
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2,9-Dibenzo[b,def]chrysene as a building block for organic electronics†
Félix
Gagnon
a,
Vicky
Tremblay
a,
Armand
Soldera
 b,
Michael U.
Ocheje
c,
Simon
Rondeau-Gagné
c,
Mario
Leclerc
a and
Jean-François
Morin
*a
b,
Michael U.
Ocheje
c,
Simon
Rondeau-Gagné
c,
Mario
Leclerc
a and
Jean-François
Morin
*a
aDépartement de chimie and Centre de Recherche sur les Matériaux Avancés (CERMA), 1045 Ave de la Médecine, Université Laval, Québec, QC G1V 0A6, Canada. E-mail: jean-francois.morin@chm.ulaval.ca
bLaboratory of Physical Chemistry of Matters, Department of chemistry, Faculty of science, 2500 Boulevard de l’Université, Université de Sherbrooke, Sherbrooke, QC J1K 2R1, Canada
cDepartment of Chemistry and Biochemistry, 401 Sunset Avenue, University of Windsor, Windsor, ON N9B 3P4, Canada
First published on 10th November 2021
Abstract
In this article, a new series of conjugated polymers based on a low-cost, easily accessible vat dye called Vat orange 1 or 2,9-dibromo-dibenzo[b,def]chrysene-7,14-dione have been prepared. This compound was made electron-donating by reducing and alkylating the ketone groups into alkoxy groups, allowing the introduction of solubilizing, branched alkyl chains at the 7 and 14 positions. Polymerization reactions using Suzuki–Miyaura coupling with 6,6′-isoindigo, 3,6-di-2-thienyl-pyrrolo[3,4-c]pyrrole-1,4-dione (DPP) and 4,7-dithieno-2,1,3-benzothiadiazole (TBT) yielded three donor–acceptor polymers with bandgap values ranging from 1.61 to 1.86 eV. Field-effect transistors (OFETs) and organic solar cells (OSCs) were fabricated and hole mobility values of up to 3.62 × 10−4 cm2 V−1 s−1 and a maximum power conversion efficiency of 1.2% were measured, respectively.
Introduction
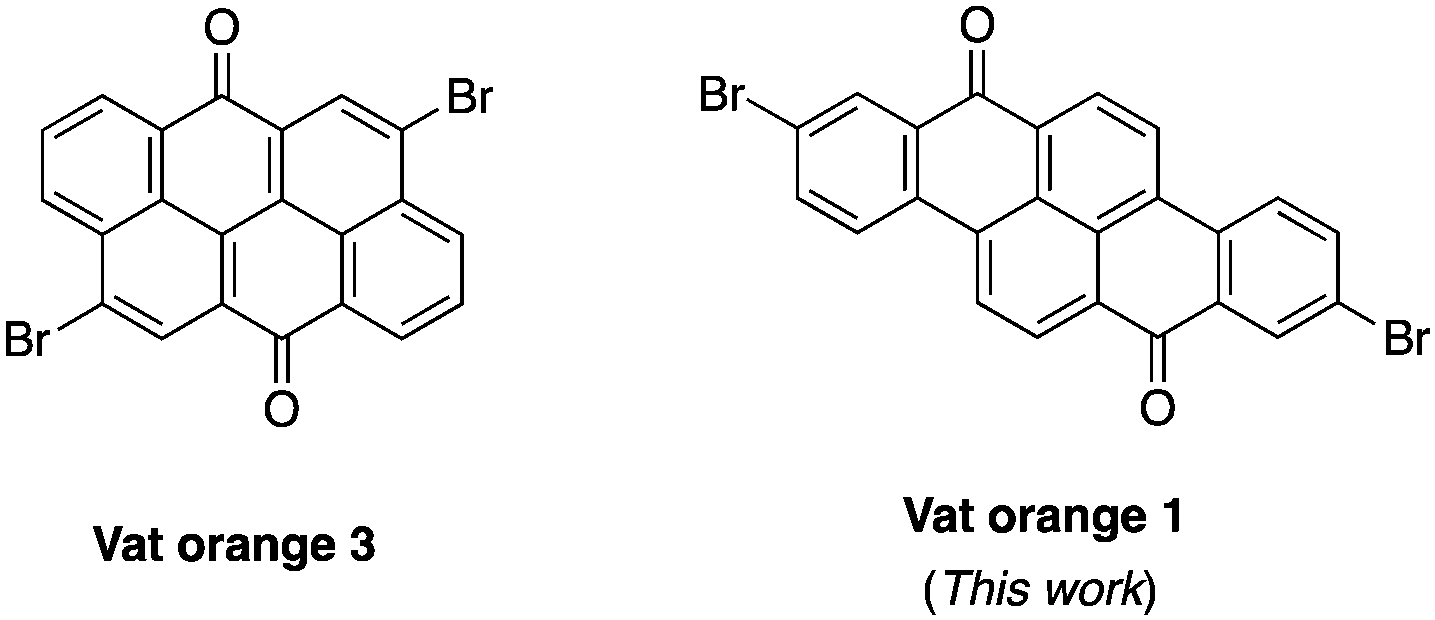
Dyes possessing a carbon-rich, extended π-conjugated core are promising building blocks for the preparation of semiconductors in organic electronics due to their relatively low cost, stability and ease of functionalization.1–3 In fact, many of them possess synthetic handles such as halogens and ketones that allow easy chemical functionalization and modifications of their electronic properties. Recently, vat orange 3 (4,10-dibromoanthanthrone) has been used as the starting material for several π-conjugated molecules and polymers for multiple applications, including field-effect transistors,4–7 light-emitting diodes,8,9 aggregation-induced emission (AIE),10 solar cells11–17 and singlet fission.18,19 This dye showed good versatility as it can be functionalized at the 4, 6, 10 and 12 positions to yield materials with a wide range of properties.20–27 However, the major drawback of this dye is the presence of two hydrogen atoms at the peri position relative to the two bromine atoms (4 and 10 positions) that cause significant steric hindrance with the neighbouring conjugated units.28 This structural flaw in regard to the effective conjugation length along with its neutral electronic nature (neither a donor nor an acceptor) makes the synthesis of low bandgap materials based on vat orange 3 very challenging. To overcome this problem, strategies such as the introduction of conjugated spacers such as alkene or alkyne moieties and rigidification through the formation of a carbon bridge to eliminate the peri proton have been explored.17,28,29 Unfortunately, none of them succeed in significantly decreasing the bandgap of anthanthrone-based molecules and polymers.Thus, other similar vat dyes have to be explored in order to prepare low bandgap materials that would be suitable for organic electronics applications. Among the commercially available dihalogenated vat dyes, vat orange 1 is particularly appealing since it possesses two bromine atoms at the 2 and 9 positions where virtually no steric hindrance could lead to a high dihedral angle. Moreover, this dye also has two ketones (like vat orange 3), allowing a wide variety of chemical modifications.
Herein, we report the synthesis, characterization and device performances of conjugated polymers based on 2,9-dibenzo[b,def]chrysene (vat orange 1). In order to ensure the solubility of the polymers, the dye was alkylated at the 7 and 14 positions to give two electron-donor alkoxy groups. This unit was then polymerized with three different electron-accepting units, namely isoindigo (P1), diketopyrrolopyrrole (P2) and bisthiophenylbenzothiadiazole (P3). The polymers were characterized and tested as semiconductors in field-effect transistors and bulk heterojunction solar cells.
Results and discussion
The synthesis of P1–P3 is described in Scheme 1. In order to ensure the solubility of P1–P3, 2,9-dibromo-dibenzo[b,def]chrysene-7,14-dione (vat orange 1) was first alkylated using a branched alkyl chain using an adapted protocol developed for vat orange 3 to obtain compound 1.20 Then, a two-fold Miyaura borylation reaction under standard conditions obtained compound 2 in 53% yield. P1–P3 were prepared by Suzuki coupling using the same procedure by coupling compound 2 with isoindigo, DPP and TTBT, respectively. After the synthesis, the polymers were purified by precipitation in methanol, followed by washing with methanol, hexanes and chloroform using a Soxhlet apparatus to remove the oligomers and the catalyst residues. The reaction yield for each polymer was calculated using the soluble fraction only. The molecular weight values (Mn and Mw) of P1–P3 were measured by size-exclusion chromatography using polystyrene standards and 1,2,4-trichlorochlorobenzene as the eluent at 110 °C (Table 1). Number average molecular weight (Mn) values ranging from 33 to 46 kg mol−1 were obtained, corresponding to the degree of polymerization (Xn) values between 21 and 28, providing good film-forming properties.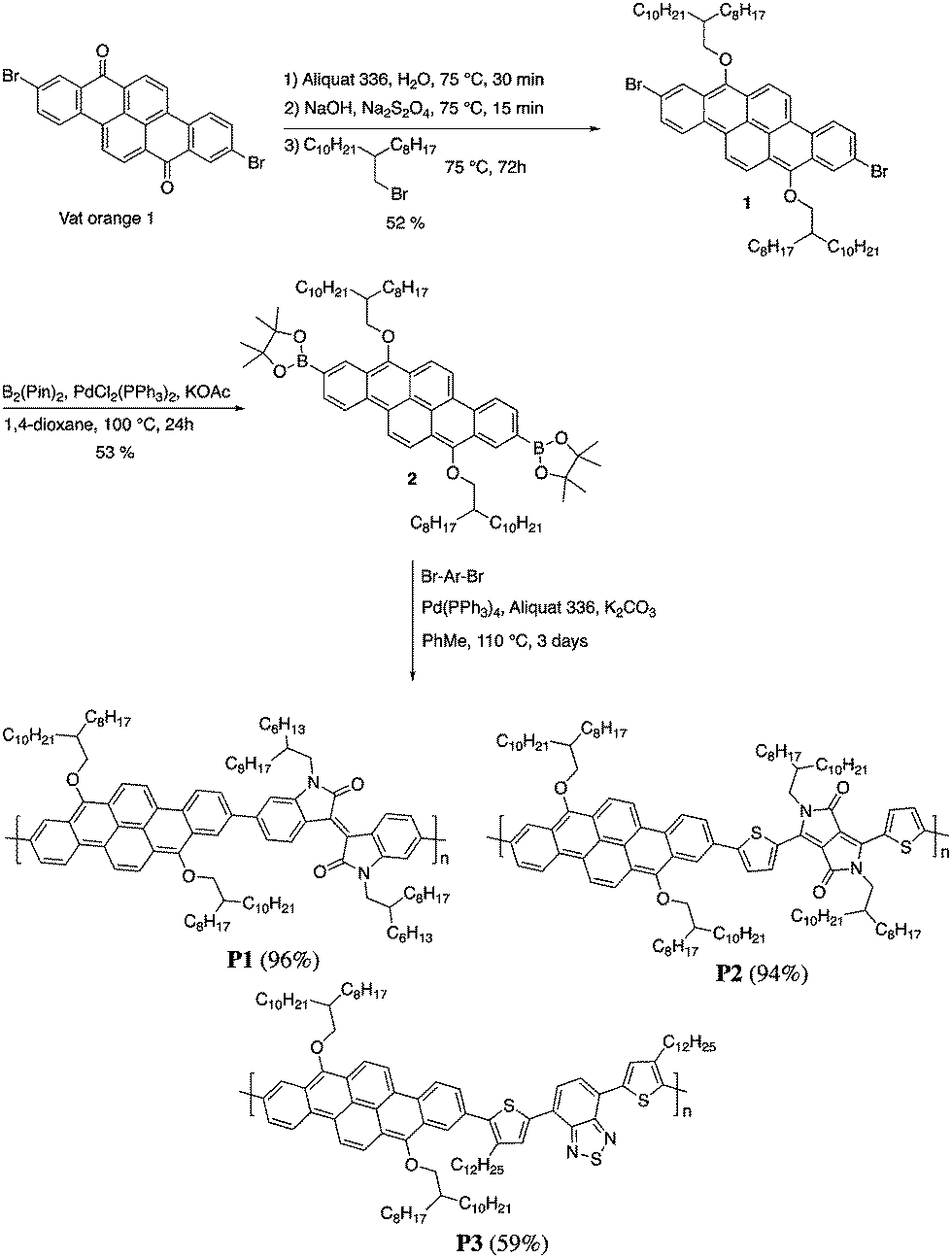 | ||
| Scheme 1 Synthesis of P1–P3. | ||
| Polymer |
|
|
Đ
|
X n |
|---|---|---|---|---|
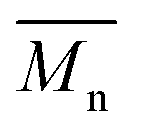 : the number average molecular weight. : the number average molecular weight.  : the weight average molecular weight. Đ: dispersity index.a Molecular weight values and Đ values of P1, P2 and P3 were measured by SEC at 110 °C using 1,2,4-trichlorobenzene as the eluent and polystyrene as the standard. : the weight average molecular weight. Đ: dispersity index.a Molecular weight values and Đ values of P1, P2 and P3 were measured by SEC at 110 °C using 1,2,4-trichlorobenzene as the eluent and polystyrene as the standard. |
||||
| P1 | 33 | 53 | 1.6 | 21 |
| P2 | 46 | 119 | 2.6 | 26 |
| P3 | 42 | 95 | 2.2 | 28 |
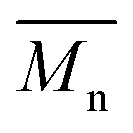
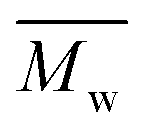
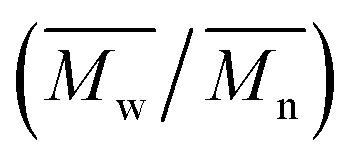
Thermal properties were measured using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) and the results are shown in Fig. S9–S12 (ESI†). In all cases, the measured glass transition temperature (Tg) values were between 100 and 120 °C and no melting transition was observed, meaning that P1–P3 are amorphous. These observations agree with the nature of the long, branched alkyl chain attached to the 2,9-dibenzo[b,def]chrysene unit.30 TGA analysis was performed under a nitrogen atmosphere at a heating rate of 10 °C per minute. Decomposition temperature (Td) values between 280 and 290 °C (taken a 5% weight) were measured for all three polymers, indicating that the most sensitive part of the polymers is likely the 2,9-dibenzo[b,def]chrysene unit. These Td values are similar to those measured for the conjugated polymers based on anthanthrene (vat orange 3).29 Although these values are lower than those of most conjugated polymers, they are high enough to ensure good thermal stability of the polymers for device fabrication and operation.
The optical properties in both the solution and solid states of P1–P3 are shown in Fig. 1 and are summarized in Table 2. All the polymers showed a broad absorption band in the UV–visible region with λmax values ranging from 478 to 700 nm in solution. For P1, the band in the high energy region centered at 478 nm can be attributed to the π–π* transition while the broad band centered at 572 nm is attributed to the presence of a charge transfer complex between the dibenzo[b,def]chrysene and isoindigo units.31 Interestingly, the solid state spectrum exhibits only a slight bathochromic shift compared to that in solution, indicating that the polymer backbone conformation is very similar in both states. The same behavior was also observed for P2 and P3.
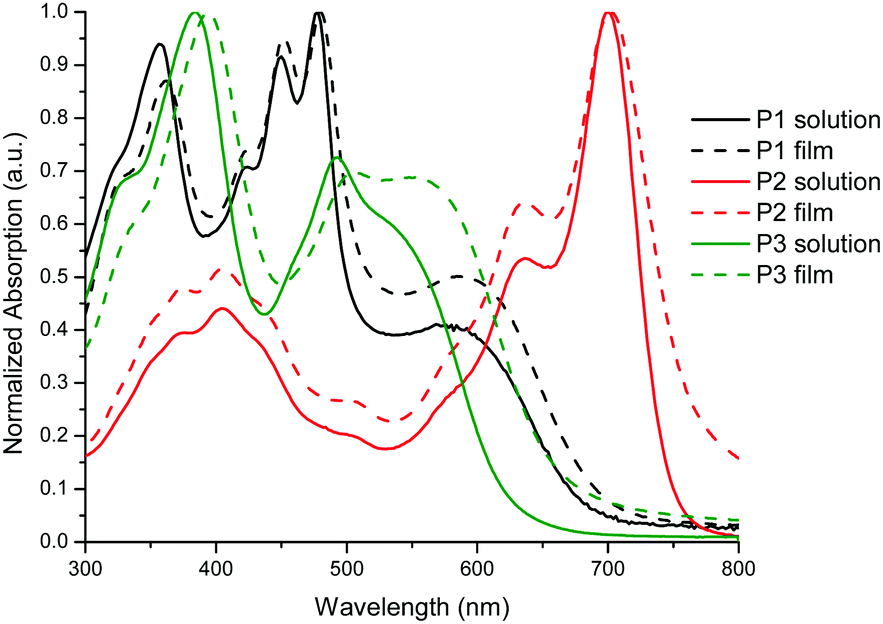 | ||
| Fig. 1 Normalized UV–vis absorption spectra of polymers P1–P3 in a solution of CHCl3 and as a thin film. | ||
| Polymer | λ solmax nm a | λ filmmax nm b | E optg (eV) c | E HOMO (eV) d | E LUMO (eV) d | E elecg (eV) |
|---|---|---|---|---|---|---|
| a Solution in chloroform. b Spin cast film from chloroform solution. c The optical bandgap values were estimated from the edge of the absorption spectra of thin films (Eoptg = 1240/λonset). d HOMO and LUMO energy levels were calculated using the formula EHOMO = −(4.71 + Eoxonset) eV and ELUMO = −(4.71 + Ereonset) eV, in which Eoxonset and Ereonset are the oxidation onset potential (the CV thin film) and the reduction onset potential (the CV thin film), respectively, versus SCE. | ||||||
| P1 | 478 | 480 | 1.77 | −5.36 | −3.49 | 1.86 |
| P2 | 700 | 702 | 1.61 | −5.31 | −3.73 | 1.58 |
| P3 | 492 | 506 | 1.86 | −5.35 | −3.40 | 1.95 |
As reported previously for BTD-based conjugated polymers,32,33P3 exhibits two sets of absorption bands centered at 384 and 492 nm. Interestingly, the shoulder at 540 nm in the solution UV–visible spectrum, associated to the presence of an intramolecular donor–acceptor complex, increased in intensity in the solid state while becoming broader towards the low energy region of the spectrum. This can be attributed to a planarization of the polymer backbone in the solid state, making the charge transfer complex between the 2,9-dibenzo[b,def]chrysene and BTD units more efficient. All the polymers exhibit a bandgap value below 1.9 eV (see Table 2), the lowest value being obtained for P2 (1.61 eV). These values correlate well with those measured using electrochemistry in thin films. As expected, the HOMO energy values for P1–P3 are very similar since they are governed by the donor unit that is the same for all three polymers (2,9-dibenzo[b,def]chrysene unit). For the LUMO, P2 exhibits the lowest energy value at −3.73 eV, which is suitable to undergo charge transfer upon excitation with most of the electron-acceptor molecules used in the fabrication of solar cells.
Theoretical calculations
DFT calculations were performed on the dimers of the three polymers to assess their conformation, the energy levels, and the frontier orbitals distribution using the Gaussian 09 software.34 Research on the conformation of the minimum energy was first carried out. For this, each part, the vat orange derivative with the bromine atoms substituted by ethyl chains, and the three groups, were first optimized. To find the minimum of energy for the final molecule, the bond between the two parts was rotated by 10 degrees, and the potential energy was computed for each dihedral angle. The density functional theory (DFT) approach was considered using 6-31G** as the basis set and B3LYP as the functional. The dihedral angle corresponds to four consecutive carbon atoms.Of the three polymers, P2 is the one presenting the lowest dihedral angle (θ = 30.3°) between the vat orange 1 and the comonomer (see Table S3 and Fig. S23 in the ESI† section). For P1 and P3, the presence of phenyl and an alkyl chain at the 3 position of thiophene induces a wider dihedral angle (θ = 40.6° and 44.9°, respectively). The low θ value in P2 can partially explain the lower bandgap value (Eg = 1.61 eV).
For all three polymers, the HOMO orbital is mainly localized on the 2,9-dibenzo[b,def]chrysene unit, except for P2 in which the HOMO is delocalized over the entire dimer (Fig. 2). As expected, the LUMO orbital is located almost exclusively on the electron-withdrawing unit. The bandgap values calculated for the dimers of P1, P2 and P3 are 2.13, 2.30 and 2.31, respectively.
 | ||
| Fig. 2 Kohn–Sham molecular orbitals of dimers of P1, P2 and P3 based on calculation at the B3LYP/6-31G(d,p) level. | ||
Field-effect transistors (OFETs)
To investigate the influence of the acceptor moiety on the field-effect mobility and charge transport, bottom-gate, top-contact organic field-effect transistors (OFETs) were fabricated. The device parameters and the extrapolated figures of merit are shown in Table 3 and fabrication procedures and device characteristics are detailed in the ESI.† The devices prepared from P1, containing the isoindigo acceptor, exhibited a relatively low mobility (2.45 × 10−5 cm2 V−1 s−1) and a low ION/OFF current ratio. The devices prepared from P1 also possess the highest threshold voltage of −14.46 V. In contrast, P2, which contains the diketopyrrolopyrrole acceptor, exhibits a higher charge mobility and better charge transport (an average mobility of 2.99 × 10−4 cm2 V−1 s−1), and overall device characteristics. For P3, which contains benzothiadiazole, an average field-effect mobility of 3.22 × 10−4 cm2 V−1 s−1, an ION/OFF of 103 and a threshold voltage of −9.53 V were measured. Despite the important differences in charge transport between the three semiconducting polymers, all devices showed typical transfer and output characteristics as depicted in Fig. S22 (ESI†).| Polymer | W L−1 | μ maxh (× 10−4 cm2 V−1 s−1) | μ avgh (× 10−4 cm2 V−1 s−1) | I ON/OFF | V th (V) |
|---|---|---|---|---|---|
| P1 | 10 | 0.635 | 0.245 ± 0.257 | 102 | −14.46 |
| P2 | 10 | 3.62 | 2.99 ± 0.624 | 103 | −11.11 |
| P3 | 10 | 3.36 | 3.22 ± 0.0912 | 103 | −9.53 |
Organic solar cells (OSCs)
P1–P3 were tested in organic solar cells (OSCs) as p-type materials and the results are summarized in Table 4. The devices were prepared in an inverted geometry using [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) as the electron acceptor in the heterojunction blend. The geometry studied was ITO/ZnO/Polymer:PC61BM/MoO3/Ag. Different ratios of diphenyl ether (DPE) as an additive in the active layer were tested (0, 1 and 2% V/V) to achieve the highest power conversion efficiency (PCE) for each polymer.35P1 obtained a PCE of 0.25% without any additive. Under these conditions, P1 achieved the highest open-circuit voltage (VOC = 0.86 V) of the series, which is in accordance with the HOMO level of the polymer compared to P2 and P3. P2 obtained a quite a bit better PCE of 0.66% using 2% V/V DPE. This polymer gave a short-circuit current density (JSC) four times better than that of P1 (2.30 mA cm−2vs. 0.577 mA cm−2), but loses its gain because of a poor fill factor (FF) of 0.387 (P1:0.51). P3 achieved the highest JSC, FF and PCE of the series with 2.8 mA cm−2, 0.53 and 1.2%, respectively, using 1% V/V DPE.| Polymer | Additive ratiob | J sc | V oc | FF | PCE |
|---|---|---|---|---|---|
| (% V/V) | (mA cm−2) | (V) | (%) | ||
| a Maximum values reached are presented here, and the average values are presented in the ESI. b Diphenyl ether as the additive. c No increase in performance observed using the additive with this polymer. | |||||
| P1 | — | 0.577 | 0.86 | 0.51 | 0.25 |
| P2 | — | 0.9 | 0.8 | 0.33 | 0.2 |
| 2 | 2.30 | 0.74 | 0.387 | 0.66 | |
| P3 | — | 2.50 | 0.84 | 0.50 | 1.05 |
| 1 | 2.8 | 0.810 | 0.53 | 1.2 | |
In the case where no additive was used, P2 performances were very low, worse than P1, probably because the LUMO of P2 (−3.73 eV) is similar to PC61BM's.36 For P1 and P3, their differences in performance may be due to the dihedral angle between the donor and acceptor units.
Conclusions
In conclusion, three donor–acceptor polymers based on vat orange 1 were synthesized using Suzuki coupling. All three polymers are highly soluble in common organic solvents and exhibit excellent film-forming properties. As shown by DFT calculations, the dialkoxy form of vat orange 1 is an electron-donor on which most of the HOMO frontier orbital is localized when coupled to the electron-deficient π-conjugated units. All polymers possess a bandgap value of under 1.9 eV, making them suitable for applications in solar cells. Moreover, they all exhibit hole-transporting properties in field-effect transistors. Thus, vat orange 1 proved to be an efficient, low-cost building block for the preparation of the conjugated polymers for organic electronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Science and Engineering Council of Canada (NSERC) for financial support. Félix Gagnon thanks the NSERC and FRQNT for a Master scholarship. We are also grateful for professional support from CQMF and CERMA. The computational resources were provided by Calcul Québec and Compute Canada, through financial support of the Canadian Foundation Innovation (CFI). Simon Rondeau-Gagné acknowledges financial support from the NSERC through a Discovery Grant (RGPIN-2017-06611) and the Canadian Foundation for Innovation (CFI). Michael U. Ocheje thanks the NSERC for a PhD scholarship.Notes and references
- D. Thetford and A. P. Chorlton, Dyes Pigm., 2004, 61, 49–62 CrossRef CAS.
- C. Aumaitre and J.-F. Morin, Chem. Rec., 2019, 19, 1142–1154 CrossRef CAS.
- J. F. Morin, J. Mater. Chem. C, 2017, 12298–12307 RSC.
- Q. Liu, Y. Wang, L. Arunagiri, M. Khatib, S. Manzhos, K. Feron, S. E. Bottle, H. Haick, H. Yan, T. Michinobu and P. Sonar, Mater. Adv., 2020, 1, 3428–3438 RSC.
- J. B. Giguère, N. S. Sariciftci and J. F. Morin, J. Mater. Chem. C, 2015, 3, 601–606 RSC.
- M. Irimia-Vladu, P. A. Troshin, M. Reisinger, G. Schwabegger, M. Ullah, R. Schwoediauer, A. Mumyatov, M. Bodea, J. W. Fergus, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Org. Electron., 2010, 11, 1974–1990 CrossRef CAS.
- E. D. Glowacki, L. Leonat, G. Voss, M. Bodea, Z. Bozkurt, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, Org. Semicond. Sensors Bioelectron. IV, 2011, 8118, 81180M Search PubMed.
- B. K. Shah, D. C. Neckers, J. Shi, E. W. Forsythe and D. Morton, Chem. Mater., 2006, 18, 603–608 CrossRef CAS.
- B. K. Shah, D. C. Neckers, J. Shi, E. W. Forsythe and D. Morton, J. Phys. Chem. A, 2005, 109, 7677–7681 CrossRef CAS.
- M. Desroches and J. F. Morin, Org. Lett., 2018, 20, 2797–2801 CrossRef CAS PubMed.
- H. D. Pham, T. T. Do, J. Kim, C. Charbonneau, S. Manzhos, K. Feron, W. C. Tsoi, J. R. Durrant, S. M. Jain and P. Sonar, Adv. Energy Mater., 2018, 8, 1–13 Search PubMed.
- H. D. Pham, K. Hayasake, J. Kim, T. T. Do, H. Matsui, S. Manzhos, K. Feron, S. Tokito, T. Watson, W. C. Tsoi, N. Motta, J. R. Durrant, S. M. Jain and P. Sonar, J. Mater. Chem. C, 2018, 6, 3699–3708 RSC.
- Y. Geng, C. Yi, M. P. Bircher, S. Decurtins, M. Cascella, M. Grätzel and S. X. Liu, RSC Adv., 2015, 5, 98643–98652 RSC.
- J. Hong, Y. J. Kim, Y. H. Kim and C. E. Park, Macromol. Chem. Phys., 2016, 217, 2116–2124 CrossRef CAS.
- L. Zhang, B. Walker, F. Liu, N. S. Colella, S. C. B. Mannsfeld, J. J. Watkins, T.-Q. Nguyen and A. L. Briseno, J. Mater. Chem., 2012, 22, 4266–4268 RSC.
- Y. J. Kim, J. S. Lee, J. Hong, Y. Kim, S. B. Lee, S. K. Kwon, Y. H. Kim and C. E. Park, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2559–2570 CrossRef CAS.
- S. V. John, V. Cimrová, C. Ulbricht, V. Pokorná, A. Ružička, J. B. Giguère, A. Lafleur-Lambert, J. F. Morin, E. Iwuoha and D. A. M. Egbe, Macromolecules, 2017, 50, 8357–8371 CrossRef CAS PubMed.
- Y. Jue Bae, M. D. Krzyaniak, M. B. Majewski, M. Desroches, J. F. Morin, Y. L. Wu and M. R. Wasielewski, ChemPlusChem, 2019, 84, 1432–1438 CrossRef.
- S. K. Sugunan, C. Greenwald, M. F. Paige and R. P. Steer, J. Phys. Chem. A, 2013, 117, 5419–5427 CrossRef.
- J. B. Giguère, Q. Verolet and J. F. Morin, Chem. – Eur. J., 2013, 19, 372–381 CrossRef.
- J. B. Giguère, J. Boismenu-Lavoie and J. F. Morin, J. Org. Chem., 2014, 79, 2404–2418 CrossRef.
- J.-B. Giguère and J.-F. Morin, J. Org. Chem., 2015, 80 Search PubMed.
- U. Koldemir, J. S. Tinkham, R. Johnson, B. Lim, H. A. Yemam, K. J. Gagnon, S. Parkin and A. Sellinger, J. Mater. Chem. C, 2017, 5, 8723–8733 RSC.
- Y. Du, H. B. Lovell, F. Lirette, J. F. Morin and K. N. Plunkett, J. Org. Chem., 2021, 86, 1456–1461 CrossRef CAS.
- P. W. Münich, M. Pfäffli, M. Volland, S. X. Liu, R. Häner and D. M. Guldi, Nanoscale, 2020, 12, 956–966 RSC.
- C. Aumaitre, D. Fong, A. Adronov and J. F. Morin, Polym. Chem., 2019, 10, 6440–6446 RSC.
- J.-B. Giguère, Q. Verolet and J.-F. Morin, Chem. – Eur. J., 2013, 19, 372–381 CrossRef.
- F. Lirette, C. Aumaitre, C. É. Fecteau, P. A. Johnson and J. F. Morin, ACS Omega, 2019, 4, 14742–14749 CrossRef CAS.
- A. Lafleur-Lambert, J. B. Giguère and J. F. Morin, Polym. Chem., 2015, 6, 4859–4863 RSC.
- R. Xie, A. R. Weisen, Y. Lee, M. A. Aplan, A. M. Fenton, A. E. Masucci, F. Kempe, M. Sommer, C. W. Pester, R. H. Colby and E. D. Gomez, Nat. Commun., 2020, 11, 893 CrossRef CAS.
- X. Xu, L. Li, B. Liu and Y. Zou, Appl. Phys. Lett., 2011, 98, 063303 CrossRef.
- J. D. Harris, M. Stihl, H. W. Schmidt and K. R. Carter, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 60–69 CrossRef CAS.
- K. W. Song, M. H. Choi, H. J. Song, S. W. Heo, J. Y. Lee and D. K. Moon, Sol. Energy Mater. Sol. Cells, 2014, 120, 303–309 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- E. F. Manley, J. Strzalka, T. J. Fauvell, T. J. Marks and L. X. Chen, Adv. Energy Mater., 2018, 8, 1–20 Search PubMed.
- X. Gong, T. Yu, Y. Cao and A. J. Heeger, Sci. China: Chem., 2012, 55, 743–748 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00799h |
| This journal is © The Royal Society of Chemistry 2022 |