DOI:
10.1039/D1MA00953B
(Paper)
Mater. Adv., 2022,
3, 592-598
Electrocatalytic nitrogen reduction directed through the p-band center of boron on BSAC@Mo2C†
Received
13th October 2021
, Accepted 6th November 2021
First published on 9th November 2021
Abstract
Greener modes of ammonia synthesis via the electrocatalytic route have been investigated on pristine and defective Mo2C based monolayers anchored with metal-free boron atom catalysts. Boron single atom catalysts (SACs) on the defective Mo2C monolayer has been found to activate N2 strongly with an adsorption energy of −1.92 eV and reduce it to NH3 efficiently with a significantly low overpotential of 0.41 eV. The exothermic adsorption of N2 and low overpotential for the nitrogen reduction reduction (NRR) appertain to the p-band center of the boron atom catalyst and charge transfer between the adsorbed N2 and the catalyst, respectively. This work brings forth the correlation between electron occupancy on the boron center and NRR catalytic efficiency on a metal-free SACs@Mo2C monolayer couple, thereby serving as a lead in designing metal free electrocatalysts for the NRR.
1 Introduction
With the rise in global temperature and greenhouse gas emissions, most industrial processes aim to achieve carbon neutrality. However, one process with an extremely high carbon footprint accounting for 6% of global CO2 emissions and the consumption of approximately 1–2% of global energy1 is the Haber–Bosch2 ammonia synthesis process. With ammonia being an irreplaceable precursor in agriculture, various industries and energy applications,3,4 there is an urgent need to develop greener techniques for NH3 synthesis through photocatalytic, electrocatalytic or photo-electrocatalytic routes to meet the current demands.5,6 The optimal goal towards achieving ammonia economy is to develop a catalyst that reduces N2 to NH3 under ambient conditions like the nitrogenase enzyme. The electrocatalytic route of nitrogen fixation is an attractive alternative owing to its efficiency and environment-friendly nature.7,8 However, with N2 being a highly stable molecule with a N–N triple bond energy of 940 kJ mol−1, the kinetics associated with the electrocatalytic nitrogen reduction reaction (NRR) are sluggish and the faradaic efficiency is low.7 As a result, the competing hydrogen evolution reaction (HER) is usually preferred over the sluggish NRR. Therefore, it is essential to design new and smart materials with high electrical conductivity that have the capability to subjugate the HER and enhance the NRR. Several scarce metals such as Ru,9,10 Au11,12 and Pd13,14 have been reported as highly efficient electrocatalysts for the NRR with faradaic efficiencies of up to 36.6%,13 their cost and availability are issues that cannot be overcome. Hence, identifying cost effective electrocatalysts based on earth abundant metals such as Mo, Fe, and Co, etc. as potential alternatives for the NRR is the need of the hour.15
Notably, Mo metal is present as an active centre in nitrogenase enzymes, and possesses the requisite electronic structure to capture N2 molecules and has been extensively studied for the NRR.16 Several other Mo-based two-dimensional (2D) materials, nanoparticles and nanoclusters, single atom catalysts (SACs) and organometallic complexes have been reported as efficient catalysts for the NRR.17–26 While Mo-containing organic and inorganic complexes efficiently adsorb and form stable dinitrogen complexes, their application as electrocatalysts or electrodes are not warranted.18 Mo clusters and SACs, on the other hand, require a conducting/semi-conducting support for further usage as electrocatalysts.19,20,22,27 In this regard, Mo-based 2D materials such as MoS2,24,28–31 MXenes (Mo2C)32–34 and MBenes (Mo2B2)35,36 are appealing electrocatalysts owing to their high electrical conductivity and the presence of active Mo metal centres. These 2D materials not only show high hydrophilicity, stability and conductivity but are interesting from the catalytic aspect of nitrogen reduction as the Mo metal is accessible to the nitrogen molecule. Compared to MBenes, MXenes have been extensively investigated experimentally for the electrocatalytic NRR.32,37–39
While computational studies on the electrochemical applications of Mo2C based 2D materials have been extensively carried out, they have rarely been explored for the electrocatalytic NRR. One notable study on the electrocatalytic application of Mo2C for the NRR was made recently by Zhang et al. wherein Mo2C monolayers were investigated for the NRR through density functional theory (DFT) calculations.34 It would be imperative to recollect that effective strategies to fine tune and improve the N2 binding efficacy on a 2D monolayer are structural modifications and incorporating active atomic centers on it.40 Thereby, pristine and defective Mo2C monolayers coupled with a metal free single atom catalyst, in particular boron, are expected to enhance the N2 adsorption and inherently improve the NRR process. Several experimental and computational studies have reported boron based metal free catalysts on 2D materials to remarkably augment the electrocatalytic NRR by promoting N2 adsorption and inhibiting the HER.41–43 The boron center behaves as a Lewis acid with empty p-orbitals, thereby resulting in a strong π–π* interaction with N2 and less energy demanding subsequent protonation steps for the NRR.44,45
The correlation between charge transfer from the catalytic center and nitrogen reduction efficacy is one aspect of paramount importance that has not been explored to date. Thus, in the present work, we conducted a comprehensive investigation to provide a complete picture concerning the fundamental understanding of nitrogen activation and reduction on active boron atoms anchored on a pristine Mo2C monolayer. Specifically, the coupling effect of boron single atom catalysts with defects on the Mo2C monolayer and its influence on nitrogen activation and reduction was thoroughly investigated. The p-band center and electronic structure of 2D Mo2C monolayers upon the anchoring of non-metallic boron atoms directly influences their NRR catalytic efficiency. Based on our results, we found that a single boron atom catalyst anchored on a defective Mo2C monolayer can effectively produce NH3 with a record low overpotential of 0.41 eV.
2 Computational details
All DFT calculations were carried out with the Vienna ab initio Simulation Package (VASP).46 The generalized gradient approximation (GGA) and the Perdew–Burke–Ernzerhof (PBE)47 functional has been employed with an energy cutoff of 532 eV to describe all electron core–interactions. A DFT-D3 correction method has been incorporated to account for the long-range van der Waals (vdW) interactions.48 All the catalyst systems sampled with a (5 × 5 × 1) Monkhorst–Pack k-point grid and a vacuum space of 20 Å along the Z-direction are relaxed until the atomic energy and forces converge to 10−5 eV per atom and 0.005 eV Å−1, respectively. For the density of states and electronic structure calculations, a higher (11 × 11 × 1) Monkhorst–Pack k-point grid has been employed.
The stability of atomic boron catalysts on Mo2C and its analogues is evaluated in terms of binding energy, Eb which is calculated by using the equation,
| | | Eb = (EBcat@Mo2C) − (EMo2C) − (EBcat) | (1) |
where,
EBcat@Mo2C and
EMo2C are the total electronic energies of the 2D Mo
2C monolayer with and without atomic boron catalysts and
EBcat is the electronic energy of atomic boron. The adsorption of N
2 on the 2D Mo
2C monolayer has been computed for the parallel and perpendicular modes and the efficiency of these materials to chemisorp N
2 molecule has been calculated in terms of the N
2 adsorption energy, (
Eads) as given below,
| | | Eads = (EBcat@Mo2C–N2) − (EBcat@Mo2C) − (EN2) | (2) |
where,
EBcatMo
2C–N
2,
EBcat@Mo2C and
EN2 are the total electronic energies of N
2 adsorbed systems, B
cat@Mo
2C and free N
2 molecules, respectively.
The Gibbs free energy change, ΔG in every protonation step of the NRR has been calculated by employing the computational SHE (standard hydrogen electrode) model proposed by Nørskov et al.49 using the equation,
where, Δ
E is the change in electronic energy,
ΔZPE is the change in zero-point energy,
T is the room temperature (298.15 K) and Δ
S is the change in entropy. The zero-point energies and entropy contributions are calculated by considering the vibrational frequencies of the adsorbed gas phase species. Furthermore, the overpotential for the electrocatalytic NRR on the Mo
2C monolayer and its analogues has been calculated as
η =
USHE −
UPDS, where
USHE = −0.16 eV, is the standard reduction potential of N
2 to NH
3 and
UPDS = Δ
Gmax/e for the NRR pathway.
3 Results and discussion
3.1 N2 adsorption on BcatMo2C
The stability of atomic boron catalysts on Mo2C is one of the fundamental prerequisites for their implementation as electrocatalysts for the NRR. Boron has been anchored on conventional pristine 1T-Mo2C as a single atom catalyst (SAC) or a diatom catalyst (DAC), respectively referred to as BSAC@Mo2C and BDAC@Mo2C from now onwards. The geometric structures of the BSAC@Mo2C and BDAC@Mo2C catalysts are presented in Fig. S2 (see the ESI†). Anchoring BDAC on Mo2C was found to be more feasible with a binding energy of −7.07 eV as compared to BSAC (−6.90 eV). The 2D Mo2C monolayer was then subjected to two major surface modifications: a Mo-vacancy and defective Mo2C with non-metal dopants as shown in Fig. 1. Mo2C with a vacant Mo-site is referred to as Mo2C–Movac and defective Mo2C obtained by replacing one Mo atom from the surface with metal-free dopants is designated as Mo2C–Xdef, where X = B, C, N, P and S. On these Mo-vacant and defective analogues of Mo2C, we next adsorbed B single atoms to create a metal-free catalyst couple for nitrogen activation and reduction. The binding energy of BSAC ranges from −5.35 eV on the Mo2C–Movac catalyst to −6.25 eV, −5.37 eV, −7.92 eV, −5.81 eV and −4.77 eV respectively, on the Mo2C–Bdef, Mo2C–Cdef, Mo2C–Ndef, Mo2C–Pdef and Mo2C–Sdef catalysts. The exothermic binding energies of the boron atom catalysts on all Mo2C analogues illustrates the stability of the boron SAC integrated on the defective Mo2C monolayers.
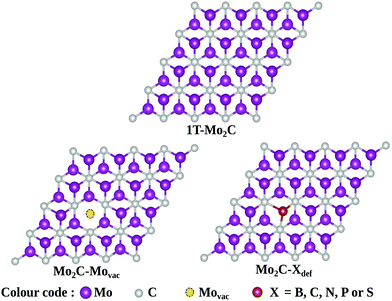 |
| | Fig. 1 The model 2D 1T-Mo2C monolayer with surface modifications considered in this study. | |
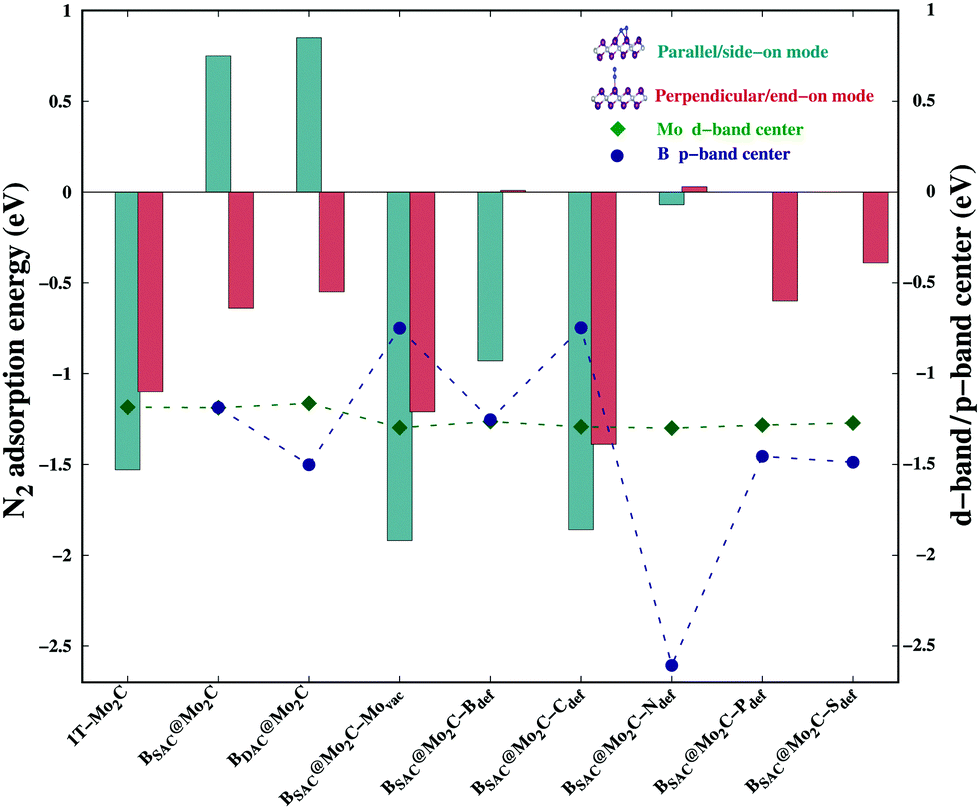
We next investigated the N2 adsorption efficacy and electronic properties of the Bcat@Mo2C catalysts through the d-band centre of Mo, the p-band centre of B and the projected density of states (PDOS) with reference to pristine 1T-Mo2C. Fig. 2 highlights the N2 adsorption energies of the Bcat@Mo2C catalysts with reference to 1T-Mo2C and the correlation between N2 adsorption with the d-band center of Mo and the p-band center of B. BSAC@Mo2C and BDAC@Mo2C exhibit a lower exothermic end-on mode of dinitrogen adsorption with Eads values of −0.64 eV and −0.55 eV, respectively with respect to −1.10 eV on 1T-Mo2C. It can also be noted that there are no significant changes in the Mo d-band center when BSAC and BDAC are anchored on Mo2C, however the B p-center on BSAC@Mo2C is more positive than that of BDAC@Mo2C. A more positive B p-band center infers that the B p-orbitals are closer to the Fermi level (EF = 0 eV), which in turn leads to the feasible adsorption of N2. Generating a Mo-vacancy on the Mo2C monolayer leads to a less positive Mo d-band center, but in contrast, the B p-band becomes more positive, which in turns leads to higher chemisorption (−1.92 eV) of N2 on BSAC@Mo2C–Movac. Moreover, on analysing the Bader charges on the Bcat centers, all the Bcat centers are found to be negatively charged, thereby implying that there is a charge transfer from the Mo2C monolayer to B. The only exception being the BSAC@Mo2C–Ndef and BSAC@Mo2C–Sdef systems with positively charged or nearly neutral Bcat centers, respectively. Interestingly, these two systems possess a more negative B p-band center as compared to the rest of the systems, and thereby show lower exothermic N2 adsorption. It is also important to note that a higher exothermic N2 adsorption is not only influenced by the B p-band center but also by a more pronounced overlap between the π orbitals of the B and N atoms. The parallel/side-on mode of N2 adsorption involves two B–N bonds, and results in two peaks in the PDOS plot (inset) with a greater electron density overlap (see Fig. 3(a)) compared to a single B–N bond for the perpendicular/end-on mode of N2 adsorption (Fig. 3(b)) on BSAC@Mo2C–Movac. The BSAC@Mo2C–Xdef catalysts follow a similar pattern of N2 adsorption. For instance, BSAC@Mo2C–Cdef with a more positive B p-band center exhibits better chemisorption of N2 when compared to BSAC@Mo2C–Ndef with a highly negative p-band center of B. It can be established that the adsorption and activation of N2 is influenced by the p-band center of Bcat on the B-anchored Mo2C catalysts. Interestingly, it was noticed that the N2 chemisorption efficacy of BSAC with a Mo-vacancy is on a par with that of BSAC with C-defective Mo2C, while the rest of the non-metals, i.e. B, P, S and N show moderate to minimal adsorption of N2. Apart from the B p-band center, the N2 adsorption trends could also be rationalized in terms of electronic properties such as charge difference density (CDD) and the PDOS plots provided in Fig. S3 of the ESI.† The PDOS plots also show the very interesting phenomenon of a synergistic effect between the C from the Mo2C and Bcat centers. The systems that show a higher exothermic N2 adsorption, in turn show a more pronounced overlap between the C p-orbitals and B p-orbitals, thus the synergistic effect of C and B enhances the N2 adsorption efficiency in the Bcat@Mo2C catalysts.
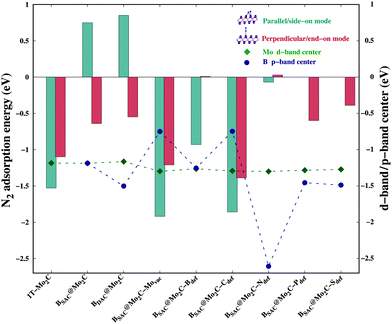 |
| | Fig. 2 N2 adsorption energies in different modes, and the Mo d-band center and B p-band center on different Bcat@Mo2C catalysts. | |
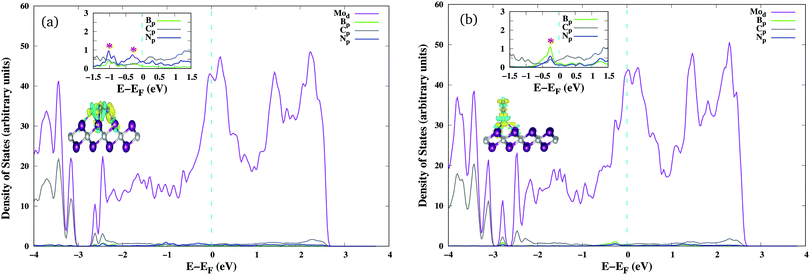 |
| | Fig. 3 PDOS of N2 adsorbed on BSAC@Mo2C–Movac with the insets showing hybridisation of the N p-orbitals and B p-orbitals and the CDD plot generated isosurface density set to 0.003 e Å−3. (a) PDOS and CDD plots of N2 adsorbed on BSAC@Mo2C–Movacvia the parallel/side-on mode. (b) PDOS and CDD plots of N2 adsorbed on BSAC@Mo2C–Movacvia the perpendicular/end-on mode. | |
3.2 N2 reduction on Bcat@Mo2C catalysts
Finally we explored the thermodynamics for the reduction of N2 to NH3 on the Bcat@Mo2C catalysts that showed strong exothermic N2 chemisorption. Also the NRR pathways on BSAC@Mo2C and BDAC@Mo2C were analysed to deduce whether the role of a second B atom enhances or inhibits the reduction process. Among the defective monolayers with surface modification, BSAC@Mo2C–Movac and BSAC@Mo2C–Cdef were considered as both showed highly exothermic chemisorption of dinitrogen in both the parallel as well as the perpendicular mode. Previous studies have revealed that the reduction of N2 can proceed via three pathways, viz. distal or alternating and enzymatic, respectively, for N2 adsorbed in the perpendicular and parallel modes. On the BSAC@Mo2C and BDAC@Mo2C catalysts with N2 adsorbed preferentially in an end-on mode, the NRR pathway was explored through the distal and alternating routes as shown in Fig. S4 and S5 of the ESI.† The last protonation step, i.e. *NH2 → *NH3 has been found to be the potential determining step (PDS) for nitrogen reduction on BSAC@Mo2C and BDAC@Mo2C for both the distal and alternating route with a ΔGmax of 1.10 eV and 0.90 eV, respectively. The corresponding overpotentials, η for reducing nitrogen to ammonia on the two catalysts are therefore, 0.94 eV and 0.74 eV, respectively. It would be interesting to note that BSAC@Mo2C, which is more exergonic towards N2 (−0.15 eV) as compared to BSAC@Mo2C (−0.05 eV) follows a more uphill first protonation step (0.21 eV) and a complementary more uphill PDS. For the BSAC@Mo2C–Movac monolayer with one Mo-atom vacancy that shows exothermic N2 adsorption in the side-on as well as end-on modes, the NRR mechanisms for all three possible routes were computed and are are presented in Fig. 4. Although, the parallel mode of N2 adsorption is more exergonic than the the perpendicular mode, and we expect the enzymatic route to be favoured over the distal or alternating route, the limiting potential for the NRR on the BSAC@Mo2C–Movac catalyst was found to be 0.57 eV (*NH2 → *NH3) for the distal, 1.05 eV (*N–NH → *NH–NH) for the alternating and 1.83 eV (*NH2–*NH2 → *NH2) for the enzymatic route. The distal route, which involves the protonation of only one nitrogen atom to form the first ammonia molecule, can be seen to show downhill reaction steps until the fourth protonation step. The preference for the distal mode could be accounted for by less steric hindrance caused by subsequent protonation on the nitrogen atoms. It is interesting to note that the BSAC@Mo2C–Movac catalyst shows a high affinity for capturing the *NH2 moiety as it can be found from the enzymatic route. The *NH2–*NH2 intermediate shows a highly exergonic adsorption with dissociation of the N–N bond and the adsorption of one *NH2 on the Mo atom of the monolayer. Dissociating the N–N bond would be the ultimate goal of the NRR, however, if the catalyst holds onto the ammonia molecule strongly, the effectiveness and applicability of the catalyst is restricted. As a result, the enzymatic route would be a highly unlikely and unfavoured route for reducing N2 molecules. A similar case of the high exergonic adsorption of the *NH2–*NH2 intermediate can be seen for the BSAC@Mo2C–Cdef catalyst (Fig. S6, ESI†), wherein the limiting potentials for nitrogen reduction were found to be 1.42 eV for the distal and alternating routes (*NH2 → *NH3) and 2.26 eV for the enzymatic route (*NH2–*NH2 → *NH2). The limiting potentials and the corresponding potential determining steps on the above mentioned Bcat@Mo2C catalysts are summarised in Table 1.
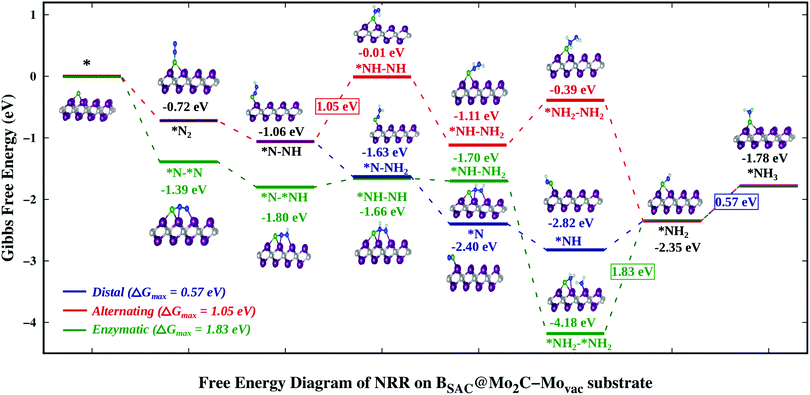 |
| | Fig. 4 Free energy profile of the nitrogen reduction reaction via the distal, alternating and enzymatic routes on the BSAC@Mo2C–Movac catalyst. | |
Table 1 Gibbs free energies, ΔG (eV) of N2 adsorption, first protonation and rate determining step (ΔGmax) and overpotential, η for all routes of the NRR on the Bcat@Mo2C catalysts
| Bcat@Mo2C catalysts |
NRR route |
ΔG*N2 (eV) |
ΔG*N2 → ΔG*N2H (eV) |
Rate determining step |
ΔGmax (eV) |
η (eV) |
| BSAC@Mo2C |
Distal |
−0.15 |
0.21 |
*NH2 → *NH3 |
1.10 |
0.94 |
|
|
Alternating |
−0.15 |
0.21 |
*NH2 → *NH3 |
1.10 |
0.94 |
| BDAC@Mo2C |
Distal |
−0.05 |
0.02 |
*NH2 → *NH3 |
0.90 |
0.74 |
|
|
Alternating |
−0.05 |
0.02 |
*NH2 → *NH3 |
0.90 |
0.74 |
| BSAC@Mo2C–Movac |
Distal |
−0.72 |
−0.34 |
*NH2 → *NH3 |
0.57 |
0.41 |
|
|
Alternating |
−0.72 |
−0.34 |
*N–NH → *NH–NH |
1.05 |
0.89 |
|
|
Enzymatic |
−1.38 |
−0.42 |
*NH2–*NH2 → *NH2 |
1.83 |
1.67 |
| BSAC@Mo2C–Cdef |
Distal |
−0.88 |
0.30 |
*NH2 → *NH3 |
1.42 |
1.26 |
|
|
Alternating |
−0.88 |
0.30 |
*NH2 → *NH3 |
1.42 |
1.26 |
|
|
Enzymatic |
−1.23 |
−0.07 |
*NH2–*NH2 → *NH2 |
2.26 |
2.10 |
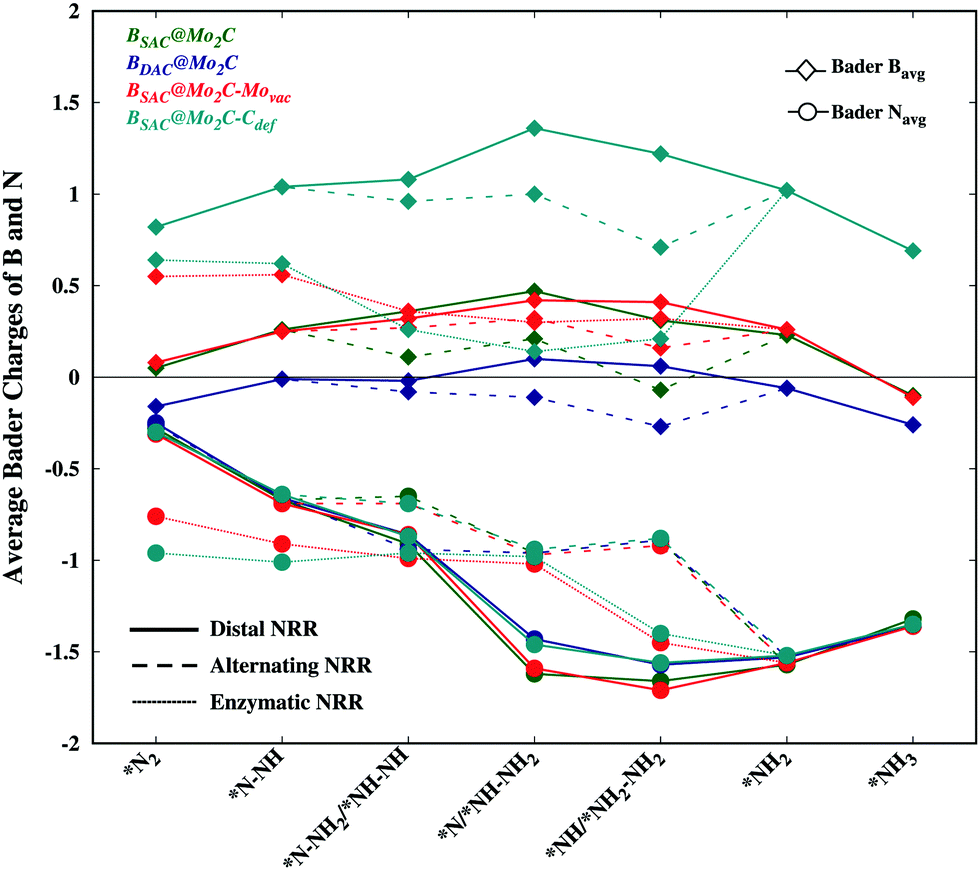
While an exergonic adsorption of N2 is essential for the feasible capture of N2, it is not the only deciding factor for successful nitrogen reduction. There have been no accounts correlating dinitrogen adsorption to the efficacy of nitrogen reduction and the corresponding limiting potential. The exergonic free energy of N2 follows the order BSAC@Mo2C–Movac > BSAC@Mo2C–Cdef > BSAC@Mo2C > BDAC@Mo2C, however the limiting potential of nitrogen reduction follows the order BSAC@Mo2C–Cdef > BSAC@Mo2C > BDAC@Mo2C > BSAC@Mo2C–Movac. In order to correlate the N2 adsorption and the limiting potential, we probe into the electronic properties of the adsorbed N2 and NxHy species via the Bader charges on the B atom and N atoms for all the Bcat@Mo2C catalysts as shown in Fig. 5. The N-atoms are seen to be negatively charged on all the NxHy intermediates and tend to be more electron rich for subsequent reaction steps thereby making the protonation facile. While the differences in Bader charge on the N-atoms are minute for different catalysts, the charges on the B-atoms are prominent with less positive B-atoms on the BDAC@Mo2C, BSAC@Mo2C–Movac and BSAC@Mo2C catalysts, followed by the BSAC@Mo2C–Cdef catalyst. A highly positive B-atom on the corresponding NxHy intermediates would deplete the negative charge on the N-atom, thereby restricting the protonation steps as seen in the case of BSAC@Mo2C–Cdef. Therefore, the BSAC@Mo2C–Movac catalyst with strong N2 chemisorption captures the dinitrogen molecule effectively and the electron deficient B-atom aids the protonation steps in the NRR, thus making it an attractive electrocatalyst with a limiting potential of 0.57 eV and an overpotential of 0.41 eV. In contrast, the relatively high limiting potential of 1.10 eV for BSAC@Mo2C in spite of the similar charges on the B-atoms can be accounted for by a less exergonic N2 adsorption as compared to BSAC@Mo2C–Movac. Additionally, BDAC@Mo2C with electron rich B-atoms shows a relatively higher limiting potential of 0.90 eV owing to its less exergonic N2 adsorption. We further correlate our analogy to the first protonation step of the NRR, which is usually considered a crucial step in the NRR. It has been reported that a less endergonic or exergonic *N2 → *N2H step leads to a lower limiting potential for nitrogen reduction. Upon comparison of the free energies of the first protonation steps, the Bcat@Mo2C catalysts with less endergonic free energies, i.e., BSAC@Mo2C–Movac (−0.34 eV) and BDAC@Mo2C (0.02 eV) exhibit lower limiting potentials as compared to BSAC@Mo2C and BSAC@Mo2C–Cdef. This analogy holds true only for the distal and alternating routes wherein N2 has been adsorbed in the end-on mode. Our previous argument correlating N2 adsorption free energies and Bader charges is in agreement with the analogy of first protonation energies, and therefore can give insightful information on the mutual correlation between the free energies of the adsorption and electronic properties that directly govern the limiting potentials of the NRR on Bcat@Mo2C catalysts. Furthermore, the BSAC@Mo2C–Movac catalyst with one Mo-vacancy is found to efficiently capture and accentuate the catalytic activity of the boron SAC with a very low overpotential of 0.41 eV for the NRR. This study provides an in-depth analysis of the electronic factors crucial for efficient N2 adsorption and reduction, and proposes the BSAC@Mo2C–Movac catalyst as a potential candidate for the NRR.
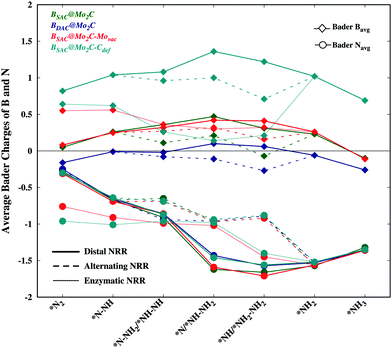 |
| | Fig. 5 Bader charges on N and B atom catalysts for NxHy intermediates involved in the free energy diagram for the NRR on all the Bcat@Mo2C catalysts. | |
4 Conclusions
In summary, a detailed and systematic DFT investigation has been carried out to analyse the efficacy of N2 adsorption and reduction on Bcat@Mo2C monolayers. Our study identifies a metal-free boron anchored defective Mo2C monolayer with superior electrocatalytic activity for the NRR at 0.41 eV on account of a more positive p-band center and the negative charge of boron SACs that implicitly stabilizes the intermediates along the energy profile of the NRR. The insights gained from this work can be implemented for further research towards the design of efficient B-atom based electrocatalysts for the nitrogen reduction reaction.
Author contributions
Ashakiran Maibam: calculation, methodology, investigation and analysis. Sailaja Krishnamurty and Manzoor Ahmad Dar: conceptualization and work design. All authors contributed equally to writing, reviewing and editing the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
A. M. acknowledges the CSIR for funding of the SRF (Senior Research Fellowship). M. A. D. acknowledges the Start-up research grant (SRG/2020/00654) from the SERB, India for financial support towards the completion of this work. The authors gratefully acknowledge the National Supercomputing Mission (NSM) for providing the computing resources of ‘PARAM Brahma’ at IISER Pune, which is implemented by C-DAC and supported by the Ministry of Electronics and Information Technology (MeitY) and the Department of Science and Technology (DST), Government of India.
Notes and references
- N. Cherkasov, O. Ibhadon and P. Fitzpatrick, Chem. Eng. Process., 2015, 90, 24–33 CrossRef CAS.
- C. J. M. Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- A. Afif, N. Radenahmad, Q. Cheok, S. Shams, J. H. Kim and A. K. Azad, Renewable Sustainable Energy Rev., 2016, 60, 822–835 CrossRef CAS.
- G. Zheng, J. M. Yan and G. Yu, Small Methods, 2019, 3, 1900070 CrossRef.
- X. Xue, R. Chen, C. Yan, P. Zhao, Y. Hu, W. Zhang, S. Yang and Z. Jin, Nano Res., 2019, 12, 1229–1249 CrossRef CAS.
- C. Tang and S.-Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 120, 5437–5516 CrossRef CAS PubMed.
- H. Tao, C. Choi, L.-X. Ding, Z. Jiang, Z. Han, M. Jia, Q. Fan, Y. Gao, H. Wang, A. W. Robertson, S. Hong, Y. Jung, S. Liu and Z. Sun, Chem, 2019, 5, 204–214 CAS.
- Y. Yao, H. Wang, X.-Z. Yuan, H. Li and M. Shao, ACS Energy Lett., 2019, 4, 1336–1341 CrossRef CAS.
- Q. Qin, T. Heil, M. Antonietti and M. Oschatz, Small Methods, 2018, 2, 1800202 CrossRef.
- K. Zhang, R. Guo, F. Pang, J. He and W. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 10214–10220 CrossRef CAS.
- H. Zhao, D. Zhang, H. Li, W. Qi, X. Wu, Y. Han, W. Cai, Z. Wang, J. Lai and L. Wang, Adv. Energy Mater., 2020, 10, 2002131 CrossRef CAS.
- G. Deng, T. Wang, A. A. Alshehri, K. A. Alzahrani, Y. Wang, H. Ye, Y. Luo and X. Sun, J. Mater. Chem. A, 2019, 7, 21674–21677 RSC.
- S. L. Foster, S. I. Perez Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 490–500 CrossRef.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS.
- X. Guo, X. Wan and J. Shui, Cell Rep. Phys. Sci., 2021, 2, 100447 CrossRef CAS.
- K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed.
- D. K. Yesudoss, G. Lee and S. Shanmugam, Appl. Catal., B, 2021, 287, 119952 CrossRef CAS.
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lü, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS PubMed.
- Q. Li, S. Qiu, C. Liu, M. Liu, L. He, X. Zhang and C. Sun, J. Phys. Chem. C, 2019, 123, 2347–2352 CrossRef CAS.
- A. Maibam, T. Govindaraja, K. Selvaraj and S. Krishnamurty, J. Phys. Chem. C, 2019, 123, 27492–27500 CrossRef CAS.
- L. R. Johnson, S. Sridhar, L. Zhang, K. D. Fredrickson, A. S. Raman, J. Jang, C. Leach, A. Padmanabhan, C. C. Price, N. C. Frey, A. Raizada, V. Rajaraman, S. A. Saiprasad, X. Tang and A. Vojvodic, ACS Catal., 2020, 10, 253–264 CrossRef CAS.
- B. H. R. Suryanto, D. Wang, L. M. Azofra, M. Harb, L. Cavallo, R. Jalili, D. R. G. Mitchell, M. Chatti and D. R. MacFarlane, ACS Energy Lett., 2019, 4, 430–435 CrossRef CAS.
- B. Zhang, J. Zhou, S. R. Elliott and Z. Sun, J. Mater. Chem. A, 2020, 8, 23947–23954 RSC.
- Q. Li, L. He, C. Sun and X. Zhang, J. Phys. Chem. C, 2017, 121, 27563–27568 CrossRef CAS.
- P. Ou, X. Zhou, F. Meng, C. Chen, Y. Chen and J. Song, Nanoscale, 2019, 11, 13600–13611 RSC.
- T. Yang, T. T. Song, J. Zhou, S. Wang, D. Chi, L. Shen, M. Yang and Y. P. Feng, Nano Energy, 2020, 68, 104304 CrossRef CAS.
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Mater., 2018, 30, 1800191 CrossRef PubMed.
- X. Zhai, L. Li, X. Liu, Y. Li, J. Yang, D. Yang, J. Zhang, H. Yan and G. Ge, Nanoscale, 2020, 12, 10035–10043 RSC.
- C. Ma, X. Yan, H. He, B. Liu and S. Yan, Sustainable Energy Fuels, 2021, 5, 2415–2418 RSC.
- K. Ba, G. Wang, T. Ye, X. Wang, Y. Sun, H. Liu, A. Hu, Z. Li and Z. Sun, ACS Catal., 2020, 10, 7864–7870 CrossRef CAS.
- K. P. Ramaiyan, S. Ozden, S. Maurya, D. Kelly, S. K. Babu, A. Benavidez, F. G. Garzon, Y. S. Kim, C. R. Kreller and R. Mukundan, J. Electrochem. Soc., 2020, 167, 044506 CrossRef CAS.
- B. Zhang, J. Zhou, S. R. Elliott and Z. Sun, J. Mater. Chem. A, 2020, 8, 23947–23954 RSC.
- S. Qi, Y. Fan, L. Zhao, W. Li and M. Zhao, Appl. Surf. Sci., 2021, 536, 147742 CrossRef CAS.
- X. Yang, C. Shang, S. Zhou and J. Zhao, Nanoscale Horiz., 2020, 5, 1106–1115 RSC.
- Y. Liu, L. Gu, X. Zhu, Q. Zhang, T. Tang, Y. Zhang, Y. Li, J. Bao, Z. Dai and J.-S. Hu, J. Mater. Chem. A, 2020, 8, 8920–8926 RSC.
- X. Ren, J. Zhao, Q. Wei, Y. Ma, H. Guo, Q. Liu, Y. Wang, G. Cui, A. M. Asiri, B. Li, B. Tang and X. Sun, ACS Cent. Sci., 2019, 5, 116–121 CrossRef CAS PubMed.
- Y. Ma, T. Yang, H. Zou, W. Zang, Z. Kou, L. Mao, Y. Feng, L. Shen, S. J. Pennycook, L. Duan, X. Li and J. Wang, Adv. Mater., 2020, 32, 2002177 CrossRef CAS PubMed.
- A. Maibam and S. Krishnamurty, J. Colloid Interface Sci., 2021, 600, 480–491 CrossRef CAS PubMed.
- X. Liu, X. Jiao, Y. Zheng and S.-Z. Qiao, ACS Catal., 2020, 10, 1847–1854 CrossRef CAS.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS.
- S. Xiao, F. Luo, H. Hua and Z. Yang, Chem. Commun., 2020, 56, 446–449 RSC.
- L. Shi, Q. Li, C. Ling, Y. Zhang, Y. Ouyang, X. Baia and J. Wang, J. Mater. Chem. A, 2019, 7, 4865–4871 RSC.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- S. Grimmea, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00953b |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Sailaja
Krishnamurty
ab,
Sailaja
Krishnamurty