DOI:
10.1039/D1MA00706H
(Paper)
Mater. Adv., 2021,
2, 6388-6402
Doped and non-doped blue organic light-emitting diodes based on AIEgens with high exciton utilization efficiency and external quantum efficiency†
Received
10th August 2021
, Accepted 24th August 2021
First published on 24th August 2021
Abstract
A novel molecular design strategy has been exploited for blue luminogens, NSPI-DVP and CNSPI-DVP consisting of triphenylethene (TPE) at the C5 and C10 positions of phenanthroimidazole with high-lying charge-transfer (CT) state to harvest dark triplet excitons. These materials show hybridised local and charge transfer (HLCT) states and aggregation-induced emission (AIE) properties. Non-doped OLEDs with NSPI-DVP and CNSPI-DVP show high exciton utilization efficiency (EUE) of 36.00 and 64.00%, power efficiency (PE) of 4.99 and 4.72 lm W−1, external quantum efficiency (EQE) of 3.2 and 5.3% and current efficiency (CE) of 5.61 and 5.03 cd A−1, respectively. The non-doped device with CNSPI-DVP shows excellent performance with negligible efficiency roll-off and high exciton utilization efficiency (EUE) than NSPI-DVP. The doped OLEDs with CBP:x wt% NSPI-DVP (EQE-7.60/8.98%) and CBP:CNSPI-DVP (EQE-8.12/9.81%) show higher efficiencies than that of non-doped devices. The improvement of C5/C10 modified phenanthrimidazole mechanofluorochromic (MFC) materials with AIE behaviour could open a new way to enhance OLEDs performances.
1. Introduction
High-performance blue organic light-emitting diodes (OLEDs) have been attracting attention constantly because of their significant application potential in displays.1,2 Blue emitters lag behind red and green emitters owing to a wide band gap, which results in inefficient carrier injection and unfavourable electrical behavior.3 Anthracene, phenanthrene and pyrene substituted phenanthroimidazole derivatives with planar and rigid structures show pure blue emission because of restricted conformational relaxation in the excited state.2 However, blue luminogens with strong π–π stacking interactions meet aggregation-caused quenching (ACQ), which lowers the device efficiency.3 Aggregation-induced emission (AIE) could be an alternative to ACQ.4–7 Mechanochromic materials exhibit AIE characteristics that show opposite behaviour to ACQ, and may be utilised as emissive materials.8 Highly efficient OLEDs with 100% exciton utilization efficiency (EUE) have been harvested from phosphorescent as well as thermally-activated delayed fluorescent (TADF) luminogens.9 However, blue phosphors suffer from poor stability and high cost, whereas blue TADF materials with donor–acceptor geometry result in poor colour purity due to intramolecular charge transfer (CT).
Luminogens with AIE characteristics inhibit concentration quenching and exciton annihilation and result in intensified emission in the film.10–13 However, the utilization of electrogenerated singlet exciton limits to 25% is a major issue in the EL process. To enhance the singlet exciton population to improve EUE, luminogens with high-lying CT are offered to harvest dark triplet excitons.14 Therefore, it is aimed to construct AIE luminogens having a high-lying CT state and low-lying LE state for pure blue emission via a reverse intersystem crossing (RISC) process.
Phenanthroimidazoles with C2 substitution develop effective conjugation and results in a deep blue emission, whereas N1 substitution does not affect the optical properties significantly because of the perpendicular configuration.15–19 Though C2 and N1 substituted phenanthroimidazoles have been studied widely, C6 and C9 substituted phenanthrimidazole derivatives have not been analysed broadly.20,21 Recently, our research group reported C6/C9, as well as C5/C10, substituted phenanthrimidazole derivatives (D–A) with excellent efficiency and low roll-off efficiency.22–25
In continuation, herein, we have designed two tailor-made blue luminogens, 1-(naphthalen-1-yl)-5,10-bis(4-(2,2-diphenylvinyl)phenyl)-2-styryl-1H-phenanthro[9,10-d]imidazole (NSPI-DVP) and (E)-4-(5,10-bis(4-(2,2-diphenylvinyl)phenyl)-2-styryl-1H-phenanthro[9,10-d]imidazol-1-yl)-1-naphthonitrile (CNSPI-DVP) consisting of AIE active triphenylethene (TPE) unit at C5 and C10 positions of phenanthrimidazole having D–A structures providing moderate conjugation when compared to C2 substitution and also alleviating the ACQ effect. These luminogens having branched naphthyl moiety at azomethine nitrogen restricted the intermolecular interactions.25 Theoretical and photophysical studies reveal that NSPI-DVP and CNSPI-DVP having HLCT behaviour with LE state (low-lying) and CT state (high-lying), enhanced the exciton utilisation efficiency (EUE). The non-doped OLEDs with NSPI-DVP and CNSPI-DVP show high EUE of 36.00 and 64.00%, PE of 4.99 and 4.72 lm W−1, EQE of 3.2 and 5.3% and CE of 5.61 and 5.03 cd A−1, respectively. The doped OLEDs with CBP:x wt% NSPI-DVP (EQE-7.60/8.98%) and CBP:CNSPI-DVP (EQE-8.12/9.81%) show higher efficiencies than those of non-doped devices.
2. Experimental section
The detailed experimental procedure (Scheme S1) and fabrication of devices are discussed in the ESI.†
3. Results and discussion
3.1. Molecular design strategy
For structure–activity analysis, frontier molecular orbital (FMO) investigations of NSPI-DVP and CNSPI-DVP were carried out using the DFT method (Fig. 1 and Table 1). The excited state analysis was performed by a TD-DFT method.26,27 The twisted conformation of NSPI-DVP and CNSPI-DVP show C–H interactions rather than π–π interactions, which leads to minimisation of the tight packing arrangement, hence, these materials show mechanochromism i.e., response to external stimuli (grinding, fuming, annealing, etc.). The twisted conformation reduced the π–π stacking interactions and reduced ACQ in the aggregated form of NSPI-DVP and CNSPI-DVP. The triphenylethylene units coupled with phenanthroimidazole core in planar mode with small torsion angles of θ1-20.2° and θ1′-22.5° (NSPI-DVP) and θ1-14.0° and θ1′-16.8°-(CNSPI-DVP) implying moderate conjugation between triphenylethylene and phenanthroimidazole fragments. Because of the strong steric interaction, the styryl ring at C2 of phenanthrimidazole shows an enlarged dihedral angle from 46.1° (NSPI-DVP) to 60.2° (CNSPI-DVP). Similarly, substituent at azomethine nitrogen shows an enlarged torsion angle from 59.6° (NSPI-DVP) to 76.9° (CNSPI-DVP). The higher energy CT state was formed in CNSPI-DVP relative to NSPI-DVP due to the electron-withdrawing ability of the p-cyano group (Fig. 1). Natural transition orbitals (NTO) of NSPI-DVP reveal that hole of S1 and S2 states distributed on phenanthrimidazole core and particle of S1 and S2 states localised on triphenylethylene unit, which are the unique characteristics of LE states. In CNSPI-DVP, the hole of S1 state distributed on phenanthrimidazole core and particle of S1 state localised on triphenylethylene unit, whereas the particle of S2 state located on styryl substituent, confirming CT dominant characteristics. The HONTOs and LUNTOs of NSPI-DVP and CNSPI-DVP have been depicted in Fig. 2 and Fig. S1, respectively (Tables S1 and S2, ESI†). From theoretical findings, the molecular design strategy of NSPI-DVP and CNSPI-DVP has been carried out. These findings are in agreement with the strategy of energy level regulation. As designed, the triphenylethylene fragment of NSPI-DVP and CNSPI-DVP are involved in the S1 → S0 transition, so their AIE activity could be anticipated.24 The theoretical energy gap of NSPI-DVP and CNSPI-DVP are 3.32 and 3.20 eV, respectively.
 |
| | Fig. 1 Molecular structure, optimized geometry and molecular electrostatic potential map (MEP) of NSPI-DVP and CNSPI-DVP. | |
Table 1 Optical and thermal properties of NSPI-DVP and CNSPI-DVP
| Properties |
NSPI-DVP |
CNSPI-DVP |
|
T
g/Td (°C) |
162/445 |
213/502 |
|
λ
ab (nm) (soln/film) |
323, 350/320, 342 |
320, 345/318, 340 |
|
λ
em (nm) (soln/film) |
412/438 |
405/428 |
|
Φ (soln/film) |
9.2/70.7 |
7.8/40.8 |
|
τ (ns) (soln/film) |
0.76/2.93 |
0.41/1.62 |
| HOMO/LUMO (eV) |
−5.56/−2.27 |
−5.43/−2.32 |
|
E
T (eV) |
2.36 |
2.81 |
|
k
r (× 108 s−1) |
1.21/2.41 |
1.90/2.51 |
|
k
nr (× 108 s−1) |
1.07/2.37 |
1.65/2.45 |
 |
| | Fig. 2 (a) Natural transition orbital pairs (HONTOs and LUNTOs) with transition character of NSPI-DVP [f-oscillator strength and % weights of hole-particle] and (b) Potential energy scan (PES) of NSPI-DVP and CNSPI-DVP. | |
3.2. Potential energy surface (PES) scan
The potential energy surface scan of NSPI-DVP and CNSPI-DVP is depicted in Fig. 2. The minimum energy conformation in which the dihedral angle between substituent at azomethine nitrogen and phenanthroimidazole (59.6°-NSPI-DVP; 76.9°-CNSPI-DVP) and the substituent at C2 with phenanthroimidazole (46.1°-NSPI-DVP; 60.2°-CNSPI-DVP) reduced the intermolecular packing. The incorporation of N-coupling and C-coupling enhanced the molecular distortion as well as suppressed the aggregation formation and π–π stacking.20 The restricted intermolecular interaction (RIR) in the non-coplanar conformation of NSPI-DVP and CNSPI-DVP results in enhanced quantum yield in film and hence shows AIE active mechanofluorochromic behaviour.20
3.3. Synthesis of mechanofluorochromic materials
The 5,10-dibromo-1-(naphthalen-1-yl)-2-styryl-1H-phenanthro[9,10-d]imidazole (DBSPI) and 4-(5,10-dibromo-2-styryl-1H-phenanthro[9,10-d]imidazol-1-yl)naphthalene-1-carbonitrile (DBSPI-NCN) intermediates were isolated by the condensation of 2,7-dibromophenanthrene-9,10-dione with respective amines, ammonium acetate and cinnmaldehyde. The Suzuki-coupling of DBSPI and DBSPI-NCN with (4-(2,2-diphenylvinyl)phenyl) boronic acid using Pd(PPh3)4 catalyst in nitrogen stream yielded phenanthroimidazoles, 1-(naphthalen-1-yl)-5,10-bis(4-(2,2-diphenylvinyl)phenyl)-2-styryl-1Hphenanthro[9,10-d]imidazole (NSPI-DVP) and 4-(5,10-bis(4-(2,2-diphenylvinyl)phenyl)-2-styryl-1H-phenanthro[9,10-d]imidazol-1-yl) naphthalene-1-carbonitrile (CNSPI-DVP), respectively (Scheme S1, ESI†). The structure of NSPI-DVP and CNSPI-DVP were confirmed by spectral studies.
3.4. Thermal stability and electrochemical properties
The thermal stability of NSPI-DVP and CNSPI-DVP was analysed using TGA (thermogravimetric analysis) and DSC (differential scanning calorimetry) at a heating rate of 10 °C min−1 in N2 protection (Fig. 3 and Table 1). The twisted geometry of NSPI-DVP and CNSPI-DVP with twist angles of 46.1° and 60.2° between styryl at C2 and phenanthrimidazole core increased the thermal stability (Fig. 1). The high thermal decomposition temperature (Td) of NSPI-DVP (445 °C) and CNSPI-DVP (502 °C) reveal that NSPI-DVP and CNSPI-DVP are thermally stable and appropriate for device fabrication. The highly-twisted geometry of CNSPI-DVP compared to NSPI-DVP exhibits high thermal stability. DSC curves of NSPI-DVP and CNSPI-DVP reveal an endothermic process with a high glass transition temperature (Tg) of 162 and 213 °C, respectively, because of bulky substituents at both C2- and azomethine nitrogen atoms. The interaction between the substituents of NSPI-DVP and CNSPI-DVP tuned the packing arrangement and improved thermal stability, which is essentially required for the device fabrication.
 |
| | Fig. 3 (a) TGA and DSC graph; (b) cyclic voltammogram; (c) normalized absorption spectra; (d) emission spectra; (e) Lippert–Mataga plot and (f) Phosphorescence spectra of NSPI-DVP and CNSPI-DVP. | |
The electrochemical properties of NSPI-DVP and CNSPI-DVP were explored by cyclic voltammetry analysis. The cyclic voltammogram of NSPI-DVP and CNSPI-DVP show quasi-reversible redox waves (Fig. 3). The EHOMO of NSPI-DVP (−5.56 eV) and CNSPI-DVP (−5.43 eV) were obtained from the equation of EHOMO (eV) = −(Eonset + 4.8) eV and ELUMO of NSPI-DVP (−2.27 eV) and CNSPI-DVP (−2.32 eV) were calculated from ELUMO (eV) = HOMO − 1239/λonset.28,29 The electrochemical energy gap (EHOMO–LUMO) of NSPI-DVP and CNSPI-DVP are of 3.29 eV and 3.11 eV, respectively.
3.5. The photophysical properties
The absorption peaks of NSPI-DVP (323 nm) and CNSPI-DVP (320 nm) could be due to the π–π* transition from aromatic chromophoric groups (Fig. 3). The absorption at 350 and 345 nm was due to high energy electronic transition from the phenanthroimidazole region. Incorporation of DVP increased the molar extinction coefficient (εmax) of the intramolecular charge transfer absorption peak: [NSPI-DVP: 323 nm (εmax – 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 960 cm−1 M−1), 320 nm (εmax – 31250 cm−1 M−1); CNSPI-DVP: 350 nm (εmax – 28571 cm−1 M−1), 345 nm (εmax – 28985 cm−1 M−1)].30–33 The NSPI-DVP and CNSPI-DVP exhibit weak emissions at 412 and 405 nm with a quantum yield (Φ) of 9.2% and 7.8%, respectively (Table 1). Meanwhile, NSPI-DVP and CNSPI-DVP could luminesce strongly in the film, and show emissions at 438 and 428 nm with a maximum quantum yield of 70.7% and 40.8%, respectively. The high triplet energy (ET) of NSPI-DVP (2.77 eV) and CNSPI-DVP (2.86 eV) may be sufficient to excite the dopants (Fig. 3).
960 cm−1 M−1), 320 nm (εmax – 31250 cm−1 M−1); CNSPI-DVP: 350 nm (εmax – 28571 cm−1 M−1), 345 nm (εmax – 28985 cm−1 M−1)].30–33 The NSPI-DVP and CNSPI-DVP exhibit weak emissions at 412 and 405 nm with a quantum yield (Φ) of 9.2% and 7.8%, respectively (Table 1). Meanwhile, NSPI-DVP and CNSPI-DVP could luminesce strongly in the film, and show emissions at 438 and 428 nm with a maximum quantum yield of 70.7% and 40.8%, respectively. The high triplet energy (ET) of NSPI-DVP (2.77 eV) and CNSPI-DVP (2.86 eV) may be sufficient to excite the dopants (Fig. 3).
The excited-state properties of NSPI-DVP and CNSPI-DVP were analysed by solvent effects (Fig. S2 and Tables S3, S4, ESI†). The NSPI-DVP and CNSPI-DVP show red-shifted emission with increasing solvent polarity due to intramolecular charge transfer interaction of solvent with solute and defined by Lippert–Mataga plot [hc(![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) abs − flu) = hc(hcvacabs − hcvacflu) + 2 (μe − μg)2/a03].34,35 The variation of Stokes shift of NSPI-DVP and CNSPI-DVP with solvent polarity exhibits double linear relationship confirmed the HLCT behaviour (Fig. 3). The linear line in the low-polarity region with a smaller slope may be due to the LE state, whereas ELE remained unchanged in moderate and high polar solvents, however, ECT was smaller than ELE (ECT < ELE). From the Lippert–Mataga plot, small dipole moment (μe) obtained in low Δf solvents (<0.17) [7.8 D-NSPI-DVP; 11.08 D-CNSPI-DVP] and in high Δf (>0.17), high μe of 19.81 D (NSPI-DVP) and 23.08 D (CNSPI-DVP) were calculated. This ICT in NSPI-DVP and CNSPI-DVP was expected to enhance the device efficiency.36–40 The ICT was supported by theoretical investigations also (Fig. S1–S4 and Tables S5–S9, ESI†).
abs − flu) = hc(hcvacabs − hcvacflu) + 2 (μe − μg)2/a03].34,35 The variation of Stokes shift of NSPI-DVP and CNSPI-DVP with solvent polarity exhibits double linear relationship confirmed the HLCT behaviour (Fig. 3). The linear line in the low-polarity region with a smaller slope may be due to the LE state, whereas ELE remained unchanged in moderate and high polar solvents, however, ECT was smaller than ELE (ECT < ELE). From the Lippert–Mataga plot, small dipole moment (μe) obtained in low Δf solvents (<0.17) [7.8 D-NSPI-DVP; 11.08 D-CNSPI-DVP] and in high Δf (>0.17), high μe of 19.81 D (NSPI-DVP) and 23.08 D (CNSPI-DVP) were calculated. This ICT in NSPI-DVP and CNSPI-DVP was expected to enhance the device efficiency.36–40 The ICT was supported by theoretical investigations also (Fig. S1–S4 and Tables S5–S9, ESI†).
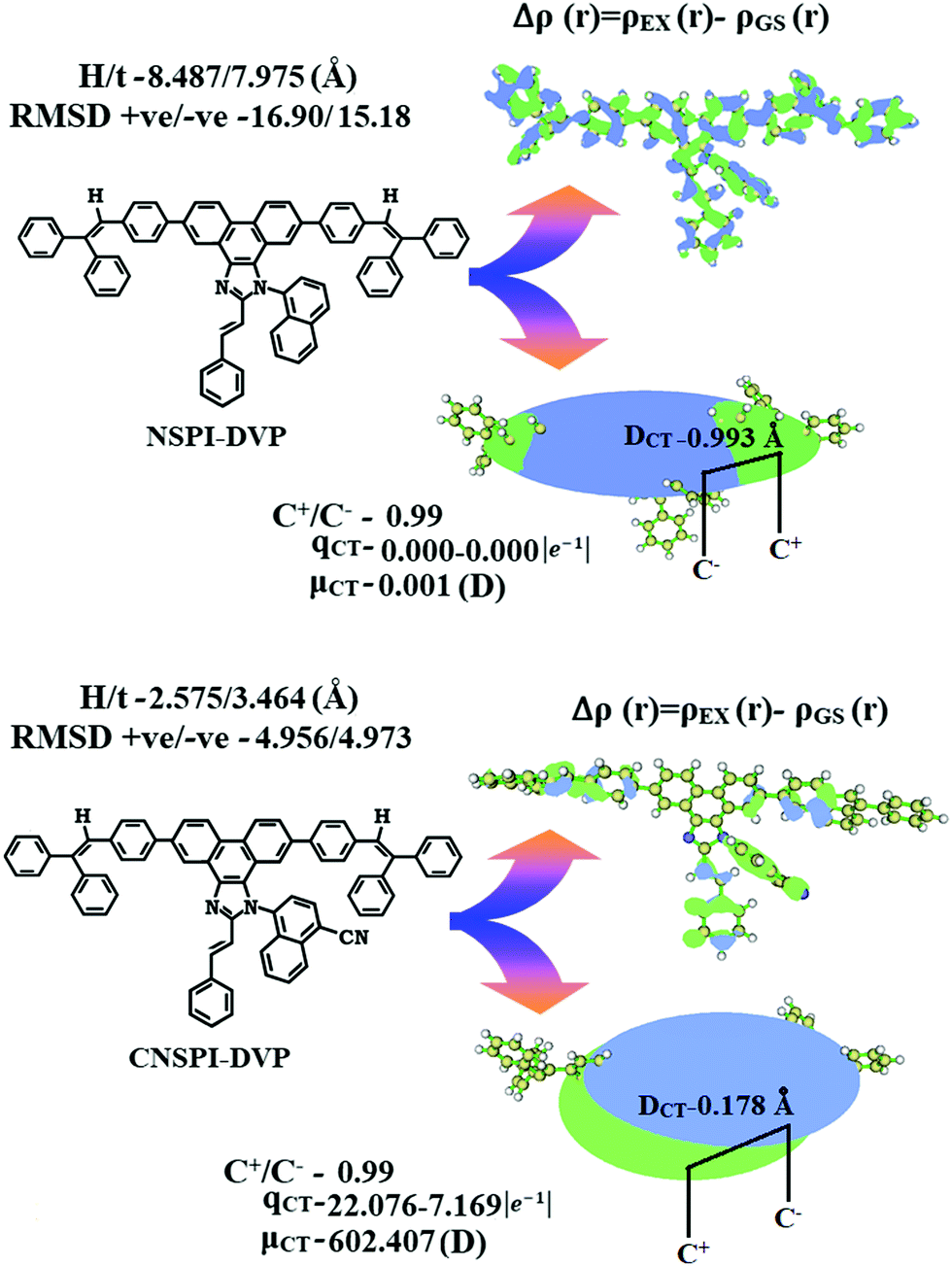
The existence of LE and CT components was analyzed using excitation energies of NSPI-DVP and CNSPI-DVP. Based on initial LE and CT state energies of NSPI-DVP and CNSPI-DVP, non-equivalent hybridization was constructed (Fig. 4 and Fig. S3, ESI†) because of a similar hole/electron wave function between S1 and S2. Small ΔEST ≈ 0 between S2 and T3 states (NSPI-DVP) and S2 and T5 states (CNSPI-DVP) assist rapid reverse intersystem crossing (RISC) with hot exciton process because of high-lying triplet CT character (hRISC). Thus, compared with NSPI-DVP, CNSPI-DVP show high EUE and high EQE due to a high-lying CT state (Tables S3 and S4, ESI†). The large ΔET1–T2 between T1 and T2 states of NSPI-DVP (1.19 eV) and CNSPI-DVP (1.60 eV) assisted the fast RISC by restriction of internal conversion, called hot-exciton mechanism and ΔET1–T2 of CNSPI-DVP is larger than NSPI-DVP due to the cyano group (Fig. 4 and Fig. S3, ESI†).20–23 The degree of hybridization of LE with CT states depends on both interstate-coupling strength and the initial ELE–ECT energy gap. The effective non-equivalent hybridization can enhance the photoluminance efficiency and EUE of fluorescent OLED materials. The HLCT behaviour of NSPI-DVP and CNSPI-DVP can be further confirmed by electron–hole distribution as well as transition density matrix (TDM) (Fig. 5 – NSPI-DVP; Fig. S4, ESI† – CNSPI-DVP). The diagonal (confined on the backbone) and off-diagonal area denote the LE component and CT component, respectively. The TDM analysis also supports LE and CT states in the emissive states. The non-zero t of NSPI-DVP and CNSPI-DVP implies the severe overlap of the hole with the electron (C+/C−: 0.99-NSPI-DVP and CNSPI-DVP; Fig. 6) and the calculated eign value (>0.99) reveals that hybridization with 96% of transition was observed in NSPI-DVP and CNSPI-DVP.
 |
| | Fig. 4 (a) Schematic representation of RISC process by high-lying CT states and (b) hybridization process of LE and CT states of CNSPI-DVP. | |
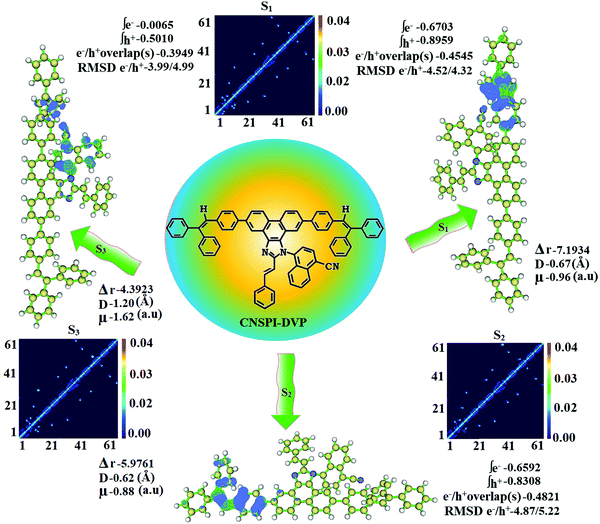 |
| | Fig. 5 Hole and particle distribution [green – increasing electron density and blue – decreasing electron density] and contour plots of transition density matrices (TDM) of CNSPI-DVP for [S1–S3 states: density = transition = n/IOp(6/8 = 3)]. | |
 |
| | Fig. 6 Graphical representation of DCT and centroid of charges [C+(r)/C−(r); isosurface for NSPI-DVP and for CNSPI-DVP (0.1 a.u.)]. | |
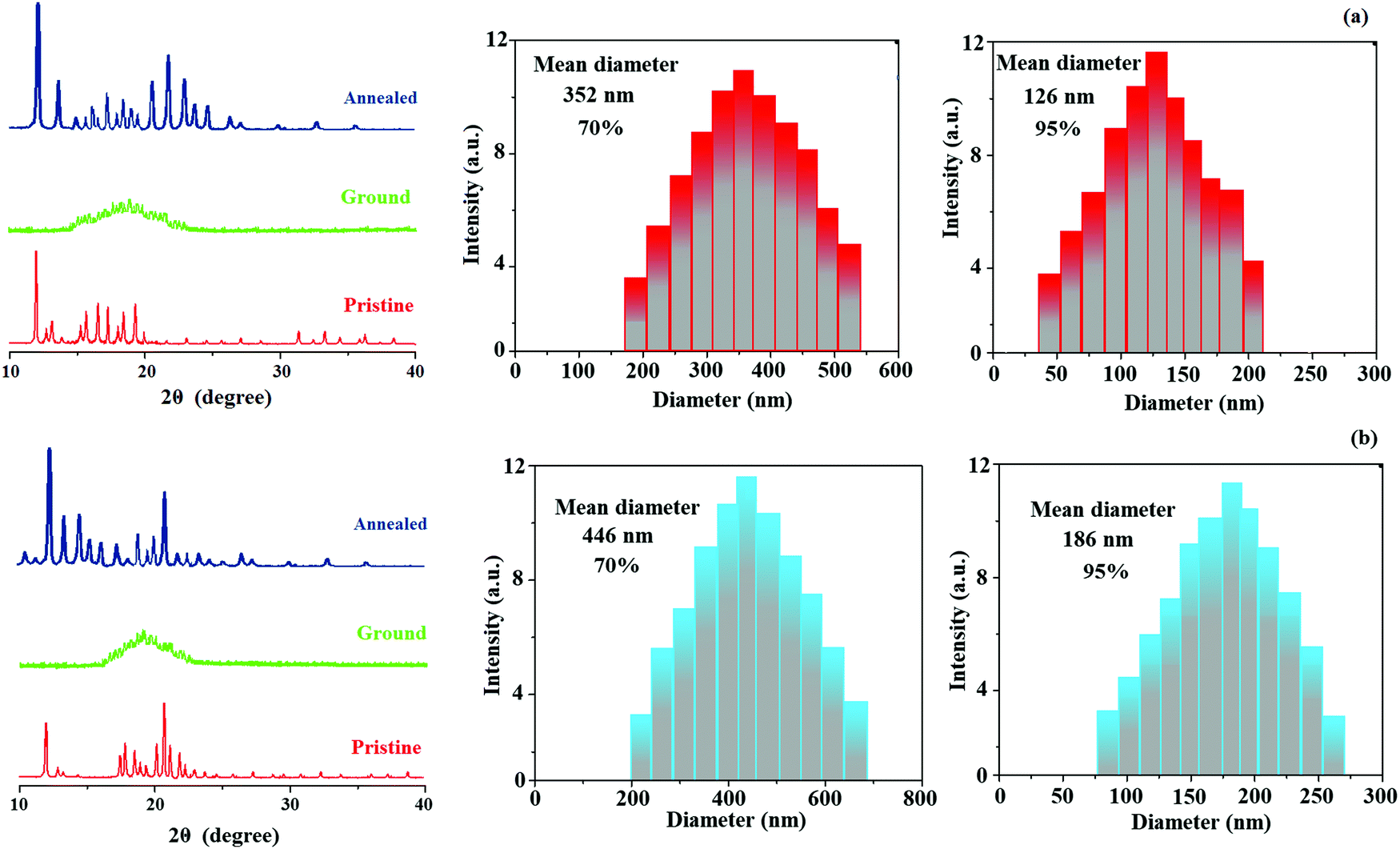
This transition among excited states suppressed the non-radiative decay, increasing the photoluminescence efficiency and oscillator strength. The propeller-shaped triphenylethylene moiety in NSPI-DVP and CNSPI-DVP initiates AIE behaviour and it is confirmed by the emission behaviour of NSPI-DVP and CNSPI-DVP (Fig. 7). These materials are completely soluble in THF and less soluble in water, hence, the AIE behaviour was analysed in THF–H2O mixtures. The gradual increase in water addition to these newborn materials in the THF solution initiates aggregate formation. The NSPI-DVP and CNSPI-DVP are feebly and highly fluorescent at low and high % water fractions, respectively. From PLQYs/f and lifetime (τ) s/f of NSPI-DVP (9.2/70.7%: 0.76 ns) and CNSPI-DVP (7.8/40.8%: 0.41/1.62 ns), the calculated kr, radiative and knr, non-radiative rate constants are of NSPI-DVP [krs/f (108 s−1) – 1.21/2.41: knrs/f (108 s−1) – 1.07/2.37 and CNSPI-DVP (krs/f (108 s−1) – 1.90/2.51: knrs/f (108 s−1) – 1.65/2.45)]. The increase of kr and decrease in knr of CNSPI-DVP and NSPI-DVP is in line with the aim of our design strategy. The decrease in the non-radiative rate (knr) of CNSPI-DVP and NSPI-DVP in solution relative to that of the film may be due to the restriction of intermolecular motion (RIM), which is the cause of the AIE behaviour of CNSPI-DVP and NSPI-DVP.33,34 The aggregates at higher % H2O addition limit the rotation of aryl moiety and intensified the emission with a bathochromic shift (Fig. 7). The emission of NSPI-DVP and CNSPI-DVP in THF solution (>55% water fraction) show a bathochromic shift with increasing intensity because of the aggregate formation. The emission spectra at 90% water fraction give an emission enhancement to a greater extent (Fig. 7). The AIE behaviour of NSPI-DVP and CNSPI-DVP has also been investigated by absorption spectra (Fig. 8). The absorption of NSPI-DVP and CNSPI-DVP in THF solution (>60% water fraction) show a bathochromic shift with increasing intensity because of the aggregate formation above which scattering of light is observed by nano-aggregate suspension under different THF–H2O combinations. The AIE behaviour of NSPI-DVP and CNSPI-DVP are depicted in photographic images under UV radiation (Fig. 7). The luminogen CNSPI-DVP emits different colors by varying water fraction, the aggregates formed at 75–80% H2O emit blue light (420 nm), whereas aggregates formed at >90% water fraction emit bluish-green light (460 nm). Similarly, aggregates of NSPI-DVP formed in 75–80% H2O emit blue light (430 nm) and aggregates formed at >90% water fraction emit bluish-green light (465 nm). The sizes of NSPI-DVP and CNSPI-DVP aggregates at various water fractions were determined by dynamic-light-scattering (DLS) and the calculated average diameter of CNSPI-DVP and NSPI-DVP aggregates are 446 nm and 352 nm (70% H2O) and 186 nm and 126 nm (95% H2O), respectively (Fig. 9). The average size of CNSPI-DVP is larger when compared to NSPI-DVP, which leads to a lower photoluminescence quantum yield (PLQY) because of more contribution of molecules on the surface of aggregates. However, the more non-radiative excited state energy loss leads to enhanced emission intensity.35
 |
| | Fig. 7 Emission spectra and UV illumination of (a) NSPI-DVP and (b) CNSPI-DVP in THF–H2O mixture with different water fractions. | |
 |
| | Fig. 8 Absorption spectra of (a) NSPI-DVP and (b) CNSPI-DVP in THF–H2O with different water fractions; (c) emission spectra of pristine, ground and annealed solids of NSPI-DVP and CNSPI-DVP and (d) comparison of operational stability of devices. | |
 |
| | Fig. 9 Powder XRD and dynamic light scattering in different water fractions of (a) NSPI-DVP and (b) CNSPI-DVP. | |
3.6. Mechanochromism
The reversible mechanochromic behaviour of NSPI-DVP and CNSPI-DVP have been studied using solid emission spectra (Fig. 8 and Table 2). The pristine NSPI-DVP and CNSPI-DVP show blue emission at 438 nm and 405 nm, which were shifted to bluish green regions of 476 nm and 463 nm, respectively, upon grinding. The reversible colour change was investigated by annealing at 230 °C (NSPI-DVP) and 200 °C (CNSPI-DVP) for 5 minutes, which results in the blue emission 436 nm (NSPI-DVP) and 440 nm (CNSPI-DVP). The emission at 476 nm (NSPI-DVP) and 463 nm (CNSPI-DVP) may be attributed to intermediate species formed by grinding the pristine NSPI-DVP and CNSPI-DVP. These findings imply that NSPI-DVP and CNSPI-DVP are highly sensitive towards mechanical stimuli due to triphenylethylene moiety. The emission spectra of CNSPI-DVP in film show hypochromatic shift to a larger extent in comparison to that of NSPI-DVP because of highly twisted molecular geometry. The grinding-induced spectral shifts are 40 nm (NSPI-DVP) and 23 nm (CNSPI-DVP). The fast color change of CNSPI-DVP with external stimuli could be attributed to the cyano group, which is involved in hydrogen bonding interactions. Also, the EHOMO–LUMO confirmed fast intramolecular interactions. The mechanochromic behaviour of these materials has also been evidenced by quantum yield measurements. The quantum yield of NSPI-DVP/CNSPI-DVP in distinct forms are of 70.7%/40.8% (pristine), 80.2%/53.66% (grinding) and 85.64%/60.32% (annealing). The powder XRD patterns of NSPI-DVP and CNSPI-DVP were recorded to investigate mechanochromic behaviour (Fig. 9). The sharp diffraction of pristine NSPI-DVP and CNSPI-DVP attributed to crystalline form which disappeared on grinding and exhibit broadband due to amorphous form. The crystallinity of NSPI-DVP and CNSPI-DVP recovered by annealing was confirmed by sharp peaks.36–40 Before and after grinding, the emission lifetime of NSPI-DVP and CNSPI-DVP was enhanced from 2.93 ns to 3.12 ns and from 1.62 ns to 1.86 ns, respectively. The enhanced lifetime reveals that the intermolecular interactions were strengthened in NSPI-DVP and CNSPI-DVP due to their more planarized conformation in the amorphous state (Fig. 10).
Table 2 Emission behaviour of NSPI-DVP and CNSPI-DVP under external stimuli
| Compounds |
λ
pristine (nm) |
λ
ground (nm) |
λ
annealed (nm) |
Δλ (nm) |
| NSPI-DVP |
438 |
476 |
436 |
40 |
| CNSPI-DVP |
405 |
463 |
440 |
23 |
 |
| | Fig. 10 Mechanochromic behaviour and life time of (a) NSPI-DVP and (b) CNSPI-DVP. | |
3.7. Electroluminescent studies
The electrical properties of NSPI-DVP and CNSPI-DVP were analyzed by the fabrication of single-carrier devices (Fig. S5, ESI†): (a) ITO/NPB (10 nm)/NSPI-DVP and CNSPI-DVP (60 nm)/NPB (10 nm)/LiF(1 nm)/Al (100 nm)41 (HOD: hole-only device): NPB (LUMO: −2.3 eV) prevents electron injection from cathode (Ef – 4.3 eV): (b) ITO/TPBi (10 nm)/NSPI-DVP and CNSPI-DVP (60 nm)/TPBi (10 nm)/LiF(1 nm)/Al (100 nm) (EOD: electron-only device): TPBi (HOMO: −6.2 eV) prevents hole injection41 (NPB-N,N′-di-1-naphthyl-N,N′-diphenylbenzidine and TPBi-1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene). The strong carrier transport properties of NSPI-DVP and CNSPI-DVP were confirmed by the current density–voltage investigation of the respective HOD and EOD (Fig. S5, ESI†). The negligible current density differences between HOD and EOD reveal that NSPI-DVP and CNSPI-DVP are efficient bipolar materials and transporting both holes and electrons effectively, which leads to balanced recombination.41–43
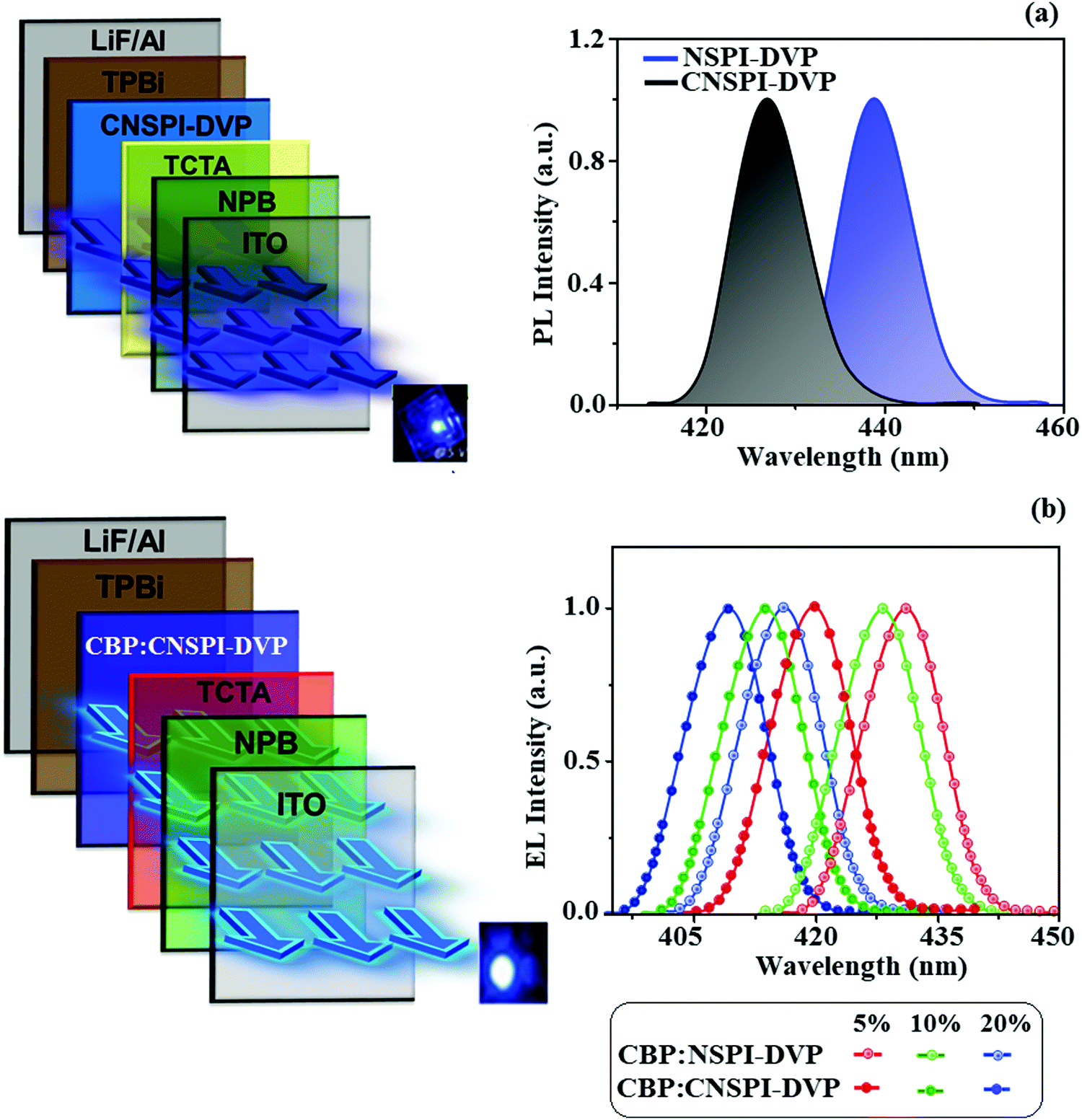
Because of high PLQY in film, excellent thermal stability and AIE character, NSPI-DVP and CNSPI-DVP have been used as an emissive layer in non-doped devices with the architecture of ITO/NPB (60 nm)/TCTA (25 nm)/NSPI-DVP or CNSPI-DVP (30 nm)/TPBi (25 nm)/LiF (1 nm)/Al (100 nm), where, NPB and TPBi have been used as electron- and hole-blocking layers, respectively (Fig. 11). The low turn-on voltage [2.7 V-NSPI-DVP and 2.6 V-CNSPI-DVP] may be due to efficient charge transportation and balanced recombination. The small energy gap (EHOMO–LUMO) of NSPI-DVP and CNSPI-DVP and the small energy barrier between the adjacent layers promotes carrier injection. Because of the large energy gap of TCTA and TPBi, exciton will be completely confined in the emitting layer and leads to higher efficiency. The linear current density–luminance graph of the fabricated non-doped devices infer typical photoelectric characteristics of NSPI-DVP and CNSPI-DVP.
 |
| | Fig. 11 Device configuration with EL spectra: (a) non-doped and (b) doped devices. | |
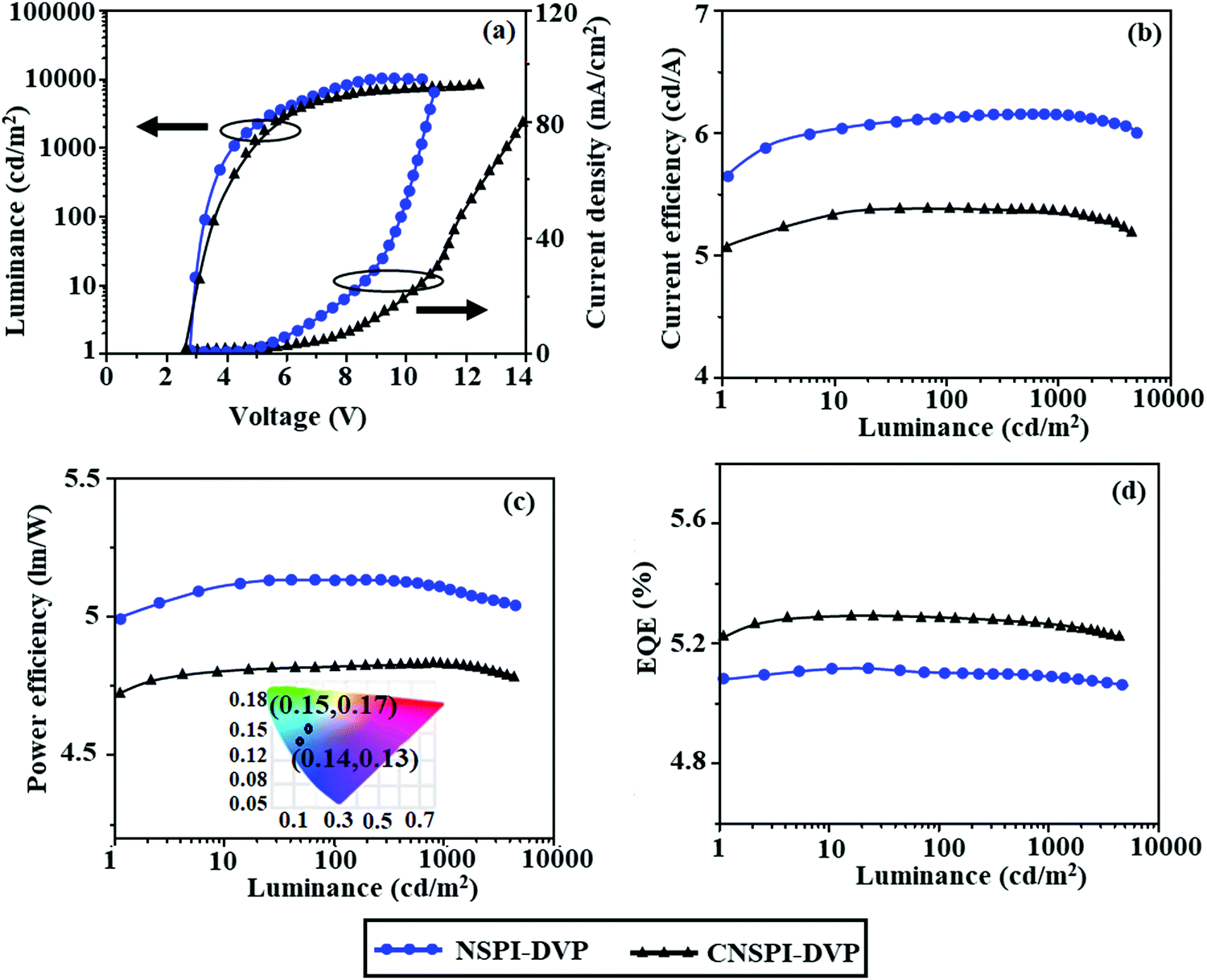
The increase in current density with the increasing voltage of the fabricated devices may be due to shallow LUMO energy and a smaller energy gap. The small hole-injection barrier (HIB) [TCTA:NSPI-DVP (0.60 V) and TCTA:CNSPI-DVP (0.47 eV)] and electron-injection barrier (EIB) [TPBi:NSPI-DVP (0.47 eV) and TPBi:CNSPI-DVP (0.28 eV)] promotes effective hole/electron injection/transportation and results in higher efficiency.44 The devices with NSPI-DVP or CNSPI-DVP exhibit blue emission at 439 nm (CIE: 0.15, 0.17) and 427 nm (CIE: 0.14, 0.13), respectively (Fig. 11 and Table 3). The EL emission wavelength is close to PL in the film, which supports the exciton confinement in the emitting layer. The absence of excimer or exciplex emission reveals that h+–e− recombination is confined within the emissive layer.45 The non-doped devices with NSPI-DVP and CNSPI-DVP exhibit high EQE of 5.09 and 5.23%, CE of 5.61 and 5.03 cd A−1 and PE of 4.99 and 4.72 lm W−1, respectively (Fig. 12). In comparison, CNSPI-DVP shows higher EQE than NSPI-DVP under the same device architecture because of the high-lying CT state. The non-doped devices with NSPI-DVP and CNSPI-DVP exhibit negligible roll-off efficiency [NSPI-DVP 1.76% and CNSPI-DVP 0.96%] due to effective h+–e− recombination and reduced exciton quenching. The rapid hRISC reduced the triplet exciton concentration when the current density was increased, hence the efficiency roll-off was negligible. The non-doped devices of NSPI-DVP and CNSPI-DVP show excellent operational stability, as proved by small efficiency roll-off as luminance increases. The negligible roll-off efficiency supports the potential of NSPI-DVP or CNSPI-DVP for industrial applications. A comparison of device efficiencies with reported efficiencies reveals that the devices based on NSPI-DVP and CNSPI-DVP are excellent emitters in terms of efficiencies (Table S10, ESI†).
Table 3 Electroluminescent performances of non-doped devices
| Properties |
NSPI-DVP |
CNSPI-DVP |
|
V
on (V) |
2.7 |
2.6 |
|
L (cd m−2) |
8932 |
7623 |
| CE (cd A−1) |
5.61/5.22 |
5.03/4.65 |
| PE (lm W−1) |
4.99/4.41 |
4.72/4.29 |
| EQE (%) |
5.09/5.00 |
5.23/5.18 |
| Roll-off (%) |
1.77 |
0.95 |
| IQE (%) |
25.45 |
26.15 |
| EUE (%) |
36.00 |
64.00 |
| EL (nm) |
439 |
427 |
| CIE (x,y) |
(0.15, 0.17) |
(0.14, 0.13) |
 |
| | Fig. 12 Non-doped device performances: (a) luminance/current density–voltage; (b) current efficiency–current density; (c) power efficiency–current density (inset: CIE) and (d) external quantum efficiency–Current density. | |
The theoretical maximum EQE of NSPI-DVP and CNSPI-DVP was found to be 2–4.8% by using light out-coupling efficiency (ηout) of 20–25%,46 which supports the dark triplet exciton harvesting in the EL process. The maximum EUE of devices with NSPI-DVP (36.00%) and CNSPI-DVP (64.0%) exceeds the 25% theoretical limit (Fig. 4 and Fig. S3, ESI,†Table 3). The contribution of triplet–triplet annihilation (TTA) was ruled out from the linear luminance–voltage curve.47 The large energy gap (ΔES1T1) between S1 and T1 of NSPI-DVP and CNSPI-DVP excluded the TADF process (Fig. 4 and Fig. S3, ESI†). Hence, high-lying CT states play a vital role in increasing EUE. The energy gap (ΔEST) between S2 and T5 (CNSPI-DVP) and S2 and T3 (CNSPI-DVP) was found to be ∼0.01 eV (Tables S1 and S2, ESI†), which promotes fast hRISC from high energy-excited states for effective triplet exciton harvesting and leads to high EUE.46 The excellent performance of the CNSPI-DVP-based device implies that efficient hole and electron injection, as well as transportation during electroluminescence, leads to balanced electrons and holes recombination in the emitting layer.
To know the potential of NSPI-DVP and CNSPI-DVP as emissive materials in efficient OLEDs for industrial applications, the operational lifetime (T50) of these devices was measured along with the reference device.48Fig. 10 depicts the L–T curve (relative brightness-L/L0vs. time) of devices with 10 mA cm−2 current density. The half-life time was calculated from both the L–T curve and the equation [L0n × T50 = constant: initial luminance (L0), half lifetime (T50), accelerated factor (n – 1.5)] and a lifetime of devices has been determined at 500 cd m−2. The devices with NSPI-DVP and CNSPI-DVP show a longer lifetime of 105 h and 135 h, respectively, which in turn reflects the stability of the devices. The longer lifetime may be ascribed to higher thermal stability and wide recombination zone as well as efficient h+–e− recombination in the NSPI-DVP and CNSPI-DVP layer of these devices.
In terms of higher radiative exciton production (ηr) and EL efficiency, NSPI-DVP and CNSPI-DVP were used to fabricate doped OLEDs. The fabricated doped device structure was: ITO/NPB (40 nm)/CBP:x wt% NSPI-DVP or CNSPI-DVP (20 nm)/TPBi (25 nm)/LiF (1 nm)/Al (100 nm) (Fig. 11), where, NSPI-DVP or CNSPI-DVP doped in 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) with doping concentration of 5 wt%, 10 wt% and 20 wt% functioned as an emissive layer. All the fabricated devices exhibit blue-shifted electroluminescent spectra with enhanced device efficiencies (Table 4). The small ΔEST can be adequate for Forster energy transfer from triplet (host) to dopants, the emissive materials were well diluted in the host and leads to improve the efficiency. The doped device based on CBP:NSPI-DVP/CBP:CNSPI-DVP show maximum efficiencies of CE – 5.23/5.06 cd A−1, PE – 4.82/4.61 lm W−1, EQE – 5.55/6.02% (5 wt%); CE – 6.29/6.10 cd A−1, PE – 5.93/5.82 lm W−1, EQE – 7.60/8.12% (10 wt%); CE – 7.65/7.56 cd A−1, PE – 6.48/6.89 lm W−1, EQE – 8.98/9.81% (20 wt%). Because of maximum exciton utilization with high ηr, deep-blue devices based on CBP:NSPI-DVP/CBP:CNSPI-DVP show high EQE with CIE of (0.15, 0.08)/(0.14, 0.06) and it is close to the National Television Standards Committee (NTSC) blue color standard. Additional triplet exciton utilized in OLEDs leads to enhanced performance, the EL efficiencies are among the state-of-the-art blue OLEDs in the relative color gamut. The efficiency roll-off may be due to exciton loss from the high triplet state of NSPI-DVP/CNSPI-DVP to the triplet state of CBP (2.6 eV) (Fig. 11). The EL spectra of doped devices CBP:NSPI-DVP/CBP:CNSPI-DVP were analogous to pristine PL spectra (NSPI-DVP or CNSPI-DVP). The similar position and shape of EL spectra of CBP:NSPI-DVP/CBP:CNSPI-DVP, when compared to PL spectra of pristine NSPI-DVP or CNSPI-DVP confirm the energy transfer from the host, CBP to NSPI-DVP or CNSPI-DVP (Fig. 11). The blue-shifted EL emission was obtained when compared to non-doped devices. The electroluminescence (EL) spectra of the devices are similar to their PL spectra, which show both EL and PL originate from the same radiative decay of the singlet excitons. The doped devices exhibit higher EQEs as well as deeper CIEs, probably attributed to the reasonable doping concentration that not only can suppress excitons concentration-quenching effect but also can effectually reduce the intermolecular charge transfer effect caused by a bathochromic shift.49,50
Table 4 Electroluminescent performances of doped devices
| CBP:NSPI-DVP/CNSPI-DVP |
5 wt% |
10 wt% |
20 wt% |
| EL (nm) |
430/420 |
428/415 |
416/409 |
|
V
on (V) |
2.9/2.7 |
2.8/2.6 |
2.7/2.6 |
|
L (cd m−2) |
9223/8237 |
9297/8412 |
9812/9967 |
| CE (cd A−1) |
5.23/5.08 |
6.29/6.10 |
7.65/7.56 |
| PE (lm W−1) |
4.82/4.61 |
5.93/5.82 |
6.48/6.89 |
|
EQE (%) |
5.55/6.02 |
7.60/8.12 |
8.98/9.81 |
| IQE (%) |
28/30 |
38/45 |
45/49 |
| EUE (%) |
40/96 |
54/96 |
64/99 |
| CIE |
(0.15, 0.08)/(0.14, 0.06) |
(0.15, 0.08)/(0.14, 0.06) |
(0.15, 0.08)/(0.14, 0.06) |
The CBP:NSPI-DVP/CBP:CNSPI-DVP-based OLEDs exhibit enhanced device efficiencies compared with that of the non-doped device (Fig. 13). The EL efficiency was increased by increasing doping concentration (20 wt%) and the absence of excimer emission may be the cause for improved efficiencies. The current density of heavily doped OLEDs increased rapidly compared to low dopant device concentration.
 |
| | Fig. 13 Doped device performances: (a) luminance/current density–voltage; (b) current efficiency–current density (inset: CIE); (c) power efficiency–current density and (d) external quantum efficiency–current density. | |
The theoretically calculated high EQE are 3.5/4.1% NSPI-DVP (neat/20 wt%) and 2.0/3.0% CNSPI-DVP (neat/20 wt%) [ηEQE = ηout × ηrc × ηγ × ΦPL, ΦPL: NSPI-DVP (70.7/82%) and CNSPI-DVP (40.8/60%), ηout – out-coupling efficiency (20%), ηrc – product of charge recombination efficiency (100%), ηγ – radiative exciton-production (25%)] and experimental EQE are of 5.09/8.98% NSPI-CVP (neat/20 wt%) and 5.23/9.81%, CNSPI-DVP (neat/20 wt%).15,16 The experimental EQE > theoretical EQE because larger triplet excitons are converted to singlet excitons in the EL process.15,16 The ηr calculated for NSPI-DVP (neat/20 wt%) (27–32%/29–34%) and CNSPI-DVP (neat/20 wt%) (29–34%/30–36%) indicates ηγ < 100% may be due to unbalanced carrier transportation in the emissive layer. The enhanced IQE of 25/28/38/49% (CBP:NSPI-DVP: neat/5/10/20 wt%); 26/30/41/49% (CBP:CNSPI-DVP: neat/20 wt%) and maximum EUE of 40/54/64% (CBP:NSPI-DVP: neat/5/10/20 wt%) and 74/96/99% (CBP:CNSPI-DVP: neat/5/10/20 wt%) [EUE = ηout × ηPL × ηres/ηEL] were maybe because of retained CT% in NSPI-DVP and CNSPI-DVP. The calculated EUE breaking the theoretical limit of 25%, i.e., triplet excitons converted to singlet excitons by RISC and leads to high-efficiency blue OLEDs. These research findings demonstrated that the non-doped and doped devices with NSPI-DVP and CNSPI-DVP show precision in our emitter design-strategy.
4. Conclusion
We have designed blue HLCT emissive materials, NSPI-DVP and CNSPI-DVP with orthogonal D–A geometry to harvest high PLQY in film and high EUE in blue OLEDs. Optical and theoretical analyses imply that their excited states having LE and CT distribution. Theoretical simulations of NSPI-DVP and CNSPI-DVP reveal that the S2 state exhibits high % CT characteristics. These materials show aggregation-induced emission (AIE) characteristics and are free of concentration quenching in the film. The NSPI-DVP and CNSPI-DVP show reversible mechanochromism with excellent color contrast between blue and green color. The fabricated non-doped devices based on CNSPI-DVP show blue emission at 427 nm with CIE of (0.14, 0.13). The special excited state of CNSPI-DVP can activate the fast hRISC with a hot exciton process and harvesting more triplet excitons, which leads to a higher EUE of 64.0% than NSPI-DVP (36.0%). Therefore, non-doped blue devices based on CNSPI-DVP exhibit excellent EL performance than NSPI-DVP-based devices with a higher EQE of 5.23% and negligible roll-off efficiency. The doped device with CBP:20 wt% CNSPI-DVP show enhanced efficiencies (EQE-9.81%, CE-7.56 cd A−1 and PE-6.81 lm W−1) compared to that of non-doped devices. The harvested efficient device performances could shed light on low-cost hot exciton materials for next-generation fluorescent OLEDs.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC;
(b) Y. Im, S. Byun, J. Kim, D. Lee, C. Oh, K. Yook and J. Lee, Adv. Funct. Mater., 2017, 27, 1603007 CrossRef;
(c) X. Cai and S. J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef;
(d) J. H. Lee, C. H. Chen, P. H. Lee, H. Y. Lin, M. K. Leung, T. L. Chiu and C. F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
-
(a) J. Ding, J. Gao, Y. Cheng, Z. Xie, L. Wang, D. Ma, X. Jing and F. Wang, Adv. Funct. Mater., 2006, 16, 575–581 CrossRef CAS;
(b) H. Sasabe, J. Takamatsu, T. Motoyama, S. Watanabe, G. Wagenblast, N. Langer, O. Molt, E. Fuchs, C. Lennartz and J. Kido, Adv. Mater., 2010, 22, 5003–5007 CrossRef CAS PubMed;
(c) T. Fleetham, G. J. Li, L. L. Wen and J. Li, Adv. Mater., 2014, 26, 7116–7121 CrossRef CAS PubMed.
-
(a) J. Huang, N. Sun, Y. Dong, R. Tang, P. Lu, P. Cai, Q. Li, D. Ma, J. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329–2337 CrossRef CAS;
(b) C. J. Tonzola, A. P. Kulkarni, A. P. Gifford, W. Kaminsky and S. A. Jenekhe, Adv. Funct. Mater., 2007, 17, 863–874 CrossRef CAS;
(c) C. H. Chien, C. K. Chen, F. M. Hsu, C. F. Shu, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2009, 19, 560–566 CrossRef CAS.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Li, T. Liu, H. Liu, M. Z. Tian and Y. Li, Acc. Chem. Res., 2014, 47, 1186–11988 CrossRef CAS PubMed.
- H. Liu, J. Xu and Y. Li, Acc. Chem. Res., 2010, 43, 1496–1508 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
-
(a) Z. Yang, Z. Mao, T. Yu, Y. Zhang, S. Liu, J. Xu and Z. Chi, ACS Symp. Ser., 2016, 1226, 221–259 CrossRef CAS;
(b) J. Li, T. Shan, M. Yao, Y. Gao, X. Han, B. Yang and P. Lu, J. Mater. Chem. C, 2017, 5, 2552–2558 RSC;
(c) T. Han, X. Gu, J. W. Y. Lam, A. C. S. Leung, R. T. K. Kwok, T. Han, B. Tong, J. Shi, Y. Dong and B. Z. Tang, J. Mater. Chem. C, 2016, 4, 10430–10434 RSC;
(d) K. Matsuoka, K. Albrecht, K. Yamamoto and K. Fujita, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed;
(e) J. Y. Sun, J. Q. Han, Y. Liu, Y. A. Duan, T. Y. Han and J. Yuan, J. Mater. Chem. C, 2016, 4, 8276–8283 RSC.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
-
(a) Y. Cao, I. D. Parker, G. Yu, C. Zhang and A. J. Heeger, Nature, 1999, 397, 414–417 CrossRef CAS PubMed;
(b) L. Yao, S. Zhang, R. Wang, W. Li, F. Shen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119–2123 CrossRef CAS PubMed;
(c) S. Zhang, L. Yao, Q. Peng, W. Li, Y. Pan, R. Xiao, Y. Gao, C. Gu, Z. Wang, P. Lu, F. Li, S. Su, B. Yang and Y. Ma, Adv. Funct. Mater., 2015, 25, 1755–1762 CrossRef CAS;
(d) X. H. Ouyang, X. L. Li, L. Ai, D. B. Mi, Z. Y. Ge and S. J. Su, ACS Appl. Mater. Interfaces, 2015, 7, 7869–7877 CrossRef CAS PubMed;
(e) Z. Gao, G. Cheng, F. Shen, S. Zhang, Y. Zhang, P. Lu and Y. Ma, Laser Photonics Rev., 2014, 8, L6–L10 CrossRef CAS.
-
(a) A. D. Gorse and M. Pesquer, J. Phys. Chem., 1995, 99, 4039–4049 CrossRef CAS;
(b) W. Y. Hung, G. C. Fang, Y. C. Chang, T. Y. Kuo, P. T. Chou, S. W. Lin and K. T. Wong, ACS Appl. Mater. Interfaces, 2013, 5, 6826–6831 CrossRef CAS PubMed;
(c) W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS.
-
(a) C. J. Kuo, T. Y. Li, C. C. Lien, C. H. Liu, F. I. Wu and M. J. Huang, J. Mater. Chem., 2009, 19, 1865–1871 RSC;
(b) Z. M. Wang, P. Lu, S. M. Chen, Z. Gao, F. Z. Shen, W. S. Zhang, Y. X. Xu, H. S. Kwok and Y. G. Ma, J. Mater. Chem., 2011, 21, 5451–5456 RSC;
(c) W. J. Li, D. D. Liu, F. Z. Shen, D. G. Ma, Z. M. Wang, T. Feng, Y. X. Xu, B. Yang and Y. G. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS;
(d) K. Wang, F. C. Zhao, C. G. Wang, S. Y. Chen, D. Chen, H. Y. Zhang, Y. Liu, D. G. Ma and Y. Wang, Adv. Funct. Mater., 2013, 23, 2672–2680 CrossRef CAS;
(e) W. C. Chen, Y. Yuan, G. F. Wu, H. X. Wei, L. Tang, Q. X. Tong, F. L. Wong and C. S. Lee, Adv. Opt. Mater., 2014, 2, 626–631 CrossRef CAS;
(f) W. C. Chen, Q. X. Tong and C. S. Lee, J. Mater. Chem. C, 2015, 3, 10957–10963 RSC;
(g) C. Li, S. Wang, W. Chen, J. Wei, G. Yang, K. Ye, Y. Liu and Y. Wang, Chem. Commun., 2015, 51, 10632–10635 RSC;
(h) W. C. Chen, Y. Yuan, G. F. Wu, H. X. Wei, J. Ye, M. Chen, F. Lu, Q. X. Tong, F. L. Wong and C. S. Lee, Org. Electron., 2015, 17, 159–166 CrossRef CAS;
(i) L. J. Bin, W. Zhao, C. Y. Luo, L. Feng, S. L. Tao and Q. X. Tong, J. Mater. Chem. C, 2016, 4, 2003–2010 Search PubMed.
-
(a) Z. M. Wang, Y. Feng, X. S. T. Zhang, Y. Cao, G. Zhao, Y. M. Chen, X. J. Zhang, P. Lu, B. Yang, P. Chen, Y. G. Ma and S. Y. Liu, Phys. Chem. Chem. Phys., 2014, 16, 20772–20779 RSC;
(b) W. C. Chen, Q. X. Tong and C. S. Lee, Sci. Adv. Mater., 2015, 7, 2193–2205 CrossRef CAS.
-
(a) S. Tang, W. J. Li, F. Z. Shen, D. D. Liu, B. Yang and Y. G. Ma, J. Mater. Chem., 2012, 22, 4401–4408 RSC;
(b) A. D. Yu, T. Kurosawa, Y. C. Lai, T. Higashihara, M. Ueda, C. L. Liu and W. C. Chen, J. Mater. Chem., 2012, 22, 20754–20763 RSC.
- J. Jayabharathi, J. Anudeebhana, V. Thanikachalam and S. Sivaraj, RSC Adv., 2020, 10, 8866–8879 RSC.
-
J. Jayabharathi, V. Thanikachalam and J. Anudeebhana, Australian Innovation Pat., 2020104413, 2021 Search PubMed.
- J. Jayabharathi, S. Sivaraj, V. Thanikachalam and B. Seransenguttuvan, RSC Adv., 2020, 10, 25059–25072 RSC.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, (Revision A.02), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. Jayabharathi, S. Sivaraj, V. Thanikachalam and J. Anudeebhana, J. Mater. Chem. C, 2021, 9, 10334–10346 RSC.
- J. Jayabharathi, J. Anudeebhana, V. Thanikachalam, S. Sivaraj and A. Prabhakaran, RSC Adv., 2020, 10, 4002–4013 RSC.
- J. Jayabharathi, S. Panimozhi and V. Thanikachalam, Sci. Rep., 2020, 10, 1–11 CrossRef PubMed.
-
(a) J. Jayabharathi, S. Panimozhi and V. Thanikachalam, Sci. Rep., 2019, 9, 1–14 CrossRef CAS PubMed;
(b) J. Jayabharathi, V. Thanikachalam and G. Abirama Sundari, RSC Adv., 2019, 9, 6658–6680 RSC.
-
(a) Y. Yuan, D. Li, X. Zhang, X. Zhao, Y. Liu, J. Zhang and Y. Wang, New J. Chem., 2011, 35, 1534–1540 RSC;
(b) Y. Zhang, S. L. Lai, Q. X. Tong, M. Y. Chan, T. W. Ng, Z. C. Wen, G. Q. Zhang, S. T. Lee, H. L. Kwong and C. S. Lee, J. Mater. Chem., 2011, 21, 8206–8214 RSC;
(c) B. Liu, J. Zhao, C. Luo, F. Lu, S. Tao and Q. Tong, J. Mater. Chem. C, 2016, 4, 2003–2010 RSC;
(d) H. Zhang, A. Li, G. Li, B. Li, Z. Wang, S. Xu, W. Xu and B. Z. Tang, Adv. Opt. Mater., 2020, 8, 1902195 CrossRef CAS.
- J. Jayabharathi, S. Thilagavathy and V. Thanikachalam, RSC Adv., 2021, 11, 8606–8618 RSC.
- V. Thanikachalam, P. Jeeva and J. Jayabharathi, ChemistrySelect, 2017, 2, 1860–1870 CrossRef CAS.
- J. Jayabharathi, P. Sujatha, V. Thanikachalam and P. Nethaji, RSC Adv., 2018, 8, 2046–2069 Search PubMed.
-
(a) Y. Pan, W. Li, S. Zhang, L. Yao, C. Gu, H. Xu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 510–515 CrossRef CAS;
(b) W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS;
(c) W. Li, Y. Pan, R. Xiao, Q. Peng, S. Zhang, D. Ma, F. Li, F. Shen, Y. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2014, 24, 1609–1614 CrossRef CAS;
(d) Y. Xu, C. Wang, X. Zhou, J. Zhou, X. Guo, X. Liang, D. Hu, F. Li, D. Ma and Y. Ma, J. Phys. Chem. Lett., 2019, 10, 6878–6884 CrossRef CAS PubMed;
(e) J. Yang, Q. Guo, J. Wang, Z. Ren, J. Chen, Q. Peng, D. Ma and Z. Li, Adv. Opt. Mater., 2018, 6, 1800342 CrossRef.
- Y. Liu, L. S. Cui, M. F. Xu, X. B. Shi, D. Y. Zhou, Z. K. Wang, Z. Q. Jiangand and L. S. Liao, J. Mater. Chem. C, 2014, 2, 2488–2495 RSC.
- Y. H. Lou, M. F. Xu, L. Zhang, Z. K. Wang, S. Naka, H. Okada and L. S. Liao, Org. Electron., 2013, 14, 2698–2704 CrossRef CAS.
- Z. Wang, Y. Lou, S. Naka and H. Okada, Appl. Phys. Lett., 2011, 98, 063302 CrossRef.
- M. J. Cho, S. J. Kim, S. H. Yoon, J. Shin, T. R. Hong and H. J. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 19808–19815 CrossRef CAS PubMed.
-
(a) H. Wang, L. Meng, X. Shen, X. Wei, X. Zheng, X. Lv, Y. Yi, Y. Wang and P. Wang, Adv. Mater., 2015, 27, 4041–4047 CrossRef CAS PubMed;
(b) K. R. Justin Thomas, M. S. Fan and K. C. Ho, ChemistrySelect, 2017, 2, 2807–2814 CrossRef.
- J. Jayabharathi, S. Thilagavathy and V. Thanikachalam, Mater. Adv., 2021, 2, 5160–5169 RSC.
-
(a) Y. Xu, X. Liang, X. Zhou, P. Yuan, J. Zhou, C. Wang, B. Li, D. Hu, X. Qiao, X. Jiang, L. Liu, S. J. Su, D. Ma and Y. Ma, Adv. Mater., 2019, 31, 1807388 CrossRef PubMed;
(b) Y. Xu, X. Liang, Y. Liang, X. Guo, M. Hanif, J. Zhou, X. Zhou, C. Wang, J. Yao, R. Zhao, D. Hu, X. Qiao, D. Ma and Y. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 31139–31146 CrossRef CAS PubMed;
(c) X. Zhang, Z. Chi, B. Xu, C. Chen, X. Zhou, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem., 2012, 22, 18505–18513 RSC.
-
(a) J. Tagare and S. Vaidyanathan, J. Mater. Chem. C, 2018, 6, 10138–10173 RSC;
(b) Z. Li, C. Li, Y. Xu, N. Xie, X. Jiao and Y. Wang, J. Phys. Chem. Lett., 2019, 10, 842–847 CrossRef CAS PubMed;
(c) C. Chao, W. C. Chen, J. X. Chen, L. Yang, X. Z. Wang, H. Yang, B. Huang, Z. L. Zhu, Q. X. Tong and C. S. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 46746–46755 CrossRef PubMed.
-
(a) W. Qin, Z. Yang, Y. Jiang, J. W. Y. Lam, G. Liang, H. S. Kwok and B. Z. Tang, Chem. Mater., 2015, 27, 3892–3901 CrossRef CAS;
(b) C. Wang, Y. Yu, Y. Yuan, C. Ren, Q. Liao, J. Wang, Z. Chai, Q. Li and Z. Li, Matter, 2020, 2, 181–193 CrossRef.
-
(a) S. Zhang, W. Li, L. Yao, Y. Pan, F. Shen, R. Xiao, B. Yang and Y. Ma, Chem. Commun., 2013, 49, 11302–11304 RSC;
(b) Y. Gao, S. Zhang, Y. Pan, L. Yao, H. Liu, Y. Guo, Q. Gu, B. Yang and Y. Ma, Phys. Chem. Chem. Phys., 2016, 18, 24176–24184 RSC.
-
(a) Z. Wang, X. Li, K. Xue, H. Li, X. Zhang, Y. Liu, Z. Yu, P. Lu and P. Chen, J. Mater. Chem. C, 2016, 4, 1886–1894 RSC;
(b) Q. Wan, B. Zhang, J. Tong, Y. Li, H. Wu, H. Zhang, Z. Wang, Y. Pan and B. Z. Tang, Phys. Chem. Chem. Phys., 2019, 21, 9837–9844 RSC;
(c) H. Zhang, J. Zeng, W. Luo, H. Wu, C. Zeng, K. Zhang, W. Feng, Z. Wang, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 6359–6368 RSC.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
-
(a) H. Yuan, K. Wang, K. Yang, B. Liu and B. Zou, J. Phys. Chem. Lett., 2014, 5, 2968–2973 CrossRef CAS PubMed;
(b) N. Li, Y. Gu, Y. Chen, L. Zhang, Q. Zeng, T. Geng, L. Wu, L. Jiang, G. Xiao, K. Wang and B. Zou, J. Phys. Chem. C, 2019, 123, 6763–6767 CrossRef CAS;
(c) Y. Liu, Q. Zeng, B. Zou, Y. Liu, B. Xu and W. Tian, Angew. Chem., Int. Ed., 2018, 57, 15670–15674 CrossRef CAS PubMed;
(d) Y. Gong, P. Zhang, Y. Gu, J. Wang, M. Han, C. Chen, X. Zhan, Z. Xie, B. Zou, Q. Peng, Z. Chi and Z. Li, Adv. Opt. Mater., 2018, 6, 1800198 CrossRef;
(e) W. Jiang, L. Duan, J. Qiao, G. Dong, L. Wang and Y. Qiu, Org. Lett., 2011, 13, 3146–3149 CrossRef CAS PubMed;
(f) Y. Tao, Q. Wang, C. Yang, C. Zhong, K. Zhang, J. Ma and D. Qin, Adv. Funct. Mater., 2010, 20, 304–311 CrossRef CAS.
-
(a) Y. Zhang, J. Zhang, J. Shen, J. Sun, K. Wang, Z. Xie, H. Gao and B. Zou, Adv. Opt. Mater., 2018, 6, 1800956 CrossRef;
(b) S. Zhang, Y. Dai, S. Luo, Y. Gao, N. Gao, K. Wang, B. Zou, B. Yang and Y. Ma, Adv. Funct. Mater., 2017, 27, 1602276 CrossRef;
(c) Q. Qi, J. Qian, X. Tan, J. Zhang, L. Wang, B. Xu, B. Zou and W. Tian, Adv. Funct. Mater., 2015, 25, 4005–4010 CrossRef CAS.
-
(a) A. Li, Z. Ma, J. Wu, P. Li, H. Wang, Y. Geng, S. Xu, B. Yang, H. Zhang, H. Cui and W. Xu, Adv. Opt. Mater., 2018, 6, 1700647 CrossRef;
(b) X. Liu, C. Ma, A. Li, W. Xu, Z. Ma and X. Jia, J. Mater. Chem. C, 2019, 7, 8398–8403 RSC;
(c) J. Zhang, A. Li, H. Zou, J. Peng, J. Guo, W. Wu, H. Zhang, J. Zhang, X. Gu, W. Xu, S. Xu, S. Liu, A. Qin, J. W. Y. Lam and B. Z. Tang, Mater. Horiz., 2020, 7, 135–142 RSC.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
-
(a) T. Nakagawa, S. Y. Ku, K. T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC;
(b) R. Leuji, K. Goushi1 and C. Adachi, Nat. Commun., 2019, 10, 1–10 CrossRef PubMed.
- M. G. Helander, Z. B. Wang, J. Qiu, M. T. Greiner, D. P. Puzzo, Z. W. Liu and Z. H. Lu, Science, 2011, 332, 944–947 CrossRef CAS PubMed.
-
(a) Y. Xu, X. Liang, X. Zhou, P. Yuan, J. Zhou, C. Wang, B. Li, D. Hu, X. Qiao, X. Jiang, L. Liu, S. J. Su, D. Ma and Y. Ma, Adv. Mater., 2019, 31, 1807388 CrossRef PubMed;
(b) Y. Xu, X. Liang, Y. Liang, X. Guo, M. Hanif, J. Zhou, X. Zhou, C. Wang, J. Yao, R. Zhao, D. Hu, X. Qiao, D. Ma and Y. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 31139–31146 CrossRef CAS PubMed.
- C. H. Shih, P. Rajamalli, C. A. Wu, W. T. Hsieh and C. H. Cheng, ACS Appl. Mater. Interfaces, 2015, 7, 10466–10474 CrossRef CAS PubMed.
- X. Du, G. Li, J. Zhao, S. Tao, C. Zheng, H. Lin, Q. Tong and X. Zhang, Adv. Opt. Mater., 2017, 5, 1700498 CrossRef.
- J. Jayabharathi, G. Goperundevi and S. Panimozhi, ACS Omega, 2018, 3, 13598–13608 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00706h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Sekar
Sivaraj
,
Venugopal
Thanikachalam
*,
Sekar
Sivaraj
,
Venugopal
Thanikachalam