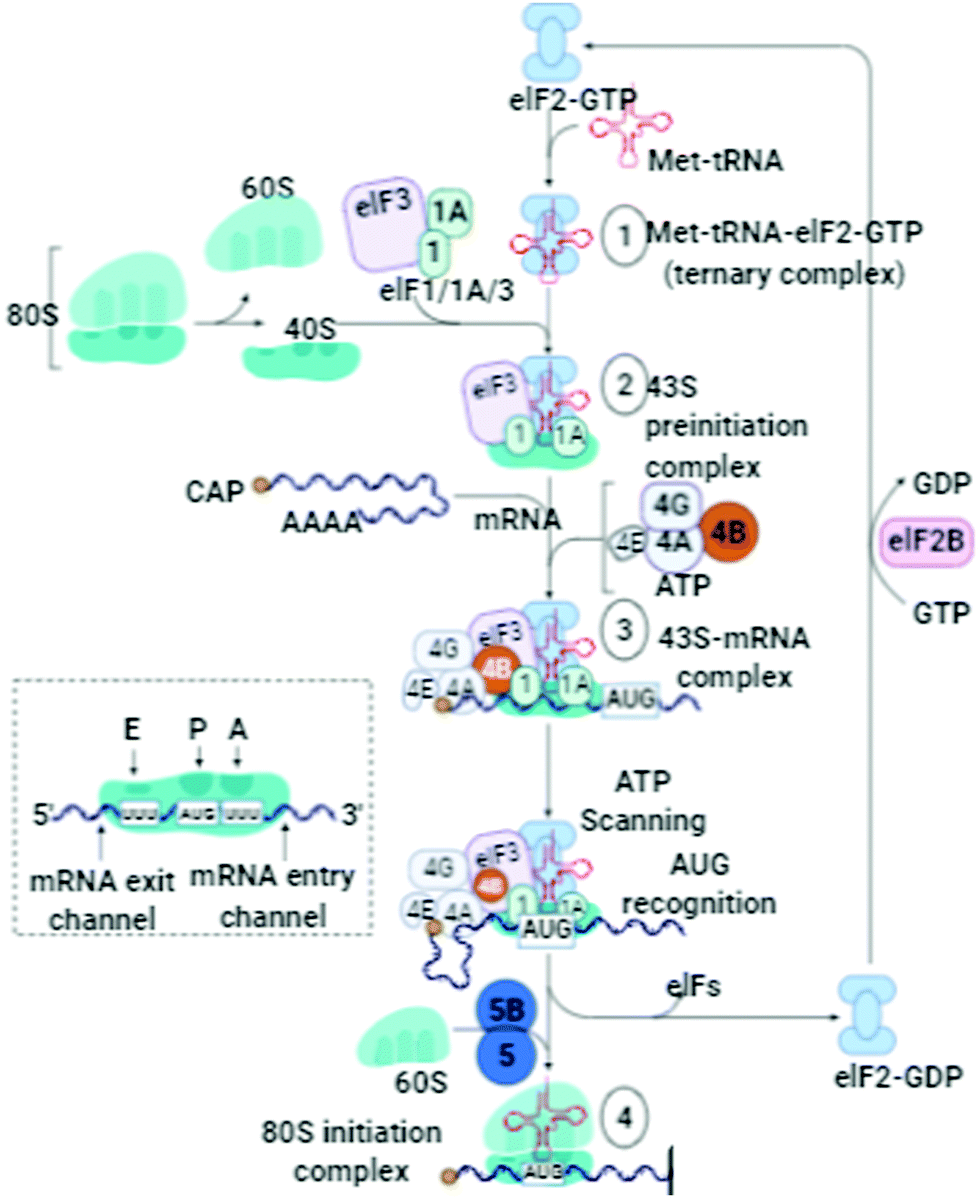 | ||
| Fig. 8 Schematic of the protein translation cascade, ER, signal sequence cleavage and link between protein-misfolding/folding and chemical instability, bond formation (oxidation) and bond cleavage (hydrolysis). “Adapted and created with permission from [http://biorender.com]. Same is referred and acknowledged as per instructions”. | ||
Thus, it inhibits Corin and is involved in peptide cell surface expression and prothrombin secretion.75 Calnexin expression controls the Corin cell surface expression and prothrombin secretion in their protease domains. Functional modification of the RING finger and the ubiquitin system, (catalyzing cofactor and amino acid biosynthesis) having adenylating E1-like enzymes and ubiquitin-like proteins (Ubls) can allow then to possess E2 enzymes, respectively. The class of RING finger E3s is operational with E1s, E2s, JAB domain peptides, and Ubls. These E1s, E2s, and RING fingers change gradually. They are involved in sequencing with the creation of an oxyanion-hole in E2s and C-terminal in the E1 enzyme existing with E2 in the active site. These RING fingers show different functions and not all of them participate in Ub-related functions. The analysis of the kinetic accessibility during the folding and refolding processes may be helpful to understand the folding patterns. To understand the protein conformation conversions and interactions, which are the central domain of the rational design of new tools in the field of interdisciplinary research (biomaterial sciences, nanotechnology76,77 and nanomedicine,78,79 and findings in biochemistry, chemistry, medicine, and physics),80,81 theoretical interpretations and analysis of proteins must be explored. Further, the role of the protein–surface binding mechanisms43,45 in stimulating the binding specificity of proteins on different surfaces must be investigated simultaneously. The obtained results can be employed to alter models with the detailed features of the thermodynamics and kinetics of adsorption.82 These models can be achieved by employing computer programming to explore the possibilities and routes followed by the dynamics of the proteins and related materials. These methods can also reveal the existing similarities and dissimilarities in proteins. Further, this highlights the role of the employed models and force-field tactics utilized for the analysis of the physical properties and the chemical/physical interactions at the material surfaces. These results can differentiate the related features of biological molecules in comparison with other materials.83 In particular, the force fields and other models employed for biomolecular simulations84 are often not directly applicable to detect the feasibility of surface simulations and vice versa.
Undiscovered pathways and mechanisms: the what, the whys, and the hows
An outline of the ER-associated degradation pathway and the mechanism of tracking pathways are highlighted for further analysis. The mechanism of tagging ubiquitin substrates by the coordinated action of cell organelles/proteins/enzyme/ATP/gene alteration/DNA/PQCPs/cell cleaning is one of the key pathways to be highlighted. The analysis of the presence of intra- and extracellular deposits of proteins, which alter conformation and post-translational modifications by prior attachment to polymers of ubiquitin, must be specifically done and applied to the interpretation of various pathways. The analyses of the pathologies of the protein misfolding underline the intra/extra cellular aggregation that is executed at the time of the origin of the diseases in proteins. One more requirement of these studies and interpretation is to determine the terminuses existing in the secretory pathway. These investigations can also discover the impact of the non-native and unassembled subunits existing in the multimeric proteins degraded through ubiquitination. These phenomena occur in the residues marked as the proteasomal degradation signal, which are the hidden areas that must be identified and underlined in the near future. The detection of diseases associated with ERAD substrates is important in the analysis of these pathways and mechanisms with emphasis on the factors that catalyze distinct activities. Therefore, the necessity of cell cleaning and interrelated cell processes under proteotoxic stress and situations must be investigated for a suitable answer. The illustration of the different routes can lead to the exploration and understanding of the mitigating mechanism involved in the translation rate. The sequestration of mRNA-encoded cleaning routes is adopted in proteins. Here, do stress granules assist the redirection of energy in the direction of crucial pro-survival pathways?85 Molecular chaperones and associated factors recognize and target substrates for retrotranslocation to the cytoplasm, wherein they are degraded by the ubiquitin–proteasome machinery. How the molecular machinery identifies the main proteins involved in the handling of mitochondrial Ca2+ traffic must be analyzed and the findings must be utilized for opening new fascinating and ambitious avenues for the development of mitochondria-based pharmacological strategies. The Ca-activated and ATP-independent proteolytic processes are correlated with the cysteine proteases, which are termed calpains.86 How are these proteases activated during cell injury and what is their role in the increase of cytosolic Ca2+ must be investigated. What to do to distinguish between the folded and out-of-register misfolded β-hairpin structures (N,N-terminus and C,C-terminus), and the side-chains of hydrophobic residues above/below both hairpins? The answer to these queries will be a key option to redefine several features of these processes. Why do the β-strands have a predisposition to self-associate to form β-sheets, which consist of several β-strands bound together by hydrogen bonds between the backbone atoms?87 There is a need to search for suitable causes responsible for them. For instance, this can be defined by a single set of (φ and Ψ) values, for both the folded (F) and misfolded (M) states. Therefore, the dihedral angles of the turn residues may be used to differentiate these two distinct states.Thereafter, in the configurations of the misfolded β-hairpin, a shift in residues occurs to regulate it. These shifts in the residue vary during the assembling of the hydrogen bonds in the pattern. One of the configurations, omega (ω), exists around the C–N bond secondary types. All the successive deposits displayed similar dihedral angle (φ and ψ) values in the identified regions.88 The main component of protein assembly is the α-helix, which can be easily identified easily by its spiral appearance. It is important to know how the structural features stabilize the process of the formation of H-bonds. These hydrogen bonds are situated between the atoms of successive residues of proteins. These descriptions analyze the origin of the misfolding occurring in β-hairpin. This can be further supported by investigations into β-hairpin folding.29 It is a challenging task to detect the impact of misfolded proteins in the creation of the β-hairpin (naturally bent arrangement) during the redesigning of experiments. There are different types of co- and post-translational modifications occurring in the lumen of the ER.
Signal sequence cleavage, N-glycosylation, glycosylphosphatidylinositol (GPI)-anchoring, and disulfide-bond formation/isomerization are the main transformation routes and pathways required to be investigated. To elucidate the link between protein-misfolding/folding and chemical instability, there is a need to explore the mechanism of bond formation (oxidation) or bond cleavage (hydrolysis), which is usually applied (Fig. 9). These mechanisms originate naturally and play an important role in tissue injury, necrosis, and autolysis, and thus these facts must be scrutinized. For further research, there are so many other unsolved mysteries persisting in these pathways, which need to be answered. For example, how does a greater percentage of Ca2+ in the lumen stimulate extracellular existence and is responsible for the stable conformation implementation? These routes govern the secretive pathways of Ca2+, which initiate the activities of the calcium/calmodulin-dependent protein kinase II. These transformations occur through biochemical changes and triggered apoptosis. Also, mitochondrial pathways and death receptors play a crucial role in them.89 Ubiquitin renews the routes and the concerned pathways to modify and alter the proteins inevitably. This may influence the alternative (non-degradative) roles of ubiquitin in the proper regulation of the routes and pathways of DNA repair and endocytosis. Thus, it is necessary to expose these processes and determine how these mechanisms play a crucial role in the regulation of events such as the recognition, unfolding, and cleavage of proteins. The process of mitochondrial Ca2+ homeostasis has been identified and applied in therapeutic strategies to treat several disorders. These approaches propose new themes to find suitable treatment options biochemically. Accordingly, identifying the molecular processes involved in mitochondrial Ca2+ traffic and their further proper utilization for opening new channels in the innovation and development of mitochondria-based pharmacological strategies are the main goal of this review.
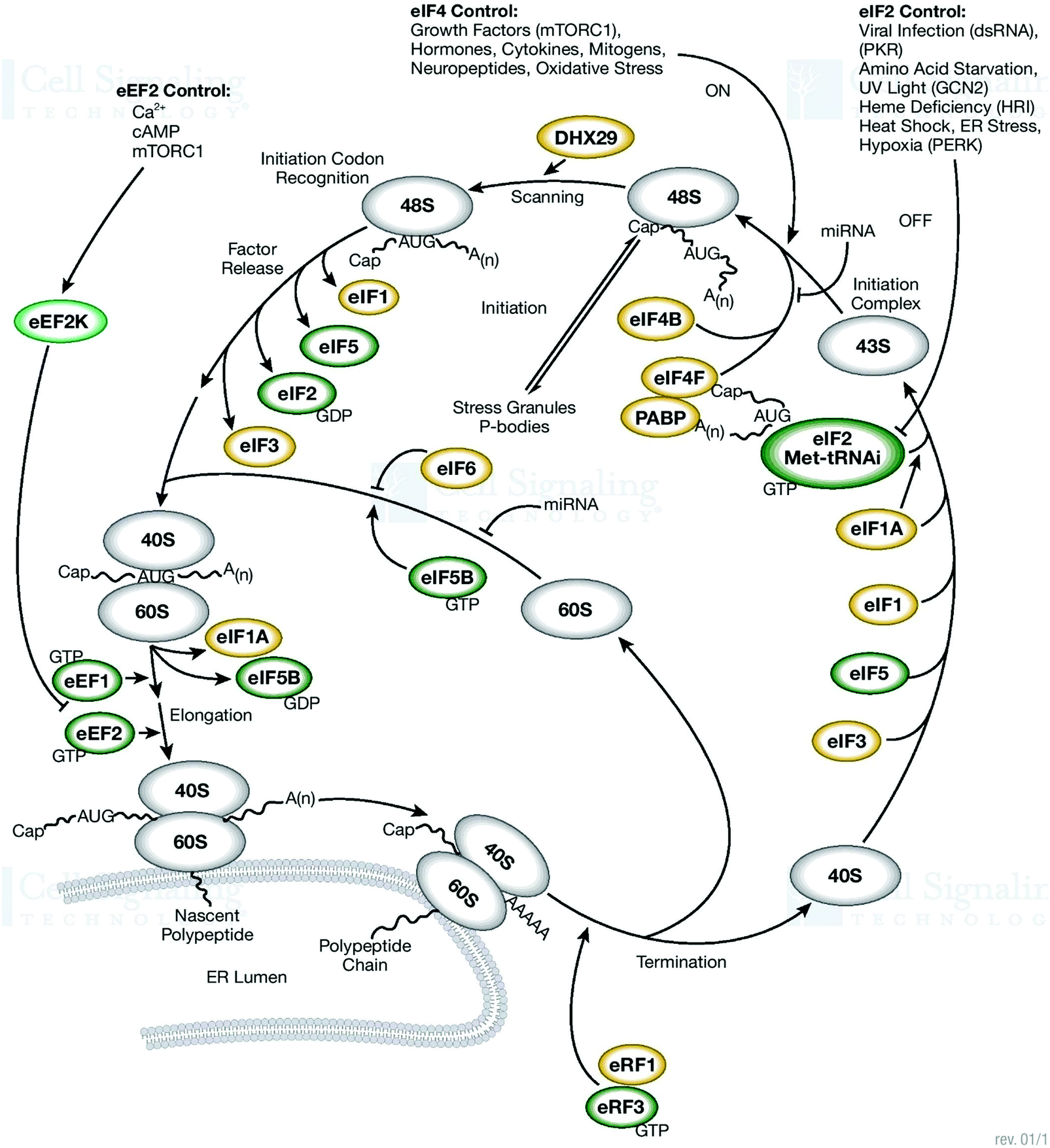 | ||
| Fig. 9 Schematic of the translation control over various co- and post-translational modifications in the lumen of the ER (signal sequence cleavage, N-glycosylation, glycosylphosphatidylinositol (GPI)-anchoring, and disulfide-bond formation/isomerization). “Adapted with permission from [http://www.cellsignal.com] and the same is acknowledged”. | ||
How to distinguish between proteins that have definite degradation signals and degrons? How to understand E3 ligase at the N-end rule pathway that has inner lysine residue? How to identify the unstructured N-terminal extension that is responsible for ubiquitination? How to determine the substrates of ubiquitination that provide a degradation signal (degron)? What are the main components of these signaling transformations? Finally, how to identify an N-terminal type 1 disrupting residue or type 2 destabilizing residues? These are the main queries to be discussed and answered urgently.90 Furthermore, similar questions such as why does the alteration of the Ca2+ signals influence the functioning of the mitochondria in various pathological conditions? Also, how does oxidative stress generate processes responsible for initiating apoptosis? These hidden mechanisms and their related facts must be revealed. These investigations can help to focus on the urgency of the development of advanced mitochondria-based therapies as a natural remedy. The ubiquitin proteasome pathway is involved in anonymous, unidentified, and hidden pathways of unconventional (non-degradative) functions for the regulation of apoptosis. The research findings on enzymatic components link chains of Ub on proteins including the E1, E2, and third enzyme, E3, which is a key factor in terms of the specificity of proteolysis at the expense of energy in the form of ATP. These interpretations will be crucial to explain the proper pathway of ubiquitin activation.91 Neurodegenerative diseases have a negative influence on heme biosynthesis, oxygen sensing, calcium homeostasis, and cell growth. Factors such as responses to stress, extracellular modulators, immune disorders, and inflammation must be considered as major risk factors during higher incidence and initiate aging. Enzymes/metal catalysis and cellular environmental stress are among the critical reasons for the destruction of cell constituents during apoptosis. To underline the needs of this discussion, in this review article, we recommended the following key points that need to be addressed (1) the boosting phenomenon of the cellular defense structures in the topology of misfolded proteins, (2) identifying strategies for survival failures of protein quality control, (3) causes of proteasome failure, (4) determining the persisting failures occurring during quality control in mitochondria, ER, nuclear and ribosomal PQCPs, and (5) identify the enzymes that can be catalyzed by a chemical mechanism, which will open doors to understanding how pathological protein quality control catastrophes reduce protein synthesis to increase folding. To detect the pathways that are not associated with protein synthesis, there is an urgent need to identify these veiled, anonymous, and unidentified causes, which are solely responsible for increasing folding.
Role of optical tweezers in protein folding
The key causes of any error occurring in the regular and natural phenomenon of protein folding require the appropriate analysis. It is very important to know how it occurs and what are the mechanisms behind it. By utilizing optical tweezers, it is easy to determine the main causes responsible for these errors in folding together with their related features.92 Therefore, the involved pathways and related parameters can be investigated and identified by interrupting the extract folding energies and single-molecule measurements. Further, optical tweezers are quite helpful in determining the topographical changes, ranging from linear to three-dimensional, originating by the disrupted sequencing in dynamic equilibrium. This high-resolution technique is based on single-molecule manipulation, and thus can exposes the protein folding of a single protein or protein complex located between two beads. Their existence can be identified by optical traps and pulled as a transition. The analysis of the changes and persist disruptions in protein topologies during conformations and energies of folding intermediates together with associated transition kinetics will be the key to gaining further insight. Optical tweezers play a crucial role in revealing the associated pathways necessary for the investigation of the folding of proteins and protein complexes, including data analysis, and folding energies from single-molecule measurements. Generally, optical tweezers detect the time-resolved extension of a single macromolecule, which is a force-dependent property within two trapped beads.93 Thus, it involves the measurement of various characteristics related to the folding intermediates, energies, and kinetics of the concerned macromolecule. Previously, the reaction coordinates were applied to identify the observed folding transitions that superficially define the folding without considering the impact of the force. However, in this methodology, the total extensions, energies, and transition rates of the proteins are considered for the calculation as a function of force to determine the best-fit conformations in related folding states. The folding of macro proteins occurs in the translation process to discontinue inter-domain misfolding. The aspect of this analysis is to observe the folding intermediates and illustrate the sequential folding pathway originating from the C-terminus. Simply, the forces govern the structure and dynamics of these biomacromolecules by enabling folding, disrupting their assembly, and determining the conformational fluctuations. Optical tweezers can analyze the features of molecular motors that translocate DNA, which are ATP-dependent chromatin and remodel to re-structure chromatin. These findings evidence the impactful role and features of high-resolution optical tweezers during the quantitative illustration of the multi-faceted and multi-scale dynamics of biomacromolecules.Conclusion
Research is necessary to determine the significant errors in systems, error correction mechanisms, and early etiological foundations of health and diseases such as protein conformational disorders (PCDs), trafficking defects, mitochondria-to-nucleus signaling pathway, cardiovascular diseases, neuronal dysfunction, endothelial dysfunction, diabetes mellitus, aqueous-deficient dry eyes (ADDE), age-related macular degeneration, cataracts, proteasome activity, neurodegenerative disease, Pick's disease, and ubiquitination in the pathogenesis of human diseases. The analysis and interpretation of the (1) mechanism of organelle-specific PQCPs, (2) cellular strategies to scrutinize proteins at the endoplasmic reticulum (ER)–Golgi interface, (3) ribosomal perspective on proteostasis at the expense of ATP hydrolysis, (4) molecular chaperones-therapeutic approaches, (5) calcium-activated potassium channels, (6) degradation signal (degron) and variations in degradation signals, (7) pathway of the stress to cellular homeostasis, (8) building blocks and molecular dissection of the secretory pathway, and (9) genetic alterations and proteostasis networks are very prolific, which were the main objectives of this review article. Further, we attempted to cover important pathways and different mechanisms of (1) metal–protein interface originating from an interaction that is similar to the Lewis acid/base reaction and the other similar regulators occurring during the interaction of metal–, ligand–, and enzyme–metal complexation, where all the induced metals act as an electron donor or acceptor according to the need of the interface/reaction/interaction. (2) Principal types of machinery maintaining proteostasis–ubiquitin proteasomal and lysosomal autophagy systems. (3) Cell survival responses to environmental stress, oxidants, metabolism, and signal transduction. (4) How cells adapt to proteasome assembly under stress conditions. (5) Boosting cellular resilience to protein misfolding. (6) Identifying strategies for proteasome activation by natural or chemical compounds. (7) Endothelial apoptosis, angiogenesis and proteasomal degradation. (8) Serine-protease inhibitors, and (9) ring-finger domains. Important suggestions were presented to identify these hidden pathways and mechanisms occurring within the cellular compartment, wherein protein misfolding and error in the mitochondria-to-nucleus signaling pathway are responsible for the degradation of whole types of machinery and deteriorated cell environmental conditions. Further, it was suggested that the findings of the organic reaction mechanism, molecular alterations, metallic/enzymatic catalyzed bio-inorganic/organic transformation, computational spectroscopy and predictions, and interdisciplinary experiences are worthwhile for these interpretations. Finally, it is believed that these efforts will be very useful to delineate the hidden cellular mechanisms.Author contributions
RK supervised and wrote this review article. BC, KG, and MC suggested revising the manuscript. All authors read and approved the manuscript.Funding
The authors thank the national institute of medical science for its financial support.Availability of data and materials
Wherever necessary, relevant citations are included in the reference section.Conflicts of interest
The authors have declared that no competing interest exists.Acknowledgements
One of the authors (Rajiv Kumar) gratefully acknowledges his younger brother Bitto for motivation. The authors acknowledge bio render for providing facility to illustrate the diagrams. The authors acknowledge the support towards the reproduction of the illustrations (Fig. 2, 4 and 7–9) as a courtesy from Cell Signaling Technology, Inc. (http://www.cellsignal.com).References
- J. Gandhi, A. C. Antonelli, A. Afridi, S. Vatsia, G. Joshi, V. Romanov, I. V. J. Murray and S. A. Khan, Rev. Neurosci., 2019, 30, 339–358 Search PubMed.
- N. Hosokawa, I. Wada, K. Hasegawa, T. Yorihuzi, L. O. Tremblay, A. Herscovics and K. Nagata, EMBO Rep., 2001, 2, 415–422 CrossRef CAS.
- K. R. Thomas, K. J. Bangen, A. J. Weigand, E. C. Edmonds, C. G. Wong, S. Cooper, L. Delano-Wood and M. W. Bondi, Neurology, 2020, 94, e397–e406 CrossRef.
- A. H. Hsieh and J. D. Twomey, J. Biomech., 2010, 43, 137–145 CrossRef.
- L. Grozinger, M. Amos, T. E. Gorochowski, P. Carbonell, D. A. Oyarzún, R. Stoof, H. Fellermann, P. Zuliani, H. Tas and A. Goñi-Moreno, Nat. Commun., 2019, 10, 5250 CrossRef.
- S. I. Nishikawa, J. L. Brodsky and K. Nakatsukasa, J. Biochem., 2005, 137, 551–555 CrossRef CAS.
- J. Lavie, H. De Belvalet, S. Sonon, A. M. Ion, E. Dumon, S. Melser, D. Lacombe, J. W. Dupuy, C. Lalou and G. Bénard, Cell Rep., 2018, 23, 2852–2863 CrossRef CAS.
- M. P. Mayer and L. M. Gierasch, J. Biol. Chem., 2019, 294, 2085–2097 CrossRef CAS.
- S. Acharyya, Chem. Biol. Lett., 2020, 7, 99–112 CAS.
- I. Moreno-Gonzalez and C. Soto, Semin. Cell Dev. Biol., 2011, 22, 482–487 CrossRef CAS.
- M. P. Jackson and E. W. Hewitt, Essays Biochem., 2016, 60, 173–180 CrossRef.
- R. Dabur, B. Sharma and A. Mittal, Chem. Biol. Lett., 2018, 5, 63–99 Search PubMed.
- D. P. de Lima, T. N. Reddy, A. Beatriz, R. da, S. Lopes, M. R. Marques, E. dos, A. dos Santos, C. I. Nantes, M. de, F. C. Matos, R. T. Perdomo, I. C. Pereira and S. S. Weber, Chem. Biol. Lett., 2018, 5, 41–54 Search PubMed.
- P. Jadiya and D. Tomar, Genes, 2020, 11, 563 CrossRef CAS.
- C. Xiang, S. Zhang, X. Dong, S. Ma and S. Cong, Front. Mol. Neurosci., 2018, 11, 153 CrossRef.
- S. Savickas, P. Kastl and U. auf dem Keller, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140392 CrossRef CAS.
- S. Pemberton, K. Madiona, L. Pieri, M. Kabani, L. Bousset and R. Melki, J. Biol. Chem., 2011, 286, 34690–34699 CrossRef CAS.
- Y. Feng, D. He, Z. Yao and D. J. Klionsky, Cell Res., 2014, 24, 24–41 CrossRef CAS.
- A. Kuma, M. Komatsu and N. Mizushima, Autophagy, 2017, 13, 1619–1628 CrossRef CAS.
- M. Ponpuak, M. A. Mandell, T. Kimura, S. Chauhan, C. Cleyrat and V. Deretic, Curr. Opin. Cell Biol., 2015, 35, 106–116 CrossRef CAS.
- L. L. Yan and H. S. Zaher, EMBO J., 2019, 38, e101633 CrossRef.
- P. Bragoszewski, M. Turek and A. Chacinska, Open Biol., 2017, 7, 170007 CrossRef.
- F. H. Ma, C. Li, Y. Liu and L. Shi, Adv. Mater., 2020, 32, e1805945 CrossRef.
- R. S. Balaban, S. Nemoto and T. Finkel, Cell, 2005, 120, 483–495 CrossRef CAS.
- K. S. Lee and B. Lu, Front. Cell. Neurosci., 2014, 8, 330 Search PubMed.
- L. Fernandez-Mosquera, K. F. Yambire, R. Couto, L. Pereyra, K. Pabis, A. H. Ponsford, C. V. Diogo, M. Stagi, I. Milosevic and N. Raimundo, Autophagy, 2019, 15, 1572–1591 CrossRef CAS.
- O. Moltedo, P. Remondelli and G. Amodio, Front. Cell Dev. Biol., 2019, 7, 172 CrossRef.
- S. Long and P. Tian, Sci. Rep., 2016, 6, 34481 CrossRef CAS.
- Q. Shao, J. Phys. Chem. B, 2015, 119, 3893–3900 CrossRef CAS.
- A. Shelar, P. Kumar and M. Bansal, Biomolecular Forms and Functions: A Celebration of 50 Years of the Ramachandran Map, 2012, pp. 116–127 Search PubMed.
- G. von Heijne, Eur. J. Biochem., 1983, 133, 17–21 CrossRef CAS.
- C. J. Bosques, S. M. Tschampel, R. J. Woods and B. Imperiali, J. Am. Chem. Soc., 2004, 126, 8421–8425 CrossRef CAS.
- L. E. Kay, M. Ikura, R. Tschudin and A. Bax, J. Magn. Reson., 1990, 89, 496–514 CAS.
- B. Panaretou, C. Prodromou, S. M. Roe, R. O’Brien, J. E. Ladbury, P. W. Piper and L. H. Pearl, EMBO J., 1998, 17, 4829–4836 CrossRef CAS.
- H. Sghaier, T. L. H. Ai, T. Horiike and T. Shinozawa, In Silico Biol., 2004, 4, 311–322 CAS.
- Y. Feng, W. Li, J. Li, J. Wang, J. Ge, D. Xu, Y. Liu, K. Wu, Q. Zeng, J. W. Wu, C. Tian, B. Zhou and M. Yang, Nature, 2012, 491, 478–482 CrossRef CAS.
- D. Akhmedov, M. Braun, C. Mataki, K. Park, T. Pozzan, K. Schoonjans, P. Rorsman, C. B. Wollheim and A. Wiederkehr, FASEB J., 2010, 24, 4613–4626 CrossRef CAS.
- H. J. Hwang, Y. R. Yang, H. Y. Kim, Y. Choi, K. S. Park, H. Lee, J. S. Ma, M. Yamamoto, J. Kim, Y. C. Chae, J. H. Choi, L. Cocco, P. O. Berggren, H. J. Jang and P. G. Suh, FASEB J., 2019, 33, 10668–10679 CrossRef CAS.
- S. Sharma and V. Bhatia, Chem. Biol. Lett., 2020, 7, 215–224 CAS.
- Y. Pan, Exp. Gerontol., 2011, 46, 847–852 CrossRef CAS.
- C. Giorgi, C. Agnoletto, A. Bononi, M. Bonora, E. de Marchi, S. Marchi, S. Missiroli, S. Patergnani, F. Poletti, A. Rimessi, J. M. Suski, M. R. Wieckowski and P. Pinton, Mitochondrion, 2012, 12, 77–85 CrossRef CAS.
- S. Voet, S. Srinivasan, M. Lamkanfi and G. Loo, EMBO Mol. Med., 2019, 11, e10248 Search PubMed.
- V. Chandel, M. Srivastava, A. Srivastava, S. Asthana and D. Kumar, Chem. Biol. Lett., 2020, 7, 47–54 CAS.
- D. K. Behera, P. M. Behera, L. Acharya and A. Dixit, Chem. Biol. Lett., 2017, 4, 1–8 CAS.
- D. Sharma, M. Pathak, R. Sharma, N. Sharma, P. Tyagi, R. Chawla, M. Basu and H. Ojha, Chem. Biol. Lett., 2017, 4, 27–32 CAS.
- M. Compiani and E. Capriotti, Biochemistry, 2013, 52, 8601–8624 CrossRef CAS.
- K. V. Chanu, D. Thakuria, R. Sood, A. K. Pateriya, S. Bhatia and S. Kumar, Chem. Biol. Lett., 2017, 4, 20–26 CAS.
- R. G. Pillai, Chem. Biol. Lett., 2020, 7, 41–46 CAS.
- X. Yuan, M. D. Johnson, J. Zhang, A. W. Lo, M. A. Schembri, L. C. Wijeyewickrema, R. N. Pike, G. H. M. Huysmans, I. R. Henderson and D. L. Leyton, Nat. Commun., 2018, 9, 1395 CrossRef.
- F. Simona, G. Tiana, R. A. Broglia and G. Colombo, J. Mol. Graphics Modell., 2004, 23, 263–273 CrossRef CAS.
- A. J. Kowaltowski, S. L. Menezes-Filho, E. A. Assali, I. G. Gonçalves, J. V. Cabral-Costa, P. Abreu, N. Miller, P. Nolasco, F. R. M. Laurindo, A. Bruni-Cardoso and O. S. Shirihai, FASEB J., 2019, 33, 13176–13188 CrossRef CAS.
- W. Y. Wang, M. S. Tan, J. T. Yu and L. Tan, Ann. Transl. Med., 2015, 3, 136 Search PubMed.
- Y. Nakazawa, Y. Hara, Y. Oka, O. Komine, D. van den Heuvel, C. Guo, Y. Daigaku, M. Isono, Y. He, M. Shimada, K. Kato, N. Jia, S. Hashimoto, Y. Kotani, Y. Miyoshi, M. Tanaka, A. Sobue, N. Mitsutake, T. Suganami, A. Masuda, K. Ohno, S. Nakada, T. Mashimo, K. Yamanaka, M. S. Luijsterburg and T. Ogi, Cell, 2020, 180, 1228–1244.e24 CrossRef CAS.
- M. P. Murphy, Biochem. J., 2009, 417, 1–13 CrossRef CAS.
- Y. Tu, C. Chen, J. Pan, J. Xu, Z. G. Zhou and C. Y. Wang, Int. J. Clin. Exp. Pathol., 2012, 5, 726–738 CAS.
- S. K. Misra, P. Moitra, B. S. Chhikara, P. Kondaiah and S. Bhattacharya, J. Mater. Chem., 2012, 22, 7985–7998 RSC.
- B. S. Chhikara, S. K. Misra and S. Bhattacharya, Nanotechnology, 2012, 23, 065101 CrossRef.
- R. Oshidari, K. Mekhail and A. Seeber, Trends Cell Biol., 2020, 30, 144–156 CrossRef CAS.
- M. O. Turgeon, N. J. S. Perry and G. Poulogiannis, Front. Oncol., 2018, 8, 15 CrossRef.
- P. J. Bindu, T. R. R. Naik, K. M. Mahadevan and G. Krishnamurthy, Chem. Biol. Lett., 2019, 6, 8–13 CAS.
- P. Sengupta, S. Dutta, A. Alahmar and U. J. A. D'souza, Chem. Biol. Lett., 2020, 7, 75–84 CAS.
- D. Branzei and M. Foiani, Nat. Rev. Mol. Cell Biol., 2008, 9, 297–308 CrossRef CAS.
- S. YAlçın, P. Mutlu, G. Unsoy, R. Khodadust and U. Gunduz, J. Mater. Nanosci., 2020, 7, 29–35 Search PubMed.
- B. S. Chhikara, S. Ashraf, S. Mozaffari, N. St Jeans, D. Mandal, R. K. Tiwari, Z. Ul-Haq and K. Parang, Molecules, 2020, 25, 2135 CrossRef CAS.
- M. Hedglin, M. Aitha, A. Pedley and S. J. Benkovic, J. Biol. Chem., 2019, 294, 5157–5168 CrossRef CAS.
- S. Devi, J. Singh, V. Kumar and V. Malik, Chem. Biol. Lett., 2019, 6, 39–45 CAS.
- P. Greulich and B. D. Simons, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7509–7514 CrossRef CAS.
- Z. Cao, D. Goyal, S. E. Meiler, Y. Zhou and W. S. Dynan, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 2196–2204 CrossRef CAS.
- B. Gooptu, J. A. Dickens and D. A. Lomas, Trends Mol. Med., 2014, 20, 116–127 CrossRef CAS.
- T. Sawada, M. Yamagami, S. Akinaga, T. Miyaji and M. Fujita, Chem. – Asian J., 2017, 12, 1715–1718 CrossRef CAS.
- W. Zhu, L. Li, M. Deng, B. Wang, M. Li, G. Ding, Z. Yang, D. Medynski, X. Lin, Y. Ouyang, J. Lin, L. Li and X. Lin, FEBS Open Bio, 2018, 8, 1711–1721 CrossRef CAS.
- B. Wu, A. J. McDonald, K. Markham, C. B. Rich, K. P. McHugh, J. Tatzelt, D. W. Colby, G. L. Millhauser and D. A. Harris, eLife, 2017, 6, e23473 CrossRef.
- J. A. Carver, A. B. Grosas, H. Ecroyd and R. A. Quinlan, Cell Stress Chaperones, 2017, 22, 627–638 CrossRef CAS.
- L. Meuris, F. Santens, G. Elson, N. Festjens, M. Boone, A. Dos Santos, S. Devos, F. Rousseau, E. Plets, E. Houthuys, P. Malinge, G. Magistrelli, L. Cons, L. Chatel, B. Devreese and N. Callewaert, Nat. Biotechnol., 2014, 32, 485–489 CrossRef CAS.
- F. Schwarz and M. Aebi, Curr. Opin. Struct. Biol., 2011, 21, 576–582 CrossRef CAS.
- R. Kumar and M. Sharma, J. Mater. Nanosci., 2018, 5, 35–58 Search PubMed.
- R. Kumar, B. S. Chhikara, K. Gulia and M. Chhilar, Theranostics DOI:10.7150/thno.49619.
- B. S. Chhikara, J. Mater. Nanosci., 2017, 4, 19–24 Search PubMed.
- B. S. Chhikara, R. Kumar, B. Rathi, S. Krishnamoorthy and A. Kumar, J. Mater. Nanosci., 2016, 3, 50–56 Search PubMed.
- M. Ozboyaci, D. B. Kokh, S. Corni and R. C. Wade, Q. Rev. Biophys., 2016, 49, e4 CrossRef.
- B. S. Chhikara, B. Rathi and K. Parang, J. Mater. Nanosci., 2019, 6, 47–66 CAS.
- E. A. Roman and F. L. G. Flecha, Biomolecules, 2014, 4, 354–373 CrossRef.
- E. Flapan, A. He and H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9360–9369 CrossRef CAS.
- W. S. El-serwy, N. A. Mohamed, E. M. M. Kassem, E. S. Nossier and A. S. G. Shalaby, Chem. Biol. Lett., 2020, 7, 197–206 CAS.
- H. Zare, A. Khodursky and V. Sartorelli, BMC Evol. Biol., 2014, 14, 74 CrossRef.
- M. Wilkowska, W. Andrzejewska, R. Zieliński and M. Kozak, Biophys. J., 2018, 114, 221a CrossRef.
- P. H. Nguyen, J. M. Campanera, S. T. Ngo, A. Loquet and P. Derreumaux, J. Phys. Chem. B, 2019, 123, 3643–3648 CrossRef CAS.
- A. Altis, P. H. Nguyen, R. Hegger and G. Stock, J. Chem. Phys., 2007, 126, 244111 CrossRef.
- S. J. Baumgart and B. Haendler, Int. J. Mol. Sci., 2017, 18 Search PubMed.
- I. E. Suka, N. F. Roslan, B. L. Chew, H. H. Goh, Z. Zainal and N. M. Isa, Sains Malays., 2018, 47, 1465–1471 CrossRef CAS.
- D. H. Wolf and R. Menssen, FEBS Lett., 2018, 592, 2515–2524 CrossRef CAS.
- D. Bauer, S. Meinhold, R. P. Jakob, J. Stigler, U. Merkel, T. Maier, M. Rief and G. Žoldák, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4666–4671 CrossRef CAS.
- A. Mashaghi, G. Kramer, P. Bechtluft, B. Zachmann-Brand, A. J. M. Driessen, B. Bukau and S. J. Tans, Nature, 2013, 500, 98–101 CrossRef CAS.
Footnote |
| † The authors dedicate this review article to Hon’ble Dr Yoganand Shastri Ji, Distinguished Professor, University of Delhi; Honorable Speaker of Delhi Legislative Assembly; Cabinet Minister for Development, Food and Civil Supplies; and Cabinet Minister for Health and Social Welfare (Delhi Legislative Assembly). |
| This journal is © The Royal Society of Chemistry 2021 |