
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Linked glycosylation profiles of therapeutic induced senescent (TIS) triple negative breast cancer cells (TNBC) and their extracellular vesicle (EV) progeny†
Emma L.
Kavanagh
a,
Melinda
Halasz
 bc,
Paul
Dowling
d,
Jo
Withers
e,
Sinéad
Lindsay
a,
Michaela J.
Higgins
f,
Jane A.
Irwin
g,
Pauline M.
Rudd
e,
Radka
Saldova‡
*ae and
Amanda
McCann‡
*ab
bc,
Paul
Dowling
d,
Jo
Withers
e,
Sinéad
Lindsay
a,
Michaela J.
Higgins
f,
Jane A.
Irwin
g,
Pauline M.
Rudd
e,
Radka
Saldova‡
*ae and
Amanda
McCann‡
*ab
aUCD Conway Institute of Biomolecular and Biomedical Research, University College Dublin (UCD), Dublin, Ireland. E-mail: amanda.mccann@ucd.ie
bUCD School of Medicine, College of Health and Agricultural Science (CHAS), University College Dublin (UCD), Dublin, Ireland
cSystems Biology Ireland (SBI), University College Dublin (UCD), Dublin, Ireland
dBiology Department, National University of Ireland (NUI) Maynooth, Co., Kildare, Ireland
eThe National Institute for Bioprocessing, Research and Training (NIBRT), Dublin, Ireland. E-mail: radka.fahey@nibrt.ie
fOncology Department, Mater Misericordiae University Hospital (MMUH), Dublin, Ireland
gUCD School of Veterinary Medicine, University College Dublin (UCD), Dublin, Ireland
First published on 16th December 2020
Abstract
Triple negative breast cancer (TNBC) has poor clinical outcomes and limited treatment options. Chemotherapy, while killing some cancer cells, can result in therapeutic-induced-senescent (TIS) cells. Senescent cells release significantly more extracellular vesicles (EVs) than non-senescent cells. Recently, N- and O-linked glycosylation alterations have been associated with senescence. We aimed to profile the N-linked glycans of whole cells, membrane, cytoplasm and EVs harvested from TIS TNBC cells and to compare these to results from non-senescent cells. TIS was induced in the Cal51 TNBC cells using the chemotherapeutic agent paclitaxel (PTX). Ultra-performance liquid chromatography (UPLC) analysis of exoglycosidase digested N-linked glycans was carried out on TIS compared to non-treated control cells. LC-Mass spectrometry (MS) analysis of the N-linked glycans and lectin blotting of samples was carried out to confirm the UPLC results. Significant differences were found in the N-glycan profile of the Cal51 membrane, cytoplasm and EV progeny of TIS compared to non-senescent cells. Protein mass spectrometry showed that the TIS cells contain different glycan modifying enzymes. The lectin, calnexin demonstrated a lower kDa size (∼58 kDa) in TIS compared to control cells (∼90 kDa) while Galectin 3 demonstrated potential proteolytic cleavage with 32 kDa and ∼22 kDa bands evident in TIS compared to non-senescent control cells with a major 32 kDa band only. TIS CAL51 cells also demonstrated a reduced adhesion to collagen I compared to control non-senescent cells. This study has shown that therapeutic-induced-senescent TNBC cells and their EV progeny, display differential N-glycan moieties compared to non-senescent Cal51 cells and their resultant EV progeny. For the future, N-glycan moieties on cancer senescent cells and their EV progeny hold potential for (i) the monitoring of treatment response as a liquid biopsy, and (ii) cancer senescent cell targeting with lectin therapies.
Introduction
Glycosylation is an important post-translational modification in which glycans are attached to proteins and lipids, contributing to their diversity. Glycosylation has many functions including assisting protein folding in the endoplasmic reticulum, enabling ligand binding, mediating cell–cell interactions, aiding in immune system evasion and protecting proteins from recognition by proteases or antibodies.1 Altered glycosylation is a feature of tumour glycoproteins.2Many technical developments have improved glycan analysis including the development of glycosylation based software and databases, enabling a greater understanding of glycan modifications3–5 and facilitating glycan biomarker discovery in the serum from pathologies such as breast cancer.6,7 Most common methods used for the characterisation of released glycans include (i) capillary electrophoresis (CE), (ii) liquid chromatography-based methods such as reversed-phase liquid chromatography (RP-LC), and hydrophilic interaction chromatography (HILIC), (iii) mass spectrometry (MS)-based methods such as matrix-assisted laser desorption ionization (MALDI), (iv) electrospray ionisation (ESI), (v) ion trap instrument (ITMS), (vi) porous graphitised carbon liquid chromatography-tandem mass spectrometry (PGC-LC-MS/MS) or (vii) nuclear magnetic resonance (NMR) spectroscopy.3,7,8 Some of these methods, such as HILIC-UPLC, CE or MALDI have been developed in a high-throughput format potentially suitable for clinical applications.6,9 The approach used of separating 2-aminobenzamide (2AB) labelled glycans with HILIC-UPLC, combined with exoglycosidase digestions and LC-MS, allowed us accurately quantify as well as separate structural isomers.7
Alterations of N-linked and O-linked glycomes are associated with the senescence phenotype.8 Senescence-associated-beta-galactosidase (SA-β-Gal) is a well-recognised marker of cellular senescence.9 SA-β-Gal functions by cleaving the disaccharide, lactose, to form glucose and galactose which can then enter glycolysis.10 β-Galactosidase is used to study N-linked glycosylation,11 suggesting its importance in senescent cell N-linked glycosylation which have increased SA-β-Gal activity.
The enzyme alpha-fucosidase (FUCA1) cleaves fucose on N-linked glycans.12 FUCA1 has been described as a novel marker for cellular senescence.13 Low FUCA1 expression is associated with a poor prognosis in TNBC compared to non-TNBC presentations,14 while positive FUCA1 expression is associated with better survival for breast cancer patients with the luminal B lymph node-positive subtype compared to the luminal B lymph node-negative subtype.15 FUCA1 is downstream of p53, with its expression induced by chemotherapeutic drugs.16 Overexpression of FUCA1 suppresses the growth of cancer cell lines,17 suggesting its importance in senescence induction and maintenance.
Lectins are proteins which can specifically bind to glycans. A novel lectin called Eucheuma Serra Agglutinin (ESA) binds specifically to altered glycans on the surface of osteosarcoma cells inducing apoptosis.18 Mannose binding lectin induces cell lysis of aged senescent fibroblast cells but not non-senescent cells in vitro.19 This suggests that aged senescent cells display a differential glycan profile, enabling their specific cell lysis. A high mannose type glycan containing nine outer arm mannose residues has been found in an in vivo mouse model of breast cancer and in sera from breast cancer patients.20
A recent study followed the N-glycans from the serum glycoproteins of breast cancer patients over the course of their diagnosis and treatment. Following chemotherapy, the glycome contained pro-inflammatory glycans with lymph node positive disease sera containing an increase in galactosylation and sialylation (inflammation associated glycans), with a concomitant increase in glycans containing the Sialyl Lewis X epitope, highly branched and sialylated glycans (cancer associated glycans).21,22N-Glycan signatures may inform on treatment response and overall patient survival.
Extracellular vesicles (EV) are released by cells and are a major interest of liquid biopsy research.23 The tetraspanin CD63 is a well-documented exosomal marker24 with three N-linked glycosylation sites.25 Ribophorin II-mediated glycosylation of CD63 leads to co-localising with the multi-drug resistance protein 1 at the cell membrane, followed by an efflux of chemotherapy in breast cancer.26N-glycan profiling of B16–F10 melanoma EV subsets: Exo-S, Exo-L and exomeres, using lectin blotting and MS analysis, have shown that all three subsets of EVs had distinct N-glycan profiles.27
The objective of this study was to profile the N-linked glycans of whole cells, membrane, cytoplasm and EVs harvested from TIS Cal51 TNBC cells compared to non-senescent Cal51 TNBC cells. This approach holds potential as N-linked glycan alterations may identify cancer senescent cells amenable to lectin-based therapies, while alterations on EVs could potentially be analysed to monitor response to treatment as a liquid biopsy.
Results
Confirmation of paclitaxel induced TIS in Cal51 TNBC cells and extracellular vesicle (EV) isolation for glycan analysis
As previously described,28 Cal51 TNBC cells were treated with 75 nM paclitaxel (PTX) for seven days to induce therapeutic-induced-senescence (TIS), recognised as positive SA-β-Gal activity, characteristic large flattened morphology (Fig. 1A) and SDS-PAGE western blot detection of increased expression of p21 in senescent compared to control non-senescent cells (Fig. 1B). EVs were isolated from the Cal51 cells by differential centrifugation and confirmed to be calnexin negative (negative control) and CD63 positive (positive control) by SDS-PAGE western blotting (Fig. 1C). The smear present for CD63 in the lanes loaded with whole cell lysates (W) reflects the glycosylation of CD63 which is known to have 3 N-linked glycosylation sites,25 while a single band of ∼58 kDa is present in the EV lanes (E). EV isolation was further confirmed using nanoparticle tracking analysis (NTA) measuring the concentration and size of the particles (Fig. 1D).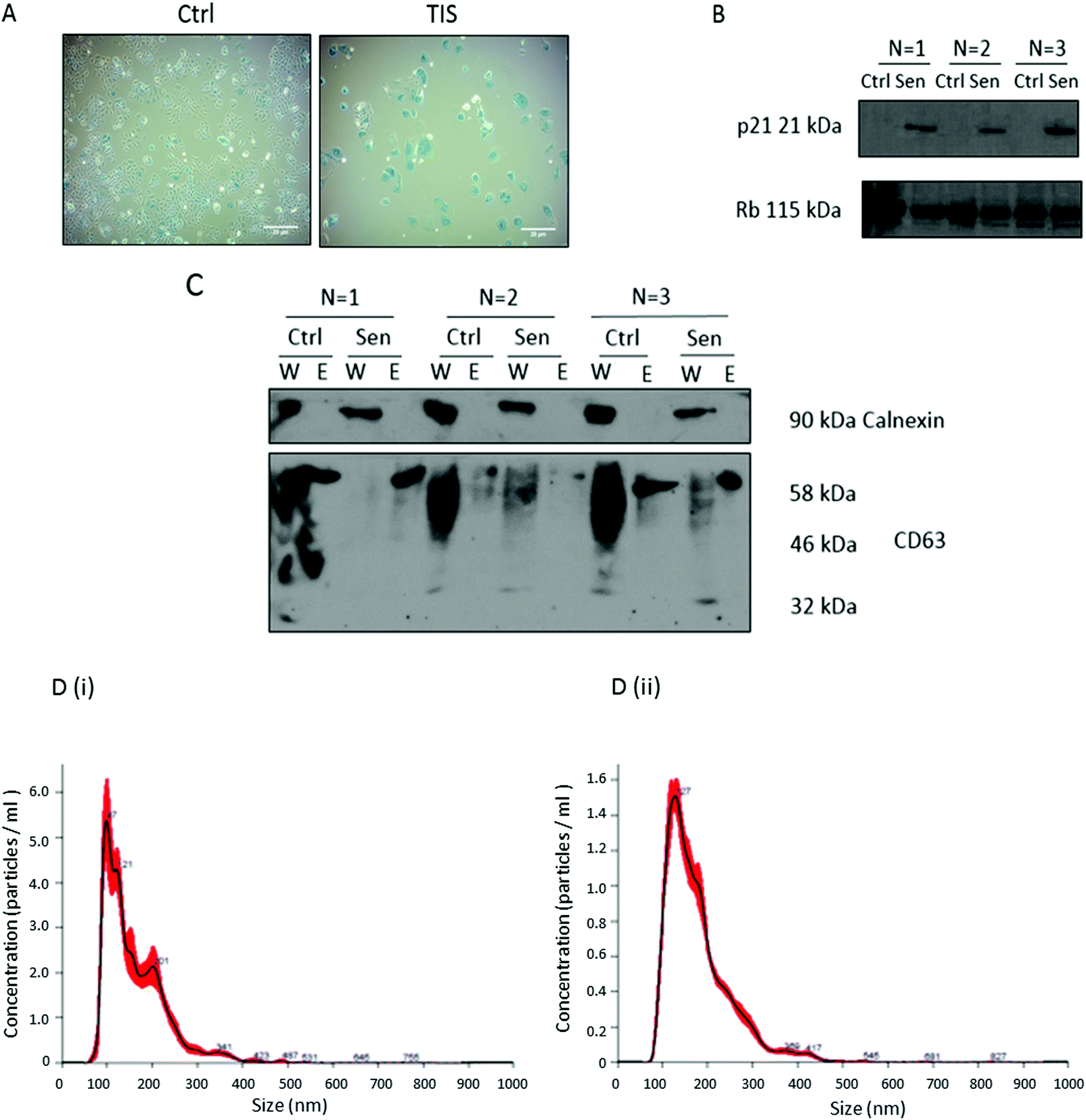 | ||
Fig. 1 Confirmation of PTX induced TIS in Cal51 cells treated with 75 nM PTX for seven days and successful isolation of EVs for glycan analysis. (A) Cal51 treated with 75 nM PTX for one week seeded at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well. Cells were stained using the SA-β-Gal staining kit (Cell Signalling) with 5 mg ml−1 X-gal. Scale bars represent 20 μm. (B) Cal51 cells were treated with 75 nM PTX for one week. SDS-PAGE western blotting was performed using a 12% gel for p21 and RB as a loading control. (C) EVs were isolated using ultracentrifugation and SDS-PAGE western blotting analyses were carried out for the EV marker CD63. Calnexin was used as a negative control as it represents an ER protein whose absence ensures EV enrichment, of endocytic origin. W stands for whole cell lysate and E stands for extracellular vesicles. The smear present for CD63 in the lanes loaded with whole cell lysates (W) reflects the glycosylation of CD63 which is known to have 3 N-linked glycosylation sites,25 while a single band of ∼58 kDa is present in the EV lanes. (D) Nanoparticle tracking analysis of EVs was carried out for both (i) control and (ii) TIS EVs using a NanoSight NS300 Malvern. This figure represents n = 3 biological replicates. 000 cells per well. Cells were stained using the SA-β-Gal staining kit (Cell Signalling) with 5 mg ml−1 X-gal. Scale bars represent 20 μm. (B) Cal51 cells were treated with 75 nM PTX for one week. SDS-PAGE western blotting was performed using a 12% gel for p21 and RB as a loading control. (C) EVs were isolated using ultracentrifugation and SDS-PAGE western blotting analyses were carried out for the EV marker CD63. Calnexin was used as a negative control as it represents an ER protein whose absence ensures EV enrichment, of endocytic origin. W stands for whole cell lysate and E stands for extracellular vesicles. The smear present for CD63 in the lanes loaded with whole cell lysates (W) reflects the glycosylation of CD63 which is known to have 3 N-linked glycosylation sites,25 while a single band of ∼58 kDa is present in the EV lanes. (D) Nanoparticle tracking analysis of EVs was carried out for both (i) control and (ii) TIS EVs using a NanoSight NS300 Malvern. This figure represents n = 3 biological replicates. | ||
HILIC-UPLC N-glycan profiles from the whole cell lysates (WCL), cytoplasm, membrane and the EV progeny from TIS and non-senescent Cal51 cells
HILIC-UPLC analysis of 2-AB labelled N-linked glycans of whole cell lysates (WCL), cytoplasm, membrane and EVs harvested from TIS Cal51 cells compared to non-senescent cells were profiled. There was no significant difference in the profile of N-glycans in the WCL of TIS cells compared to the WCL of non-senescent TNBC cells (ESI,† Fig. S1A). There were significant differences in the N-glycans in (i) the cytoplasm (ESI,† Fig. S1B), (ii) the membrane (ESI,† Fig. S1C) and in (iii) the EVs from TIS compared to non-senescent cells (ESI,† Fig. S1D). Statistics was carried out on three biological replicates (ESI,† Fig S2).Identification and determination of significant peak differences by HILIC-UPLC glycan analysis of 2-AB labelled N-glycans digested with exoglycosidases and their confirmation by LC/MS.
Digestions with exoglycosidase arrays followed by HILIC-UPLC analysis of the digest is a well described method to identify N-glycan peaks.7 The exoglycosidases used were sialidase A (ABS), ABS + β1-4-galactosidase (SPG), ABS + β1-3,4-galactosidase (BTG), ABS + BTG + β-N-acetylglucosaminidase (GUH), ABS + BTG + α1-3,4-fucosidase (AMF), ABS + BTG + α1-2,3,4,6-fucosidase (BKF), α1-2,3,6-mannosidase (JBM) and sialidase S (NAN1). HILIC-UPLC analysis of digestions using exoglycosidase arrays of 2-AB labelled N-linked glycans was carried out on WCLs (ESI,† Fig. S3A), cytoplasm (ESI,† Fig. S3C), membrane (ESI,† Fig. S3E) and EVs (ESI,† Fig. S3G), from TIS TNBC cells compared to non-senescent cells. The chromatograms from these digests were integrated using Empower™ software. Specific peak areas were assigned to N-glycans by analysing the shifts in the peak areas following digestions. Digestions with ABS and NAN1, identified linkage of sialic acids, as ABS digests all linkages of sialic acids, but NAN1 only digests alpha2–3 linked sialic acids. Digestions with ABS + BTG and ABS + SPG identified linkage of galactose, as BTG digests both beta1–3 and beta1–4 galactoses, but SPG only digests beta1–4 galactose. JBM identified high mannosylated and hybrid glycans and AMF and BKF identified outer arm fucosylated glycans and BKF identified core fucosylated glycans. GUH digested the final glucosamine residues (ESI,† Table S13 and Supplemental UPLC file).LC/MS analysis of undigested 2-AB labelled N-linked glycans was carried out on WCLs (ESI,† Fig. S3B), cytoplasm (ESI,† Fig. S3D), membrane (ESI,† Fig. S3F) and EVs (ESI,† Fig. S3H) from TIS TNBC cells compared to non-senescent cells. The masses of the assigned N-glycans from the HILIC-UPLC analysis of digestions with exoglycosidase arrays were determined using GlycoWorkbench 2 software. The chromatograms from the LC/MS analysis were checked for these mass values to confirm the HILIC-UPLC analysis of digestions with exoglycosidase arrays, using the MassLynx™ V4.1 software (ESI,† MS file). Significant N-linked glycan peak differences identified in ESI,† Fig. S1 were assigned via HILIC-UPLC glycan analysis of 2-AB labelled N-glycans digested with exoglycosidases, and all N-linked glycan assignments from the HILIC-UPLC digestion data were confirmed in the LC/MS data (Table 1).
| Sample | Increase or decrease | Peak | Main glycans in TIS peaks | UPLC ctrl peak area (%) ± SD | UPLC TIS Peak area (%) ± SD | UPLC average GU | MS ctrl experimental mass (m/z) | MS TIS experimental mass (m/z) | MS theoretical mass (m/z) | MS ion | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TIS Membrane | Increase | 13 | M5 |

|
1.02 ± 0.2 | 1.75 ± 0.2 | 6.11 | 1353.4824 | 1353.4824 | 1353.4949 | [M − H]−1 |
| 676.2487 | 676.2433 | 676.2438 | [M − 2H]−2 | ||||||||
| 17 | A3G1 |
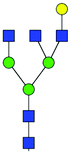
|
0.38 ± 0.02 | 0.88 ± 0.29 | 6.60 | 899.8213 | 899.8521 | 899.8365 | [M − 2H]−2 | ||
| 33 | A2G2S(6)1 |
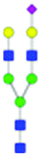
|
1.55 ± 0.32 | 2.68 ± 0.42 | 8.29 | Not present | 1024.8640 | 1024.8709 | [M − 2H]−2 | ||
| 41 | A2G2S(3)2 |
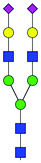
|
0.46 ± 0.09 | 1.62 ± 0.40 | 9.26 | 1170.3973 | 1170.4043 | 1170.4186 | [M − 2H]−2 | ||
| 45 | A2G2S(6)2 |
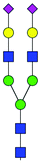
|
1.04 ± 0.78 | 7.45 ± 3.73 | 9.56 | 1170.4183 | 1170.4324 | 1170.4186 | [M − 2H]−2 | ||
| 47 | FA2G2S(3)2 |
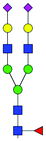
|
0.85 ± 0.14 | 1.47 ± 0.31 | 10.02 | 1243.4425 | 1243.4352 | 1243.4476 | [M − 2H]−2 | ||
| 54 | FA2F3G2Lac1S(3)1 |
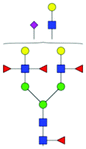
|
0.77 ± 0.22 | 1.45 ± 0.13 | 11.08 | 999.3527 | 999.3527 | 999.3661 | [M − 3H]−3 | ||
| 59 | A4G4Lac2S(3)1 |
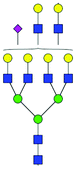
|
0.29 ± 0.10 | 0.81 ± 0.41 | 12.28 | 1169.7378 | 1169.7378 | 1169.7544 | [M − 3H]−3 | ||
| TIS cytoplasm | Decrease | 16 | M6 |
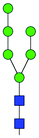
|
7.02 ± 0.17 | 6.29 ± 0.28 | 7.07 | 1515.5175 | 1515.5493 | 1515.5477 | [M − H]−1 |
| 757.2662 | 757.2662 | 757.2702 | [M − 2H]−2 | ||||||||
| TIS EVs | Increase | 30 | FA2BG2 |
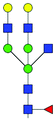
|
0.54 ± 0.19 | 1.44 ± 0.31 | 7.83 | 1053.8970 | 1053.8770 | 1053.8918 | [M − 2H]−2 |
| 35 | A2G2S(6)1 |
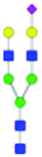
|
0.35 ± 0.12 | 0.93 ± 0.24 | 8.55 | 1024.8903 | 1024.8771 | 1024.8709 | [M − 2H]−2 | ||
| TIS EVs | Decrease | 33 + 34 | FA2G2S(6)1 |
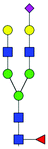
|
3.10 ± 0.41 | 1.97 ± 0.22 | 8.33 | 1097.9178 | 1097.8906 | 1097.8998 | [M − 2H]−2 |
| 42 + 43 | M9 |
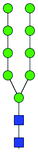
|
8.06 ± 2.73 | 3.14 ± 1.41 | 9.63 | 2001.6893 | 2001.7168 | 2001.7062 | [M − H]−1 | ||
| 1000.3579 | 1000.3579 | 1000.3495 | [M − 2H]−2 | ||||||||
| 47 | FA3G3S(6)2 |
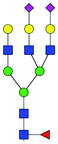
|
1.37 ± 0.19 | 0.96 ± 0.05 | 10.21 | 1426.0114 | 1426.0267 | 1426.0137 | [M − 2H]−2 | ||
The following glycan structures were increased in the TIS membrane compared to control membrane: M5, A3G1, A2G2S(6)1, A2G2S(3)2, A2G2S(6)2, FA2G2S(3)2, FA2F3G2Lac1S(3)1 and A4G4Lac2S(3)1. M6 was decreased on the TIS cytoplasm. FA2BG2 and A2G2S(6)1 were increased on TIS EVs, whereas FA2G2S(6)1, M9 and FA3G3S(6)2 were decreased on TIS EVs compared to control EVs. The percentage areas of N-linked glycan structures identified in the WCL, membrane, cytoplasm and in the EV progeny of TIS vs. non-senescent TNBC cells are presented in Table 2. The ratio of α2–3/2–6 sialylated N-linked glycans increased in TIS cells and TIS membrane compared to controls but decreased in TIS EVs and TIS cytoplasm compared to controls. The EVs are highly sialylated at 52% in the controls compared to 28% in the TIS cells (Table 2). The total area of galactosylated N-linked glycans decreased in TIS EVs and TIS cytoplasm compared to controls.
| WCL Control | WCL TIS | Membrane control | Membrane TIS | Cytoplasm control | Cytoplasm TIS | EV control | EV TIS | |
|---|---|---|---|---|---|---|---|---|
| High mannose | 25.74 | 26.15 | 29.74 | 22.59 | 22.90 | 21.71 | 23.30 | 18.56 |
| Monoantennary | 0.00 | 0.88 | 0.33 | 0.92 | 0.45 | 0.87 | 0.83 | 0.72 |
| Biantennary | 10.32 | 8.82 | 6.63 | 14.79 | 10.30 | 7.76 | 14.32 | 9.52 |
| Triantennary | 17.29 | 15.91 | 7.03 | 12.20 | 7.11 | 7.03 | 33.01 | 14.79 |
| Tetranantennary | 2.64 | 2.46 | 2.44 | 3.17 | 1.41 | 1.70 | 1.65 | 0.89 |
| Lac | 0.97 | 1.59 | 1.56 | 3.46 | 0.94 | 2.76 | 1.19 | 1.42 |
| Outer arm fucose | 5.43 | 14.58 | 9.04 | 12.18 | 9.47 | 9.43 | 9.10 | 8.14 |
| Core fucose | 14.58 | 20.28 | 14.76 | 20.84 | 13.32 | 13.83 | 16.29 | 14.28 |
| Bisects | 10.38 | 7.83 | 6.39 | 13.89 | 7.75 | 8.54 | 6.98 | 5.66 |
| Agalactosylated glycans (G0) | 22.88 | 22.09 | 24.94 | 21.55 | 21.56 | 20.35 | 16.08 | 15.57 |
| Monogalactosylated glycans (G1) | 4.06 | 2.47 | 4.27 | 7.02 | 2.43 | 4.52 | 2.99 | 2.84 |
| Digalactosylated glycans (G2) | 14.71 | 20.31 | 12.25 | 18.14 | 15.18 | 12.67 | 19.04 | 13.47 |
| Trigalactosylated glycans (G3) | 16.49 | 16.93 | 10.66 | 13.31 | 9.12 | 8.15 | 36.01 | 17.18 |
| Tetragalactosylated glycans (G4) | 3.97 | 3.04 | 2.60 | 2.64 | 2.52 | 1.98 | 1.11 | 1.83 |
| Galactosylation beta 1–3 | 21.31 | 20.01 | 9.70 | 11.85 | 15.74 | 14.01 | 29.62 | 22.54 |
| Galactosylation beta 1–4 | 6.96 | 2.13 | 6.24 | 14.03 | 2.56 | 3.28 | 7.16 | 3.33 |
| Galactosylation both | 5.37 | 6.48 | 1.66 | 2.32 | 0.20 | 0.50 | 9.5 | 0.00 |
| Total galactosylation | 39.23 | 42.87 | 28.26 | 39.81 | 28.52 | 24.47 | 54.45 | 32.80 |
| Asialylated glycans (S0) | 34.92 | 35.87 | 34.32 | 33.57 | 31.85 | 31.56 | 24.71 | 23.82 |
| Monosialylated glycans (S1) | 7.62 | 11.16 | 9.40 | 15.88 | 8.43 | 9.68 | 10.91 | 7.13 |
| Disialylated glycans (S2) | 6.19 | 7.84 | 6.80 | 9.60 | 7.56 | 4.20 | 14.59 | 7.36 |
| Trisilylated glycans (S3) | 7.25 | 8.86 | 2.79 | 3.14 | 2.63 | 1.69 | 26.40 | 13.10 |
| Tetrasialylated glyadns (S4) | 4.85 | 2.46 | 0.54 | 0.28 | 1.24 | 1.28 | 0.00 | 0.62 |
| Sialylation alpha2–3 | 4.03 | 17.07 | 7.37 | 13.77 | 8.95 | 5.09 | 16.85 | 2.58 |
| Sialylation alpha2–6 | 13.55 | 11.86 | 11.45 | 15.13 | 10.21 | 11.22 | 15.54 | 11.39 |
| Sialylation both | 8.34 | 1.39 | 0.71 | 0.00 | 0.82 | 0.55 | 19.51 | 14.24 |
| Total sialylation | 25.92 | 30.32 | 19.53 | 28.90 | 19.99 | 16.85 | 51.90 | 28.21 |
SDS-PAGE western blot validation of the protein mass spectrometry of TIS and control WCL, membrane, cytoplasm and EV protein samples
Protein mass spectrometry was carried out on TIS and control TNBC WCL, membrane, cytoplasm and EV protein samples isolated from the gels which were remaining following the isolation of the N-linked glycans (ESI,† Tables S1–S12). Subsequent western blot analysis found that in 2 of 3 biological replicates, there was (i) an increased expression of CD44 (ii) cleavage of Galectin 3 and the presence of a lower (∼58 kDa) band for calnexin compared to non-senescent control cells where calnexin appears at 90 kDa (Fig. 2). There was also a decrease in GRP78 in TIS cells compared to controls (Fig. 2) by SDS-PAGE confirming the MS results for the protein in higher or lower abundance in TIS WCL, membrane, cytoplasm and EVs compared to control samples (ESI,† Tables S1–S12).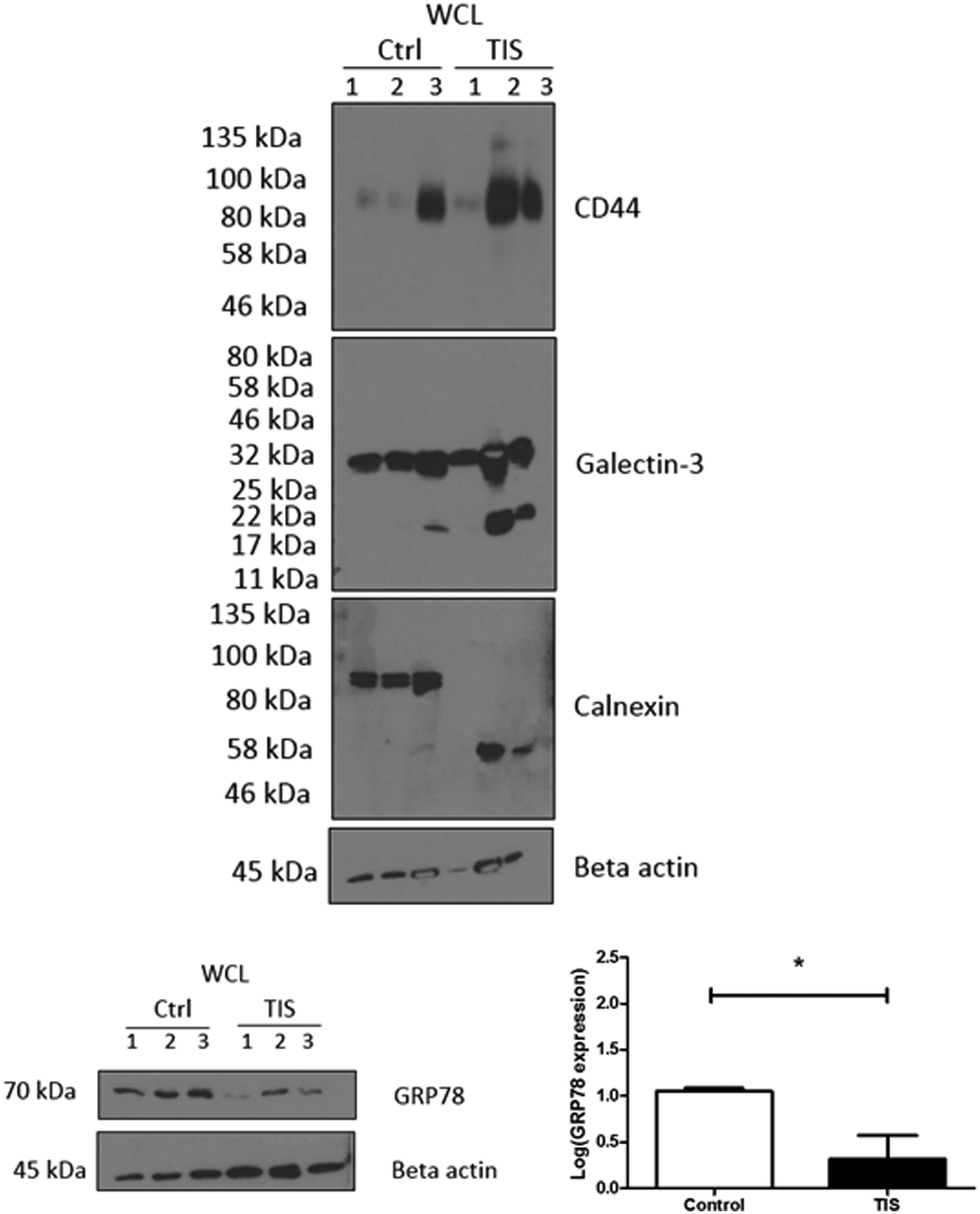 | ||
| Fig. 2 SDS-PAGE western blot validation of the MS results showing an increase in CD44 and Galectin-3 in PTX induced TIS and also a decrease in GRP78 and calnexin. Cal51 cells were treated with 75 nM Paclitaxel for 7 days to induce senescence. Control Cal51 whole cell lysates (WCL) and senescent Cal51 WCL were subsequently blotted for CD44, calnexin, Galectin-3, GRP78 and beta actin. This figure represents n = 3 biological replicates. Densitometry was completed using Image J software (Fiji). All values are expressed as the mean of three independent experiments ±SEM. The limit of ±1 skewness was set as normal distribution. Skewed data were transformed to Y = log(Y) to fit a normal distribution. Results were analysed using the Student's t-test with significant differences having a p ≤ 0.05 *, p ≤ 0.01 **, p ≤ 0.001 ***. | ||
Key glycosyltransferases, glycosidases and glycoproteins identified from the MS analysis which are significantly increased or decreased in TIS or control WCL, membrane, cytoplasm and EV protein samples
The TIS WCL, membrane, cytoplasm and EV protein samples contain different glycan modifying enzymes and glycoproteins compared to control non-senescent samples (Table 3).| Glycosyltransferases, glycosidases and glycoproteins | Accession no. | p-Value | Increase or decrease |
|---|---|---|---|
| Lysosomal alpha-mannosidase | O00754 | N/A | Exclusive to TIS WCL |
| 0.0393853 | Lower in TIS membrane | ||
| Alpha-mannosidase2 | Q16706 | 0.00820896 | Lower in TIS membrane |
| Basement membrane-specific heparan sulfate proteoglycan core protein | P98160 | 0.0308949 | Higher in TIS cytoplasm |
| 0.0297909 | Higher in TIS membrane | ||
| 0.0412032 | Lower in TIS EVs | ||
| Mannosyl-oligosaccharide glucosidase | Q13724 | N/A | Exclusive to ctrl WCL |
| 0.00546559 | Lower in TIS cytoplasm | ||
| Beta-galactosidase | P16278 | 0.00748672 | Higher in TIS cytoplasm |
| Glycogenin-1 | P46976 | 0.00674984 | Higher in TIS cytoplasm |
| Dolichyl-diphosphooligosaccharide—protein glycosyltransferase 48 kDa subunit | P39656 | 0.00687461 | Lower in TIS cytoplasm |
| N/A | Exclusive to TIS EVs | ||
| UDP-glucose: glycoprotein glucosyltransferase1 | Q9NYU2 | 0.0274089 | Lower in TIS cytoplasm |
| N/A | Exclusive to TIS EVs | ||
| Beta-hexosaminidase subunit alpha | P06865 | 0.0260433 | Higher in TIS WCL |
| N-Acetylgalactosaminyltransferase 7 | Q86SF2 | 0.0459031 | Lower in TIS membrane |
| CD44 | P16070 | 0.00528984 | Higher in TIS membrane |
| Calnexin | P27824 | 0.0156759 | Lower in TIS membrane |
| Galectin-3 | P17931 | 0.0393726 | Higher in TIS membrane |
| N/A | Exclusive to ctrl EVs |
Top five significant pathways which change comparing proteins from TIS and control WCL, membrane, cytoplasm and EV protein samples from MS analysed using Ingenuity® Pathway Analysis
Pathway analysis of the WCL, membrane, cytoplasm and EV protein samples was carried out using Ingenuity® Pathway Analysis (IPA), and identified the top five pathways which changed comparing proteins from TIS and control (Table 4).| Top altered canonical pathways in TIS WCL | p-Value |
|---|---|
| EIF2 signalling | 4.6 × 10−43 |
| Regulation of eIF4 and p70S6K | 1.77 × 10−20 |
| mTOR signalling | 1.16 × 10−16 |
| Remodelling of epithelial adherans junctions | 3.59 × 10−14 |
| Epithelial adherans junction signalling | 2.57 × 10−13 |
| Top altered canonical pathways in TIS membrane | p-Value |
|---|---|
| Remodelling of epithelial adherans junctions | 1.99 × 10−9 |
| Germ cell-sertoli cell junction signalling | 6.49 × 10−8 |
| Epithelial adherans junction signalling | 3.70 × 10−7 |
| Phagosome maturation | 4.06 × 10−7 |
| Sertoli cell-sertoli cell junction signalling | 1.41 × 10−6 |
| Top altered canonical pathways in TIS cytoplasm | p-Value |
|---|---|
| EIF2 signalling | 2.03 × 10−74 |
| Regulation of eIF4 and p70S6K | 1.28 × 10−24 |
| mTOR signalling | 1.53 × 10−20 |
| Remodelling of epithelial adherans junctions | 4.72 × 10−18 |
| Epithelial adherans junction signalling | 1.11 × 10−12 |
| Top altered canonical pathways in TIS EVs | p-Value |
|---|---|
| Protein ubiquitination pathway | 1.39 × 10−10 |
| Sirtuin signalling pathway | 5.60 × 10−7 |
| tRNA charging | 9.34 × 10−7 |
| EIF2 signalling | 2.59 × 10−6 |
| Regulation of eIF4 and p70S6K | 6.75 × 10−5 |
Lectin blotting demonstrates a differential profile for LCA blotting in WCL and for PHA-E in membranes from PTX induced TIS compared to controls. PHA-E lectin treatment of Cal51 TNBC cells reduces cell viability
The lectins Phaseolus vulgaris Erythroagglutinin (PHA-E), and Lens culinaris agglutinin (LCA) were blotted in the WCL from control and senescent Cal51 cells (Fig. 3A). A differential profile for LCA (a lectin which recognises mannose glycan residues) was evident in the WCL from PTX induced TIS cells compared to controls. Specifically, there was an increase in binding of LCA to mannosylated glycoproteins at ∼100 kDa in the TIS WCL compared to control samples. The lectin PHA-E was blotted with the membrane from control and senescent Cal51 cells. A differential profile for PHA-E (a lectin which recognises bisected glycan residues) was evident in the membranes from PTX induced TIS cells compared to controls (Fig. 3A). Subsequently, both control and TIS cells were treated with buffer, PHA-E or LCA (1–50 μg μl−1) for 48 h (Fig. 3B). An MTT assay revealed that control cells were sensitive to PHA-E at 50 μg μl−1 whereas TIS cells remained resistant (Fig. 3B).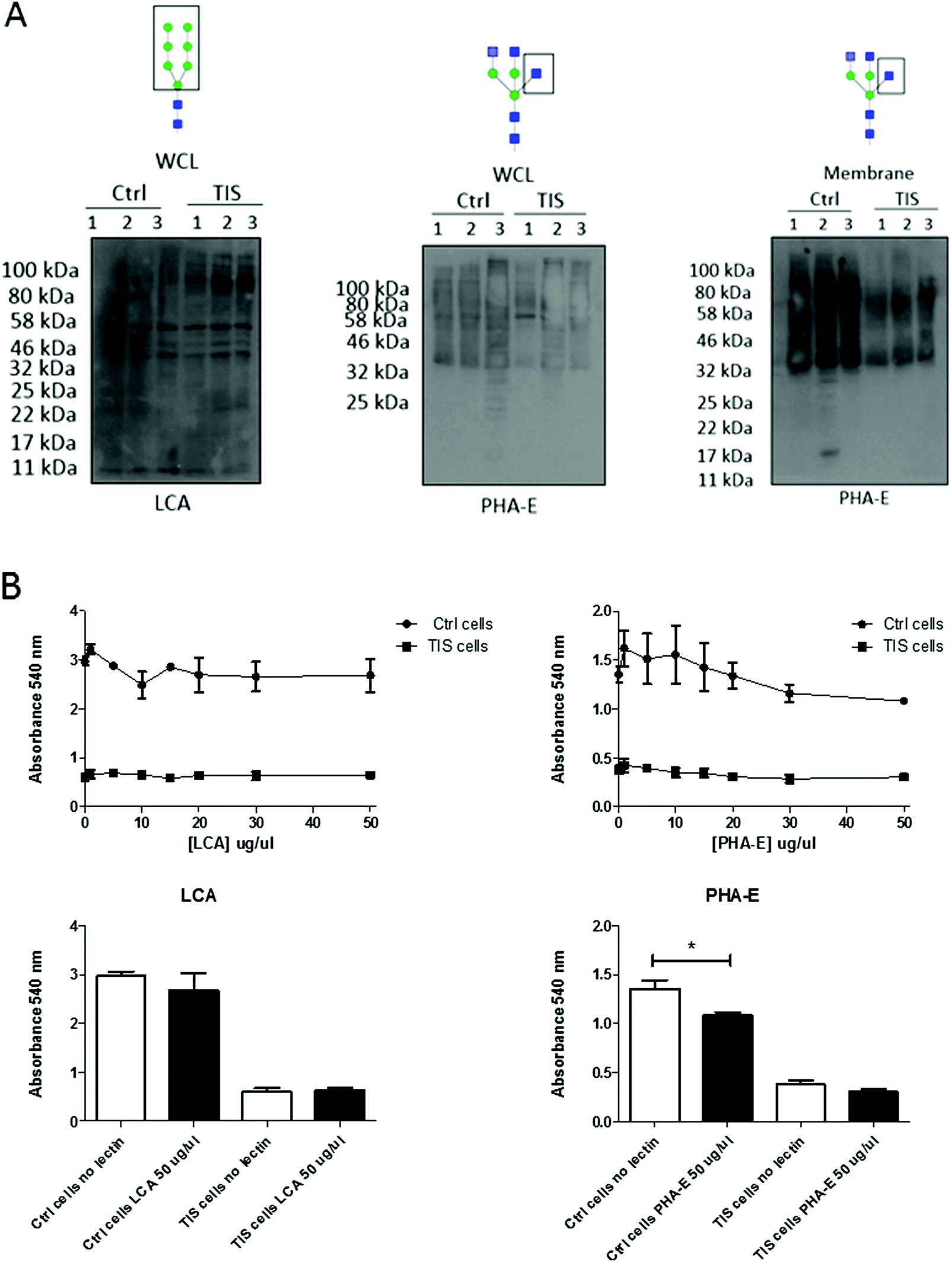 | ||
| Fig. 3 Lectin blotting demonstrates a differential profile for LCA blotting in WCL and for PHA-E in membranes from PTX induced TIS compared to controls. PHA-E lectin treatment of Cal51 TNBC cells reduces the viability. (A) Cal51 cells were treated with 75 nM Paclitaxel for 7 days to induce senescence. Control Cal51 whole cell lysates (WCL) and senescent Cal51 whole cell lysates (WCL) were ran on an 12% gel and blotted for PHA-E and LCA. Control Cal51 membrane and senescent Cal51 membrane were run on an 12% gel and blotted for PHA-E. (B) Both control and TIS cells were treated with buffer, PHA-E or LCA (1–50 μg μl−1) for 48 h. Cells were incubated with MTT (5 mg ml−1) reagent for 6 h at 37 °C. Crystals were resuspended by adding 200 μl of DMSO. The absorbance was measured on a plate reader at 540 nm. All values are expressed as the mean of three independent experiments ±SEM. Results were analysed using the Student's t-test with significant differences having a p ≤ 0.05 *, p ≤ 0.01 **, p ≤ 0.001 ***. | ||
PHA-E lectin treatment of Cal51 TNBC cells affects morphology. TIS cells have a significant reduction in their adhesion to collagen I compared to control cells, but PHA-E treatment had no effect
PTX induced TIS and control cells were treated with buffer or 50 μg μl−1PHA-E for 48 h. The morphology of both control and TIS cells changed to a more rounded shape upon treatment with PHA-E (Fig. 4A), whereas there was no morphological change when cells were treated with LCA (data not shown). The ECM Cell Adhesion Array Kit adhesion assay measured the adhesion of control and TIS cells either treated or untreated with PHA-E 50 μg μl−1 for 48 h to extracellular matrix proteins, for example fibronectin, laminin and collagen) (Fig. 4B). TIS cells induced with PTX had significantly less adhesion to collagen I when compared to control Cal51 cells.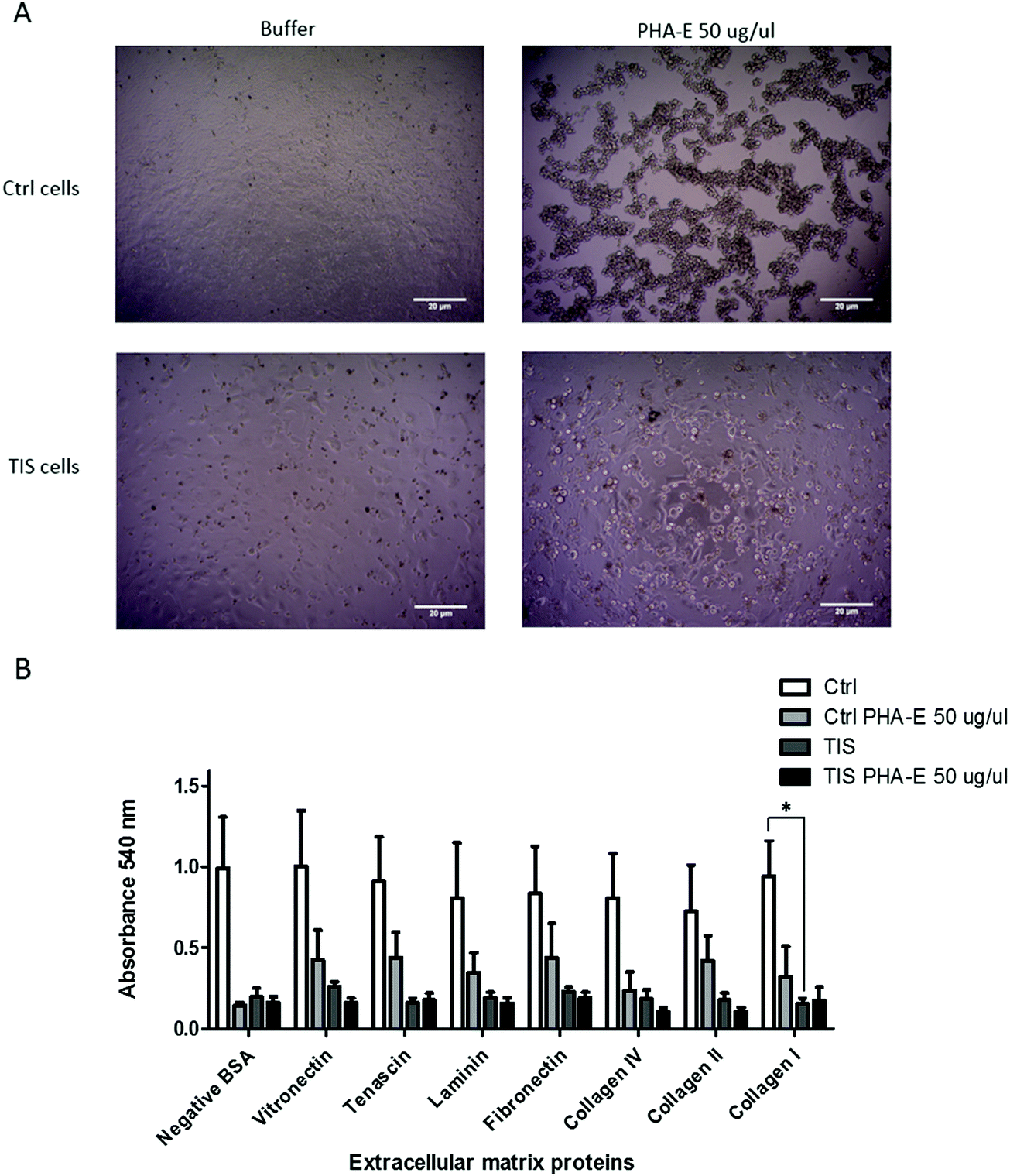 | ||
| Fig. 4 PHA-E lectin treatment of Cal51 TNBC cells affects morphology. TIS cells have a significant reduction in their adhesion to collagen I compared to control cells, but PHA-E treatment had no effect. (A) Cal51 cells were treated with 75 nM paclitaxel for 7 days to induce TIS. Control and TIS Cal51 cells treated with buffer or PHA-E for 48 h. Scale bars represent 20 μm. (B) Both control and TIS cells were treated with buffer or 50 μg μl−1PHA-E for 48 h. The ECM Cell Adhesion Array Kit adhesion assay was carried out on the cells. The absorbance was measured on a plate reader at 540 nm. All values are expressed as the mean of three independent experiments ±SEM. Results were analysed using the Student's t-test with significant differences having a p ≤ 0.05 *, p ≤ 0.01 **, p ≤ 0.001 ***. | ||
Discussion
The HILIC-UPLC and LC-MS results of N-linked glycosylation profiling demonstrated that the mannose structure M5 (peak 13) was significantly increased in TIS compared to control membranes. The mannose structure M9 (peak 42 and 43) was significantly decreased in the TIS EVs and the M6 (peak 16) was significantly decreased in the TIS cytoplasm. A previous study has found that the high mannose binding lectin ESA, bound to specific glycans on the surface of osteosarcoma cells and induced apoptosis of these cells.18 In a study on age-induced senescence, the mannose binding lectin MBL induced cell lysis of aged senescent fibroblast cells in vitro.19 Moreover, profiling the N-linked glycans of the serum from patients with metastatic breast cancer identified a high mannose (nine mannoses) glycan.20 This combined data suggests that high mannose glycans may be a feature of cancer senescent membranes, and this differential glycan profile could potentially enable their specific cell lysis.The HILIC-UPLC and LC-MS results demonstrated that the fucosylated glycan structures FA2F3G2Lac1S1 (peak 54) and FA2G2S2 (peak 47) were significantly increased in TIS cell membranes. FA2G2S1 (peak 33 + 34) was decreased, and its non-fucosylated form A2G2S1 (peak 35) increased in the EVs derived from TIS cells. This suggests that FUCA1 activity may be membrane bound on TIS cells and its non-fucosylated form within EVs. FUCA1 is an enzyme which cleaves fucose from N-glycans,12 and is a novel marker of cellular senescence.13 Clinically, its decreased expression is a marker of poor prognosis in TNBC patients.14 Potentially, FUCA1 may be more active in TIS cells potentially explaining the decreased fucosylated glycan structure FA2G2S1 in the TIS EVs and the increased non-fucosylated form A2G2S1 in TIS EVs.
The HILIC-UPLC and LC-MS results demonstrated that the biantennary, galactose and sialic acid containing N-linked glycan structure A2G2S1 is significantly increased in both the TIS membrane (peak 33) and TIS EVs (peak 35), suggesting retention of this post-translational modification on the membrane of the parent cell and its EV progeny.
The N-glycan profile of the EV subsets has been analysed previously using lectin blotting and MS analyses.27 All three subsets of EVs while displaying distinct N-glycan profiles, commonly shared complex N-glycans and high levels of sialylation.27 Similarly, in our results, the HILIC-UPLC and LC-MS results show an increase in the sialylated glycan structure A2G2S1 (peak 35) in TIS EVs compared to control EVs. There was also a decrease in the sialylated glycan structures FA2G2S1 (peak 33 + 34) and FA3G3S2 (peak 47) in TIS EVs compared to control EVs, possibly reflecting enhanced FUCA1 activity in TIS cells.
To identify a potential mechanism of how the differential N-linked glycan profiling occurs in TIS, a review of the MS data identified key glycosyltransferases, glycosidases and glycoproteins in differential abundance in TIS or control WCL, membrane, cytoplasm and EV protein samples. Lysosomal alpha-mannosidase, an enzyme that digests N-linked glycoproteins by degradation of high mannose glycans29 was exclusive to TIS WCL, and lower in the TIS membrane fraction.
From the literature, it is known that Alpha-D-mannosidase activity increases with senescence in human fibroblasts.30 In our study, alpha-mannosidase 2 was lower in TIS membranes. The Golgi alpha-mannosidase 2 removes two mannose residues from GlcNAcMan5GlcNAc2; a step in the synthesis of complex N-linked glycans.31 Swainsonine, an inhibitor of Golgi alpha-mannosidase 2,32 reduces metastasis and improves outcome in colon, breast, and skin cancer,33 potentially identifying a mechanism for the observed increase in the high mannose structure M5 (peak 13) in the TIS membranes.
Beta-galactosidase was higher in the TIS cytoplasm, confirmed by increased SA-β-gal activity in TIS compared to controls. SA-β-Gal is a well-recognised marker of cellular senescence.9 The total area of galactosylated N-linked glycans decreased in TIS EVs and TIS cytoplasm compared to controls, suggesting that SA-β-Gal is potentially important in the N-linked glycosylation of senescent cells due to their increased activity of SA-β-Gal which leads to a decrease of galactosylation in the TIS EVs and cytoplasm.
Other key glycosyltransferases and glycosidases involved in glycosylation processing were differentially abundant in TIS samples compared to controls (Table 3)34–39 which may partially explain differential glycosylation in TIS cells.
CD44, a stem cell marker which is known to be increased in senescence,40 was identified as being higher in the TIS membrane by MS, and confirmed by western blotting in 2/3 biological replicates. Glycosylation of CD44 is crucial to its role in cell adhesion.41 Galectin-3 which is present in higher abundances in TIS cells is known to upregulate CD44 expression.42 The increased abundance of CD44 on the TIS membrane suggests that it may have a role in TIS cell adhesion. CD44 plays a role in breast cancer cell metabolism with CD44 knockdown in breast cancer cells decreasing glucose uptake, ATP and lactate production.43 The increase in CD44 in TIS membrane may potentially enable survival by aiding the metabolism of TIS cells.
In TIS cells, calnexin demonstrated a different size (∼58 kDa) compared to control cells (∼90 kDa). Calnexin is a lectin which binds N-linked glycans in the ER and facilitates the protein folding process. Interestingly, in the literature it has been demonstrated that calnexin is involved in apoptosis induced by ER stress and in a fission yeast (S. pombe) model,44 it was found that calnexin could be physiologically cleaved under normal growth conditions when cells approach stationary phase. This cleavage suggested that the two naturally produced calnexin fragments are needed to continue growth into stationary phase and to prevent cell death. This may be a possible explanation for the lower calnexin band in TIS cells whose feature is retained viability and not apoptosis.
Basement membrane-specific heparan sulfate proteoglycan core protein was higher in the TIS cytoplasm and TIS membrane but lower in TIS EVs. Heparan sulfate proteoglycans are crucial in the tumour microenvironment as they function as receptors for the internalisation of cancer cell EVs.45 This is evidence that the TIS cells may have selective control of EV uptake, as the basement membrane-specific heparan sulfate proteoglycan core protein was higher in the TIS membrane fraction and lower in the TIS harvested EVs.
Galectin-3 was increased on the membrane of TIS cells in 2 of 3 biological replicates mirroring the increased levels of CD44. This lectin has roles in invasion, immune suppression and apoptosis,46 is expressed at high levels in TNBC and is thought to be a potential therapeutic target.47 It is known that the bisected GlcNAc of N-glycans may alter the function of glycoproteins by changing the structure of the N-glycan leading to reduced binding to Galectins.48 The reduced binding of Galectin-3 to bisected glycans may attenuate the possibility of TIS cells being able to take up their own EVs, as these possess the bisected FA2BG2 (peak 30) present in significantly higher abundances. This may represent a potential mechanism for selective uptake of EVs. The presence of the lower kDa band in the Galectin-3 profiles of TIS cells compared to controls possibly reflects the fact that Gal-3 can be modulated by proteolytic processing, yielding different Gal-3 forms of 27, 22, 20.2, 18.9, 16, 15.5, and 14 kDa.49–53 Moreover, additional truncated forms have also been detected in cancer.54 Importantly, Galectin-3 proteolysis has also been implicated in tumour progression in growing breast cancers55 and in prostate cancer progression.56
IPA revealed that remodelling of epithelial adherans junctions was the highest pathway involving proteins upregulated in TIS membranes. Galectin-3, which is higher on the TIS membranes, recognises N-linked β-galactosides and is known to regulate epithelial intercellular adhesion.57 This suggests that Galectin-3 may be an important lectin on the membrane of TIS cells for adherence. The relevance of the lower Galectin 3 isoform/truncated protein in the biology of therapeutic induced senesce is not clear.
An MTT assay and adherence assay was carried out on control and TIS cells, treated or not with PHA-E. PHA-E is a lectin which specifically recognises bisected glycans. The morphology of both the control and TIS cells changed with PHA-E treatment. The control cells were sensitive to PHA-E, whereas the TIS cells remained resistant to PHA-E. Control and TIS cells demonstrated differential adherence to collagen I with TIS cells demonstrating less adherance Galectin-3 regulates adhesion to collagen I via integrins with data showing that cells depleted in Galectin-3 were more likely to be in an enhanced adhesion state.58 Similarly, control Cal51 cells displayed decreased Galectin-3 and greater adhesion to collagen I compared to TIS cells potentially affording the latter an enhanced migratory potential. The potential role of proteolytic cleavage of Galectin 3 and its association with the reduced adherence of TIS cells to collagen warrants further investigation.
The ratio of α2–3/2–6 sialylated N- and O-linked intracellular glycoproteins increases in an aging cellular senescence model.59 The ratio of α2–3/2–6 sialylated N-linked glycans is also increased in TIS cells and in the TIS membrane compared to controls while the ratio of α2–3/2–6 sialylated N-linked glycans is decreased in TIS EVs and TIS cytoplasm compared to controls. Previously, it was shown that EVs have high levels of sialylation.27 We found that the EVs were highly sialylated control EVs at 52% and TIS EVs at 28%.
In summary, this study has demonstrated that therapeutic-induced-senescent (TIS) TNBC cells and their EV progeny, display differential N-glycan moieties compared to non-senescent Cal51 cells and their resultant EV progeny. The acknowledged histological and molecular heterogeneity of TNBC is complex, as is the tumour microenvironment of the epithelial, stromal and infiltrative lymphocyte components. Comparative studies on the findings from this in vitro study with TNBC patient plasma and matched tumour samples from patients with known treatment response and outcome is warranted.
Further investigations will also establish if the alterations in glycosylation are due to changes in glycoprotein levels or in the expression/activity levels of the relevant glycan modifying enzymes. Unique N-glycan moieties on cancer senescent cells and their EV progeny holds potential for specific cancer senescent cell targeting with lectin therapies and the potential monitoring of treatment response as a liquid biopsy respectively.
Materials and methods
Cell culture
The Cal51 TNBC cell line (p53 wild type) (DSMZ, ACC 302) was cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Lonza, BE12-614F) supplemented with 10% (v/v) foetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific, 10270-106) and 1% (v/v) sodium pyruvate (Gibco, Thermo Fisher Scientific, S8636-100ML) at 37 °C in a humidified atmosphere of 5% CO2. Cal51 cells were authenticated by DDC Medical in 2016 and routinely tested negative for mycoplasma contamination.Chemotherapy induced senescence
Cal51 cells were treated for 7 days with 75 nM paclitaxel (PTX) (Sigma-Aldrich, T7402-1MG). Senescence was confirmed through appreciation of SA-β-Gal activity and p21 protein expression using western blot analyses.Senescence associated β-galactosidase (SA-β-Gal) staining assay
TIS cells were seeded at 1 × 105 cells per ml, cultured for 24 h, fixed and stained using the SA-β-Gal staining kit, 5 mg ml−1 (Cell Signalling Technology, 9860), incubated for 16 h, imaged using a light microscope (Olympus model CKX41) and enumerated for SA-β-Gal positivity using ImageJ™ software (FIJI).SDS-PAGE western blotting
Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting were performed as described previously.60,61 The Bradford assay was carried out to quantify the amount of protein loaded and to ensure equal loading into each lane. Antibodies used were: calnexin (Santa Cruz Biotechnology, sc-80645), CD63 (System Biosciences, EXOAB-CD63A-1), p21 (Cell Signaling Technology, 2947), Rb (Cell Signaling Technology), Galectin-3 (Abcam, ab76245), GRP78 (Abnova, PAB2462), beta actin (Sigma Aldrich, A5441) and CD44 (Cell Signaling Technology, 3570T).SDS-PAGE lectin blotting
SDS-PAGE and lectin blotting were performed as described previously ensuring equal loading in each lane using the Bradford assay.62 The reagents used were Lectin kit II biotinylated and streptavidin horseradish peroxidase conjugated (Vector Laboratories).Extracellular vesicle (EV) isolation
Cells grown for EV harvesting purposes were cultured prior to isolation in media supplemented with bovine EV-depleted FBS for 48 h and isolated as previously described.28NanoSight NS300 EV profiling
EV samples were run using the standard measurement procedure at 25 °C with a constant syringe infusion rate as per the NanoSight NTA 3.1 Software (Malvern). The data for each sample were obtained from 10 independent 60 s video captures on the NS300, as previously described.28Glycoprotein isolation
Glycoproteins were isolated from whole cell lysates (WCLs). A cell pellet was resuspended in 250 μl of sample buffer and left on ice for 20 min. The samples were moved to Eppendorf tubes, the 15 ml Falcon tube was washed with 250 μl of sample buffer (62.5 mM Tris pH 6.8, 2% SDS) drawn up/down 10 times through a 21-gauge needle and then pelleted. Supernatants were recovered and dried in a speed vacuum.Glycoproteins were isolated from the cytoplasm and the membrane using the Triton X-114 phase partitioning of membrane proteins method as previously published.63 The upper aqueous layer, containing the cytoplasmic fraction, was removed carefully and stored at −20 °C. The lower layer was mixed with 1 ml of ice-cold acetone and left overnight at −20 °C. The membrane proteins were precipitated by centrifugation at 1000 g for 3 min.
Glycoproteins were isolated from EVs. EVs were confirmed as enriched for exosomes using the exosome positive marker CD63, negative marker calnexin and nanoparticle tracking analysis. For glycoprotein isolation, the EVs were sonicated for 3 × 5 min in radioimmunoprecipitation assay lysis buffer (RIPA). Protein was precipitated by adding 50% ice cold acetone to the protein solution. Samples were mixed and incubated in ice-cold acetone overnight and pelleted, the supernatant was discarded, and the pellet dried for 5 min using a speed vacuum. The pellet was dissolved in 2 μl of sample buffer (62.5 mM Tris pH 6.8, 2% SDS) and 7 μl water.
In-gel-block high throughput release and processing of total N-glycans
N-Glycans were released from glycoproteins in samples by in situ digestion with N-glycosidase F (PNGaseF) in-gel blocks as described previously.4 Briefly, N-linked glycan samples were reduced and alkylated, and set into SDS-gel blocks. N-Glycans were released by adding 50 μL of 0.1 U ml−1 PNGase F (Prozyme) in 20 mM NaHCO3, pH 7.2.Fluorescent 2-aminobenzamide (2AB) labelling
2AB labelling mixture (5 μl) was added to each well and agitated for 5 min, to ensure the labelling mixture was mixed with the sample. The samples were then incubated at 65 °C for 30 min, agitated for another 5 min and incubated at 65 °C for a further 1.5 h. The samples were then placed in the −20 °C freezer overnight. Excess 2AB was removed using 3 MM Whatman chromatography paper.Digestion protocol for N-linked Glycans
Exoglycosidase enzymes, 10× buffer (diluted to 50 mM Na acetate pH 5.5), zinc buffer (JBM digest), water and sample were added to a volume of 10 μl. The sample was dried in a speed vacuum. Samples were vortexed, centrifuged and incubated for 16 h at 37 °C. Following digestion, samples were filtered using a microcentrifuge filter (Pall 10 kDa MWCO microcentrifuge filtration device). The glycans were resuspended in MilliQ water for UPLC (for HILIC-UPLC, samples were resuspended in 6 μl MilliQ water, transferred to the UPLC vial and 14 μl acetonitrile was added).HILIC-UPLC (hydrophilic interaction ultra-performance liquid chromatography) glycan analysis
Samples were prepared in a volume of 20 μl (14 μl acetonitrile + 6 μl (including 10% sample + water). UPLC was carried out as previously described7 using BEH Glycan 1.7 μm particles in 2.1 × 150 mm column on an Acquity UPLC with a temperature control module and Acquity fluorescence detector (Waters). Solvent A was 50 mM formic acid adjusted to pH 4.4 with ammonia solution. Solvent B was acetonitrile. The column temperature was set to 40 °C. The 30 min method was used with a linear gradient of 30–47% with buffer A at 0.56 ml min−1 flow rate for 23 min followed by 47–70% A and finally reverting to 30% A to complete the run. Samples were injected in 70% acetonitrile. Fluorescence was measured at 420 nm with excitation at 330 nm. The system was calibrated using an external standard of hydrolysed and 2AB-labeled glucose oligomers to create a dextran ladder, as previously described.64Liquid chromatography mass spectrometry (LC/MS) analysis of N-linked glycans
Acquity®UPLC-FLD-QTof, is a liquid chromatography mass spectrometry (LC/MS) system which enables the acquisition and processing of mass data from chromatographic analyses. The components of the system were: Waters Xevo-G2-QTof mass spectrometer and Waters ACQUITY®UPLC system, operated using MassLynx software. Calibration was performed in sensitivity and negative mode. Excess 2AB label was removed using Phynexus PhyTips. Samples were resuspended in 3 μl water and 9 μl acetonitrile and 10 μl was injected into the LC/MS.Quantitative proteomic profiling by label-free LC-MS/MS analysis
These experiments were performed as previously described.28Pathway analysis
Pathway analyses were performed using Ingenuity® Pathway Analysis (IPA, www.ingenuity.com) software (Ingenuity Systems, Qiagen). P values reported for IPA results were calculated by IPA using a right-sided Fisher exact test for over-representation analysis and Benjamini–Hochberg correction for multiple hypothesis testing correction.Glycan structure abbreviations
All N-glycans have two core GlcNAcs ; F at the start of the abbreviation indicates a core-fucose
; F at the start of the abbreviation indicates a core-fucose  α1,6-linked to the inner GlcNAc; Mx, number of mannose
α1,6-linked to the inner GlcNAc; Mx, number of mannose  on core GlcNAcs; A(x), number of antenna (GlcNAc) on trimannosyl core; A2, biantennary with both GlcNAcs as β1,2-linked; A3, triantennary with a GlcNAc linked β1,2 to both mannose and the third GlcNAc linked β1,4 to the α1,3 linked mannose; A4, GlcNAcs linked as A3 with additional GlcNAc β1,6 linked to α1, 6 mannose; B, bisecting GlcNAc linked β1,4 to β1,3 mannose; G(x), number of β1,4 linked galactose
on core GlcNAcs; A(x), number of antenna (GlcNAc) on trimannosyl core; A2, biantennary with both GlcNAcs as β1,2-linked; A3, triantennary with a GlcNAc linked β1,2 to both mannose and the third GlcNAc linked β1,4 to the α1,3 linked mannose; A4, GlcNAcs linked as A3 with additional GlcNAc β1,6 linked to α1, 6 mannose; B, bisecting GlcNAc linked β1,4 to β1,3 mannose; G(x), number of β1,4 linked galactose  on antenna; F(x), number of fucose linked α1,2 or α1,3 to antenna GlcNAc; S(x), number of sialic acids
on antenna; F(x), number of fucose linked α1,2 or α1,3 to antenna GlcNAc; S(x), number of sialic acids  linked to galactose; Lac(x), number of lactosamine
linked to galactose; Lac(x), number of lactosamine  (Galβ1–4GlcNAc) extensions.
(Galβ1–4GlcNAc) extensions.
Statistical analysis
Statistical analysis of the UPLC glycan data was transformed using the following formula: log(peak/(100-peak)). Data were checked for normality using the Kolmogorov Smirnov test. Normally distributed data were then analysed using the multivariate analysis of variance (MANOVA) test. The post hoc and Tukey tests were utilised to test for significance between the groups. Levene's test, tested for homogeneity of variance. Significant differences are defined as p ≤ 0.05*, p ≤ 0.01**, p ≤ 0.001***.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
Funding is acknowledged from the UCD Wellcome Institutional Strategic Support Fund, which was financed jointly by University College Dublin and the SFI-HRB-Wellcome Biomedical Research Partnership (ref. 204844/Z/16/Z) (AMcC and SL). Funding is also acknowledged from The Mater Foundation, Mater Misericordiae University Hospital (MMUH) Dublin Ireland (EK). RS acknowledges funding from the Science foundation Ireland Starting Investigator Research grant (SFI SIRG) under grant number 13/SIRG/2164. PD acknowledges LC-MS facilities funded by competitive awards from Science Foundation Ireland (12/RI/2346 (3)) and the Irish Higher Education Authority.References
- Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2nd edn, 2009 Search PubMed.
- T. Feizi, Cancer Surv., 1985, 4(1), 245–269 CAS.
- K. Marino, J. Bones, J. J. Kattla and P. M. Rudd, Nat. Chem. Biol., 2010, 6(10), 713–723 CrossRef CAS PubMed.
- L. Royle, M. P. Campbell, C. M. Radcliffe, D. M. White, D. J. Harvey, J. L. Abrahams, Y. G. Kim, G. W. Henry, N. A. Shadick, M. E. Weinblatt, D. M. Lee, P. M. Rudd and R. A. Dwek, Anal. Biochem., 2008, 376(1), 1–12 CrossRef CAS PubMed.
- H. Stockmann, R. M. Duke, S. Millan Martin and P. M. Rudd, Anal. Chem., 2015, 87(16), 8316–8322 CrossRef CAS.
- H. Stockmann, R. O'Flaherty, B. Adamczyk, R. Saldova and P. M. Rudd, Integr. Biol., 2015, 7(9), 1026–1032 CrossRef CAS PubMed.
- R. Saldova, A. Asadi Shehni, V. D. Haakensen, I. Steinfeld, M. Hilliard, I. Kifer, A. Helland, Z. Yakhini, A. L. Borresen-Dale and P. M. Rudd, J. Proteome Res., 2014, 13(5), 2314–2327 CrossRef CAS PubMed.
- Y. Itakura, N. Sasaki, D. Kami, S. Gojo, A. Umezawa and M. Toyoda, Cell Biosci., 2016, 6(1), 1–11 CrossRef PubMed.
- G. P. Dimri, X. Lee, G. Basile, M. Acosta, G. Scott, C. Roskelley, E. E. Medrano, M. Linskens, I. Rubelj and O. Pereira-Smith, et al. , Proc. Natl. Acad. Sci. U. S. A., 1995, 92(20), 9363–9367 CrossRef CAS PubMed.
- D. H. Juers, B. W. Matthews and R. E. Huber, Protein Sci., 2012, 21(12), 1792–1807 CrossRef CAS PubMed.
- P. M. Rudd, T. Endo, C. Colominas, D. Groth, S. F. Wheeler, D. J. Harvey, M. R. Wormald, H. Serban, S. B. Prusiner, A. Kobata and R. A. Dwek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(23), 13044–13049 CrossRef CAS PubMed.
- G. A. Levvy and A. McAllan, Biochem. J., 1961, 80(2), 435–439 CrossRef CAS PubMed.
- D. G. Hildebrand, S. Lehle, A. Borst, S. Haferkamp, F. Essmann and K. Schulze-Osthoff, Cell Cycle, 2013, 12(12), 1922–1927 CrossRef CAS PubMed.
- T. C. Cheng, S. H. Tu, L. C. Chen, M. Y. Chen, W. Y. Chen, Y. K. Lin, C. T. Ho, S. Y. Lin, C. H. Wu and Y. S. Ho, Oncotarget, 2015, 6(25), 21283–21300 CrossRef PubMed.
- S. Bonin, A. Parascandolo, C. Aversa, R. Barbazza, N. Tsuchida, M. D. Castellone, G. Stanta and G. Vecchio, Oncotarget, 2018, 9(20), 15228–15238 CrossRef PubMed.
- A. D. Baudot, D. Crighton, J. O'Prey, J. Somers, P. Sierra Gonzalez and K. M. Ryan, Cell Cycle, 2016, 15(17), 2299–2308 CrossRef CAS PubMed.
- I. Ezawa, Y. Sawai, T. Kawase, A. Okabe, S. Tsutsumi, H. Ichikawa, Y. Kobayashi, F. Tashiro, H. Namiki, T. Kondo, K. Semba, H. Aburatani, Y. Taya, H. Nakagama and R. Ohki, Cancer Sci., 2016, 107(6), 734–745 CrossRef CAS PubMed.
- K. Hayashi, P. Walde, T. Miyazaki, K. Sakayama, A. Nakamura, K. Kameda, S. Masuda, H. Umakoshi and K. Kato, J. Drug Delivery, 2012, 2012, 842785 Search PubMed.
- R. Tomaiuolo, A. Ruocco, C. Salapete, C. Carru, G. Baggio, C. Franceschi, A. Zinellu, J. Vaupel, C. Bellia, B. Lo Sasso, M. Ciaccio, G. Castaldo and L. Deiana, Aging Cell, 2012, 11(3), 394–400 CrossRef CAS PubMed.
- M. L. A. de Leoz, L. J. T. Young, H. J. An, S. R. Kronewitter, J. Kim, S. Miyamoto, A. D. Borowsky, H. K. Chew and C. B. Lebrilla, Mol. Cell. Proteomics, 2011, 10, 1 CrossRef PubMed.
- R. Saldova, V. D. Haakensen, E. Rodland, I. Walsh and H. Stockmann, Mol. Oncol., 2017, 11(10), 1361–1379 CrossRef CAS PubMed.
- A. Varki, R. Kannagi and B. Toole, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 3rd edn, 2017, ch. 47 Search PubMed.
- M. G. Amorim, R. Valieris, R. D. Drummond, M. P. Pizzi, V. M. Freitas, R. Sinigaglia-Coimbra, G. A. Calin, R. Pasqualini, W. Arap, I. T. Silva, E. Dias-Neto and D. N. Nunes, Sci. Rep., 2017, 7(1), 14395 CrossRef PubMed.
- http://exocarta.org/exosome_markers .
- http://www.uniprot.org .
- N. Tominaga, K. Hagiwara, N. Kosaka, K. Honma, H. Nakagama and T. Ochiya, Mol. Cancer, 2014, 13, 134 CrossRef PubMed.
- H. Zhang, D. Freitas, H. S. Kim, K. Fabijanic, Z. Li, H. Chen, M. T. Mark, H. Molina, A. B. Martin, L. Bojmar, J. Fang, S. Rampersaud, A. Hoshino, I. Matei, C. M. Kenific, M. Nakajima, A. P. Mutvei, P. Sansone, W. Buehring, H. Wang, J. P. Jimenez, L. Cohen-Gould, N. Paknejad, M. Brendel, K. Manova-Todorova, A. Magalhães, J. A. Ferreira, H. Osório, A. M. Silva, A. Massey, J. R. Cubillos-Ruiz, G. Galletti, P. Giannakakou, A. M. Cuervo, J. Blenis, R. Schwartz, M. S. Brady, H. Peinado, J. Bromberg, H. Matsui, C. A. Reis and D. Lyden, Nat. Cell Biol., 2018, 20(3), 332–343 CrossRef CAS PubMed.
- E. L. Kavanagh, S. Lindsay, M. Halasz, L. C. Gubbins, K. Weiner-Gorzel, M. H. Z. Guang, A. McGoldrick, E. Collins, M. Henry, A. Blanco-Fernández, P. O'Gorman, P. Fitzpatrick, M. J. Higgins, P. Dowling and A. McCann, Oncogenesis, 2017, 6, e388 CrossRef CAS PubMed.
- T. Beccari, S. Stinchi and A. Orlacchio, Biosci. Rep., 1999, 19(3), 157–162 CrossRef CAS PubMed.
- M. Knas, A. Zalewska, R. Kretowski, M. Niczyporuk, N. Waszkiewicz, M. Cechowska-Pasko, D. Waszkiel and K. Zwierz, Folia Histochem. Cytobiol., 2012, 50(2), 220–227 CrossRef CAS PubMed.
- N. Shah, D. A. Kuntz and D. R. Rose, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(28), 9570–9575 CrossRef CAS PubMed.
- A. D. Elbein, R. Solf, P. R. Dorling and K. Vosbeck, Proc. Natl. Acad. Sci. U. S. A., 1981, 78(12), 7393–7397 CrossRef CAS PubMed.
- P. E. Goss, C. L. Reid, D. Bailey and J. W. Dennis, Clin. Cancer Res., 1997, 3(7), 1077–1086 CAS.
- https://www.ncbi.nlm.nih.gov/gene/7841 .
- P. Roboti and S. High, J. Cell Sci., 2012, 125(Pt 14), 3474–3484 CAS.
- S. E. Trombetta and A. J. Parodi, J. Biol. Chem., 1992, 267(13), 9236–9240 CrossRef CAS.
- N. Fukuishi, S. Murakami, A. Ohno, N. Yamanaka, N. Matsui, K. Fukutsuji, S. Yamada, K. Itoh and M. Akagi, J. Immunol., 2014, 193(4), 1886–1894 CrossRef CAS PubMed.
- L. Warren and H. Felsenfeld, J. Biol. Chem., 1962, 237(5), 1421–1431 CrossRef CAS.
- E. P. Bennett, H. Hassan, M. A. Hollingsworth and H. Clausen, FEBS Lett., 1999, 460(2), 226–230 CrossRef CAS.
- B. Ritschka, M. Storer, A. Mas, F. Heinzmann, M. C. Ortells, J. P. Morton, O. J. Sansom, L. Zender and W. M. Keyes, Genes Dev., 2017, 31(2), 172–183 CrossRef CAS PubMed.
- A. Bartolazzi, A. Nocks, A. Aruffo, F. Spring and I. Stamenkovic, J. Cell Biol., 1996, 132(6), 1199–1208 CrossRef CAS PubMed.
- Z. Cao, Z. Hao, M. Xin, L. Yu, L. Wang, Y. Zhang, X. Zhang and X. Guo, Lab. Invest., 2018, 98(12), 1642–1656 CrossRef CAS PubMed.
- K. Nam, S. Oh and I. Shin, Biochem. J., 2016, 473(19), 3013–3030 CrossRef CAS PubMed.
- R. Guerin, P. B. Beauregard, A. Leroux and L. A. Rokeach, PLoS One, 2009, 4(7), e6244 CrossRef PubMed.
- H. C. Christianson, K. J. Svensson, T. H. van Kuppevelt, J.-P. Li and M. Belting, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(43), 17380–17385 CrossRef CAS PubMed.
- M. Farhad, A. S. Rolig and W. L. Redmond, OncoImmunology, 2018, 7(6), e1434467 CrossRef PubMed.
- H. Zhang, M. Luo, X. Liang, D. Wang, X. Gu, C. Duan, H. Gu, G. Chen, X. Zhao, Z. Zhao and C. Liu, PLoS One, 2014, 9(9), e103482 CrossRef PubMed.
- H. E. Miwa, Y. Song, R. Alvarez, R. D. Cummings and P. Stanley, Glycoconjugate J., 2012, 29(8-9), 609–618 CrossRef CAS PubMed.
- J. Dumic, S. Dabelic and M. Flogel, Biochim. Biophys. Acta, 2006, 1760(4), 616–635 CrossRef CAS.
- M. Guevremont, J. Martel-Pelletier, C. Boileau, F. T. Liu, M. Richard, J. C. Fernandes, J. P. Pelletier and P. Reboul, Ann. Rheum. Dis., 2004, 63(6), 636–643 CrossRef CAS.
- J. Ochieng, B. Green, S. Evans, O. James and P. Warfield, Biochim. Biophys. Acta, 1998, 1379(1), 97–106 CrossRef CAS.
- J. Ochieng, R. Fridman, P. Nangia-Makker, D. E. Kleiner, L. A. Liotta, W. G. Stetler-Stevenson and A. Raz, Biochemistry, 1994, 33(47), 14109–14114 CrossRef CAS PubMed.
- I. Pelletier and S. Sato, J. Biol. Chem., 2002, 277(20), 17663–17670 CrossRef CAS PubMed.
- P. Subhasitanont, C. Srisomsap, P. Punyarit and J. Svasti, Cancer Genomics Proteomics, 2006, 3(6), 389–394 CAS.
- P. Nangia-Makker, T. Raz, L. Tait, V. Hogan, R. Fridman and A. Raz, Cancer Res., 2007, 67(24), 11760–11768 CrossRef CAS PubMed.
- Y. Wang, P. Nangia-Makker, L. Tait, V. Balan, V. Hogan, K. J. Pienta and A. Raz, Am. J. Pathol., 2009, 174(4), 1515–1523 CrossRef CAS PubMed.
- K. Jiang, C. R. Rankin, P. Nava, R. Sumagin, R. Kamekura, S. R. Stowell, M. Feng, C. A. Parkos and A. Nusrat, J. Biol. Chem., 2014, 289(15), 10510–10517 CrossRef CAS PubMed.
- J. Friedrichs, A. Manninen, D. J. Muller and J. Helenius, J. Biol. Chem., 2008, 283(47), 32264–32272 CrossRef CAS PubMed.
- Y. Itakura, N. Sasaki and M. Toyoda, Aging, 2018, 10(8), 2190–2208 CrossRef CAS.
- K. Weiner-Gorzel, E. Dempsey, M. Milewska, A. McGoldrick, V. Toh, A. Walsh, S. Lindsay, L. Gubbins, A. Cannon, D. Sharpe, J. O'Sullivan, M. Murphy, S. F. Madden, M. Kell, A. McCann and F. Furlong, Cancer Med., 2015, 4(5), 745–758 CrossRef CAS PubMed.
- F. Furlong, P. Fitzpatrick, S. O'Toole, S. Phelan, B. McGrogan, A. Maguire, A. O'Grady, M. Gallagher, M. Prencipe, A. McGoldrick, P. McGettigan, D. Brennan, O. Sheils, C. Martin, W. K. E. J. O'Leary and A. McCann, J. Pathol., 2012, 226(5), 746–755 CrossRef CAS PubMed.
- J. Cao, S. Guo, K. Arai, E. H. Lo and M. M. Ning, Methods Mol. Biol., 2013, 1013, 227–233 CrossRef CAS PubMed.
- M. Anugraham, F. Jacob, S. Nixdorf, A. V. Everest-Dass, V. Heinzelmann-Schwarz and N. H. Packer, Mol. Cell. Proteomics, 2014, 13(9), 2213–2232 CrossRef CAS PubMed.
- L. Royle, C. M. Radcliffe, R. A. Dwek and P. M. Rudd, Methods Mol. Biol., 2006, 347, 125–143 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0mo00017e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |