
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReview on green carbon dot-based materials for the photocatalytic degradation of dyes: fundamentals and future perspective
Nivetha
Basavaraj
 ,
Anithadevi
Sekar
* and
Rakhi
Yadav
,
Anithadevi
Sekar
* and
Rakhi
Yadav
Department of Chemistry, Madras Christian College, Affiliated to the University of Madras, East Tambaram, Chennai, Tamil Nadu, India. E-mail: rakhiyadav@mcc.edu.in
First published on 4th November 2021
Abstract
Water pollution has become more prevalent as the discharge of dyes from industries into water bodies has increased in modern life. Photocatalytic approaches are among the most effective dye degradation methods to address these issues. In this regard, carbon nanodots (CDs), well-known as quasi-spherical zero-dimensional nanomaterials, have replaced semiconductor quantum dots in recent years. CDs derived from green sources have appealed to the interest of scientists due to their tuneable fluorescence behavior, biocompatibility, large-scale production, excellent photostability, and eco-friendliness compared to CDs derived from chemical precursors. Moreover, a colossal of works reported CDs as an outstanding photocatalyst candidate for efficient dye degradation. Therefore, the study first focuses on the green synthetic routes employed for CDs and metal/non-metal infused CDs synthesis in this review article. Then, the structural, optical, and morphological properties of CDs and metal/non-metal infused CDs synthesis are discussed. The next objective discusses the fundamental motive of the review article, i.e., the potential roles of CDs and metal/non-metal infused CDs in light-harvesting, charge transfer dynamics, and surface catalytic reaction for dye degradation. It also emphasizes the various factors influencing photodegradation, such as pH, dopant, dye concentration, photocatalyst loading, light intensity, irradiation time, and temperature. Furthermore, the role of several radical scavengers on the photocatalyst in responsibly identifying active species and improving the photodegradation mechanism of dyes are briefly discussed. Besides, the reusability, stability, and cytotoxicity aspects are emphasized. To conclude, the drawbacks and the probable solutions to overcome the research gap in future routes of CDs as photocatalysts are discussed.
 Nivetha Basavaraj | Nivetha Basavaraj pursued her Master's degree in chemistry at Madras Christian College, Tamil Nadu, India (2021). She obtained her BSc degree from Kongunadu Arts and Science College, Coimbatore, Tamil Nadu, India (2019). Her area of interest in research focuses on carbon nanodots in fluorescence sensing and photocatalysis applications. |
 Anithadevi Sekar | Anithadevi Sekar is currently a PhD scholar at the Madras Christian College, Tamil Nadu, India. She received her Master's degree from the same college in the year 2014. She obtained her BSc degree from the Anna Adarsh College for Women, Tamil Nadu, India (2012). Her research interests include the green synthesis of fluorescent carbon nanomaterials and their applications in sensing, photocatalysis, and cell imaging. |
 Rakhi Yadav | Rakhi Yadav is an Assistant professor at the Madras Christian College, Tamil Nadu, India. She has been serving this institute for over a decade, integrating teaching and research to meet innovations in science. Her interest lies in strengthening undergraduate and postgraduate students’ research and writing skills to make the teaching –learning process more visible. She received her PhD degree in Chemistry in the year 2005. She has worked on tribal medicinal plants to explore natural therapeutic agents. At present, she is engaged in the development of bio-mediated nanomaterials for catalysis, biosensing, bioimaging, and cancer theranostics. |
1. Introduction
Water is one of the most powerful resources, yet inappropriate water treatment exists globally, leading to scarcity. A few examples of ill-treatment of water are the mixing of industrial waste that contains toxic chemicals into the water bodies and improper disposal of school/colleges and hospital's solid or chemical waste that ultimately contaminate water.1–3 Using such water in agriculture kills or poisons and inhibits the growth of individual plants/trees. Significantly, the irregular outlet of industrial waste containing organic dyes into the water bodies is the primary reason for severe health issues of living beings.4 Consuming water that contains unsafe contaminants leads to several health issues, such as affecting the nervous and reproductive systems, gastrointestinal problems, sometimes also causing chronic diseases including cancer, while the physical contact leads to skin problems such as allergies, hair falls, and psoriasis. Water contains several impurities such as toxic chemicals, biological and radiological contaminants, and total suspended solids (TSS). One such anthropogenic pollution arises due to the high level of dyes in water, which leads to a rise in biological and chemical oxygen demand (BOD, COD), damages photosynthesis, weakens the growth of plants, affects animals, and aqua systems, supplies recalcitrance, and bioaccumulation.5 Dyes are classified into acid, base, disperse, azo, anthraquinone, cationic, and anionic, depending upon their chemical structures. The presence of dyes such as Congo Red (CR), Rhodamine-B (RhB), Methylene Blue (MB), Methyl Orange (MO), Crystal Violet (CV), Eriochrome Black-T (EBT), Malachite Green (MG), Eosin Y (EY), azo dyes, and other textile dyes increases toxicity of the water and cause severe health issues such as eye irritation, nausea, vomiting, and diarrhea.5Several physical, chemical, and biological techniques, namely sedimentation, aeration, chlorination, coagulation, aerobic, lagoons, and many more, are employed to process contaminated water and maintain the water cycle balance. The most commonly known and trending wastewater treatment that has ruled out other methods is photocatalysis. This method is preferred over others due to low cost, reusability, complete degradation, eco-friendliness, and no formation of secondary pollutants. In this photoreaction, there occurs oxidation and reduction from photogenerated holes and electrons, respectively.6 Researchers spotted that the heterogeneous photocatalysis process is advantageous over methods like coagulation and flocculation due to the complete elimination of dye molecules rather than transforming from one form to another. This process makes use of the photon energy to convert itself to chemical energy. It effectively degrades a wide range of organic dyes and mineralizes them into CO2 and H2O.7 The photocatalytic reaction depends purely on the structure of the electronic band and band energy. It has unique disinfectant and decontamination activity on water due to its good photochemical stability with solar irradiation for water treatment. It is known that the photocatalytic reaction rate is increased with the aid of a catalyst by decreasing the activation energy (Ea). An excellent photocatalyst should possess a high surface area, good crystallinity, cost efficiency, free from toxicity, ease of handling, and especially prevent the formation of toxic or hazardous by-products.8 These photocatalytic dye degradations are often driven by solar irradiation, UV-Visible light, visible irradiation, and NIR light.
In the world of nanomaterials, carbon-based nanoparticles exhibit a wide range of applications such as bio-imaging, biosensors, cancer therapy, tissue engineering, drug and gene delivery, energy storage, sorbents, antimicrobial agents, pollution preventers, depth filters, high flux membranes, hydrogen evolution, and light-emitting diodes.9–14 Carbon exists in various forms such as diamond, graphite, carbon fibres, carbon black, and activated carbon. Nano carbons exist in several forms, such as Buckminsterfullerene, carbon nanotubes (single/multi-walled CNTs), carbon nanofibers (CNF), carbon quantum dots (CQDs), carbon nanodots (CDs), etc.15 Herein we seek ultra-size quasi-spherical CDs due to their unique properties such as nontoxicity, excellent photostability, cost-efficiency, ease of synthesis, good solvent dispersion, tuneable fluorescent excitation, and emission properties.16–21 It shows better results than other fluorescent organic dyes and quantum dots.22–26 In addition, CDs exhibit high photobleaching resistance, chemical stability, stable colloidal solution, and inertness.27–30 Researchers have studied the CDs synthesis by both top-down and bottom-up approaches with the help of both chemical and green precursors.31
The photocatalytic dye degradation is often performed using metals or complexes, non-metals, semiconducting materials, metal oxides such as ZnS, WO2, ZnO, SrO2, CdS, and TiO2 doped with CDs as catalysts.32,33 To improve the PL properties and thus enhance the catalytic, sensing, and biological applications, heteroatoms such as P, S, N, and B are doped with the prepared CDs during synthesis.34 Comparing all the atoms, N/CDs have unique FL probes and are used frequently. Single and few-layered nanocomposites improvise photocatalytic dye degradation due to high charge carrier mobility and increased surface area.35 Carbon-based nanomaterials have also been kindling the interest of researchers in photocatalytic dye degradation.
Although there exists a hand full of research on the synthesis of CDs from chemical precursors,36,37 CDs from green precursors are found to be advantageous since it is environmentally friendly, low cost, facile and rapid synthesis, short synthesis duration, and non-toxicity. Several leaves, plants, tree bark, vegetables, fruits, juice, and biodegradable wastes are used as precursors to synthesise CDs by eco-friendly methods. Synthetic routes such as hydrothermal, solvothermal, microwave, microwave-assisted hydrothermal, and pyrolysis are followed.38–42 CDs from green precursors have also been probed to detect dyes such as RhB, MB, MO, CR, CV, and many other azo dyes in aqueous solutions.43–47 The objectives of the current review are compared with specific review articles reported on CDs derived from chemical and green precursors for photocatalytic dye degradation in Table 1.
| Title | Synthesis | Properties | Factors influencing photocatalytic activity | Light-harvesting | Charge transfer dynamics | Surface catalytic reaction | Role of radical scavenging activity | Mechanism | Reusability/Stability | Toxicity | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Application of carbon quantum dots to increase the activity of conventional photocatalysts: a systematic review | ✓ | ✓ | ✓ | 48 | |||||||
| Recent progress of carbon dot precursors and photocatalysis applications | ✓ | ✓ | ✓ | 49 | |||||||
| Multifaceted applications of green carbon dots synthesized from renewable sources | ✓ | 50 | |||||||||
| Band gap tuning and surface modification of carbon dots for sustainable environmental remediation and photocatalytic hydrogen production – a review | ✓ | ✓ | 51 | ||||||||
| Recent progress on the photocatalysis of carbon dots: classification, mechanism and applications | ✓ | ✓ | ✓ | 52 | |||||||
| A minireview on doped carbon dots for photocatalytic and electrocatalytic applications | ✓ | ✓ | ✓ | 53 | |||||||
| Recent developments in synthesis and photocatalytic applications of carbon dots | ✓ | ✓ | ✓ | 54 | |||||||
| Green carbon dots based materials for the catalytic degradation of dyes: ground rules and operating parameters | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | This work |
Herein, we discuss the synthesis of CDs from green precursors doped with metals and their complexes, non-metals, metal oxides, and semiconductors. It is used in photocatalytic dye degradation with three major principles: light-harvesting, charge transfer dynamics, and surface catalytic reactions. The optimum condition with which a photocatalytic activity takes place opens the gate to better results. Further, the employment of radical scavengers to detect the active species responsible for the efficient degradation of dyes enables us to understand the mechanism feasibly. The stability, reusability, and toxicity of the as-synthesized CDs are labelled for further practical applications. We tend to overcome all other review works by highlighting the utilization of green precursors, thus addressing the problems and research gaps.
2. Synthesis
CDs/dopants composites are either synthesized via a simple one-pot or two-step synthetic process or even multi-step methods.44,55,56 Dopants help in increasing the photoluminescent properties of CDs. It helps in synthesizing CDs with desired size and high quantum yield. Heteroatoms such as N, P, S, and B are doped during the synthesis of CDs to improve photoluminescent properties and attain better photocatalytic activity.34,57 The enhanced properties of N/CDs lead to the application of fluorescent probes and catalysts.In the first case, bare CDs are achieved by any of the bottom-up approaches, namely, hydrothermal, carbonization, microwave-assisted, ultrasonication, and pyrolysis.45,58,59 Among these, hydrothermal methods are considered facile processes due to their soft chemical route resulting in a uniform particle size that serves as an excellent candidate for photocatalytic activity. For instance, Xue et al.59 synthesized fluorescent CDs with lychee as the precursor with a quantum yield of 10.6% and particle size ∼1.12 nm. It follows pyrolysis, where the starting material was heated at a rate of 10 °C min−1 in a ceramic crucible at 300 °C for 2 h. The product was then powdered into fine particles. A black solution was obtained by adding 0.1 g of this powder in 10 ml water followed by ultrasound dispersion. Finally, it was filtered and maintained at 4 °C for future use.
Similarly, the one-pot approach includes mixing together CDs, photocatalyst, and composite and processing. In that way, Prasannan et al.60 synthesized CDs from orange peels by hydrothermal carbonization at 180 °C for 12 h, whose quantum yield was 12.3% and particle size ∼4.5 nm. Later, the prepared metal oxide, ZnO, was loaded on the as-prepared CDs by the solution dispersion method. 2-Propanol and water were used to disperse 0.9 g of ZnO and 0.1 g of CDs and treated at 100 °C for 3 h. Likewise, metal oxides such as TiO261 and SiO262 are doped on the prepared CDs. Heterogeneous metals such as N, S, P, and non-metals like Cl are also doped with the prepared CDs.63–65 Atchudan et al.66 reported the one-step synthesis of TiO2 NP@N/CDs where TiO2 nanoparticles were synthesized using the sol–gel method with titanium(IV)isopropoxide as a starting material. N/CDs were prepared from peach fruit as the green precursor and NH4OH as the source of N dopant. Embedding heteroatoms on CDs also fall under a single-step process such as N/CDs, where ammonia acts as a source of nitrogen dopant.66 Likewise, ammonium bicarbonate63 and urea67 are used as a source of nitrogen.
Anand et al.55 reported the two-step synthesis of red-emitting Mg–N/CDs extracted from the leaves of the Bougainvillea plant. This involved (i) the green precursor extract from the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of ethanol and water and (ii) microwave charring. In the same way, a two-step synthesis of highly fluorescent CNFs/CQDs prepared from Pine fruit by ball milling followed by a hydrothermal process was reported.68
:1 solution of ethanol and water and (ii) microwave charring. In the same way, a two-step synthesis of highly fluorescent CNFs/CQDs prepared from Pine fruit by ball milling followed by a hydrothermal process was reported.68
The multistep process either involves treating CDs and dopants prepared prior and incubated or processing the solution of a photocatalyst precursor containing CDs prepared earlier. For example, Dadigala et al.56 reported synthesizing AgNPs/CDs/CNNs composites where BCN was prepared from melamine by thermal polymerization at 600 °C for 2 h followed by processing CNN by liquid exfoliation. CDs were prepared from onion waste as a precursor by an autoclave. The as-prepared CNNs were impregnated by hydrothermal method at 100 °C for 2 h for various amounts of CDs. Later, AgNPs were loaded on multiple concentrations of CCNNs by photochemical reduction at 60 °C overnight with AgNO3 as the source for Ag.
The synthesis of CDs from green precursors has been widely done through a bottom-up approach due to the formation of a small particle size that possesses a large surface area. The most commonly followed methods include hydrothermal, microwave, pyrolysis, and carbonization. By tuning the reaction conditions, we can achieve CDs with small and uniform particle sizes. Moreover, heteroatoms and metal oxide-supported CDs open the way for efficient photocatalytic activity.
3. Characterization
3.1. Surface morphology
CDs are known for their well dispersed, nano-dimension, and quasi-spherical material, and these morphologies are verified by SEM Fig. 1(a). N-CQDs derived from EFB fibre at 50× magnification exhibit a smooth, uniform, and continuous nature that arises due to the purity level with unidentified compounds. They possess high surface energy that can lead to particle aggregation while observing SEM.67 At the same time, the SEM Fig. 1(b) of CQDs 500× magnification exhibits spherical shape and avoidable agglomeration of the particles due to the good particle size and large crystalline surfaces.67 Typically, CDs from the extracts of Calocasia esculenta leaf, Mesua ferrea Linn leaf, tea leaf, and flower bud of Syzygium aromaticum were reduced, which displayed a spherical shape recorded in HRTEM Fig. 1(c).44 | ||
| Fig. 1 SEM images of (a) N/CQDs at 50× magnification (b) CQDs at 500× magnification; reproduced with permission from ref. 67 Copyright 2021 Elsevier; (c) HRTEM of reduced CDs; (d) particle distribution of reduced CDs (inset: SAED image); FFT of reduced CDs (e) before & (f) after masking; (g) IFFT of reduced CDs exhibiting lattice fringes; reproduced with permission from ref. 44 Copyright 2017 American Chemical Society. | ||
Further, the Fast Fourier Transform (FFT) image Fig. 1(e) and (f) image and its inverse (IFFT) Fig. 1(g) of Reduced CDs (R-CDs) phase exhibited lattice fringes with 0.20 nm of interplanar spacing, and the average diameter was determined to be ∼3 nm. While calculating the interplanar spacing with Bragg's equation, the lattice spacing was found to be 0.15 nm and is attributed to the dilute solution of R-CDs samples used during TEM analysis and powdered form of R-CDs for XRD analysis.44 The poor crystalline nature of the prepared R-CDs was investigated with selected area electron diffraction (SAED) Fig. 1(d inset) by blazing spots and diffuse rings in the designated area. The corresponding technique for HR-TEM is the energy dispersive X-ray spectrum (EDX), which delivers details of the chemical composition existing in the synthesized R-CDs Fig. 2(a).44 The results exhibit the atomic percentage corresponding to carbon and oxygen in which there is an increase in the ratio of carbon-to-oxygen (C/O) of RCD to 11.2 while the proportion of carbon-to-oxygen of bare CDs was 1.59. Zhu's group reported CDs derived from palm powder doped with sulphur and chlorine.65 The average diameter of S, Cl/CDs was ∼3.54 nm. It also displayed a well-resolved lattice fringe with 0.21 nm interplanar spacing, indicating a graphene plane near 100 facets. The homogeneous size distribution of S, Cl/CDs ranged between 2.5 nm and 5.5 nm Fig. 2(b).65 Moreover, the average diameter of Bio-CDs was calculated to be 3.54 ± 0.82 nm.
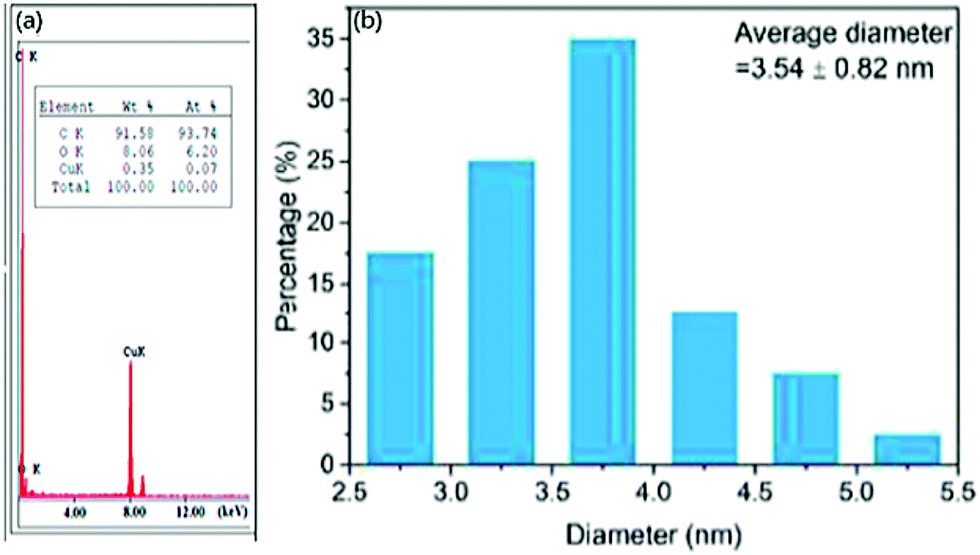 | ||
| Fig. 2 (a) EDX spectrum of reduced CDs; reproduced with permission from ref. 44 Copyright 2017 American Chemical Society. (b) Particle size distribution of CDs derived from Palm powder; reproduced with permission from ref. 63 Copyright 2020 Elsevier. | ||
3.2. Structural properties
Fig. 3(a) displays the FT-IR spectrum of red-emitting Mg–N/CDs derived from the Bougainvillea plant as a carbon source.55 To determine the surface functional group, FT-IR is carried out in which O–H,![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H, and –C–H stretching vibrations were observed at 3401–3200 cm−1, 3035 cm−1, 2923 cm−1, and 2859 cm−1, respectively. Stretching vibrations of CO and CC were observed as merged peaks at 1728 cm−1 and 1624 cm−1, respectively.55 Unsymmetrical stretching vibrations showed merged peaks at 1400 cm−1 corresponding to –CH2 and C–N attributed to the presence of nitrogen embedded on CDs. The stretching and bending vibration merged peaks of –C–O– and –C–H was observed at 1114 cm−1 and 1067 cm−1. Bending vibration peaks of substituted alkenes were observed at 824 cm−1 and 634 cm−1, respectively.55 The peak at ∼516 cm−1 confirmed the Mg atom due to the bending vibrations of the metal–ligand bond.70 Atchudan et al.66 reported the FT-IR spectrum of TiO2, N/CDs and observed vibrations at 1438 cm−1, 1368 cm−1, 1026 cm−1, and 580 cm−1, denoting –CN–, phenolic C–OH, epoxy C–O–C/C–N, and TiO/TiO–Ti frameworks apart from the ones discussed above. Heng et al.62 reported the FT-IR spectrum of CDs/SiO2 where peaks at 460 cm−1 and 900 cm−1 correspond to Si–O–Si, 1100 cm−1 and 3500 cm−1 corresponded to Si–OH bending and stretching vibrations, respectively, and at 1140 cm−1 for Si–O–C vibration. Also, the appearance of thionyl chloride in the CDs displayed peaks at 2488 cm−1, 1068 cm−1, and 1174 cm−1, indicating S–H, CS, and C–S vibrations and 1010 cm−1, 1105 cm−1, and 500–900 cm−1 indicating C–S–C, C–Cl in aromatic hydrocarbons and aliphatic hydrocarbons, respectively.65
C–H, and –C–H stretching vibrations were observed at 3401–3200 cm−1, 3035 cm−1, 2923 cm−1, and 2859 cm−1, respectively. Stretching vibrations of CO and CC were observed as merged peaks at 1728 cm−1 and 1624 cm−1, respectively.55 Unsymmetrical stretching vibrations showed merged peaks at 1400 cm−1 corresponding to –CH2 and C–N attributed to the presence of nitrogen embedded on CDs. The stretching and bending vibration merged peaks of –C–O– and –C–H was observed at 1114 cm−1 and 1067 cm−1. Bending vibration peaks of substituted alkenes were observed at 824 cm−1 and 634 cm−1, respectively.55 The peak at ∼516 cm−1 confirmed the Mg atom due to the bending vibrations of the metal–ligand bond.70 Atchudan et al.66 reported the FT-IR spectrum of TiO2, N/CDs and observed vibrations at 1438 cm−1, 1368 cm−1, 1026 cm−1, and 580 cm−1, denoting –CN–, phenolic C–OH, epoxy C–O–C/C–N, and TiO/TiO–Ti frameworks apart from the ones discussed above. Heng et al.62 reported the FT-IR spectrum of CDs/SiO2 where peaks at 460 cm−1 and 900 cm−1 correspond to Si–O–Si, 1100 cm−1 and 3500 cm−1 corresponded to Si–OH bending and stretching vibrations, respectively, and at 1140 cm−1 for Si–O–C vibration. Also, the appearance of thionyl chloride in the CDs displayed peaks at 2488 cm−1, 1068 cm−1, and 1174 cm−1, indicating S–H, CS, and C–S vibrations and 1010 cm−1, 1105 cm−1, and 500–900 cm−1 indicating C–S–C, C–Cl in aromatic hydrocarbons and aliphatic hydrocarbons, respectively.65
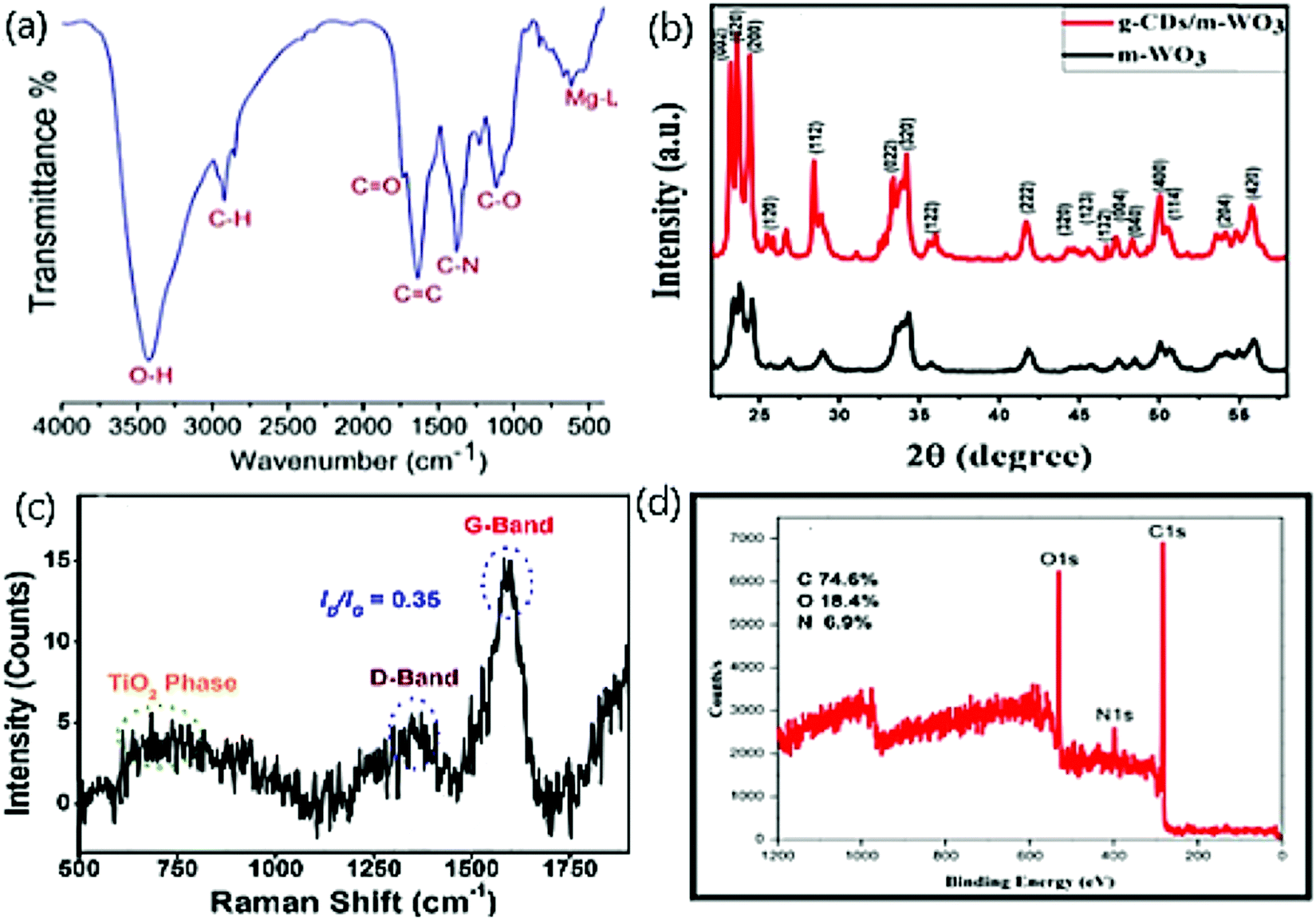 | ||
| Fig. 3 (a) FT-IR spectra of red emitting Mg–N-CDs; reproduced with permission from ref. 55 Copyright 2018 American Chemical Society. (b) XRD pattern of bare WO3 and CDs/WO3; reproduced with permission from ref. 71 Copyright 2020 Elsevier. (c) Raman spectra of TiO2/CDs; reproduced with permission from ref. 66 Copyright 2018 Elsevier. (d) XPS spectrum of N/CDs; reproduced with permission from ref. 43 Copyright 2018 Elsevier. | ||
The reduction of the amorphous nature of CDs was verified by observing two new sharp peaks at 19.5° (002) and a broad peak at 21.6–22.1° range after the successful embedding of CDs on WO3 nano structure Fig. 3(b).71,72 The incorporation of CDs on WO3 did not affect the stability of WO3 and aided the crystalline growth, which was studied by XRD patterns. The decline in amorphous nature is noticed with less intense broader planes of CDs doped with WO3 than bare CDs, and it is due to the change from amorphous nature to polycrystalline nature owing to the presence of WO3 on CDs.73 The formation of fused polymeric fragments occurs due to the carbonization of reactant materials. The polycrystalline order was attained by connecting such fragments into multiple layers on groups embedded on CDs.73 The growth of the polycrystalline nature was also supported by our work that displayed the XRD patterns of bare CDs, ZnO, and four composites of Zn/CDs with varying amounts of Zn.74
The Raman spectra display the higher degree of graphitization and crystallization of TiO2/N/CDs with a relative intensity ratio (ID/IG) ca. 0.35.66 Further, the spectrum of defected carbon −1350 cm−1 (D band), graphitic carbon −1597 cm−1 (G band)75,76 and a broad spectrum for TiO2 at 600–800 cm−1 were observed as in Fig. 3(c).
The chemical composition of the prepared N/CDs from Actinidia deliciosa is characterized and elucidated by X-ray photoelectron spectroscopy (XPS).43Fig. 3(d) displays the XPS survey scan spectrum at three peaks for C 1s (285 eV), N 1s (400 eV), and O 1s (532 eV). The high-resolution XPS spectrum displays the π–π* transitions and C–H, CC (sp2), C–N/O–CO bonds in C1s,77–79 C–NH2 and OC–NH2 bonds in N1s76,80 and CO, –C–O/–N–O and C–OH/C–O–C bonds on O1s.77,78,81 In addition, the XPS technique reveals the atomic percentage of carbon, oxygen, and nitrogen to be 74.6, 18.5, and 6.9, respectively.82
From the perspective of structure and morphology, CDs act as an excellent photocatalyst due to their high surface area. Furthermore, due to the presence of heteroatoms and metal oxides, highly functionalized CDs display better interaction between the surface of the catalyst and dye molecules than bare CDs or single atom doped materials. Therefore, by setting the above structural properties on photocatalyst, we can attain efficient photocatalytic activity.
3.3. Optical properties
In general, two peaks in the range of 250–350 nm usually occur in the optical absorption spectrum for doped CDs.83 Usually, these peaks are labelled as sp2 C atoms and groups at the edge of CDs. The optical absorption spectrum for bare CDs near the UV region at 350 nm peak is yet unresolved. Typically, our work with ZnO fabricated on CDs derived from gum ghatti displayed the UV-Vis absorption spectrum of bare CDs at 263 nm whose tail attains visible range, ascribed to π–π* transition of CC aromatic bonds.74 Pure ZnO nanoparticles display a broad spectrum at ∼357 nm.84 Further the CDs were doped with different proportions of ZnO NPs such as 0.5 wt% Fig. 4(a), 1 wt% Fig. 4(b), 1.5 wt% Fig. 4(c), and 2 Fig. 4(d) wt%. The absorption peak in the range 350–360 nm denotes the occurrence of crystalline and impurity embedded on ZnO. The change in absorption peaks verified the increase in the rate of cyclization with the progression of the reaction. The increase in ZnO proportion in CDs eventually increases the absorption intensity. The intensity of visible light increases due to the presence of CDs in the composite. This is attributed to the rise in electrical charge at the surface of the oxides on CDs that ultimately increases the photocatalytic activity in the visible range.85 Likewise, the presence of ZnO in CDs increases the performance of the photocatalytic activity.
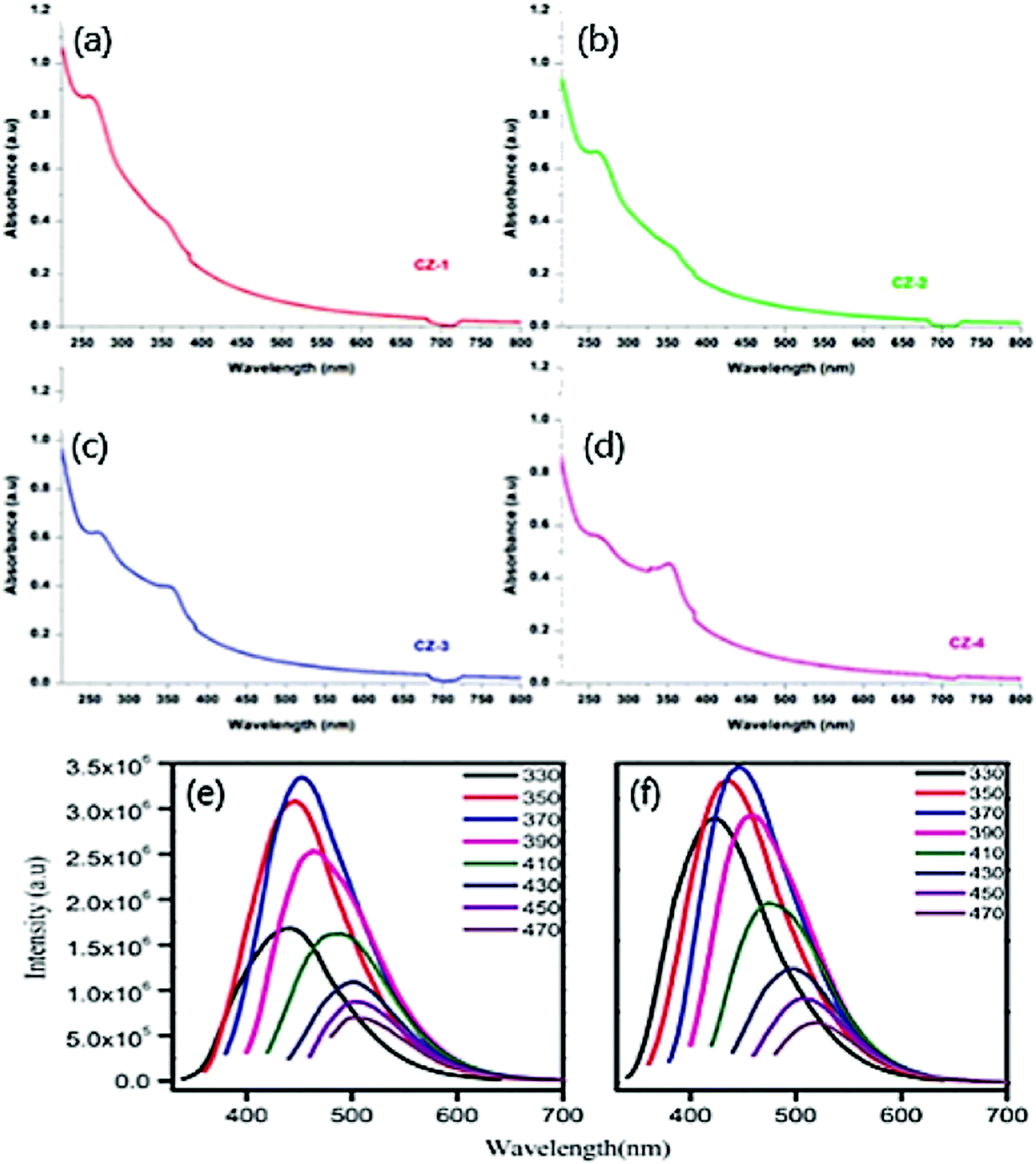 | ||
| Fig. 4 UV-vis absorption spectra of CDs with (a) 0.5 wt% (b) 1 wt% (c) 1.5 wt%, (d) 2 wt% of ZnO; reproduced with permission from ref. 74 Copyright 2021 Elsevier. Excitation-dependent PL spectra of (e) UCQD; (f) NCQD; reproduced with permission from ref. 63 Copyright 2018 Elsevier. | ||
The bandgap was calculated using Tauc plot to be 3.30 eV and 3.12 eV for bare CDs and pure ZnO. Similarly, the bandgap of CDs with various proportions of ZnO was calculated to be 2.93 eV, 2.92 eV, 2.98 eV, and 3.03 eV. The bandgap reduction is credited to forming the Zn–O–C chemical bond and eventually absorbs added light from a longer wavelength.86 Typically, Shahba et al.68 obtained UV spectra with a peak at 359 nm corresponding to n–π* transition of carbonyl groups and band gap at 3.2 eV obtained from UV-Vis spectra. The band gap thus obtained was higher than that of PL results, attributing that temperature plays a crucial role.
A strong emission band in the blue region (450 nm) contests the excitation absorption peak of n–π transition, whereas a weak emission band in the far UV region contests the π–π* excitation absorption peak.83 Such a specific property observed by CDs is called an excitation-dependent emission that involves a bathochromic shift. Absorption in the visible region is low, which is responsible for the weak intensity. In the same way, when excitation wavelength rises from 380–400 nm, the emission band remains unchanged, following which it experiences a red shift. In contrast, the emission band experiences a decline with an increase in excitation wavelength.83
Optical emission is responsible for the above-mentioned properties; for instance, the PL spectra of bare CDs and N/CDs exhibit excitation-dependent emission behaviour.63Fig. 4(e) displays the variation of excitation wavelength (330–470 nm) with emission variation (420–510 nm). Undoped CDs showed strong emission at 452 nm and excitation wavelength at 370 nm. Likewise, N/CDs Fig. 4(f) displays a strong emission at 447 nm and excitation wavelength at 370 nm.63 A remarkable decline in the emission intensity of both CDs was noticed when the excitation wavelength crossed 370 nm and eventually experienced a red shift at a longer excitation wavelength. The plausible mechanism for the PL emission is attributed to the characteristic size of the particle and the presence of various surface energy traps on the CDs.63 Therefore, the particle excites at a lower wavelength when it is smaller in size, and the particle excites at a higher wavelength when larger in size. It is inferred that the number of excited particles by the specific excitation wavelength reflects the PL emission intensity.63 The excitation wavelength at 370 nm due to the high number of excited particles at a particular excitation wavelength corresponds to CDs with the highest PL emission intensity. On modifying the surface properties, multi-PL emission sites were observed to be responsible for the excitation-dependent PL emission of CDs.63
Further, the plausible PL mechanism of CDs is attributed to two reasons. First, the surface state of CDs is one of the crucial factors to determine the electronic properties of CDs.69 Groups such as oxygen and amine on the surface of the CDs affect the optical band gap, thereby reasoning the starting material and its synthesis method for the alteration of electronic properties. Next, the quantum confinement of CDs is responsible for the PL mechanism.63
4. Potential roles of CDs as photocatalyst
The three primary steps for a photocatalytic reaction are light-harvesting, generation and separation of charge, and catalytic reaction.87 The equilibrium between the thermodynamics and the kinetics of the above three primary steps determines the photocatalytic reaction's total efficiency.87 Researchers have made intensive advancements in photocatalysts using the excellent ability of light-harvesting, high efficiency of charge carrier separation, and effectual catalytic interactions.87 With light being the core need for a successive photocatalytic reaction, it is necessary to meet all the requirements of the emerging research in which CDs derived from green precursors act as catalysts. However, the need for a suitable material that satisfies the above discussed criteria is yet to be determined due to the lack of efficient conversion of chemical energy from solar power. For any photocatalytic system, dopants act as both an active catalytic site and the light-harvesting unit. Hence the photocatalytic performance is closely associated with the interaction between the CDs and their dopants. The potential role of CDs that affects light-harvesting, charge transfer dynamics, and surface catalytic reactions in photocatalytic systems are demonstrated in Fig. 5 and discussed here.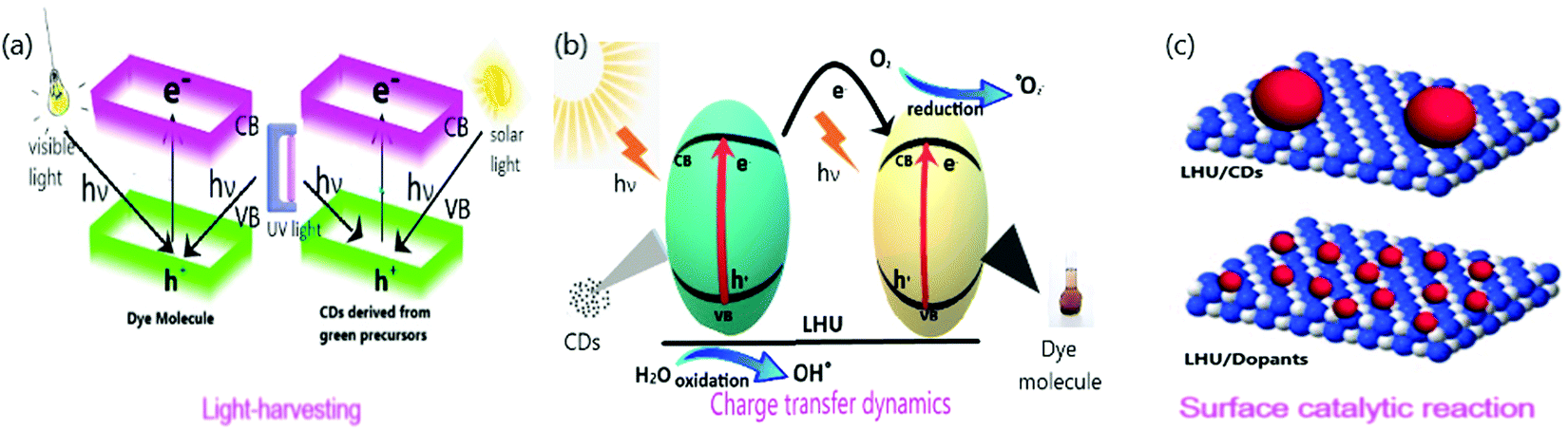 | ||
| Fig. 5 Systematic representation of (a) light-harvesting (b) charge transfer dynamics (c) surface catalytic reaction. | ||
4.1. Light-harvesting
Among the three primary steps, light-harvesting is the first step that determines the capacity of the system to generate electron–hole pairs by absorbing photons for a successive catalytic reaction.88 The spectrum light, whose source is naturally occurring sunlight, generally consists of 43% visible, 52% infrared, and only 5% ultraviolet light. The absorption of incident light shifts, especially in the visible/infrared region, due to the modification of energy or electron band structure when CDs are embedded on the dopants. The utilization of visible and infrared light absorption is predominant in solar energy.89 The energy band structure is utilized to determine light-harvesting capacity in semiconductors that are doped to CDs.Previous works have reported the effect of material on light-harvesting. (i) The characterization of spectral range on which light is absorbed, (ii) the optical bandgap, (iii) the position of conduction and valence band edges in the case of inorganic semiconductors, or (iv) energy levels of HOMO and LUMO in the case of organic semiconductors must be determined.80 UV-diffuse reflectance spectroscopy (DRS) and the Tauc plot from its spectra are employed to determine the spectral range90 and band gap.91 To further determine the electronic band structures, (i) the Mott–Schottky plot is used to determine the conduction band and valence band edge,92–95 and (ii) Ultraviolet Photoelectron Spectroscopy (UPS) is employed to determine the energy levels of HOMO and LUMO.96 In the case of coordination structures, broader light-harvesting is observed and validated by density functional theory (DFT) stimulation to determine the HOMO and LUMO energy levels.97 Also, photocatalytic activity with CDs as the catalyst is sensitized by solar, UV, and Visible light and is discussed elaborately.
| α = −C(hυ − Ebulk)1/2 | (1) |
Remli et al.98 reported the synthesis of CQDs that degraded MO and observed an absorption peak at 318 nm elongated to a visible range like a tail. The photocatalytic degradation was enhanced by narrowing the respective semiconductors' bandgap, which eventually led to a chemical bond between the semiconductor and CQDs. The increase in energy bandgap between the valence band and the conduction band decreased the particle size. Zhu et al.65 prepared CDs from palm powder and embedded them with sulphur and chlorine and degraded RhB (71.7%) and MB (94.2%) under visible light. Metal oxides have been accepted as efficient photocatalysts due to their suitable band gap, making them an appropriate light-responsive photocatalyst. The structural defect influences the formation of the band gap in the CDs microstructure. CDs hold the properties of electron-accepting, transferring, and transporting, turning them into efficient light-harvesting agents. CDs have not been explored much in photocatalytic degradation under visible light due to the limited activity.58
Thakur et al.99 determined the bandgap by Tauc plot for PAg-CQD/TiO2 and PAu-CQD/TiO2 NCs to be 2.4 eV and 2.6 eV, respectively, which coordinates with the surface plasmon resonance (SPR) peak of Ag and Au nanoparticles. TiO2 is absorbed in the UV region (3.2 eV) in contrast to CQDs absorbed in the visible region (2.4–2.6 eV) and enhanced due to the distinct effect of SPR. The excited electrons are injected into the conduction band of metal oxide when embedded on PM-CQDs and are responsible for the efficient harvesting of visible light. Shahba et al.68 obtained the optical band gap at 3.2 eV, which deviated from the UV-Vis absorption spectra from 359 nm and successively degraded EBT, methyl, MO, MB, acid blue (AB), and EY dyes.
Although we notice efficient degradation of dyes in sunlight, the influence of dopant on the catalyst shows better photocatalytic activity. Mathew et al.100 reported the synthesis of CDs from anhydrous d-glucose doped with phosphorus showing a rise in electrostatic potential on the surface, which assists in the movement of holes from the valence band to the surface. Therefore, heteroatoms can be embedded on the CDs due to increased potential at the surface level, which eventually improves the photocatalytic activity sensitized by sunlight. The potential role of utilizing sunlight in effective light-harvesting activity is its maximum absorption of visible and infrared light. Although photocatalytic dye degradation has been carried out with CDs as a catalyst derived from biomass, there is no detailed working mechanism for modifying CDs on light absorption and its efficiency on light-harvesting capability. The structure of electronic and energy bands is more important to understand the light-harvesting capacity of CDs. To accomplish all the criteria necessary to understand the effect of light-harvesting, detailed investigations such as utilizing UV-DRS, UPS, DFT stimulations, Tauc plot, and Mott–Schottky plot can be extended on photocatalytic activity of CDs and their dopants.
4.2. Charge transfer dynamics
Apart from the potential light-harvesting activity of the dopant, charge transfer dynamics plays a vital role in modifying the photocatalytic activity's efficiency.101,102 To attain high performance of the photocatalytic activity, the charge separation and transfer of electrons and holes that are photogenerated at the CDs and dopant interface should be efficient. A short charge migration distance should exist between the CDs and their dopants. There is an electronic equilibrium between the metal and its semiconductor, which helps transfer photogenerated electrons by the Schottky barrier.103 It is inevitable that higher the recombination of photogenerated charge transporters lesser is the photocatalytic performance reflected the non-feasible applications in commercial platforms.104,105 High-efficiency charge separation carriers and a wide range of photo absorption are the two main properties for an ideal photocatalyst. The dopants embedded on the CDs should be modified to improvise the photocatalytic performance. Coupling with noble metal nanoparticles (Au, Ag, Pt), heterogeneous atoms (N, P, S), or metal oxides (TiO2, ZnO) helps in potential photogenerated charge carriers along with transportation and hinders charge recombination.106–110 The ability of CDs to experience photogenerated charge transfer on dye species in its interface is ruled by the levels of the surface energy trap.111,112 The change influences the surface energy trap levels of the CDs with pH. Hu et al.113 reported the increase in degradation of MB dye with an increase in pH. Compared to bare CDs, its reduced form holds developed photocatalytic efficiency that arises due to its superior surface adsorption capability to hydroxyl groups and mild recombination rate of charge carrier.44CDs doped with nitrogen atoms diminish the energy barrier among the reactants and products due to the charge transfer mediator nature of N/CDs.114–116 Then, the reduction is completed within 10 min since the reaction rate is restored.44 Arul et al.43 reported N/CQDs that degraded 97–98% of MB and MG dyes in 120–180 min under sunlight but only degraded 59% of the same dyes in the absence of N/CQDs. This verifies that N/CDs generated potential photocatalytic degradation activity, indicating efficient utilization of sunlight to produce more photogenerated electrons and holes.109 In the same way, Mg incorporated in N/CDs showed an absorbance peak ∼674 nm and reasoned out to be due to the charge transfer between the bond of metal and ligands.55
The presence of metal oxide embedded on CDs shows the complete degradation of dyes when compared with bare metal oxide or CDs. Thakur et al.117 fabricated CDs on TiO2 nanofiber and exhibited complete degradation of MB but only 75% and 82% in the case of bare CDs and TNF, respectively. To understand the charge transfer dynamics, this was further characterized by cyclic voltammetry and electrochemical impedance spectroscopy. Charge transfer resistance (Rct) was calculated with the aid of Randel's equivalent circuit fit and obtained lower Rct for CDs/TNF than bare TNF, implying that unsaturated carbons on CDs are responsible for charge transfer. The hetero-structures of TiO2 on CDs improve charge separation and transportation. It was reported that TiO2/CQDs degraded dyes ∼2.5 times more than bare TNF.110 Decorating noble metal nanoparticles on CDs shows an improved activity of photocatalytic degradation. Incorporating such noble metal nanoparticles on CDs/TNFs helps in electron transfer and generates oxygen vacancies on the surface of TNFs. These composites' longer lifetime and potential charge separation ability are observed in time-dependent photoluminescence spectra.99 Likewise, ZnO is embedded on CDs and verified that the band alignment is the primary property that enhances the catalytic activity.64 Tungsten trioxide was decorated on CDs, and charge carrier separation was achieved successfully.71 A suitable material is necessary to migrate charge carriers across interlayers apart from quick charge migration distance. This can be achieved by a single metal atom and CDs decorated on nanosheets. Dadigala et al.56 reported the synthesis of Ag and CDs embedded on g-C3N4 nanosheets that formed heterojunctions, thereby promoting the efficiency of the charge carrier transfer and improving photocatalytic activity. Up-conversion of the CDs is attributed to enhance the photocatalytic activity.
The above works show that CDs hold good in improvising the charge carrier dynamics by which the photocatalytic activity is remarkably promoted. To further devise the charge carrier transfer efficiency, the selection of the dopants should be worked out intensively. Selecting the dopant with a less energetic valence band compared to that of CDs is encouraged. Up-conversion property is the most important factor since it increases the amount of light absorbed and influences its spectrum, which is still unclear, and more understanding of the mechanism should be achieved. More specific research is necessary for CDs that absorb NIR radiation and emits visible light that helps in potential charge transfer dynamics. Since the length, geometry, and electronic structure of the bridging ligand is essential for charge carrier dynamics but still unclear in CDs, organic porphyrin linkers can be utilized. To further elucidate the mechanism and track photogenerated charge carrier dynamics, investigations such as Electron Spin Resonance (ESR), Transient absorption spectroscopy (TAS), steady-state PL spectroscopy, and Time-resolved PL spectroscopy can be employed.
4.3. Surface catalytic reaction
The surface reaction of any system determines the photocatalytic activity and its selectivity. Geometry and electronic structure are the two properties that administer the surface reaction in photocatalytic reactions. These properties are modified depending upon the structure of the dopant's surface.118–120 The governable surface structure of various dopants offers access to regulate the geometric and electronic structure through the interaction between the metal and the support, thereby altering (i) the reaction pathway, (ii) the strength of the reactant, (iii) product, and (iv) intermediate, and (v) adsorption mode.87 Therefore, choosing a dopant that possess particular, coordinatively unsaturated sites and unique electronic structures helps generate enormous surface-active sites that increase the catalytic reaction. CDs have been attracted to their most common photo-physical property of excitation-dependent emission due to their varied size and surface state distribution. The presence of several organic functional groups on the surface of the CDs is ascribed for the same.80,121 CDs are known for their efficient photocatalytic activity because of their greater capacity towards surface adsorption on hydroxyl groups and decline in charge carrier recombination rate.44 N/CQDs improve the quantum yield, stability, and active sites of the CQDs, which eventually increases the photocatalytic activity due to numerous functional groups. Due to the enormous number of active surface sites on N/CQDs, rapid dye degradation occurs.67 For example, N/CDs have been credited with an excellent catalytic activity due to the high efficiency of electron-accepting potential and large surface area.43The increase in active sites on the surface of the catalyst increases the activity of photocatalysis. For instance, the increase in the amount of catalyst (1 mg mL−1) increases the degradation rate of MB dye, which is attributed to the rise in active surface sites.99 Surface molecular states are responsible for tuning the emission of CDs and were investigated by Dhenadhayalan et al.122 Lesser the band gap of the product higher is the photocatalytic activity; therefore, the intensity of the product's PL is improved by modifying the surface, thus decreasing the surface defects with EDTA, AcAc, PEG, and PEGVP.68 Surface charge is also one of the critical factors that determine the adsorption and photocatalytic activity. Owing to the negative surface charge, g-CDs/m-WO3 exhibited better catalytic activity under visible light.71
In CDs, there should arise more understanding of estimating the role of surface catalytic reaction towards potential photocatalytic activity. The incorporation of dopants that improves the adsorption of CO2 considerably lowers the activation-energy barrier, which increases photocatalytic activity performance. The role of dopants must be in such a way to stabilize the intermediate formed during photocatalysis, thus altering the reaction pathway to attain high selectivity of the product during the process. The ability of the dopant to tune the binding energy of the product ensures quick release of the product, resulting in high reaction activity. All the above can be achieved by incorporating single atoms such as Co or Pt in the CDs and observing the results of surface catalytic reactions.
Several factors govern these three main principles that influence the potential role of CDs as photocatalysts. According to photocatalysts, embedded dopants123,124 and surface structure87 (geometry and electronic structure) of the material play a crucial role in harvesting light and surface catalytic reactions, respectively. But charge transfer kinetics is altered by several parameters like embedding single atom, lifetime, bridging ligands, work function, bandgap, quick charge transfer distance, and strong interaction between CDs and dopants.87,91,125 These parameters remain a research gap in CDs compared to other literature available on different photocatalysts. Regardless of diverse factors that alter the light-harvesting, charge transfer kinetics, and surface catalytic reaction separately, they contribute equally as potential candidates for CDs as photocatalysts.
5. Factors influencing the photocatalytic dye degradation
The photocatalytic dye degradation rate and its efficiency are influenced by specific functional parameters that administer the dye degradation of the photocatalytic system. Numerous studies have been reported on the operational parameters that rule the photocatalytic activity and its significance. Effect of pH, the concentration of dopants or CDs, photocatalyst loading, light intensity and irradiation time, the impact of temperature, and initial concentration of dyes are some of the basic parameters that impact the photocatalytic activity of dye degradation. The photodegradation of dyes in an aqueous medium with the aid of CDs has been studied widely, and the optimum conditions of the operational parameters that influence dyes' photocatalytic degradation have been derived. It is to be noted that these conditions differ system to system and are highly dependent on the surface charge of the photocatalyst, nature of the photocatalyst and substrate, metal/non-metal dopants, and structure of the photocatalyst and substrates.5.1. Effect of pH
One of the crucial factors that affect photocatalytic dye degradation is the pH of the solution. The potential of the catalytic reaction is shifted due to modification of the pH of the solution, which changes the surface charge on the metal or metal oxide/CDs and the alteration in conduction and valence bands position.126,127 Thus, there is an alteration in the reaction rate due to the change in surface adsorption of the dye. As a result, the metal or metal oxide/CDs are either protonated or deprotonated under acidic or basic conditions.128 The following reaction can be an example:| XOH + H+ → XOH2+ | (2) |
| XOH + OH− → XO− + H2O | (3) |
Zaib et al.137 reported the optimum pH to potentially degrade CR due to the capability of the dye to form R-SO3−, thereby, the electrostatic interaction between CDs and dye turns to be positive in acidic and negative in basic conditions. On lowering the pH, the molecular form of the dye predominates over the anionic form while vice versa with an increased pH.138 On further increasing the pH, a decline in degradation occurred, attributed to the deprotonation of functional groups on CDs, which acquires a negative charge. Eventually, the electrostatic interaction between the CDs and anionic dye is decreased.139 But MB dye being a cationic dye, showed potential degradation at pH 8, below which less electrostatic interaction was observed between the CDs and dye. Below a pH of 3, neutral form predominates, and at pH 3.8, the neutral and cationic form of dye is in equilibrium. Above pH 6, the cationic nature of the dye prevails that suits the photocatalytic dye degradation at its best.140 Typically, our work has reported the optimum pH of 8 where 94% of MG dye was degraded, which owes to the same reason as above where dye is cationic in nature as depicted in Fig. 6(a).74 Therefore, it is vital to identify the optimum pH for a better performance of the photocatalytic activity.
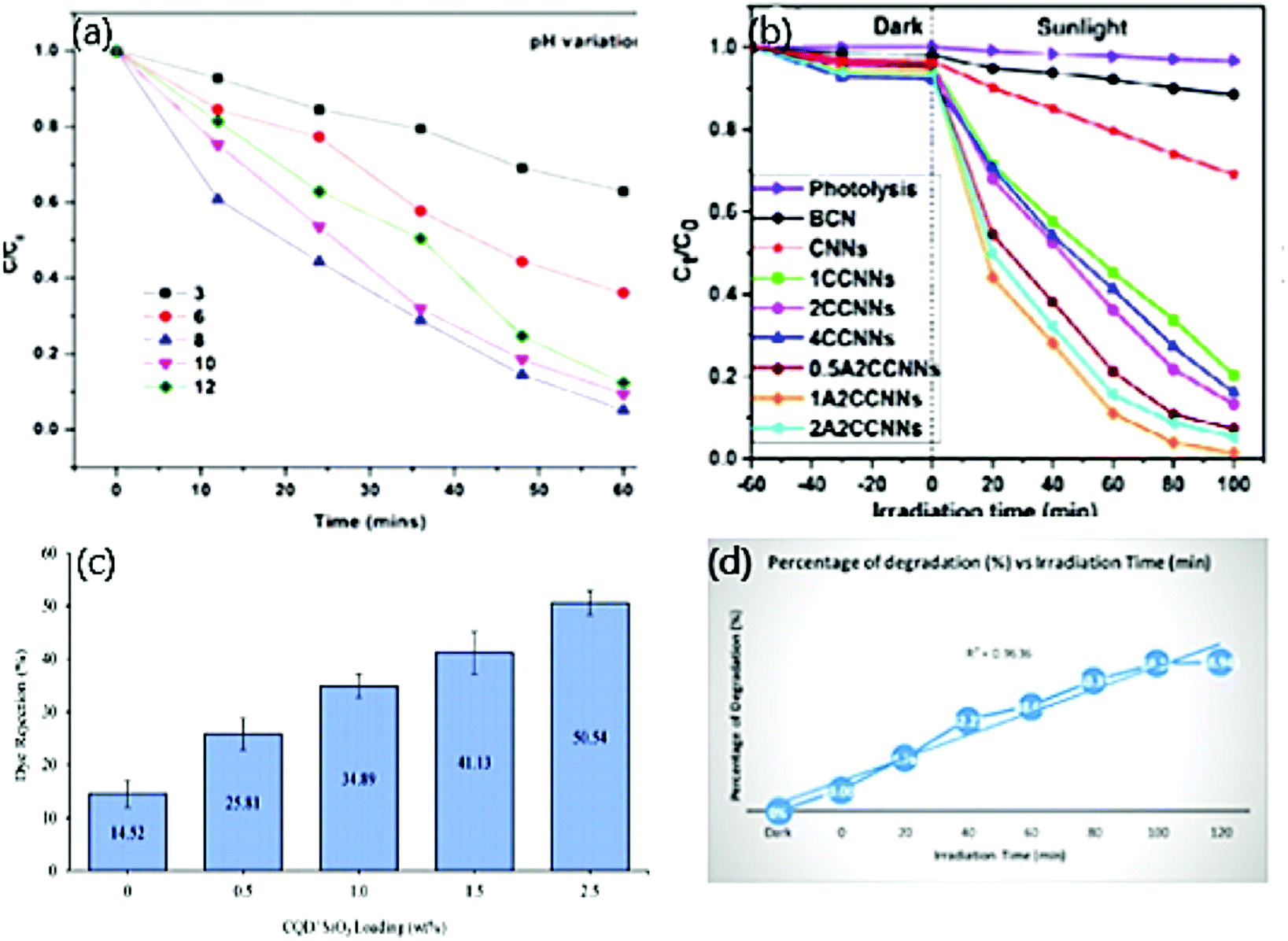 | ||
| Fig. 6 (a) Effect of pH on photocatalytic degradation of MG dye; reproduced with permission from ref. 74 Copyright 2021 Elsevier. (b) Effect of volume of CNN, CDs, and Ag on the photocatalytic degradation of MO; reproduced with permission from ref. 56 Copyright 2017 Elsevier. (c) Effect of photocatalyst loading on photocatalytic degradation of Tartrazine dye; reproduced with permission from ref. 62 Copyright 2021 Elsevier. (d) Effect of time on photocatalytic degradation of MO dye; reproduced with permission from ref. 98 Copyright 2020 IOP Conference Series: Materials Science and Engineering. | ||
5.2. Effect of concentration of dopant or CDs
Apart from the effect of photocatalyst loading, the effect of concentration of dopant or CDs in particular also affects photocatalytic dye degradation. The concentration of dopants and CDs plays a dynamic role in the potential degradation of both anionic and cationic dyes. To determine the effective volume of dopants and CDs for the active performance of the photocatalytic dye degradation, different quantity of dopants and CDs is taken each time separately and investigated with a constant dye concentration. The performance of the photocatalytic activity fades out when the volume of dopants and CDs deviates from the optimum volume. In the first case, varying the volume of dopants embedded on CDs exhibits a varied performance in photocatalytic activity. The improved catalytic activity is observed on increasing the volume of dopants due to the following reasons: (i) formation of vacancies to occupy an additional amount of oxygen (ii) provides photoinduced electron/hole recombination centres.126 For instance, in our work, we derived CDs from gum ghatti and doped them with 0.5 wt%, 1 wt%, 1.5 wt%, and 2 wt% ZnO. Here, CDs with 1wt% Zn showed 94.8% degradation of MG dye, which was observed to be the maximum when compared with another composition.74Unlike the above case by changing the volume of dopants alone, researchers have proposed works in which both CDs and dopants have been altered separately, and the photocatalytic activity is monitored. Dadigala et al.56 reported the photocatalytic dye degradation of MO dye by CDs/Ag nanoparticles embedded on g-C3N4 as depicted in Fig. 6(b). At first, 1 wt%, 2 wt%, 4 wt% CDs were impregnated on 100 mg CNNs. Maximum degradation of 86% was observed in 2 wt% of CDs on CNNs and is credited to the presence of several CDs on graphene sheets (CNNs), which cover the CNNs surface that eventually prevents light absorption. The surface further promotes recombination by increasing its sites for charge carriers.141 Later, 100 mg of 2 wt% CDs/CNNs composite were embedded with AgNPs by adding 0.5 wt%, 1 wt%, and 2 wt% of AgNO3. CDs, Ag/CNNs composite prepared by 1wt% AgNO3 showed maximum efficiency of 98.6% photocatalytic degradation of MO. On further increasing the volume of AgNO3, the photocatalytic activity decreased, which might be due to the weak light-harvesting capacity of the respective composite because of the shielding effect. Thus, the particular composite acts as a recombination centre.142 Therefore, it is noticed that increasing the volume of dopants exhibits effective photocatalytic dye degradation, but an improvement of the same by ∼4% is observed while identifying the optimum volume of both dopants and CDs. Hence, it is inferred to follow the same to modify and determine the optimum level of dopants and CDs individually to improve the photocatalytic activity.
5.3. Effect of photocatalyst loading
The amount of photocatalysts influences photocatalytic dye degradation. Usually, in heterogeneous catalysis, the degradation of the dye increases when the amount of photocatalyst increases. At the same time, the number of active sites increases on the surface of the photocatalyst, followed by an increase in ˙OH and ˙O2 radicals’ formation that ultimately plays a major role in discoloring the dye solution.143 Further increasing the volume of photocatalysts from the optimum level reflects the decline in photocatalytic activity due to the excess volume of the catalyst. It increases the turbidity of the suspension that eventually shields the irradiation of light and decreases the photo-activated volume of the respective dye.144 Next, there appears a very low degradation performance below the optimum level and is credited to less availability of reactive species.145 At the optimum level, a photocatalyst absorbs more photons, thereby generating a significant number of reactive species that eventually reflect maximum degradation of respective dyes.146 The second reason for reducing dye degradation by increasing the photocatalyst is attributed to the occurrence of nanoparticle agglomeration that consequently minimizes the surface area of the respective photocatalyst.147 The above reasons were defensible in our work where CDs/ZnO from gum ghatti showed maximum degradation of 94% when loaded with 1.5 mg of photocatalyst.74 Below and above the optimum concentration of CDs/ZnO, the photocatalytic activity declined to 88% and 89%, respectively.74 The proportionality between the photocatalyst and degradation depends upon the geometry, working mechanism of the photoreactor, and the complete proportion of the photocatalyst entirely irradiated. Therefore, the overall cost of the activity can be reduced by preventing an extra dosage of photocatalyst and identifying its optimum concentration.148–150 When an extra dosage of the photocatalyst is loaded, unfavourable light scattering and penetration of minimum light into the solution are observed.151 For instance, Zaib et al.137 derived CDs from Elettaria cardamomum leaves and varied its concentration from 0.5 to 10.5 mL to degrade 10 ppm CR and MB dye concentration, respectively. The degradation of CR dye increased while increasing the volume of CDs from 0.5–5 ml. On further improving the volume, the photocatalytic activity decreased due to mild penetration of light that arises because of a turbid suspension. The degradation of MB dye was maximum at 5 ml volume of CDs and below which the generation of reactive species was less in number. Similarly, CDs derived from sugarcane bagasse showed maximum degradation of MO, EBT, Methyl Red (MR), and Fast green (FG) dye when CDs were loaded with 0.003 g (81.25%), 0.003 g (49.3), 0.005 g (87.5%), and 0.010 g (90.25%), respectively.152 The absorption peak was observed at 0.3, 1.1, 0.2, and 0.2, respectively, which exhibited a linear increase in photocatalytic degradation with an increase in photocatalysts.152Heng et al.62 observed maximum dye rejection when the percentage weight of the respective photocatalyst was 2.5 Fig. 6(c). This was attributed to the ability of dye molecules to repel themselves due to the negative charge on the surface of the photocatalyst and due to the ability of dye molecules to retain themselves due to the pore size of the catalyst being relatively small.153 Therefore, from the above inference, it is necessary to identify the optimum concentration of photocatalytic loading in every activity to attain a potential photocatalytic activity.
5.4. Effect of initial concentration of dye
Photocatalytic dye degradation is significantly influenced by the amount of dye adsorbed on the surface of the photocatalyst. It is to be noted that only the quantity of dye adsorbed on the photocatalyst surface contributes to the photocatalytic activity and not the bulk solution of the dye. The initial concentration of the dye is an important factor that should be considered for photocatalytic dye degradation since the adsorption of dye depends on this factor. Generally, by keeping the photocatalyst concentration constant and increasing dye concentration, one can observe a decline in degradation percentage.154,155 The decline in degradation percentage is ascribed to the increase in adsorption of dye molecules on the surface of the photocatalyst when the concentration of the dye increases. The availability of photons to reach the catalyst's surface is less, which decreases the formation of ˙OH radical.156 It is stated that the increase in initial dye concentration demands high catalyst volume since the requirement of ˙OH and ˙O2− radicals species becomes high to degrade dye molecules.144 The demand for dye molecules for excitation and energy transfer rises with an increase in the initial concentration of the dye. Zaib et al.137 studied the effect of dye concentration by increasing the volume of CR Fig. 7(a) from 5 ppm to 75 ppm, where the lowest degradation was observed at 75 ppm and indicated the above reason that the dye molecules act as a barrier to irradiation light. Meanwhile, at a low dye concentration, a controversial effect of reaction suppression occurs by capturing the photons on their catalyst that leads to a high number of OH radical formation. Sometimes, fixed amounts of catalysts offer a particular number of reactive species, which is not sufficient to decline the higher dye concentration.157,158 The enhancement of CR dye is accomplished by effective usage of reactive species because there is sufficient interaction between the dye and reactive species at a high dye concentration.143,159 Meanwhile, maximum degradation of MB dye occurs at 5 ppm Fig. 7(b). Typically, our work carried out the effect of initial concentrations varying from 0.04–0.08 g L−1 with a constant volume of Zn/CDs (1.5 mg) at pH 8 and observed maximum efficiency at 0.05 g L−1 dye concentration.74 The dye adsorbed on the catalyst's surface forms multiple monolayers favouring high initial dye concentration.154 The efficiency of degradation remains constant since the surface of the catalyst is not entirely covered before the attainment of optimum concentration of the dye, and the activity is reversed on further increasing the dye concentration.126,154,160 Therefore, it is necessary to study the effect of initial dye concentration and identify the optimum level to perform photocatalytic activity effectively.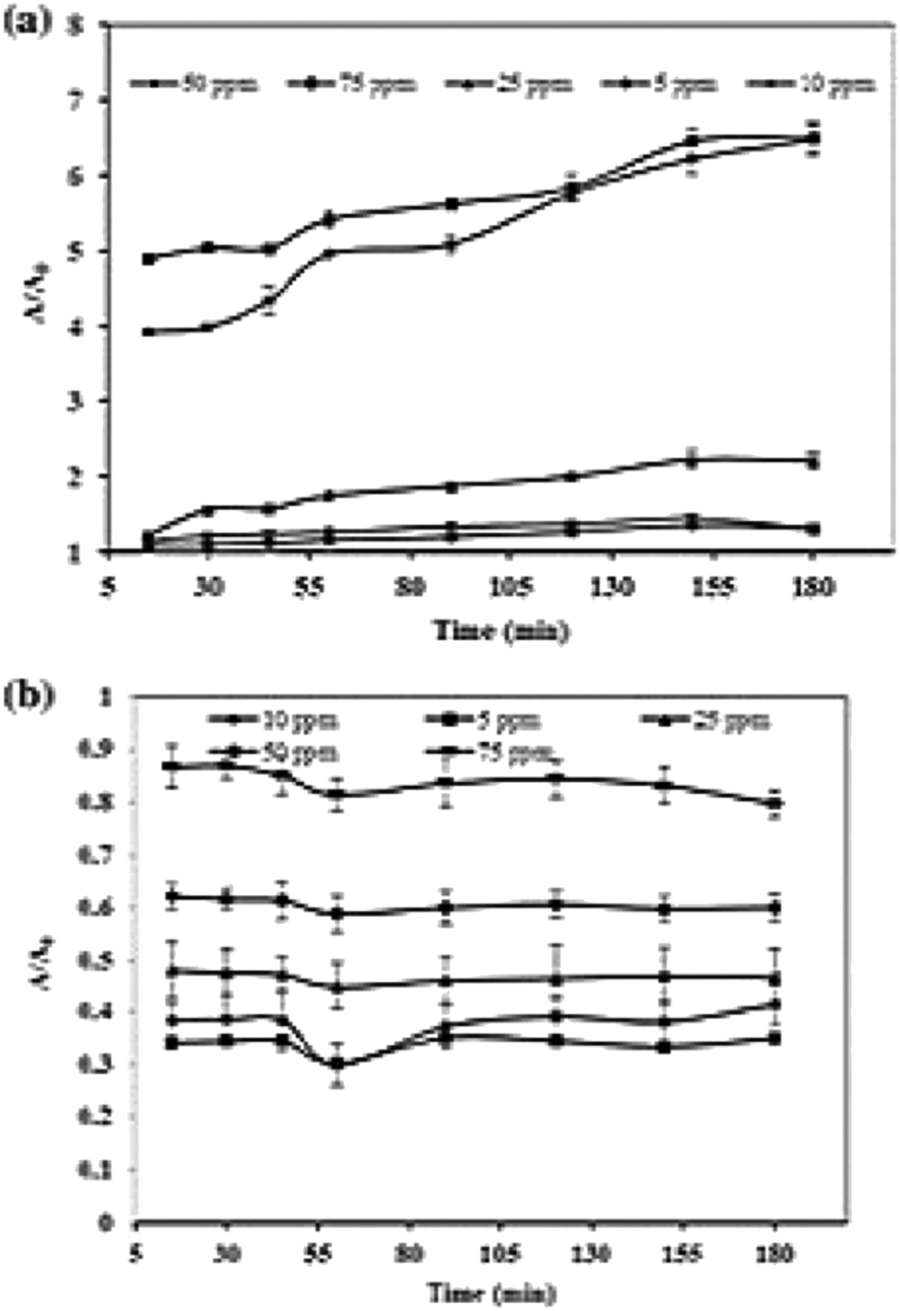 | ||
| Fig. 7 Effect of initial concentration on photocatalytic degradation of (a) CR dye and (b) MB dye; reproduced with permission from ref. 137 Copyright 2020 Springer. | ||
5.5. Effect of light intensity
The adsorption of light by the photocatalyst influences photocatalytic activity.131,161 The literature explains that the increased rate of the reaction at higher light intensity is due to the irradiation light falling on the surface of the photocatalyst directly, which opens the way for hydroxyl radical formation and controls the electron–hole recombination.162–164 The path of the photodegradation reaction is not affected by the light's nature, or its form, or the mechanism of band-gap sensitization.165 When the intensity of the light is low (0–20 mW cm−2), the reaction rate increases linearly with an increase in light intensity, and at intermediate light intensity (25 mW cm−2), the rate of the reaction might depend on the square root of the light intensity.166 This is because the formation of electron-holes is predominant, and electron–hole separation is insignificant. On further increasing the intensity of the light from the optimum level, the reaction rate is independent of its intensity since there is competition between the formation and recombination of electron-holes.127,155 This explanation was supported with the experimental setup in our work to degrade MG dye with namely four conditions;(i) dark + without CDs/ZnO (ii) dark + with CDs/ZnO (iii) MG + visible light + without CDs/ZnO (iv) MG + visible light + with CDs/ZnO. There was no significant progress in photocatalytic degradation under dark conditions. In comparison, 2% decolorization was observed in the third case, which verified that increasing the light intensity in a suitable photocatalyst favours the dye degradation.745.6. Effect of irradiation time
Similar to light intensity, the effect of irradiation time also influences photocatalytic activity.161 As the irradiation time increases, there is a decline in the reaction rate (first-order kinetics), and there is competition between reactant and intermediate products while degradation occurs. There are two reasons for the decline in kinetics after optimum level, which is: (i) the disability of the reaction to progress due to short-chain aliphatic present along with hydroxyl radicals and (ii) the lifetime of the photocatalyst being short since there is a deposition of strong by-products, which causes deactivation of active sites.155,167 This was illustrated in CDs derived from Elettaria cardamomum to degrade CR and MB dyes, whose photodegradation activity increases with an increase in contact time and maximum degradation observed within 50 and 55 min, respectively.137 Atchudan et al.168 reported the degradation of MB dye under UV irradiation with ZnO,N/C sheet nanocomposites whose maximum dye decolorization of 95% was observed within 60 min, which followed pseudo-first-order kinetics. Remli et al.98 exhibited the increase in degradation percentage from 0–68.94% with an increase in time from 0 to 120 min and observed improvised outcomes compared to other works Fig. 6(d).5.7. Effect of temperature
It is a very commonly known fact that an increase in reaction temperature increases catalytic activity. Further elevation of temperature from the optimum level promotes electron–hole recombination and disfavours the decolorization of dyes.169 There occurs an elevation in activation energy when the temperature falls to 0 °C.170,171 The optimum temperature at which maximum dye adsorption occurs with CDs as a photocatalyst would be from 20–80 °C. Unfortunately, no such experimental work is carried out that compares photocatalytic dye degradation by modifying the temperature with the aid of CDs photocatalysts derived from biomass precursors.6. Radical scavenging activity
Photocatalytic dye degradation is a simple oxidative reaction where the participation of active radical species such as holes (h+), electrons (e−), superoxide anions (˙O2−), and hydroxyl (OH˙) radicals takes place.172–174 Generally, to determine the significant participation and the role of these radicals, trap experiments are carried out with the aid of scavengers. Photocatalytic activity of various dyes with CDs as a catalyst has utilized a handful of scavengers in various works. The structure of the CDs stands advantageous for its high density on surface traps and notable absorption of illumination of light that leads to efficient photocatalytic dye degradation.58 The most frequently used scavengers to determine the active species in the field of CDs derived from green precursors are (i) Na2-EDTA and ammonium oxalate (AO) (to trap h+), (ii) para-benzoquinone (p-BZQ), and ascorbic acid (AA) (to trap superoxide anion radicals) and (iii) tertiary butyl alcohol (t-BA), iso-propyl alcohol (IPA), and ethanol (to trap hydroxyl radicals), which will be discussed in the latter part. Aliphatic alcohols such as ethanol, t-BA, IPA, etc., are used to trap OH˙ radicals since the intermediate products generated by these alcohols turn out to be highly inert on the illumination of light. It triggers the reaction between the hydroxyl radical and the organic dye molecule.175–177 Trap agents used for holes are mostly EDTA due to their oxidizing property and can decompose themselves to glyoxylic acid and ethylenediamine- N, N’ diacetic acid without reversing.178,179 p-BZQ is frequently used as a scavenger to trap ˙O2− radicals from CDs since it readily reduces BZQ˙− radical (1.1 × 109 L mol−1 s−1) with the aid of steady light irradiation. The product derived after the interaction between superoxide anion radicals and p-BZQ is hydroquinone.180To identify the active species responsible for potential photocatalytic degradation, a trapping experiment has been carried out, and several works have been reported. Aggarwal et al.45 performed a trapping experiment with scavengers such as Na2-EDTA, p-BZQ, and t-BA to trap holes, superoxide, and hydroxyl radicals, respectively. The photocatalytic activity rate decreases when electrons and holes are trapped by the Na2-EDTA complex, while the activity of other trapping agents is less distinct. The electron–hole generated reacts with water molecules absorbed to produce either OH˙ radicals or oxidize the dye molecules to organic radicals. Subsequently, the reactive oxygen species (ROS) reacts with the dye molecule and degrades into smaller fragments.45 Anand and the group carried out similar work with CDs derived from the Bougainvillea plant with 0.5 to 5 mM of the same scavengers.55 This exhibited a 55% reduction in photocatalytic activity at 0.5 mM of p-BZQ and a 25% reduction in photocatalytic activity efficiency at 0.5 mM of Na2-EDTA and t-BA because superoxide radicals influence the photodegradation activity strongly. Das et al.58 proposed a similar experiment again with the same scavengers (10 mM) on CDs derived from pear juice to degrade MB dye. They concluded that the active species that participate majorly in photocatalytic dye degradation to be Na2-EDTA and t-BA to trap h+ and OH˙ radicals that exhibited 25% and 33% of suppression, respectively, but 50% reduction with p-BZQ to trap ˙O2−. Other scavengers like 1 mM of AO, BZQ, and IPA were employed to trap h+, ˙O2−, and OH˙ radicals, respectively, Fig. 8(a) and resolved that h+ and ˙O2− exhibited photocatalytic degradation of MO by 37% reduction. At the same time, IPA did not influence the rate of the reaction.56 Smrithi et al.71 carried out the same experiment with BZQ (0.1 mM), AO (1 mM), and IPA (1 mM) and rejected the participation of h+ and ˙O2− radicals as there was no significant change in the addition of AO and BZQ. At the same time, IPA exhibited a more significant decline in the photocatalytic degradation rate of CV dye and confirmed the participation of OH˙ radicals, which is a powerful oxidizing agent that helps degrade organic dyes. Diverse from the above scavengers used, our work has performed the radical scavenging activity with the help of 1 mM of NaCl, K2Cr2O7, (AA), and ethanol to trap h+, e−, ˙O2−, and OH˙ radicals, respectively. We exhibited efficient photocatalytic degradation of MG dye with a reduction of 53.85%, 16.24%, 8.55%, and 69.23%.67 These results confirmed that e− and ˙O2− were the predominant active species to degrade MG dye under visible light. On further interpreting the datum from the above, it is to be noted that when bare CDs are used as a photocatalyst for dye degradation, the primary active species responsible for efficient degradation is h+. While for metal oxide and heteroatom doped CDs, superoxide anion radicals and hydroxyl radicals are primarily accountable for efficient dye degradation. Therefore, it is necessary to identify the active species responsible for productive photocatalytic dye degradation.
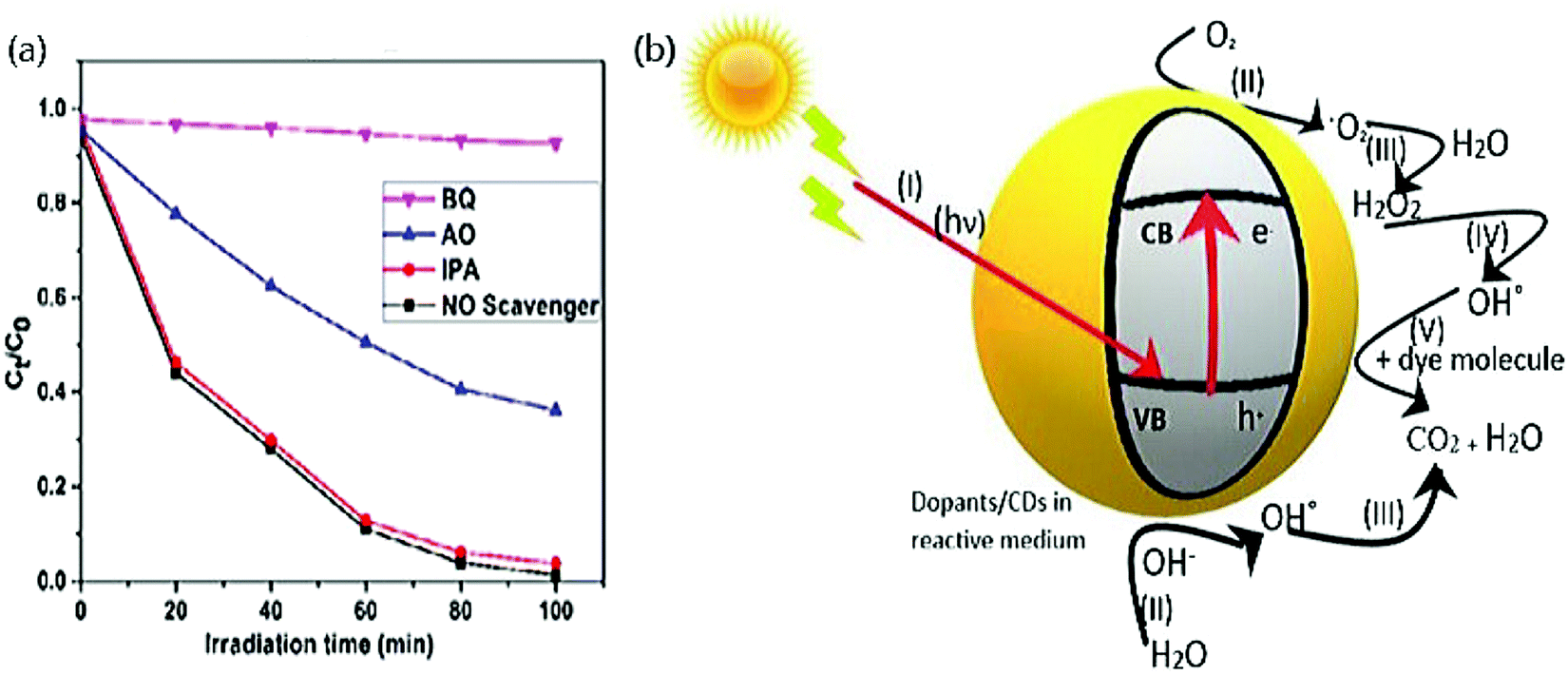 | ||
| Fig. 8 (a) Photocatalytic degradation of MO dye with various scavengers by trap experiment; reproduced with permission from ref. 56 Copyright 2017 Elsevier. (b) Systematic representation of the photocatalytic degradation mechanism. | ||
Despite a handful of experiments, the mechanism between the scavengers and radicals should be explored in detail. A detailed mechanism can be understood by carrying out investigations like EPR intensity signal and understanding the charge carrier separation.181 In addition, the stability of the hydrogen bond corresponding to the holes can be investigated by Scan diffuse-reflectance infrared Fourier transform spectroscopy (DRIFTS) studies.181
7. Mechanism
Apart from other common photocatalysts such as ZnO, TiO2, CdS, etc., CDs derived from green precursors show favourable photocatalytic activity as tabulated in Table 2. Various mechanisms have been proposed based on the interaction of CDs and dopants with the respective dye. The principle of the photocatalytic activity of dye with the CDs is as follows: (i) the ability of CDs to bind with the organic dyes with the aid of π–π* interaction that eventually increases the possibility of degradation50,182 (ii) the ability of CDs that owes up-conversion photoluminescence to absorb light in the visible region and emit UV light, which is later absorbed by dopants that open the path for photo-excited electron formation.50 The energy gap between the HOMO (valence band) and LUMO (conduction band) decreases due to extensive π-conjugation in the aromatic rings of the CDs, displaying independent photocatalysis reaction of CDs. Further, the visible light-sensitivity and the capacitance of the photocatalytic activity are increased by embedding suitable dopants like heteroatoms on the CDs183,184 (iii) the comparatively low bandgap energy and excitation wavelength help in forming e−/h+ pair that supports the participation of OH˙ and ˙O2− radicals.65,185 The interaction between the functional groups on the CDs and dye molecules under the irradiation light generates e−/h+ pairs, and these holes either convert to hydroxyl radicals or directly oxidize dye molecules.186 This ROS helps in the degradation of dye molecules into smaller fragments.45 Studies prove that compared to any other source of light, sunlight can convert the CDs to catalysts since the absorption band of the respective dye decreases. The observation of CO2 reduction helps to study the photocatalytic activity of CDs.| S. no | Precursor | Size (nm) | Doping | Type | Dye | Light | Ref. |
|---|---|---|---|---|---|---|---|
| HC – hydrothermal carbonization; RhB – rhodamine-B; C – carbonization; CV – crystal violet; MW – microwave; MB – methylene blue; HT – hydrothermal; MO – methyl orange; P – pyrolysis; SR – solar radiation; BM – ball milling; MG – malachite green; US – ultra sonication; CR – congo red; AB – acid blue; AR – acid red; EY – eosin Y; EBT – eriochrome black T; AC – autoclave; NBB – naphthol blue black azo dye; M – methyl; MR – methyl red; FG – fast green; Vis – visible light; CDs – carbon nanodots; CQDs – carbon quantum dots; CNF – carbon nanofiber. | |||||||
| 1 | Actinidia deliciosa/HC | ∼3.59 | N/CDs | Non metal | RhB | UV | 43 |
| 2 | Bitter apple/C | CDs | — | CV | SR | 45 | |
| 3 | Bougainvillea/MW | ∼10.9 | r-Mg–N/CDs | Hybrid | MB | SR | 55 |
| 4 | Calocasia esculenta. l, mesua ferrea Linn.l, tea. l, syzygium aromaticum bud/HT | ∼3 | CDs | — | MB, MO and MB + MO | SR | 44 |
| 5 | Citrus grandis juice/HT | ∼3 | CDs & N/CDs | Non metal | MB | SR | 63 |
| 6 | Citrus limetta waste pulp/P | ∼5.5 | CDs | — | MB | UV-Vis | 117 |
| 7 | Citrus limetta waste/P | ∼10 | PM-CQDs/TiO2 (M–Au&Ag) | Metal/metal oxide | MB | UV-Vis | 99 |
| 8 | Coal/BM | ∼2 | CDs | — | MB, MO | Vis | 113 |
| 9 | EFB fibre/HT | ∼3.6 | N/CQDs | Non metal | MB | SR | 67 |
| MG | |||||||
| 10 | Elettaria cardamomum/US | — | CDs | — | CR, MB | Vis | 137 |
| 11 | Frying Oil | — | CDs | — | MB | SR | 189 |
| 12 | Grass/HT | ∼10 | N/CQDs | Non metal | AB, AR, EY, EBT, MO, MB | UV and Vis | 46 |
| 13 | Gum Ghatti/MW-P | ∼2.6 | ZnO/CDs | Metal oxide | MG | Vis | 74 |
| 14 | Instant coffee | ∼3.5 | ZnO/CDs | Metal oxide | MB | Vis-NIR | 64 |
| 15 | Lemon juice/HT | ∼7 | N, S/CDs (PVP) | Non metal | CV, MG, EY | SR | 193 |
| 16 | Lemon peel/HT | ∼2 | TiO2/CQDs | Metal oxide | MB | UV | 110 |
| 17 | Lychee seed/P | ∼1.12 | CDs | — | MB | UV Vis | 59 |
| 18 | Musk melon | ∼7 | CDs | — | MB | UV | 194 |
| 19 | Onion waste/AC | ∼7 | AgNPs, CDs/CNNs | Hybrid | MO | SR | 56 |
| 20 | Orange waste/HC | ∼4.5 | CDs/ZnO | Metal oxide | NBB | UV | 60 |
| 21 | Palm fronds/C | ∼35 | CDs | — | MO | SR | 182 |
| 22 | Palm powder/HT | ∼3.54 | S, Cl/CDs | Non metal | RhB | Vis | 65 |
| MB | |||||||
| 23 | Peach fruit/HT | ∼4 | TiO2, N/CDs | Metal oxide | MB | UV | 66 |
| 24 | Pear juice/HT | ∼4.5 | CQDs | — | MB | Vis | 168 |
| 25 | Pine fruit/HT | <10 | CQDs/CNF | — | AB, EY, EBT, MB, MO, M | — | 68 |
| 26 | Rice husk/HT | CDs/SiO2 | Metal oxide | Tartrazine | 62 | ||
| 27 | Sugarcane bagasse | — | CDs | — | MO, EBT, MR | SR | 152 |
| FG | |||||||
| 28 | T. cucumerina | ∼30 | WO3/CDs | Metal oxide | CV | Vis | 71 |
| 29 | Turmeric powder/HT | ∼65 | CoFe2O4/CQDs | Metal oxide | Acid black, brown, red | UV | 47 |
| 30 | Water melon rinds/HT | ∼3 | CQDs | — | MO | Vis | 98 |
| 31 | Yerba mate | ∼5.1, ∼2.8, ∼4.2 | CDs | — | MB | — | 185 |
| Avocado seed | |||||||
| Orange peel | |||||||
A typical reaction during photocatalysis of CDs are as follows;
The molecular oxygen absorbed on the surface of the catalyst is scavenged by photo-induced e− to produce ˙O2− radicals and H2O2.187
| CDs + hν → CDs (ecb− + hvb+) | (4) |
| O2 + ecb− → O2− | (5) |
| 2H+ + O2 + 2e− → H2O2 | (6) |
| H2O + hvb+ → OH˙ + H+ | (7) |
| H2O2 + O2− → OH˙ + O2 + OH− | (8) |
| dye + hvb+ → Oxidation | (9) |
| dye + ecb− → Reduction | (10) |
| dye + OH˙ → Degradation | (11) |
| dye + O2− → CO2 + H2O | (12) |
| dye + hvb+ → CO2 + H2O | (13) |
Typically, Konstantinou et al.187 reported the mechanism of N/CDs from Citrus grandis to be both photooxidation and photosensitization.63 Further, Thakur et al.99 observed complete MB dye degradation with proton NMR, which exhibited a chemical shift of 6 aliphatic and 6 aromatic rings. Aji et al.189 inferred the degradation of MB dye with CDs as a proportionality relation between the dye concentration and diffusion current density and concluded that it follows a diffusion mechanism. As mentioned earlier, Omer et al.64 studied the up-conversion photoluminescence by ZnO/CDs derived from instant coffee attributing the explanation to charge separation.190,191 Along with the up-conversion photoluminescence property of CDs, SPR effect of metals such as Ag induces local electric field enhancement.56,192
The above explanation is to be noted that conducting a trapping experiment offers a more precise idea to elucidate the mechanism. As there is a need to know the active species responsible for efficient photocatalytic dye degradation to interpret the mechanism, it is necessary to figure it out with the aid of radical scavengers. Further flash radiolysis can indicate trapped charged carriers that have not been done in this work. It is highly convenient to carry out photocatalytic dye degradation in air, water, respective photocatalyst, and contaminant.131 The deposition of dopants on the CDs can be observed with the help of XPS and differential thermal analysis (DTA) to get a clearer picture of the mechanism.
8. Reusability
Focussing on the practical applications, the stability and reusability of CDs along with dopants when employed in photocatalytic dye degradation is vibrant in treating wastewater. Several studies stand proof that CDs derived from green precursors have promising catalytic performance even after several successive cycles. For instance, N/CDs exhibited excellent photocatalytic activity of about 80% dye degradation even up to 10 processes Fig. 10(a) because N/CQDs undergo protonation and deprotonation due to surface charge modifications.67,195 Anand et al.55 reported the utility and stability of red-emitting Mg–N/CDs by carrying out a recycling study that disclosed a decline in photocatalytic efficiency by 38% after four cycles. Typically, our work reported the recycling study after centrifugation that exhibited efficient photocatalytic activity up to 3 cycles, after which there is a decline in the activity due to the loss of catalyst material during centrifugation.74 With the same process, WO3/CDs exhibited a negligible change in the degradation up to 4 cycles suggesting the low degree of intermediate product deposition on the surface of the WO3/CDs.71 The same reason of material loss was credited for the decline of photocatalytic dye degradation with TiO2/CDs, yet displaying excellent performance until 3 cycles.66 CDs and Ag on g-C3N4 showed excellent catalytic activity after 4 cycles where the catalyst is separated, washed, and dried after every run over 100 min.56 From the above works, we can conclude that the reusability of the catalyst is based upon the interaction between the catalyst and the dye molecules in each cycle. The process in which the catalyst is separated after each turn of the degradation process also accounts for higher reusability. The dopant plays a significant role in the interaction between the corresponding catalyst and dye, thereby modifying the utility and reusability carried out with the help of recycling reactions under the same experimental conditions. The centrifugation recovery has a drawback, such as the complication of installing a centrifuge with a huge capacity. The usage of the electromagnetic system for magnetic recovery can be utilized to detect the efficiency, but they are still large systems to be installed. In recent works, catalytic membranes such as Mn3O4/g-C3N4@PTFE have been utilized where there is no need to recover catalyst since it can show efficient photocatalytic activity up to 5 cycles.196 Therefore, catalytic membranes suitable for CDs can be fabricated for better results for the reusability of catalysts.9. Stability
The stability of CDs catalysts in photocatalytic dye degradation is confirmed with a series of characterization, and the results are compared with the first set of degradations whose experimental setup are the same. These investigations help to understand the modification of physicochemical properties that includes structural and chemical composition of the photocatalyst before and after recycling. The characterizations include Scanning electron microscopy (SEM), Transmission electron microscopy (TEM), Fourier transforms infrared spectroscopy (FTIR), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS). Atchudan et al.66 reported XRD Fig. 9(b), FESEM Fig. 9(c), and TEM Fig. 9(d) image of TiO2, N/CDs photocatalyst after one stand degradation under irradiation light and also carried out the same characterization after 3 cycles Fig. 9(a). The results indicate no change in the structure or the chemical composition and conclude it to be suitable for industrial applications. Dadigala et al.56 carried out XRD and FT-IR images of CDs/Ag on g-C3N4 before and after 4 cycles and proved the outstanding stability of the photocatalyst even after several cycles. Apart from this, fluorescence (FL) spectroscopy and UV light irradiation photograph analysis was carried out with N/CDs at the instance of preparation and after 6 months. The excitation wavelength was observed at 340 nm before and after 6 months, which explained the fluorescent stability of N/CDs. The bright blue fluorescence under UV illumination still exists after a prolonged period of 6 months.168 These results exposed the excellent stability in detail. Rani et al.67 revealed the fascinating stability of N/CDs even after 8 months of storage and 10 cycles of reusability with the constant fluorescence emission. The reason was credited to rich oxygenated functional groups present in the catalyst that can increase the hydrophilicity and dispersibility of the respective photocatalyst. The stability also relies on the photocatalyst's zeta potential in an aqueous medium that consists of stable water suspension.197 The colloidal form of the N/CDs, even after 8 months, is probably due to the covalent bonds between nitrogen and CDs surface, which is attributed to exceptional stability. A relaxation phenomenon of photocatalyst is necessary for the polar medium that contributes to good stability even after long-term storage of the photocatalyst.198 The high stability of the photocatalyst subsidizes outstanding electronic conductivity due to e− donor-receptor properties. All these are evaluated based on dopants chosen to be embedded on the CDs. Further, the relative chemical stability of the CDs can be figured out by carrying out EPR spectra that boons as a gap.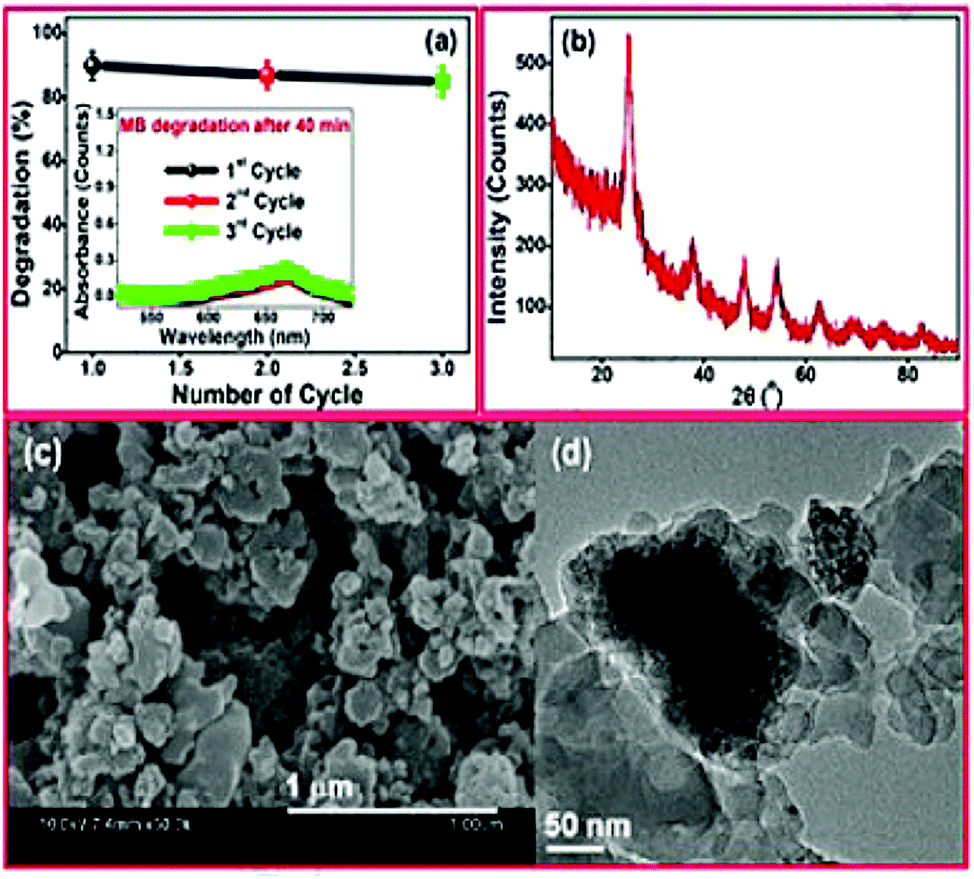 | ||
| Fig. 9 (a) Reusability study of TiO2/CDs showing efficient photocatalytic activity for 3 cycles; stability study by employing characterization such as (b) XRD spectrum; (c) FESEM and (d) TEM; reproduced with permission from ref. 66 Copyright 2018 Elsevier. | ||
 | ||
| Fig. 10 (a) Reusability study of MB dye by photocatalytic activity up to 80% until 10 cycles; reproduced with permission from ref. 67 Copyright 2021 Elsevier. (b) In vitro cytotoxicity valuation of CDs using MTT assay on fibroblast cells; reproduced with permission from ref. 182 Copyright 2018 Nature. | ||
10. Toxicity
Apart from the reusability and stability that determines the practical application, another important feature is the cytotoxicity of CDs and its corresponding catalyst, which further kindles the practical in vitro and in vivo applications. Generally, the cytotoxicity of CDs is highly dependent on its structural and morphological aspects along with chemical composition and physicochemical properties that include dispersion, surface status, and hydrophilicity.199,200 CDs hold good for materials possessing low toxicity and excellent biocompatibility that encounter the necessities to explore practical applications. The cytotoxicity activity of a few CDs derived from green precursors is tabulated for improved view in Table 3. For instance, Atchudan et al.168 reported N/CDs owing to non-toxicity and good biocompatibility by measuring the cell viability of Clone 9 hepatocytes to be greater than 97% by incubating N/CDs with a maximum concentration of 100 μg mL−1via MTT assay. The cellular images before and after treatment with the cell are observed by confocal laser scanning microscopy and considered appropriate material for multicolour cell imaging and biological fields.201,202 Kavitha et al.182 found the toxicity of mesoporous CDs insignificant even after 5 days and observed cell viability higher than 95% until 100 mg L−1 of CDs with fibroblast cells Fig. 10(b). This showed promising application in bioimaging for both in vitro and in vivo studies and drug delivery due to the mesoporous structure of CDs. Typically, the cell viability of HepG2 was observed to be greater than 90%, with the concentration of CDs being higher up to 1000 μg mL−1, and its availability was determined by CLSM images where blue emissions were detected in the intracellular region. This showed that CDs could mark both cell membrane and cytoplasm of the respective cell and remained a probe for respectable optical imaging apart from its biocompatibility.59 Arul et al.43 determined the plausible mechanism of cytotoxicity activity of N/CDs of L-929 cells to be passive diffusion and the anticancer activity of MCF-7 cells to be receptor-mediated endocytosis mechanism whose cell viability was less than 80% and 90%, respectively.203 Although CDs stand excellent for non-toxicity and biocompatibility and are determined by MTT assay, the later part to stop the growth of cells by resolubilizing to formazan and measuring it with spectrophotometer can be carried out.204 Further, the influence of dopants on toxicity and biocompatibility activity can be detected by experimental works.| Precursors | Doping | Conc. range | Cell | Cell viability (%) | Ref. |
|---|---|---|---|---|---|
| Actinidia deliciosa | N/CDs | 0–25 μL | L-929 | <90 | 43 |
| MCF-7 | <80 | ||||
| Citrus limetta waste | CDs | 0.01–1.0 mg mL−1 | U373 | — | 117 |
| U87MG | |||||
| Lychee seed | CDS | 0–1000 μg mL−1 | HepG2 | >90 | 59 |
| Date palm fronds | CDs | 0–100 mg L−1 | Fibroblast | >95 | 182 |
| Peach fruit juice | N/CDs | 0–100 μg mL−1 | Clone 9 | >97 | 168 |
| Hepatocytes | |||||
| Peach fruit | N/CDs | 10 μg mL−1 | C. albicans | — | 66 |
11. Conclusion and future perspective
This review mainly covered CDs as photocatalysts derived from renewable green sources doped with metal/metal oxide and heteroatoms. Nowadays, it is conceivable to modify catalysts to precise applications, and there is the possibility to enhance photocatalytic activity and improve its performance. The eco-friendly nature of this method makes the CDs a promising applicant for photocatalytic degradation. However, this method has mainly been studied in the research laboratory and not significantly employed in industries. Researchers have estimated the preparation of CDs derived from green precursors using advanced approaches that deliver better catalytic performances. Herein, we discussed the three main steps for effective photocatalytic reactions: light-harvesting, charge transfer dynamics, and surface catalytic reaction. Later, the factors influencing the catalytic reaction were examined for CDs derived from green precursors to estimate the optimum condition of pH, concentration of dopants or CDs, loading of photocatalyst, dye concentration, light intensity, irradiation time, and temperature. Next, we highlighted the role of radical scavengers in increasing the efficiency of the photocatalytic activity, which in turn gave a clear picture of the mechanism. Further, the practical application of CDs as a photocatalyst to degrade dyes was evaluated in terms of reusability, stability, and toxicity studies. Although there is a better understanding of CDs' working as photocatalysts in the degradation of dyes, there are few research gaps on every aspect discussed in this review. These problems can be resolved with solutions. A few of them are discussed in comparison with previous works that consist of various other materials as photocatalysts. Below is the explanation of every objective in this review, truths about particular misconceptions in every objective, and possible solutions to overcome the problems.(i) The three main criteria that set a foundation for potential roles of photocatalytic activities are light-harvesting, charge transfer dynamics, and surface catalytic reaction, which is discussed in detail. In a nutshell, the efficiency of these three principles can be increased by choosing and modifying the suitable material as dopants on CDs. Further, the workings of the catalytic reaction can be viewed and understood by carrying out more investigations that are relevant to these three terms.
(ii) When approaching the factors influencing the photocatalytic dye degradation, it is imperative to set the boundary with optimum pH, the concentration of dopants/CDs, photocatalyst and dye molecules, light intensity, irradiation time, and temperature. Unfortunately, although a handful of research works satisfy the photocatalytic activity by setting the boundaries, no work identifies the catalytic system's optimum temperature.
(iii) Identifying the optimum concentration of both dopants and CDs exhibiting excellent catalytic performance rather than identifying the optimum level of dopants alone.
(iv) Further, there is a misconception between dopants/CDs and the photocatalyst effect. The effect of dopants/CDs is the concentration in individuals, whereas the effect of photocatalyst loading is the concentration of the whole composite to carry out the catalytic activity. Choosing the optimum level of both in a sequence further kindles the efficiency of the photocatalytic activity.
(v) Radical scavengers are employed to identify the active species responsible for effective catalytic performance. There are possibilities for low selectivity of certain agents during this action, and other active radical species forms formed. Therefore, the usage of radical scavengers must be sensibly evaluated. It is significant to relate the degradation percentage with the influence of radicals in the degradation of dye molecules using scavengers. Results that rely on this method alone open the way to create misunderstanding when scrutinizing the reaction mechanism.
(vi) So far, the general catalytic mechanism has been explained in CDs derived from green precursors. To better understand the mechanism, the trapped charged carriers should be identified by employing flash radiolysis. Further, a comparison of investigations before and after the photocatalytic dye degradation is highly encouraged to understand the mechanism.
(vii) Fabrication of catalytic membranes suitable for CDs can be employed for better reusability studies results since other methods have drawbacks. The nature of the dopants is responsible for efficient catalytic activity even when stored for a long period. Therefore, high importance should be given to choosing the dopants for excellent stability.
(viii) Evaluation of cytotoxicity on various cells can be employed to determine the same viability. This can be extended to in vivo applications in research laboratories for identifying its practical applications.
(ix) Apart from the objectives set in this review, the most common issue identified is applying the as-prepared CDs photocatalysts in the degradation of dyes from industrial effluents. Therefore, this work must be extended to industrial applications outside the laboratory.
Therefore, by examining CDs derived from green sources as an excellent tool for photocatalytic dye degradation, it can be further nurtured by tuning the material into a better one by filling the gaps. This opens the path for environmentally friendly and non-toxic outcomes.
Abbreviations
| CDs | Carbon nanodots |
| CQDs | Carbon quantum dots |
| CNF | Carbon nanofiber |
| CNT | Carbon nanotube |
| HC | Hydrothermal carbonization |
| RhB | Rhodamine-B |
| C | Carbonization |
| CV | Crystal violet |
| MW | Microwave |
| MB | Methylene blue |
| HT | Hydrothermal |
| MO | Methyl orange |
| P | Pyrolysis |
| SR | Solar radiation |
| BM | Ball milling |
| MG | Malachite green |
| US | Ultrasonication |
| CR | Congo red |
| AB | Acid blue |
| AR | Acid red |
| EY | Eosin Y |
| EBT | Eriochrome black T |
| AC | Autoclave |
| NBB | Naphthol blue black |
| M | Methyl |
| MR | Methyl red |
| FG | Fast green |
| Vis | Visible light |
| AO | Ammonium oxalate |
| AA | Ascorbic acid |
| EDTA | Ethylenediaminetetraacetic acid |
| t-BA | Tertiary butyl alcohol |
| p-BZQ | para-Benzoquinone |
| IPA | Iso propyl alcohol |
| SPR | Surface plasmon resonance |
| ROS | Reactive oxygen species |
Author contributions
Nivetha Basavaraj – data curation, formal analysis, writing – original draft writing – review & editing, Anithadevi Sekar – conceptualization, supervision writing – review & editing, Rakhi Yadav – supervision, validation, writing – review & editing.Funding
No funds, grants, or other support were received.Conflicts of interest
There is no conflicts to declare.Acknowledgements
The authors acknowledge the Principal Dr P. Wilson and Dr E. Iyyappan, Head of Chemistry department, Madras Christian College for their encouragement and support during this work.References
- R. Khan, V. Patel and Z. Khan, Abatement Environ. Pollut., 2020, 107–125 CrossRef.
- M.-A. Armour, J. Chem. Educ., 1988, 65(3), A64 CrossRef CAS.
- M. Magdy and A. El-Salam, J. Environ. Manage., 2010, 91, 618–629 CrossRef PubMed.
- S. Thakur and M. S. Chauhan, Water Qual. Manage., 2018, 117–129 Search PubMed.
- B. Lellis, C. Z. Fávaro-Polonio, J. A. Pamphile and J. C. Polonio, Biotechnol. Res. Innovation, 2019, 3, 275–290 CrossRef.
- A. Yamakata, T. A. Ishibashi and H. Onishi, J. Phys. Chem. B, 2001, 05, 7258–7262 CrossRef.
- A. Di Mauro, M. Cantarella, G. Nicotra, G. Pellegrino, A. Gulino, M. V. Brundo, V. Privitera and G. Impellizzeri, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- P. Singh, P. Shandilya, P. Raizada, A. Sudhaik, A. Rahmani-Sani and A. Hosseini-Bandegharaei, Arabian J. Chem., 2020, 13, 3498–3520 CrossRef CAS.
- D. Maiti, X. Tong, X. Mou and K. Yang, Front. Pharmacol., 2019, 9, 1401 CrossRef PubMed.
- S. Wang, J. Lin, T. Wang, X. Chen and P. Huang, Theranostics, 2016, 6, 2394 CrossRef CAS PubMed.
- M. S. Mauter and M. Elimelech, Environ. Sci. Technol., 2008, 42, 5843–5859 CrossRef CAS PubMed.
- M. Sharon and M. Sharon, Def. Sci. J., 2008, 58, 460 CrossRef CAS.
- L. Ai, Y. Yang, B. Wang, J. Chang, Z. Tang, B. Yang and S. Lu, Sci. Bull., 2021, 66, 839–856 CrossRef CAS.
- W. Li, Y. Liu, B. Wang, H. Song, Z. Liu, S. Lu and B. Yang, Chin. Chem. Lett., 2019, 30, 2323–2327 CrossRef CAS.
- S. Chen, Y.-L. Yu and J.-H. Wang, Anal. Chim. Acta, 2018, 999, 13–26 CrossRef CAS PubMed.
- Y. Zheng, D. Zhang, S. N. A. Shah, H. Li and J.-M. Lin, Chem. Commun., 2017, 53, 5657–5660 RSC.
- M. L. Liu, B. B. Chen, C. M. Li and C. Z. Huang, Green Chem., 2019, 21, 449–471 RSC.
- Y. Wang, Y. Zhu, S. Yu and C. Jiang, RSC Adv., 2017, 7, 40973–40989 RSC.
- X. Sun and Y. Lei, TrAC, Trends Anal. Chem., 2017, 89, 163–180 CrossRef CAS.
- H. N. Abdelhamid, Z. Y. Chen and H. F. Wu, Anal. Bioanal. Chem., 2017, 409, 4943–4950 CrossRef CAS PubMed.
- X. Yang, L. Sui, B. Wang, Y. Zhang, Z. Tang, B. Yang and S. Lu, Sci. China Chem., 2021, 64, 1547–1553 CrossRef CAS.
- Z. Y. Chen, H. N. Abdelhamid and H. F. Wu, Rapid Commun. Mass Spectrom., 2016, 30, 1403–1412 CrossRef CAS PubMed.
- H. N. Abdelhamid and H.-F. Wu, RSC Adv., 2015, 5, 76107–76115 RSC.
- H. N. Abdelhamid and H.-F. Wu, RSC Adv., 2015, 5, 50494–50504 RSC.
- H. N. Abdelhamid and H.-F. Wu, RSC Adv., 2014, 4, 53768–53776 RSC.
- W. Wang, C. Damm, J. Walter, T. J. Nacken and W. Peukert, Phys. Chem. Chem. Phys., 2016, 18, 466–475 RSC.
- S. Lu, L. Sui, J. Liu, S. Zhu, A. Chen, M. Jin and B. Yang, Adv. Mater., 2017, 29, 1603443 CrossRef PubMed.
- Y. Liu, Y. Liu, S.-J. Park, Y. Zhang, T. Kim, S. Chae, M. Park and H.-Y. Kim, J. Mater. Chem. A, 2015, 3, 17747–17754 RSC.
- T. Fiuza, G. Gomide, A. F. C. Campos, F. Messina and J. Depeyrot, C, 2019, 5, 74 CAS.
- M. Perikala and A. Bhardwaj, ACS Omega, 2019, 4, 21223–21229 CrossRef CAS PubMed.
- P. Zuo, X. Lu, Z. Sun, Y. Guo and H. He, Microchim. Acta, 2016, 183, 519–542 CrossRef CAS.
- S. A. Rub Pakkath, S. S. Chetty, P. Selvarasu, A. Vadivel Murugan, Y. Kumar, L. Periyasamy, M. Santhakumar, S. R. Sadras and K. Santhakumar, ACS Biomater. Sci. Eng., 2018, 4, 2581–2596 Search PubMed.
- J. Jana, J. S. Chung and S. H. Hur, ACS Omega, 2019, 4, 17031–17038 CrossRef CAS PubMed.
- A. B. Bourlinos, G. Trivizas, M. A. Karakassides, M. Baikousi, A. Kouloumpis, D. Gournis, A. Bakandritsos, K. Hola, O. Kozak, R. Zboril, I. Papagiannouli, P. Aloukos and S. Couris, Carbon, 2015, 83, 173–179 CrossRef CAS.
- M. Xiong, Q. Rong, H. Min Meng and X. Bing Zhang, Biosens. Bioelectron., 2017, 89, 212–223 CrossRef CAS PubMed.
- F. Yan, D. Kong, Y. Fu, Q. Ye, Y. Wang and L. Chen, J. Colloid Interface Sci., 2016, 466, 268–274 CrossRef CAS PubMed.
- C. Wang, K. Yang, X. Wei, S. Ding, F. Tian and F. Li, Ceram. Int., 2018, 44, 22481–22488 CrossRef CAS.
- Z. Zhang, B. Hu, Q. Zhuang, Y. Wang, X. Luo, Y. Xie and D. Zhou, Anal. Lett., 2020, 53, 1704–1718 CrossRef CAS.
- N. K. Sahoo, G. C. Jana, M. N. Aktara, S. Das, S. Nayim, A. Patra, P. Bhattacharjee, K. Bhadra and M. Hossain, Mater. Sci. Eng., C, 2020, 108, 110429 CrossRef CAS PubMed.
- Q. Huang, Q. Li, Y. Chen, L. Tong, X. Lin, J. Zhu and Q. Tong, Sens. Actuators, B, 2018, 276, 82–88 CrossRef CAS.
- R. Purbia and S. Paria, Biosens. Bioelectron., 2016, 79, 467–475 CrossRef CAS PubMed.
- N. Pourreza and M. Ghomi, Mater. Sci. Eng., C, 2019, 98, 887–896 CrossRef CAS PubMed.
- V. Arul and M. G. Sethuraman, Opt. Mater., 2018, 78, 181–190 CrossRef CAS.
- R. Duarah and N. Karak, ACS Sustainable Chem. Eng., 2017, 5, 9454–9466 CrossRef CAS.
- R. Aggarwal, D. Saini, B. Singh, J. Kaushik, A. K. Garg and S. K. Sonkar, Sol. Energy, 2020, 197, 326–331 CrossRef CAS.
- M. Sabet and K. Mahdavi, Appl. Surf. Sci., 2019, 463, 283–291 CrossRef CAS.
- S. Ahmadian-Fard-Fini, M. Salavati-Niasari and H. Safardoust-Hojaghan, J. Mater. Sci.: Mater. Electron., 2017, 28, 16205–16214 CrossRef CAS.
- M. Pirsaheb, A. Asadi, M. Sillanpää and N. Farhadian, J. Mol. Liq., 2018, 271, 857–871 CrossRef CAS.
- K.-W. Chu, S. L. Lee, C.-J. Chang and L. Liu, Polymers, 2019, 11, 689 CrossRef CAS PubMed.
- N. Tejwan, S. K. Saha and J. Das, Adv. Colloid Interface Sci., 2020, 275, 102046 CrossRef CAS PubMed.
- A. Mehta, A. Mishra, S. Basu, N. P. Shetti, K. R. Reddy, T. A. Saleh and T. M. Aminabhavi, J. Environ. Manage., 2019, 250, 109486 CrossRef CAS PubMed.
- M. Han, S. Zhu, S. Lu, Y. Song, T. Feng, S. Tao, J. Liu and B. Yang, Nano Today, 2018, 19, 201–218 CrossRef CAS.
- Z. Zhang, G. Yi, P. Li, X. Zhang, H. Fan, Y. Zhang, X. Wang and C. Zhang, Nanoscale, 2020, 12, 13899–13906 RSC.
- C. Sakdaronnarong, A. Sangjan, S. Boonsith, D. C. Kim and H. S. Shin, Catalysts, 2020, 10, 320 CrossRef CAS.
- S. R. Anand, G. Kumar and A. K. Garg, ACS Publ., 2018, 6, 9246–9256 Search PubMed.
- R. Dadigala, R. K. Bandi, B. R. Gangapuram and V. Guttena, J. Photochem. Photobiol., A, 2017, 342, 42–52 CrossRef CAS.
- Q. Xu, P. Pu, J. Zhao, C. Dong, C. Gao, Y. Chen, J. Chen, Y. Liu and H. Zhou, J. Mater. Chem. A, 2015, 3, 542–546 RSC.
- G. S. Das, J. P. Shim, A. Bhatnagar, K. M. Tripathi and T. Y. Kim, Sci. Rep., 2019, 9, 1–9 Search PubMed.
- M. Xue, M. Zou, J. Zhao, Z. Zhan and S. Zhao, J. Mater. Chem. B, 2015, 3, 6783–6789 RSC.
- A. Prasannan and T. Imae, Ind. Eng. Chem. Res., 2013, 52, 15673–15678 CrossRef CAS.
- A. Thakur, P. Kumar, D. Kaur, N. Devunuri, R. K. Sinha and P. Devi, RSC Adv., 2020, 10, 8941–8948 RSC.
- Z. W. Heng, Y. Y. Tan, W. C. Chong, E. Mahmoudi, A. W. Mohammad, H. C. Teoh, L. C. Sim and C. H. Koo, J. Water Process Eng., 2021, 40, 101805 CrossRef.
- V. Ramar, S. Moothattu and K. Balasubramanian, Sol. Energy, 2018, 169, 120–127 CrossRef CAS.
- K. M. Omer, N. N. Mohammad and S. O. Baban, Catal. Lett., 2018, 148, 2746–2755 CrossRef CAS.
- Z. Zhu, P. Yang, X. Li, M. Luo, W. Zhang, M. Chen and X. Zhou, Spectrochim. Acta, Part A, 2020, 227, 117659 CrossRef CAS PubMed.
- R. Atchudan, T. N. J. I. Edison, S. Perumal, R. Vinodh and Y. R. Lee, J. Alloys Compd., 2018, 766, 12–24 CrossRef CAS.
- U. A. Rani, L. Y. Ng, C. Y. Ng, E. Mahmoudi, Y. S. Ng and A. W. Mohammad, J. Water Process Eng., 2021, 40, 101816 CrossRef.
- H. Shahba and M. Sabet, J. Fluoresc., 2020, 30, 927–938 CrossRef CAS PubMed.
- S. Ahmadian-Fard-Fini, M. Salavati-Niasari and H. Safardoust-Hojaghan, J. Mater. Sci.: Mater. Electron., 2017, 28, 16205–16214 CrossRef CAS.
- R. Choudhary, S. Koppala and S. Swamiappan, J. Asian Ceram. Soc., 2015, 3, 173–177 CrossRef.
- S. P. Smrithi, N. Kottam, V. Arpitha, A. Narula, G. N. Anilkumar and K. R. V. Subramanian, J. Sci.: Adv. Mater. Devices, 2020, 5, 73–83 Search PubMed.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Simple one-step synthesis of highly luminescent carbon dots from orange juice: application as excellent bio-imaging agents, Chem. Commun., 2012, 48, 8835–8837 RSC.
- A. Pal, G. Natu, K. Ahmad and A. Chattopadhyay, J. Mater. Chem. A, 2018, 6, 4111–4118 RSC.
- S. Anithadevi and R. Yadav, Optik, 2021, 167311 Search PubMed.
- B. Wang, H. Xin, X. Li, J. Cheng, G. Yang and F. Nie, Sci. Rep., 2014, 4, 1–7 Search PubMed.
- R. Atchudan, A. Pandurangan and K. Subramanian, J. Porous Mater., 2012, 19, 797–805 CrossRef CAS.
- B. B. Campos, R. Contreras-Cáceres, T. J. Bandosz, J. Jiménez-Jiménez, E. Rodríguez-Castellón, J. C. G. Esteves da Silva and M. Algarra, Carbon, 2016, 106, 171–178 CrossRef CAS.
- R. Atchudan, T. N. J. I. Edison, D. Chakradhar, S. Perumal, J.-J. Shim and Y. R. Lee, Sens. Actuators, B, 2017, 246, 497–509 CrossRef CAS.
- Y. J. Li, S. G. Harroun, Y. C. Su, C. F. Huang, B. Unnikrishnan, H. J. Lin, C. H. Lin and C. C. Huang, Adv. Healthcare Mater., 2016, 5, 2545–2554 CrossRef CAS PubMed.
- G. Zorn, L. H. Liu, L. Árnadóttir, H. Wang, L. J. Gamble, D. G. Castner and M. Yan, J. Phys. Chem. C, 2014, 118, 376–383 CrossRef CAS PubMed.
- T. N. J. I. Edison, R. Atchudan, J.-J. Shim, S. Kalimuthu, B.-C. Ahn and Y. R. Lee, J. Photochem. Photobiol., B, 2016, 158, 235–242 CrossRef CAS PubMed.
- T. N. J. I. Edison, R. Atchudan, M. G. Sethuraman, J. J. Shim and Y. R. Lee, J. Photochem. Photobiol., B, 2016, 161, 154–161 CrossRef CAS PubMed.
- A. Sekar, R. Yadav and N. Basavaraj, New J. Chem., 2021, 45, 2326–2360 RSC.
- H. Bozetine, Q. Wang, A. Barras, M. Li, T. Hadjersi, S. Szunerits and R. Boukherroub, J. Colloid Interface Sci., 2016, 465, 286–294 CrossRef CAS PubMed.
- M. Ahmad, E. Ahmed, Z. L. Hong, J. F. Xu, N. R. Khalid, A. Elhissi and W. Ahmed, Appl. Surf. Sci., 2013, 274, 273–281 CrossRef CAS.
- M. M. Hossain, B.-C. Ku and J. R. Hahn, Appl. Surf. Sci., 2015, 354, 55–65 CrossRef CAS.
- C. Gao, J. Low, R. Long, T. Kong, J. Zhu and Y. Xiong, Chem. Rev., 2020, 120, 12175–12216 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- X. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS PubMed.
- Y. Li, Z. Wang, T. Xia, H. Ju, K. Zhang, R. Long, Q. Xu, C. Wang, L. Song, J. Zhu, J. Jiang and Y. Xiong, Adv. Mater., 2016, 28, 6959–6965 CrossRef CAS PubMed.
- C. Gao, J. Wang, H. Xu and Y. Xiong, Chem. Soc. Rev., 2017, 46, 2799–2823 RSC.
- Y. Nakabayashi, M. Nishikawa and Y. Nosaka, Electrochim. Acta, 2014, 125, 191–198 CrossRef CAS.
- T. Baran, S. Wojtyła, A. Dibenedetto, M. Aresta and W. Macyk, ChemSusChem, 2016, 9, 2933–2938 CrossRef CAS PubMed.
- S. Saha, G. Das, J. Thote and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 14845–14851 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, J. Phys. Chem. Lett., 2016, 7, 431–434 CrossRef CAS PubMed.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li and H. Zhao, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS PubMed.
- T. Tong, B. Zhu, C. Jiang, B. Cheng and J. Yu, Appl. Surf. Sci., 2018, 433, 1175–1183 CrossRef CAS.
- U. R. R. P. Remli and A. A. Aziz, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 736, 042038 CAS.
- A. Thakur, P. Kumar, D. Kaur, N. Devunuri, R. K. Sinha and P. Devi, RSC Adv., 2020, 10, 8941–8948 RSC.
- R. M. Mathew, J. John, E. S. Zachariah, J. Jose, T. Titus, R. Abraham, A. Joseph and V. Thomas, React. Kinet., Mech. Catal., 2020, 129, 1131–1143 CrossRef CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- Y. Okada and K. Chiba, Chem. Rev., 2018, 118, 4592–4630 CrossRef CAS PubMed.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- A. Habibi-Yangjeh and A. Akhundi, J. Mol. Catal. A: Chem., 2016, 415, 122–130 CrossRef CAS.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- G. Zhang, H. Miao, X. Hu, J. Mu, X. Liu, T. Han, J. Fan, E. Liu, Y. Yin and J. Wan, Appl. Surf. Sci., 2017, 391, 345–352 CrossRef CAS.
- H. Liu, Z. Chen and Y. Wang, J. Photochem. Photobiol., A, 2016, 326, 30–40 CrossRef CAS.
- S. Cao, J. Jiang, B. Zhu and J. Yu, Phys. Chem. Chem. Phys., 2016, 18, 19457–19463 RSC.
- Y. Qu, X. Xu, R. Huang, W. Qi, R. Su and Z. He, Chem. Eng. J., 2020, 382, 123016 CrossRef CAS.
- A. Tyagi, K. M. Tripathi, N. Singh, S. Choudhary and R. K. Gupta, RSC Adv., 2016, 6, 72423–72432 RSC.
- S. Hu, Chem. Rec., 2016, 16, 219–230 CrossRef CAS PubMed.
- S. Hu, W. Zhang, Q. Chang, J. Yang and K. Lin, Carbon, 2016, 103, 391–393 CrossRef CAS.
- S. Hu, Z. Wei, Q. Chang, A. Trinchi and J. Yang, Appl. Surf. Sci., 2016, 378, 402–407 CrossRef CAS.
- H. Huang, J. Zhang, L. Jiang and Z. Zang, J. Alloys Compd., 2017, 718, 112–115 CrossRef CAS.
- S. L. D'souza, B. Deshmukh, J. R. Bhamore, K. A. Rawat, N. Lenka and S. K. Kailasa, RSC Adv., 2016, 6, 12169–12179 RSC.
- K. Mallick, M. Witcomb and M. Scurrell, Mater. Chem. Phys., 2006, 97, 283–287 CrossRef CAS.
- A. Thakur, P. Devi, S. Saini, R. Jain, R. K. Sinha and P. Kumar, ACS Sustainable Chem. Eng., 2019, 7, 502–512 CrossRef CAS.
- S. Bai, L. Wang, Z. Li and Y. Xiong, Adv. Sci., 2017, 4, 1600216 CrossRef PubMed.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- C. Zhu, J. Zhai and S. Dong, Chem. Commun., 2012, 48, 9367–9369 RSC.
- N. Dhenadhayalan, K. C. Lin, R. Suresh and P. Ramamurthy, J. Phys. Chem. C, 2016, 120, 1252–1261 CrossRef CAS.
- X. Wu, H. Zhang, J. Dong, M. Qiu, J. Kong, Y. Zhang, Y. Li, G. Xu, J. Zhang and J. Ye, Nano Energy, 2018, 45, 109–117 CrossRef CAS.
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. Chan Lee, E. Jung, W. Je Chang, K. S. Lee, J. H. Kim, S. P. Cho and H. Kim, Nat. Mater., 2019, 18, 620–626 CrossRef CAS PubMed.
- Y. Li, Z. Wang, T. Xia, H. Ju, K. Zhang, R. Long and Q. Xu, et al. , Adv. Mater., 2016, 28, 6959–6965 CrossRef CAS PubMed.
- L. A. Chanu, W. Joychandra Singh, K. Jugeshwar Singh and K. Nomita Devi, Results Phys., 2019, 12, 1230–1237 CrossRef.
- K. M. Lee, C. W. Lai, K. S. Ngai and J. C. Juan, Water Res., 2016, 88, 428–448 CrossRef CAS PubMed.
- R. J. Davis, J. L. Gainer, G. O’Neal and I.-W. Wu, Water Environ. Res., 1994, 66, 50–53 CrossRef CAS.
- S. Mozia, W. M. Antoni, M. Toyoda and M. Inagaki, Desalination, 2009, 241, 97–105 CrossRef CAS.
- Z. Shourong, H. Qingguo, Z. Jun and W. Bingkun, J. Photochem. Photobiol., A, 1997, 108, 235–238 CrossRef.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS.
- Y. Yang, X. Li, C. Zhou, W. Xiong, G. Zeng, D. Huang, C. Zhang, W. Wang, B. Song, X. Tang and X. Li, Water Res., 2020, 116200 CrossRef CAS PubMed.
- C. Lu, Y. Wu, F. Mai, W. Chung, C. Wu, W. Lin and C. Chen, J. Mol. Catal. A: Chem., 2009, 310, 159–165 CrossRef CAS.
- L. Saikia, D. Bhuyan, M. Saikia, B. Malakar, D. K. Dutta and P. Sengupta, Applied Catalysis A: General, 2015, 490, 42–49 CrossRef CAS.
- F. Zhang, J. Chen, X. Zhang, W. Gao, R. Jin, N. Guan and Y. Li, Langmuir, 2004, 20, 9329–9334 CrossRef CAS PubMed.
- M. Qamar and A. K. Ganguli, Bull. Mater. Sci., 2013, 36, 945–951 CrossRef CAS.
- M. Zaib, A. Akhtar, F. Maqsood and T. Shahzadi, Arabian J. Sci. Eng., 2021, 46, 437–446 CrossRef CAS.
- A. Elaziouti and B. Ahmed, J. Chem. Eng. Process Technol., 2011, 2, 1–9 Search PubMed.
- S. Erdemoğlu, S. K. Aksu, F. Sayılkan, B. Izgi, M. Asiltürk, H. Sayılkan, F. Frimmel and Ş. Güçer, J. Hazard. Mater., 2008, 155, 469–476 CrossRef PubMed.
- J. J. Salazar-Rabago, R. Leyva-Ramos, J. Rivera-Utrilla, R. Ocampo-Perez and F. J. Cerino-Cordova, Sustainable Environ. Res., 2017, 27, 32–40 CrossRef CAS.
- K. Li, F.-Y. Su and W.-D. Zhang, Appl. Surf. Sci., 2016, 375, 110–117 CrossRef CAS.
- L. Ge, C. Han, J. Liu and Y. Li, Appl. Catal., A, 2011, 409, 215–222 CrossRef.
- N. Daneshvar, D. Salari and A. R. Khataee, J. Photochem. Photobiol., A, 2003, 157, 111–116 CrossRef CAS.
- A. Fadllan, P. Marwoto, M. P. Aji, Susanto and R. S. Iswari, AIP Conf. Proc., 1788, 2017, 30003 Search PubMed.
- N. Soltani, E. Saion, M. Z. Hussein, M. Erfani, A. Abedini, G. Bahmanrokh, M. Navasery and P. Vaziri, Int. J. Mol. Sci., 2012, 13, 12242–12258 CrossRef CAS PubMed.
- A. Franco, M. C. Neves, M. M. L. Ribeiro Carrott, M. H. Mendonça, M. I. Pereira and O. C. Monteiro, J. Hazard. Mater., 2009, 161, 545–550 CrossRef CAS PubMed.
- S. Sharma, S. K. Mehta and S. K. Kansal, J. Alloys Compd., 2017, 699, 323–333 CrossRef CAS.
- S. Malato, P. Fernández-Ibáñez, M. I. Maldonado, J. Blanco and W. Gernjak, Catal. Today, 2009, 147, 1–59.
- S. Malato, I. M. Manuel, P. Fernandez-Ibanez, I. Oller, I. Polo and R. Sánchez-Moreno, Mater. Sci. Semicond. Process., 2016, 42, 15–23 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- H. Chun, W. Yizhong and T. Hongxiao, Chemosphere, 2000, 41, 1205–1209 CrossRef CAS PubMed.
- C. M. Sharma, L. J. Barthakur and G. K. Gogoi, Int. Educ. Res. J., 2019, 5, 5–7 Search PubMed.
- C. P. Vun, A. W. Mohammad, T. Y. Haan and E. Mahmoudi, J. Teknol., 2017, 79 Search PubMed.
- K. M. Reza, A. Kurny and F. Gulshan, Appl. Water Sci., 2017, 7, 1569–1578 CrossRef CAS.
- A. Kumar and G. Pandey, Mater. Sci. Eng. Int. J., 2017, 1, 1–10 Search PubMed.
- K. Azad and P. Gajanan, Chem. Sci. J., 2017, 8, 164 Search PubMed.
- P. Jantawasu, T. Sreethawong and S. Chavadej, Chem. Eng. J., 2009, 155, 223–233 CrossRef CAS.
- R. A. Prates, A. M. Yamada Jr, L. C. Suzuki, M. C. E. Hashimoto, S. Cai, S. Gouw-Soares, L. Gomes and M. S. Ribeiro, J. Photochem. Photobiol., B, 2007, 86, 70–76 CrossRef CAS PubMed.
- M. A. Rauf, M. A. Meetani and S. Hisaindee, Desalination, 2011, 276, 13–27 CrossRef CAS.
- U. G. Akpan and H. H. Bassim, J. Hazard. Mater., 2009, 170, 520–529 CrossRef CAS PubMed.
- D. Curcó, J. Giménez, A. Addardak, S. Cervera-March and S. Esplugas, Catal. Today, 2002, 76, 177–188 CrossRef.
- A. Shafaei, M. Nikazar and M. Arami, Desalination, 2010, 252, 8–16 CrossRef CAS.
- M. Mahalakshmi, B. Arabindoo, M. Palanichamy and V. Murugesan, J. Hazard. Mater., 2007, 143, 240–245 CrossRef CAS PubMed.
- M. A. Behnajady, N. Modirshahla and R. Hamzavi, J. Hazard. Mater., 2006, 133, 226–232 CrossRef CAS PubMed.
- M. Stylidi, I. K. Dimitris and X. E. Verykios, Appl. Catal., B, 2004, 47, 189–201 CrossRef CAS.
- M. Asahi, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef PubMed.
- V. Augugliaro, M. Litter, L. Palmisano and J. Soria, J. Photochem. Photobiol., C, 2006, 7, 127–144 CrossRef CAS.
- R. Atchudan, T. N. J. I. Edison, S. Perumal, N. Karthik, D. Karthikeyan, M. Shanmugam and Y. R. Lee, J. Photochem. Photobiol., A, 2018, 350, 75–85 CrossRef CAS.
- K. Hashimoto, H. Irie and A. Fujishima, Jpn. J. Appl. Phys., Part 1, 2005, 44, 8269–8285 CrossRef CAS.
- J. Peral, X. Domènech and D. F. Ollis, J. Chem. Technol. Biotechnol., 1997, 70, 117–140 CrossRef CAS.
- G. Mamba, M. A. Mamo, X. Y. Mbianda and A. K. Mishra, Ind. Eng. Chem. Res., 2014, 53, 14329–14338 CrossRef CAS.
- H. Liu, Y. Hu, Z. Zhang, X. Liu, H. Jia and B. Xu, Appl. Surf. Sci., 2015, 355, 644–652 CrossRef CAS.
- K-i. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, J. Photochem. Photobiol., A, 2000, 134, 139–142 CrossRef CAS.
- C. S. A. Antunes, M. Bietti, M. Salamone and N. Scione, J. Photochem. Photobiol., A, 2004, 163, 453–462 CrossRef CAS.
- R. Terzian, N. Serpone, R. B. Draper, M. A. Fox and E. Pelizzetti, Langmuir, 1991, 7, 3081–3089 CrossRef CAS.
- Y. Nosaka, S. Komori, K. Yawata, T. Hirakawa and A. Y. Nosaka, Phys. Chem. Chem. Phys., 2003, 5, 4731–4735 RSC.
- S. H. Szczepankiewicz, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. B, 2000, 104, 9842–9850 CrossRef CAS.
- L. Zang, P. Qu, J. Zhao, T. Shen and H. Hidaka, J. Mol. Catal. A: Chem., 1997, 120, 235–245 CrossRef.
- Q. Lu, Y. Zhang and S. Liu, J. Mater. Chem. A, 2015, 3, 8552–8558 RSC.
- M. Zhu, J. Lu, Y. Hu, Y. Liu, S. Hu and C. Zhu, Environ. Sci. Pollut. Res., 2020, 27, 31289–31299 CrossRef CAS PubMed.
- F. Parrino, S. Livraghi, E. Giamello, R. Ceccato and L. Palmisano, ACS Catal., 2020, 10, 7922–7931 CrossRef CAS.
- T. Kavitha and S. Kumar, Sci. Rep., 2018, 8, 1–10 CAS.
- Z. Ma, H. Ming, H. Huang, Y. Liu and Z. Kang, New J. Chem., 2012, 36, 861–864 RSC.
- Y. Zhou, M. Z. Elsayed, B. A. Quiroga, J. Perez, K. J. Mintz, Z. Peng, P. Y. Liyanage, R. R. Pandey, C. C. Chusuei and R. M. Leblanc, Appl. Catal., B, 2019, 248, 157–166 CrossRef CAS PubMed.
- D. S. Monje, K. M. Chacon, I. C. Galindo, C. Castaño, L. M. Ballesteros-Rueda, G. C. Valencia, M. C. Gonzalez and D. F. Mercado, Mater. Today Chem., 2021, 20, 100445 CrossRef CAS.
- E. A. Asl, M. Haghighi and A. Talati, Sol. Energy, 2019, 184, 426–439 CrossRef.
- I. K. Konstantinou and A. A. Triantafyllos, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
- A. Sharma, M. Varshney, J. Park, T.-K. Ha, K.-H. Chae and H.-J. Shin, RSC Adv., 2015, 5, 21762–21771 RSC.
- M. P. Aji, P. A. Wiguna, N. Rosita and S. Aisyah, Am. J. Appl. Sci., 2016, 13, 432–438 CrossRef CAS.
- C. Zhou, C. Lai, P. Xu, G. Zeng, D. Huang, C. Zhang, M. Cheng, L. Hu, J. Wan, Y. Liu, W. Xiong, Y. Deng and M. Wen, ACS Sustainable Chem. Eng., 2018, 6, 4174–4184 CrossRef CAS.
- C. Zhou, C. Lai, P. Xu, G. Zeng, D. Huang, Z. Li, C. Zhang, M. Cheng, L. Hu, J. Wan, F. Chen, W. Xiong and R. Deng, ACS Sustainable Chem. Eng., 2018, 6, 6941–6949 CrossRef CAS.
- Y. Yang, Y. Guo, F. Liu, X. Yuan, Y. Guo, S. Zhang, W. Guo and M. Huo, Appl. Catal., B, 2013, 142–143, 828–837 CrossRef CAS.
- S. Nayak, S. R. Prasad, D. Mandal and P. Das, J. Hazard. Mater., 2020, 392, 122287 CrossRef CAS PubMed.
- R. Mahajan, A. S. Bhadwal, N. Kumar, M. Madhusudanan, R. N. Pudake and R. M. Tripathi, IET Nanobiotechnol., 2017, 11, 360–364 CrossRef PubMed.
- J. Zhu, H. Chu, T. Wang, C. Wang and Y. Wei, Microchem. J., 2020, 158, 105142 CrossRef CAS.
- C. Chen, M. Xie, L. Kong, W. Lu, Z. Feng and J. Zhan, J. Hazard. Mater., 2020, 390, 122146 CrossRef CAS PubMed.
- A. Singh, P. K. Mohapatra, D. Kalyanasundaram and S. Kumar, Mater. Chem. Phys., 2019, 225, 23–27 CrossRef CAS.
- S. Zhao, X. Song, X. Chai, P. Zhao, H. He and Z. Liu, J. Cleaner Prod., 2020, 263, 121561 CrossRef CAS.
- P. Miao, K. Han, Y. Tang, B. Wang, T. Lin and W. Cheng, Nanoscale, 2015, 7, 1586–1595 RSC.
- R. Knoblauch and C. D. Geddes, Materials, 2020, 13, 4004 CrossRef CAS PubMed.
- S. A. A. Vandarkuzhali, V. Jeyalakshmi, G. Sivaraman, S. Singaravadivel, K. R. Krishnamurthy and B. Viswanathan, Sens. Actuators, B, 2017, 252, 894–900 CrossRef CAS.
- V. Borse, M. Thakur, S. Sengupta and R. Srivastava, Sens. Actuators, B, 2017, 248, 481–492 CrossRef CAS.
- N. Gupta, H. P. Singh and R. K. Sharma, J. Mol. Catal. A: Chem., 2011, 335, 248–252 CrossRef CAS.
- P. Kumar, A. Nagarajan and P. D. Uchil, Cold Spring Harbor Protocols, 2018, 6, pdb-prot095505 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |